
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 18 July 2019
Sec. Fungi and Their Interactions
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.01647
This article is part of the Research Topic Invasive Fungal Infections: Diagnosis And Treatments View all 12 articles
 Minh Thuy Vi Hoang1,2
Minh Thuy Vi Hoang1,2 Laszlo Irinyi1,2,3
Laszlo Irinyi1,2,3 Sharon C. A. Chen1,3,4
Sharon C. A. Chen1,3,4 Tania C. Sorrell1,2,3the ISHAM Barcoding of Medical Fungi Working Group
Tania C. Sorrell1,2,3the ISHAM Barcoding of Medical Fungi Working Group Wieland Meyer1,2,3,5*
Wieland Meyer1,2,3,5*Invasive fungal infections, such as aspergillosis, candidiasis, and cryptococcosis, have significantly increased among immunocompromised people. To tackle these infections the first and most decisive step is the accurate identification of the causal pathogen. Routine identification of invasive fungal infections has progressed away from culture-dependent methods toward molecular techniques, including DNA barcoding, a highly efficient and widely used diagnostic technique. Fungal DNA barcoding previously relied on a single barcoding region, the internal transcribed spacer (ITS) region. However, this allowed only for 75% of all fungi to be correctly identified. As such, the translational elongation factor 1α (TEF1α) was recently introduced as the secondary barcode region to close the gap. Both loci together form the dual fungal DNA barcoding scheme. As a result, the ISHAM Barcoding Database has been expanded to include sequences for both barcoding regions to enable practical implementation of the dual barcoding scheme into clinical practice. The present study investigates the impact of the secondary barcode on the identification of clinically important fungal taxa, that have been demonstrated to cause severe invasive disease. Analysis of the barcoding regions was performed using barcoding gap analysis based on the genetic distances generated with the Kimura 2-parameter model. The secondary barcode demonstrated an improvement in identification for all taxa that were unidentifiable with the primary barcode, and when combined with the primary barcode ensured accurate identification for all taxa analyzed, making DNA barcoding an important, efficient and reliable addition to the diagnostic toolset of invasive fungal infections.
While AIDS-associated Pneumocystis jirovecii pneumonia (Pjp) and cryptococcosis have declined in developed countries due to widespread use of highly active antiretroviral treatment (Dromer et al., 2004; Morris et al., 2004; Rajasingham et al., 2017), the overall burden of invasive fungal diseases (IFDs), especially candidemia and invasive aspergillosis has increased worldwide (Patterson, 2005; Maschmeyer, 2006; Pfaller et al., 2006b; Warnock, 2007; Pappas et al., 2010; Benedict et al., 2017). IFDs alone cause about 1.6 million deaths/year (Brown et al., 2012). The rise in incidence of IFDs is largely due to an increase in at-risk populations, especially immunocompromised individuals, such as recipients of solid organs or hematopoietic stem cell transplants, and patients with underlying chronic diseases (Kontoyiannis et al., 2010; Brown et al., 2012; Armstrong-James et al., 2014; Bitar et al., 2014; Schelenz et al., 2015). It is paramount that the management of invasive mycoses must be improved through advancements in prevention, diagnosis, treatment and surveillance (Denning, 2016; Kneale et al., 2016; Cole et al., 2017).
The majority of the current fungal diagnostic techniques are inadequate for the identification of all pathogenic fungi, which is a pre-requisite for timely initiation of appropriate antifungal therapy (Schelenz et al., 2015; Irinyi et al., 2016; Cole et al., 2017). Culture-based identification techniques rely on morphological and phenotypic characteristics, are often inaccurate, and lack in most cases species-specific features. Phenotypic traits also fail to differentiate between closely related species or species complexes with near-identical morphological characteristics but distinguishable genetic traits, such as the casual agents of many mold infections, including aspergillosis and scedosporiosis (Tavanti et al., 2005). Further, not all pathogenic fungi grow under laboratory conditions. Morphology based diagnostics are also time-consuming (7–14 days), highly laborious, and heavily dependent on the level of mycological expertise of the microscopist, making them unsuitable for rapid and reliable diagnosis (Irinyi et al., 2016).
Serological tests or fungal biomarkers, such as the Aspergillus antigen test or the Cryptococcus lateral flow assay (CrAg® LFA, IMMY, Norman, OK, United States), are available, but they are designed to identify specific pathogens (Chen et al., 2014; Schelenz et al., 2015; Cadena et al., 2016).
To overcome the limitations of standard phenotypic diagnosis and identification methods, culture- and “expert-free” methods capable of identifying fungi directly from biological specimens are needed. Sequence-based identification has proven to be more accurate than conventional methods in diagnostic clinical mycology (Ciardo et al., 2006; Balajee et al., 2007). Among the applied molecular techniques, DNA barcoding is one of the most promising and efficient methods, as it enables rapid identification of species and recognition of cryptic species across all fungal genera. As such, DNA barcoding has recently been established as the gold standard identification technique for fungal species and has been proven to be more accurate than conventional identification techniques (Ciardo et al., 2006; Balajee et al., 2007). Barcodes are standardized, easily amplified, universal short DNA sequences (500–800 bp), which are divergent at the species level enabling rapid identification by comparison with a validated reference sequence collection. To ensure consistency of identification, barcodes should be unique to a single species, and stable within each species (Hebert et al., 2003). Additionally, interspecies variation must exceed the intraspecies variation, generating a “break” in the distribution of distances, which is referred to as the “barcoding gap” (Meyer and Paulay, 2005).
DNA barcoding and its associated references databases [BOLD (Hebert et al., 2003), UNITE (Kõljalg et al., 2013), RefSeq at Genbank (Schoch et al., 2014), and the “ISHAM Barcoding Database” (Meyer et al., 2018)], plays a central role in the identification landscape, as it is the basis for all future methods, either culture dependent or culture independent (Figure 1).
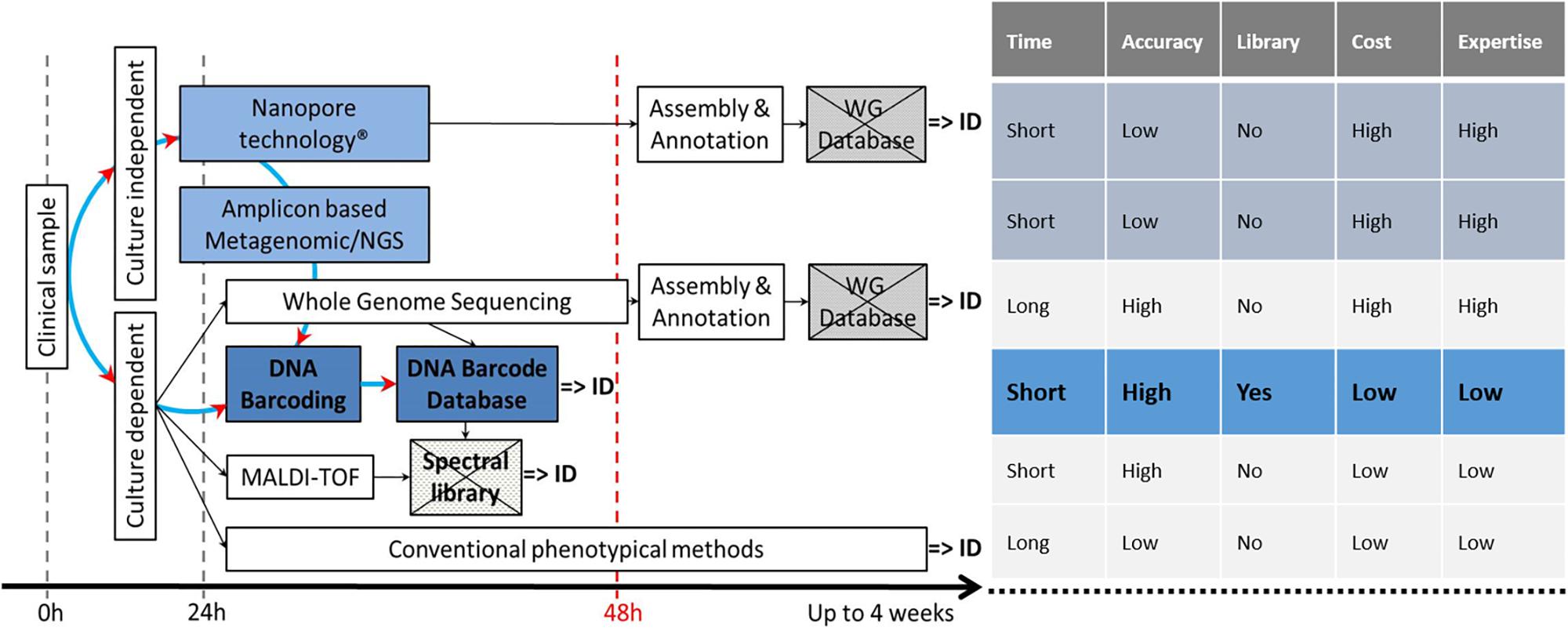
Figure 1. Current and potentially available techniques for fungal ID, showing the central role of the reference DNA barcode database and the pros and cons of all techniques. Crossed boxes indicate missing databases. DNA barcoded reference strains are fundamental to build up a spectral library.
After numerous candidate genetic loci were evaluated, with varying success rates, the internal transcribed spacer (ITS) region was established as the primary fungal DNA barcode by Schoch et al. (2012). The ITS region is composed of two non-coding and variable regions, ITS1 and ITS2, flanking the highly conserved 5.8S gene. They are located between the 18S [small subunit (SSU)] and 28S [large subunit (LSU)] genes in the nrDNA repeat (White et al., 1990). The advantage of the ITS region is, that it can be easily amplified from most fungal taxa, using universal primers, with the most commonly used ones being the ITS1, ITS2, ITS3, ITS4, and ITS5 (White et al., 1990). Fungal-specific primers were also designed to avoid cross reactivity with plant or animal DNA, such as SR6R and LR1 (Vilgalys and Hester, 1990), V9D, V9G, and LS266 (Gerrits van den Ende and de Hoog, 1999), IT2 (Beguin et al., 2012), ITS1F (Gardes and Bruns, 1993) and NL4b (O’Donnell, 1993; Figure 2 and Table 1). To provide quality controlled reference primary fungal DNA barcode sequences the International Society for Human and Animal Mycology (ISHAM) “ITS DNA barcode database” was established in 2015 (Irinyi et al., 2015). The primary fungal DNA barcode region identifies up to 75% of the estimated ∼700 pathogenic fungal species (de Hoog et al., 2014).
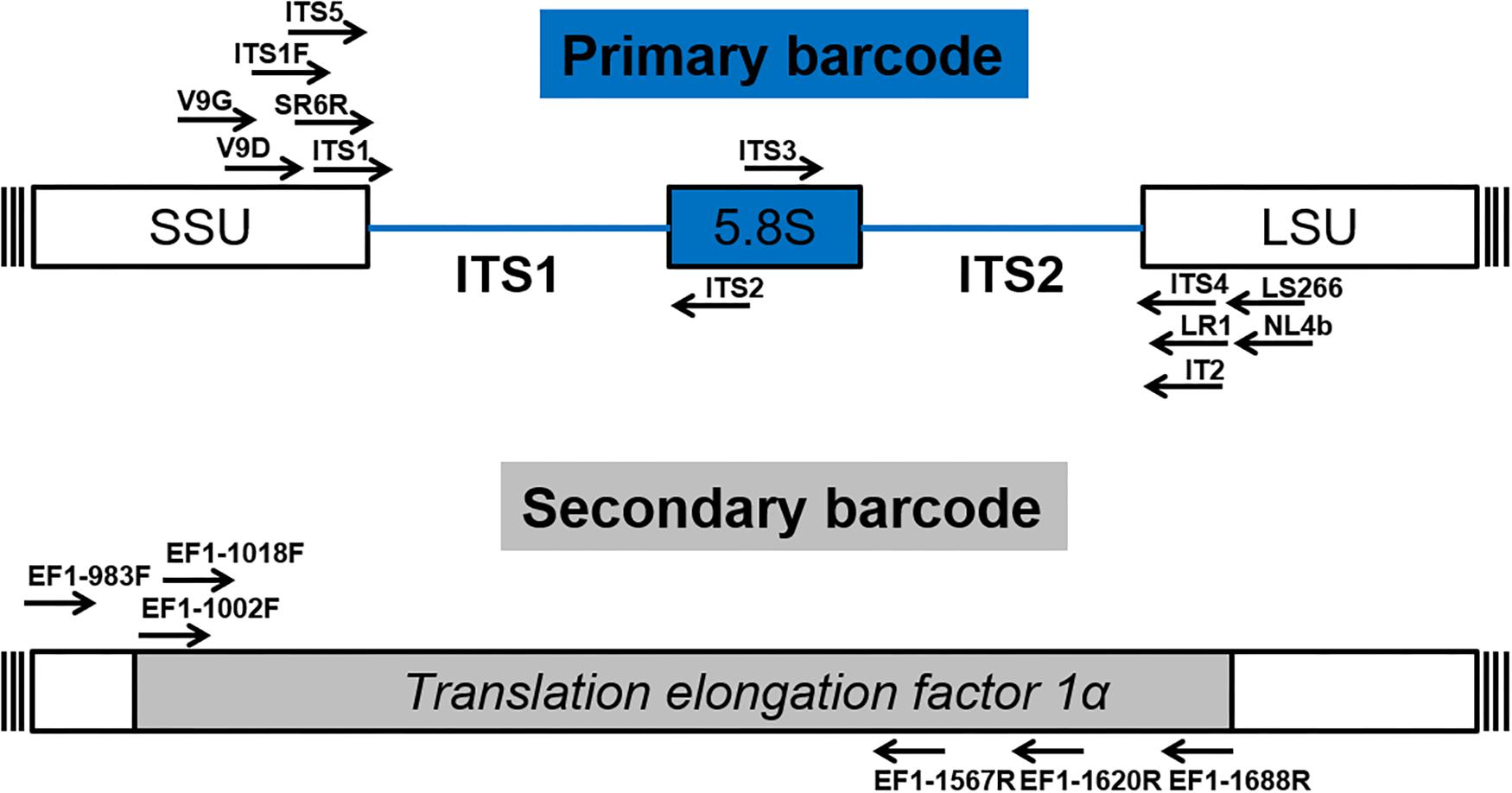
Figure 2. Schematic structure of the primary (ITS) and secondary (TEF1α) fungal DNA barcode regions indicating universal primers for their amplification.
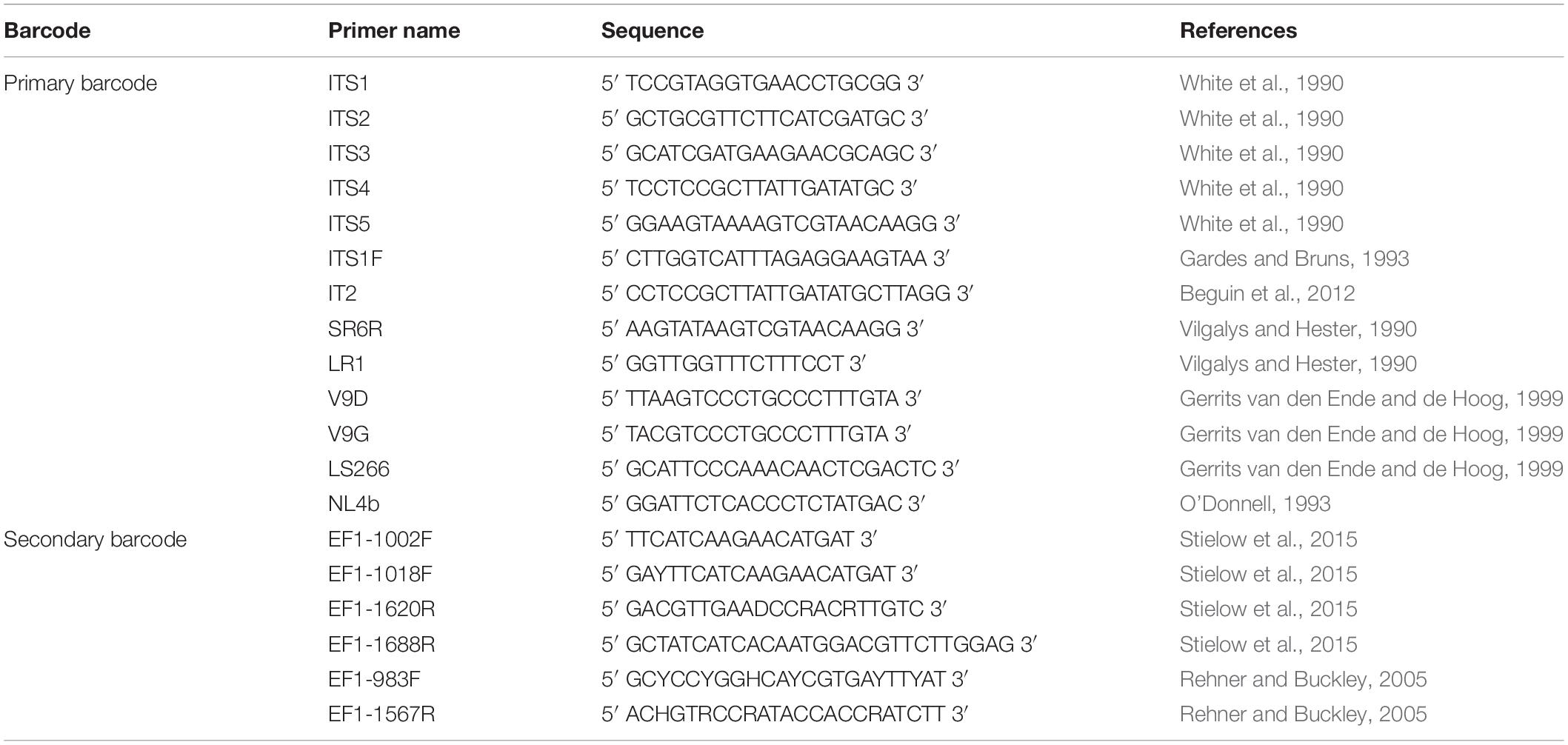
Table 1. Universal and fungal specific primers for the dual DNAbarcoding scheme.
To address the shortcomings of the ITS region and hence close this identification gap, a secondary barcode was proposed in 2015 (Stielow et al., 2015). The translational elongation factor 1α (TEF1α) was selected due to its high species discrimination across fungal taxa and the ability to design universal primers, such as EF1-1018F (Al33F)/EF1-1620R (Al33R), EF1-1002F (Al34F)/EF1-1688R (Al34R) (Stielow et al., 2015), or EF1-983F/EF1-1567R (Rehner and Buckley, 2005; Figure 2 and Table 1). To accommodate the secondary fungal DNA barcode a dedicated database that would eventually include all medically relevant fungal species was established to complement the ISHAM-ITS database, which contains only quality-controlled TEF1α sequences obtained from taxonomically verified fungal cultures (Meyer et al., 2018). Both databases have been combined within the “ISHAM Barcoding Database”,1 which was launched in November 2017 at the 7th International DNA barcoding conference at the Kruger National Park in South Africa. The combined dual barcode database provides the medical and veterinary community with quality-controlled primary and secondary fungal DNA barcodes reference sequences. This database is publicly available, and its quality-controlled sequences are shared with other major databases, including RefSeq within GenBank (Benson et al., 2014) and UNITE (Kõljalg et al., 2013).
To date, the clinical utility and accuracy of the dual DNA barcoding system for identification of pathogenic fungi has not been assessed. This study aimed to increase the number of reference secondary barcode sequences for pathogenic fungal species and to compare the accuracy and resolution of the primary and secondary fungal DNA barcodes separately or in combination, focusing on fungi causing invasive fungal infections.
To generate quality controlled TEF1α sequences 270 strains, representing 90 human/animal pathogenic fungal species, were used (see Supplementary Table 1).
DNA was isolated and purified from cultures using either previously described manual method (Ferrer et al., 2001) or the Quick-DNA Fungal/Bacterial Kit (D6007, Zymo Research) according to the manufacturer’s instructions.
The secondary fungal DNA barcoding region (TEF1α) was amplified using the primers described by Stielow et al. (2015), including: EF1-1018F (Al33F) (5′ GAYTTCATCAAGAACATGAT 3′) and EF1-1620R (Al33R) (5′ GACGTTGAADCCRACRTTGTC 3′) being used together and EF1-1002F (Al34F) (5′ TTC ATCAAGAACATGAT 3′) and EF1-1688R (Al34R) (5′ CTATCATCACAATGG ACGTTCTTGGAG 3′) being used together (Stielow et al., 2015). The primer set Al33F-Al33R was first used to amplify the TEF1α region. If amplification was unsuccessful then it was repeated using the second primer set Al34F-Al34R. Both primer pairs used the following PCR amplification protocol: 5 min initial denaturing at 94°C, followed by 40 of 50 s at 94°C, 50 s annealing at 48°C, 50 s at 72°C and 7 min final extension at 72°C (Stielow et al., 2015). Amplification success was visualized through gel electrophoresis in a 1.5% agarose gel containing ethidium bromide (EtBr) with ultra-violet illumination. Successfully amplified PCR products were sent for commercial sequencing, e.g., Macrogen Inc., South Korea in both forward and reverse directions. Bidirectional sequenced were assembled and edited using Sequencher® ver. 5.3. (Gene Codes Corporation, Ann Arbor, MI, United States). Sequences were manually checked to resolve ambiguous bases on the forward and reverse trace files considering the PHRED scores received.
The sequences for each taxon were aligned with the program CLUSTALW (Thompson et al., 1994) part of the software MEGA ver. 7 (Larkin et al., 2007). Resulting alignments were checked visually and edited when needed.
The intraspecies diversity was estimated by calculating the average nucleotide diversity (π) within species, where there were sequences available from more than three strains. The proportion of nucleotide differences in all haplotypes in the sample was derived using the software DnaSP ver. 5.10.01 (Librado and Rozas, 2009).
Individual analysis of the primary and secondary barcoding regions was performed through barcoding gap analysis. Additionally, analysis was performed on the combined barcoding regions. Taxonomic groups were selected for analysis if represented by three or more species, and if those species were represented by three or more strains. Furthermore, taxonomic groups were required to include at least one species that has been demonstrated to cause invasive fungal infections. Only strains with both primary and secondary barcodes in the database where included in the analysis. When available, more specific taxonomic groups such as clades were selected over genera. Sequences for each genus were aligned and cut to equal length using CLUSTALW as present in MEGA ver. 7 (Larkin et al., 2007; Kumar et al., 2016). Genetic distances between each strain were calculated using the Kimura 2-parameter model (K2P) and the intraspecies and interspecies genetic distances were compared (Kimura, 1980). Intra- and interspecies genetic distances were graphed against frequency and analyzed for barcoding gaps. Barcoding gaps were defined by the presence of a distinct difference between the largest intraspecies genetic distance and smallest interspecies genetic distance (Irinyi et al., 2015), i.e., there was no common x value between the intra- and interspecies groups.
The introduction of the secondary fungal DNA barcode was accessed for each genus on the basis that there would be an improvement in DNA barcoding if the secondary barcode generated a barcoding gap when the primary barcode did not, or if the overlap between the intra- and interspecies genetic distances was reduced.
The study produced 270 new quality controlled secondary fungal barcode sequences, covering 90 pathogenic fungal species (Supplementary Table 1). Overall, the PCR success rate was high within the 270 TEF1 sequences. 220 secondary barcodes were generated using the Al33F–Al33R primer pair and 50 were amplified with the Al34F–Al34R primer pair. There was no trend in amplification success in different fungal species. However, the Al34F–Al34R primer set was required to amplify all strains of Aspergillus niger, Candida albicans, Candida dubliniensis, Kluyveromyes marxianus, and Pichia kudriavzevii. There was unsuccessful amplification with both primer sets for some strains of Cladosporium spp., Rhodotorula spp., and Trichosporon spp.
All sequences were submitted to the ISHAM Barcoding Database (see footnote 1). The length of the ITS and partial TEF1α sequences in the database ranges 285–791 and 534–1002 bp, respectively.
The analysis of the nucleotide diversity (π) of 43 fungal species with more than three strains in the ISHAM barcoding database showed that the TEF1α region is less diverse than the ITS region in most species (Figure 3). The intraspecies variation of TEF1α was for most species below 1.5%, confirming the secondary barcode as a more discriminator marker. According to the selection criteria four different taxonomic groups were selected as proof of principle for the dual barcoding system. These included the two genera, Diutina and Scedosporium, and the two taxonomic clades, Lodderomyces and Pichia. The Diutina genus was selected for analysis despite one of the species, Diutina rugosa, being represented by only two strains as this genus is newly established that causes rare disease (Table 2). The species and number of strains included in these analyses are outlined in Table 2.
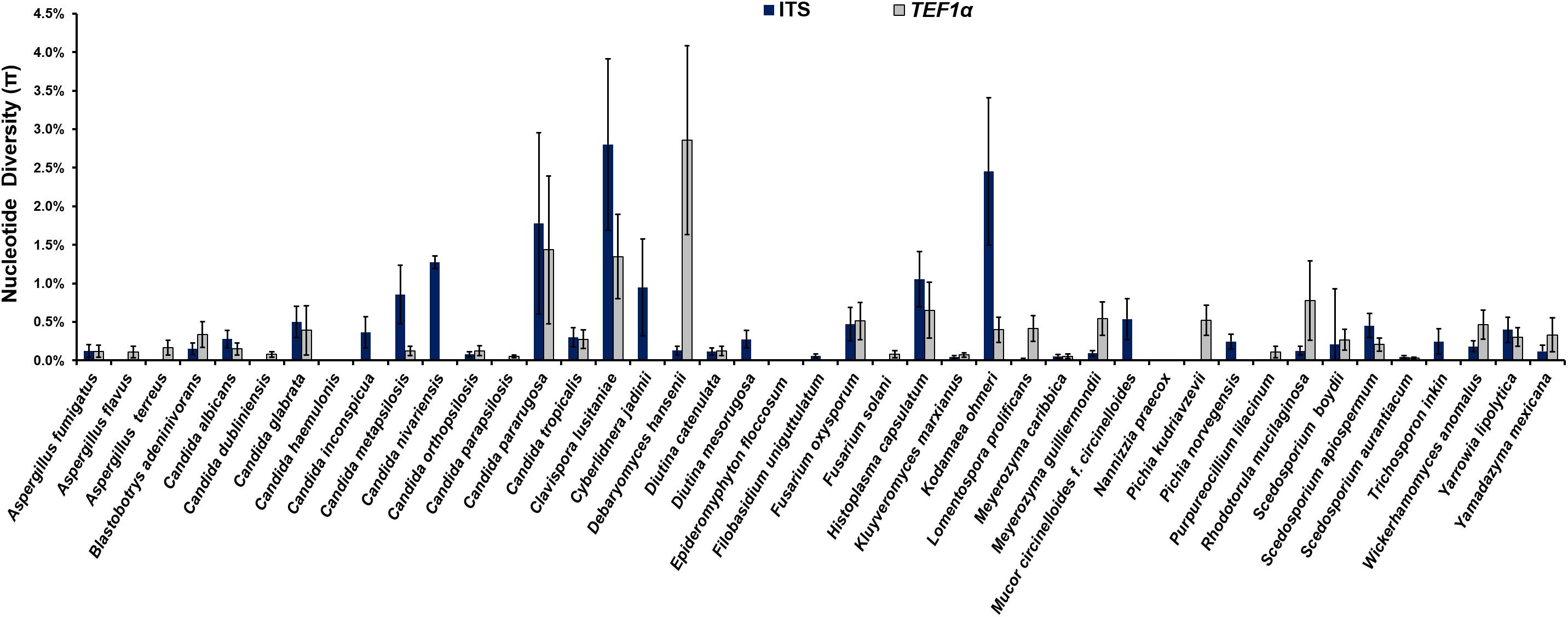
Figure 3. Intraspecies variation for species, which are represented by more than three strains, in the internal transcribed spacer (ITS) (blue bars) primary fungal DNA barcode compared with the translation elongation factor 1α (TEF1α) (gray bars) secondary fungal DNA barcode.
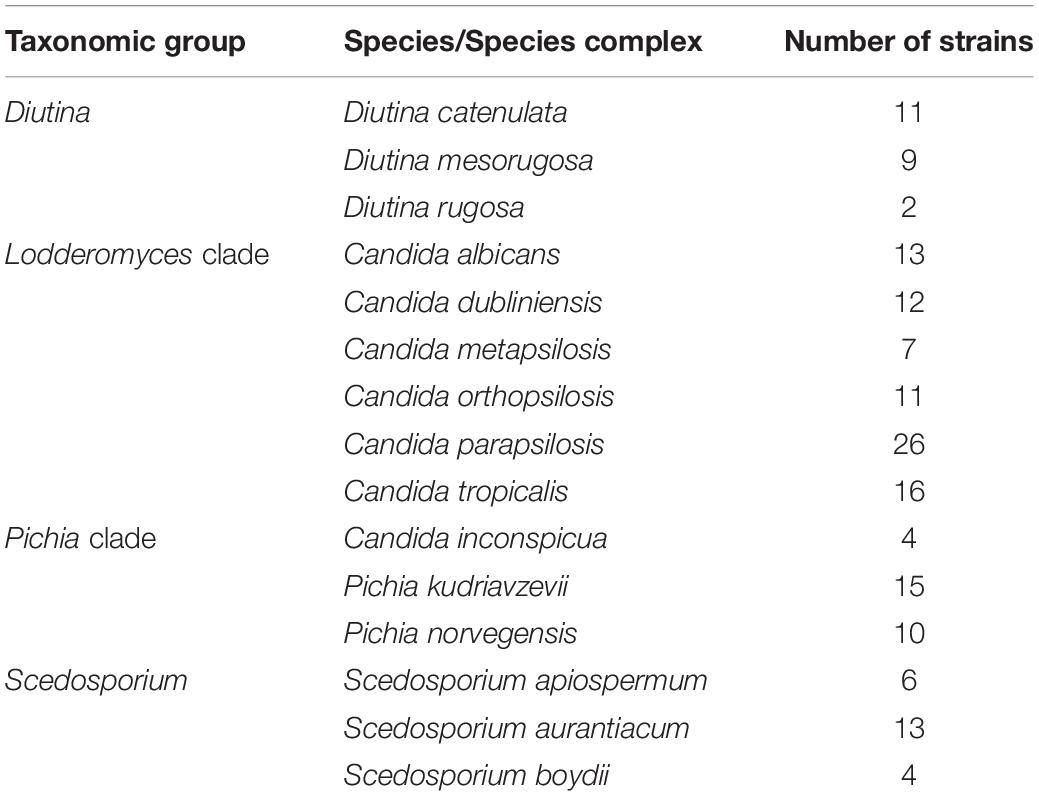
Table 2. Number of fungal strains with primary and secondarybarcodes present in the ISHAM Barcoding Databaseanalyzed in this study.
Barcoding gap analysis of the genus Diutina demonstrated no overlap between the intraspecies and interspecies genetic distances with either the primary or secondary fungal DNA barcodes (Figure 4). There was similarly no overlapping region for the barcoding gap analysis of the combination of the primary and secondary barcodes (Figure 4). As such, both fungal barcodes and the combination of the barcodes generated appropriate barcoding gaps.
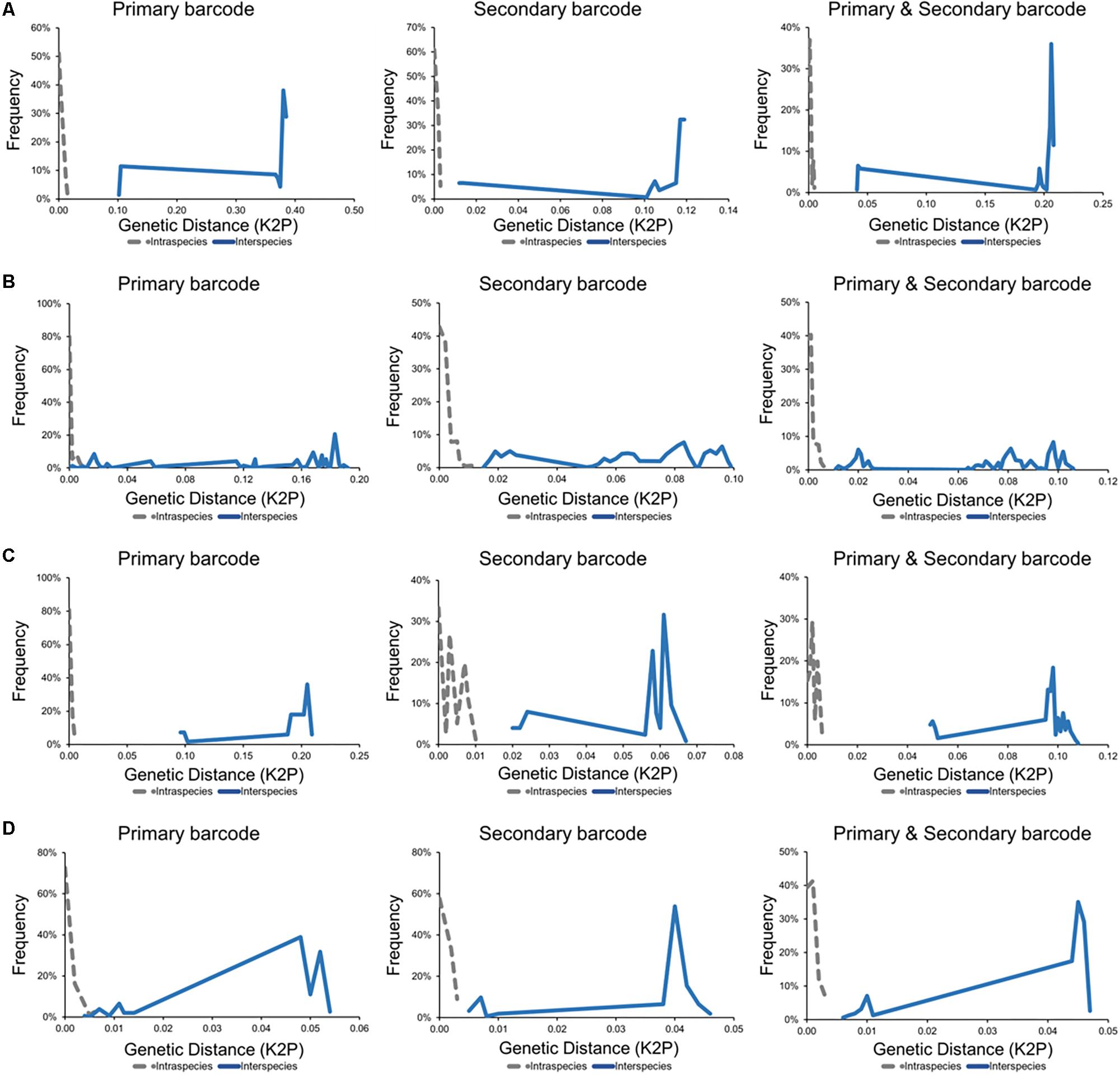
Figure 4. Barcoding gap analyses for fungal taxa including causative agents of invasive fungal disease with intraspecies (gray) and interspecies (blue) genetic distances calculated using the Kimura 2-parameter model. (A) Diutina genus (Diutina catenulata, Diutina mesorugosa, and Diutina rugosa). (B) Lodderomyces-clade (Candida albicans, Candida dublinsiensis, Candida metapsilosis, Candida orthopsilosis, Candida parapsilosis, and Candida tropicalis). (C) Pichia-clade (Candida inconspicua, Pichia kudriavzevii, and Pichia norvegensis). (D) Scedosporium genus (Scedosporium apiospermum, Scedosporium aurantiacum, and Scedosporium boydii).
For the Lodderomyces clade, the intraspecies and interspecies genetic distances, generated by the primary fungal barcode produced an overlapping region thereby indicating that there was no barcoding gap (Figure 4). The barcoding gap analysis for the secondary DNA barcode (TEF1α) demonstrated no overlap in the intraspecies and interspecies genetic distances for Lodderomyces thus showing a clear barcoding gap (Figure 4). The Lodderomyces clade analysis using the combination of both the ITS and TEF1α regions resulted in the generation of a barcoding gap being generated (Figure 4).
Barcoding gap analysis of the Pichia clade did not reveal any overlapping regions in the genetic distances for either the ITS or TEF1α regions (Figure 4). As there were no overlapping regions this demonstrated that there are barcoding gaps for both the primary and secondary barcode. These results were similar when both barcoding regions were combined as there was a barcoding gap produced (Figure 4).
Barcoding gap analysis of the primary fungal DNA barcode showed an overlapping region (Figure 4). For the secondary barcode, the barcoding gap analysis revealed that there was no overlap between the intraspecies and interspecies genetic distances and so a barcoding gap was present (Figure 4). The barcoding gap analysis of the combination of both barcodes, produced a barcoding gap as there was no overlap between the intraspecies and interspecies genetic distances (Figure 4).
Accurate and rapid routine diagnostic tools are essential to reduce the burden of fungal disease. DNA barcoding requires reliable quality-controlled reference sequences for its implementation. As such, fungal DNA Barcoding databases need to have high taxonomic coverage and reliable sequences to ensure users can accurately match sample sequences to the correct reference sequences. Additionally, the more sequences from different strains for each species are represented, the higher is the accuracy of the identification, as this reflects more realistically the existing species variation.
In this study, we generated 270 secondary barcodes, which could be amplified reliably using either the primer set Al33F–Al33R or the primer set Al34f–A134R (Figure 2), with a few exceptions where no amplifications where obtained for some strains of Cladosporium spp., Rhodotorula spp., or Trichosporon spp. In those cases, modification of the amplification conditions, such as touchdown PCR, may be appropriate.
As a result of this study the ISHAM Barcoding Database contains, as of the May 10, 2019, 4091 primary (ITS) and 868 secondary barcode (TEF1α) sequences covering 640 and 129 pathogenic fungal species, respectively. From those 2681 ITS and 613 TEF1α sequences representing 252 species cause invasive fungal diseases. There are an estimated 700 pathogenic fungal species, with the majority rarely causing infections. As such, the taxonomic coverage of ITS sequences within the “ISHAM Barcoding Database” is close to covering all pathogenic fungal species, whilst the TEF1α sequences cover less than one fifth. In addition, 59 species are currently only represented by one TEF1α sequence, which limits identification due to underrepresented intraspecies variation. As the “ISHAM Barcoding Database” has only been recently expanded to include TEF1α sequences, it is expected that as more TEF1α sequences are submitted, these issues will be resolved. Currently the “ISHAM Barcoding Database” has enabled pragmatic implementation of the dual barcode scheme for the identification of pathogenic fungi.
Since the establishment of the secondary fungal DNA barcode the improvement of the identification accuracy for pathogenic fungi has not been assessed. In this study, we compared for the first time the primary and secondary fungal DNA barcodes via barcoding gap analysis based on intraspecies and interspecies genetic distance values as calculated using the K2P model (Kimura, 1980). In addition, the combination of both barcodes was tested and compared to the individual barcodes.
The genus Diutina was established only recently. This group of fungal species was previously part of the genus Candida (Kurtzman et al., 2011; Khunnamwong et al., 2015). Diutina species are uncommon agents of disease, but are associated with nosocomial infections, with fatality rates of up to 70% (Padovan et al., 2013). Diutina catenulata and Diutina rugosa are well known causes of fungemia in immunocompromised patients and the seriously ill (Radosavljevic et al., 1999; Minces et al., 2009; Behera et al., 2010; Ha et al., 2018). D. rugosa is notable for its increasing resistance against multiple antifungal agents worldwide, thereby increasing the need for early detection and identification to enable timely initiation of effective treatment (Pfaller et al., 2006a).
An exception was made for the genus Diutina to be included into this analysis, as D. rugosa is only represented by two strains in the analysis (Table 2). The inclusion of the species was important as Diutina mesorugosa and D. rugosa have been proposed as synonyms with the variability of ITS sequences used as supporting evidence (Ming et al., 2019).
Barcoding gap analysis indicates that these species can be clearly identified as separate species. The Diutina species are representative of the 75% of pathogenic fungi that are accurately identified by the primary barcode (Irinyi et al., 2016). The secondary barcode and the combination of both barcodes equally identified these species (Figure 4), demonstrating the same level of resolution as the primary barcode (Stielow et al., 2015).
The Lodderomyces clade contains 21 Candida species, including some of the major pathogenic fungal species (Kurtzman et al., 2011). This clade contains three of the five most common pathogenic Candida species: Candida albicans, Candida dubliniensis, and Candida parapsilosis (Spampinato and Leonardi, 2013). These species cause a wide spectrum of diseases, from cutaneous infections to fatal disseminated septicemia (Tavanti et al., 2005). Candida species are predominantly commensals in the body and as such these infections largely target immunocompromised patients and are commonly the cause of nosocomial outbreaks (Brown et al., 2012).
Candida is a polyphyletic genus with a highly complex taxonomic history (Kurtzman et al., 2011). In the latest edition of The Yeasts A Taxonomic Study, 314 different Candida species were described excluding species with anamorph-teleomorph linkages (Kurtzman et al., 2011). The genus Candida was initially intended to include undifferentiated yeasts that cannot be identified by phenotype (Berkhout, 1923). As such there is little stability in the genus with some species being linked to others as anamorphs and the introduction of newly found species. With the advanced use of genomics in taxonomic studies the composition of species and the Lodderomyces clade are expected to change.
The barcoding gap analysis of the primary barcode was unable to generate a barcoding gap and so was unable to accurately identify all species of the clade (Figure 4). Upon closer inspection, the lack of a barcoding gap was due to the close relationships between Candida orthopsilosis and Candida parapsilosis, which form part of the Candida parapsilosis species complex. This species complex was previously representing a single species, Candida parapsilosis, with three subgroups that were then revised to be separate species based on various molecular techniques, including sequencing of the ITS region. The barcoding gap analysis of this study, however, did not reflect this differentiation and so ITS was unable to identify these different species. Barcoding gap analysis of the secondary barcode did produce a barcoding gap and thereby demonstrated that TEF1α could accurately identify all species of the Lodderomyces clade (Figure 4). As such, the introduction of the dual barcoding scheme resulted in an accurate identification of these pathogenic fungal species.
The Pichia clade is composed of 20 species. In our study, Candida inconspicua was also included in the analysis, as it is thought to be the anamorph for Pichia cactophila (Kurtzman and Robnett, 1998; Kurtzman et al., 2008, 2011). Pichia kudriavzevii and Pichia norvegensis are predominantly linked to nosocomial pathogens and may cause outbreaks of infections (Nagarathnamma et al., 2017). Candida inconspicua has also been found in clinical samples whilst the other species of the Pichia clade have not yet been found to be medically relevant (Kurtzman et al., 2011; Guitard et al., 2013). These species also cause invasive infections in patients with a highly compromised immune systems, and are often found under previous names in the literature (Kurtzman et al., 2011; Guitard et al., 2013; Schuster et al., 2013; Sanclemente et al., 2015; Douglass et al., 2018).
Similarly, to the genus Diutina, barcoding gap analysis of the Pichia clade revealed barcoding gaps for the primary barcode, secondary barcode and the combination of both barcodes (Figure 4), resulting in an accurate identification of all species of the Pichia clade.
Fungi belonging to the genus Scedosporium cause a wide variety of diseases from localized infections to invasive diseases (Cortez et al., 2008). Scedosporium species mainly cause infections in patients with a compromised immune systems largely due to underlying illnesses such as solid organ transplantation, cystic fibrosis, leukemia and bone marrow transplantation (Tamm et al., 2001; Allen et al., 2013; Kubisiak-Rzepczyk et al., 2013; Yu et al., 2013; Johnson et al., 2014). Scedosporium spp. can also cause infection in immunocompetent patients (Ceccarelli et al., 2012; Agatha et al., 2014; Cruysmans et al., 2015).
The taxonomy of the genus Scedosporium is complex and has changed multiple times over the last decade. Previously, the nomenclature of the genus ruled Scedosporium to be the anamorph name whilst Pseudallescheria and Petriella were the teleomorph names (Rainer and de Hoog, 2006; Cortez et al., 2008; Lu et al., 2011). The two major clinical species were Scedosporium prolificans and Scedosporium apiospermum and its teleomorph form, Pseudallescheria boydii (Cortez et al., 2008). Multilocus sequencing, morphological analysis and physiological testing later demonstrated that S. apiospermum and P. boydii are separate species and not anamorphs teleomorph pairs (Gilgado et al., 2008). In 2011, the new nomenclature rules dictating “one fugus = one name” led to the fact that Scedosporium took precedence over Pseudallescheria as the genus name (Hawksworth, 2011; Lackner et al., 2014). The composition of the genus was also changed with S. prolificans being removed from the genus and renamed to Lomentospora prolificans (Lackner et al., 2014). The separation between S. apiospermum and S. boydii was reinforced, however, it was noted that there was no clinical difference between the species, as such all species are referred to the S. apiospermum species complex (Lackner et al., 2014).
In this study, there was no barcoding gap present for the primary barcode indicating that Scedosporium species cannot be accurately identified using this region alone (Figure 4). The predominant reason for the overlap in intraspecies and interspecies genetic distances was the high similarity between S. apiospermum and S. boydii. This was reflective of the nomenclature history as these species were previously thought to be an anamorph – teleomorph pair. Barcoding gap analysis of the secondary barcoding region introduced a barcoding gap and hence allowed for the accurate identification of all Scedosporium species (Figure 4). As the separation of S. apiospermum and S. boydii was established via multilocus sequence analysis, it is reassuring that TEF1α can resolve all Scedosporium species to the same degree (Gilgado et al., 2008). With the introduction of the dual barcoding scheme, all Scedosporium species can now be accurately identified.
Meaningful implementation of the dual DNA barcoding scheme depends on demonstration of an improvement in the identification accuracy of fungal species causing infections when adding the secondary barcode, TEF1α. To demonstrate this the two fungal barcodes were analyzed separately and in combination from selected species, which had sequences from both barcodes being in the “ISHAM Barcoding Database,” using barcoding gap analysis through the generation of genetic distances by the K2P model (Kimura, 1980).
Although K2P has been used in all DNA barcoding studies since its introduction by Hebert et al. (2003), the accuracy of this model has been questioned (Kimura, 1980; Hebert et al., 2003). In the selection of ITS region as the primary fungal DNA barcode, the uncorrected p-distances were used for barcoding gap analysis whereas in the case of the secondary fungal DNA barcode (TEF1α) analysis the K2P method was applied (Schoch et al., 2012; Stielow et al., 2015). The K2P region was selected to generate barcoding gaps over uncorrected p-distances as K2P has been found to generate larger barcoding gaps for most data sets (Srivathsan and Meier, 2012). Additionally, in datasets where uncorrected p-distances were preferred, the performance of K2P was found to be similar (Srivathsan and Meier, 2012). More complicated models have been applied in some studies in an attempt to improve the accuracy of the K2P model, however, the differences were minimal, prompting us to use K2P in our study (Collins et al., 2012; Barley and Thomson, 2016).
Comparison of the number of pathogenic fungal species and strains represented in the “ISHAM Barcoding Database” highlighted the deficit in the number of TEF1α reference sequences. Therefore, the value of this study is limited as only strains with both barcodes available were used for the analysis. More species represented by more sequences would give a more exact assessment about the value and resolution power of the dual barcoding concept.
DNA barcode gab analysis showed that two of the groups (Diutina and Pichia) tested were accurately identified using the primary barcode and the secondary barcode on its own, demonstrating that both barcodes had similar resolution. For the remaining two groups (Lodderomyces and Scedosporium) the primary barcode was not able to produce barcoding gaps whilst the secondary did, demonstrating that the secondary barcode had a higher resolution power. In addition, the combination of both barcodes increased the discriminatory power enabling a more accurate identification. These results indicate, that the application of the dual barcoding system drastically improves species identification in cases where a single barcoding system is unable to do so.
We envisage that the proposed dual barcoding scheme can be applied in the routine diagnostic setting in a stepwise procedure. After a clinical specimen is obtained, the unknown fungal isolate is first assessed based on its morphologic and/or biochemical characteristics. Then those unknown fungal isolates which lack obvious morphological characteristics or result in unclear biochemical profiles should be subjected to DNA isolation and primary fungal DNA barcoding (ITS1/2 region). If the obtained sequence shows less than 98.5% identity to a given ITS reference sequence in the database, the secondary fungal DNA barcode (TEF1α) should be obtained to achieve a final species identification (Figure 5).
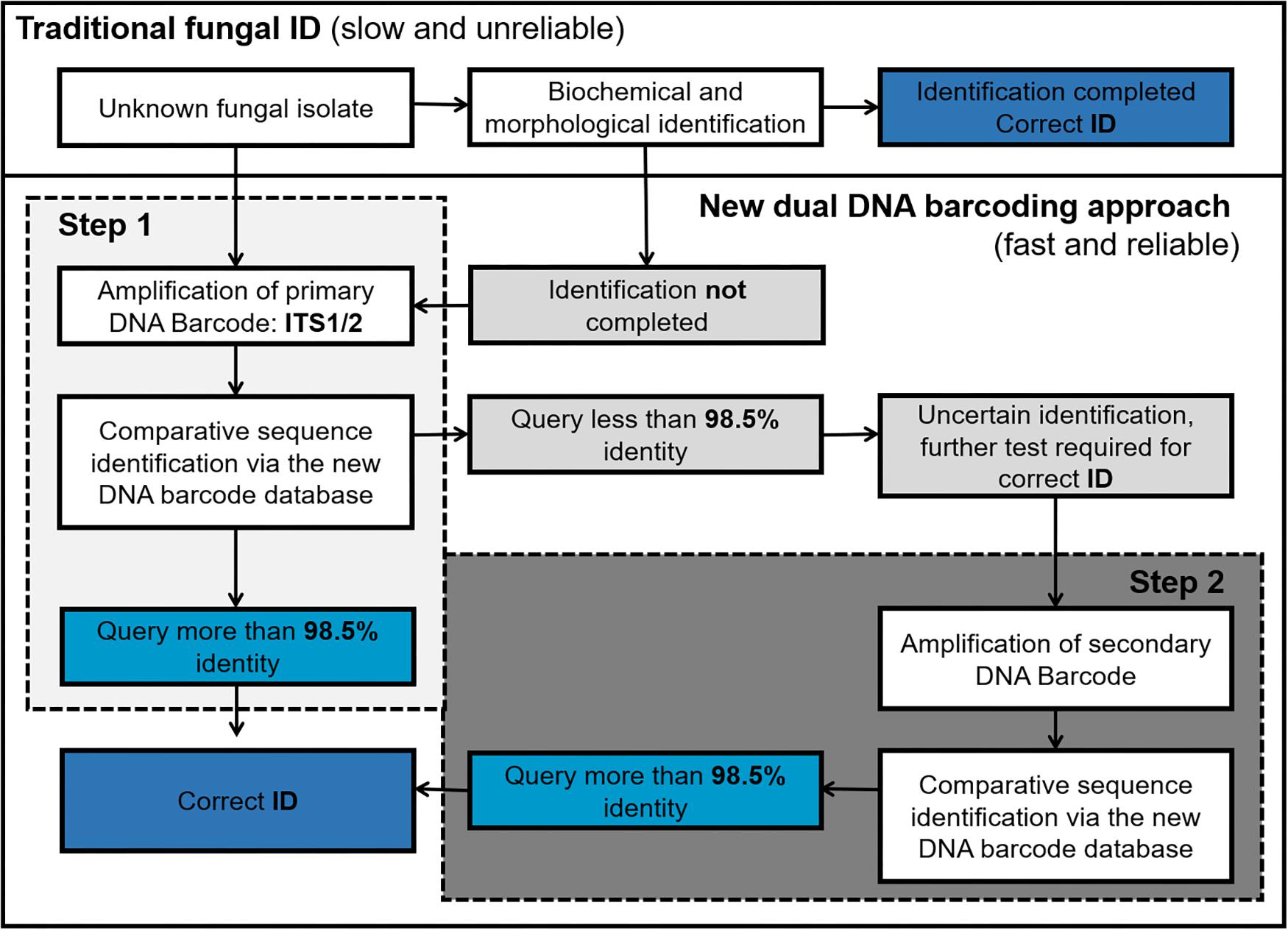
Figure 5. Workflow of DNA barcoding based fungal ID (Steps 1 and 2 can be done in parallel to speed up the process).
The dual fungal DNA barcoding scheme is an efficient diagnostic tool for the identification of agents of invasive mycoses and can be confidently implemented into routine diagnostics. Its implementation along with the expansion of the “ISHAM Barcoding Database” will lead to a paradigm shift in fungal disease diagnostics, enabling a highly accurate identification of the agents of fungal disease. The timely initiation of appropriate therapy will improve patient outcomes through the reduction in morbidity and mortality, prevent the use of unnecessary or inappropriate antifungal drugs, minimizing drug toxicity and resistance development and greatly reduce the associated health care cost.
To achieve a 100% coverage of all pathogenic fungi and reflect the intraspecies variation for both the primary and secondary fungal DNA barcode we call for the submission of quality-controlled reference sequences of the ITS region and the TEF1α for inclusion into the “ISHAM Barcode Database.” Please contact the curators of the database WM at d2llbGFuZC5tZXllckBzeWRuZXkuZWR1LmF1 and LI at bGFzemxvLmlyaW55aUBzeWRuZXkuZWR1LmF1.
Michael Arabatzis, National and Kapodistrian University of Athens, Athens, Greece; Ian Arthur, PathWest Laboratory Medicine, Nedlands, WA, Australia; Jose F. Cano-Lira, Universitat Rovira i Virgili, Reus, Spain; Gianluigi Cardinali, Università degli Studi di Perugia, Perugia, Italy; Laura Rosio Castañón, Universidad Nacional Autónoma de México, Mexico City, Mexico; Sharon Chen, Westmead Hospital, University of Sydney, Sydney, NSW, Australia; Wen Chen, Agriculture and Agri-Food Canada, Ottawa, ON, Canada; Ariya Chindamporn, Chulalongkorn University, Bangkok, Thailand; Arnaldo L. Colombo, Universidade Federal de São Paulo, São Paulo, Brazil; Marie Desnos-Ollivier, Institut Pasteur, Paris, France; Wilhelm de Beer, FABI, Pretoria, South Africa; Sybren de Hoog, Westerdijk Fungal Biodiversity Institute, Utrecht, Netherlands; Françoise Dromer, Institut Pasteur, Paris, France; Dea Garcia-Hermoso, Institut Pasteur, Paris, France; Marieka Gryzenhout, University of the Free State, Bloemfontein, South Africa; Josep Guarro, Universitat Rovira i Virgili, Reus, Spain; Catriona Halliday, University of Sydney/Westmead Hospital, Sydney, NSW, Australia; Marijke Hendrickx, BCCM/IHEM, Scientific Institute of Public Health, Brussels, Belgium; Sabine Huhndorf, The Field Museum, Chicago, IL, United States; Laszlo Irinyi, Westmead Hospital, University of Sydney, Sydney, NSW, Australia; C. Andre Levesque, Agriculture and Agri-Food Canada, Ottawa, ON, Canada; Wieland Meyer, Westmead Hospital, University of Sydney, Sydney, NSW, Australia; Maria Luiza Moretti, University of Campinas, Campinas, Brazil; Mauro de Medeiros Muniz, Instituto de Pesquisa Clínica Evandro Chagas (IPEC), Fundação Oswaldo Cruz (FIOCRUZ), Rio de Janeiro, Brazil; Analy Salles de Azevedo Melo, Universidade Federal de São Paulo, São Paulo, Brazil; Angela Satie Nishikaku, Universidade Federal de São Paulo, São Paulo, Brazil; Anne-Cécile Normand, Aix-Marseille University, Marseille, France; Célia Pais, University of Minho, Braga, Portugal; Renaud Piarroux, Aix-Marseille University, Marseille, France; Stéphane Ranque, Aix-Marseille University, Marseille, France; Barbara Robbertse, National Center for Biotechnology Information, Bethesda, MD, United States; Vincent Robert, Westerdijk Fungal Biodiversity Institute, Utrecht, Netherlands; Conrad L. Schoch, National Center for Biotechnology Information, Bethesda, MD, United States; Keith A. Seifert, Agriculture and Agri-Food Canada, Ottawa, ON, Canada; Célia Maria de Almeida Soares, Universidade Federal de Goiás, Goiânia, Brazil; Tania C. Sorrell, Westmead Hospital, University of Sydney, Sydney, NSW, Australia; John L. Spouge, National Center for Biotechnology Information, Bethesda, MD, United States; Dirk Stubbe, BCCM/IHEM, Scientific Institute of Public Health, Brussels, Belgium; Maria Lucia Taylor, Universidad Nacional Autónoma de México, Mexico City, Mexico; Conchita Toriello, Universidad Nacional Autónoma de México, Mexico City, Mexico; Aristea Velegraki, National and Kapodistrian University of Athens, Athens, Greece; Chompoonek Yurayart, Kasetsart University, Bangkok, Thailand; Rosely Maria Zancopé-Oliveira, Instituto de Pesquisa Clínica Evandro Chagas (IPEC), Fundação Oswaldo Cruz, FIOCRUZ, Rio de Janeiro, Brazil.
The primary and secondary fungal barcodes generated and analyzed for this study are available in the ISHAM Barcoding Database (http://its.mycologylab.org/).
WM, SC, and TS conceived the study. WM coordinated the study and supervised the molecular and data analysis. All members of the ISHAM working group for “Barcoding of Medical Fungi” performed the sample collections. MH and LI conducted the sample processing, all molecular studies, and data analysis. MH, LI, and WM performed the data interpretation. All authors wrote and corrected the manuscript.
This study was supported by the National Health and Medical Research Council of Australia (NH & MRC) grant (#APP1121936) to WM, SC, and TS, and Western Sydney Local Health District Research and Education Network Research Grant Scheme to WM, TS, SC, and LI. TS is a Sydney Medical School Foundation Fellow.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank all members of the ISHAM working group for “Barcoding of Medical Fungi” for the contribution of strains and barcode sequences.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01647/full#supplementary-material
Agatha, D., Krishnan, K. U., Dillirani, V. A., and Selvi, R. (2014). Invasive lung infection by Scedosporium apiospermum in an immunocompetent individual. Indian J. Pathol. Microbiol. 57, 635–637. doi: 10.4103/0377-4929.142716
Allen, P., Koka, R., Kleinberg, M. E., and Baer, M. R. (2013). Scedosporium apiospermum soft tissue infection as the initial presentation of acute myeloid leukemia: a case report. J. Clin. Oncol. 31, e98–e100. doi: 10.1200/jco.2012.43.9489
Armstrong-James, D., Meintjes, G., and Brown, G. D. (2014). A neglected epidemic: fungal infections in HIV/AIDS. Trends Microbiol. 22, 120–127. doi: 10.1016/j.tim.2014.01.001
Balajee, S. A., Sigler, L., and Brandt, M. E. (2007). DNA and the classical way: identification of medically important molds in the 21st century. Med. Mycol. 45, 475–490. doi: 10.1080/13693780701449425
Barley, A. J., and Thomson, R. C. (2016). Assessing the performance of DNA barcoding using posterior predictive simulations. Mol. Ecol. 25, 1944–1957. doi: 10.1111/mec.13590
Beguin, H., Pyck, N., Hendrickx, M., Planard, C., Stubbe, D., and Detandt, M. (2012). The taxonomic status of Trichophyton quinckeanum and T. interdigitale revisited: a multigene phylogenetic approach. Med. Mycol. 50, 871–882. doi: 10.3109/13693786.2012.684153
Behera, B., Singh, R., IXess, I., Mathur, P., Hasan, F., and Misra, M. C. (2010). Candida rugosa: a possible emerging cause of candidaemia in trauma patients. Infection 38, 387–393. doi: 10.1007/s15010-010-0044-x
Benedict, K., Richardson, M., Vallabhaneni, S., Jackson, B. R., and Chiller, T. (2017). Emerging issues, challenges, and changing epidemiology of fungal disease outbreaks. Lancet Infect. Dis. 17, e403–e411. doi: 10.1016/S1473-3099(17)30443-7
Benson, D. A., Clark, K., Karsch-Mizrachi, I., Lipman, D. J., Ostell, J., and Sayers, E. W. (2014). GenBank. Nucleic Acids Res. 42, D32–D37. doi: 10.1093/nar/gkw1070
Berkhout, C. M. (1923). De Schimmelgeslachten Monilia, Oidium, Oospora en Torula. Utrecht: Rijksuniversiteit Utrecht.
Bitar, D., Lortholary, O., Le Strat, Y., Nicolau, J., Coignard, B., Tattevin, P., et al. (2014). Population-based analysis of invasive fungal infections, France, 2001–2010. Emerg. Infect. Dis. 20, 1149–1155. doi: 10.3201/eid2007.140087
Brown, G. D., Denning, D. W., Gow, N. A., Levitz, S. M., Netea, M. G., and White, T. C. (2012). Hidden killers: human fungal infections. Sci. Transl. Med. 4:165rv113. doi: 10.1126/scitranslmed.3004404
Cadena, J., Thompson, G. R. III, and Patterson, T. F. (2016). Invasive Aspergillosis: current strategies for diagnosis and management. Infect. Dis. Clin. North Am. 30, 125–142. doi: 10.1016/j.idc.2015.10.015
Ceccarelli, L., Calisti, G., Delle Rose, D., Ricciardi, A., Maffongelli, G., Sordillo, P., et al. (2012). Dapsone hypersensitivity syndrome complicated by scedosporium apiospermum pneumonia in an immunocompetent patient. Infection 40, 459–462. doi: 10.1007/s15010-011-0225-2
Chen, S. C., Meyer, W., and Sorrell, T. C. (2014). Cryptococcus gattii infections. Clin. Microbiol. Rev. 27, 980–1024. doi: 10.1128/CMR.00126-13
Ciardo, D. E., Schar, G., Bottger, E. C., Altwegg, M., and Bosshard, P. P. (2006). Internal transcribed spacer sequencing versus biochemical profiling for identification of medically important yeasts. J. Clin. Microbiol. 44, 77–84. doi: 10.1128/JCM.44.1.77-84.2006
Cole, D. C., Govender, N. P., Chakrabarti, A., Sacarlal, J., and Denning, D. W. (2017). Improvement of fungal disease identification and management: combined health systems and public health approaches. Lancet Infect. Dis. 17, e412–e419. doi: 10.1016/S1473-3099(17)30308-0
Collins, R. A., Boykin, L. M., Cruickshank, R. H., and Armstrong, K. F. (2012). Barcoding’s next top model: an evaluation of nucleotide substitution models for specimen identification. Methods Ecol. Evol. 3, 457–465. doi: 10.1111/j.2041-210X.2011.00176.x
Cortez, K. J., Roilides, E., Quiroz-Telles, F., Meletiadis, J., Antachopoulos, C., Knudsen, T., et al. (2008). Infections caused by Scedosporium spp. Clin. Microbiol. Rev. 21, 157–197. doi: 10.1128/CMR.00039-07
Cruysmans, C., Rodriguez-Villalobos, H., Fomekong, E., Dumitriu, D., Nassogne, M. C., and Van der Linden, D. (2015). Epidural abscess caused by Scedosporium apiospermum in an immunocompetent child. Pediatr. Infect. Dis. J. 34, 1277–1278. doi: 10.1097/INF.0000000000000864
de Hoog, G. S., Guarro, J., Gené, J., and Figueras, A. A. (2014). Atlas of Clinical Fungi. Netherlands: Centraalbureau voor Schimmelcultures (CBS).
Denning, D. W. (2016). Minimizing fungal disease deaths will allow the UNAIDS target of reducing annual AIDS deaths below 500 000 by 2020 to be realized. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371:0468. doi: 10.1098/rstb.2015.0468
Douglass, A. P., Offei, B., Braun-Galleani, S., Coughlan, A. Y., Martos, R., Ortiz-Merino, R. A., et al. (2018). Population genomics shows no distinction between pathogenic Candida krusei and environmental Pichia kudriavzevii: One species, four names. PLoS Pathog. 14:e1007138. doi: 10.1371/journal.ppat.1007138
Dromer, F., Mathoulin-Pelissier, S., Fontanet, A., Ronin, O., Dupont, B., and Lortholary, O. (2004). Epidemiology of HIV-associated cryptococcosis in France (1985-2001): comparison of the pre- and post-HAART eras. Aids 18, 555–562. doi: 10.3201/eid2007.140087
Ferrer, C., Colom, F., Frases, S., Mulet, E., Abad, J., and Alio, J. (2001). Detection and identification of fungal pathogens by PCR and by ITS2 and 5.8S ribosomal DNA typing in ocular infections. J. Clin. Microbiol. 39, 2873–2879. doi: 10.1128/JCM.39.8.2873-2879.2001
Gardes, M., and Bruns, T. D. (1993). ITS primers with enhanced specificity for basidiomycetes - application to the identification of mycorrhizae and rusts. Mol. Ecol. 2, 113–118. doi: 10.1111/j.1365-294X.1993.tb00005.x
Gerrits van den Ende, A. H. G., and de Hoog, G. S. (1999). Va*riability and molecular diagnostics of the neurotropic species Cladophialophora bantiana. Stud. Mycol. 43, 151–162.
Gilgado, F., Cano, J., Gene, J., Sutton, D. A., and Guarro, J. (2008). Molecular and phenotypic data supporting distinct species statuses for Scedosporium apiospermum and Pseudallescheria boydii and the proposed new species Scedosporium dehoogii. J. Clin. Microbiol. 46, 766–771. doi: 10.1128/JCM.01122-07
Guitard, J., Angoulvant, A., Letscher-Bru, V., L’Ollivier, C., Cornet, M., Dalle, F., et al. (2013). Invasive infections due to Candida norvegensis and Candida inconspicua: report of 12 cases and review of the literature. Med. Mycol. 51, 795–799. doi: 10.3109/13693786.2013.807444
Ha, M. V., Choy, M. S., McCoy, D., Fernandez, N., and Suh, J. S. (2018). Candida catenulata candidaemia and possible endocarditis in a cirrhotic patient successfully de-escalated to oral fluconazole. J. Clin. Pharm. Ther. 43, 910–913. doi: 10.1111/jcpt.12728
Hawksworth, D. L. (2011). A new dawn for the naming of fungi: impacts of decisions made in Melbourne in July 2011 on the future publication and regulation of fungal names. IMA Fungus 2, 155–162. doi: 10.5598/imafungus.2011.02.02.06
Hebert, P. D., Cywinska, A., Ball, S. L., and deWaard, J. R. (2003). Biological identifications through DNA barcodes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 270, 313–321. doi: 10.1098/rspb.2002.2218
Irinyi, L., Lackner, M., de Hoog, G. S., and Meyer, W. (2016). DNA barcoding of fungi causing infections in humans and animals. Fungal Biol. 120, 125–136. doi: 10.1016/j.funbio.2015.04.007
Irinyi, L., Serena, C., Garcia-Hermoso, D., Arabatzis, M., Desnos-Ollivier, M., Vu, D., et al. (2015). international society of human and animal mycology (ISHAM)-ITS reference DNA barcoding database-the quality controlled standard tool for routine identification of human and animal pathogenic fungi. Med. Mycol. 53, 313–337. doi: 10.1093/mmy/myv008
Johnson, L. S., Shields, R. K., and Clancy, C. J. (2014). Epidemiology, clinical manifestations, and outcomes of Scedosporium infections among solid organ transplant recipients. Transpl Infect Dis. 16, 578–587. doi: 10.1111/tid.12244
Khunnamwong, P., Lertwattanasakul, N., Jindamorakot, S., Limtong, S., and Lachance, M. A. (2015). Description of Diutina gen. nov., Diutina siamensis, f.a. sp. nov., and reassignment of Candida catenulata, Candida mesorugosa, Candida neorugosa, Candida pseudorugosa, Candida ranongensis, Candida rugosa and Candida scorzettiae to the genus Diutina. Int. J. Syst. Evol. Microbiol. 65, 4701–4709. doi: 10.1099/ijsem.0.000634
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Kneale, M., Bartholomew, J. S., Davies, E., and Denning, D. W. (2016). Global access to antifungal therapy and its variable cost. J. Antimicrob. Chemother. 71, 3599–3606. doi: 10.1093/jac/dkw325
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F. S., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Kontoyiannis, D. P., Marr, K. A., Park, B. J., Alexander, B. D., Anaissie, E. J., Walsh, T. J., et al. (2010). Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001-2006: overview of the transplant-associated infection surveillance network (TRANSNET) database. Clin. Infect. Dis. 50, 1091–1100. doi: 10.1086/651263
Kubisiak-Rzepczyk, H., Gil, L., Zawirska, A., Kubisiak-Michalska, A., Mol, A., Reich, A., et al. (2013). Scedosporium prolificans fungaemia in a patient with acute lymphoblastic leukaemia. J. Mycol. Med. 23, 261–264. doi: 10.1016/j.mycmed.2013.08.003
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtzman, C., and Robnett, C. (1998). Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Van Leeuwenhoek 73, 331–371. doi: 10.1023/A:1001761008817
Kurtzman, C. P., Fell, J. W., and Boekhout, T. (2011). The Yeasts: A Taxonomic Study. Amsterdam: Elsevier.
Kurtzman, C. P., Robnett, C. J., and Basehoar-Powers, E. (2008). Phylogenetic relationships among species of Pichia, Issatchenkia and Williopsis determined from multigene sequence analysis, and the proposal of Barnettozyma gen. nov., Lindnera gen. nov. and Wickerhamomyces gen. nov. FEMS Yeast Res. 8, 939–954. doi: 10.1111/j.1567-1364.2008.00419.x
Lackner, M., Hagen, F., Meis, J. F. Gerrits van den Ende, A. H., Vu, D., Robert, V., et al. (2014). Susceptibility and diversity in the therapy-refractory genus scedosporium. Antimicrob. Agents Chemother. 58, 5877–5885. doi: 10.1128/AAC.03211-14
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lu, Q., Gerrits van den Ende, A. H., Bakkers, J. M., Sun, J., Lackner, M., Najafzadeh, M. J., et al. (2011). Identification of Pseudallescheria and Scedosporium species by three molecular methods. J. Clin. Microbiol. 49, 960–967. doi: 10.1128/JCM.01813-10
Maschmeyer, G. (2006). The changing epidemiology of invasive fungal infections: new threats. Int. J. Antimicrob. Agents 27(Suppl. 1), 3–6. doi: 10.1016/j.ijantimicag.2006.03.006
Meyer, C. P., and Paulay, G. (2005). DNA barcoding: error rates based on comprehensive sampling. PLoS Biol. 3:e422. doi: 10.1371/journal.pbio.0030422
Meyer, W., Irinyi, L., Hoang, M. T. V., Robert, V., Garcia-Hermoso, D., Desnos-Ollivier, M., et al. (2018). Database establishment for the secondary fungal DNA barcode-translational elongation factor 1α (TEF1α). Genome 62, 160–169. doi: 10.1139/gen-2018-0083
Minces, L. R., Ho, K. S., Veldkamp, P. J., and Clancy, C. J. (2009). Candida rugosa: a distinctive emerging cause of candidaemia. A case report and review of the literature. Scand. J. Infect Dis. 41, 892–897. doi: 10.3109/00365540903161531
Ming, C., Huang, J., Wang, Y., Lv, Q., Zhou, B., Liu, T., et al. (2019). Revision of the medically relevant species of the yeast genus Diutina. Med. Mycol. 57, 226–233. doi: 10.1093/mmy/myy001
Morris, A., Lundgren, J. D., Masur, H., Walzer, P. D., Hanson, D. L., Frederick, T., et al. (2004). Current epidemiology of Pneumocystis pneumonia. Emerg. Infect. Dis. 10, 1713–1720. doi: 10.3201/eid1010.030985
Nagarathnamma, T., Chunchanur, S. K., Rudramurthy, S. M., Vineetha, K. R., Ramamurthy, K., Joseph, J., et al. (2017). Outbreak of Pichia kudriavzevii fungaemia in a neonatal intensive care unit. J. Med. Microbiol. 66, 1759–1764. doi: 10.1099/jmm.0.000645
O’Donnell, K. (1993). “Fusarium and its near relatives,” in The Fungal Holomorph: Mitotic, Meiotic and Pleomorphic Speciation in Fungal Systematics, eds D. R. Reynolds and J. W. Taylor (Wallingford: CAB International), 225–233.
Padovan, A. C., Melo, A. S., and Colombo, A. L. (2013). Systematic review and new insights into the molecular characterization of the Candida rugosa species complex. Fungal Genet. Biol. 61, 33–41. doi: 10.1016/j.fgb.2013.10.007
Pappas, P. G., Alexander, B. D., Andes, D. R., Hadley, S., Kauffman, C. A., Freifeld, A., et al. (2010). Invasive fungal infections among organ transplant recipients: results of the transplant-associated infection surveillance network (TRANSNET). Clin. Infect. Dis. 50, 1101–1111. doi: 10.1086/651262
Patterson, T. F. (2005). Advances and challenges in management of invasive mycoses. Lancet 366, 1013–1025. doi: 10.1016/S0140-6736(05)67381-3
Pfaller, M. A., Diekema, D. J., Colombo, A. L., Kibbler, C., Ng, K. P., Gibbs, D. L., et al. (2006a). Candida rugosa, an emerging fungal pathogen with resistance to azoles: geographic and temporal trends from the ARTEMIS DISK antifungal surveillance program. J. Clin. Microbiol. 44, 3578–3582. doi: 10.1128/JCM.00863-06
Pfaller, M. A., Pappas, P. G., and Wingard, J. R. (2006b). Invasive fungal pathogens: current epidemiological trends. Clin. Infect. Dis. 43(Suppl. 1), S3–S14. doi: 10.1086/504490
Radosavljevic, M., Koenig, H., Letscher-Bru, V., Waller, J., Maloisel, F., Lioure, B., et al. (1999). Candida catenulata fungemia in a cancer patient. J. Clin. Microbiol. 37, 475–477.
Rainer, J., and de Hoog, G. S. (2006). Molecular taxonomy and ecology of Pseudallescheria, Petriella and Scedosporium prolificans (Microascaceae) containing opportunistic agents on humans. Mycol. Res. 110(Pt 2), 151–160. doi: 10.1016/j.mycres.2005.08.003
Rajasingham, R., Smith, R. M., Park, B. J., Jarvis, J. N., Govender, N. P., Chiller, T. M., et al. (2017). Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysis. Lancet Infect. Dis. 17, 873–881. doi: 10.1016/S1473-3099(17)30243-8
Rehner, S. A., and Buckley, E. (2005). A Beauveria phylogeny inferred from nuclear ITS and EF1-alpha sequences: evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia 97, 84–98. doi: 10.3852/mycologia.97.1.84
Sanclemente, G., Marco, F., Cervera, C., Hoyo, I., Colmenero, J., Pitart, C., et al. (2015). Candida norvegensis fungemia in a liver transplant recipient. Rev. Iberoam. Micol. 32, 115–117. doi: 10.1016/j.riam.2013.11.005
Schelenz, S., Barnes, R. A., Barton, R. C., Cleverley, J. R., Lucas, S. B., and Kibbler, C. C. (2015). British society for medical mycology best practice recommendations for the diagnosis of serious fungal diseases. Lancet Infect. Dis. 15, 461–474. doi: 10.1016/S1473-3099(15)70006-X
Schoch, C., Seifert, K., Huhndorf, S., Robert, V., Spouge, J., Levesque, C., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Schoch, C. L., Robbertse, B., Robert, V., Vu, D., Cardinali, G., Irinyi, L., et al. (2014). Finding needles in haystacks: linking scientific names, reference specimens and molecular data for Fungi. Database 2014:bau061. doi: 10.1093/database/bau061
Schuster, M. G., Meibohm, A., Lloyd, L., and Strom, B. (2013). Risk factors and outcomes of Candida krusei bloodstream infection: a matched, case-control study. J. Infect. 66, 278–284. doi: 10.1016/j.jinf.2012.11.002
Spampinato, C., and Leonardi, D. (2013). Candida infections, causes, targets, and resistance mechanisms: traditional and alternative antifungal agents. BioMed. Res. Int. 2013:204237. doi: 10.1155/2013/204237
Srivathsan, A., and Meier, R. (2012). On the inappropriate use of Kimura-2-parameter (K2P) divergences in the DNA-barcoding literature. Cladistics 28, 190–194. doi: 10.1111/j.1096-0031.2011.00370.x
Stielow, J. B., Lévesque, C. A., Seifert, K. A., Meyer, W., Irinyi, L., Smits, D., et al. (2015). One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia 35, 242–263. doi: 10.3767/003158515X689135
Tamm, M., Malouf, M., and Glanville, A. (2001). Pulmonary Scedosporium infection following lung transplantation. Transpl. Infect. Dis. 3, 189–194. doi: 10.1034/j.1399-3062.2001.30402.x
Tavanti, A., Davidson, A. D., Gow, N. A. R., Maiden, M. C. J., and Odds, F. C. (2005). Candida orthopsilosis and Candida metapsilosis spp. nov. to replace Candida parapsilosis groups II and III. J. Clin. Microbiol. 43, 284–292. doi: 10.1128/JCM.43.1.284-292.2005
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Vilgalys, R., and Hester, M. (1990). Rapid genetic identification and mapping of enzymatically amplified ribosomal DNA from several Cryptococcus species. J. Bacteriol. 172, 4238–4246. doi: 10.1128/jb.172.8.4238-4246.1990
Warnock, D. W. (2007). Trends in the epidemiology of invasive fungal infections. Nippon Ishinkin Gakkai Zasshi 48, 1–12. doi: 10.3314/jjmm.48.1
White, T. J., Bruns, T., Lee, S., and Taylor, J. W. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: a Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfandm, J. J. Sninsky, and T. J. White (New York: Academic Press), 315–322. doi: 10.1016/b978-0-12-372180-8.50042-1
Keywords: identification, fungal DNA barcoding, dual barcoding system, internal transcribed spacer region, translational elongation factor 1α, ISHAM Barcoding Database, invasive fungal diseases
Citation: Hoang MTV, Irinyi L, Chen SCA, Sorrell TC, the ISHAM Barcoding of Medical Fungi Working Group and Meyer W (2019) Dual DNA Barcoding for the Molecular Identification of the Agents of Invasive Fungal Infections. Front. Microbiol. 10:1647. doi: 10.3389/fmicb.2019.01647
Received: 10 May 2019; Accepted: 03 July 2019;
Published: 18 July 2019.
Edited by:
Saad J. Taj-Aldeen, Hamad Medical Corporation, QatarReviewed by:
Zheng Wang, Yale University, United StatesCopyright © 2019 Hoang, Irinyi, Chen, Sorrell, the ISHAM Barcoding of Medical Fungi Working Group and Meyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wieland Meyer, d2llbGFuZC5tZXllckBzeWRuZXkuZWR1LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.