 Caroline Eve Dubé
Caroline Eve Dubé Chin-Long Ky2,3,4
Chin-Long Ky2,3,4
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 05 July 2019
Sec. Microbial Symbioses
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.01548
Elucidating the role of prokaryotic symbionts in mediating host physiology has emerged as an important area of research. Since oysters are the world’s most heavily cultivated bivalve molluscs, numerous studies have applied molecular techniques to understand the taxonomic and functional diversity of their associated bacteria. Here, we expand on this research by assessing the composition and putative functional profiles of prokaryotic communities from different organs/compartments of the black-lipped pearl oyster Pinctada margaritifera, a commercially important shellfish valued for cultured pearl production in the Pacific region. Seven tissues, in addition to mucous secretions, were targeted from P. margaritifera individuals: the gill, gonad, byssus gland, haemolymph, mantle, adductor muscle, mucus, and gut. Richness of bacterial Operational Taxonomic Units (OTUs) and phylogenetic diversity differed between host tissues, with mucous layers displaying the highest richness and diversity. This multi-tissues approach permitted the identification of consistent microbial members, together constituting the core microbiome of P. margaritifera, including Alpha- and Gammaproteobacteria, Flavobacteriia, and Spirochaetes. We also found a high representation of Endozoicimonaceae symbionts, indicating that they may be of particular importance to oyster health, survival and homeostasis, as in many other coral reef animals. Our study demonstrates that the microbial communities and their associated predicted functional profiles are tissue specific. Inferred physiological functions were supported by current physiological data available for the associated bacterial taxa specific to each tissue. This work provides the first baseline of microbial community composition in P. margaritifera, providing a solid foundation for future research into this commercially important species and emphasises the important effects of tissue differentiation in structuring the oyster microbiome.
Microbial symbionts play key roles in the survival, homeostasis and development of eukaryotic organisms (McFall-Ngai et al., 2013; Bang et al., 2018). Symbiotic associations between bacteria (among other microbes) and macroorganisms are ubiquitous in nature and have been the focus of extensive research. For instance, bacterial communities have been found to have profound impact on host nutrition, pathogen resistance and development of immune function, as well as other beneficial processes (Taylor et al., 2007; Kamada et al., 2013; Mueller and Sachs, 2015; Vandenkoornhuyse et al., 2015; Foster et al., 2017), while the eukaryotic host provides a stable environment and a constant nutrient supply for their associated bacteria. Previous studies have demonstrated that host-associated microbial community compositions are not stochastic, but mostly determined by host phylogeny and habitat (Cardenas et al., 2014; Pantos et al., 2015; Brooks et al., 2016; Carrier and Reitzel, 2018), and can be unevenly distributed within an individual host (Hernández-Agreda et al., 2016). Understanding the mechanics and importance of host-microbiome systems thus requires prior knowledge of both the distribution of microbial communities at the scale of the individual, and some assessment of the influence that microbiome composition may have on host functioning.
Rapid advances in sequencing technology and a decline in associated costs have spurred an increase in microbiome research, targeting a multitude of organisms and environments (Caporaso et al., 2010). Of these, the microbiota of marine invertebrates, including crustaceans, polychaetes, echinoderms, tunicates, sponges and corals, have been investigated extensively over the last two decades. These studies have demonstrated that many marine invertebrates host diverse, core microbial communities that are phylogenetically distinct from those of surrounding waters (Moitinho-Silva et al., 2014; Lokmer et al., 2016b; Zhang et al., 2016; Lu et al., 2017), and that these microbial associations can be host-specific regardless of geography (Reveillaud et al., 2014; van de Water et al., 2016; Brener-Raffalli et al., 2018). Nonetheless, there are several biological factors that can contribute to specific variation in microbial communities, such as host genetics and physiology (Jaenike, 2012; Wegner et al., 2013; Kohl and Carey, 2016; Amato et al., 2018), host health (Cárdenas et al., 2012; Lu et al., 2013; Sweet and Bulling, 2017), diet (Carrier et al., 2018), life stages (Trabal et al., 2012; Lema et al., 2014), and host–tissue differentiation (Meisterhans et al., 2016; Engelen et al., 2018; Høj et al., 2018).
Studies on microbiota of bivalves are of particular interest because of their importance in ecosystem functioning, but also due to their economic value and consumption by humans. Oysters are one of the most widely consumed and most valuable of all bivalve molluscs exploited commercially (Dumbauld et al., 2009), making them a relevant model for investigating host–microbiome systems. As such, microbial associations in oysters have been well documented, though the vast majority of the literature focuses on the role of associated microbial communities in preventing colonisation and establishment of pathogens (Lokmer and Wegner, 2015; Green et al., 2018; King et al., 2018; Laroche et al., 2018). Bacterial communities can vary as a function of tissue types in many organisms (e.g., bivalves, sponges, corals, plants, and humans) due, among others, to differences in host cell functions and/or composition of cellular components (Costello et al., 2009; Antunes et al., 2010; Schmitt et al., 2012; Ainsworth et al., 2015; Coleman-Derr et al., 2016). As a consequence, tissue differentiation is thought to strongly influence the microbiome of oysters. Yet, most microbial surveys of oyster species have been performed on whole body homogenates (Beleneva et al., 2007) or single tissues (Trabal et al., 2012; Trabal-Fernández et al., 2014; Banker and Vermeij, 2018). Understanding oyster-microbial associations requires an understanding of organ compartmentalisation, since different oyster compartments (hereafter named tissues) may harbour unique microbial assemblages that are linked to the physiological role of each host compartment. To date, variations in the composition of microbial assemblages between oyster tissues has rarely been considered (but see King et al., 2012; Lokmer et al., 2016b). To fill this gap, we investigated and compared the prokaryote communities of different tissues in the black-lipped pearl oyster Pinctada margaritifera. This shellfish species is highly valued for cultured pearl production and its industry represents the second most valuable economic resource in French Polynesia after tourism (Ky et al., 2019). This oyster species can produce valuable black pearls by biomineralisation, following the insertion of a mantle graft from a donor into the gonad of a recipient oyster, together with a nucleus (Gervis and Sims, 1992; Taylor and Strack, 2008). While recent studies have investigated the genome, transcriptome (Le Luyer et al., 2019) and phenome (Ky et al., 2018) to identify potential links to cultured pearl quality traits in P. margaritifera, no data relating to its microbiome exists at present. Characterising the microbial composition of symbiont assemblages in P. margaritifera is critical, since these microbial communities likely play a role in maintaining oyster fitness (e.g., Lokmer et al., 2016b) and by extension, may subsequently impact upon pearl quality.
The aim of this study, therefore, is to provide a microbial baseline for P. margaritifera and determine the balance between a stable core microbiome and transient microbial components between pearl oyster tissue types. To do so, we characterised the composition, diversity and predicted functional roles of bacterial communities of eight tissues and secretions, including the gill, gonad, byssus gland, haemolymph, mantle, adductor muscle, mucus, and gut. Our study shows that bacterial community composition and underlying functional profiles can be readily distinguished across P. margaritifera tissues. Although microbial communities of the gill, gut and haemolymph have been well studied in oysters (among other bivalves), to our knowledge the present study provides the first description of the microbiota associated with oyster mucus, muscle and byssus gland. Our multi-tissues approach also led to the identification of consistent microbial members, including several tissue-specific bacterial taxa. Unexpectedly, it was found that many tissue microbiomes (e.g., gills, gonad and mantle, among others) were dominated by the symbiont Endozoicomonas, a bacterial genus that is also prominent in many coral reef animals, although nearly absent in other oyster species.
Wild Pinctada margaritifera individuals (N = 12) were collected in January–February 2016 in the lagoon of Takapoto atoll (14°32′ S, 145°14′ W, Tuamotu Archipelago, French Polynesia) during the natural spawning season. After 20 months of subsurface rearing (3–5 m below the surface) the pearl oysters (shell dorso-ventral measurements of 8.4 ± 1.8 cm) were removed from the spat collectors, pierced and tied together onto chaplets rearing systems, where they remained until their transfer to Ifremer facilities (Vairao, Tahiti, French Polynesia) in May 2018. Individuals were kept for one month in the lagoon at Vairao prior to the sampling of eight selected tissues and secretions: the gill, the gonad, the byssus gland, the haemolymph, the mantle, the adductor muscle, the mucus, and the gut (Supplementary Figure S1). To avoid differences in microbial community compositions as a function of oyster size, 12 individuals of similar sizes were selected to investigate the composition and functional characterisation of P. margaritifera microbiome. Pearl oysters were scrubbed and rinsed to remove any remaining debris on the outer shell and then shucked using shucking knives. For each oyster, 500 μL of haemolymph was collected in the byssus using a sterile syringe and the mucus via a sterile swap and preserved in absolute ethanol. These two aqueous tissues were collected prior to entirely opening the shells of each sampled animal. By doing so, we aimed to collect the mucous layer before inducing acute physiological stress, that typically results in the secretion of mucus in large amounts. Then, we opened the shells and dissected the animal to collect the remaining six tissues using sterile scalpel blades. Prior to transfer the samples in absolute ethanol, each sample was rinsed with 70% ethanol to reduce the possibility of cross-contamination between tissues from the large amounts of mucus secreted during dissection.
Prior to DNA extraction, all samples, with the exception of the haemolymph and mucus, were crushed using mortar and pestle until the tissue was completely homogenised and poured back in 15 ml falcons. Before processing the samples for homogenisation, each sample was once again rinsed with 70% ethanol to reduce the amount of mucus still present in the absolute ethanol that served to preserve the tissue. Each homogenised sample was centrifuged at 1,500 g for at least 45 s to separate the particulate matter from the ethanol and kept on ice until DNA extraction. For the mucus samples, swaps were soaked directly in PowerBeads tubes (included in PowerSoil DNA Kit, see below) for 10 min and gently vortexed before being removed. Remnant mucus and ethanol were centrifuged at 1,500 g for 10 min. Haemolymph samples were directly centrifuged at 1,500 g for 10 min. Pellets from centrifuged mucus and haemolymph samples were kept on ice until DNA extraction. DNA was extracted from homogenised tissues using a modified protocol of the Qiagen DNeasy PowerSoil Kit. About 0.5 g of homogenised tissues were added into the PowerBead tubes, while the entire sample was used for the gonad, mucus and haemolymph. A succession of three thermal shocks was performed prior to DNA extraction to facilitate the breakage of the bacterial outer membrane, each including three steps: (1) −80°C for 10 min, (2) 65°C for 5 min, and (3) 2 min of vortexing at maximum speed. Once all thermal shocks were completed, each sample were kept at room temperature for 10 min. Next, DNA extraction was processed according to manufacturer’s intructions with a 10 min heating step at 70°C as recommended in the troubleshouting guide. Extracted DNA was quantified using a Qubit dsDNA HS Kit (Invitrogen, Carlsbad, CA, United States). DNA was then shipped to the Génome Québec Innovation Centre (McGill University, Canada) for the preparation and sequencing of the 16S rRNA amplicon library. The V4 region of the 16S rRNA gene was amplified using Polymerase Chain Reactions (PCRs) of the primers 515F and 806R (Caporaso et al., 2010) with added sequencing adapters CS1 and CS2 (Ison et al., 2016) (forward: 5′–ACACTGACGACATGGTTCTACAGTGCCAGCMGCCGCGG TAA–3′; reverse: 5′–TACGGTAGCAGAGACTTGGTCTGGACT ACHVGGGTWTCTAAT–3′; CS1 and CS2 adaptor sequences are underlined). The library was prepared following three successive thermocycler runs for tagging the 16S rRNA gene and another run for barcoding the fragments. Fragments were then pooled and purified using AMPure XP beads (Beckman Coulter, Inc.) (Bronner et al., 2014), and library quality control was performed. The library was run on a MiSeq sequencing system using paired end read of 250 base pairs (PE250).
Raw sequences were processed with mothur v.1.39.5 (Schloss et al., 2009). After paired end reads were assembled into contigs, sequences longer than 298 bp (97.5% cutoff) were removed for further analyses, as well as sequences containing ambiguously called bases. We filtered the dataset for singletons and aligned the remaining sequences for the V4 region of the SILVA reference database, allowing for up to a 2 nt difference between the sequences. Chimera reads were removed using the UCHIME program (Edgar et al., 2011) as implemented in mothur. Sequences were then classified taxonomically according to the Greengenes database1 with a minimum bootstrap value of 60%. Any sequences belonging to mitochondria, chloroplasts, and eukaryotes were removed. Bacterial sequences were subsequently subsampled and rarefied based on the sample having the smallest number of sequences, i.e., to 2,687 (as described in the MiSeq SOP protocol, Kozich et al., 2013). Subsampled sequences were clustered into Operational Taxonomic Units (OTUs) using the average neighbour algorithm method with minimum 97% of similarity (cutoff = 0.03) and 16S rRNA reference amplicon sequences were determined for each OTU (Supplementary Data S1).
Phylogenetically annotated 16S sequences were used to characterise bacterial community composition of each tissue type at the family level. Stacked-column barplots were generated using the package ‘ggplot2’ in R based on total abundances of OTU counts for mean abundances of a given bacterial family within each tissue type. Then, relative abundances of the 22 bacterial families, including the “Other” category, were calculated from these mean abundances. Potential differences in community composition as a function of tissue type were assessed at both the family and OTU level by a permutational multivariate analysis of variance (PERMANOVA) and analysis of similarities (ANOSIM) with the adonis and anosim functions in the package ‘vegan’ integrated in R. For those tissues and secretions that showed significant dissimilarity in bacterial community composition (pairwise adonis function in R, P-values with Bonferroni corrections), similarity percentage (SIMPER) analyses were then conducted in the R package ‘vegan’ to determine which families contributed most to the degree of dissimilarity between host tissues. Community diversity and richness, as described respectively by the Shannon diversity index and Chao-1 richness, were inferred at the OTU level using mothur. Differences in alpha diversity between tissues were assessed by an analysis of variance (ANOVA) for the Shannon diversity index and a Kruskal–Wallis test for Chao richness (since normality of residuals and homogeneity of variances were rejected, i.e., Shapiro and Bartlett tests, respectively). Similarity in the assemblage of bacterial OTUs (based on the 4,085 OTUs rather than their family assignments) between host tissues was also assessed by PERMANOVA and ANOSIM tests and results were visualised using a non-metric multidimensional scaling (nMDS) ordination plot. All statistical analysis were performed on Bray Curtis distances of log (x + 1) transformed OTU counts using R version 3.4.4. The core microbiome of P. margaritifera was identified by plotting the abundance of OTUs by the percentage of sample representation at 2% intervals (from 0 to 100%, as in Ainsworth et al., 2015). Based on this distribution frequency across samples (see Supplementary Figure S2), OTUs present in at least 70% of samples were chosen as members of the core microbiome and their sequences were BLASTed against GenBank database [nr/nt and 16S rRNA sequences (Bacteria and Archaea)] for closely related matches.
Next, we performed an indicator species analysis using the R package IndicSpecies (De Cáceres and Legendre, 2009) to identify bacterial taxa that significantly associate with distinct host tissues. The analysis was conducted on OTU counts with host tissues as the grouping variable. Only OTUs that displayed highly significant (P < 0.01) associations with one tissue type were considered as indicator OTUs. The heatmap was compiled based on abundances of OTU counts that were previously identified as being tissue specific using the IndicSpecies analysis. Graphical representation of these tissue-specific OTUs was compiled in R using ‘ggplot2’, as well as the stacked-column plots representing relative abundances of tissue-specific bacterial taxa grouped at the family level.
Finally, to predict the functionality of the metagenomic content of each tissue type, we applied a computational approach using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States software (PICRUSt version 1.0) (Langille et al., 2013) as implemented in the Galaxy web application2. Briefly, the input file of the OTU table was created in mothur using the command ‘make.biom’. The command ‘normalise_by_copy_number.py’ was applied to account for differences in genomic copy number and metagenome predictions were conducted using ‘predict_metagenomes.py’. Individual KEGG Ortholog groups (KOs) were summarised at KEGG pathway level 1, 2, and 3 with the command ‘categorise_by_function.py’. This table was in turn used as input for LEfSe (Segata et al., 2011). Using LEfSe method, we tested data for statistical significance, biological consistency, and effect size relevance of predicted KEGG pathways of bacterial communties between tissue types, based on an LDA threshold of 2.5 for levels 1–3 of individual KOs and significant cutoff < 0.05. Graphical representations of LEfSe results were generated using ‘ggplot2’ R package and are presented hierarchically using barplots for individual KOs that displayed highly significant (LDA > 2.5) associations with one tissue type. A weighted Nearest Sequenced Taxon Index (NSTI) score was also calculated for each sample to confirm the accuracy of this computational approach, which mostly depends on the availability of reasonably related reference genomes (Langille et al., 2013).
Bacterial DNA was isolated from eight tissues and secretions (gill, gonad, byssus gland, haemolymph, mantle, muscle, mucus, and gut) collected from twelve individuals of P. margaritifera, totalling 95 samples (the muscle was not analysed for one individual). 16S rRNA gene sequencing yielded to 4,331,391 sequences that were further clustered into 4,085 distinct OTUs (Supplementary Data S1). P. margaritifera hosted 35 unique bacterial phyla, 103 classes, and 134 orders. The most abundant phylum recovered across all samples was the Proteobacteria (56.5%), followed by the Bacteroidetes (11.4%), Firmicutes (5.4%) and Planctomycetes (4.6%), and many other unclassified bacteria (13.4%). BLAST searches for representative sequences showed that the most abundant unassigned OTU (OTU0002, 10.2% of relative abundance across all samples) is related to Spirochaetes previously detected in marine invertebrates (Table 1).
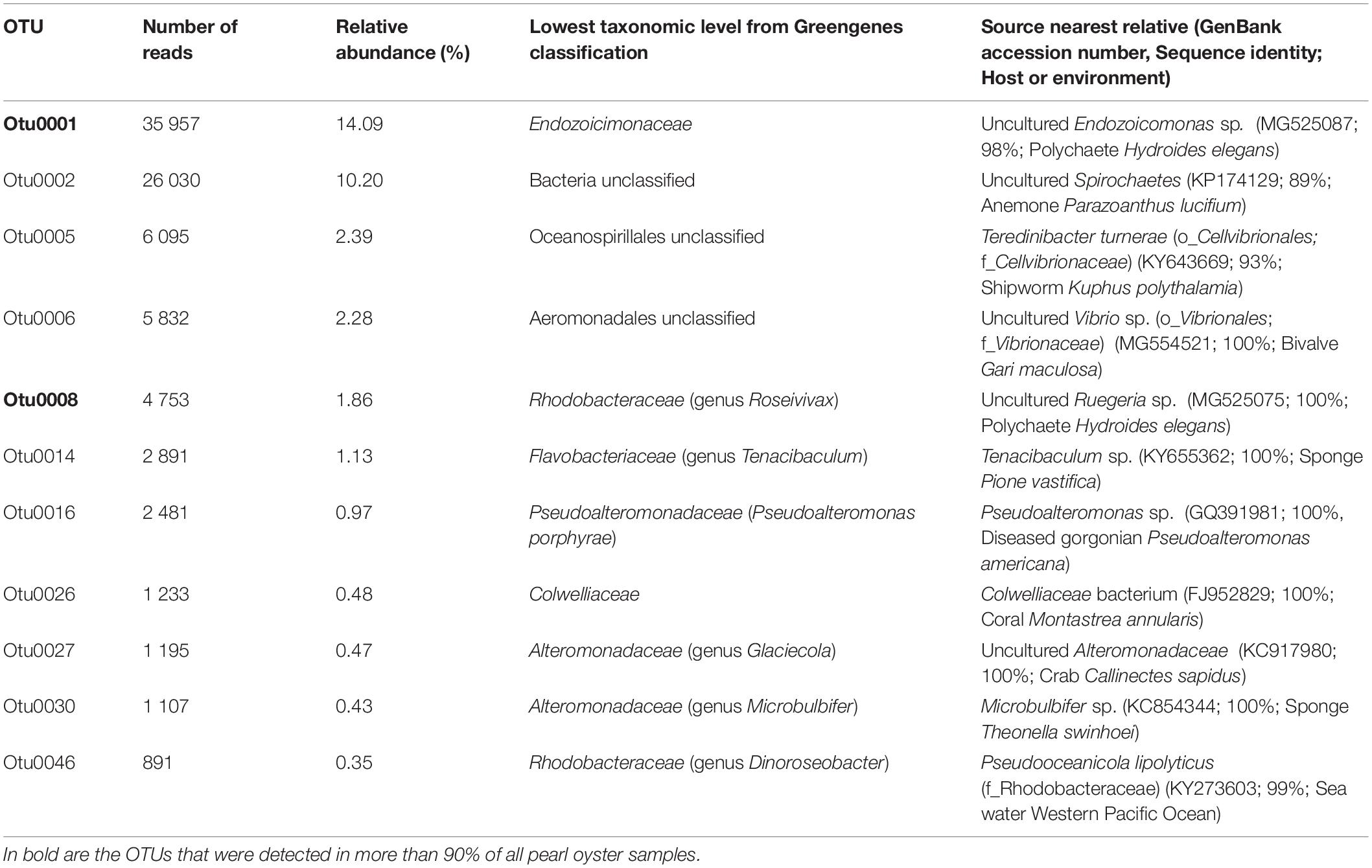
Table 1. Summary of bacterial OTUs corresponding to the core microbiome of the pearl oyster Pinctada margaritifera.
Although no OTU was found across all samples (100% sampling coverage), eleven conserved bacterial OTUs (present in at least 70% of all samples) were identified as ubiquitous members of the putative core microbiome, regardless of their relative abundances and tissue association (Table 1). Gammaproteobacteria was the most dominant taxa, representing 63.6% of the core microbiome. Two highly conserved core members were recovered across 90% of all pearl oyster samples, one was of the Endozoicimonaceae family and another of the genus Roseiviax (OTUs 0001 and 0008 respectively, Table 1). The majority of bacterial taxa identified as members of the core microbiome were present at low abundances (<5%).
The composition of tissue-specific microbiomes in P. margaritifera displayed significant differences between tissue types, based on relative abundances of bacterial families (Figure 1; PERMANOVA: pseudo-F = 4.6471, p = 0.0001; ANOSIM: R = 0.3646, p = 0.0001), individual OTUs (Figure 2; PERMANOVA: pseudo-F = 3.6285, p = 0.0001; ANOSIM: R = 0.4407, p = 0.0001) and community diversity and richness (Figure 3; ANOVA and Kruskal–Wallis: p < 0.0001, respectively). However, only OTU compositions in the gill, byssus gland and gut differed significantly from all other tissues based on the number of individual OTUs reads (PERMANOVA: p < 0.05; Supplementary Data S2).
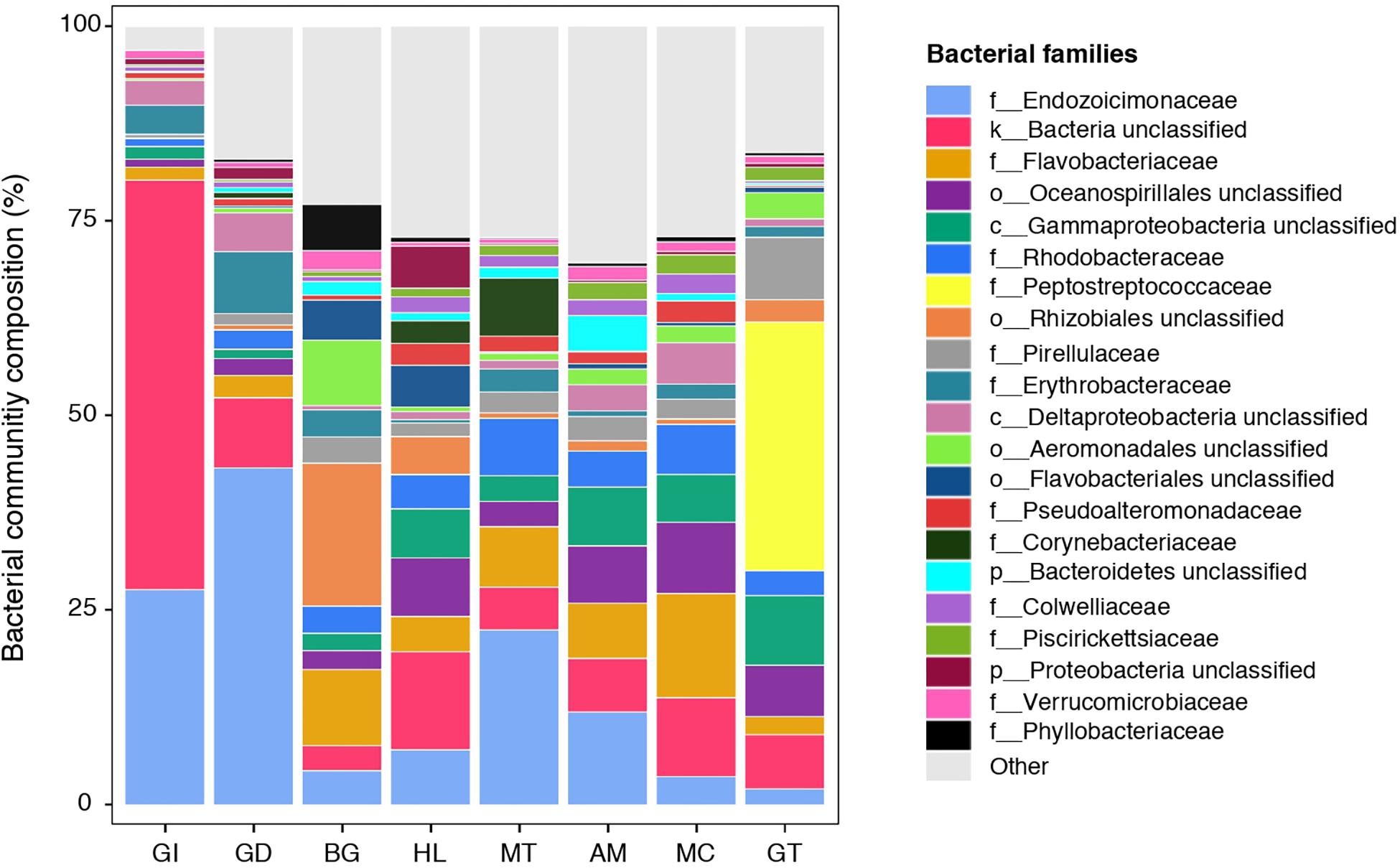
Figure 1. Bacterial community composition of the pearl oyster Pinctada margaritifera. Average relative abundances of the most represented families are shown for each of the eight tissue types; gill (GI), gonad (GD), byssus gland (BG), haemolymph (HL), mantle (MT), adductor muscle (AM), mucus (MC), and gut (GT). Each colour represents a distinct family level OTU or lowest level available. The 21 dominant families are indicated. Less commun families were grouped as ‘Other’ and are shown in light grey.
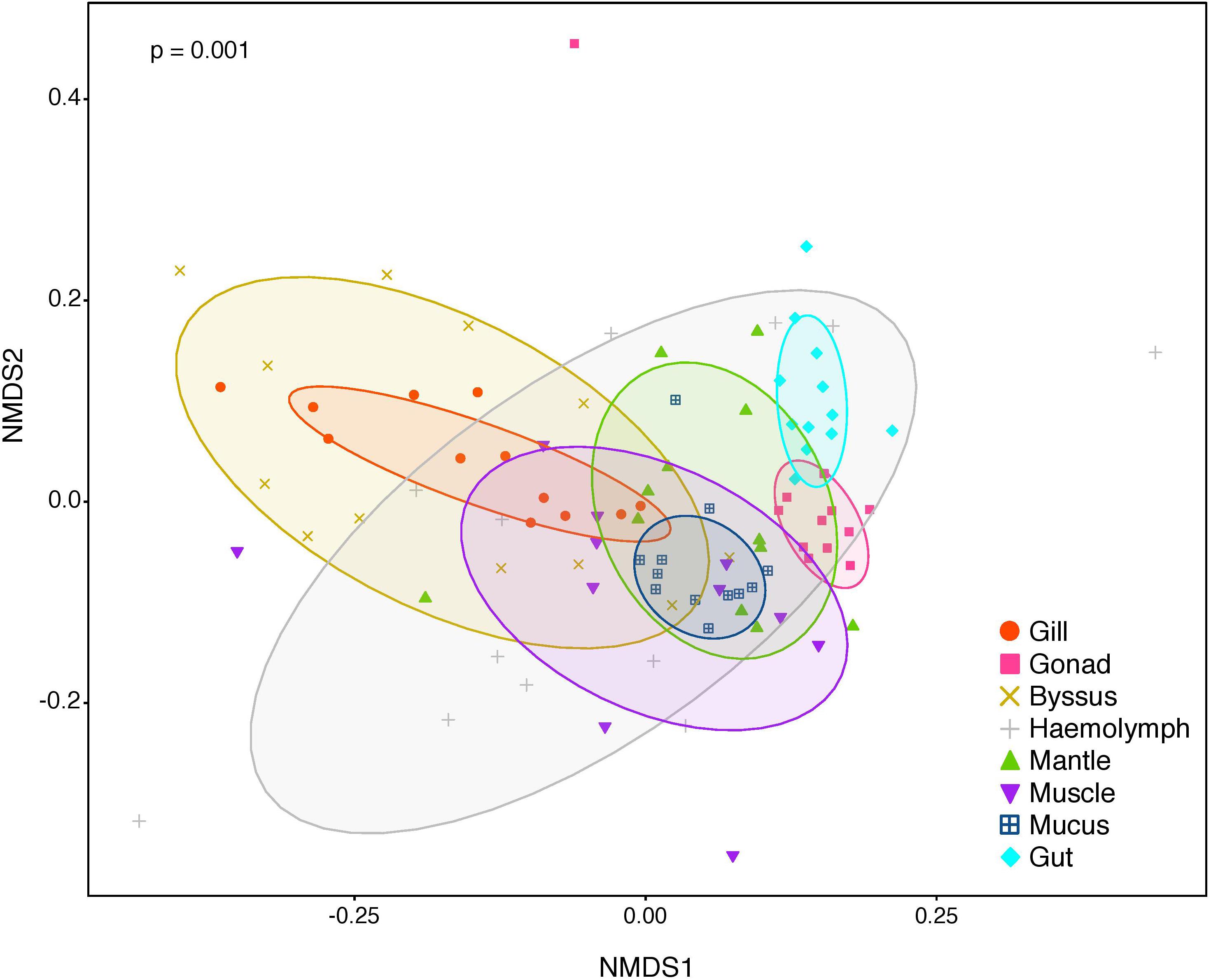
Figure 2. Non-metric multidimensional scaling (nMDS) of bacterial community composition of P. margaritifera at the OTU-level. Each symbol colour denotes one tissue (Gill = red, Gonad = pink, Byssus gland = gold, Haemolymph = grey, Mantle = green, Muscle = purple, Mucus = blue, Gut = cyan), ellipses are drawn around each group’s centroid (95%). P-values indicate significant differences in bacterial community composition between tissues (PERMANOVA and ANOSIM tests).
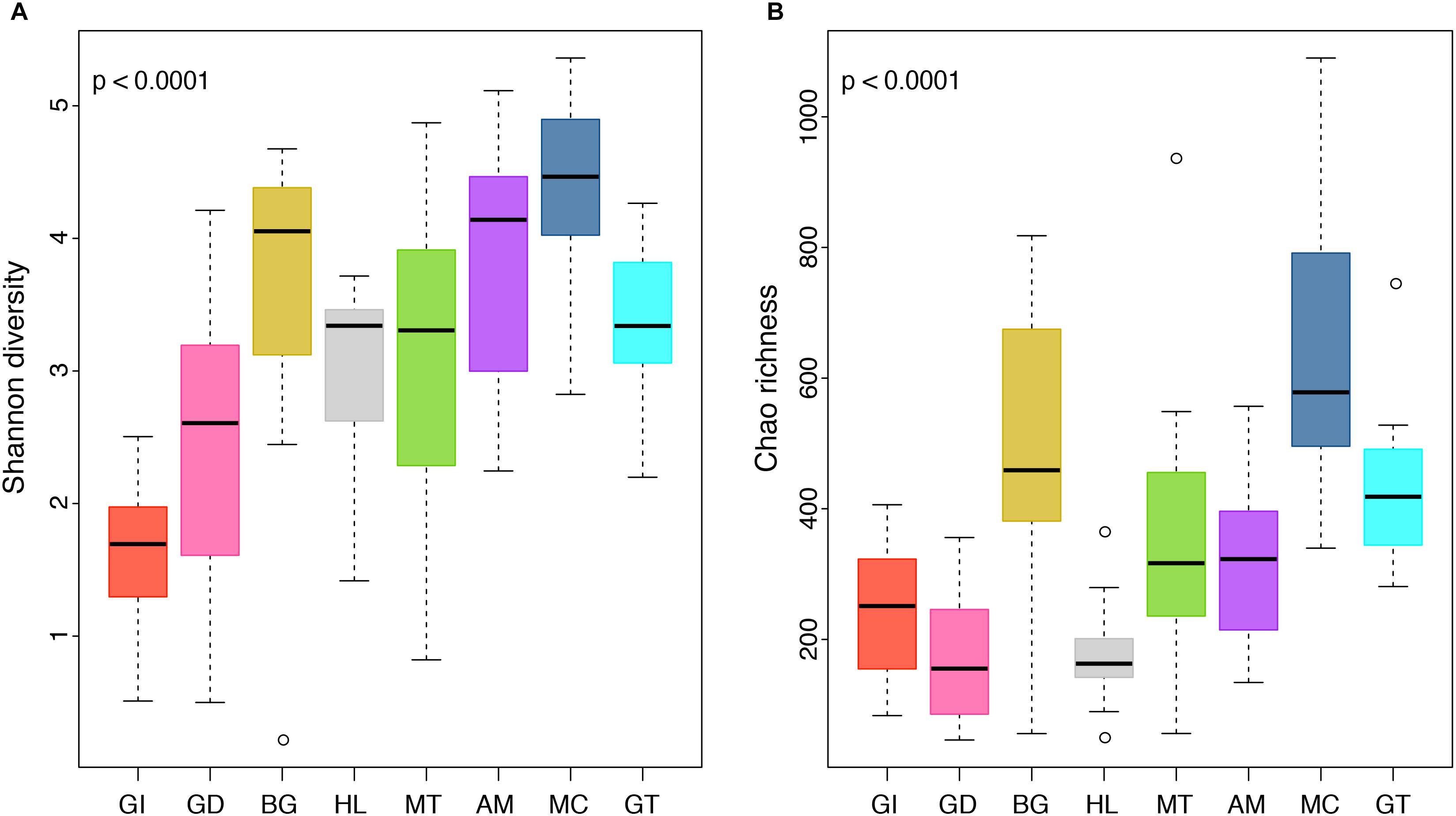
Figure 3. Boxplot displaying differences in community diversity between P. margaritifera tissues. Shannon diversity (A) and Chao richness (B) values are shown for each of the eight tissue types; gill (GI), gonad (GD), byssus gland (BG), haemolymph (HL), mantle (MT), adductor muscle (AM), mucus (MC), and gut (GT). Boxes display the first and third quartile spread of the data, with the line in the box indicating the median, the whiskers denoting the minimum and maximum values and open dots as outliners of the data. P-values indicate significant differences in community diversity between tissues (ANOVA and Kruskal–Wallis tests).
We further analysed our data for the presence of tissue-specific bacterial OTUs that characterised microbial variations between P. margaritifera tissues. Each tissue was consistently associated with a set of tissue-specific bacterial OTUs (Figure 4), most of which were detected in low abundances (<1%; Figure 5). The number of tissue-specific OTUs ranged from 2 to 32 (mean: 9.5 and median: 4.0) and their phylogenetic membership varied between tissues (Figure 5 and Supplementary Data S4). Tissue-specific bacterial OTUs represented less than 2% of the microbiomes associated with the gill, gonad, haemolymph, mantle, muscle, and mucus, while they represented nearly 10% of the byssus gland bacterial community and over 40% for the gut (Figure 5).
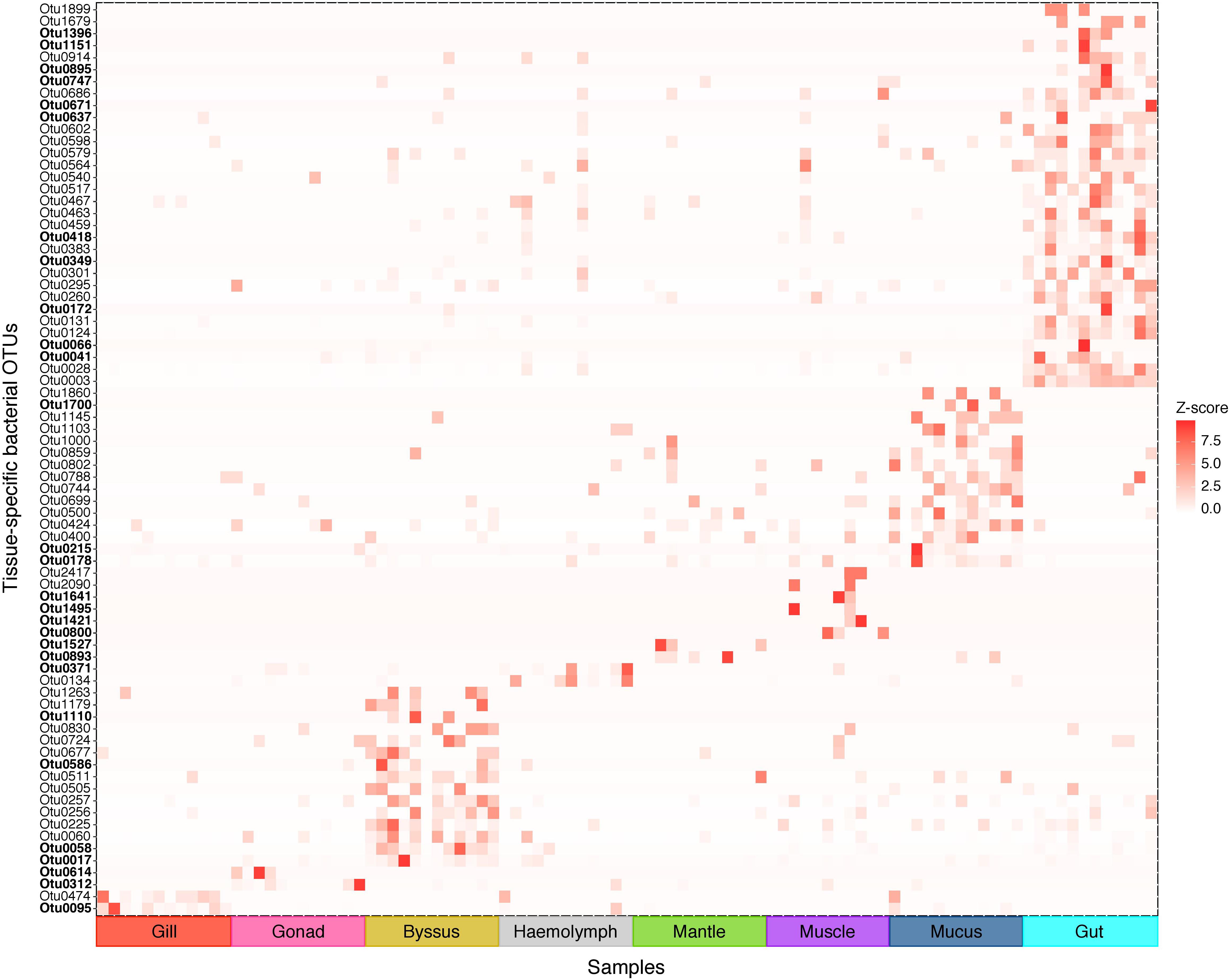
Figure 4. Indicator bacterial OTUs for variation in bacterial communities between P. margaritifera tissues. Heatmap based on IndicSpecies analysis shows specific bacterial OTUs that characterise each tissue. Each cell represents the standard transformation of the counts for each specific OTU per tissue. Bold OTUs indicate highly tissue-specific OTUs with Z-scores > 7.5. See Supplementary Data S4 for details on IndicSpecies analysis and Z-score values.
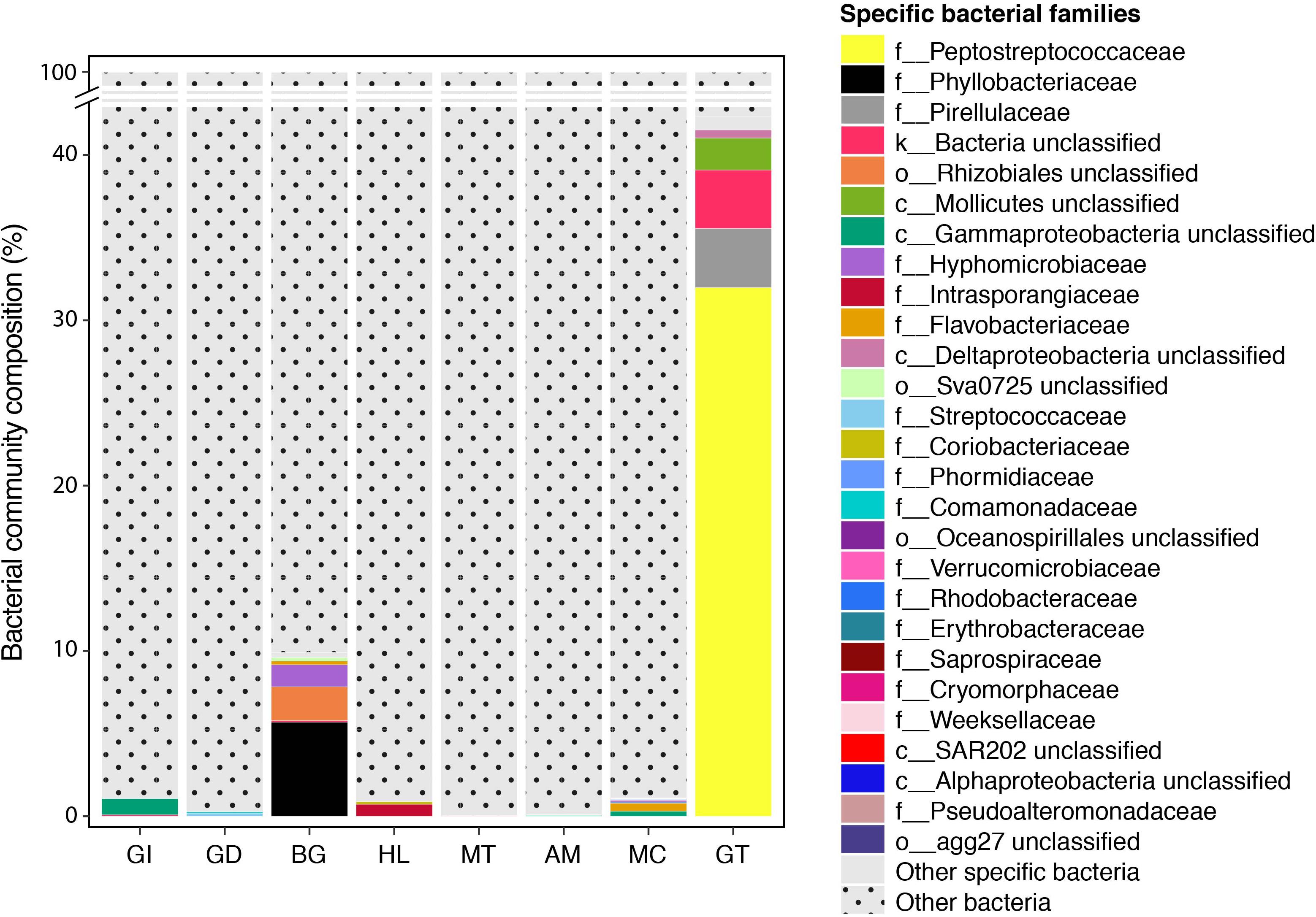
Figure 5. Indicator bacterial families for variation in bacterial communities between P. margaritifera tissues. Specific bacterial families associated with each tissue. Bar plots represent the average relative abundance of each identified specific bacterial OTU at the family level; dotted bars are none specific OTUs.
The gill microbiome displayed a higher relative abundance of unclassified bacteria (52.6%; Figure 1) (SIMPER: p < 0.05; Supplementary Data S3), of which nearly all reads (99.0%) were identified as Spirochaetes based on BLAST searches. OTUs classified as belonging to Endozoicimonaceae were notably abundant in the gill (27.6%; Figure 1). This bacterial family was detected in all oyster tissues, although at lower relative abundances, apart from the gonad in which Endozoicimonaceae dominated the microbial community (43.3%; Figure 1). Two tissue-specific OTUs of unclassified Gammaproteobacteria and bacteria (Figures 4, 5) were identified in the gill corresponding to Endozoicomonas (NCBI accession number MG525087; 92% sequence identity) and Arcobacter (KF721482; 79% sequence identity) based on BLAST searches.
In the gonad, the bacterial dominance by OTUs belonging to Endozoicimonaceae resulted in low values of evenness (Shannon) and species richness (Chao) (Figure 3), as well as in a tight cluster in the nMDS plot (Figure 2). The gonad also had higher relative abundance of Erythrobacteraceae-related sequences (7.9%; Figure 1), as well as many other rarer families (<1%), including the Streptococcaceae, Moraxellaceae, Bacillaceae, and Brevibacteriaceae (Supplementary Data S3). The gonad microbiome displayed two tissue-specific OTUs classified as Streptococcus and Alicycliphilus (Comamonadaceae) (Figures 4, 5 and Supplementary Data S4).
The byssus gland had a microbiome with relatively high alpha diversity (Figure 3). This was reflected in high proportions of bacterial families with relative abundances < 1% (25.6%; Figure 1). Many of these rare bacterial families were found in higher abundances within this tissue, including Hyphomicrobiaceae, Sphingomonadaceae, Microthrixaceae (Supplementary Data S3), as well as abundant OTUs belonging to Phyllobacteriaceae (5.9%) and unclassified Rhizobiales (18.3%; Figure 1). About seven percent of the byssus gland microbiome were represented by one tissue-specific OTU that belonged to the family Phyllobacteriaceae, while the remaining fourteen were mostly of unclassified Rhizobiales and Hyphomicrobiaceae (Figure 5).
The haemolymph microbiome displayed a low value of species richness (Figure 3), in addition to increased abundances of many rare bacterial families, such as Intrasporangiaceae and Coriobacteriaceae (including two tissue-specific OTUs belonging to these bacterial families; Figures 4, 5), as well as Dermabacteraceae and Prevotellaceae (Supplementary Data S3, S4).
The gut microbiome was dominated by a single OTU classified as Peptostreptococcaceae (32.0%; Figure 1), resulting in a tight cluster in the nMDS plot (Figure 2). Despite this bacterial dominance, the gut had relatively high values of alpha diversity (Figure 3). This was reflected in the high proportions of Pirellulaceae (8.0%; Figure 1) and many other rare families (e.g., Synechococcaceae, Mycobacteriaceae, Desulfobulbaceae, and unclassified Mollicutes) (Supplementary Data S3). Thirty-one percent of the gut microbiome were represented by a single tissue-specific OTU related to Peptostreptococcaceae, about 8% by unclassified bacteria and Pirellulaceae (5 and 8 OTUs, respectively), and 2% by two OTU related to unclassified Mollicutes (Figure 5 and Supplementary Data S4).
Microbiomes associated to the mucus, mantle and muscle were highly similar based on relative abundances of bacterial families and individual OTUs (PERMANOVA: p > 0.05), with high proportions of bacterial families with relative abundances < 1% (30.3, 29.9, and 32.7% for the mucus, mantle, and muscle respectively; Figure 1). Of these rare families, Oceanospirillaceae, Hyphomonadaceae and Haliangiaceae, among others, had significant higher proportions in the mucus compared to all other tissue types, while Syntrophobacteraceae, Kordiimonadaceae, and Spirochaetaceae had significant higher proportions within the muscle (Supplementary Data S3). The mucus microbiome also had the highest values for both evenness and species richness (Figure 3), as well as the highest relative abundance of OTUs belonging to Flavobacteriaceae (13.4%; Figure 1). The mucous layer had a tissue-specific microbiome of fifteen OTUs dominated by the Flaovobacteriaceae (Figure 5), including the OTU belonging to unclassified Gammaproteobacteria with the closest sequence matching Ascidianibacterium (AB377123; 98% of sequence identity). The mantle microbiome displayed relatively high abundances of Endozoicimonaceae and Corynebacteriaceae (22.5 and 7.8%, respectively; Figure 1), and only four rare families had higher proportions within this tissue (HTCC2089, Ellin6075, Oscillatoriophycideae and unclassified BPC015) (Supplementary Data S3). Two tissue-specific OTUs belonging to the bacterial families Verrucomicrobiaceae and Saprospiraceae were also identified in the mantle (Figures 4, 5), while the muscle microbiome had six tissue-specific OTUs, among which the most abundant was an unclassified Gammaproteobacteria identified as Oceanospirillaceae (EU167355; 96% sequence identity) based on BLAST searches.
Functional profiles of the 4,085 OTUs were determined using a predictive metagenomic analysis to identify putative functions and processes underlying distinct microbial communities associated with specific host tissues. Mean NSTI scores varied from 0.11 (SD ± 0.03) and 0.15 (SD ± 0.02) depending on the tissue types (Supplementary Data S5). These values were within the range of mammal microbiomes that have been previously predicted with reasonable accuracy, though well covered microbiome samples are generally below 0.05 (Langille et al., 2013). After metagenomic predictions were summarised at the level 2 of KOs, LEfSe analysis showed significant differences in predicted functional profiles with 21 distinguishing traits, identified in the microbiomes of particular host tissues (Figure 6 and Supplementary Data S6). Generally, PICRUSt and LEfSE analyses suggested significant differences in several predicted metabolic pathways of distinct bacterial community composition characterising the gonad, mantle and mucus. These differences were mostly through enrichment in genes encoding for pathways related to different metabolisms (e.g., terpenoids and polyketides, nucleotides, cofactors and vitamins, amino acids, and enzyme families). Metagenomic predictions also suggested that genes associated with the endocrine system, cell motility and signalling functions were the most abundant metabolic categories for the gill microbiome, while the byssus gland microbiome was enriched in genes affiliated to xenobiotic, amino acid and secondary-metabolite metabolisms, together with membrane transport and cell growth and death. The gut was mostly enriched in genes related to genetic information processing, including translation and transcription. No predicted functional trait was identified for the bacterial community associated with the haemolymph.
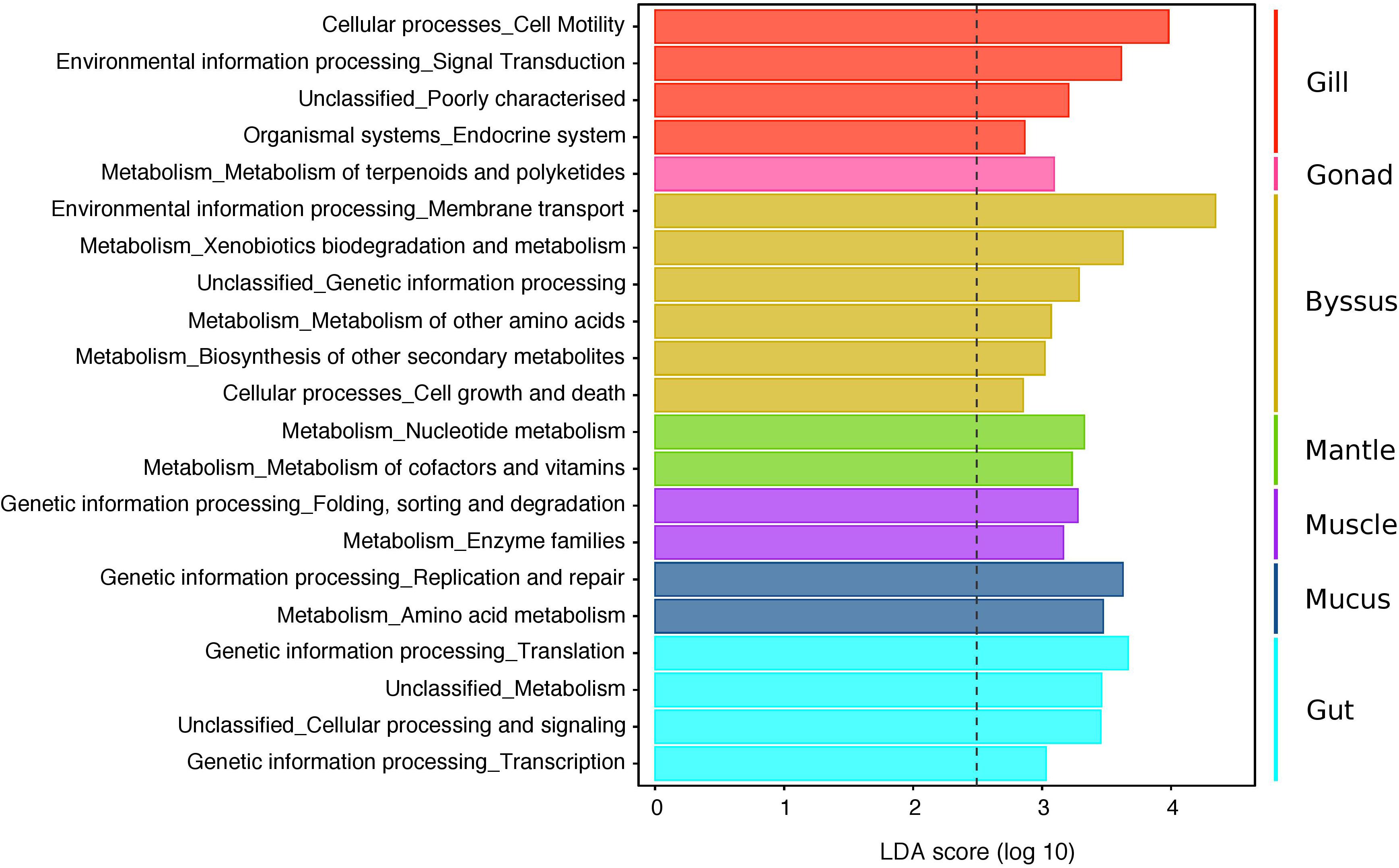
Figure 6. LEfSe analysis displaying predicted functional profiles of the different tissue types in P. margaritifera. The barplot shows the 21 differentially abundant KEGG pathways (level 2) identified in the microbiomes of particular host tissues, based on a Linear Discriminant Analysis (LDA, p < 0.05 for factorial Kruskal–Wallis and pairwise Wilcoxon tests). Each colour denotes the tissue type, wherein the distinguishing functional traits were identified (Gill = red, Gonad = pink, Byssus gland = gold, Mantle = green, Muscle = purple, Mucus = blue, Gut = cyan). Only KEGG pathways meeting an LDA significant threshold > 2.5 are shown. The threshold of the logarithmic LDA score is represented by the dotted line. See Supplementary Data S6 for abundances of predicted gene counts of KEGG pathways (level 1 to 3).
The present investigation is the first report that characterises the microbiome and tissue-specific bacterial composition of the black-lipped pearl oyster Pinctada margaritifera. Specifically, the objectives underpinning this study were two-fold: (1) providing the first description of consistent and dominant microbial members of P. margaritifera that can then be targeted in future function-based investigations of bacterial community composition for the potential improvement of cultured pearl quality, and (2) testing the hypothesis that different functional host compartments (i.e., different tissues and secretions) of P. margaritifera harbour different microbial communities and display distinct inferred functional profiles.
Similar to the microbiome of many oysters (Najiah et al., 2008; Green and Barnes, 2010; Fernández-Piquer et al., 2012; Cleary et al., 2015; Asmani et al., 2016; Pierce et al., 2016; Banker and Vermeij, 2018; Laroche et al., 2018), P. margaritifera microbial communities were dominated by the largest and most phenotypically diverse bacterial phylum, the Proteobacteria. The prevalence of this phylum was mostly attributable to Alpha- and Gammaproteobacteria. Alphaproteobacteria were identified as members of the P. margaritifera core microbiome, in particular OTUs related to aerobic heterotrophs from the Rhodobacteraceae family (Roseivivax_OTU0008 and Dinoroseobacter_OTU0046). Roseivivax and Dinoroseobacter are bacterial genera that are known to metabolise organic sulphur compounds, including dimethylsulphoniopropionate (DMSP), as well as carbon monoxide, a pathway often coupled with sulphate respiration processes (Gonzalez et al., 1999, 2000; Ansede et al., 2001; Zubkov et al., 2001; Rabus et al., 2006). These bacteria are also thought to have a great diversity of physiological properties, ranging from the degradation of lignin, aerobic oxidation of sulphites and production of antibiotics to aerobic anoxygenic photosynthesis (Buchan et al., 2005; Dang et al., 2008; Tang et al., 2010). Recovered Endozoicimonaceae (Gammaproteobacteria) sequences were in high abundances in the black-lipped pearl oyster (from 2.0 to 43.3% depending on the tissue) and present in at least 90% of all samples irrespective of the tissue (OTU0001). These bacterial symbionts have not previously been reported from other oyster species, but are dominant members of the core microbiome of several marine invertebrates (Neave et al., 2016). Endozoicomonas have been suggested to have important functional roles in their hosts related to nutrient acquisition, including host-associated protein and carbohydrate transport and cycling, and the production of amino acids and antimicrobial compounds (Neave et al., 2017). Other Gammaproteobacteria-related OTUs were identified as core members, including Alteromonadaceae and Pseudoalteromonadaceae, although observed at lower abundances across all tissues (<1%). Alteromonadaceae (e.g., Glaciecola and Microbulbifer genera) have been reported to play a role in oyster larvae development (Laroche et al., 2018), while Pseudoalteromonas porphyrae, and more specifically its associated oxidoreducting enzymes, are involved in plant growth and stress tolerance (Dimitrieva et al., 2006). Further investigations are thus needed to determine whether these bacteria play similar roles in P. margaritifera.
A high representation of Bacteroidetes in P. margaritifera, and more specifically Flavobacteriia, is in accordance with previous studies on oyster-associated bacterial community composition (Hernández-Zárate and Olmos-Soto, 2006; Zurel et al., 2011; Fernández-Piquer et al., 2012; Fernández-Gómez et al., 2013). These marine bacteria have been implicated in nutrient uptake via the production and/or remineralisation of organic matter ingested or produced by their hosts (Agostini et al., 2012), but also in biofilm formation (Ríos-Castillo et al., 2018), algal polysaccharide degradation (Mann et al., 2013), and infectious diseases (Nematollahi et al., 2003). Of note is the identification of the genus Tenacibaculum (Flavobacteriaceae) as a member of P. margaritifera core and stable microbial community. Tenacibaculum species are known pathogens reported in diseased oysters, and to have caused considerable economic losses in mollusc aquaculture (Collado et al., 2014; Burioli et al., 2018). These bacteria do not seem to express pathogenic capacity for infecting the French Polynesian pearl oyster (at least for the twelve individuals collected in our study, as they appeared healthy and showed no sign of infection), suggesting that Tenacibaculum may switch from being a commensal species to an opportunistic pathogen only under particular conditions (such as the presence of parasites; see Burioli et al., 2018). Furthermore, Tenacibaculum sp. have been pinpointed as being indicative of the reef flat environment (van Oppen et al., 2018), the habitat in which P. margaritifera oysters are grown and kept for pearl production. Oyster samples also returned high relative abundances of one core bacterial OTU identified via BLAST searches as belonging to the phylum Spirochaetes (OTU0002). Members of this phylum can play an important role in host nutrition, as well as in the production of antimicrobials in some gorgonian corals (van de Water et al., 2016; Wessels et al., 2017), but can also be causative agents of Akoya oyster disease (Pinctada fucata martensii; Matsuyama et al., 2017).
As in all suspension-feeding bivalves, P. margaritifera uses its gills to pump water into the pallial cavity to capture, process and transport food particles (Beninger et al., 1997; Beninger and Veniot, 1999). Microbial analysis and BLAST searches revealed that the gill microbiome was dominated by Endozoicomonas- and Spirochaetes-related OTUs (Endozoicimonaceae_OTU0001 and Unassigned_OTU0002), a bacterial genus and phylum known for their roles in host nutrient acquisition and antimicrobial production (van de Water et al., 2016; Neave et al., 2017; Wessels et al., 2017). Inferred functional profile of the gill was characterised by higher relative abundances in genes encoding for pathways related to the endocrine system, cell motility and signal transduction, and those encoding for protein kinases (Supplementary Data S6), a key cytoplasmic signal transducer in immune cell signalling (Lim et al., 2015). Furthermore, BLAST searches identified a gill-specific bacterial OTU belonging to Arcobacter, a genus that has been implicated in pathogen defence and acclimation (Defer et al., 2013; Lokmer and Wegner, 2015). Together, these findings suggest that P. margaritifera has the potential to select specific bacteria through filtration by the gills. This selection process may target bacteria that are able to produce enzymes and antimicrobials involved in oyster immunity, functions that have been previously documented for bacterial communities associated to the gills of other oyster species (Zurel et al., 2011; Roterman et al., 2015) and marine bivalves (Antunes et al., 2010; Meisterhans et al., 2016).
Similar to the gills, the mucus is involved in the transport of particles during feeding and cleansing processes. In marine invertebrates, the mucus also serves as a physical and biological barrier against pathogens (Espinosa et al., 2016), and play a role in locomotion (Davies and Beckwith, 1999), adhesion (Smith et al., 1999) and nutrition (Ward and Shumway, 2004). In the present study, the mucus of P. margaritifera was characterised by the highest bacterial richness and diversity, and was dominated by Flavobacteriaceae and Oceanospirillales (including six tissue-specific OTUs), both reported as major bacterial components in coral mucus (Taniguchi et al., 2015; Badhai et al., 2016). The mucus microbiome was predicted to be enriched in genes encoding for pathways related to amino acid metabolism and carbon fixation, together with DNA replication and repair. These predicted functions are in accordance with the results of a previous study on the composition and functional characterisation of microbiome associated with coral mucus (Badhai et al., 2016). Using a true functional analysis of the metagenome, the authors accurately deciphered the role of the mucus microbiota in host nutrient uptake and protection against physicochemical injuries and virulence. The mantle, muscle and mucus microbiomes were highly similar despite a reduced abundance of Endozoicimonaceae in the mucus. Mantle tissues also returned significantly higher abundances of OTUs belonging to the genus Corynebacterium compared to all other tissue types. The role of these bacteria in marine invertebrates is unknown, but they have been reported to use urea as a nitrogen source (Siewe et al., 1998) and to produce metabolites, especially antibiotics (Dickschat et al., 2010).
In marine invertebrates, the haemolymph is a critical site in host immune response. Similar to other oysters, P. margaritifera haemolymph was characterised by a low bacterial richness, albeit still phylogenetically diverse (Potgieter et al., 2015) comprising relatively high abundances of Spirochaetes, Flavobacteriia, Alpha- and Gammaproteobacteria (Lokmer and Wegner, 2015; Lokmer et al., 2016a). Although Vibrio, Acinetobacter, and Aeromonas pathogens have been identified as prevalent groups of the haemolymph in some aquatic invertebrates (Zhang et al., 2018), including oysters (Vezzuli et al., 2018), these bacteria occurred in low abundances in P. margaritifera microbiome. In the present study, we were not able to confirm that the presence of the core members Pseudoalteromonas, Endozoicomonas and Rhodobacterales (among others) were involved in the production of antimicrobials against these pathogenic bacteria (as reported by Boöhringer et al., 2017; Vezzuli et al., 2018; Zhang et al., 2018), but such discrepancies warrant further exploration. The persistence of relatively rare, presumably resident bacteria, such as Intrasporangiaceae and Coriobacteriaceae, indicates that these microorganisms can resist the host immune system and might provide benefits to their host functioning or be commensal symbionts.
Gonad tissues of non-grafted P. margaritifera (i.e., no biomineralisation for pearl production) were largely dominated by Endozoicimonaceae, suggesting that these bacteria also play a role in oyster reproduction. Erythrobacteraceae, among other Proteobacteria, were prominent members of the gonad microbiome. These bacteria are facultative photoheterotrophs, metabolising a variety of organic carbon compounds, including glucose, pyruvate, acetate, butyrate, and glutamate (Tonon et al., 2014). They are also thought to be involved in coral reproduction (Ceh et al., 2012; Liang et al., 2017) and may play a similar role in oysters. Other gonad-specific bacteria, such as Streptococcaceae and Comamonadaceae, are involved in nutrient metabolism, and specifically in sulphur cycling (Tian et al., 2014; Gauthier et al., 2016). Predicted analysis of metagenome revealed that the gonad microbiome was enriched in genes encoding for pathways related to carbon cycling through aerobic mineralisation of some aromatic compounds (e.g., benzoate and naphthalene) and metabolisms of terpenoids, polyketides, and beta-alanine (Supplementary Data S6). These findings suggest that the gonad microbiome can be taxonomically and functionally diverse, and provide a first survey on the distribution of various predicted metabolic pathways previously unexplored in this reproductive tissue. Future investigations of grafted gonads would be highly valuable to elucidate the effects of the grafting process on microbial community composition. For instance, some bacteria can be introduced into the gonad during the grafting operation and depreciate the commercial value of pearls by negatively affecting the biomineralisation process and pearl quality traits (e.g., the occurrence of depressed rings and superficial depressed spots) (Cuif et al., 2018).
In the alimentary tissue, our microbial analysis revealed that P. margaritifera gut was dominated by Peptostreptococcaceae (phylum Firmicutes), Pirellulaceae (Planctomycetes) and Mollicutes (Tenericutes). These bacterial phyla are commonly associated with animal guts, including oysters (Green and Barnes, 2010; King et al., 2012; Lokmer et al., 2016b). For instance, species of the genus Tepidibacter (OTU0003) have been implicated in the decomposition of organic matter produced by organisms colonising deepsea hydrothermal vents, while other members of Peptostreptococcaceae and related genera were found in human intestines (Slobodkin, 2015). Pirellulaceae species have also been reported to exploit sulphated algal polysaccharides (Glockner et al., 2003), compounds that are commonly ingested by oysters as a consequence of phytoplankton consumption. Although Mollicutes have been previously reported in oyster guts (King et al., 2012), as well as in other invertebrate and fish guts (Kellogg et al., 2009; Nechitaylo et al., 2009; Huang et al., 2010), relatively little is known about their role in digestive systems. In this study, we observed an enrichment of predicted genes encoding for pathways related to various metabolisms, including pyrimidine, methane and sphingolipid, as well as high abundances of those encoding for the production of amino acids and vitamins, degradation of polysaccharides, and other genetic information processing, such as transcription and translation (Supplementary Data S6). Such an inferred functional profile could align with the contribution of gut microbial symbionts to heterotrophic metabolism and nutrient uptake, though ground-truthed metagenomic and/or metatranscriptomic data are needed to confirm these predicted metabolic functions.
The byssus gland, a compartment involved in oyster adhesion through secretions that form the byssal threads, had microbiomes mostly dominated by Rhizobiales, including tissue-specific Phyllobacteriaceae and Hyphomicrobiaceae species. Bacteroidetes, and particularly Flavobacteriaceae, were also prominent microbial members of the byssus gland. These bacteria have been implicated in organic matter degradation, such as cellulose and pectin (Thomas et al., 2011). Consequently, Bacteroidetes are often found in the guts of animals, where the secretion of a range of extracellular enzymes help the degradation of the decomposed organic matter. Strikingly, however, Flavobacteriia were found in low abundance in P. margaritifera gut, although prevalent in the byssus gland and mucus. This result highlights the need for more detailed studies to elucidate the role of these marine bacteria in oysters. Nonetheless, the predicted enrichment in genes involved in various metabolic pathways, including amino acids, metabolites and xenobiotics, as well as genes encoding for membrane transport, cell growth and death, may suggest that bacterial communities of the byssus gland are not only taxonomically diverse, but also display a variety of functional roles.
The present study provides a snapshot of the microbial community composition and predicted functions in eight distinct tissues and secretions of the pearl oyster P. margaritifera. Several bacteria of the phylum Proteobacteria, Bacteroidetes, and Spirochaetes were identified as members of the core microbiome of the black-lipped pearl oyster P. margaritifera, regardless of their tissue affiliation. Such a stability in microbial communities between tissues implies that there is no tissue-specific condition that favours the growth of these microbial species. Above all, it is important to mention that core OTUs identified in the current study may also be ubiquitous due to the presence of overlying mucous layer, acting as a vector for bacterial transfer between the mucus and the tissues sampled, and this despite our efforts to minimise cross-contamination during dissection. Among these core members, many have been previously identified as prevalent microbial symbionts of several oyster species, including bacterial families of Rhodobacteraceae, Alteromonadaceae, Pseudoalteromonaceae, and Flavobacteriaceae (Asmani et al., 2016; Banker and Vermeij, 2018; Laroche et al., 2018). Conversely, Endozoicimonaceae and Spirochetes were identified for the first time as core members of oysters, in addition to being further prevalent in P. margaritifera gills and gonads. Such a novelty may be linked to host geography and habitat rather than host specificity. In fact, these bacteria are often dominating microbiomes of marine invertebrates inhabiting coral reef environments (van de Water et al., 2016; Neave et al., 2017; Wessels et al., 2017). Given the changeable state of the microbiome, meta-microbiome changes with time, seasons, locations and host age would be required to validate those core members identified in the current study (as described in Hernández-Agreda et al., 2017). One striking result is the identification of Streptococcaceae and Comamonadaceae as gonad-specific bacterial taxa, where both are known to be involved in sulphur cycling. Given that some bacteria are contributing to the formation of biochemical blends that can further impact the biomineralisation process of grafted pearl oysters (Cuif et al., 2018), function-based studies will be of great interest to investigate whether and how these sulphuric bacteria may depreciate pearl quality. Futhermore, our study highlights that, in addition to a ubiquitous core microbiome, bacterial community compositions vary between some tissue types, with certain bacterial taxa that seem to support their respective physiological functions. Consequently, the eight tissues surveyed in this study represent bacterial niches, with the gill, byssus gland and gut having the most distinct microbial assemblages, thus revealing a tissue-specific microbial consortium as reported in humans (Turnbaugh et al., 2007; Costello et al., 2009; Faust et al., 2015) and several marine invertebrates (King et al., 2012; Lokmer et al., 2016b; Høj et al., 2018). Overall, this study provides new perspectives on the largely understudied black-lipped pearl oyster-associated microbiome and directions for future studies.
Raw sequencing data determined in this study are available under the NCBI BioProject ID PRJNA544968 (https://www.ncbi.nlm.nih.gov/bioproject/544968) for 16S rRNA gene data. Other data are available in the Supplementary Information.
The collection of P. margaritifera followed institutional and national guidelines. The study was approved by the ethics committee of Ifremer.
CD, C-LK, and SP designed the research. CD performed the sampling, laboratory work, data analysis, and wrote the manuscript. C-LK and SP funded the research and reviewed draughts of the manuscript.
This work was supported by a grant from the ‘Direction des Ressources Marines’ of French Polynesia, through the AmeliGEN project (#10065/MEI/DRMM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the Pahai Poe Pearl farm for providing animals for the sampling, and A. Mercière, L. Bisch, G. Le Moullac, V. Quillien, and D. Potin for their technical assistance. We also thank Génome Québec Innovation Centre for the preparation and sequencing of the 16S rRNA amplicon library. We gratefully acknowledge the reviewers for their thorough reviews that have significantly improved an earlier version of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01548/full#supplementary-material
FIGURE S1 | P. margaritifera compartments/tissues. Photo of each of the two shell valves showing, on the right, the collected tissues: GI, the gills; GD, the gonad; BG, the byssus gland; MT, the mantle; AM, the adductor muscle; and GT, the gut (shown partially on the photo). The haemolymph was collected from the byssus using a sterile syringe, while the mucus was collected on the mantle using a sterile swap.
FIGURE S2 | Plotted bacterial OTU abundance in core microbiome’s of 0 to 100% of samples of the pearl oyster P. margaritifera.
DATA S1 | Overview over sequence counts, taxonomic classification, and 16S rRNA reference amplicon sequence for all 4,085 OTUs.
DATA S2 | Overview of significant tissue differences in microbiome composition based on relative abundances of bacterial families, individual OTUs and community diversity and richness.
DATA S3 | Similarity percentage (SIMPER) analysis of bacterial community composition associated with different tissues of P. margaritifera.
DATA S4 | IndicSpecies analysis of indicator OTUs/bacterial taxa associated with P. margaritifera tissues.
DATA S5 | Overview of weighted Nearest Sequenced Taxon Index (NSTI) scores over all oyster samples.
DATA S6 | PICRUSt and LEfSe analyses, an overview of differentially abundant KEGG pathways identified in the microbiomes of particular host tissues (per sample data provided).
Agostini, S., Suzuki, Y., Higuchi, T., Casareto, B., Yoshinaga, K., Nakano, Y., et al. (2012). Biological and chemical characteristic of the coral gastric cavity. Coral Reefs. 31, 147–156. doi: 10.1007/s00338-011-0831-6
Ainsworth, T. D., Krause, L., Bridge, T., Torda, G., Raina, J. B., Zakrzewski, M., et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9:2261. doi: 10.1038/ismej.2015.39
Amato, K. R., Sanders, J. G., Song, S. J., Nute, M., Metcalf, J. L., Thompson, L. R., et al. (2018). Evolutionary trends in host physiology outweigh dietary niche in structuring primate gut microbiomes. ISME J. 13:576. doi: 10.1038/s41396-018-0175-0
Ansede, J. H., Friedman, R., and Yoch, D. C. (2001). Phylogenetic analysis of culturable dimethyl sulfide-producing bacteria from a spartina-dominated salt marsh and estuarine water. Appl. Environ. Microbiol. 67, 1210–1217. doi: 10.1128/aem.67.3.1210-1217.2001
Antunes, F., Hinzmann, M., Lopes-Lima, M., Machado, J., and Da Costa, P. M. (2010). Association between environmental microbiota and indigenous bacteria found in hemolymph, extrapallial fluid and mucus of Anodonta cygnea (Linnaeus, 1758). Microb. Ecol. 60, 304–309. doi: 10.1007/s00248-010-9649-y
Asmani, K., Petton, B., Le Grand, J., Mounier, J., Robert, R., and Nicolas, J. L. (2016). Establishment of microbiota in larval culture of pacific oyster. Crassostrea gigas. Aquaculture 464, 434–444. doi: 10.1016/j.aquaculture.2016.07.020
Badhai, J., Ghosh, T. S., and Das, S. K. (2016). Composition and functional characterization of microbiome associated with mucus of the coral Fungia echinata collected from Andaman Sea. Front. Microbiol. 7:936. doi: 10.3389/fmicb.2016.00936
Bang, C., Dagan, T., Deines, P., Dubilier, N., Duschl, W. J., Fraune, S., et al. (2018). Metaorganisms in extreme environments: do microbes play a role in organismal adaptation? Zoology 127, 1–19. doi: 10.1016/j.zool.2018.02.004
Banker, R., and Vermeij, G. J. (2018). Oyster microbial communities and implications for chalky deposit formation. Hydrobiologia 816, 121–135. doi: 10.1007/s10750-018-3569-0
Beleneva, I. A., Zhukova, N. V., Lan, H., and Nguyen Tran, D. H. (2007). Taxonomic composition of bacteria associated with cultivated mollusks Crassostrea lugubris and Perna viridis and with the water of the gulf of nha trang lagoon, Vietnam. Microbiology 76, 220–228. doi: 10.1134/s0026261707020142
Beninger, P. G., Dufour, S. C., and Bourque, J. (1997). Particle processing mechanisms of the eulamellibranch bivalves Spisula solidissima and Mya arenaria. Mar. Ecol. Prog. Ser. 150, 157–169. doi: 10.3354/meps150157
Beninger, P. G., and Veniot, A. (1999). The oyster proves the rule: mechanisms of pseudofeces transport and rejection on the mantle of Crassostrea virginica and C. gigas. Mar. Ecol. Prog. Ser. 190, 179–188. doi: 10.3354/meps190179
Böhringer, N., Fisch, K. M., Schillo, D., Bara, R., Hertzer, C., Grein, F., et al. (2017). Antimicrobial potential of bacteria associated with marine sea slugs from North Sulawesi, Indonesia. Front. Microbiol. 8:1092. doi: 10.3389/fmicb.2017.01092
Brener-Raffalli, K., Clerissi, C., Vidal-Dupiol, J., Adjeroud, M., Bonhomme, F., Pratlong, M., et al. (2018). Thermal regime and host clade, rather than geography, drive Symbiodinium and bacterial assemblages in the scleractinian coral Pocillopora damicornis sensu lato. Microbiome 6:39. doi: 10.1186/s40168-018-0423-6
Bronner, I. F., Quail, M. A., Turner, D. J., and Swerdlow, H. (2014). Improved protocols for illumina sequencing. Curr. Protoc. Hum. Genet. 80, 11–42.
Brooks, A. W., Kohl, K. D., Brucker, R. M., van Opstal, E. J., and Bordenstein, S. R. (2016). Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PLoS Biol. 14:e2000225. doi: 10.1371/journal.pbio.2000225
Buchan, A., González, J. M., and Moran, M. A. (2005). Overview of the marine roseobacter lineage. Appl. Environ. Microbiol. 71, 5665–5677. doi: 10.1128/aem.71.10.5665-5677.2005
Burioli, E. A. V., Varello, K., Trancart, S., Bozzetta, E., Gorla, A., Prearo, M., et al. (2018). First description of a mortality event in adult Pacific oysters in Italy associated with infection by a Tenacibaculum soleae strain. J. Fish. Dis. 41, 215–221. doi: 10.1111/jfd.12698
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2010). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Cárdenas, A., Rodriguez-r, L. M., Pizarro, V., Cadavid, L. F., and Arévalo-Ferro, C. (2012). Shifts in bacterial communities of two caribbean reef-building coral species affected by white plague disease. ISME J. 6:502. doi: 10.1038/ismej.2011.123
Cardenas, C. A., Bell, J. J., Davy, S. K., Hoggard, M., and Taylor, M. W. (2014). Influence of environmental variation on symbiotic bacterial communities of two temperate sponges. FEMS Microbiol. Ecol. 88, 516–527. doi: 10.1111/1574-6941.12317
Carrier, T. J., and Reitzel, A. M. (2018). Convergent shifts in host-associated microbial communities across environmentally elicited phenotypes. Nat. Commun. 9:952. doi: 10.1038/s41467-018-03383-w
Carrier, T. J., Wolfe, K., Lopez, K., Gall, M., Janies, D. A., Byrne, M., et al. (2018). Diet-induced shifts in the crown-of-thorns (Acanthaster sp.) larval microbiome. Mar. Biol. 165:157.
Ceh, J., Raina, J. B., Soo, R. M., van Keulen, M., and Bourne, D. G. (2012). Coral-bacterial communities before and after a coral mass spawning event on ningaloo reef. PLoS One 7:e36920. doi: 10.1371/journal.pone.0036920
Cleary, D. F., Becking, L. E., Polónia, A. R., Freitas, R. M., and Gomes, N. C. (2015). Composition and predicted functional ecology of mussel-associated bacteria in indonesian marine lakes. Antonie Van Leeuwenhoek 107, 821–834. doi: 10.1007/s10482-014-0375-1
Coleman-Derr, D., Desgarennes, D., Fonseca-Garcia, C., Gross, S., Clingenpeel, S., Woyke, T., et al. (2016). Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol. 209, 798–811. doi: 10.1111/nph.13697
Collado, L., Jara, R., Vaìsquez, N., and Telsaint, C. (2014). Antimicrobial resistance and virulence genes of Arcobacter isolates recovered from edible bivalve molluscs. Food Control 46, 508–512. doi: 10.1016/j.foodcont.2014.06.013
Costello, E. K., Lauber, C. L., Hamady, M., Fierer, N., Gordon, J. I., and Knight, R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
Cuif, J. P., Perez-Huerta, A., Lo, C., Belhadj, O., and Dauphin, Y. (2018). On the deep origin of the depressed rings on pearl surface illustrated from Polynesian Pinctada margaritifera (Linnaeus 1758). Aquacult. Res. 49, 834–1847.
Dang, H., Li, T., Chen, M., and Huang, G. (2008). Cross-ocean distribution of Rhodobacterales bacteria as primary surface colonizers in temperate coastal marine waters. Appl. Environ. Microbiol. 74, 52–60. doi: 10.1128/aem.01400-07
Davies, M. S., and Beckwith, P. (1999). Role of mucus trails and trail-following in the behaviour and nutrition of the periwinkle Littorina littorea. Mar. Ecol. Prog. Ser. 179, 247–257. doi: 10.3354/meps179247
De Cáceres, M., and Legendre, P. (2009). Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574. doi: 10.1890/08-1823.1
Defer, D., Desriac, F., Henry, J., Bourgougnon, N., Baudy-Floc’h, M., Brillet, B., et al. (2013). Antimicrobial peptides in oyster hemolymph: the bacterial connection. Fish Shellfish Immunol. 34, 1439–1447. doi: 10.1016/j.fsi.2013.03.357
Dickschat, J. S., Wickel, S., Bolten, C. J., Nawrath, T., Schulz, S., and Wittmann, C. (2010). Pyrazine biosynthesis in Corynebacterium glutamicum. Eur. J. Org. Chem. 14, 2687–2695. doi: 10.1002/ejoc.201000155
Dimitrieva, G. Y., Crawford, R. L., and Yüksel, G. U. (2006). The nature of plant growth- promoting effects of a pseudoalteromonad associated with the marine algae Laminaria japonica and linked to catalase excretion. J. Appl. Microbiol. 100, 1159–1169. doi: 10.1111/j.1365-2672.2006.02831.x
Dumbauld, B. R., Ruesink, J. L., and Rumrill, S. S. (2009). The ecological role of bivalve shellfish aquaculture in the estuarine environment: a review with application to oyster and clam culture in West Coast (USA) estuaries. Aquaculture 290, 196–223. doi: 10.1016/j.aquaculture.2009.02.033
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Engelen, A. H., Aires, T., Vermeij, M. J. A., Herndl, G. J., Serrão, E. A., and Frade, P. R. (2018). Host differentiation and compartmentalization of microbial communities in the azooxanthellate cupcorals Tubastrea coccinea and Rhizopsammia goesi in the Caribbean. Front. Mar. Sci. 5:391. doi: 10.3389/fmars.2018.00391
Espinosa, E. P., Koller, A., and Allam, B. (2016). Proteomic characterization of mucosal secretions in the eastern oyster, Crassostrea virginica. J. Proteomics 132, 63–76. doi: 10.1016/j.jprot.2015.11.018
Faust, K., Lima-Mendez, G., Lerat, J. S., Sathirapongsasuti, J. F., Knight, R., Huttenhower, C., et al. (2015). Cross-biome comparison of microbial association networks. Front. Microbiol. 6:1200. doi: 10.3389/fmicb.2015.01200
Fernaìndez-Goìmez, B., Richter, M., Schüler, M., Pinhassi, J., Acinas, S. G., Gonzaìlez, J. M., et al. (2013). Ecology of marine bacteroidetes: a comparative genomics approach. ISME J. 7:1026. doi: 10.1038/ismej.2012.169
Fernaìndez-Piquer, J., Bowman, J. P., Ross, T., and Tamplin, M. L. (2012). Molecular analysis of the bacterial communities in the live Pacific oyster (Crassostrea gigas) and the influence of postharvest temperature on its structure. Appl. Microbiol. 112, 1134–1143. doi: 10.1111/j.1365-2672.2012.05287.x
Foster, K. R., Schluter, J., Coyte, K. Z., and Rakoff-Nahoum, S. (2017). The evolution of the host microbiome as an ecosystem on a leash. Nature 548:43. doi: 10.1038/nature23292
Gauthier, M. E. A., Watson, J. R., and Degnan, S. M. (2016). Draft genomes shed light on the dual bacterial symbiosis that dominates the microbiome of the coral reef sponge Amphimedon queenslandica. Front. Mar. Sci. 3:196. doi: 10.3389/fmars.2016.00196
Gervis, M. H., and Sims, N. (1992). The biology and culture of pearl oysters (Bivalvia: Pteriidae). ICLARM Stud. Rev. 21, 1–49.
Glockner, F. O., Kube, M., Bauer, M., Teeling, H., Lombardot, T., Ludwid, W., et al. (2003). Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc. Natl. Acad. Sci. U.S.A. 100, 8298–8303.
Gonzalez, J. M., Kiene, R. P., and Moran, M. A. (1999). Transformation of sulfur compounds by an abundant lineage of marine bacteria in the alpha-subclass of the class Proteobacteria. Appl. Environ. Microbiol. 65, 3810–3819.
Gonzalez, J. M., Simo, R., Massana, R., Covert, J. S., Casamayor, E. O., Pedros-Alio, C., et al. (2000). Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl. Environ. Microbiol. 66, 4237–4246. doi: 10.1128/aem.66.10.4237-4246.2000
Green, T. J., and Barnes, A. C. (2010). Bacterial diversity of the digestive gland of Sydney rock oysters, Saccostrea glomerata infected with the paramyxean parasite, Marteilia Sydney. Appl. Microbiol. 109, 613–622. doi: 10.1111/j.1365-2672.2010.04687.x
Green, T. J., Siboni, N., King, W. L., Labbate, M., Seymour, J. R., and Raftos, D. (2018). Simulated marine heat wave alters abundance and structure of Vibrio populations associated with the Pacific oyster resulting in a mass mortality event. Microb. Ecol. 77, 736–747. doi: 10.1007/s00248-018-1242-9
Hernández-Agreda, A., Gates, R. D., and Ainsworth, T. D. (2017). Defining the core microbiome in corals’ microbial soup. Trends Microbiol. 25, 125–140. doi: 10.1016/j.tim.2016.11.003
Hernández-Agreda, A., Leggat, W., Bongaerts, P., and Ainsworth, T. D. (2016). The microbial signature provides insight into the mechanistic basis of coral success across reef habitats. mBio 7:e00560-16. doi: 10.1128/mBio.00560-16
Hernández-Zárate, G., and Olmos-Soto, J. (2006). Identification of bacterial diversity in the oyster Crassostrea gigas by fluorescent in situ hybridization and polymerase chain reaction. J. Appl. Microbiol. 100, 664–672. doi: 10.1111/j.1365-2672.2005.02800.x
Høj, L., Levy, N., Baillie, B. K., Clode, P. L., Strohmaier, R. C., Siboni, N., et al. (2018). Crown-of-thorns sea star, Acanthaster cf. solaris, have tissue-characteristic microbiomes with potential roles in health and reproducti&on. Appl. Environ. Microbiol. 84:e00181-18. doi: 10.1128/AEM.00181-18
Huang, Z. B., Guo, F., Zhao, J., Li, W. D., and Ke, C. H. (2010). Molecular analysis of the intestinal bacterial flora in cage-cultured adult small abalone, Haliotis diversicolor. Aquacult. Res. 41, e760–e769. doi: 10.1111/j.1365-2109.2010.02577.x
Ison, S. A., Delannoy, S., Bugarel, M., Nagaraja, T. G., Renter, D. G., den Bakker, H. C., et al. (2016). Targeted amplicon sequencing for single-nucleotide-polymorphism genotyping of attaching and effacing Escherichia Coli O26:H11 cattle strains via a high-throughput library preparation technique. Appl. Environ. Microbiol. 82, 640–649. doi: 10.1128/AEM.03182-15
Jaenike, J. (2012). Population genetics of beneficial heritable symbionts. Trends Ecol. Evol. 27, 226–232. doi: 10.1016/j.tree.2011.10.005
Kamada, N., Chen, G. Y., Inohara, N., and Nunez, G. (2013). Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 14, 685–690. doi: 10.1038/ni.2608
Kellogg, C. A., Lisle, J. T., and Galkiewicz, J. P. (2009). Culture independent characterization of bacterial communities associated with the cold-water coral Lophelia pertusa in the northern Gulf of Mexico. Appl. Environ. Microbiol. 75, 2294–2303. doi: 10.1128/aem.02357-08
King, G. M., Judd, C., Kuske, C. R., and Smith, C. (2012). Analysis of stomach and gut microbiomes of the eastern oyster (Crassostrea virginica) from Coastal Louisiana, USA. PLoS One 7:e51475. doi: 10.1371/journal.pone.0051475
King, W. L., Jenkins, C., Go, J., Siboni, N., Seymour, J. R., and Labbate, M. (2018). Characterisation of the Pacific oyster microbiome during a summer mortality event. Microb. Ecol. 77, 502–512. doi: 10.1007/s00248-018-1226-9
Kohl, K. D., and Carey, H. V. (2016). A place for host-microbe symbiosis in the comparative physiologist’s toolbox. J. Exp. Biol. 219, 3496–3504. doi: 10.1242/jeb.136325
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Ky, C. L., Broustal, F., Potin, D., and Lo, C. (2019). The pearl oyster (Pinctada margaritifera) aquaculture in french polynesia and the indirect impact of long-distance transfers and collection-culture site combinations on pearl quality traits. Aquacult. Rep. 13:100182. doi: 10.1016/j.aqrep.2019.100182
Ky, C. L., Quillien, V., Broustal, F., Soyez, C., and Devaux, D. (2018). Phenome of pearl quality traits in the mollusc transplant model Pinctada margaritifera. Sci. Rep. 8:2122. doi: 10.1038/s41598-018-20564-1
Langille, M. G., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Laroche, O., Symonds, J. E., Smith, K. F., Banks, J. C., Mae, H., Bowman, J. P., et al. (2018). Understanding bacterial communities for informed biosecurity and improved larval survival in Pacific oysters. Aquaculture 497, 164–173. doi: 10.1016/j.aquaculture.2018.07.052
Le Luyer, J., Auffret, P., Quillien, V., Leclerc, N., Reisser, C., Vidal-Dupiol, J., et al. (2019). Whole transcriptome sequencing and biomineralization gene architecture associated with cultured pearl quality traits in the pearl oyster, Pinctada margaritifera. BMC Genomics 20:111. doi: 10.1186/s12864-019-5443-5
Lema, K. A., Bourne, D. G., and Willis, B. L. (2014). Onset and establishment of diazotrophs and other bacterial associates in the early life history stages of the coral Acropora millepora. Mol. Ecol. 23, 4682–4695. doi: 10.1111/mec.12899
Liang, J., Yu, K., Wang, Y., Huang, X., Huang, W., Qin, Z., et al. (2017). Distinct bacterial communities associated with massive and branching scleractinian corals and potential linkages to coral susceptibility to thermal or cold stress. Front. Microbiol. 8:979. doi: 10.3389/fmicb.2017.00979
Lim, P. S., Sutton, C. R., and Rao, S. (2015). Protein kinase C in the immune system: from signalling to chromatin regulation. Immunology 146, 508–522. doi: 10.1111/imm.12510
Lokmer, A., Goedknegt, M. A., Thieltges, D. W., Fiorentino, D., Kuenzel, S., Baines, J. F., et al. (2016a). Spatial and temporal dynamics of Pacific oyster hemolymph microbiota across multiple scales. Front. Microbiol. 7:1367. doi: 10.3389/fmicb.2016.01367
Lokmer, A., Kuenzel, S., Baines, J. F., and Wegner, K. M. (2016b). The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environ. Microbiol. 18, 970–987. doi: 10.1111/1462-2920.13163
Lokmer, A., and Wegner, K. M. (2015). Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 9:670. doi: 10.1038/ismej.2014.160
Lu, F., Liu, M., Simister, R., Webster, N. S., and Thomas, T. (2013). Marine microbial symbiosis heats up: the phylogenetic and functional response of a sponge holobiont to thermal stress. ISME J. 7:991. doi: 10.1038/ismej.2012.165
Lu, G., Wang, F., Yu, Z., Lu, M., Wang, Y., Liu, C., et al. (2017). Bacterial communities in gills and intestines of yesso scallop (Patinopecten yessoensis) and its habitat waters in changhai (Dalian, China). Invertebrate Surviv. J. 14, 340–351.
Mann, A. J., Hahnke, R. L., Huang, S., Werner, J., Xing, P., Barbeyron, T., et al. (2013). The genome of the alga-associated marine flavobacterium Formosa agariphila KMM 3901T reveals a broad potential for degradation of algal polysaccharides. Appl. Environ. Microbiol. 79, 6813–6822. doi: 10.1128/AEM.01937-13
Matsuyama, T., Yasuike, M., Fujiwara, A., Nakamura, Y., Takano, T., Takeuchi, T., et al. (2017). A Spirochaete is suggested as the causative agent of Akoya oyster disease by metagenomic analysis. PLoS One 12:e0182280. doi: 10.1371/journal.pone.0182280
McFall-Ngai, M., Hadfield, M. G., Bosch, T. C., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A 110, 3229–3236.
Meisterhans, G., Raymond, N., Girault, E., Lambert, C., Bourrasseau, L., Montaudouin, X. D., et al. (2016). Structure of manila clam (Ruditapes philippinarum) microbiota at the organ scale in contrasting sets of individuals. Microb. Ecol. 71, 194–206. doi: 10.1007/s00248-015-0662-z
Moitinho-Silva, L., Bayer, K., Cannistraci, C. V., Giles, E. C., Ryu, T., Seridi, L., et al. (2014). Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol. Ecol. 23, 1348–1363. doi: 10.1111/mec.12365
Mueller, U. G., and Sachs, J. L. (2015). Engineering microbiomes to improve plant and animal health. Trends Microbiol. 23, 606–617. doi: 10.1016/j.tim.2015.07.009
Najiah, M., Nadirah, M., Lee, K. L., Lee, S. W., Wendy, W., Ruhil, H. H., et al. (2008). Bacteria flora and heavy metals in cultivated oysters Crassostrea iredalei of Setiu Wetland, East Coast Peninsular Malaysia. Vet. Res. Commun. 32, 377–381. doi: 10.1007/s11259-008-9045-y
Neave, M. J., Apprill, A., Ferrier-Pagès, C., and Voolstra, C. R. (2016). Diversity and function of prevalent symbiotic marine bacteria in the genus Endozoicomonas. Appl. Microbiol. Biotechnol. 100, 8315–8324. doi: 10.1007/s00253-016-7777-0
Neave, M. J., Michell, C. T., Apprill, A., and Voolstra, C. R. (2017). Endozoicomonas genomes reveal functional adaptation and plasticity in bacterial strains symbiotically associated with diverse marine hosts. Sci. Rep. 7:40579. doi: 10.1038/srep40579
Nechitaylo, T. Y., Timmis, K. N., and Golyshin, P. N. (2009). Candidatus Lumbricincola, a novel lineage of uncultured Mollicutes from earthworms of family Lumbricidae. Environ. Microbiol. 11, 1016–1026.
Nematollahi, A., Decostere, A., Pasmans, F., and Haesebrouck, F. (2003). Flavobacterium psychrophilum infections in salmonid fish. J. Fish Dis. 26, 563–574. doi: 10.1046/j.1365-2761.2003.00488.x
Pantos, O., Bongaerts, P., Dennis, P. G., Tyson, G. W., and Hoegh-Guldberg, O. (2015). Habitat-specific environmental conditions primarily control the microbiomes of the coral Seriatopora hystrix. ISME J. 9:1916. doi: 10.1038/ismej.2015.3
Pierce, M. L., Ward, J. E., Holohan, B. A., Zhao, X., and Hicks, R. E. (2016). The influence of site and season on the gut and pallial fluid microbial communities of the eastern oyster, Crassostrea virginica (Bivalvia, Ostreidae): community-level physiological profiling and genetic structure. Hydrobiologia 765, 97–113. doi: 10.1007/s10750-015-2405-z
Potgieter, M., Bester, J., Kell, D. B., and Pretorius, E. (2015). The dormant blood microbiome in chronic, inflammatory diseases. FEMS Microbiol. Rev. 39, 567–591. doi: 10.1093/femsre/fuv013
Rabus, R., Hansen, T. A., and Widdel, F. (2006). “Dissimilatory sulfate- and sulfur-reducing prokaryotes,” in The Prokaryotes – a Handbook on the Biology of Bacteria, eds M. Dworkin, S. Falkow, H. Rosen-berg, K.-H. Schleifer, and E. Stackebrandt. (New York, NY: Springer), 659–768. doi: 10.1007/0-387-30742-7_22
Reveillaud, J., Maignien, L., Eren, A. M., Huber, J. A., Apprill, A., Sogin, M. L., et al. (2014). Host-specificity among abundant and rare taxa in the sponge microbiome. ISME J. 8:1198. doi: 10.1038/ismej.2013.227
Ríos-Castillo, A. G., Thompson, K. D., Adams, A., Marín de Mateo, M., and Rodríguez-Jerez, J. J. (2018). Biofilm formation of Flavobacterium psychrophilum on various substrates. Aquacult. Res. 49, 3830–3837. doi: 10.1111/are.13849
Roterman, Y. R., Benayahu, Y., Reshef, L., and Gophna, U. (2015). The gill microbiota of invasive and indigenous Spondylus oysters from the Mediterranean Sea and northern Red Sea. Environ. Microbiol. Rep. 7, 860–867. doi: 10.1111/1758-2229.12315
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schmitt, S., Tsai, P., Bell, J., Fromont, J., Ilan, M., Lindquist, N., et al. (2012). Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6:564. doi: 10.1038/ismej.2011.116
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Siewe, R. M., Weil, B., Burkovski, A., Eggeling, L., Krämer, R., and Jahns, T. (1998). Urea uptake and urease activity in Corynebacterium glutamicum. Arch. Microbiol. 169, 411–416. doi: 10.1007/s002030050591
Slobodkin, A. (2015). “Tepidibacter”. Bergey’s Manual of Systematics of Archaea and Bacteria. Hoboken, NJ: Wiley.
Smith, A. M., Quick, T. J., and St Peter, R. L. (1999). Differences in the composition of adhesive and non-adhesive mucus from the limpet Lottia limatula. Biol. Bull. 196, 34–44. doi: 10.2307/1543164
Sweet, M. J., and Bulling, M. T. (2017). On the importance of the microbiome and pathobiome in coral health and disease. Front. Mar. Sci. 4:9. doi: 10.3389/fmars.2017.00009
Tang, K., Huang, H., Jiao, N., Wu, C. H., and Ch, W. (2010). Phylogenomic analysis of marine Roseobacters. PLoS One 5:e11604. doi: 10.1371/journal.pone.0011604
Taniguchi, A., Yoshida, T., Hibino, K., and Eguchi, M. (2015). Community structures of actively growing bacteria stimulated by coral mucus. J. Exp. Mar. Biol. Ecol. 469, 105–112. doi: 10.1016/j.jembe.2015.04.020
Taylor, J., and Strack, E. (2008). “Pearl production,” in The Pearl Oyster, eds P. C. Southgate and J. S. Lucas (Amsterdam: Elsevier).
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/mmbr.00040-06
Thomas, F., Hehemann, J.-H., Rebuffet, E., Czjzek, M., and Gurvan, M. (2011). Environmental and gut Bacteroidetes: the food connection. Front. Microbiol. 2:93. doi: 10.3389/fmicb.2011.00093
Tian, R. M., Wang, Y., Bougouffa, S., Gao, Z. M., Cai, L., Bajic, V., et al. (2014). Genomic analysis reveals versatile heterotrophic capacity of a potentially symbiotic sulfur-oxidizing bacterium in sponge. Environ. Microbiol. 16, 3548–3561. doi: 10.1111/1462-2920.12586
Tonon, L. A. C., Moreira, A. P. B., and Thompson, F. (2014). “The family erythrobacteraceae,” in The Prokaryotes, eds E. Rosenberg, E. F. DeLong, S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer), 213–235. doi: 10.1007/978-3-642-30197-1_376
Trabal, N., Mazón-Suástegui, J. M., Vázquez-Juárez, R., Ascencio-Valle, F., Morales-Bojórquez, E., and Romero, J. (2012). Molecular analysis of bacterial microbiota associated with oysters (Crassostrea gigas and Crassostrea corteziensis) in different growth phases at two cultivation sites. Microb. Ecol. 64, 555–569. doi: 10.1007/s00248-012-0039-5
Trabal-Fernández, N., Mazón-Suástegui, J. M., Vázquez-Juárez, R., Ascencio-Valle, F., and Romero, J. (2014). Changes in the composition and diversity of the bacterial microbiota associated with oysters (Crassostrea corteziensis, Crassostrea gigas and Crassostrea sikamea) during commercial production. FEMS Microbiol. Ecol. 88, 69–83. doi: 10.1111/1574-6941.12270
Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007). The human microbiome project. Nature 449:804.
van de Water, J. A., Melkonian, R., Junca, H., Voolstra, C. R., Reynaud, S., Allemand, D., et al. (2016). Spirochaetes dominate the microbial community associated with the red coral Corallium rubrum on a broad geographic scale. Sci. Rep. 6:27277. doi: 10.1038/srep27277
van Oppen, M. J. H., Bongaerts, P., Frade, P., Peplow, L. M., Boyd, S. E., Nim, H. T., et al. (2018). Adaptation to reef habitats through selection on the coral animal and its associated microbiome. Mol. Ecol. 27, 2956–2971. doi: 10.1111/mec.14763
Vandenkoornhuyse, P., Quaiser, A., Duhamel, M., Le Van, A., and Dufresne, A. (2015). The importance of the microbiome of the plant holobiont. New Phytol. 206, 1196–1206. doi: 10.1111/nph.13312
Vezzuli, L., Stagnaro, L., Grande, C., Tassistro, G., Canesi, L., and Pruzzo, C. (2018). Comparative 16SrDNA gene-based microbiota profiles of the Pacific oyster (Crassostrea gigas) and the Mediterranean mussel (Mytilus galloprovincialis) from a shellfish farm (Ligurian Sea, Italy). Microb. Ecol. 75, 495–504. doi: 10.1007/s00248-017-1051-6
Ward, J. E., and Shumway, S. E. (2004). Separating the grain from the chaff: particle selection in suspension-and deposit-feeding bivalves. J. Exp. Mar. Biol. Ecol. 300, 83–130. doi: 10.1016/j.jembe.2004.03.002
Wegner, K. M., Volkenborn, N., Peter, H., and Eiler, A. (2013). Disturbance induced decoupling between host genetics and composition of the associated microbiome. BMC Microbiol. 13:252. doi: 10.1186/1471-2180-13-252
Wessels, W., Sprungala, S., Watson, S. A., Miller, D. J., and Bourne, D. G. (2017). The microbiome of the octocoral Lobophytum pauciflorum: minor differences between sexes and resilience to short-term stress. FEMS Microbiol. Ecol. 93:fix013. doi: 10.1093/femsec/fix013
Zhang, M., Sun, Y., Chen, L., Cai, C., Qiao, F., Du, Z., et al. (2016). Symbiotic bacteria in gills and guts of chinese mitten crab (Eriocheir sinensis) differ from the free-living bacteria in water. PLoS One 11:e0148135. doi: 10.1371/journal.pone.0148135
Zhang, X., Sun, Z., Zhang, X., Zhang, M., and Li, S. (2018). Hemolymph microbiomes of three aquatic invertebrates as revealed by a new cell extraction method. Appl. Environ. Microbiol. 84:e0282417. doi: 10.1128/AEM.02824-17
Zubkov, M. V., Fuchs, B. M., Archer, S. D., Kiene, R. P., Amann, R., and Burkill, P. H. (2001). Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ. Microbiol. 3, 304–311. doi: 10.1046/j.1462-2920.2001.00196.x
Keywords: pearl oyster, microbiome, tissue-specific bacterial communities, 16S rRNA gene sequencing, functional profiling prediction
Citation: Dubé CE, Ky C-L and Planes S (2019) Microbiome of the Black-Lipped Pearl Oyster Pinctada margaritifera, a Multi-Tissue Description With Functional Profiling. Front. Microbiol. 10:1548. doi: 10.3389/fmicb.2019.01548
Received: 20 March 2019; Accepted: 20 June 2019;
Published: 05 July 2019.
Edited by:
Sébastien Duperron, Muséum National d’Histoire Naturelle (France), FranceReviewed by:
Konstantinos Ar. Kormas, University of Thessaly, GreeceCopyright © 2019 Dubé, Ky and Planes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Eve Dubé, Y2Fyb2xpbmUuZHViZS5xY0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.