
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Microbiol., 10 September 2019
Sec. Evolutionary and Genomic Microbiology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.02094
 Luisa K. Hallmaier-Wacker1,2
Luisa K. Hallmaier-Wacker1,2 Simone Lüert1,2Sabine Gronow3Cathrin Spröer3
Simone Lüert1,2Sabine Gronow3Cathrin Spröer3 Jörg Overmann3,4Nicky Buller5Rebecca J. Vaughan-Higgins6
Jörg Overmann3,4Nicky Buller5Rebecca J. Vaughan-Higgins6 Sascha Knauf1*†
Sascha Knauf1*†The genus Treponema contains a number of human and animal pathogenic as well as symbiotic bacteria that are found in vastly different anatomical and environmental habitats. Our understanding of the species range, evolution, and biology of these important bacteria is still limited. To explore the diversity of treponemes, we established, validated, and tested a novel metataxonomic approach. As the informative nature of the hypervariable regions of the 16S rRNA gene differ, we first analyzed each variable region independently. Considering the in silico results obtained, we established and validated the sequencing of the V4-region of the 16S rRNA gene using known mixtures of Treponema species as well as a selected number of clinical samples. The metataxonomic approach was able to identify Treponema to a near-species level. We demonstrate that using a spirochete-specific enrichment, our method is applicable to complex microbial communities and large variety of biological samples. The metataxonomic approach described provides a useful method to unravel the full diversity and range of Treponema in various ecosystems.
Spirochaetes, a phylum of spiral-shaped bacteria, range from pathogenic (e.g., Treponema pallidum) to symbiotic (e.g., Sphaerochaeta coccoides) to free-living (e.g., Exilispira thermophile) species (Paster, 2001). The ability of spirochetes to inhabit vastly anatomical and ecological habitats is remarkable and indicates a high diversity of the bacterial members of this phylum (Paster, 2001). Until recently, spirochetes were predominantly discovered and subsequently characterized using cultivation, microscopical, or serological approaches. These techniques make it difficult and sometimes impossible to characterize not-yet-cultivated species, to identify species in multiple-spirochete infections, or to discover commensal microbes. The advent of cultivation-independent molecular techniques (e.g., nucleic acid amplification technology) has allowed for a broader detection of Treponema in different biological niches. To date, the 16S rRNA phylogenetic marker gene has been particularly instrumental in the detection of Treponema diversity (Pace, 1997). Based on defined similarity thresholds the 16S rRNA sequences can be grouped into phylotypes. For example, using a clonal 16S rRNA gene library and subsequent Sanger sequencing, the termite (Reticulitermes flavipes) gut was found to harbor more than 67 different treponemal phylotypes (Lilburn et al., 1999) and the human oral cavity up to 23 different treponemal clusters (Choi et al., 1994).
More recently, 16S rRNA gene-based metataxonomic studies have moved from clonal libraries to high-throughput sequencing approaches. Single hypervariable regions of the 16S rDNA have been used to examine different microbiomes (Kozich et al., 2013) and have identified treponemes in many ecological niches (Hong et al., 2012; Klitgaard et al., 2014; Rodriguez-R et al., 2015; Clayton et al., 2018; Hicks et al., 2018). For example, the gut microbiome of wild western lowland gorillas (Gorilla gorilla gorilla) (Hicks et al., 2018) and other nonhuman primates (Clayton et al., 2018) harbors multiple operational taxonomic units (OTUs) corresponding to the genus Treponema. Yet, conventional data analysis pipelines used in microbiome studies still do not allow for species-level characterization (Schloss et al., 2009; Caporaso et al., 2010). Taxonomic classification for many bacterial genera is restricted by the limited sequence differences in the 16S rRNA gene (Wang et al., 2007). Rossi-Tamisier et al. (2015) showed that spirochetes, in particular Treponema and Spirochaeta, have an exceptionally large variability in the 16S rRNA gene. For Treponema, only 2.1% of the analyzed 16S rRNA sequences fell within the recommended similarity threshold (95–98.7%) (Rossi-Tamisier et al., 2015).
To explore the range and diversity of Treponema, we established, validated, and tested a newly designed spirochete-specific metataxonomic approach that utilizes the 16S rRNA gene. Based on the known variability of the 16S rRNA gene, we hypothesized that a single hypervariable region of this gene provides a good target for a metagenomics-based assay to examine the diversity of Treponema in various biological sample types.
Based on the nomenclature of Bergey’s Manual of Systematic Bacteriology, we selected all bacterial species that are classified within the phylum Spirochaetes (Paster, 2001). Subsequently, a representative 16S rRNA gene sequence corresponding to each Spirochaetes bacterial species was retrieved from the GenBank database1. Where possible, sequences were chosen with maximal length and no ambiguous bases. Sequences shorter than 1,250 base pairs and/or containing more than two ambiguous bases were not included in the dataset even if no other sequence of the bacterial species was available (Supplementary Table S1).
The Perl-based high-throughput software tool V-Xtractor was used to locate the hypervariable regions (V2–V8) of the 16S rRNA sequences using Hidden Markov Models (Hartmann et al., 2010). Subsequently, the sequences of each variable region were analyzed using the mothur software package (v.1.41.1) (Schloss et al., 2009). In an initial step, identical sequences were removed using the unique.seq command. Then, the SILVA bacterial reference database (Quast et al., 2012) was utilized to align the sequences [align.seqs command using kmer searching (8mers) and Needleman–Wunsch pairwise alignment method]. OTU clustering was performed for distance threshold ranging from 0.01 to 0.10 at increments of 0.01 (cluster.split command with the OptiClust algorithm) (Westcott and Schloss, 2017).
The spirochete mock community comprised an equal mixture of 19 strains of the phylum Spirochaetes. Single bacterial DNA isolates were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ). DNA from rabbit inoculated T. pallidum subsp. pertenue strain Gauthier (referred to as T. pallidum throughout the manuscript) was obtained from David Šmajs, Department of Biology, Faculty of Medicine, Masaryk University, Brno, Czech Republic. The 19 Spirochaetes species which were used in this study, including the cultivation method, DSMZ reference number, 16S rRNA gene copy number, genome size, and NCBI reference, are shown in Supplementary Table S2.
The DNA of the cultured spirochetes obtained from DSMZ was quantified using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific). T. pallidum DNA, due to the rabbit background DNA from in vivo inoculation experiments, was quantified using an established TaqMan PCR (qPCR) targeting the polA gene with slight modifications as described previously (Knauf et al., 2018). Based on the DNA content, genome size and 16S rRNA gene copy number, the 19 spirochetes were mixed together at equimolar (even) ribosomal RNA operon counts per organism. The final spirochete mock community contained 100,000 16S rDNA copies/μl of each species. All dilutions were made using Microbial DNA-Free water (Qiagen GmbH). Suitable precautions were taken during all sample handling and processing to avoid microbial contamination.
In addition to the spirochete mock community, we created three bacterial DNA validation sets to evaluate the intra-metagenomic assays performance. T. pallidum DNA was quantified using TaqMan PCR as described above. For the first validation set, the stock of T. pallidum (50,000 16S rRNA copies) was used to make a 10-fold dilution series. The dilutions of T. pallidum DNA were subsequently mixed with bacterial DNA contained no Spirochaetes [Microbial Mock Community, HM-280, Biodefense and Emerging Infectious Research (BEI) Resources, Manassas, VA, United States] (Supplementary Table S3). The second validation set was a mixture of T. pallidum and T. denticola in different ratios (Supplementary Table S4). The final ratios of the T. pallidum to T. denticola were 1:100, 1:10, 1:1, 10:1, and 100:1. The third validation set was a 10-fold serial dilution series of T. pallidum starting at 50,000 copies of 16S rRNA gene. Dilutions for all validation sets were made using Microbial DNA-Free water (Qiagen GmbH).
Spirochete-selective primers were used to enrich spirochetal DNA (Dewhirst et al., 2010). The primers F24 (5′-GAGTTTGATYMTGGCTCAG-3′) and M98 (5′-GTTACGACTTCACCCYCCT-3′) were used to amplify a ∼1,450 bp fragment of the 16S rRNA gene covering the V1–V9 region. This first PCR step was performed in triplicates using the Phusion Hot Start II High-Fidelity DNA Polymerase (Thermo Fisher Scientific), which has been validated for the use in microbiome studies (Hallmaier-Wacker et al., 2018). PCR reactions consisted of 12.5 μl of 2× PCR master mix, 9.5 μl of Microbial DNA-Free water (Qiagen GmbH), 1.0 μl of each primer (0.5 mM each, Metabion), and 1 μl of template in a total reaction volume of 25 μl. PCR cycling conditions comprised of a pre-denaturation step of 30 s at 98°C, followed by either 20 or 35 cycles of 98°C for 10 s, 57°C for 15 s and 72°C for 120 s, and a final 10 min extension step at 72°C. A 16S rRNA amplification control sample (blank controls; Microbial DNA-Free water) was included. Subsequently, PCR triplicates were pooled before library preparation.
A pre-test to re-amplify the V3, V4, and V6 regions was performed to identify the most suitable variable regions. The V4 region was selected, as the V3 and V6 region primers demonstrated technical issues to evenly amplify the variable regions. A modular, two-step PCR process was used to specifically re-amplify the V4-region of the 16S rRNA gene and prepare the samples for sequencing on the MiSeq platform. In the first step, the V4 region of the 16S rRNA gene was amplified using TruSeq adaptor-tailed universal primers 515F and 806R. The primers 515F-TruSeq (5′-ACACTCT TTCCCTCCACGACGCTCTTCCGCTCTGTGTGCCAGCMGC CGCGGTAA-3′) and 806R-TruSeq (5′-GTGACTGGAGTTCA GACGTGTGCTCTTCCGATCCCGGACTACHVGGGTWTCT AAT-3′) were composed of the universal primer targeting the V4 region (Caporaso et al., 2011) followed by a linker and the TruSeq adaptor (Illumina, Inc.). Amplification was performed in triplicates and each 25.0 μl reaction contained 1.0 μl of PCR product of the enrichment step, 12.5 μl of 2× Phusion Hot Start II High-Fidelity PCR Master Mix (Thermo Fisher Scientific), 9.5 μl of Microbial DNA-Free water (Qiagen GmbH), and 1.0 μl of each V4-targeting 16S primer (0.5 mM each, Metabion). The cycling conditions were as follows: a pre-denaturation step of 30 s at 98°C, followed by 20 cycles of 98°C for 10 s, 55°C for 15 s and 72°C for 60 s, and a final 10 min extension step at 72°C. To monitor contamination, the blank control of the enrichment step was included as a 16S rDNA amplification control.
In the second-step PCR reaction, sample-specific Illumina indices and flow cell adapters were added in an indexing reaction. Illumina i7 and i5 indices were added to each amplicon using the indexing primer P5 (5′-AATGATACGGCGACCACCGAG ATCTACAC-[i5-INDEX] -ACACTCTTTCCCTACACGACGCTC-3′) and indexing primer P7 (5′-CAAGCAGAAGACGGCATACGAGAT-[i7-INDEX]-GTGACTGGAGTTCAGAC GTGT-3′). Amplification was performed in a 50.0 μl reaction containing 2.0 μl of PCR product from the first-step, 25.0 μl of 2× KAPA HiFi HotStart ReadyMix (KAPA Biosystems), 21.0 μl of Microbial DNA-Free water (Qiagen GmbH), and 1.0 μl of each Truseq index primer (0.5 mM each, Metabion). The cycling conditions were as follows: a pre-denaturation step of 3 min at 98°C, followed by eight cycles of 98°C for 20 s, 62°C for 30 s and 72°C for 30 s, and a final 5 min extension step at 72°C. To monitor overall contamination, the blank control of the first-step PCR reaction was included as a 16S rRNA gene amplification control.
For comparison the initial enrichment PCR was not performed on a sample of the spirochete mock community and a sample of validation set 1 (5,000 16S rDNA copies of T. pallidum). For these two samples the first-step V4-targeting PCR reaction was performed for additional 15 cycles (total of 35 cycles). A 16S rRNA gene amplification control was included for this altered procedure. All other conditions were kept identical.
The applicability of the metataxonomic approach was tested on extracted DNA from genital swabs of Gilbert’s potoroo (Potorous gilbertii), a small marsupial found in Western Australia (Vaughan et al., 2009). For more information on sample processing see the Supplementary Materials.
After the indexing reaction, all amplicons were purified using 0.7× AMPure XP beads (Beckman Coulter), and quantified using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific) (Supplementary Table S5). The amplicon integrity was verified for a representative number of four samples using the BioAnalyzer 2000 (Agilent). Equimolar amounts (2 nM) of sample amplicons were pooled. For samples with <2 nM concentration, the maximum volume (5 μl) was pooled prior to sequencing. The Transcriptome and Genome Analysis Laboratory at the University of Goettingen performed the Illumina MiSeq 2 × 250 bp paired-end sequencing (Illumina V2 chemistry) run.
Raw reads were processed using the mothur software package (version 1.41.1) (Schloss et al., 2009). Initial pre-processing and quality control were performed in accordance with the MiSeq SOP (Schloss et al., 2009). Briefly, paired-end reads were assembled using the make.contigs command. Subsequently, the screen.seqs command was used to trim sequences and filter out any sequences with ambiguous base calls. Identical trimmed sequences (unique.seq command) were aligned (align.seqs command) to the SILVA bacterial reference database (Quast et al., 2012). Poorly aligned sequences, chimeras [chimera.uchime command; UCHIME algorithm (Edgar et al., 2011)], and other erroneous non-bacterial sequences (remove.lineage command) were removed. The remaining sequences were classified using a Bayesian classifier implemented in mothur and OTUs were assigned based on a distance threshold of 0.03.
For the species-level classification, Treponema-classified sequences were extracted from the dataset using the get.lineage command. Using the Treponema sequence data in the in silico fasta file (Supplementary Table S1), a database was created using the create.database command. Using this database, the taxonomy of the filtered sequences was assigned using the classify.otu command.
All generated read files have been deposited in the NCBI Sequence Read Archive under the accession number PRJNA541286.
We analyzed each hypervariable region (V2–V8) of the 16S rRNA gene for its potential to distinguish nine bacterial genera that make up the phylum Spirochaetes. In total, we analyzed the information content of the variable regions of 114 representative sequences in silico (Supplementary Table S1). Hypervariable regions V2–V8 were able to distinguish the nine bacterial genera at a similarity threshold of 97% (Table 1). The V2 region identified the largest total number of OTUs (n = 69) compared to all other tested regions (Table 1). Overall, the least number of OTUs were identified in the genera Leptospira, Borrelia, and Brachyspira. For the genus Treponema on the other hand, all variable regions with the exception of V7 were able to detect a high number of distinct OTUs (Table 1). Regions V2 and V3 were both able to detect 25 OTUs at a threshold of 97% in the in silico dataset containing 28 unique representative sequences. To examine the robustness of the in silico results for Treponema, we examined the identifiable OTUs at threshold cutoffs ranging from 90 to 99% (Supplementary Figure S1). For V2, V3, and V4 regions at 90% similarity threshold, >15 OTUs are distinguishable in the Treponema genus (Supplementary Figure S1).
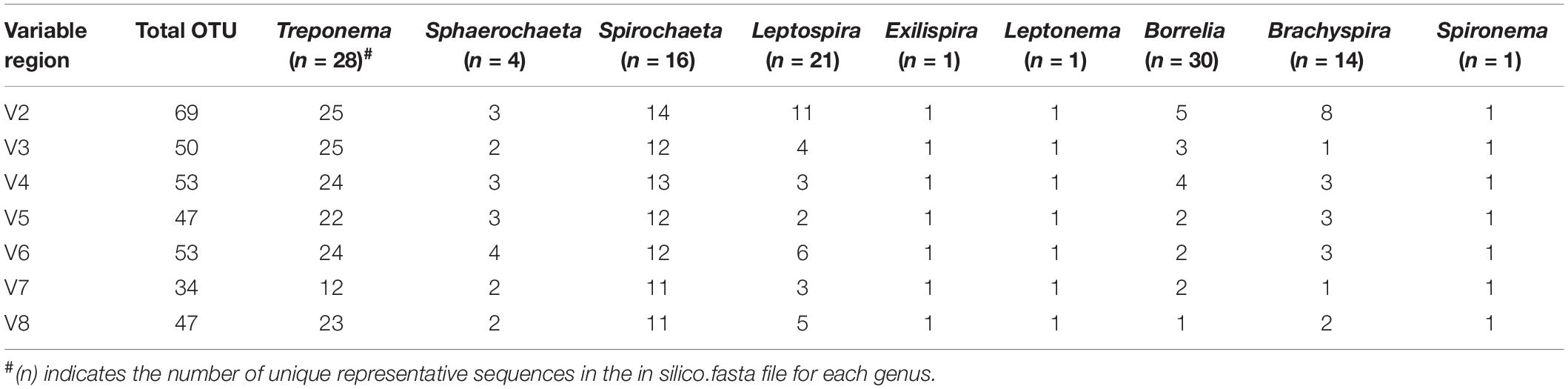
Table 1. Identifiable in silico OTUs for the different genus within the phylum of Spirochaetes at a 97% threshold.
We tested three different amplification conditions targeting the V4-region of the 16S rRNA gene (Figures 1, 2). All 16S rRNA gene amplification conditions were able to identify all seven genera that were included in the mock community samples (Figure 2). The amplification condition without spirochete-specific 16S rRNA gene enrichment differed less from the actual mixing proportion than the samples which were enriched (Figure 2). In all conditions, Treponema, Leptonema, and Sphaerochaeta were preferentially detected. For the spirochete enriched samples (20 cycles and 35 cycles), Borrelia, Leptospira, and Brachyspira made up <1% of the detected sequence reads (Figure 2). In addition to the genus-level identification, we classified the Treponema sequences on a species-level using a Treponema-specific database of the V4-region. At 97% similarity threshold, the database contains 24 OTUs of which 21 OTUs correspond to single species and 3 OTUs correspond to species-clusters (denticola-, medium-, and the pallidum-cluster) (Supplementary Table S6). All 16S rDNA amplification conditions were able to identify all seven species of Treponema in the mock community (Figure 2). Independent of amplification conditions, sequences corresponding to T. pallidum-cluster were amplified less efficiently.
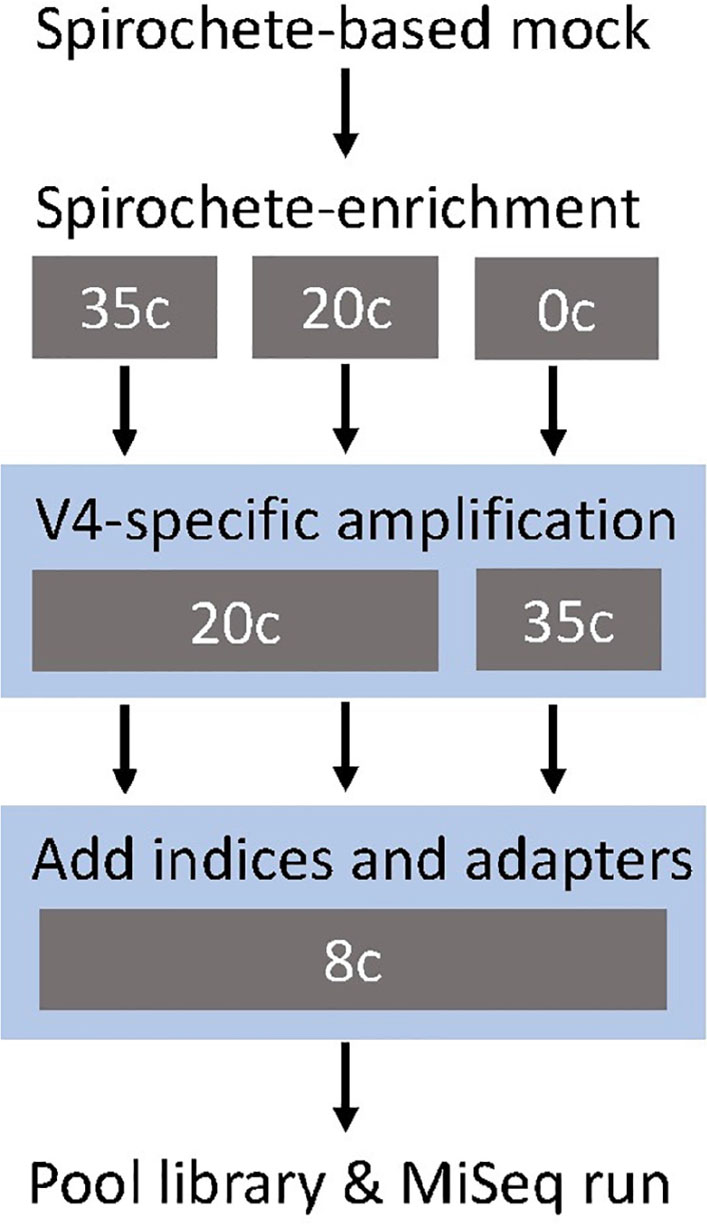
Figure 1. Study design of the metataxonomic assay targeting the V4-region of the 16S rRNA gene. The gray boxes show the tested cycle (c) conditions for each step. The blue shading indicates the modular two-step library preparation.
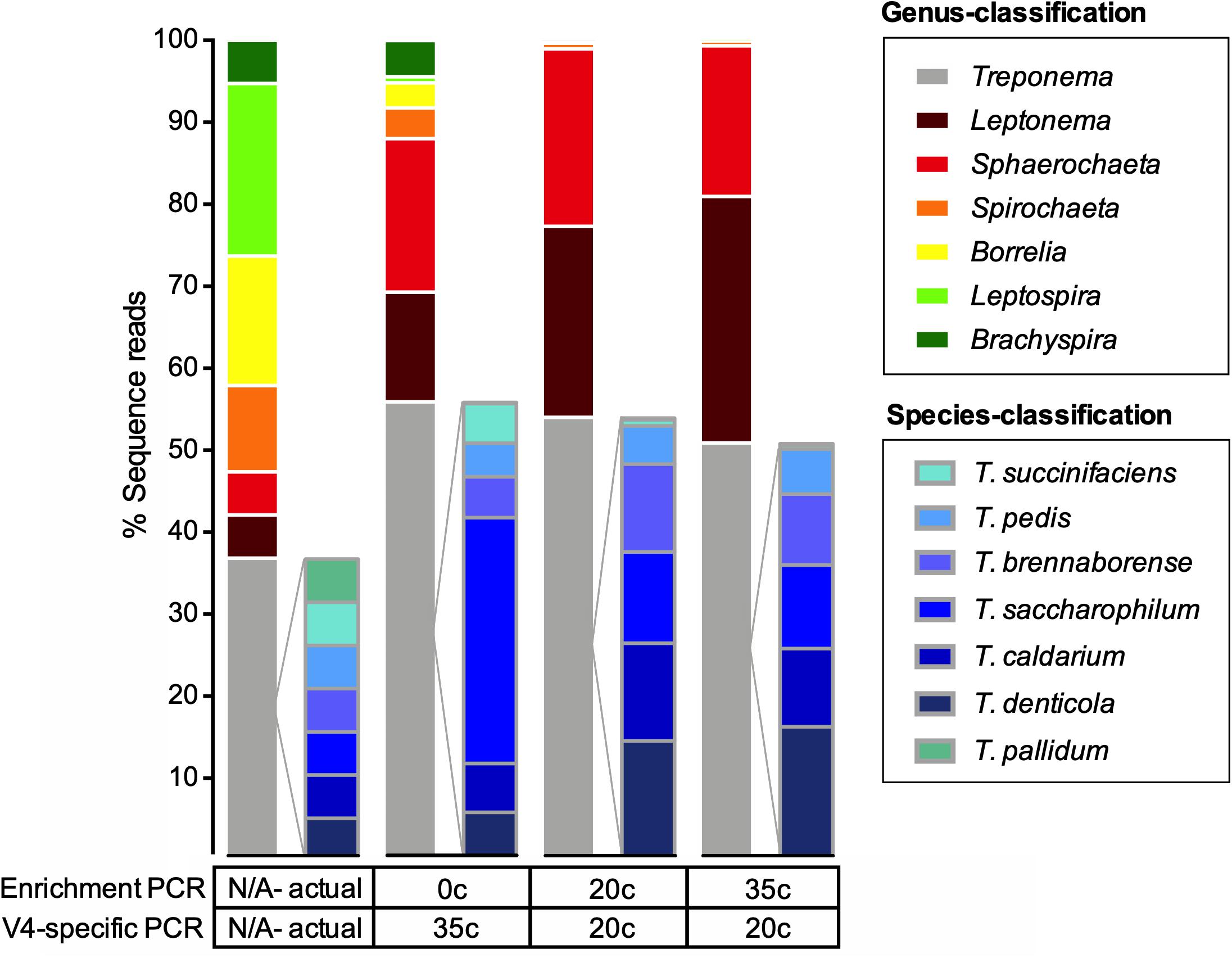
Figure 2. Actual and observed proportions of a spirochete mock community to a genus and species-level. Relative abundance of OTUs in percentage of reads for a spirochete microbial mock community amplified using multiple methods. The spirochete enrichment and V4-region-specific PCR cycle numbers are shown below the taxa plots. The expected strain proportion (actual) of the spirochete mock community represents the theoretical composition and was corrected for differential copy number of the 16S rRNA gene. Species-level classification only shown for the genus Treponema.
Amplification control samples (blank samples) were included for each amplification method to test for contamination during the amplification process. The blank sample from the 20-cycle enrichment had the lowest amplicon quantity before sequencing as well as the lowest overall corresponding number of sequences reads (Supplementary Figure S2 and Supplementary Table S5). Compared to the blank control enriched for 20 cycles, the control of the enrichment for 35 cycles had a 100× fold increase in sequence reads (Supplementary Figure S2). Despite an overall lower cycle count, the sequence reads corresponding to the nonenriched sample were as high as for the control enriched for 35 cycles. Unlike the enriched samples (20 cycle and 35 cycle), which detected minimal Treponema in the blank sample (<10 sequence reads), the non-enriched control sample detected 6,364 reads of Treponema (Supplementary Figure S2).
The validation sample sets were used to assess the efficiency of the spirochete enrichment amplification, the effect of competition between two species, and the detection limit of the amplicon sequencing approach. The first validation set was a mixture of different concentrations of T. pallidum with a microbial mock community (HM-280). Figure 3 shows that both 20 cycle- and 35 cycle-enrichment steps significantly improve the detection of T. pallidum compared to the unenriched sample at 5,000 16S rRNA gene copies of T. pallidum. As the input DNA of T. pallidum decreases, the dilution effect between T. pallidum to microbial mock community HM-280 can be visualized clearly (Figure 3). Four to six 16S rRNA gene copies of T. pallidum were detected for 35 cycle- (44,141 sequence reads) and 20 cycle-enrichment (2,224 sequence reads).
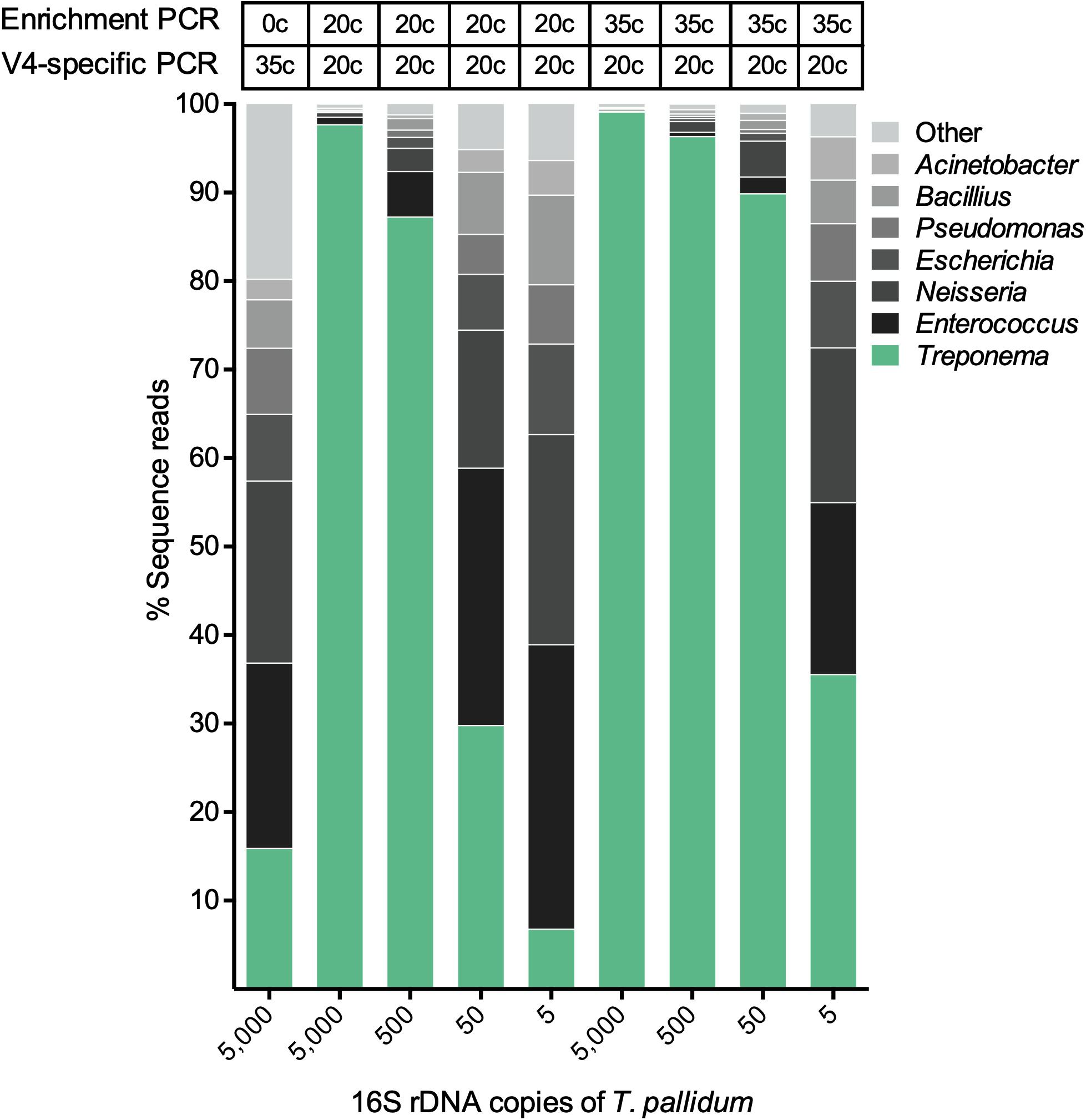
Figure 3. The effect of spirochete enrichment on the detection limit of T. pallidum. Relative abundance of OTUs in percentage of reads for a T. pallidum-spiked microbial mock community (HM-280). The theoretically calculated amount of spike in T. pallidum ranging from 5 to 5,000 16S rDNA copies is shown along the x-axis. The biologically effective range of T. pallidum may differ from theoretically calculated amount (e.g., 5 copies = 4–6 copies). Cycle number for the spirochete enrichment and V4-region-specific PCR is shown above the taxa plots.
The second validation set was a mixture of T. pallidum and T. denticola in different ratios (see the section “Materials and Methods” for details). For all ratios, T. denticola outcompeted T. pallidum in detected sequence reads (Table 2). However, both species of Treponema were detected at all tested ratios (Table 2). The third validation set was a 10-fold serial dilution series of T. pallidum. Sequence reads were >9,000 reads down to 500 16S rRNA gene copies of T. pallidum (Figure 4). At 50 16S rRNA gene copies, T. pallidum was detectable but overall sequence reads were markedly decreased (13,453 sequence reads). For the final two dilutions, total read numbers were <1,500 and T. pallidum sequence detection was analogous to the blank control (<10 sequence reads) (Figure 4).

Table 2. Relative abundance of OTUs in percentage of reads for different proportions of T. pallidum and T. denticola 16S rRNA gene.
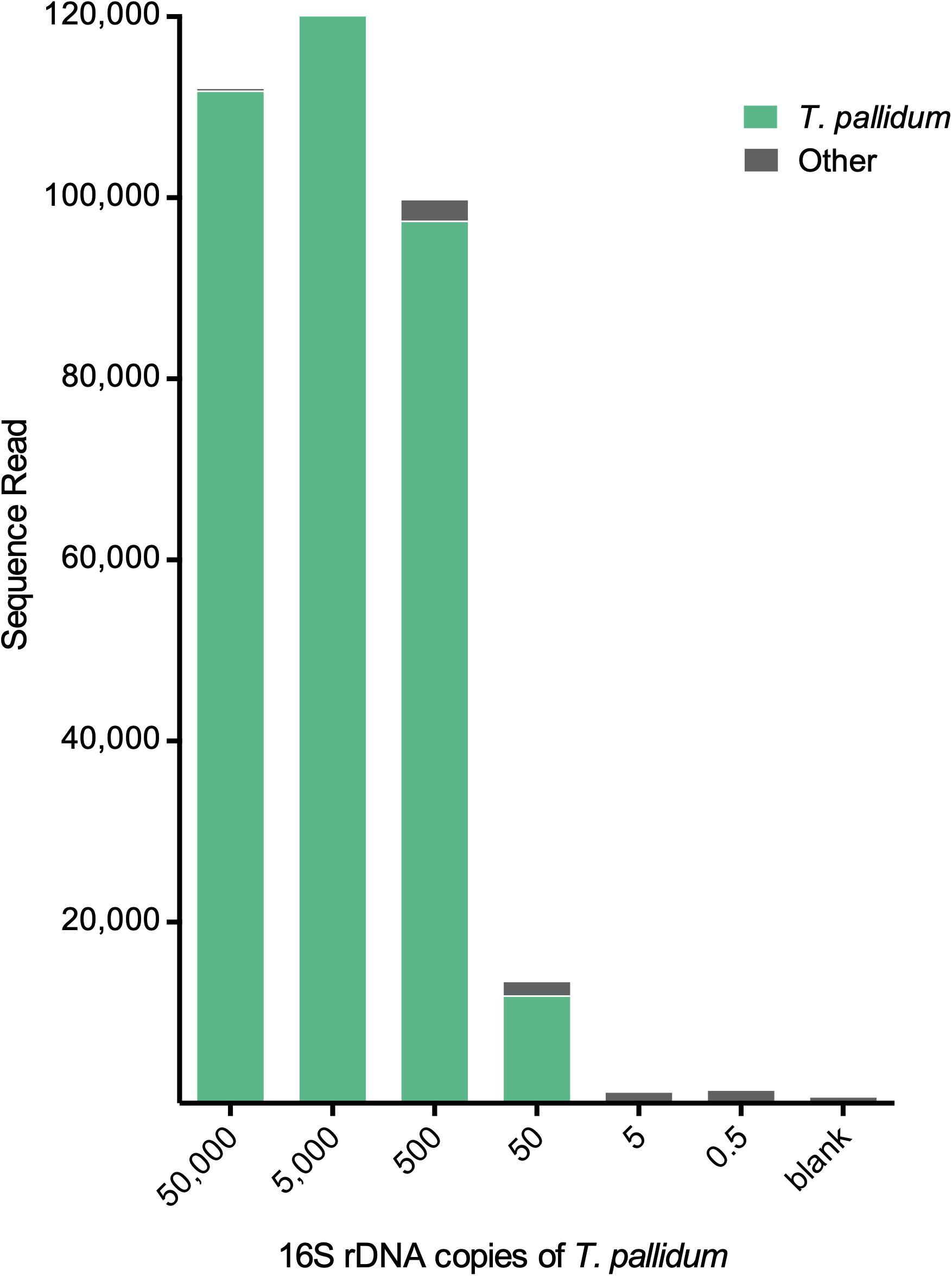
Figure 4. Detection limit of the metagenomic approach for Treponema. Total sequence reads resulting from different input amounts of T. pallidum. Displayed amounts of T. pallidum (50,000–0.5 16S rDNA copies) represent theoretically calculated amounts. Biologically effective range may differ from theoretically calculated amount (e.g., 0.5 copies = 0–2 copies). Blank control represents the 16S rDNA amplification control using microbial DNA-free water as input. For these samples 20 cycles of enrichment PCR was followed by 20 cycles of V4-region-specific PCR (for more detail see the section “Materials and Methods”).
We examined samples from four Gilbert’s potoroo which had been found to harbor a Treponema infection (Vaughan et al., 2009). Using the amplicon sequencing technique, we identified a Treponema species in all four clinical samples (Supplementary Figure S3). Sequence reads corresponding to the Treponema made up >75% of the total read count for Gilbert’s potoroo samples No. 2–4 (Supplementary Figure S3). The Treponema sequences clustered into a single OTU, which cannot be identified using the Treponema-specific V4-region database at a 97% threshold identity.
We used the in silico analysis to predict the informative nature of each 16S rDNA hypervariable region for different spirochetes. Our findings expand on the results of Rossi-Tamisier et al. (2015), indicating that three genera of spirochetes have low interspecies sequence similarity with the 16S rRNA gene, which makes it a suitable gene target for identification (Table 1). The V3 and V4 region have been previously described for their discriminatory power (Chakravorty et al., 2007; Yang et al., 2016; Graspeuntner et al., 2018). In a study of 110 bacterial species, V2 and V3 were the most suitable candidates (Chakravorty et al., 2007). Considering phylogenetic resolution, the variable regions 4, 5, and 6 have been previously identified as prime targets (Yang et al., 2016; Graspeuntner et al., 2018). Overall, the in silico analysis provided good initial data to efficiently design the in vitro experiments. It is, however, important to note that technical caveats of NGS sequencing must be considered prior to the selection of the most appropriate region (Kozich et al., 2013). For example, for the Illumina MiSeq Platform, paired-end sequencing can currently cover 300 base pairs. Considering the overall error rate of this platform [∼0.1–0.01% per base, depending on the data-filtering scheme (Meacham et al., 2011; Loman et al., 2012)], the ideal read length for a metataxonomic approach allows for full overlap of the two pair-end reads (Kozich et al., 2013). Based on our in silico results and pre-test using different primers, we selected the V4 region of the 16S rRNA gene for further in vitro testing.
A spirochete mock community of known species composition allowed for the systematic comparison between the different amplification methods (Figure 1; Brooks et al., 2015). Independent of the amplification method, our metagenomic approach was able to detect all seven genera of spirochetes in the mock community, as well as all species of Treponema (Figure 2). However, not all spirochetes were detected equally well with all amplification procedures (Figure 2). The spirochete-specific enrichment step, which was included for a better detection of spirochetes, led to the distortion of the actual proportions and favored Treponema, Sphaerochaeta, and Leptonema (Figure 2). The distortion of the bacterial profiles due to preferential amplification of multi-template PCR is a known phenomenon and a major limitation of 16S rRNA gene amplification that results from sub-optimal primer binding (Polz and Cavanaugh, 1998; Brooks et al., 2015; Hallmaier-Wacker et al., 2018). It has been previously shown that this distortion effect is not significantly influenced with decreasing the number of amplification cycles (Acinas et al., 2005; Sipos et al., 2007; Wu et al., 2010; Brooks et al., 2015). Similarly, our results did not remarkably change with an increased number of enrichment cycles (20 cycles vs. 35 cycles; Figure 2). Nevertheless, the use of unnecessary cycles should be avoided as it can lead to formation of unwanted side products such as chimeras (Ahn et al., 2012), as well as a higher risk of overamplifying reads that originate from contamination (blank controls; Supplementary Figure S2) (Salter et al., 2014).
To examine the benefits of the spirochete enrichment PCR (20 cycles and 35 cycles) on the detection limit of analysis, we tested the metataxonomic approach on mock communities that simulate bacterial proportions found in clinical samples (Supplementary Table S3). For these samples, the enrichment step critically improved the detection of Treponema at low copy numbers, thus indicating that enrichment is a useful tool for samples with low spirochete numbers (<5,000 16S rDNA copies) (Figure 3). We showed that five 16S rRNA gene copies of T. pallidum were detectable in a sample with 20 other bacterial species (even bacterial mock HM-280). Using serial dilutions, we were able to detect as little as 50 16S rRNA gene copies of T. pallidum using 20 cycle enrichment amplification (Figure 4). These data indicate a sensitivity of our assay that is comparable to standard TaqMan qPCR and which outcompetes the conventional 16S rRNA clonal approach (Leslie et al., 2007). Obtaining a high detection limit using a clonal approach is both time consuming and resource intensive (Leigh et al., 2010). On the other hand, Sanger sequence analysis of clone libraries provide greater phylogenetic resolution due to an increased read length, covering the full 16S rRNA gene (Leigh et al., 2010). The complex microbial communities present in many clinical samples is a frequent challenge in diagnostics and in these sample the occurrence of multi-Treponema species is not uncommon. For example, in oral syphilis patients, T. pallidum can be found in combination with T. denticola (Scott and Flint, 2005). We therefore tested the effect of competing species by simulating a co-infection of T. pallidum and T. denticola (Supplementary Table S4). Overall, the metataxonomic approach underestimated the ratio of T. pallidum in the samples (Table 2). Amplicon sequencing was, however, sufficient to identify both species of Treponema at all tested ratios (Table 2). We note here that the metataxonomic approach does not accurately represent absolute abundance of different species (Widder et al., 2016). The used primers may have a significant effect in distorting tested ratios and thus alternative primer should be designed and evaluated for specific research questions. Additionally, quantitative techniques such as qPCR, flow cytometry, or fluorescence in situ hybridization (FISH) may be superior for evaluating known competing species (e.g., T. pallidum and T. denticola) (Props et al., 2017). Moreover, the metataxonomic approach should not be used for defining novel bacterial species even if species-level clustering is possible using the 16S rRNA sequence information (e.g., Treponema) (Tindall et al., 2010). Instead, 16S rRNA gene amplicon sequencing provides a qualitative view on the diversity of treponemes within a given DNA sample. For example, we used the metataxonomic approach to examine clinical samples of the Gilbert’s potoroo that have been previously found to harbor a Treponema infection (Vaughan et al., 2009). As the potoroo clinical samples were associated with a polymicrobial environment and infection was believed to be chronic (Vaughan et al., 2009), we performed a 35 cycle-enrichment in order to detect low concentrations of spirochetes. We identified a single Treponema species in all tested samples of the four Gilbert’s potoroos, which currently remains unclassified at a species level. The high percentage of Treponema in the detected samples (>75%; Supplementary Figure S3) indicates that the amplicon method is applicable for clinical samples and guides subsequent approaches that aim to fully characterize the discovered Treponema species. The results from the metataxonomic approach can be used to select most promising samples for whole-genome analysis (WGS), as well as provide a preliminary understanding of the possible phylogeny, which may assist in reference-based assembly (Wyres et al., 2014). Importantly, further WGS sequences of known and unknown Treponema are crucial to study the evolution and epidemiology of this ancient group of bacteria and to enhance future shotgun metagenomic studies. Currently, there is only a limited number of whole-genome sequences of Treponema, in particular the non-pathogenic species, due to the difficulty to culture many of the species [e.g., from the termite gut (Paster et al., 1996)].
We showed that the V4 region of the 16S rRNA gene is a valuable target to explore the diversity of Treponema in various biological sample types. To monitor the quality of each sequencing run, it is essential to including relevant controls with all clinical samples. When applied appropriately, the presented modular metataxonomic approach is broadly applicable as it requires only small amounts of bacterial DNA for the detection of a broad range of Treponema species.
The datasets generated for this study can be found in the NCBI Sequence Read Archive accession number PRJNA541286.
LH-W and SK conceived and designed the study. LH-W, SL, SG, CS, and SK performed the experiments in the laboratory. LH-W, SL, and SK analyzed the data. NB and RV-H contributed DNA from the Gilbert’s potoroos. SG, CS, and JO contributed DNA samples of spirochetes for the mock sample. All authors contributed to the writing of the manuscript, read, reviewed, and approved the final manuscript.
SK received funding to conduct parts of this study by the German Research Foundation (DFG KN1097/3-2 and KN1097/7-1). The funders had no role in any part of this study.
SG, CS, and JO were employed by the Leibniz Institute DSMZ – German Collection of Microorganisms and Cell Cultures.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Biodefense and Emerging Infectious Research (BEI) Resources, NIAID, NIH for providing the cells from Microbial Mock Community (Even, HM-280) as part of the Human Microbiome Project. We thank David Šmajs of the Department of Biology, Faculty of Medicine at the Masaryk University for providing DNA from the T. pallidum subsp. pertenue strain Gauthier. Additionally, we thank Christian Roos (German Primate Center) and Fabian Ludewig (Transcriptome and Genome Analysis Laboratory at the University of Göttingen) for their assistance in optimizing the sequencing run. Finally, we thank Simone Severitt and Carola Berg (both DSMZ) for excellent technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02094/full#supplementary-material
Acinas, S. G., Sarma-Rupavtarm, R., Klepac-Ceraj, V., and Polz, M. F. (2005). PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 71, 8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005
Ahn, J.-H., Kim, B. Y., Song, J., and Weon, H. Y. (2012). Effects of PCR cycle number and DNA polymerase type on the 16S rRNA gene pyrosequencing analysis of bacterial communities. J. Microbiol. 50, 1071–1074. doi: 10.1007/s12275-012-2642-z
Brooks, J. P., Edwards, D. J., Harwich, M. D., Rivera, M. C., Fettweis, J. M., Serrano, M. G., et al. (2015). The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 15:66. doi: 10.1186/s12866-015-0351-6
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chakravorty, S., Helb, D., Burday, M., Connell, N., and Alland, D. (2007). A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 69, 330–339. doi: 10.1016/j.mimet.2007.02.005
Choi, B. K., Paster, B. J., Dewhirst, F. E., Göbel, U. B. (1994). Diversity of cultivable and uncultivable oral spirochetes from a patient with severe destructive periodontitis. Infect. Immun. 62, 1889–1895.
Clayton, J. B., Gomez, A., Amato, K., Knights, D., Travis, D. A., Blekhman, R., Knight, R., et al. (2018). The gut microbiome of nonhuman primates: lessons in ecology and evolution. Am. J. Primatol. 30:e22867. doi: 10.1002/ajp.22867
Dewhirst, F. E., Chen, T., Izard, J., Paster, B. J., Tanner, A. C., Yu, W.-H., et al. (2010). The human oral microbiome. J. Bacteriol. 192, 5002–5017. doi: 10.1128/JB.00542-10
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Graspeuntner, S., Loeper, N., Künzel, S., Baines, J. F., and Rupp, J. (2018). Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci. Rep. 8:9678. doi: 10.1038/s41598-018-27757-8
Hallmaier-Wacker, L. K., Lueert, S., Roos, C., and Knauf, S. (2018). The impact of storage buffer, DNA extraction method, and polymerase on microbial analysis. Sci. Rep. 8:6292. doi: 10.1038/s41598-018-24573-y
Hartmann, M., Howes, C. G., Abarenkov, K., Mohn, W. W., and Nilsson, R. H. (2010). V-Xtractor: an open-source, high-throughput software tool to identify and extract hypervariable regions of small subunit (16S/18S) ribosomal RNA gene sequences. J. Microbiol. Methods 83, 250–253. doi: 10.1016/j.mimet.2010.08.008
Hicks, A. L., Lee, K. J., Couto-Rodriguez, M., Patel, J., Sinha, R., Guo, C., et al. (2018). Gut microbiomes of wild great apes fluctuate seasonally in response to diet. Nat. Commun. 9:1786. doi: 10.1038/s41467-018-04204-w
Hong, P. Y., Li, X., Yang, X., Shinkai, T., Zhang, Y., Wang, X., et al. (2012). Monitoring airborne biotic contaminants in the indoor environment of pig and poultry confinement buildings. Environ. Microbiol. 14, 1420–1431. doi: 10.1111/j.1462-2920.2012.02726.x
Klitgaard, K., Nielsen, M. W., Ingerslev, H.-C., Boye, M., and Jensen, T. K. (2014). Discovery of bovine digital dermatitis-associated Treponema spp. in the dairy herd environment by a targeted deep-sequencing approach. Appl. Environ. Microbiol. 80, 4427–4432. doi: 10.1128/AEM.00873-14
Knauf, S., Lüert, S., Šmajs, D., Strouhal, M., Chuma, I. S., Frischmann, S., et al. (2018). Gene target selection for loop-mediated isothermal amplification for rapid discrimination of Treponema pallidum subspecies. PLoS Negl. Trop. Dis. 12:e0006396. doi: 10.1371/journal.pntd.0006396
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Leigh, M. B., Taylor, L., Neufeld, J. D. (2010). “Clone libraries of ribosomal RNA gene sequences for characterization of bacterial and fungal communities,” in Handbook of hydrocarbon and Lipid Microbiology, ed. K. N. Timmis (Berlin: Springer) 3969–3993. doi: 10.1007/978-3-540-77587-4_310
Leslie, D. E., Azzato, F., Karapanagiotidis, T., Leydon, J., and Fyfe, J. (2007). Development of a real-time PCR assay to detect Treponema pallidum in clinical specimens and assessment of the assay’s performance by comparison with serological testing. J. Clin. Microbiol. 45, 93–96. doi: 10.1128/JCM.01578-06
Lilburn, T. G., Schmidt, T. M., Breznak, J. A. (1999). Phylogenetic diversity of termite gut spirochaetes. Environ. Microbiol. 1, 331–345. doi: 10.1046/j.1462-2920.1999.00043.x
Loman, N. J., Misra, R. V., Dallman, T. J., Constantinidou, C., Gharbia, S. E., Wain, J., et al. (2012). Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 30, 434–439. doi: 10.1038/nbt.2198
Meacham, F., Boffelli, D., Dhahbi, J., Martin, D. I., Singer, M., and Pachter, L. (2011). Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics 12:451. doi: 10.1186/1471-2105-12-451
Pace, N. R. (1997). A molecular view of microbial diversity and the biosphere. Science 276, 734–740. doi: 10.1126/science.276.5313.734
Paster, B. J. (2001). “Phylum XV. Spirochaetes,” in Bergey’s Manual of Systematic Bacteriology, 2nd Edn, Vol. 4. eds N. R. Krieg, J. T. Staley, D. R. Brown, B. P. Hedlund, B. J. Paster, N. L. Ward, et al. (New York, NY: Springer) 471–566.
Paster, B. J., Dewhirst, F. E., Cooke, S. M., Fussing, V., Poulsen, L. K., and Breznak, J. A. (1996). Phylogeny of not-yet-cultured spirochetes from termite guts. Appl. Environ. Microbiol. 62, 347–352.
Polz, M. F., and Cavanaugh, C. M. (1998). Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 64, 3724–3730.
Props, R., Kerckhof, F.-M., Rubbens, P., De Vrieze, J., Sanabria, E. H., Waegeman, W., et al. (2017). Absolute quantification of microbial taxon abundances. ISME J. 11, 584–587. doi: 10.1038/ismej.2016.117
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rodriguez-R, L. M., Overholt, W. A., Hagan, C., Huettel, M., Kostka, J. E., and Konstantinidis, K. T. (2015). Microbial community successional patterns in beach sands impacted by the Deepwater Horizon oil spill. ISME J. 9, 1928–1940. doi: 10.1038/ismej.2015.5
Rossi-Tamisier, M., Benamar, S., Raoult, D., and Fournier, P. E. (2015). Cautionary tale of using 16S rRNA gene sequence similarity values in identification of human-associated bacterial species. Int. J. Syst. Evol. Microbiol. 65, 1929–1934. doi: 10.1099/ijs.0.000161
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Scott, C., and Flint, S. R. (2005). Oral syphilis—re-emergence of an old disease with oral manifestations. Int. J. Oral Maxillofac. Surg. 34, 58–63. doi: 10.1016/j.ijom.2004.01.029
Sipos, R., Székely, A. J., Palatinszky, M., Révész, S., Márialigeti, K., and Nikolausz, M. (2007). Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol. Ecol. 60, 341–350. doi: 10.1111/j.1574-6941.2007.00283.x
Tindall, B. J., Rosselló-Móra, R., Busse, H. J., Ludwig, W., and Kämpfer, P. (2010). Notes on the characterization of prokaryote strains for taxonomic purposes. Int. J. Syst. Evol. Microbiol. 60, 249–266. doi: 10.1099/ijs.0.016949-0
Vaughan, R. J., Warren, K. S., Mills, J. S., Palmer, C., Fenwick, S., Monaghan, C. L., et al. (2009). Hematological and serum biochemical reference values and cohort analysis in the Gilbert’s potoroo (Potorous gilbertii). J. Zoo Wildl. Med. 40, 276–288. doi: 10.1638/2008-0058.1
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Westcott, S. L., and Schloss, P. D. (2017). OptiClust, an improved method for assigning amplicon-based sequence data to operational taxonomic units. mSphere 2:e00073-17. doi: 10.1128/mSphereDirect.00073-17
Widder, S., Allen, R. J., Pfeiffer, T., Curtis, T. P., Wiuf, C., Sloan, W. T., et al. (2016). Challenges in microbial ecology: building predictive understanding of community function and dynamics. ISME J. 10, 2557–2568. doi: 10.1038/ismej.2016.45
Wu, J. Y., Jiang, X. T., Jiang, Y. X., Lu, S. Y., Zou, F., and Zhou, H. W. (2010). Effects of polymerase, template dilution and cycle number on PCR based 16 S rRNA diversity analysis using the deep sequencing method. BMC Microbiol. 10:255. doi: 10.1186/1471-2180-10-255
Wyres, K. L., Conway, T. C., Garg, S., Queiroz, C., Reumann, M., Holt, K., et al. (2014). WGS analysis and interpretation in clinical and public health microbiology laboratories: what are the requirements and how do existing tools compare? Pathogens 3, 437–458. doi: 10.3390/pathogens3020437
Keywords: metagenomics, metataxonomics, one health, spirochete, 16S rRNA, Treponema, marsupial, Potorous
Citation: Hallmaier-Wacker LK, Lüert S, Gronow S, Spröer C, Overmann J, Buller N, Vaughan-Higgins RJ and Knauf S (2019) A Metataxonomic Tool to Investigate the Diversity of Treponema. Front. Microbiol. 10:2094. doi: 10.3389/fmicb.2019.02094
Received: 13 June 2019; Accepted: 26 August 2019;
Published: 10 September 2019.
Edited by:
Yasir Muhammad, King Abdulaziz University, Saudi ArabiaReviewed by:
Naruo Nikoh, The Open University of Japan, JapanCopyright © 2019 Hallmaier-Wacker, Lüert, Gronow, Spröer, Overmann, Buller, Vaughan-Higgins and Knauf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sascha Knauf, c2tuYXVmQGRwei5ldQ==
†Present address: Sascha Knauf, Division of Microbiology and Animal Hygiene, University of Göttingen, Göttingen, Germany
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.