 Di Sun1,2,3†
Di Sun1,2,3† Xingjian Wen1,2,3†
Xingjian Wen1,2,3† Mingshu Wang1,2,3†
Mingshu Wang1,2,3† Sai Mao1,2,3
Sai Mao1,2,3 Anchun Cheng1,2,3*
Anchun Cheng1,2,3* Xiaoyao Yang1,2,3
Xiaoyao Yang1,2,3 Renyong Jia1,2,3
Renyong Jia1,2,3 Shun Chen1,2,3Qiao Yang1,2,3Ying Wu1,2,3
Shun Chen1,2,3Qiao Yang1,2,3Ying Wu1,2,3 Dekang Zhu2,3
Dekang Zhu2,3 Mafeng Liu1,2,3Xinxin Zhao1,2,3
Mafeng Liu1,2,3Xinxin Zhao1,2,3 Shaqiu Zhang1,2,3Yin Wang1,2,3Zhiwen Xu1,2,3
Shaqiu Zhang1,2,3Yin Wang1,2,3Zhiwen Xu1,2,3 Zhengli Chen1,2,3Ling Zhu1,2,3
Zhengli Chen1,2,3Ling Zhu1,2,3 Qihui Luo1,2,3Yunya Liu1,2,3Yanling Yu1,2,3Ling Zhang1,2,3
Qihui Luo1,2,3Yunya Liu1,2,3Yanling Yu1,2,3Ling Zhang1,2,3 Xiaoyue Chen2,3
Xiaoyue Chen2,3- 1Institute of Preventive Veterinary Medicine, Sichuan Agricultural University, Chengdu, China
- 2Key Laboratory of Animal Disease and Human Health of Sichuan Province, Sichuan Agricultural University, Chengdu, China
- 3Avian Disease Research Center, College of Veterinary Medicine, Sichuan Agricultural University, Chengdu, China
Cell death is a fundamental process in maintaining cellular homeostasis, which can be either accidental or programed. Programed cell death depends on the specific signaling pathways, resulting in either lytic or non-lytic morphology. It exists in two primary forms: apoptosis and autophagic cell death. Apoptosis is a non-lytic and selective cell death program, which is executed by caspases in response to non-self or external stimuli. In contrast, autophagy is crucial for maintaining cellular homeostasis via the degradation and recycling of cellular components. These two mechanisms also function in the defense against pathogen attack. However, picornaviruses have evolved to utilize diverse strategies and target critical components to regulate the apoptotic and autophagic processes for optimal replication and the release from the host cell. Although an increasing number of investigations have shown that the apoptosis and autophagy are altered in picornavirus infection, the mechanism by which viruses take advantage of these two processes remains unknown. In this review, we discuss the mechanisms of picornavirus executes cellular apoptosis and autophagy at the molecular level and the relationship between these interactions and viral pathogenesis.
Introduction
Picornaviruses are divided into 47 genera and are known for causing a wide variety of diseases, including numerous human and animal pathogens (Zell et al., 2017). The genus Enterovirus (EV) of the Picornaviridae family contains poliovirus (PV), coxsackievirus (CV), rhinovirus (RV), and numerous enteroviruses (bovine enterovirus, porcine enterovirus, and simian enterovirus), and most of picornaviruses are well-studied viruses. These viruses cause a broad spectrum of diseases, ranging from hand-foot-and-mouth infections and diarrhea to paralysis and encephalitis. Except for enteroviruses, other picornaviruses are causative agents of diseases and have a substantial impact on health care and economy, for example, encephalomyocarditis virus (EMCV), foot-and-mouth disease virus (FMDV) and hepatitis A virus (HAV).
Picornaviruses are small non-enveloped RNA viruses containing a positive-stranded 7–8 kb RNA genome. The genome consists of an integral ORF, a 5′ UTR, and a 3′ UTR with a poly(A) tail. The ORF is translated into a polyprotein, which is proteolytically processed into individual viral proteins containing VP1, VP2, VP3, VP4, leader proteinase (in the genus Aphthovirus), 2A, 2B, 3A, 3B, 3C, and 3D. These mature and functional proteins participate in viral translation, transcription, replication, assembly, and release. Due to their limited proteins, picornaviruses rely on the host cellular factors for efficient replication, and release. Hence, some of the most pivotal host mechanisms that typically affected by virus infection are pathways involved in cellular responses against environmental stress and cell death. Known as two critical processes contributing to the maintenance of cellular homeostasis, autophagy controls the turnover of organelles and proteins within cells, and apoptosis is the principal mechanism by which unwanted or abnormal cells are physiologically eliminated from organisms. Picornaviruses can subvert autophagy and apoptosis by selectively exploiting some key cellular factors, which are indispensable pathways to cell fate and normal cellular functions. This article discusses the possible molecular mechanism by which picornaviruses modulate apoptosis and autophagy, and the potential role of these two pathways in viral replication and spread.
The Life Cycle of Picornaviruses
The life cycle of picornaviruses starts when viruses bind to cell surface receptors, leading to receptor-mediated endocytosis. However, different picornaviruses utilize different mechanisms to induce the endocytic process. Although a member of the genus Enterovirus, infection of HRV14 requires clathrin-mediated endocytosis for infection, poliovirus does not depend on the clathrin-mediated pathway (DeTulleo and Kirchhausen, 1998). Virus uncoating releases the viral genome via a pore in the endosomal membrane into the cytoplasm. Afterward, viral RNA is delivered to the cytosol, it is translated into a large polyprotein and subsequently processed by viral proteinases into single functional proteins including capsid proteins (VP0, VP1, and VP3) and non-structural proteins (2A–2C and 3A–3D). Viral genome replication relies on the membranous replication organelles (ROs) (van der Schaar et al., 2016). During infection, EMCV manipulates PI4KA, while enteroviruses depend on PI4KB for the formation of ROs (Dorobantu et al., 2015). Viral RNA-dependent RNA polymerase (3Dpol) is required for genome replication, which synthesizes a negative-stranded RNA that serves as a template for the synthesis of new positive-stranded RNA. After that, newly synthesized viral RNA serves as a template for translation and replication. Viral capsid proteins assemble into viral particles and then form virions with nascent viral RNA. To favor viral replication and propagation, picornaviruses selectively alter cellular pathways, such as, by inducing the shutoff of host protein translation, inhibiting the immune response and modulating cell death processes. These processes are mostly achieved by viral non-structural proteins, especially proteinase, including 2A protease and 3C protease. Picornaviruses are typically considered as cytolytic viruses as host cells are lysed for virus release. Nonetheless, an increasing of investigations has demonstrated that non-enveloped picornaviruses can also adopt a non-lytic transmission strategy via vesicles, which require autophagy to form double-membrane autophagosomes carrying virions to the extracellular environment (Feng et al., 2013).
Picornavirus and Apoptotic Pathway
Introduction to Apoptosis
Apoptosis is a non-inflammatory type of programed cell death (PCD) and requires a cascade of signaling proteins to respond to the activation of a death signal. This process is characterized by facilitating morphological changes including cell shrinkage, chromatin condensation, and plasma membrane blebbing (Galluzzi et al., 2012). The intrinsic mitochondrial pathways and the extrinsic pathway (also called death receptor) are two distinct but ultimately converging pathways for apoptosis. Caspases (a family of cysteine aspartyl-specific protease) are involved in the intrinsic and the extrinsic pathways, and the activation of caspase 3 is an executioner to induce apoptotic cell death. Host cells utilize apoptosis as a defense strategy to respond to invading pathogens. Simultaneously, viruses have evolved to subvert cell death signaling pathways. For example, PV was reported to suppress apoptosis for efficient virus production in infected cells (Tolskaya et al., 1995).
Apoptosis Machinery
In broad terms, there are two distinct signaling cascades trigger the apoptotic pathway, the extrinsic cell apoptotic pathway and the intrinsic apoptotic pathway. The initiation of the extrinsic pathway is induced by the recognition of cognate ligands, such as the FAS-ligand receptor and tumor necrosis factor receptor (TNFR). Subsequently, the death receptors recruit adaptor proteins, including TNFR-associated death domain (TRADD) and FAS-associated death domain (FADD) to form complexes in the mitochondria or cytoplasm. After recruitment, these complexes activate intracellular caspase 8, leading to activation and translocation of caspase 3 and execution of apoptotic cell death. In contrast, intracellular stresses (DNA mutation or ER stress) activate the intrinsic pathway by inducing a loss of mitochondrial outer membrane permeability (MOMP). This process leads to the efflux of pro-apoptotic proteins into the cytoplasm, such as cytochrome c, apoptosis-inducing factor (AIF), and Smac/DIABLO (Er et al., 2006). Many studies have revealed that MOMP leads to apoptosis dependent on caspase activity; however, MOMP could contribute to cell death in a caspase-independent manner (Tait and Green, 2010). The permeabilization of the outer mitochondrial membrane relies on the activation, translocation, and oligomerization of the multidomain B cell lymphoma 2 (Bcl-2) family proteins. As essential regulators, this family proteins can be divided into two classes: pro-apoptotic (Bax, Bak, Bad, and Bid) and anti-apoptotic (Bcl-2 and Bcl-xL), which dynamically regulate apoptosis. When caspase 8 cleaves the Bcl-2 protein Bid, truncated Bid (tBid) is formed and can activate two Bcl-2 proteins (BAX/BAK), resulting in their translocation to the mitochondria to induce the intrinsic apoptotic pathway (Yoshida et al., 1998; Zhang et al., 2000). BAX/BAK is required for caspase activation, regarded as a critical gateway in the intrinsic apoptotic pathway (Ruiz-Vela et al., 2005). The Bcl-2 proteins oligomerize on the mitochondrial membrane forming a pore and inducing the release of cytochrome c into the cytoplasm. The binding of cytochrome c and APAF-1 recruits and catalyzes procaspase 9 into caspase 9, leading to the activation of caspase 3 and apoptosis (Slee et al., 1999).
The Role of Apoptosis in Picornavirus Replication
The host cells have distinct mechanisms to detect the presence of pathogens and then defense against them. To survive within cells, picornaviruses have evolved diverse strategies and targeted critical components in the apoptotic cascade, to disrupt the induction of apoptosis process. For example, picornaviral proteases operate the cleavage of caspases and pro-apoptotic proteins and subvert nuclear-cytoplamic trafficking to suppress the host apoptosis. Several picornaviruses, such as PV, CVB3, FMDV, and EMCV, have been reported to trigger host apoptosis but also subvert execution of apoptosis to promote viral replication. Each of these viruses has shared specific targets or distinct mechanisms to modulate the host apoptotic response. The initiation of the antiviral response induces an antiviral transcriptional program that is composed of the expression of cytokines, chemokines, interferons (IFNs) and the activation of cell death pathways (apoptosis and necroptosis). IFN pathways, which are composed of viral sensors (RIG-I, MDA-5, TLR-3), adaptor proteins (MAVS, TRIF, and STING), kinases, and transcription factors (IRF3 and IRF7), antagonize viral infection by facilitating the expression of ISGs (Schoggins, 2014). In picornavirus infection, endosomal Toll-like receptor (TLR-3), a PRRs, recognizes and binds to double-stranded viral RNA (dsRNA), inducing the exposure of the Toll-IL-1 receptor (TIR) domain (Alexopoulou et al., 2001; de Bouteiller et al., 2005). TIR domain-containing adaptor inducing beta interferon (TRIF) binds to TLR-3 activating IRF-3, TANK binding kinase-1 (TBK-1) and NF-κB, which are transcription factors essential for IFN- β production. The second set of PRRs includes RIG-I and melanoma differentiation-associated gene-5 (MDA5), which recruit adaptor MAVS (also known as IPS-1 and VISA) via their intracellular RNA helicase activity and subsequently activate TBK-1, IRF-3 and NF-κB, leading to the phosphorylation and dimerization of IRF-3 (Kawai et al., 2005; Slater et al., 2010). Then, IRF-3 homodimers translocate to the mitochondrial membrane and complex with Bax, resulting in the formation of a pore and efflux of cytochrome c. TRIF was found to induce cell death and the initiation of this process relies on the FADD-caspase 8 axis (Kaiser and Offermann, 2005). Previous studies have shown that the TRIF/interacting protein (RIP)-1/caspase-8 complex is necessary for dsRNA-mediated TLR-3-dependent apoptosis, the interaction of TRIF with RIPK1 through its C-terminal RIP homotypic interaction motif (RHIM), and the trigger of apoptosis was in a caspase-8 dependent manner (Kaiser and Offermann, 2005; Estornes et al., 2012). Furthermore, the activation of IRF-3 by RIG-I is regulated by the caspase8-mediated cleavage of the RIPK1 protein (Rajput et al., 2011).
Picornaviral 2B Protein and Apoptosis
The picornaviral 2B protein is a small hydrophobic membrane protein, which exhibits viroporin or viroporin-like activity. Viroporins are divided into two major groups, class I (containing one transmembrane domain) and class II (containing two transmembrane domains). The viroporins of class IIA have lumenal N and C termini, whereas viroporins of class IIB have cytosolic N and C termini (Figure 1) (Nieva et al., 2012). The 2B protein of CVB3 belongs to class IIA and consists of a hydrophobic helix and a cationic amphiphilic helix (van Kuppeveld et al., 1996; de Jong et al., 2004). However, 2B proteins of PV and FMDV belong to class IIB (Agirre et al., 2002; Ao et al., 2015; Gladue et al., 2018). HAV 2B protein has a viroporin-like region that interacts with cellular membranes (Shukla et al., 2015). Picornaviral 2B protein involves in increasing membrane permeability, altering Ca2+ homeostasis and modulating host apoptosis and autophagy.
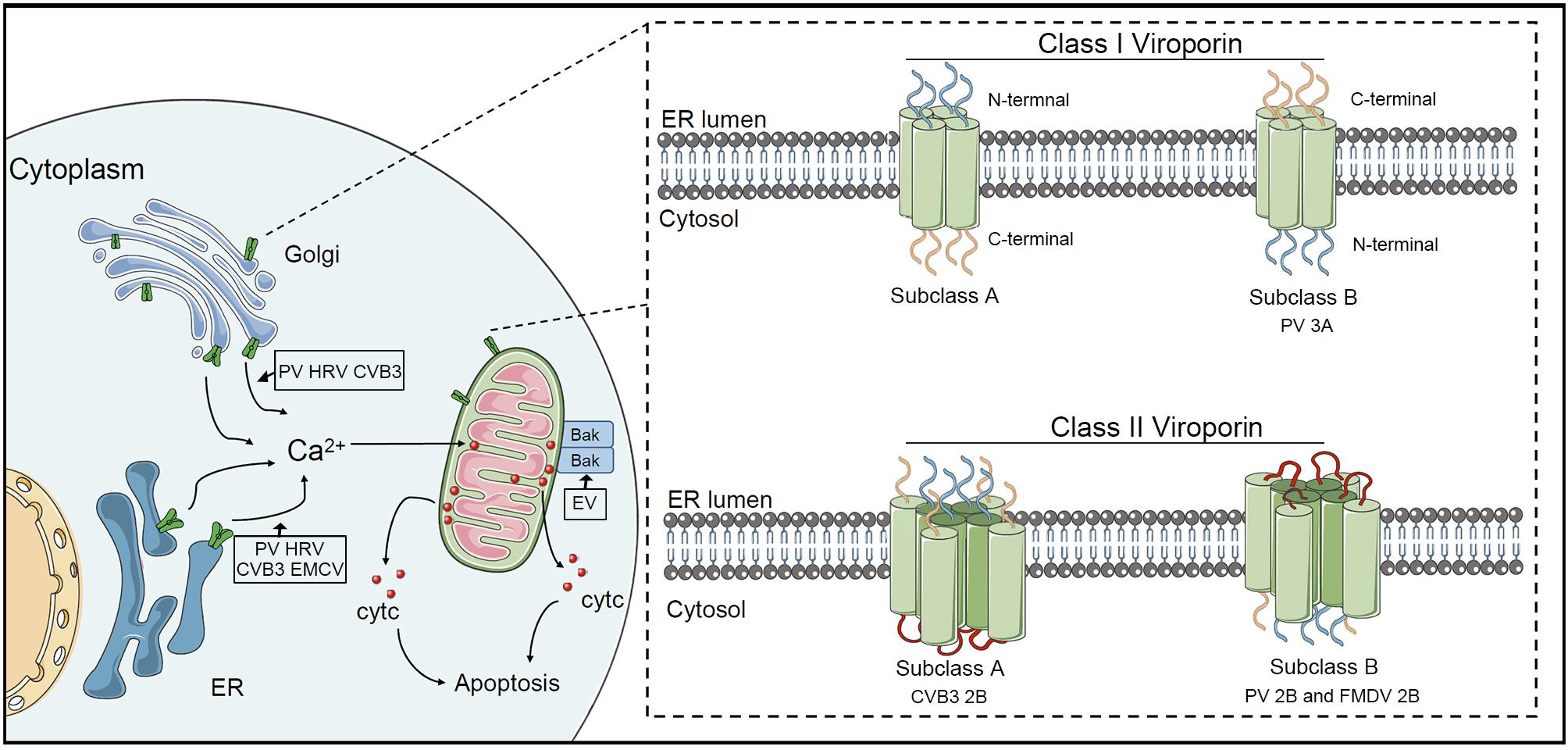
Figure 1. Picornaviral 2B protein regulates apoptosis. The 2B protein of PV, HRV, and CVB3 directly targets Golgi and ER membrane and assembles pores in the Golgi membrane and ER membrane, which induces the release of Ca2+ from organelles into the cytosol. The mitochondrion takes up Ca2+, leading to the release of cytochrome c and the formation of apoptosome. Different from these three picornaviruses, EMCV 2B protein only alters the Ca2+ content in the ER. EV71 2B protein targets the pro-apoptotic protein Bax to trigger the activation of the mitochondrial cell death pathway. There are two major groups viroporin, class I (containing one transmembrane domain) and class II (containing two transmembrane domains). CVB3 2B protein belongs to class IIA, while PV 2B and FMDV 2B belong to class IIB.
ER stress induces calcium (Ca2+) release from ER stores and Ca2+ accumulation in the mitochondrial matrix, which requires the 1,4,5-triphosphate receptor (IP3R) and voltage-dependent anion channel (VADC) as mediators. This increases in the mitochondrial Ca2+ concentration contributes to the increase in MMP; therefore, the movement of mitochondrial pro-apoptotic proteins is promoted, which triggers cell death (Deniaud et al., 2008). An early study showed that a notable increase in intracellular Ca2+ concentration was observed in PV infection (Irurzun et al., 1995). Fluorescent Ca2+ indicators were used to determine that the CVB3 2B protein manipulated intracellular Ca2+ homeostasis, including by decreasing Ca2+ content in the ER and Golgi, which failed to suppress apoptosis (Campanella et al., 2004). Notably, subsequent studies revealed that the expression of 2B protein in PV and HRV resulted in a significant decrease in Ca2+ in the ER and Golgi complex; however, EMCV 2B protein decreased only in the ER [Ca2+] while HAV 2B protein did not alter the Ca2+ content in the ER and Golgi (de Jong et al., 2008) (Figure 1). The release and translocation of Ca2+ from the ER have been demonstrated to be involved in PV-induced apoptosis (Brisac et al., 2010). Increased cytosolic Ca2+ not only leads to the activation of calcium-dependent cell death pathways, but also induces autophagy. In BHK-21 cells, FMDV 2B protein disrupted the Ca2+ concentration, which was implicated in the induction of autophagy (Ao et al., 2015). Recent studies have revealed the potential functions of picornaviral 2B, identified as a viroporin, which oligomerizes to form aqueous pores on the host cellular membrane (Fung et al., 2015). A previous study demonstrated that EV71 infection induces apoptosis by the mitochondrial pathway, in which the release of cytochrome c from the mitochondria and cleavage of caspase 9 can be observed in multiple cell lines (Chang et al., 2004). A subsequent study revealed that EV71 2B protein could induce conformational changes in the pro-apoptotic protein Bax to trigger the activation of the mitochondrial cell death pathway (Cong et al., 2016).
Picornaviral 3C Protease and Apoptosis
Picornaviral 3C protease, a chymotrypsin-like cysteine protease, exists in all picornaviruses. Despite cooperating with leader protease and 2A protease in some genera, 3C protease and its precursor are responsible for the most primary cleavages and secondary cleavages of polyproteins. 3C protease is capable of regulating the apoptotic process via protease activity toward multiple cellular factors. To attenuate the innate response, the RNA sensor RIG-I is cleaved by 3C protease in cells infected with PV, HRV, EV1, EV71, EMCV and CVB3, while adaptor MAVS is degraded in PV, EV71, HRV, CVB3, and HAV (Table 1). Notably, MAVS proteolysis in HAV requires both mitochondrial targeting property of 3A and the cysteine protease activity of 3C protease (Yang et al., 2007). In contrast to RIG-I, MDA-5 can be cleaved in PV-infected cells in proteasome and caspase-dependent manner instead of by viral proteases (Barral et al., 2007). Subsequent studies reported that MDA-5 was a target of enteroviral 2A protease (Feng Q. et al., 2014). EV 71 3C protease cleaves IRF7 to compromise the host innate immune response (Lei et al., 2013). The formation of the caspase 8/RIPK1/FADD/IPS-1 complex is essential for the induction of extrinsic apoptosis. Previous researches reported that RIPK1 could be cleaved by caspase 8 to strengthen the interaction of this complex in a pro-apoptotic manner (Lin et al., 1999). Interestingly, RIPK1 is also a target for HRV 3C protease, which might inhibit extrinsic apoptosis (Croft et al., 2018; Lötzerich et al., 2018) (Figure 2). EMCV 3C protease can cleave Ras-Gap SH3 domain-binding protein 1 (G3BP1) disrupting the formation of SGs, which are involved in regulating IFN-β gene expression (Ng et al., 2013). Similar disruption was described in PV- and CV-infected cells (White et al., 2007; Langereis et al., 2013). This observation was also reported in FMDV-infected cells, which G3BP1 could be cleaved by 3C protease of FMDV (Galan et al., 2017). Moreover, subsequent research revealed that G3BP1 is a target of leader protease of FMDV and ERAV as well (Visser et al., 2019). Accumulating evidence suggests that different picornaviruses adopt multiple mechanisms to disrupt the immune response.
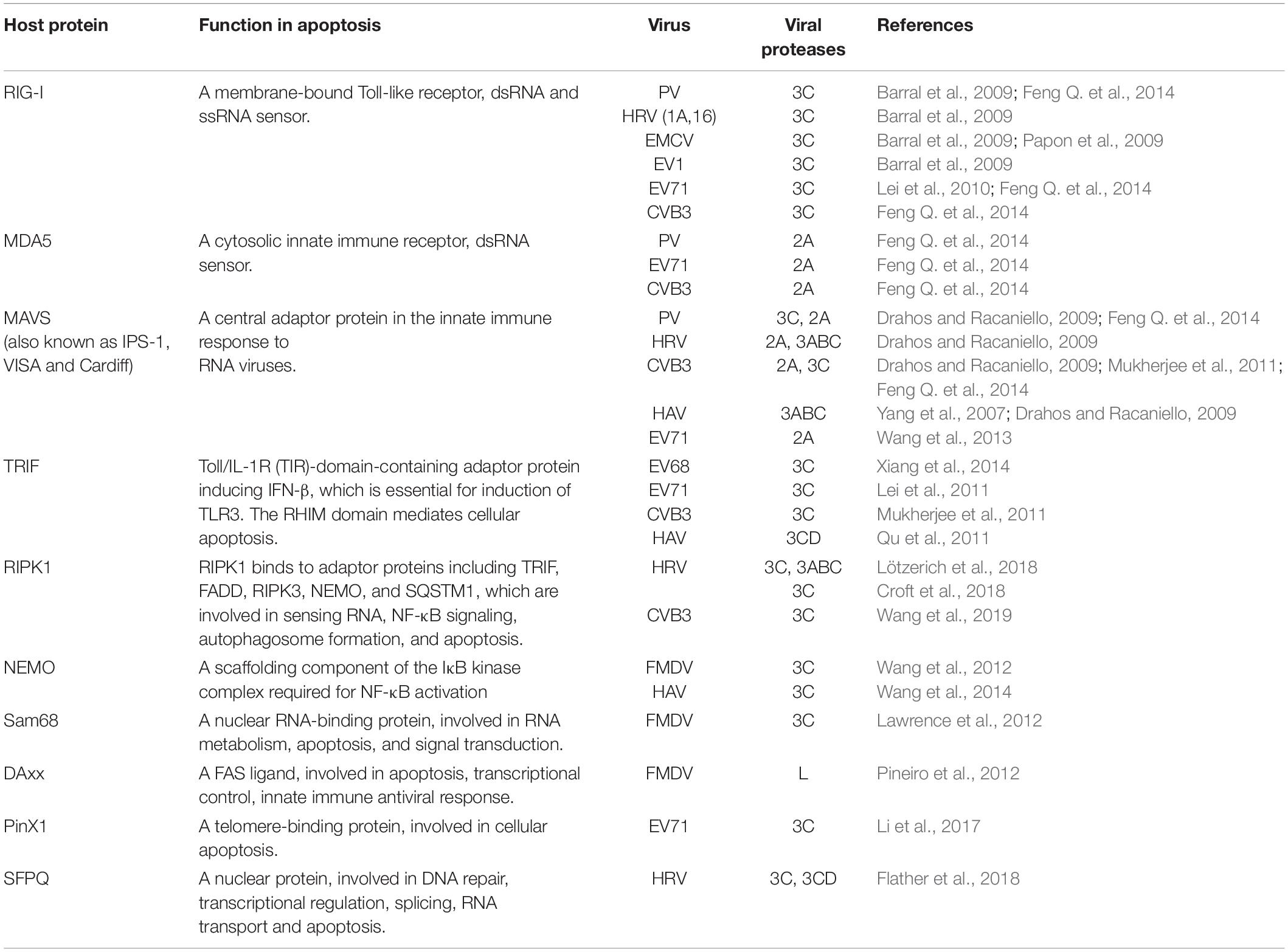
Table 1. The host proteins associated with apoptosis are cleaved by picornaviral proteases.
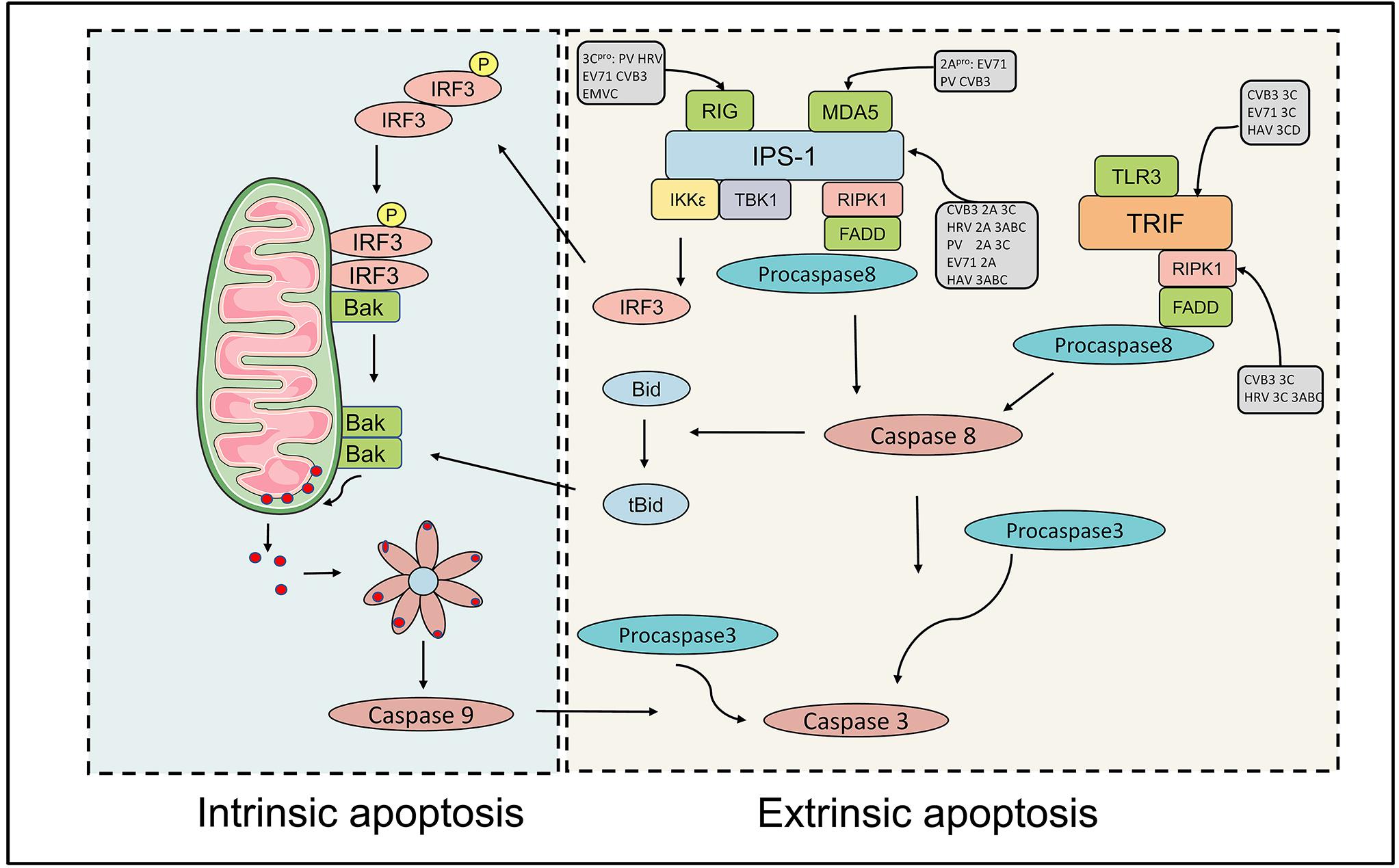
Figure 2. Effects of picornavirus infections on those intrinsic and extrinsic apoptotic pathways. Caspase 9-dependent intrinsic apoptosis is initiated by the release of mitochondrial cytochrome c into the cytoplasm. When activated, the RNA sensors RIG-I and MDA-5 associate with IPS-1 and recruit TBK1 and IKKγ a death receptor complex in mitochondria. TBK1 and IKKγ phosphorylate IRF-3 dimers, which then induce a pore in the mitochondrial membrane, allowing the release of cytochrome c into the cytoplasm, which induces intrinsic apoptosis. RIG-I is cleaved by 3C protease in cells infected with PV, HRV, EV1, EV71, EMCV and CVB3, while MDA-5 is a target of enteroviral 2A protease. The adaptor MAVS is degraded by viral proteases of PV, EV71, HRV, CVB3, and HAV. The extrinsic apoptosis requires the formation of the caspase 8/RIPK1/FADD/IPS-1 complex. RIPK1 is also a target for enteroviral 3C protease, which disrupts extrinsic apoptosis.
As an IRES trans-acting factor, polypyrimidine tract-binding protein (PTB) is present in a complex containing RNA-binding proteins NONO/p54nrb, hnRNPA1, hnRNPC1/C2, hnRNPA2/B1 and PSF/SFPQ, which is central in apoptotic cells. The alteration in a single protein with overexpression or siRNA knockdown is sufficient to modulate apoptosis rates, while the depletion of SFPQ or PTB resulted in reduced apoptosis (King et al., 2014). Interestingly, PTB has been reported to be cleaved by viral protease in PV, HRV, and HAV (Back et al., 2002; Kanda et al., 2010). Similarly, the proteolytic processing of PTB was observed in FMDV-infected cells (Rodriguez Pulido et al., 2007). Recently, it was found that SFPQ is characterized as a pro-viral factor, which is specifically targeted by HRV 3C protease for proteolysis (Flather et al., 2018). The cleavage of the far-upstream element (FUSE) binding protein 2 (FBP2) is observed in EV71-infected cells, and this cleavage is executed by 3C protease (Chen et al., 2013). EV 71 3C protease cleaves PinX1 at the Q51-G52 pair, a telomere-binding protein, disrupting telomere maintenance, which has been suggested to induce apoptosis for promoting virus release (Li et al., 2017).
The src-associated substrate in mitosis of 68 kDa (Sam68) has been characterized in multiple cellular processes associated with RNA usage and gene expression, including transcription, translation, RNA splicing and export (Li et al., 2002; Matter et al., 2002; Coyle et al., 2003). It was revealed that Sam68 could induce apoptosis via its RNA-binding ability. The specific-RNA binding-defective protein Sam68 G178E (Gly178 was substituted by Glu) could not promote apoptosis, which indicated that the induction of apoptosis required a fully-functional RNA-binding domain (Taylor et al., 2004). An alteration in the nuclear localization of Sam68 to the cytoplasm, which was related to the disruption of the NPC, was observed in the infection of PV, HRV2, HRV16, EV71, and FMDV (McBride et al., 1996; Lawrence et al., 2012; Zhang et al., 2014; Walker et al., 2015). However, the relocalization of Sam68 occurs before the complete disruption of the NPC during HRV infection, and redistribution of Sam68 is involved in PI3K/Akt activation in EV71 infection. In contrast to enteroviruses, FMDV 3C protease cleaves Sam68, resulting in the diffusion of the C-terminal portion into the cytoplasm and the attachment of viral IRES for translation (Lawrence et al., 2012). Remarkably, one of these predicted cleavage sites is Q179/G, which is close to the mutation site within RNA-binding-defective Sam68. It seems that different viruses utilize multiple strategies to disrupt the function of Sam68 including RNA binding or pro-apoptotic abilities for promoting viral infection, but the mechanisms remain unknown.
Other Viral Proteases and Apoptosis
In addition to 3C protease, leader protease in Aphthovirus and 2A protease in Enterovirus are pivotal non-structural proteins in the viral life cycle. FMDV leader protease is a papain-like cysteine protease, which exists in two forms (Laboratory and Lb) (Kirchweger et al., 1994). Different from FMDV leader protease, leader protein encoded in the Cardiovirus genus lacks the protease activity. According to structures and functions, picornaviral 2A proteins can be divided into five types (A) chymotrypsin-related protease, (B) Aphthovirus-like 2A, (C) hepatitis-A-virus-like 2A, (D) Parechovirus-like 2A, and (E) the 2A sequence of the genus Cardiovirus (Yang et al., 2017). The chymotrypsin-like 2A encoded in Enterovirus genus, service as a proteolytic enzyme processing viral polyprotein and host proteins, whose function is similar to FMDV leader protease. Here, we mainly discussed the potential mechanisms of viral proteases counteract the host immune response.
In some picornavirus infections, the induction and suppression of apoptosis regulate cell lysis and viral spread. It has been revealed that PV infection induces apoptosis early in infection and inhibits apoptosis during late-stage infection. In late-stage infection, PV 2A protease can suppress apoptosis by inducing the aberrant processing of procaspase-9 (Belov et al., 2003; Burgon et al., 2009). In contrast, CVB3 2A protease could induce caspase 8-mediated activation of caspase 3 and the release of cytochrome c, activating the caspase 9-mediated the intrinsic mitochondria-mediated apoptotic pathway (Chau et al., 2007). CVB3 2A protease is responsible for cleavage of death-associated protein 5 (DAP5), a structural homolog of eIF4G, resulting in the promotion of viral replication and host apoptosis (Hanson et al., 2016). EVA71 modulates the mitochondrial apoptotic pathway via the conformational activation of Bax (Han and Cong, 2017). Enteroviruses can induce apoptosis, and disruption of the NPC was observed during infection. However, there is little evidence that explains the link between alterations in NPC and apoptotic cell death. The PV 2A protease was responsible for degrading Nup 62, Nup 98 and Nup 153, while HRV 2A protease cleaved endogenous Nup 98 and recombinant Nup 62 (Park et al., 2008; Castello et al., 2009). The cleavage of Nups leads to diverse consequences in trafficking pathways, including the lack of nuclear import receptor cargo complexes and nuclear envelope leakiness. Transient expression of the 2A protease of EV 71 is sufficient to trigger apoptosis (Kuo et al., 2002).
The FMDV leader protease inhibits host innate immune response by multiple mechanisms. Death-domain associated protein (Daxx), a Fas ligand, involves multiple cellular mechanisms, including apoptosis, transcriptional control, and innate immune response (Yang et al., 1997). In FMDV-infected cells, Daxx is reported to be proteolysed by leader protease, which evades the host anti-viral response (Pineiro et al., 2012). Picornaviral proteases target the host innate immune sensors subverting the specific recognition of viral RNA. Similar to enteroviral 3C protease and 2A protease cleaving RIG-I and MDA-5 respectively, FMDV leader protease can cleave laboratory of genetics and physiology 2 (LGP2), which is regarded as another RIG-I-like receptor in recognizing viral RNA in antiviral immunity (Rodriguez Pulido et al., 2018).
Autophagy in Virus Infection
Overview of Autophagy
The ubiquitin-proteasome system (UPS) and the autophagy-lysosome system are the two major pathways that participate in cellular protein quality control in eukaryotic cells and are crucial for maintaining cellular homeostasis via the degradation and recycling of cellular components (Lilienbaum, 2013). Dysfunctions in autophagy cause a wide range of illnesses, including neurodegenerative disorders, cancer, cardiovascular, and inflammatory bowel disease. The degradation of autophagy is considered to be non-apoptotic cell death. Three types of autophagy have been identified in mammalian cells (macroautophagy, microautophagy, and chaperone-mediated autophagy). Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved process, in which the cytoplasmic components are sequestered within cytosolic double-membrane vesicles and then delivered to lysosomes for degradation (Mizushima and Klionsky, 2007; Klionsky et al., 2011). Autophagy is a five-step preprocess, induction, nucleation, elongation (expansion), fusion, and cargo degradation/metabolic recycling. More than 30 autophagy-related genes (Atg) participate in different stages of the execution of autophagy (Mizushima et al., 2002; Klionsky, 2007). Autophagy is initiated by the formation of crescent-shaped isolated-membrane vesicles sequestering dysfunctional organelles, misfolded protein and invasive microbes, which are known as isolation membranes or phagophores. Subsequently, the phagophore grows and the phagophore edge fuses to form an enclosed double-membrane vesicle called the autophagosome. Following elongation, the outer membrane of the autophagosome finally fuses with lysosomes to form autolysosomes, while lysosomal enzymes degrade the contents and inner membrane of the autophagosome (Figure 3). The sequestration of cargo within cytosolic double-membrane vesicles has been regarded as the most identifiable feature of macroautophagy.
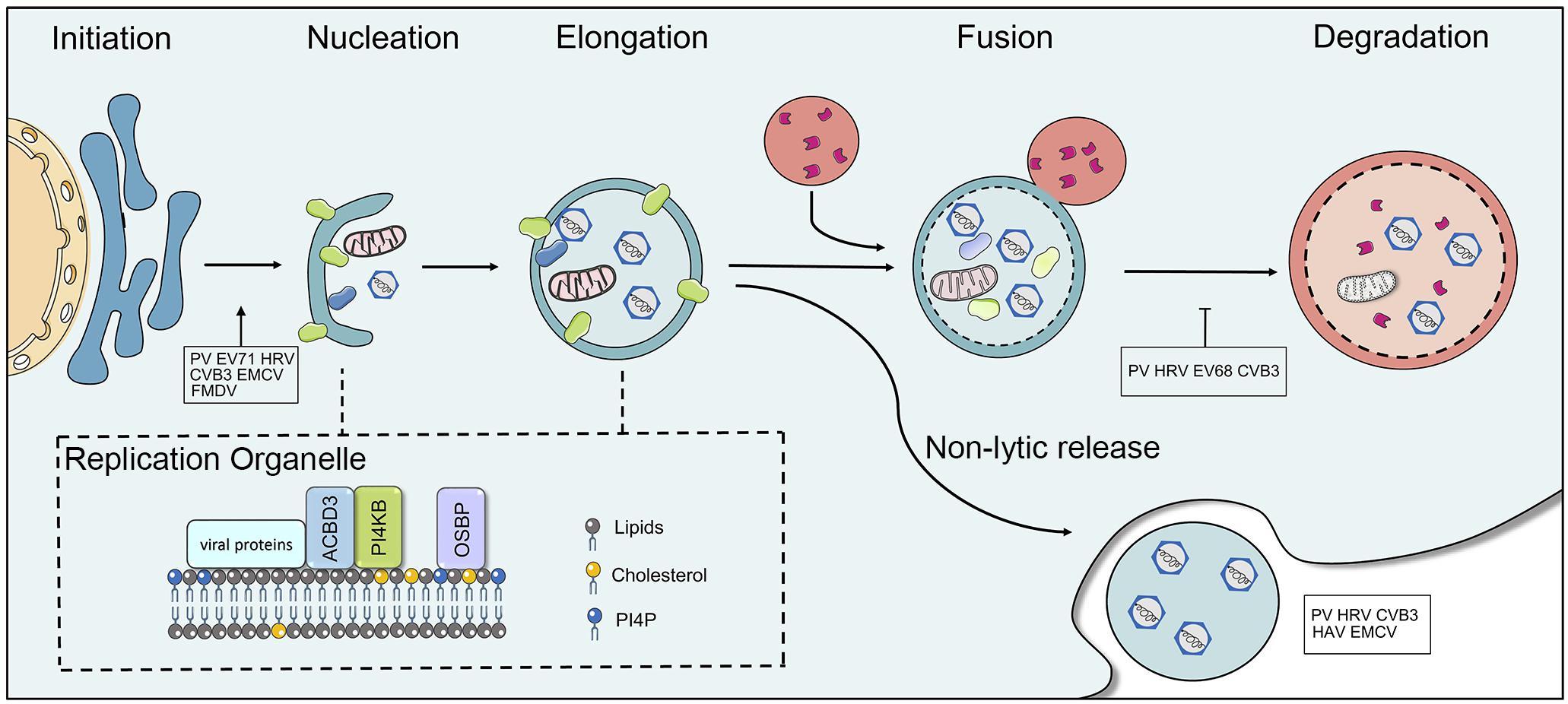
Figure 3. The interaction between picornaviruses and the host autophagy pathway. Autophagy is a five-step preprocess, including induction, nucleation, expansion (elongation), fusion, and cargo degradation/metabolic recycling. Picornaviruses have been shown to subvert the host autophagy machinery for viral replication and non-lytic release. Some viruses, including PV, CVB3, EV71, HRV, FMDV, and EMCV have been reported to utilize the autophagy for optimal infection. Picornaviruses replicate on membranous replication organelles, which require a complex containing viral (2B and 3A), ACBD3, OSBP, and PI4KB (or PI4KA). The fusion of autophagosome with lysosomes is inhibited by picornaviruses. HAV, CVB3, PV, HRV, and EMCV can trigger autophagy and release virions in single-membrane vesicles.
Autophagic Machinery
Autophagic, proteasome and cytosolic degradation achieve the maintenance of cellular activity and viability via balancing biosynthesis and turnover. Autophagy responds to various conditions of stress through regulatory factors composed of different sets of Atg proteins (Feng Y. et al., 2014). The first autophagy-specific gene, Atg1, was identified as a Ser/Thr protein kinase and is required for initiation (Matsuura et al., 1997). The ULK1 (uncoordinated 51-like autophagy activating kinase) and FIP200 are two mammalian homologs of Atg1 (Jung et al., 2009). The process of autophagy is achieved by several functional complexes including the Atg/Ulk1 complex, the PtdIn3K complex, and two ubiquitin-like conjugation systems (Wong et al., 2013), the Atg12-Atg5 conjugation system and microtubule-associated protein light chain 3 (LC3-Atg8) conjugation system (Ravikumar et al., 2010). Two essential protein complexes involved the formation of autophagosomes are Ulk1 and PI3KC3-C1. In response to intracellular or extracellular stress and signals, including starvation, ER stress, and pathogen infection, autophagy can be induced. In yeast and Drosophila, the target of rapamycin (TOR) can negatively regulate Atg1 (Chang and Neufeld, 2009). Rapamycin treatment and starvation could induce TOR inhibition in yeast. Then, the kinase activity of Atg1 is stimulated and the binding affinity to Atg13 and Atg 17 is increased, which facilitates the initiation of autophagosomes by increasing the scaffold formation of Atg1-Atg13-Atg17 complex and the recruitment of Atg proteins (Kamada et al., 2000; Kabeya et al., 2005). The inhibition of the mammalian target of rapamycin (mTOR) is inhibited when starvation induces autophagy, which is an essential regulator integrating nutrient signals and growth factors (Kuma et al., 2004). The mTORC1 negatively regulates a complex formed of ULK1, Atg13, Atg101, and FIP200 in response to growth factors that trigger the PI3K-AKT pathway or other nutrient-related signals.
The PtdIns3K complex, comprising Vps34, Vps15, Vps30, and Atg14, is involved in the vesicle nucleation and the recruitment of PtdIns3P-binding proteins. Beclin 1, the mammalian homolog of yeast Vps30/Atg6, forms a complex with PtdIn3K and localizes on the trans-Golgi network (TGN) membrane that is involved in autophagic vesicle nucleation (Kihara et al., 2001). The connection between the molecular mechanism of autophagy and human disease was first identified regarding the autophagy-promoting function of Beclin 1 in inhibition of tumorigenesis (Liang et al., 1999). Furthermore, a series of studies revealed that protein expression of Beclin 1 is linked to cell growth and death, neurodegeneration, including Alzheimer’s disease and Huntington’s disease, and pathogen infection (Liang et al., 1998; Yu et al., 2004; Boya et al., 2005; Rubinsztein et al., 2005; Orvedahl et al., 2007; Levine and Kroemer, 2008).
For Ub1 protein conjugation, there are eight homologs of Atg8 at least in mammalian cells. LC3 and GATE-16/GABARAP are two subfamilies for these homologs. They are both essential in autophagosome biogenesis; however, LC3s participate in the elongation of the phagophore membrane whereas the GABARAP subfamily is involved in later steps in autophagosome maturation (Weidberg et al., 2010). The C-terminal fragment of proLC3 is proteolytically cleaved by Atg4 to generate cytosolic LC3-I with an exposed lipid conjugation site, followed by its subsequent activation and transformation by Atg7 and Atg3, respectively. Consequently, LC3 conjugates with the cellular lipid phosphatidylethanolamine (PE), forming LC3-II (PE-conjugated form) (Kabeya et al., 2004). The formation of discrete puncta, which are indicative of the conversion of LC3-I to LC3-II and can be observed via immunofluorescence analysis, is considered a critical regulatory step of autophagosome formation. The control of the initiation of autophagosome formation by modulating the mTOR pathway and LC3 lipid conjugation and the control of processing from the maturation of autophagosomes into degradative autolysosomes are two main strategies to regulate autophagy.
Initially, autophagy was described as a random process balancing cellular components in response to different types of stress. However, there is accumulating evidence that autophagy can regulate and degrade protein aggregates and organelles in a selective manner (Glick et al., 2010; Gatica et al., 2018). Instead of taking up and degrading random cytoplasm into phagophores for non-selective autophagy, the membrane-bound vesicle removes specific components via selective autophagy (Jin et al., 2013). The well-characterized molecule, sequestosome 1 (SQSTM1)/p62 molecule is a crutial autophagy receptor protein in bridging ubiquitylated substrates to the nascent autophagic vesicles in mediating selective autophagy (Johansen and Lamark, 2011; Stolz et al., 2014). SQSTM1 mutations were identified as being linked to Paget’s diseases, in which increased and disorganized bone turnover lead to focal abnormalities (Ralston and Layfield, 2012).
Role of Autophagy in Viral Replication
Autophagy is a self-degradative process to maintain cellular homeostasis. In Picornavirus family, many viruses have been reported to utilize the autophagic machinery for optimal infection, including PV, CVB3, EV71, HRV, FMDV, and EMCV (Wong et al., 2008; Huang et al., 2009; Kemball et al., 2010; Klein and Jackson, 2011; O’Donnell et al., 2011; Zhang et al., 2011). The increased autophagosome formation was observed at the early stage of CVB3 infection, while the decreased autophagosome formation along with increased apoptosis was observed at the late stage of infection. It was presumed that the replication of CVB3 depended on the autophagosome, and the viral release relied on apoptosis (Xin et al., 2014). Studies on enteroviruses showed that autophagy was inhibited at the stage of autolysosomal degradation (Jackson et al., 2005). During CVB3 infection, increased protein polyubiquitination and the accumulation of protein-ubiquitin conjugates were observed in cells and tissues, revealing that CVB3 might take advantage of the UPS for pathogenesis (Luo et al., 2010). Moreover, it was suggested that CVB3 infection could block autolysosomal degradation due to the accumulation of autophagosomes, which causes the protein levels of LC3-II and p62 to increase (Wong et al., 2008; Kemball et al., 2010). Subsequent studies have revealed that CVB3 infection leads to the cleavage of SQSTM1 at amino acid 241 within the tumor necrosis-associated factor 6 binding (TB) domain and viral 2A protease is responsible for this cleavage (Shi et al., 2013). After that, SQSTM1 was later found to regulate viral replication, and the knockdown of SQSTM1 caused an increase in the production of viral protein and viral titers. Increasing studies have shown that SQSTM1 can interact with the CVB3 capsid protein VP1. The cleavage of SQSTM1 has been observed in multiple enteroviruses, including infection of PV, HRV, and EV D68, and it has been suggested that this is a common strategy to interact with cargo adaptors (Corona et al., 2018). The soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins are required for the fusion of the autophagosome with the endosome/lysosome (Itakura et al., 2012). One of these SNARE proteins, syntaxin 17 (STX17), could interact with the synaptosomal-associated protein 29 (SNAP29), which mediates fusion between the outer autophagosomal membrane and lysosomal membrane (Itakura and Mizushima, 2013). CVB3 infection was shown to decrease the transcription and translation of STX17, impairing the autophagic flux. It was also found that the overexpression of STX17 could restore lysosomal function and blocked apoptosis induced by CVB3 infection (Tian et al., 2018). Lang proposed that the blockage of autophagosome-lysosome fusion is the reason for the accumulation of autophagosomes in CVB3-infected cells and that SNARE complexes are potential targets. Subsequently, it was determined that CVB3 infection inhibits autophagic flux via impairing the formation of SNARE complexes and that SNAP29 and adaptor protein PLEKHM1 could be cleaved by viral 3C protease (Mohamud et al., 2018).
It has been shown that PV replication is associated with the formation of autophagosomes and independent of autophagic degradation (Richards and Jackson, 2012). Both viral proteins 2BC and 3A are required for the modification of LC3, which create double-membraned vesicles for viral replication (Taylor and Kirkegaard, 2007). Autophagy induction is promoted by two different upstream signaling pathways, AMP activated protein kinase (AMPK) activation, which leads to phosphorylation of ULK1, and mTOR inactivation, which causes the loss of the capability to phosphorylate ULK1 and disrupts the ULK1-AMPK interaction (Kim et al., 2011). From studies with a cell line of knockouts of autophagy-related genes, it’s determined that PV utilized ATG9 and LC3 for initiation of autophagy to promote viral intracellular growth (Abernathy et al., 2019).
It has been well-documented that the eukaryotic translation initiation factor 2 (EIF2S1) regulates protein synthesis by being phosphorylated under stress conditions (Kedersha et al., 1999). As a member of the activating transcription factor (ATF) subfamily, ATF4 is required for the transcriptional activation of a large number of autophagy genes in response to amino acid starvation and ER stress (B’chir et al., 2013). In the case of FMDV, viral protein VP2 was found to induce host autophagy via interacting with heat shock protein family B member 1 (HSPB1) to activate the EIF2S1-ATF4 pathway and inhibit the AKT-mTOR pathway, which could facilitate FMDV replication (Sun et al., 2018). In particular, expression of mutant VP2 protein reduced autophagy when this mutation affected viral infection and virulence. Besides, FMDV 2B protein can induce the host autophagy, which is associated with disrupting of Ca2+ content via viroporin activity (Ao et al., 2015).
Oxysterol binding protein (OSBP) regulates contracts of membranes containing phosphatidylinositol 4-phosphate (PI4P) (Mesmin et al., 2013). At the final stage of autophagy, PI4P is essential to the fusion of the autophagosome with lysosomes (Wang et al., 2015). Picornaviruses require specialized vesicular membranes (ROs) to replicate viral genomes by utilizing host OSBP/PI4P pathway. Several years ago, CVB3 was found to replicate on PI4P-riched organelles, and viral 3D polymerases could specifically bind to PI4P lipids (Hsu et al., 2010). The formation of RO membranes was later observed during RV, EV11, EV71, Achi virus (AiV), and EMCV infections (Spickler et al., 2013; Ishikawa-Sasaki et al., 2014; Roulin et al., 2014; Dorobantu et al., 2015; Strating and van Kuppeveld, 2017). Although the exact mechanisms how viruses utilized ROs for viral replication remains unclear, the formation of ROs requires viral 2BC and 3A protein to remodel intracellular membranes. Enterovirus 3A protein selectively manipulates the recruitment of PI4KB and GBF1, which provides a PI4P lipid-enriched membrane microenvironment to recruit 3Dpol from the cytosolic pool (Hsu et al., 2010). AiV requires PI4KB, Golgi protein acyl-coenzyme A binding domain containing 3 (ACBD3), and viral non-structural proteins 2B, 2BC, 2C, 3A, and 3AB forming a complex to enhance the generation of PI4P for viral replication (Ishikawa-Sasaki et al., 2014). Similarly, PV recruits PI4KB via 2BC and HRV 2B protein recruits PI4KB to replication membranes (Arita, 2014; Roulin et al., 2018). EMCV utilizes 3A protein to recruit PI4KA, another PI4K isoform, for promoting the generation of PI4P-enriched membranes. EMCV ROs depend on the activities of PI4KA and OSBP (Figure 3) (Dorobantu et al., 2015). HAV and ERAV replicate independently of both PI4KA and PI4KB. A detailed mechanism was reported in which the picornaviral 3CD protein was sufficient to induce PI4P synthesis, which indicates that this single protein has the capability to activate and commandeer the normal phospholipid biosynthetic pathway that is required for the viral life cycle (Banerjee et al., 2018). The autophagy-inducer increased PV infection, while the absence of essential genes for autophagy decreased viral infection (Jackson et al., 2005). The inhibitors of autophagosome maturation increased virion production of CVB3 (Wong et al., 2008). Thus far, growing evidence suggested that picornaviruses subverted the host autophagy pathway to promote replication. Identification of the mechanism used by picornaviruses for modulating the host membrane network and subverting autophagy will provide insights into novel mechanism of viral replication.
Role of Autophagy in Viral Dissemination
Dismantling the cell membrane (cell lysis) has traditionally been considered a prerequisite way for the exit and transmission of picornaviruses. In recent years, however, increasing data have shown picornaviruses, including HAV, CVB3, PV, HRV and EMCV, that trigger autophagy and release in vesicles, referred to as non-lytic spread (Figure 3) (Feng et al., 2013; Bird et al., 2014; Robinson et al., 2014). Secretory autophagy is a non-degradative pathway in which autophagosomes fuse with the plasma membrane instead of lysosomes to release single-membrane vesicular structures filled with virions to the extracellular environment. The secretory autophagosomes and canonical degradative autophagosomes share conventional machinery; for example, the depletion of any autophagic genes including Atg12, LC3 and Beclin-1, results in decreasing viral release. In CVB3-infected cells, extracellular microvesicles (EMVs) containing infectious virions were released from infected cells (Robinson et al., 2014). A similar phenomenon, autophagosome-mediated exit without lysis (AWOF), was observed during poliovirus infection. Subsequent studies demonstrated via quantitative time-lapse microscopy that PV could spread to another cell without lysis, which was dependent upon components of the autophagy pathway (Bird et al., 2014). The non-enveloped viruses also harness non-lytic intercellular spread for pathogenesis These LC3-II positive autophagosomes containing virus did not fuse with the lysosomes and trafficked to the cell periphery where the outer membrane of DMVs fused with the plasma membrane. Moreover, it was found that the clustered PV particles were packaged into PS lipid-enriched vesicles and then transmitted from cell-to-cell via non-lytic release (Chen et al., 2015). This mode of transmission among cells could provide greater infection efficiency. Notably, the infection of vesicles containing PV particles is PS lipid- and PV receptor-dependent and not simply membrane-membrane fusion. However, a recent report showed that CV could trigger the release in an autophagosome-bound mitochondrion-virus complex, which targeted the DRP1-mediated fragmentation of mitochondria (a precursor to mitophagy). The suppression of the mitophagy pathway resulted in a marked decrease in virus production, suggesting that CVB subverts host mitophagy machinery to support viral transmission via releasing EMVs (Sin et al., 2017).
As an unusual member of the Picornaviridea, HAV has two mature infectious forms, enveloped (cloaked in host cell membranes) and non-enveloped. The enveloped viruses (eHAV), resembling exosomes to protect virions from antibody-mediated neutralization, are released from infected cells (Feng et al., 2013; Longatti, 2015). This process is dependent upon components of the cellular endosomal sorting complex required for transport (ESCRT). Interestingly, eHAV is the main form found in the blood of infected humans and animals; however, non-enveloped HAV is primarily found in the feces. This suggested that HAV utilized the host ESCRT machinery to escape from host immune responses and promote viral spread in the liver (Feng et al., 2013). A subsequent study revealed that eHAV, and not viral RNA exosomes or non-enveloped virus, was responsible for the activation of plasmacytoid dendritic cells (pDC) to produce type IFN- α at a limited level, suggesting that viral exosomes played a central role in immune evasion (Feng et al., 2015). Autophagy and other mechanisms of cytoplasmic transfer provide a link between viral egress and compromising host immune response.
Conclusion
The researches on picornavirus and cellular factors have the potential to contribute strongly to increase our understanding of viral replication and pathogenesis. In this review, we described the current knowledge of the mechanisms of picornaviruses utilize to subvert the host apoptosis and autophagy for viral immune evasion, replication, and transmission by broad mechanisms. Several picornaviruses, such as PV, CVB3, HRV, and EMCV, can trigger the host apoptosis and subvert execution of apoptosis for their optimal replication via the cleavage of caspases and pro-apoptotic proteins, the disruption of Ca2+ homeostasis, and the suppression of nuclear-cytoplamic trafficking. Picornaviruses also share significantly conserved mechanisms of replication, for example, the requirement of remodeling the cellular membranes for viral replication. Depending on the source of cytoplasmic membranes, double-membrane autophagosomes following infection affect viral replication, and also serve as an essential player for the non-lytic release. Recently, more researches reveal the role of host pathways are subverted to build viral ROs. However, the exact relationship between autophagosomes and virus-induced membrane alteration is still being elucidated. Where picornavirus-induced ROs origin from? The contribution of apoptosis and autophagy to the different stages in the life cycle of picornavirus remains unclear. Currently, oncolytic virotherapy is regarded as a promising treatment approach, which exploited viral specific tissue tropisms to infect and kill tumor cells. The treatment of picornavirus has been developed to have greater tumor specificity in clinical trials. In this regard, the increased insights into picornavirus-host interactions can provide new opportunities to prevent viral infection and to develop oncolytic virotherapy.
Author Contributions
DS conceived, designed, and wrote the manuscript. XW, MW, SM, AC, and XY revised the manuscript. RJ, SC, QY, YWa, DZ, ML, XZ, SZ, YWu, ZX, ZC, LZhu, QL, YL, YY, LZha, and XC helped with the manuscript. All authors read and approved the final manuscript for publication.
Funding
This work was supported by grants from the National Natural Science Foundation of China (31472223), China Agricultural Research System (CARS-42-17), and Sichuan Veterinary Medicine and Drug Innovation Group of China Agricultural Research System (CARS-SVDIP).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank many staff members who have helped with this manuscript. We apologize to the authors of articles reporting relevant research that we were not able to cite in this manuscript due to space limitations. In addition, we thank AJE editing company for editing this manuscript.
Abbreviations
ACBD3, acyl-coenzyme A binding domain containing 3; AIF, apoptosis-inducing factor; Aiv, Aichi virus; ATF4, activating transcription factor 4; AWOF, autophagosome-mediated exit without lysis; Bcl-2, B cell lymphoma 2; CV, coxsackievirus; dsRNA, double-stranded RNA; EMCV, encephalomyocarditis virus; EMV, extracellular microvesicle; ERAV, equine rhinitis A virus; EV1, echovirus 1; EV11, echovirus11; EV71, enterovirus 71; FADD, FAS-associated death domain; FIP2000, the focal adhesion kinase family-interacting protein of 200 kD; FMDV, foot-and-mouth disease virus; GSDMD, gasdermin D; HAV, hepatitis A virus; HSPB1, heat shock protein family B member 1; IFN, interferon; IPS, interferon promoter stimulator; IP3R, 1,4,5-triphosphate receptor; IRF, interferon regulatory factor; ISG, IFN-stimulated gene; MAVS, mitochondrial antiviral signaling proteins; MDA5, melanoma differentiation associated gene-5; MMP, mitochondrial membrane permeability; MOMP, mitochondrial outer membrane permeability; mTOR, mammalian target of rapamycin; NPC, nuclear pore complex; ORF, open reading frame; OSBP, oxysterol binding protein; PCD, programed cell death; PE, phosphatidylethanolamine; PI4P, phosphatidylinositol 4-phosphate; PI4KA, phosphatidylinositol 4-kinase III alpha; PI4KB, phosphatidylinositol 4-kinase III beta, PRR, pattern recognition receptor; PTB, polypyrimidine tract-binding protein; PtdIn3K, phosphatidylinositol 3-kinase; PS, phosphatidylserine; PV, poliovirus; RIG, retinoic acid inducible gene; RV, rhinovirus; SFPQ, splicing factor proline-and glutamine-rich; SG, stress granule; SNAP29, synaptosomal-associated protein 29; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; STX17, syntaxin 17; TGN, trans-Golgi network; TLR-3, Toll-like receptor 3; TNFR, tumor necrosis factor receptor; TOR, target of rapamycin; TRADD, TNFR-associated death domain; ULK1, uncoordinated 51-like autophagy activating kinase; UPS, ubiquitin-proteasome system; UTR, untranslated region; VDAC, voltage-dependent anion channel.
References
Abernathy, E., Mateo, R., Majzoub, K., van Buuren, N., Bird, S. W., Carette, J. E., et al. (2019). Differential and convergent utilization of autophagy components by positive-strand RNA viruses. PLoS Biol. 17:e2006926. doi: 10.1371/journal.pbio.2006926
Agirre, A., Barco, A., Carrasco, L., and Nieva, J. L. (2002). Viroporin-mediated membrane permeabilization. Pore formation by nonstructural poliovirus 2B protein. J. Biol. Chem. 277, 40434–40441. doi: 10.1074/jbc.M205393200
Alexopoulou, L., Holt, A. C., Medzhitov, R., and Flavell, R. A. (2001). Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738. doi: 10.1038/35099560
Ao, D., Guo, H. C., Sun, S. Q., Sun, D. H., Fung, T. S., Wei, Y. Q., et al. (2015). Viroporin activity of the foot-and-mouth disease virus non-structural 2B protein. PLoS One 10:e0125828. doi: 10.1371/journal.pone.0125828
Arita, M. J. M. (2014). Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 58, 239–256. doi: 10.1111/1348-0421.12144
Back, S. H., Kim, Y. K., Kim, W. J., Cho, S., Oh, H. R., Kim, J. E., et al. (2002). Translation of polioviral mRNA is inhibited by cleavage of polypyrimidine tract-binding proteins executed by polioviral 3C(pro). J. Virol. 76, 2529–2542. doi: 10.1128/jvi.76.5.2529-2542.2002
Banerjee, S., Aponte-Diaz, D., Yeager, C., Sharma, S. D., Ning, G., Oh, H. S., et al. (2018). Hijacking of multiple phospholipid biosynthetic pathways and induction of membrane biogenesis by a picornaviral 3CD protein. PLoS Pathog. 14:e1007086. doi: 10.1371/journal.ppat.1007086
Barral, P. M., Morrison, J. M., Drahos, J., Gupta, P., Sarkar, D., Fisher, P. B., et al. (2007). MDA-5 is cleaved in poliovirus-infected cells. J. Virol. 81, 3677–3684. doi: 10.1128/jvi.01360-06
Barral, P. M., Sarkar, D., Fisher, P. B., and Racaniello, V. R. J. V. (2009). RIG-I is cleaved during picornavirus infection. Virology 391, 171–176. doi: 10.1016/j.virol.2009.06.045
B’chir, W., Maurin, A.-C., Carraro, V., Averous, J., Jousse, C., Muranishi, Y., et al. (2013). The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699. doi: 10.1093/nar/gkt563
Belov, G. A., Romanova, L. I., Tolskaya, E. A., Kolesnikova, M. S., Lazebnik, Y. A., and Agol, V. I. (2003). The major apoptotic pathway activated and suppressed by poliovirus. J. Virol. 77, 45–56. doi: 10.1128/jvi.77.1.45-56.2003
Bird, S. W., Maynard, N. D., Covert, M. W., and Kirkegaard, K. (2014). Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. U.S.A. 111, 13081–13086. doi: 10.1073/pnas.1401437111
Boya, P., Gonzalez-Polo, R. A., Casares, N., Perfettini, J. L., Dessen, P., Larochette, N., et al. (2005). Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 25, 1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005
Brisac, C., Téoulé, F., Autret, A., Pelletier, I., Colbère-Garapin, F., Brenner, C., et al. (2010). Calcium flux between the endoplasmic reticulum and mitochondrion contributes to poliovirus-induced apoptosis. J. Virol. 84, 12226–12235. doi: 10.1128/JVI.00994-10
Burgon, T. B., Jenkins, J. A., Deitz, S. B., Spagnolo, J. F., and Kirkegaard, K. (2009). Bypass suppression of small-plaque phenotypes by a mutation in poliovirus 2A that enhances apoptosis. J. Virol. 83, 10129–10139. doi: 10.1128/JVI.00642-09
Campanella, M., de Jong, A. S., Lanke, K. W., Melchers, W. J., Willems, P. H., Pinton, P., et al. (2004). The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem. 279, 18440–18450. doi: 10.1074/jbc.m309494200
Castello, A., Izquierdo, J. M., Welnowska, E., and Carrasco, L. (2009). RNA nuclear export is blocked by poliovirus 2A protease and is concomitant with nucleoporin cleavage. J. Cell Sci. 122(Pt 20), 3799–3809. doi: 10.1242/jcs.055988
Chang, S.-C., Lin, J.-Y., Lo, L. Y.-C., Li, M.-L., and Shih, S.-R. (2004). Diverse apoptotic pathways in enterovirus 71-infected cells. J. Neurovirol. 10, 338–349. doi: 10.1080/13550280490521032
Chang, Y.-Y., and Neufeld, T. P. (2009). An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 20, 2004–2014. doi: 10.1091/mbc.E08-12-1250
Chau, D. H., Yuan, J., Zhang, H., Cheung, P., Lim, T., Liu, Z., et al. (2007). Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 12, 513–524. doi: 10.1007/s10495-006-0013-0
Chen, L. L., Kung, Y. A., Weng, K. F., Lin, J. Y., Horng, J. T., and Shih, S. R. (2013). Enterovirus 71 infection cleaves a negative regulator for viral internal ribosomal entry site-driven translation. J. Virol. 87, 3828–3838. doi: 10.1128/JVI.02278-12
Chen, Y. H., Du, W., Hagemeijer, M. C., Takvorian, P. M., Pau, C., Cali, A., et al. (2015). Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 160, 619–630. doi: 10.1016/j.cell.2015.01.032
Cong, H., Du, N., Yang, Y., Song, L., Zhang, W., and Tien, P. (2016). Enterovirus 71 2B induces cell apoptosis by directly inducing the conformational activation of the proapoptotic protein Bax. J. Virol. 90, 9862–9877. doi: 10.1128/JVI.01499-16
Corona, A. K., Saulsbery, H. M., Velazquez, A. F. C., and Jackson, W. T. (2018). Enteroviruses remodel autophagic trafficking through regulation of host SNARE proteins to promote virus replication and cell exit. Cell Rep. 22, 3304–3314. doi: 10.1016/j.celrep.2018.03.003
Coyle, J. H., Guzik, B. W., Bor, Y.-C., Jin, L., Eisner-Smerage, L., Taylor, S. J., et al. (2003). Sam68 enhances the cytoplasmic utilization of intron-containing RNA and is functionally regulated by the nuclear kinase Sik/BRK. Mol. Cell. Biol. 23, 92–103. doi: 10.1128/mcb.23.1.92-103.2003
Croft, S. N., Walker, E. J., and Ghildyal, R. (2018). Human Rhinovirus 3C protease cleaves RIPK1, concurrent with caspase 8 activation. Sci. Rep. 8:1569. doi: 10.1038/s41598-018-19839-4
de Bouteiller, O., Merck, E., Hasan, U. A., Hubac, S., Benguigui, B., Trinchieri, G., et al. (2005). Recognition of double-stranded RNA by human toll-like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J. Biol. Chem. 280, 38133–38145. doi: 10.1074/jbc.M507163200
de Jong, A. S., de Mattia, F., Van Dommelen, M. M., Lanke, K., Melchers, W. J., Willems, P. H., et al. (2008). Functional analysis of picornavirus 2B proteins: effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 82, 3782–3790. doi: 10.1128/JVI.02076-07
de Jong, A. S., Melchers, W. J., Glaudemans, D. H., Willems, P. H., and van Kuppeveld, F. J. (2004). Mutational analysis of different regions in the coxsackievirus 2B protein: requirements for homo-multimerization, membrane permeabilization, subcellular localization, and virus replication. J. Biol. Chem. 279, 19924–19935. doi: 10.1074/jbc.M314094200
Deniaud, A., Maillier, E., Poncet, D., Kroemer, G., Lemaire, C., and Brenner, C. J. O. (2008). Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27, 285–299. doi: 10.1038/sj.onc.1210638
DeTulleo, L., and Kirchhausen, T. (1998). The clathrin endocytic pathway in viral infection. EMBO J. 17, 4585–4593. doi: 10.1093/emboj/17.16.4585
Dorobantu, C. M., Albulescu, L., Harak, C., Feng, Q., van Kampen, M., Strating, J. R., et al. (2015). Modulation of the host lipid landscape to promote RNA virus replication: the picornavirus encephalomyocarditis virus converges on the pathway used by hepatitis C virus. PLoS Pathog. 11:e1005185. doi: 10.1371/journal.ppat.1005185
Drahos, J., and Racaniello, V. R. (2009). Cleavage of IPS-1 in cells infected with human rhinovirus. J. Virol. 83, 11581–11587. doi: 10.1128/JVI.01490-09
Er, E., Oliver, L., Cartron, P.-F., Juin, P., Manon, S., and Vallette, F. M. (2006). Mitochondria as the target of the pro-apoptotic protein Bax. Biochim. Biophys. Acta 1757, 1301–1311. doi: 10.1016/j.bbabio.2006.05.032
Estornes, Y., Toscano, F., Virard, F., Jacquemin, G., Pierrot, A., Vanbervliet, B., et al. (2012). dsRNA induces apoptosis through an atypical death complex associating TLR3 to caspase-8. Cell Death Differ. 19, 1482–1494. doi: 10.1038/cdd.2012.22
Feng, Q., Langereis, M. A., Lork, M., Nguyen, M., Hato, S. V., Lanke, K., et al. (2014). Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 88, 3369–3378. doi: 10.1128/JVI.02712-13
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi: 10.1038/cr.2013.168
Feng, Z., Hensley, L., McKnight, K. L., Hu, F., Madden, V., Ping, L., et al. (2013). A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496, 367–371. doi: 10.1038/nature12029
Feng, Z., Li, Y., McKnight, K. L., Hensley, L., Lanford, R. E., Walker, C. M., et al. (2015). Human pDCs preferentially sense enveloped hepatitis A virions. J. Clin. Invest. 125, 169–176. doi: 10.1172/JCI77527
Flather, D., Nguyen, J. H. C., Semler, B. L., and Gershon, P. D. (2018). Exploitation of nuclear functions by human rhinovirus, a cytoplasmic RNA virus. PLoS Pathog. 14:e1007277. doi: 10.1371/journal.ppat.1007277
Fung, T. S., Torres, J., and Liu, D. X. (2015). The emerging roles of viroporins in ER stress response and autophagy induction during virus infection. Viruses 7, 2834–2857. doi: 10.3390/v7062749
Galan, A., Lozano, G., Pineiro, D., and Martinez-Salas, E. (2017). G3BP1 interacts directly with the FMDV IRES and negatively regulates translation. FEBS J. 284, 3202–3217. doi: 10.1111/febs.14184
Galluzzi, L., Vitale, I., Abrams, J. M., Alnemri, E. S., Baehrecke, E. H., Blagosklonny, M. V., et al. (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120. doi: 10.1038/cdd.2011.96
Gatica, D., Lahiri, V., and Klionsky, D. J. (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242. doi: 10.1038/s41556-018-0037-z
Gladue, D. P., Largo, E., de la Arada, I., Aguilella, V. M., Alcaraz, A., Arrondo, J. L. R., et al. (2018). Molecular characterization of the viroporin function of foot-and-mouth disease virus nonstructural protein 2B. J. Virol. 92:e01360-18. doi: 10.1128/JVI.01360-18
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Han, X., and Cong, H. (2017). Enterovirus 71 induces apoptosis by directly modulating the conformational activation of pro-apoptotic protein Bax. J. Gen. Virol. 98, 422–434. doi: 10.1099/jgv.0.000705
Hanson, P. J., Ye, X., Qiu, Y., Zhang, H. M., Hemida, M. G., Wang, F., et al. (2016). Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes. Cell Death Differ. 23, 828–840. doi: 10.1038/cdd.2015.145
Hsu, N. Y., Ilnytska, O., Belov, G., Santiana, M., Chen, Y. H., Takvorian, P. M., et al. (2010). Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141, 799–811. doi: 10.1016/j.cell.2010.03.050
Huang, S. C., Chang, C. L., Wang, P. S., Tsai, Y., and Liu, H. S. (2009). Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 81, 1241–1252. doi: 10.1002/jmv.21502
Irurzun, A., Arroyo, J., Alvarez, A., and Carrasco, L. (1995). Enhanced intracellular calcium concentration during poliovirus infection. J. Virol. 69, 5142–5146.
Ishikawa-Sasaki, K., Sasaki, J., and Taniguchi, K. (2014). A complex comprising phosphatidylinositol 4-kinase IIIβ, ACBD3, and Aichi virus proteins enhances phosphatidylinositol 4-phosphate synthesis and is critical for formation of the viral replication complex. J. Virol. 88, 6586–6598. doi: 10.1128/jvi.00208-14
Itakura, E., Kishi-Itakura, C., and Mizushima, N. (2012). The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269. doi: 10.1016/j.cell.2012.11.001
Itakura, E., and Mizushima, N. (2013). Syntaxin 17: the autophagosomal SNARE. Autophagy 9, 917–919. doi: 10.4161/auto.24109
Jackson, W. T., Giddings, T. H. Jr., Taylor, M. P., Mulinyawe, S., Rabinovitch, M., Kopito, R. R., et al. (2005). Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3:e156. doi: 10.1371/journal.pbio.0030156
Jin, M., Liu, X., and Klionsky, D. J. (2013). SnapShot: selective autophagy. Cell 152, 368–368.e2. doi: 10.1016/j.cell.2013.01.004
Johansen, T., and Lamark, T. (2011). Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296. doi: 10.4161/auto.7.3.14487
Jung, C. H., Jun, C. B., Ro, S. H., Kim, Y. M., Otto, N. M., Cao, J., et al. (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003. doi: 10.1091/mbc.E08-12-1249
Kabeya, Y., Kamada, Y., Baba, M., Takikawa, H., Sasaki, M., and Ohsumi, Y. (2005). Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 16, 2544–2553. doi: 10.1091/mbc.e04-08-0669
Kabeya, Y., Mizushima, N., Yamamoto, A., Oshitani-Okamoto, S., Ohsumi, Y., and Yoshimori, T. (2004). LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117(Pt 13), 2805–2812. doi: 10.1242/jcs.01131
Kaiser, W. J., and Offermann, M. K. (2005). Apoptosis induced by the toll-like receptor adaptor TRIF is dependent on its receptor interacting protein homotypic interaction motif. J. Immunol. 174, 4942–4952. doi: 10.4049/jimmunol.174.8.4942
Kamada, Y., Funakoshi, T., Shintani, T., Nagano, K., Ohsumi, M., and Ohsumi, Y. (2000). Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 150, 1507–1513. doi: 10.1083/jcb.150.6.1507
Kanda, T., Gauss-Muller, V., Cordes, S., Tamura, R., Okitsu, K., Shuang, W., et al. (2010). Hepatitis A virus (HAV) proteinase 3C inhibits HAV IRES-dependent translation and cleaves the polypyrimidine tract-binding protein. J. Viral Hepat. 17, 618–623. doi: 10.1111/j.1365-2893.2009.01221.x
Kawai, T., Takahashi, K., Sato, S., Coban, C., Kumar, H., Kato, H., et al. (2005). IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988. doi: 10.1038/ni1243
Kedersha, N. L., Gupta, M., Li, W., Miller, I., and Anderson, P. (1999). RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 147, 1431–1442. doi: 10.1083/jcb.147.7.1431
Kemball, C. C., Alirezaei, M., Flynn, C. T., Wood, M. R., Harkins, S., Kiosses, W. B., et al. (2010). Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 84, 12110–12124. doi: 10.1128/JVI.01417-10
Kihara, A., Kabeya, Y., Ohsumi, Y., and Yoshimori, T. (2001). Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2, 330–335. doi: 10.1093/embo-reports/kve061
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
King, H. A., Cobbold, L. C., Pichon, X., Poyry, T., Wilson, L. A., Booden, H., et al. (2014). Remodelling of a polypyrimidine tract-binding protein complex during apoptosis activates cellular IRESs. Cell Death Differ. 21, 161–171. doi: 10.1038/cdd.2013.135
Kirchweger, R., Ziegler, E., Lamphear, B. J., Waters, D., Liebig, H. D., Sommergruber, W., et al. (1994). Foot-and-mouth disease virus leader proteinase: purification of the Lb form and determination of its cleavage site on eIF-4 gamma. J. Virol. 68, 5677–5684.
Klein, K. A., and Jackson, W. T. (2011). Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J. Virol. 85, 9651–9654. doi: 10.1128/JVI.00316-11
Klionsky, D. J. (2007). Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937. doi: 10.1038/nrm2245
Klionsky, D. J., Baehrecke, E. H., Brumell, J. H., Chu, C. T., Codogno, P., Cuervo, A. M., et al. (2011). A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 7, 1273–1294. doi: 10.4161/auto.7.11.17661
Kuma, A., Hatano, M., Matsui, M., Yamamoto, A., Nakaya, H., Yoshimori, T., et al. (2004). The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036. doi: 10.1038/nature03029
Kuo, R.-L., Kung, S.-H., Hsu, Y.-Y., and Liu, W.-T. (2002). Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J. Gen. Virol. 83(Pt 6), 1367–1376. doi: 10.1099/0022-1317-83-6-1367
Langereis, M. A., Feng, Q., and van Kuppeveld, F. J. (2013). MDA5 localizes to stress granules, but this localization is not required for the induction of type I interferon. J. Virol. 87, 6314–6325. doi: 10.1128/JVI.03213-12
Lawrence, P., Schafer, E. A., and Rieder, E. J. V. (2012). The nuclear protein Sam68 is cleaved by the FMDV 3C protease redistributing Sam68 to the cytoplasm during FMDV infection of host cells. Virology 425, 40–52. doi: 10.1016/j.virol.2011.12.019
Lei, X., Liu, X., Ma, Y., Sun, Z., Yang, Y., Jin, Q., et al. (2010). The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 84, 8051–8061. doi: 10.1128/JVI.02491-09
Lei, X., Sun, Z., Liu, X., Jin, Q., He, B., and Wang, J. (2011). Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 85, 8811–8818. doi: 10.1128/JVI.00447-11
Lei, X., Xiao, X., Xue, Q., Jin, Q., He, B., and Wang, J. (2013). Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 87, 1690–1698. doi: 10.1128/JVI.01855-12
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27–42. doi: 10.1016/j.cell.2007.12.018
Li, J., Liu, Y., Kim, B. O., and He, J. J. (2002). Direct participation of Sam68, the 68-kilodalton Src-associated protein in mitosis, in the CRM1-mediated Rev nuclear export pathway. J. Virol. 76, 8374–8382. doi: 10.1128/jvi.76.16.8374-8382.2002
Li, J., Yao, Y., Chen, Y., Xu, X., Lin, Y., Yang, Z., et al. (2017). Enterovirus 71 3C promotes apoptosis through cleavage of PinX1, a telomere binding protein. J. Virol. 91:e02016-16. doi: 10.1128/JVI.02016-16
Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., et al. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676. doi: 10.1038/45257
Liang, X. H., Kleeman, L. K., Jiang, H. H., Gordon, G., Goldman, J. E., Berry, G., et al. (1998). Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 72, 8586–8596.
Lilienbaum, A. (2013). Relationship between the proteasomal system and autophagy. Int. J. Biochem. Mol. Biol. 4, 1–26.
Lin, Y., Devin, A., Rodriguez, Y., and Liu, Z. G. (1999). Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 13, 2514–2526. doi: 10.1101/gad.13.19.2514
Longatti, A. (2015). The dual role of exosomes in hepatitis A and C virus transmission and viral immune activation. Viruses 7, 6707–6715. doi: 10.3390/v7122967
Lötzerich, M., Roulin, P. S., Boucke, K., Witte, R., Georgiev, O., Greber, U. F., et al. (2018). Rhinovirus 3C protease suppresses apoptosis and triggers caspase-independent cell death. Cell Death Dis. 9:272. doi: 10.1038/s41419-018-0306-6
Luo, H., Wong, J., and Wong, B. (2010). Protein degradation systems in viral myocarditis leading to dilated cardiomyopathy. Cardiovasc. Res. 85, 347–356. doi: 10.1093/cvr/cvp225
Matsuura, A., Tsukada, M., Wada, Y., and Ohsumi, Y. (1997). Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 192, 245–250. doi: 10.1016/s0378-1119(97)00084-x
Matter, N., Herrlich, P., and König, H. J. N. (2002). Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420, 691–695. doi: 10.1038/nature01153
McBride, A., Schlegel, A., and Kirkegaard, K. (1996). Human protein Sam68 relocalization and interaction with poliovirus RNA polymerase in infected cells. Proc. Natl. Acad. Sci. U.S.A. 93, 2296–2301. doi: 10.1073/pnas.93.6.2296
Mesmin, B., Bigay, J., von Filseck, J. M., Lacas-Gervais, S., Drin, G., and Antonny, B. J. C. (2013). A four-step cycle driven by PI (4) P hydrolysis directs sterol/PI (4) P exchange by the ER-Golgi tether OSBP. Cell 155, 830–843. doi: 10.1016/j.cell.2013.09.056
Mizushima, N., and Klionsky, D. J. (2007). Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19–40. doi: 10.1146/annurev.nutr.27.061406.093749
Mizushima, N., Ohsumi, Y., and Yoshimori, T. (2002). Autophagosome formation in mammalian cells. Cell Struct. Funct. 27, 421–429. doi: 10.1247/csf.27.421
Mohamud, Y., Shi, J., Qu, J., Poon, T., Xue, Y. C., Deng, H., et al. (2018). Enteroviral infection inhibits autophagic flux via disruption of the SNARE complex to enhance viral replication. Cell Rep. 22, 3292–3303. doi: 10.1016/j.celrep.2018.02.090
Mukherjee, A., Morosky, S. A., Delorme-Axford, E., Dybdahl-Sissoko, N., Oberste, M. S., Wang, T., et al. (2011). The coxsackievirus B 3Cpro protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 7:e1001311. doi: 10.1371/journal.ppat.1001311
Ng, C. S., Jogi, M., Yoo, J. S., Onomoto, K., Koike, S., Iwasaki, T., et al. (2013). Encephalomyocarditis virus disrupts stress granules, the critical platform for triggering antiviral innate immune responses. J. Virol. 87, 9511–9522. doi: 10.1128/JVI.03248-12
Nieva, J. L., Madan, V., and Carrasco, L. (2012). Viroporins: structure and biological functions. Nat. Rev. Microbiol. 10, 563–574. doi: 10.1038/nrmicro2820
O’Donnell, V., Pacheco, J. M., LaRocco, M., Burrage, T., Jackson, W., Rodriguez, L. L., et al. (2011). Foot-and-mouth disease virus utilizes an autophagic pathway during viral replication. Virology 410, 142–150. doi: 10.1016/j.virol.2010.10.042
Orvedahl, A., Alexander, D., Talloczy, Z., Sun, Q., Wei, Y., Zhang, W., et al. (2007). HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1, 23–35. doi: 10.1016/j.chom.2006.12.001
Papon, L., Oteiza, A., Imaizumi, T., Kato, H., Brocchi, E., Lawson, T. G., et al. (2009). The viral RNA recognition sensor RIG-I is degraded during encephalomyocarditis virus (EMCV) infection. Virology 393, 311–318. doi: 10.1016/j.virol.2009.08.009
Park, N., Katikaneni, P., Skern, T., and Gustin, K. E. (2008). Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 82, 1647–1655. doi: 10.1128/jvi.01670-07
Pineiro, D., Ramajo, J., Bradrick, S. S., and Martinez-Salas, E. (2012). Gemin5 proteolysis reveals a novel motif to identify L protease targets. Nucleic Acids Res. 40, 4942–4953. doi: 10.1093/nar/gks172
Qu, L., Feng, Z., Yamane, D., Liang, Y., Lanford, R. E., Li, K., et al. (2011). Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 7:e1002169. doi: 10.1371/journal.ppat.1002169
Rajput, A., Kovalenko, A., Bogdanov, K., Yang, S.-H., Kang, T.-B., Kim, J.-C., et al. (2011). RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity 34, 340–351. doi: 10.1016/j.immuni.2010.12.018
Ralston, S. H., and Layfield, R. (2012). Pathogenesis of Paget disease of bone. Calcif. Tissue Int. 91, 97–113. doi: 10.1007/s00223-012-9599-0
Ravikumar, B., Sarkar, S., Davies, J. E., Futter, M., Garcia-Arencibia, M., Green-Thompson, Z. W., et al. (2010). Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90, 1383–1435. doi: 10.1152/physrev.00030.2009
Richards, A. L., and Jackson, W. T. (2012). Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 8:e1003046. doi: 10.1371/journal.ppat.1003046
Robinson, S. M., Tsueng, G., Sin, J., Mangale, V., Rahawi, S., McIntyre, L. L., et al. (2014). Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 10:e1004045. doi: 10.1371/journal.ppat.1004045
Rodriguez Pulido, M., Sanchez-Aparicio, M. T., Martinez-Salas, E., Garcia-Sastre, A., Sobrino, F., and Saiz, M. (2018). Innate immune sensor LGP2 is cleaved by the Leader protease of foot-and-mouth disease virus. PLoS Pathog. 14:e1007135. doi: 10.1371/journal.ppat.1007135
Rodriguez Pulido, M., Serrano, P., Saiz, M., and Martinez-Salas, E. (2007). Foot-and-mouth disease virus infection induces proteolytic cleavage of PTB, eIF3a,b, and PABP RNA-binding proteins. Virology 364, 466–474. doi: 10.1016/j.virol.2007.03.013
Roulin, P. S., Lotzerich, M., Torta, F., Tanner, L. B., van Kuppeveld, F. J., Wenk, M. R., et al. (2014). Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 16, 677–690. doi: 10.1016/j.chom.2014.10.003
Roulin, P. S., Murer, L. P., and Greber, U. F. (2018). A single point mutation in the rhinovirus 2B protein reduces the requirement for phosphatidylinositol 4-kinase class III beta in viral replication. J. Virol. 92:e01462-18. doi: 10.1128/JVI.01462-18
Rubinsztein, D. C., DiFiglia, M., Heintz, N., Nixon, R. A., Qin, Z. H., Ravikumar, B., et al. (2005). Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1, 11–22. doi: 10.4161/auto.1.1.1513
Ruiz-Vela, A., Opferman, J. T., Cheng, E. H., and Korsmeyer, S. J. (2005). Proapoptotic BAX and BAK control multiple initiator caspases. EMBO Rep. 6, 379–385. doi: 10.1038/sj.embor.7400375
Schoggins, J. W. (2014). Interferon-stimulated genes: roles in viral pathogenesis. Curr. Opin. Virol. 6, 40–46. doi: 10.1016/j.coviro.2014.03.006
Shi, J., Wong, J., Piesik, P., Fung, G., Zhang, J., Jagdeo, J., et al. (2013). Cleavage of sequestosome 1/p62 by an enteroviral protease results in disrupted selective autophagy and impaired NFKB signaling. Autophagy 9, 1591–1603. doi: 10.4161/auto.26059
Shukla, A., Dey, D., Banerjee, K., Nain, A., and Banerjee, M. (2015). The C-terminal region of the non-structural protein 2B from Hepatitis A Virus demonstrates lipid-specific viroporin-like activity. Sci. Rep. 5:15884. doi: 10.1038/srep15884
Sin, J., McIntyre, L., Stotland, A., Feuer, R., and Gottlieb, R. A. (2017). Coxsackievirus B escapes the infected cell in ejected mitophagosomes. J. Virol. 91:e01347-17. doi: 10.1128/JVI.01347-17
Slater, L., Bartlett, N. W., Haas, J. J., Zhu, J., Message, S. D., Walton, R. P., et al. (2010). Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 6:e1001178. doi: 10.1371/journal.ppat.1001178
Slee, E. A., Harte, M. T., Kluck, R. M., Wolf, B. B., Casiano, C. A., Newmeyer, D. D., et al. (1999). Ordering the cytochrome c–initiated caspase cascade: hierarchical activation of caspases-2,-3,-6,-7,-8, and-10 in a caspase-9–dependent manner. J. Cell Biol. 144, 281–292. doi: 10.1083/jcb.144.2.281
Spickler, C., Lippens, J., Laberge, M. K., Desmeules, S., Bellavance, E., Garneau, M., et al. (2013). Phosphatidylinositol 4-kinase III beta is essential for replication of human rhinovirus and its inhibition causes a lethal phenotype in vivo. Antimicrob. Agents Chemother. 57, 3358–3368. doi: 10.1128/AAC.00303-13
Stolz, A., Ernst, A., and Dikic, I. (2014). Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 16, 495–501. doi: 10.1038/ncb2979
Strating, J. R., and van Kuppeveld, F. J. (2017). Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr. Opin. Cell Biol. 47, 24–33. doi: 10.1016/j.ceb.2017.02.005
Sun, P., Zhang, S., Qin, X., Chang, X., Cui, X., Li, H., et al. (2018). Foot-and-mouth disease virus capsid protein VP2 activates the cellular EIF2S1-ATF4 pathway and induces autophagy via HSPB1. Autophagy 14, 336–346. doi: 10.1080/15548627.2017.1405187
Tait, S. W., and Green, D. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632. doi: 10.1038/nrm2952
Taylor, M. P., and Kirkegaard, K. (2007). Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 81, 12543–12553. doi: 10.1128/jvi.00755-07
Taylor, S. J., Resnick, R. J., and Shalloway, D. (2004). Sam68 exerts separable effects on cell cycle progression and apoptosis. BMC Cell Biol. 5:5. doi: 10.1186/1471-2121-5-5
Tian, L., Yang, Y., Li, C., Chen, J., Li, Z., Li, X., et al. (2018). The cytotoxicity of coxsackievirus B3 is associated with a blockage of autophagic flux mediated by reduced syntaxin 17 expression. Cell Death Dis. 9:242. doi: 10.1038/s41419-018-0271-0
Tolskaya, E. A., Romanova, L. I., Kolesnikova, M. S., Ivannikova, T. A., Smirnova, E. A., Raikhlin, N. T., et al. (1995). Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 69, 1181–1189.
van der Schaar, H. M., Dorobantu, C. M., Albulescu, L., Strating, J. R., and van Kuppeveld, F. J. M. (2016). Fat (al) attraction: picornaviruses usurp lipid transfer at membrane contact sites to create replication organelles. Trends Microbiol. 24, 535–546. doi: 10.1016/j.tim.2016.02.017
van Kuppeveld, F. J., Galama, J. M., Zoll, J., van den Hurk, P. J., and Melchers, W. J. (1996). Coxsackie B3 virus protein 2B contains cationic amphipathic helix that is required for viral RNA replication. J. Virol. 70, 3876–3886.
Visser, L. J., Medina, G. N., Rabouw, H. H., de Groot, R. J., Langereis, M. A., de Los Santos, T., et al. (2019). Foot-and-mouth disease virus leader protease cleaves G3BP1 and G3BP2 and inhibits stress granule formation. J. Virol. 93:e00922-18. doi: 10.1128/JVI.00922-18
Walker, E. J., Jensen, L. M., Croft, S., and Ghildyal, R. (2015). Variation in the nuclear effects of infection by different human rhinovirus serotypes. Front. Microbiol. 6:875. doi: 10.3389/fmicb.2015.00875
Wang, B., Xi, X., Lei, X., Zhang, X., Cui, S., Wang, J., et al. (2013). Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 9:e1003231. doi: 10.1371/journal.ppat.1003231
Wang, C., Fung, G., Deng, H., Jagdeo, J., Mohamud, Y., Xue, Y. C., et al. (2019). NLRP3 deficiency exacerbates enterovirus infection in mice. FASEB J. 33, 942–952. doi: 10.1096/fj.201800301RRR
Wang, D., Fang, L., Li, K., Zhong, H., Fan, J., Ouyang, C., et al. (2012). Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 86, 9311–9322. doi: 10.1128/JVI.00722-12
Wang, D., Fang, L., Wei, D., Zhang, H., Luo, R., Chen, H., et al. (2014). Hepatitis A virus 3C protease cleaves NEMO to impair induction of beta interferon. J. Virol. 88, 10252–10258. doi: 10.1128/JVI.00869-14
Wang, H., Sun, H. Q., Zhu, X., Zhang, L., Albanesi, J., Levine, B., et al. (2015). GABARAPs regulate PI4P-dependent autophagosome:lysosome fusion. Proc. Natl. Acad. Sci. U.S.A. 112, 7015–7020. doi: 10.1073/pnas.1507263112
Weidberg, H., Shvets, E., Shpilka, T., Shimron, F., Shinder, V., and Elazar, Z. (2010). LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 29, 1792–1802. doi: 10.1038/emboj.2010.74
White, J. P., Cardenas, A. M., Marissen, W. E., and Lloyd, R. E. (2007). Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2, 295–305. doi: 10.1016/j.chom.2007.08.006
Wong, J., Zhang, J., Si, X., Gao, G., Mao, I., McManus, B. M., et al. (2008). Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82, 9143–9153. doi: 10.1128/JVI.00641-08
Wong, P. M., Puente, C., Ganley, I. G., and Jiang, X. (2013). The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9, 124–137. doi: 10.4161/auto.23323
Xiang, Z., Li, L., Lei, X., Zhou, H., Zhou, Z., He, B., et al. (2014). Enterovirus 68 3C protease cleaves TRIF to attenuate antiviral responses mediated by Toll-like receptor 3. J. Virol. 88, 6650–6659. doi: 10.1128/JVI.03138-13
Xin, L., Xiao, Z., Ma, X., He, F., Yao, H., and Liu, Z. (2014). Coxsackievirus B3 induces crosstalk between autophagy and apoptosis to benefit its release after replicating in autophagosomes through a mechanism involving caspase cleavage of autophagy-related proteins. Infect. Genet. Evol. 26, 95–102. doi: 10.1016/j.meegid.2014.05.005
Yang, X., Cheng, A., Wang, M., Jia, R., Sun, K., Pan, K., et al. (2017). Structures and corresponding functions of five types of picornaviral 2A proteins. Front. Microbiol. 8:1373. doi: 10.3389/fmicb.2017.01373
Yang, X., Khosravi-Far, R., Chang, H. Y., and Baltimore, D. (1997). Daxx, a novel Fas-binding protein that activates JNK and apoptosis. Cell 89, 1067–1076. doi: 10.1016/s0092-8674(00)80294-9
Yang, Y., Liang, Y., Qu, L., Chen, Z., Yi, M., Li, K., et al. (2007). Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U.S.A. 104, 7253–7258. doi: 10.1073/pnas.0611506104
Yoshida, H., Kong, Y.-Y., Yoshida, R., Elia, A. J., Hakem, A., Hakem, R., et al. (1998). Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94, 739–750. doi: 10.1016/s0092-8674(00)81733-x
Yu, L., Alva, A., Su, H., Dutt, P., Freundt, E., Welsh, S., et al. (2004). Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502. doi: 10.1126/science.1096645
Zell, R., Delwart, E., Gorbalenya, A., Hovi, T., King, A., Knowles, N., et al. (2017). ICTV virus taxonomy profile: picornaviridae. J. Gen. Virol. 98, 2421–2422. doi: 10.1099/jgv.0.000911
Zhang, H., Cong, H., Song, L., and Tien, P. (2014). The nuclear protein Sam68 is redistributed to the cytoplasm and is involved in PI3K/Akt activation during EV71 infection. Virus Res. 180, 1–11. doi: 10.1016/j.virusres.2013.11.020
Zhang, L., Yu, J., Park, B. H., Kinzler, K. W., and Vogelstein, B. J. S. (2000). Role of BAX in the apoptotic response to anticancer agents. Science 290, 989–992. doi: 10.1126/science.290.5493.989
Keywords: picornavirus, apoptosis, autophagy, cellular signaling, viral replication
Citation: Sun D, Wen X, Wang M, Mao S, Cheng A, Yang X, Jia R, Chen S, Yang Q, Wu Y, Zhu D, Liu M, Zhao X, Zhang S, Wang Y, Xu Z, Chen Z, Zhu L, Luo Q, Liu Y, Yu Y, Zhang L and Chen X (2019) Apoptosis and Autophagy in Picornavirus Infection. Front. Microbiol. 10:2032. doi: 10.3389/fmicb.2019.02032
Received: 11 June 2019; Accepted: 19 August 2019;
Published: 03 September 2019.
Edited by:
Fatah Kashanchi, George Mason University, United StatesReviewed by:
Richard Yuqi Zhao, University of Maryland, Baltimore, United StatesEncarna Martinez-Salas, Severo Ochoa Molecular Biology Center (CSIC-UAM), Spain
Copyright © 2019 Sun, Wen, Wang, Mao, Cheng, Yang, Jia, Chen, Yang, Wu, Zhu, Liu, Zhao, Zhang, Wang, Xu, Chen, Zhu, Luo, Liu, Yu, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anchun Cheng, Y2hlbmdhbmNodW5AdmlwLjE2My5jb20=
†These authors have contributed equally to this work