 Yuxin Hu
Yuxin Hu Weiyue Xing1,2
Weiyue Xing1,2 Guoxiang Liu
Guoxiang Liu- 1Key Laboratory of Algal Biology, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences, Qingdao, China
- 4State Key Laboratory of Freshwater Ecology and Biotechnology, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China
This study is the first determination of six chloroplast genomes of colonial volvocine algae, Colemanosphaera charkowiensis, Volvulina compacta, Pandorina colemaniae, Pandorina morum, Colemanosphaera angeleri, and Yamagishiella unicocca. Based on 55 chloroplast protein-coding genes, we compared the nonsynonymous (dN) and synonymous (dS) substitution rates between colonial volvocine algae and the other unicellular Chlamydomonadales species. When refer to the dN, we found 27 genes were significantly different, among them, 19 genes were significant higher in unicellular species (FDR-adjusted P < 0.05). When refer to the dS, we found 10 genes were significantly different, among them, 6 genes were significant higher in unicellular species (FDR-adjusted P < 0.05). Then we identified 14 putative fast-evolving genes and 11 putative positively selected genes of unicellular species, we analyzed the function of positively selected sites of the overlap genes of putative fast-evolving and positively selected genes, and found some sites were close to the important functional region of the proteins. Photosynthesis is the process to transform and store solar energy by chloroplast, it plays a vital role in the survival of algae, this study is the first to use the chloroplast genomes to analysis the evolutionary relationship between colonial and unicellular species in Chlamydomonadales. We found more genes have higher substitution rates in unicellular species and proposed that the fast-evolving and positively selected two genes, psbA and psbC, may help to improve the photosynthetic efficiency of unicellular species in Chlamydomonadales.
Introduction
The volvocine algae belong to Chlamydomonadales (Chlorophyta, Chlorophyceae). This group of algae span the full range of organizational complexity, from unicellular species to colonial species, thus these algae are ideal model organisms to study the fundamental issues related to the transition to multicellularity. In recent years, many chloroplast genomes of Chlamydomonadales species have been sequenced using the application of next generation sequencing technology, this provided massive data for us to study the nucleotide substitution rates based on the chloroplast protein-coding genes data sets (Smith and Lee, 2009; Smith and Lee, 2010; Hamaji et al., 2013; Smith et al., 2013; Del et al., 2015; Lemieux et al., 2015; Featherston et al., 2016; Hu et al., 2019). However, due to the limited number of chloroplast genomes of colonial volvocine algae, little is known about the evolutionary relationship between colonial and unicellular species in Chlamydomonadales based on chloroplast genomes.
The nucleotide substitution rates are often used as the criterion to reflect the selection pressure. The nonsynonymous substitution rates (dN) can cause an amino acid change, the synonymous substitution rates (dS) do not cause an amino acid change. The dN/dS is the ratio of nonsynonymous substitution and synonymous substitution, the ratio of dN/dS is the measure of natural selection acting on the protein. According to Yang (Yang, 2007), dN/dS < 1 means negative purifying selection, dN/dS = 1 means neutral evolution, dN/dS > 1 means positive selection. Generally, the chloroplast protein-coding genes need to maintain the photosynthetic function, so these genes are conserved and have a low dN/dS ratio (Smith, 2015). Meanwhile, a relatively high dN/dS ratio could be interpreted as the positive or relaxed selection (Guisinger et al., 2008), the evolutionary analysis can reveal how species adaptive under selection pressure. For example, Guisinger et al. (2008) calculated the nucleotide substitution rates of chloroplast protein-coding genes of angiosperm, and found unprecedented accumulation of nucleotide substitutions in Geraniaceae, then a model was proposed to illustrate this phenomenon. Zhang et al. (2018) studied the molecular evolution of chloroplast protein-coding genes of an Antarctic sea ice alga Chlamydomonas sp. and revealed the adaptive mechanism of sed-ice environment.
In this study, we determined six chloroplast genomes of colonial volvocine algae, these genomes provide valuable opportunity for us to conduct the evolutionary study in Chlamydomonadales. Based on the chloroplast protein-coding genes, we examined the nucleotide substitution rates of Chlamydomonadales species and found that more genes have higher substitution rates in unicellular species when compared with colonial species. Then we identified the putative fast-evolving genes and positively selected genes of unicellular species, based on our analysis, we proposed that the positively selected sites of specific chloroplast protein-coding genes may improve the photosystem efficiency, and our photosynthetic experiment further support our conclusion.
Materials and Methods
Sampling, Culture Conditions, DNA Extraction, and Species Identification
The strains described in this study were isolated from water samples and deposited in the Freshwater Algae Culture Collection at the Institute of Hydrobiology (FACHB collection), Wuhan, Hubei Province, China. The Colemanosphaera charkowiensis (strain FACHB-2326) were collected from a river in Weihe (36°19′7″ N, 119°28′54″ E), Weifang, Shandong Province, China, in September 2017. The Volvulina compacta (strain FACHB-2337) were collected from a river in Tangxihe (31°7′13″ N, 108°49′10″ E), Chongqing, China, in June 2017. The Pandorina colemaniae (strain FACHB-2361) were collected from a river in Wuhe (36°22′25″ N, 119°24′52″ E), Weifang, Shandong Province, China, in September 2017. The Pandorina morum (strain FACHB-2362) were collected from a river in Weihe (36°8′1″ N, 119°25′59″ E), Weifang, Shandong Province, China, in September 2017. The Colemanosphaera angeleri (strain FACHB-2363) were collected from a river in Weihe (36°30′2″ N, 119°24′44″ E), Weifang, Shandong Province, China, in September 2017. The Yamagishiella unicocca (strain FACHB-2364) were collected from a pool in Dichi (47°18′20″ N, 120°28′38″ E), Aershan, Inner Mongolia, China, in August 2017. The strains were grown in a conical flask containing artificial freshwater-6 (AF-6) (Kato, 1982) at 20–25°C under a 14 h light: 10 h dark schedule under cool-white fluorescent lamps at an intensity of 1000–2000 lux. Total genomic DNA was extracted using a Universal DNA Isolation Kit (AxyPrep, Suzhou, China) following the manufacturer’s instructions. Species identification was based on morphological observation and phylogenetic analysis based on five chloroplast genes (rbcL, atpB, psaA, psaB, and psbC), and the sequence data of related species were chosen according to Nozaki et al. (2014). The sequence matrix was aligned by MAFFT v7.394 (Katoh and Standley, 2013), and the ambiguously aligned regions were further manually edited and adjusted by eye using MEGA7 (Kumar et al., 2016). jModelTest v.2.1.7 (Darriba et al., 2012) was used to determine the evolutionary model, which was then analyzed using Bayesian inference (BI) with MrBayes v3.2.6 (Ronquist et al., 2012) and maximum likelihood (ML) with RAxML v8.2.10 (Stamatakis, 2014). Microphotographs were taken by using an Olympus BX53 (Tokyo, Japan) light microscope with an Olympus DP80 digital camera and cellSens standard image analysis software (Tokyo, Japan).
Library Preparation, Sequencing, Genome Assembly, and Annotation
A sequencing library was prepared using an NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs, United States) and sequenced with an Illumina NovaSeq6000 at Novogene (Beijing, China). The data were trimmed using SOAPnuke v1.3.0 (Chen et al., 2017) and then assembled with SPAdes v3.10.1 (Bankevich et al., 2012). The resulting assembly contigs were determined to be form the chloroplast genome based on the following criteria: (1) blast searches of publicly available chloroplast genomes of Chlorophyta algae species with significant e-values (1e-5); (2) the GC content of the contig is less than 45% (the GC content of green algae chloroplast genomes that have been sequenced to date is normally less than 45%); and (3) the sequencing depth is higher than 100×. Then, the trimmed reads were aligned to the resulting assembly contigs by BWA-MEM v0.7.12 (Li, 2013). If reads mapped two contigs at the same time, we determined the order of contigs, and after confirming the orders of the contigs, the sequence we produced was then rechecked by Sanger dideoxy sequencing technology and synteny analysis with related species. The chloroplast genomes were initially annotated using CpGAVAS (Liu et al., 2012). Protein-coding genes were further polished using Blast with genes from the available colonial volvocine chloroplast genes. All chloroplast genome sequences have been submitted to GenBank, the accession number was listed in Table 1.
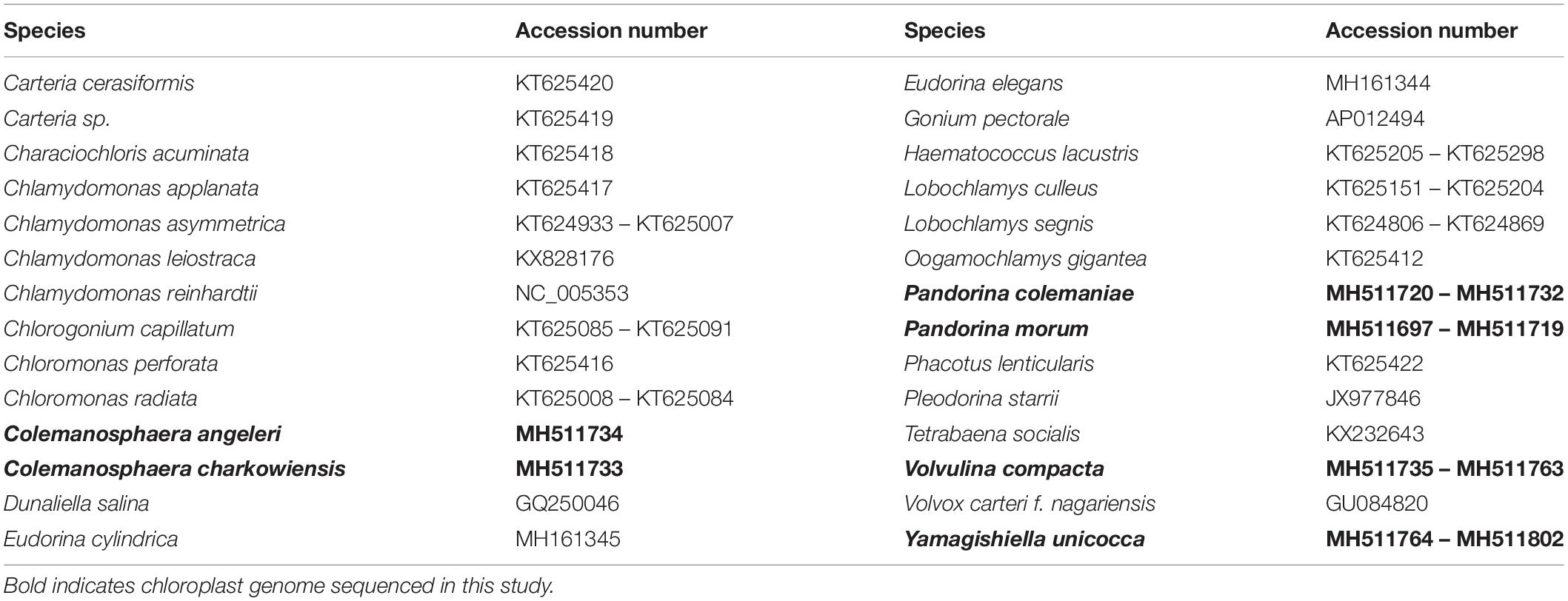
Table 1. GenBank accession numbers of Chlamydomonadales species used in this study.
Phylogenomic Analysis
Our study aims to reveal the evolutionary relationship between colonial and unicellular species in Chlamydomonadales based on the protein-coding genes of chloroplast genomes, we tried to ensure the gene we analyzed are exist in all species, however, we found some species may have limited number of protein-coding genes and some genes of specific species have poor alignment with other species, to keep a balance between the number of genes and the number of species, 12 colonial species and 16 unicellular species were chosen and species information was listed in Table 1. The data set was assembled from the following 55 protein-coding genes: atpA, atpB, atpE, atpF, atpH, atpI, ccsA, cemA, chlB, chlL, chlN, clpP, petA, petB, petD, petG, petL, psaB, psaC, psaJ, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbK, psbL, psbM, psbN, psbT, psbZ, rbcL, rpl14, rpl16, rpl2, rpl20, rpl23, rpl36, rpl5, rps11, rps12, rps14, rps18, rps19, rps2, rps3, rps4, rps7, rps8, rps9, tufa, and ycf4. The method of phylogenomic analysis was mainly refer to Lemieux et al. (2015). All genes were aligned using MUSCLE v3.8 (Edgar, 2004), and the alignments of all genes were converted into a codon alignment by TranslatorX (Abascal et al., 2010). The ambiguously aligned regions in alignment were excluded using Gblocks0.91b (Castresana, 2000) with the options −t = c, −b3 = 5, −b4 = 5, and −b5 = half. All alignments were concatenated using Phyutility v2.2.6 (Smith and Dunn, 2008), and then the Degen1.pl 1.2 script (Regier et al., 2010) was applied to the concatenated alignment. jModeltest v.2.1.7 (Darriba et al., 2012) was used to determine the evolutionary model. The data was partitioned by gene, with the model applied to each partition. Phylogenies were inferred using ML and BI methods. ML analyses were carried out using RAxML v8.2.10 (Stamatakis, 2014) and the GTRGAMMA model of sequence evolution. Bootstrap analysis with 1,000 replicates of the dataset for ML was performed to estimate statistical reliability. Bayesian analyses were performed with MrBayes v3.2.6 (Ronquist et al., 2012) with the GTR + I + G model. Markov chain Monte Carlo (MCMC) analyses were run with four Markov chains (three heated, one cold) for 1,000,000 generations, with trees sampled every 500 generations. Each time the diagnostics were calculated, a fixed proportion of samples (burninfrac = 0.25) were discarded from the beginning of the chain. A stationary distribution was assumed when the average standard deviation of the split frequencies was lower than 0.01.
Evolutionary Analysis
The CODEML program of PAML v4.9 (Yang, 2007) with the ML model (runmode = −2, CodonFreq = 2) was used to measure the values of dS and dN, the analysis was based on 55 chloroplast protein-coding genes. As Chloromonas radiata belongs, with Carteria cerasiformis and Carteria sp., to a clade that is sister to all the other species, so C. radiata was used as reference. Comparisons of the evolutionary rates were conducted using the two-tailed Wilcoxon rank sum test. The multiple testing was corrected by applying the false discovery rate method (FDR) (Benjamini and Hochberg, 1995) as implemented in R.1 The phylogenetic tree was used as a constraint tree, but branch lengths were inferred by using PAML.
The ML method is a pairwise approach to estimate the dN/dS ratio, a dN/dS ratio may indicate in one or both species, and some specific sites under positive selection may remain undetected (Dussert et al., 2018). So, two precise assessments were used to detected the difference of dN/dS and positive selection.
We use the branch model to test whether unicellular species have a different dN/dS ratio relative to the colonial species. The unicellular species were labeled as the foreground branch. A null model (model = 0), where one dN/dS ratio was fixed across all species, was compared with an alternative model (model = 2), where the unicellular species was allowed to have a different dN/dS. Likelihood ratio tests (LRT) were used to test model fit and the Chi-square test was applied for testing P values. The multiple testing was corrected by FDR. Genes were considered putative fast-evolving genes if they had an FDR-adjusted P < 0.05 and a higher dN/dS ratio in the foreground branch than in the background branches.
We use the branch-site model to find genes that potentially experienced positive selection. The improved branch-site model (model = 2, Nsites = 2) was used to detect signatures of positive selection on individual codons in a specific branch (Zhang et al., 2005). The unicellular species were set as the foreground branch. The null model assumed no positive selection occurred on the foreground branch (fix_omega = 1, omega = 1), and the alternative model assumed that sites on the foreground branch were under positive selection (fix_omega = 0, omega = 2). LRT were used to test model fit and the Chi-square test was applied for testing P values. We performed a correction for multiple testing using an FDR criterion, and Bayes empirical Bayes (BEB) method was used to statistically identify sites under positive selection. Genes were considered putative selected genes if they had an FDR-adjusted P < 0.05.
The overlap gene represent a group of genes that are not only putative fast-evolving genes but also putative positive selected genes, we analyzed the function of positive selected sites of overlap genes, the functional information were derived from the Uniprot.2 The three-dimensional (3D) structures were predicted using Phyre2 (Kelley et al., 2015). The 3D structures were visualized by ePlant Web server (Fucile et al., 2011).
Results
Species Identification
Vegetative colonies of strain FACHB-2326 were ellipsoidal in shape, composed by 16 cells of approximately identical sizes embedded by gelatinous matrix forming a hollow colonial structure. Cells spherical, the chloroplast contained more than two pyrenoids of almost identical size (Figure 1A) and prominent longitudinal striations (Figure 1B). Only two contractile vacuoles distributed near the base of the flagella (Figure 1B). Based on the morphology of colony, the number and size of pyrenoids, the number of contractile vacuoles, we identified strain FACHB-2326 as C. charkowiensis. Vegetative colonies of strain FACHB-2337 were ellipsoidal in shape, cells were embedded in a gelatinous matrix forming a hollow sphere. Cells were more or less contiguous and appeared nearly tetragonal by mutual compression. There were no prominent spaces between adjoining cells. Based on the spaces between adjoining cells and the shape of cells, we identified strain FACHB-2337 as V. compacta (Figures 1C,D). Vegetative colonies of strain FACHB-2361 were ellipsoidal in shape and contained eight cells compactly arranged in a gelatinous matrix. The chloroplast had more than two pyrenoids. Based on the shape of cells and the number of pyrenoids, we identified strain FACHB-2361 as P. colemaniae (Figures 1E,F). Vegetative colonies of strain FACHB-2362 resembled strain FACHB-2361, but the chloroplast contained a single, basal pyrenoid, so we can identify strain FACHB-2362 as P. morum (Figures 1G,H). Vegetative colonies of strain FACHB-2363 resembled strain FACHB-2326, but the chloroplast striations were not prominent and contained a large basal pyrenoid and small pyrenoids, so we identified strain FACHB-2363 as C. angeleri (Figures 1I,J). Vegetative colonies of strain FACHB-2364 resembled strain FACHB-2363, but the chloroplast only contained a single, basal pyrenoid, so we identified strain FACHB-2364 as Y. unicocca (Figures 1K,L).
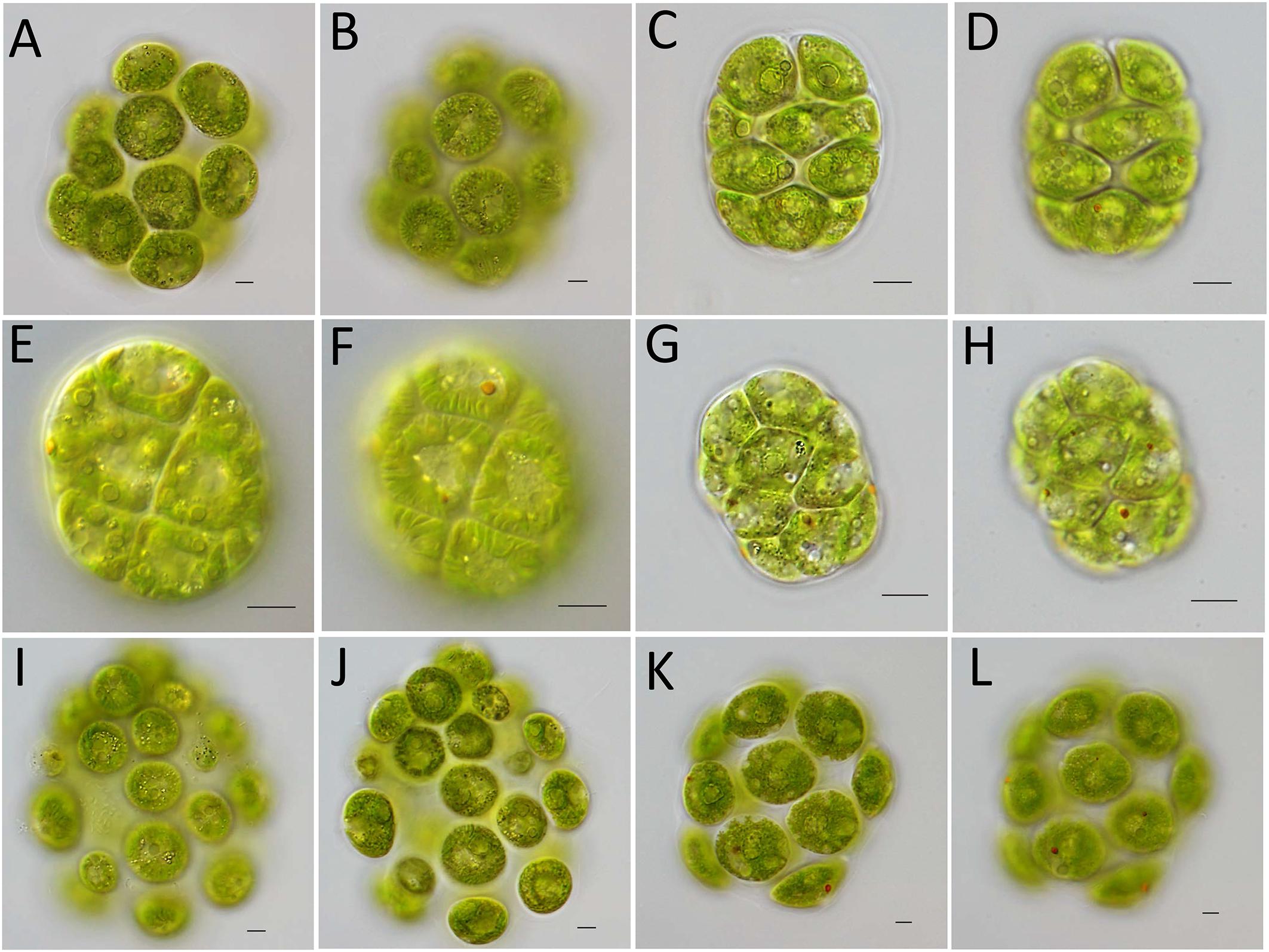
Figure 1. Light microscopy of vegetative colonies of six species of colonial volvocine algae. (A,B) Colemanosphaera charkowiensis, strain FACHB-2326. (A) The chloroplast had more than two pyrenoids of almost identical size. (B) Only two contractile vacuoles distributed near the base of the flagella, the chloroplast had prominent longitudinal striations. (C,D) Volvulina compacta, strain FACHB-2337. (C) Colony ellipsoidal in shape, cells were embedded in a gelatinous matrix formed a hollow sphere. (D) Cells were more or less contiguous and appeared nearly tetragonal by mutual compression. (E,F) Pandorina colemaniae, strain FACHB-2361. (E) The chloroplast had more than two pyrenoids. (F) Colony were ellipsoidal in shape and contained 8 cells compactly arranged in a gelatinous matrix. (G,H) Pandorina morum, strain FACHB-2362. (G) The chloroplast contained a single, basal pyrenoid. (H) Colony were ellipsoidal in shape and contained 8 cells compactly arranged in a gelatinous matrix. (I,J) Colemanosphaera angeleri, strain FACHB-2363. (I) Only two contractile vacuoles distributed near the base of the flagella. (J) The chloroplast contained a large basal pyrenoid and small pyrenoids. (K,L) Yamagishiella unicocca, strain FACHB-2364. (K) The chloroplast only contained a single, basal pyrenoid. (L) Only two contractile vacuoles distributed near the base of the flagella. Scale bars: 10 μm.
The phylogenetic tree based on five genes (Figure 2) supported our morphological identification with high bootstrap value (both 100) and Bayesian posterior probability (both 1.00), except the phylogenetic position of strain FACHB-2337. We noticed that strain FACHB-2337 clustered with Volvulina pringsheimii form a lineage. The cells of V. pringsheimii are multiangular or circular and not contiguous in surface view, and the colony of V. pringsheimii is a hollow sphere more resembles with Eudorina. But the colony of strain FACHB-2337 was more compact and resembled with Pandorina, and the cells were contiguous in surface view, the morphological observation strongly supported strain FACHB-2337 as V. compacta rather than V. pringsheimii (Starr, 1962; Nozaki and Kuroiwa, 1990). So, we still considered strain FACHB-2337 as V. compacta. In our phylogenetic tree, the phylogenetic position of most species was consisting with the study of Nozaki et al. (2014), except Volvulina steinii. The bootstrap value and posterior probability of V. steinii clade were relatively low in the study of Nozaki et al. (2014), we found the phylogenetic position of V. steinii in our study was consisting with Nakada et al. (2010), and both studies showed high bootstrap value and posterior probability of V. steinii clade. Meanwhile, recent study both show polyphyletic of genera Pandorina and Volvulina (Coleman, 2001; Nakada et al., 2010; Nozaki et al., 2014), so the phylogenetic position of these species may need further study.
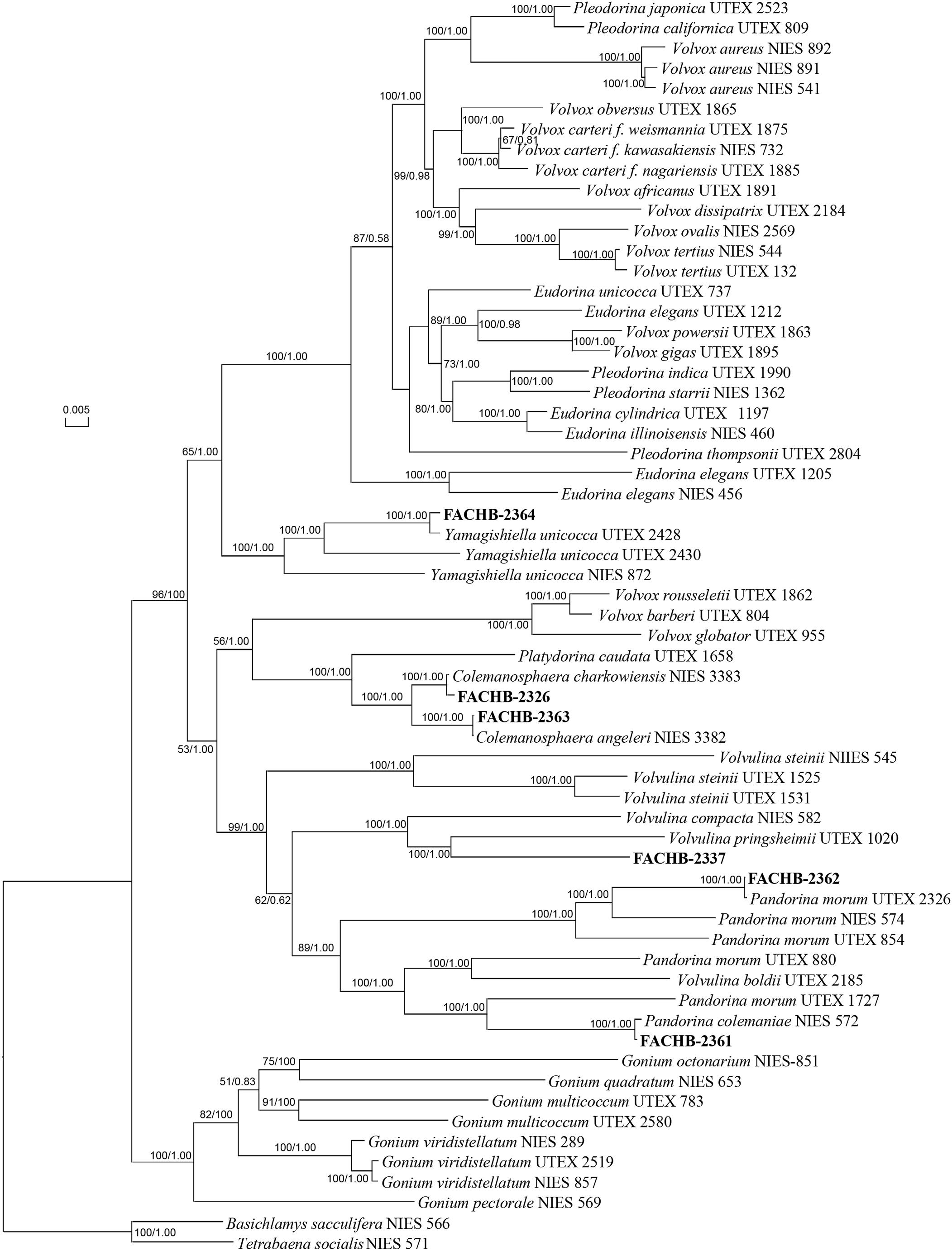
Figure 2. Phylogenetic tree of the colonial volvocine algae based on five chloroplast genes. Numbers on the left and right side at the branches represent bootstrap values and Bayesian posterior probabilities, respectively. Scale bar indicates substitutions per site. Our strains were shown in bold.
Phylogenomic Analysis and Evolutionary Rate Estimation
We conducted our phylogenomic analysis based on the nucleotide sequence of 55 chloroplast protein-coding genes, ML was carried out using RAxML, Bayesian analyses was performed with MrBayes, the phylogenetic position of most species inferred from both methods are the same except Dunaliella salina. The phylogenetic position of D. salina inferred from both method have high bootstrap value (87) and posterior probability (1.00), but the result of ML method was in accordance with previous study (Lemieux et al., 2015), so the ML tree was used to represent the result (Figure 3). The phylogenetic position of other unicellular species were consistent with previous study with high bootstrap values and posterior probability values (Yumoto et al., 2013; Lemieux et al., 2015). The phylogenetic position of most colonial species were consistent with previous study (Nozaki et al., 2014) except P. morum, we noticed that the P. morum together with V. compacta formed a lineage instead of P. colemaniae, this situation have been reported before (Coleman, 2001), this may mainly due to the polyphyletic of Pandorina (Herron, 2016). In our study, this tree was used as the constraint tree for our evolutionary analysis.
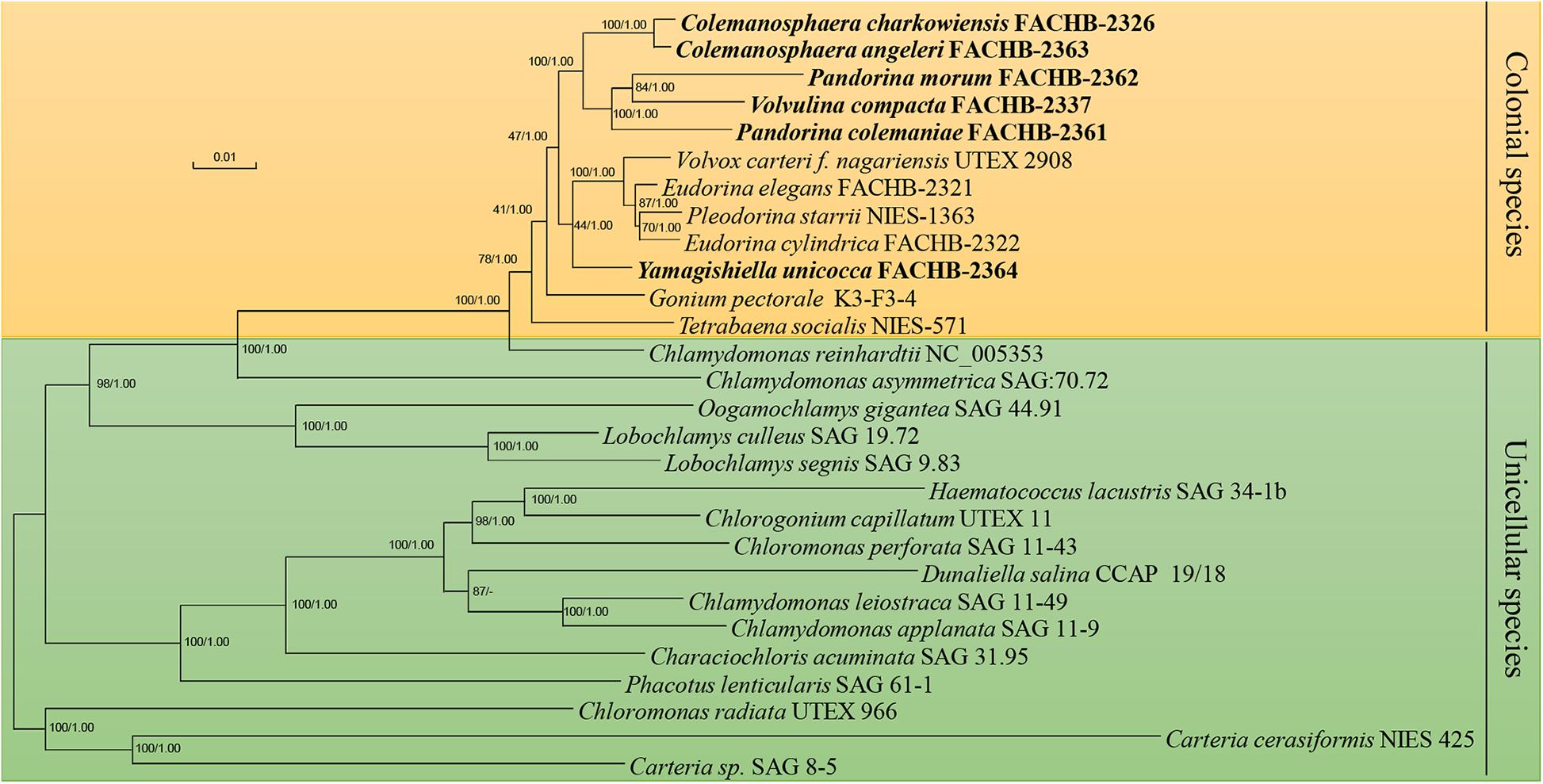
Figure 3. Phylogenetic tree of the Chlamydomonadales species based on the 55 chloroplast genes. Numbers on the left and right side at the branches represent bootstrap values and Bayesian posterior probabilities, respectively. Scale bar indicates substitutions per site. Our strains were shown in bold. The orange background indicated the colonial volvocine algae, and the green background indicated the unicellular species.
Based on the ML method of 55 chloroplast protein-coding genes, the value of dN and dS were compared between colonial and unicellular species in Chlamydomonadales (Table 2 and Supplementary Table S1). When refer to dN, 27 genes were significantly different between the two group of algae, among these genes, we found 19 genes significantly higher in unicellular species. When refer to dS, 10 genes were significantly different between the two group of algae, among these genes, we found six genes significantly higher in unicellular species. Among genes with statistical significance, both comparisons show more genes have higher substitution rates in the unicellular species.
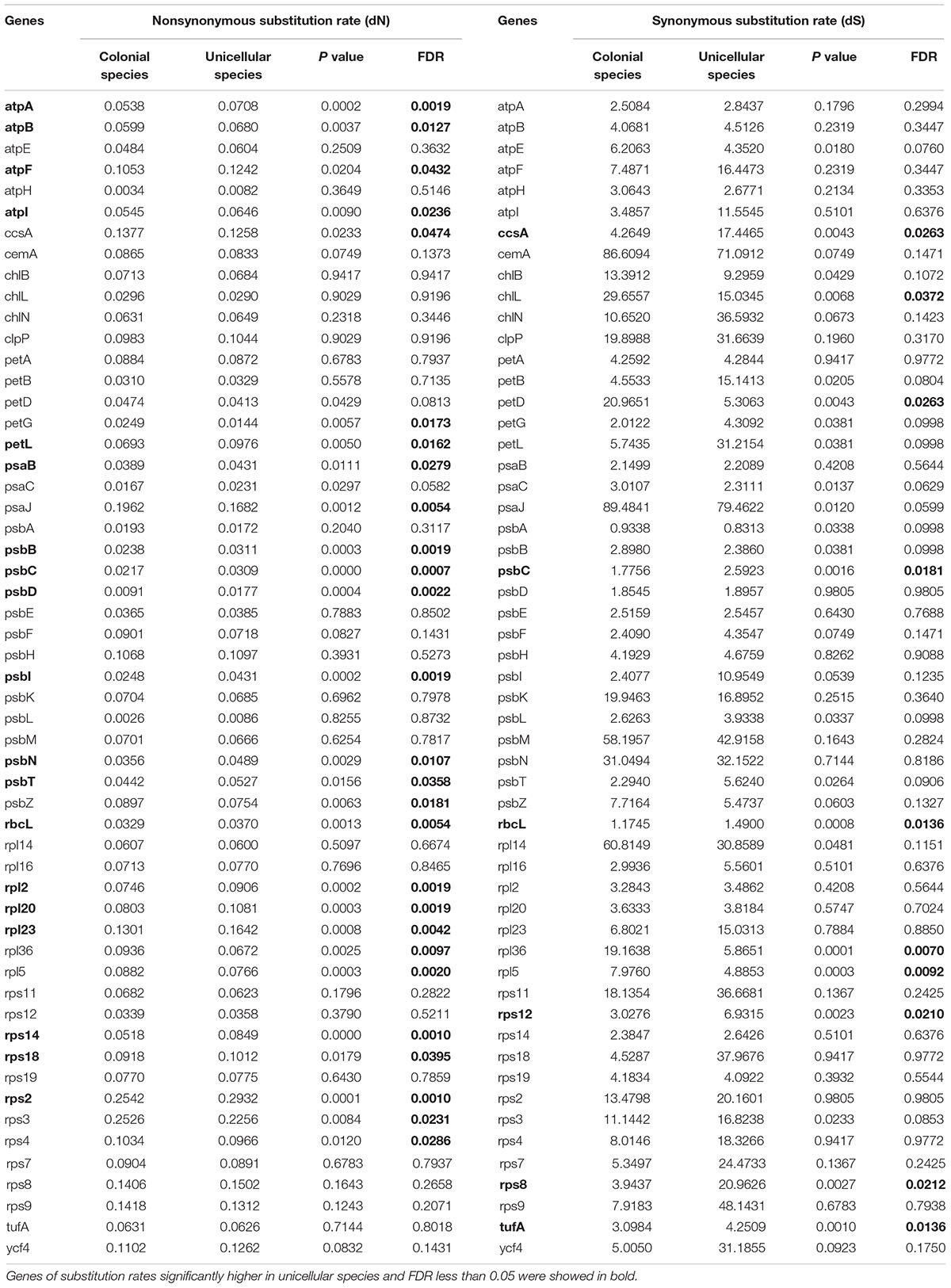
Table 2. Substitution rates in the chloroplast protein coding genes of Chlamydomonadales species.
The branch model was used to compare the dN/dS ratio between colonial and unicellular species based on the chloroplast protein-coding genes, the LRT was used to compare the fit of two models. The null model (H0) assumed that all tree branches evolved at the same rate (the same dN/dS ratio), the alternative model assumed that the foreground branch (the unicellular species) could evolved at a different rate (different dN/dS ratio). We found the FDR-adjusted P value of 16 genes were less than 0.05, this indicates the dN/dS ratio of these genes were significantly different among the unicellular and colonial species (Figure 4 and Supplementary Table S2). Among these genes, the dN/dS ratio of 14 genes (atpA, rpl16, psaB, psbC, atpB, psbE, psaJ, psbA, rps8, rpl2, rps12, rps14, psbN, atpI) were higher in the unicellular species compared with the colonial species, so these genes were considered putative fast-evolving genes.
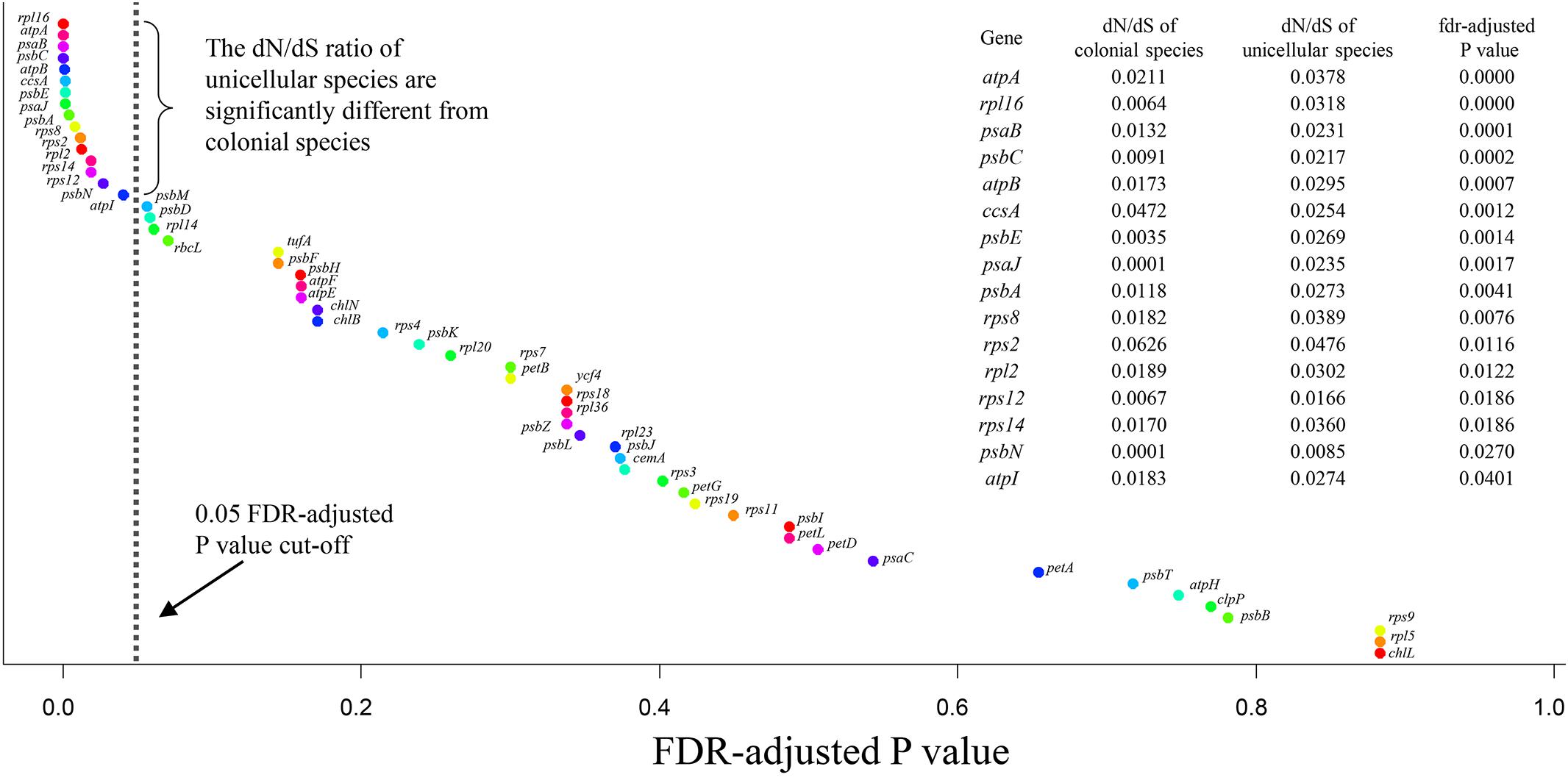
Figure 4. Plot showing ranked FDR-adjusted P values for 55 chloroplast protein-coding genes. P value were obtained from the branch model likelihood ratio tests, were applied by the false discovery rate method.
The positive selection analysis was performed based on the branch-site model, and we also conducted the comparison between the null and alternative models. The null model considered the foreground branch only have dN/dS = 1, and the alternative model considered sites on the foreground branch have dN/dS > 1 (positive selection). We used the Chi-square test to testing P values, after the FDR correction, we found 11 genes (psaB, psbB, psbC, rbcL, tufA, psbA, rps4, rpl5, rpl16, rps12, atpF) have the FDR-adjusted P value lower than 0.05 (Supplementary Table S3), and we considered these genes as the putative positively selected genes. The overlapped genes between the putative fast-evolving genes and positively selected genes were psaB, psbC, psbA, rpl16, and rps12. Based on the BEB method, the positively selected sites for each gene were shown in Table 3. We found the psaB, psbC and psbA have sites may likely under positive selection. For psaB, 134GLN have posterior probability higher than 95%, 253GLN have posterior probability higher than 90%, but there is no related functional sites information of Chlamydomonas reinhardtii in Uniprot, so the positively selected sites of psaB reminds further study. For psbA, site 237ARG (posterior probability higher than 90%) was close to the 215HIS and 272HIS of C. reinhardtii, the 215HIS is the metal binding site of iron and binding site of Quinone (B), the 272HIS is also the metal binding site of iron. For psbC, site 409SER (posterior probability higher than 95%) was close to the 355GLU of C. reinhardtii which was the metal binding site of calcium-manganese-oxide (Figure 5).
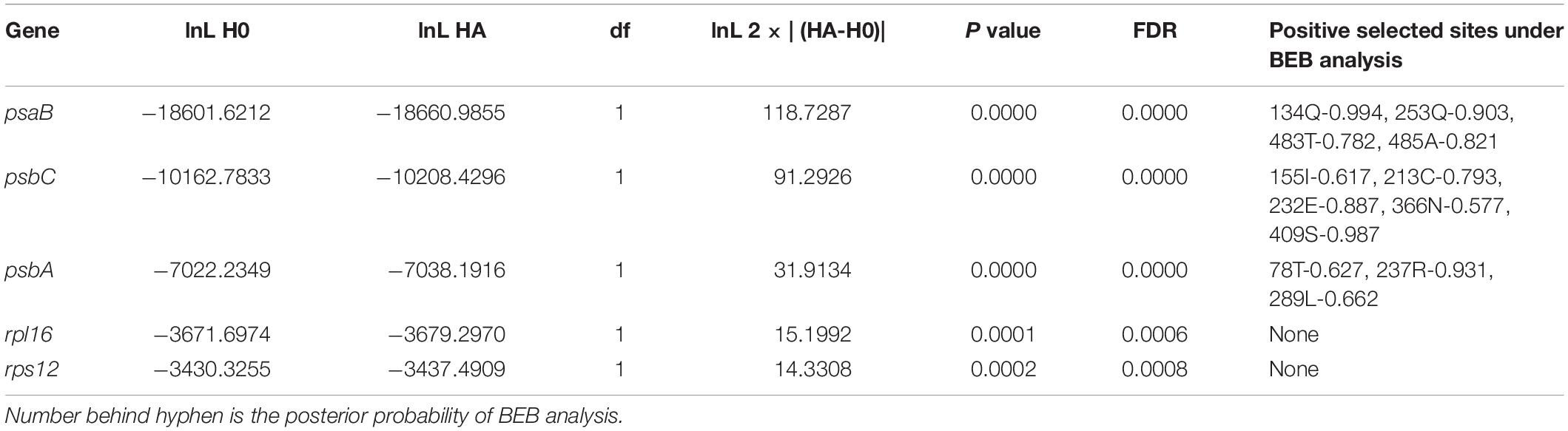
Table 3. Positively selected sites in overlapped genes of unicellular species.
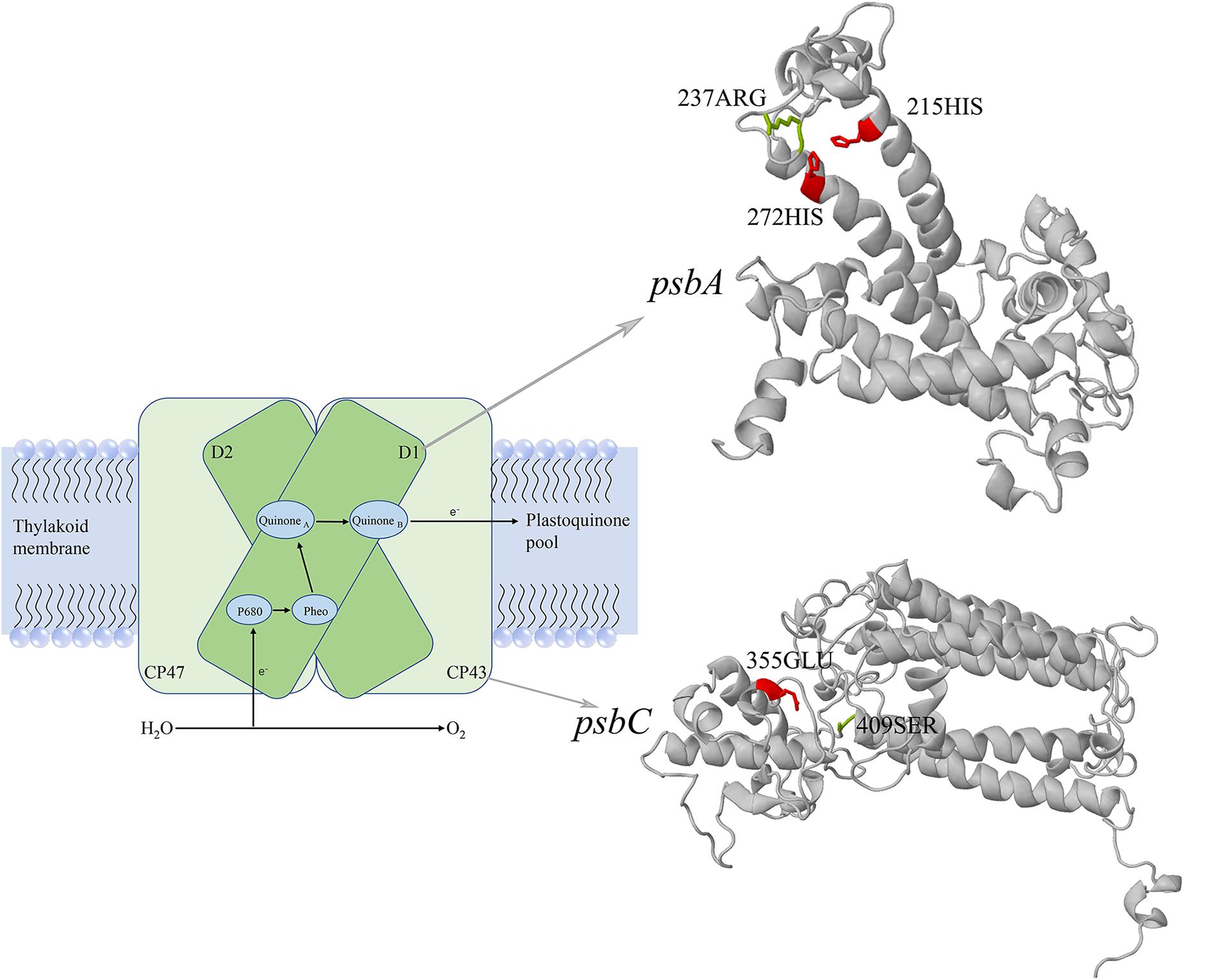
Figure 5. The three-dimensional structures of psbA and psbC. The psbA encodes photosystem II reaction center protein D1, the psbC encodes photosystem II CP43 chlorophyll apoprotein. The positively selected sites were showed in green, and the functional sites of Chlamydomonas reinhardtii were showed in red. The schematic model of photosystem II was drawn with reference from Yamamoto (2001).
Discussion
In this study, we determined six chloroplast genomes of colonial volvocine algae, this provided opportunity for us to reveal the different evolutionary rate between unicellular and colonial species in Chlamydomonadales. Our analysis was based on the protein-coding genes of 12 colonial volvocine algae and 16 unicellular species, we used the ML method to calculate the value of dN and dS of each gene and each species. The rate of synonymous substitutions and nonsynonymous substitutions of more genes were higher in unicellular species; the nonsynonymous substitution can modify the produced amino acid sequence, among the 27 significantly different genes, we found the nonsynonymous substitution of 19 genes were significantly higher (FDR-adjusted P < 0.05) in unicellular species than colonial species. More genes also have higher synonymous substitutions in the unicellular species (6 gene higher in unicellular species among 10 significantly different). All this analysis indicated more genes have higher substitution rates in unicellular species.
Guisinger et al. (2008) have found the increased substitution rates in Geraniaceae, and they proposed that the mutations in chloroplast-targeted genes could leading to increased substitution rates in chloroplast genes. Such explanation would expect rate increased for all chloroplast genes (Wang et al., 2015), since we observed the increased of dN and dS in limited number of genes in this study, the plastid DNA repair mechanism could only partly be one of the reasons responsible for the higher substitution rates in unicellular species. Shen et al. (2009) found higher substitution rates in weakly locomotive species when compared with strongly locomotive species, they associated such phenomenon with the different demand for energy. Likewise, based on our photosynthetic experiment (Supplementary Tables S4, S5), we found that the unicellular species may have lower demand for light when compared with colonial species, here, we speculate that the lower demand for light could indicate a relaxation of constraint on unicellular chloroplast genes compared with colonial species (Björnerfeldt et al., 2006), and the relaxation of constraint allow for more substitutions in the chloroplast genes of unicellular species.
To explore the substitution happens in the unicellular species whether harboring an advantageous that increased individual adaptability, we used the branch model to test whether genes were under fast-evolving in unicellular species, and we used the branch-site model to test whether sites on the unicellular branch were under positive selection. Based on our analysis, 14 genes were considered as the putative fast-evolving genes and 11 genes were considered as the putative positively selected genes, five genes were overlapped among these two group of genes. The overlap genes have higher dN/dS ratio in unicellular species (fast-evolving), meanwhile, they were undergone adaptive molecular changes (positively selection), so the positively selected sites of overlap genes may closely relate to the adaptive of unicellular species. Among the five overlap genes, we analyzed the positively selected sites for each gene by refer to the functional sites of C. reinhardtii in Uniprot, and we found two genes (psbA, psbC) may play an import role in adaption. The psbA encodes photosystem II reaction center protein D1, it is one of the two reaction center proteins of photosystem II, the function of this protein is associated with the electron transfer. The psbC encodes photosystem II CP43 chlorophyll apoprotein, it is one of the components of the core complex of photosystem II, it binds chlorophyll and helps catalyze the primary light-induced photochemical processes of photosystem II. According to the sites information in Uniprot, the positive selected sites of psbA and psbC gene were close to the functional sites of the homologous protein in C. reinhardtii. One positively selected site of psbA was close to the iron binding site and Quinone (B) binding site, this is associated with the formation of the iron-quinone complex (Wydrzynski and Satoh, 2005; Umena et al., 2011). One positively selected site of psbC was close to the binding site of calcium-manganese-oxide possibly contributed to the oxidation of water (Govindjee et al., 2010; Najafpour, 2011). In general, these two genes were all act an important role in photosynthesis, the molecular evidence show that their substitutions may contributed to the efficiency of photosystem. Our photosynthetic experiment showed the limitation of iron or calcium have lower impact on unicellular species compared with colonial species (Supplementary Tables S4, S5). We speculate that the lower impact could due to the positive selection sites in the psbA and psbC gene, the positive selection sites help these two gene have a better binding efficiency with iron or calcium, then allow unicellular species could better utilize the trace amount of iron or calcium left in the culture medium than colonial species. Our experiment further supported our conclusion.
Our study is the first determination of the chloroplast genomes of six colonial volvocine algae, by compared with the chloroplast genomes of colonial volvocine algae, we reveled more genes have higher substitution rates in unicellular species of Chlamydomonadales. We identified the fast-evolving and positively selected genes in unicellular species and found the psbA and psbC might improve the photosynthetic efficiency of unicellular species. This study not only increased the chloroplast genome information of volvocine algae but also provided useful information to understand the evolutionary relationship between unicellular and colonial species in Chlamydomonadales.
Author Contributions
YH and WX performed the experiments. YH, WX, and HS analyzed and interpreted the data. YH wrote the manuscript. All authors revised, read, and approved the final version of the manuscript.
Funding
This work was supported by the Featured Institute Service Project from the Institute of Hydrobiology, the Chinese Academy of Sciences (Grant No. Y85Z061601), the Special Foundment of Science and Technology Basic Work of China (Grant No. 2014FY120200), the National Natural Science Foundation of China (Grant No. 31670202), and the China Agriculture Research System (CARS-50).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Yonghong Bi’s assistance with the Handy PEA. We thank the Freshwater Algae Culture Collection at the Institute of Hydrobiology for the offer of unicellular species. We also thank Shuyin Li’s help in the sample collection. This research was supported by the Wuhan Branch, Supercomputing Center, Chinese Academy of Sciences, China.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01351/full#supplementary-material
Footnotes
References
Abascal, F., Zardoya, R., and Telford, M. J. (2010). TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic. Acids. Res. 38, W7–W13. doi: 10.1093/nar/gkq291
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Björnerfeldt, S., Webster, M. T., and Vilà, C. (2006). Relaxation of selective constraint on dog mitochondrial DNA following domestication. Genome Res. 16, 990–994. doi: 10.1101/gr.5117706
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, Y., Chen, Y., Shi, C., Huang, Z., Zhang, Y., Li, S., et al. (2017). SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7:gix120. doi: 10.1093/gigascience/gix120
Coleman, A. W. (2001). Biogeography and speciation in the Pandorina/Volvulina (Chlorophyta) superclade. J. Phycol. 37, 836–851. doi: 10.1046/j.1529-8817.2001.01043.x
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Del, V. M., Figueroa-Martinez, F., Featherston, J., González, M. A., Reyes-Prieto, A., Durand, P. M., et al. (2015). Massive and widespread organelle genomic expansion in the green algal genus Dunaliella. Genome Biol. Evol. 7, 656–663. doi: 10.1093/gbe/evv027
Dussert, Y., Mazet, I. D., Couture, C., Gouzy, J., Piron, M. C., Kuchly, C., et al. (2018). A high-quality grapevine downy mildew genome assembly reveals rapidly evolving and lineage-specific putative host adaptation genes. bioRxiv
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic. Acids. Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Featherston, J., Arakaki, Y., Nozaki, H., Durand, P. M., and Smith, D. R. (2016). Inflated organelle genomes and a circular-mapping mtDNA probably existed at the origin of coloniality in volvocine green algae. Eur. J. Phycol. 51, 369–377. doi: 10.1080/09670262.2016.1198830
Fucile, G., Biase, D. D., Nahal, H., La, G., Khodabandeh, S., Chen, Y., et al. (2011). ePlant and the 3D data display initiative: integrative systems biology on the world wide web. PLoS One 6:e15237. doi: 10.1371/journal.pone.0015237
Govindjee, K. J., Messinger, J., and Whitmarsh, J. (2010). Photosystem II Encyclopedia of Life Sciences (ELS). Chichester: Wiley.
Guisinger, M. M., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2008). Genome-wide analyses of Geraniaceae plastid DNA reveal unprecedented patterns of increased nucleotide substitutions. Proc. Natl. Acad. Sci. U.S.A. 105, 18424–18429. doi: 10.1073/pnas.0806759105
Hamaji, T., Smith, D. R., Noguchi, H., Toyoda, A., Suzuki, M., Kawaitoyooka, H., et al. (2013). Mitochondrial and plastid genomes of the colonial green Alga Gonium pectorale give insights into the origins of organelle DNA architecture within the Volvocales. PLoS One 8:e57177. doi: 10.1371/journal.pone.0057177
Herron, M. D. (2016). Origins of multicellular complexity: volvox and the volvocine algae. Mol. Ecol. 25, 1213–1223. doi: 10.1111/mec.13551
Hu, Y., Xing, W., Song, H., Liu, G., and Hu, Z. (2019). Analysis of mitochondrial and chloroplast genomes in two volvocine algae: Eudorina elegans and Eudorina cylindrica (Volvocaceae, Chlorophyta). Eur. J. Phycol. 54, 193–205. doi: 10.1080/09670262.2018.1539526
Kato, S. (1982). Laboratory culture and morphology of Colacium vesiculosum Ehrb. (Euglenophyceae). Jpn. J. Phycol. 30, 63–67.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N., and Sternberg, M. J. E. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lemieux, C., Vincent, A. T., Labarre, A., Otis, C., and Turmel, M. (2015). Chloroplast phylogenomic analysis of chlorophyte green algae identifies a novel lineage sister to the Sphaeropleales (Chlorophyceae). BMC Evol. Biol. 15:264. doi: 10.1186/s12862-015-0544-5
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, 1–3.
Liu, C., Shi, L., Zhu, Y., Chen, H., Zhang, J., Lin, X., et al. (2012). CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genomics 13:715. doi: 10.1186/1471-2164-13-715
Najafpour, M. M. (2011). Calcium-manganese oxides as structural and functional models for active site in oxygen evolving complex in photosystem II: lessons from simple models. J. Photochem. Photobiol. B Biol. 104, 111–117. doi: 10.1016/j.jphotobiol.2010.12.009
Nakada, T., Tomita, M., and Nozaki, H. (2010). Volvulina compacta (Volvocaceae, Chlorophyceae), new to Japan, and its phylogenetic position. J. Jpn. Bot. 85, 364–369.
Nozaki, H., and Kuroiwa, T. (1990). Volvulina compacta sp. nov. (Volvocaceae, Chlorophyta) from Nepal. Phycologia 29, 410–417. doi: 10.2216/i0031-8884-29-4-410.1
Nozaki, H., Yamada, T. K., Takahashi, F., Matsuzaki, R., and Nakada, T. (2014). New “missing link” genus of the colonial volvocine green algae gives insights into the evolution of oogamy. BMC Evol. Biol. 14:37. doi: 10.1186/1471-2148-14-37
Regier, J. C., Shultz, J. W., Zwick, A., Hussey, A., Ball, B., Wetzer, R., et al. (2010). Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 463, 1079–1083. doi: 10.1038/nature08742
Ronquist, F., Teslenko, M., Van, D. M. P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Shen, Y. Y., Shi, P., Sun, Y. B., and Zhang, Y. P. (2009). Relaxation of selective constraints on avian mitochondrial DNA following the degeneration of flight ability. Genome Res. 19, 1760–1765. doi: 10.1101/gr.093138.109
Smith, D. R. (2015). Mutation rates in plastid genomes: they are lower than you might think. Genome Biol. Evol. 7, 1227–1234. doi: 10.1093/gbe/evv069
Smith, D. R., Hamaji, T., Olson, B. J. S. C., Durand, P. M., Ferris, P., Michod, R. E., et al. (2013). Organelle genome complexity scales positively with organism size in volvocine green algae. Mol. Biol. Evol. 30, 793–797. doi: 10.1093/molbev/mst002
Smith, D. R., and Lee, R. W. (2009). Nucleotide diversity of the Chlamydomonas reinhardtii plastid genome: addressing the mutational-hazard hypothesis. BMC Evol. Biol. 9:120. doi: 10.1186/1471-2148-9-120
Smith, D. R., and Lee, R. W. (2010). Low nucleotide diversity for the expanded organelle and nuclear genomes of Volvox carteri supports the mutational-hazard hypothesis. Mol. Biol. Evol. 27, 2244–2256. doi: 10.1093/molbev/msq110
Smith, S. A., and Dunn, C. W. (2008). Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24, 715–716. doi: 10.1093/bioinformatics/btm619
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Starr, R. C. (1962). A new species of volvulina playfair. Arch. F. Mikrobiol. 42, 130–137. doi: 10.1007/bf00408169
Umena, Y., Kawakami, K., Shen, J. R., and Kamiya, N. (2011). Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9?Å. Nature 473, 55–60. doi: 10.1016/j.bbabio.2012.02.005
Wang, B., Jiang, B., Zhou, Y., Su, Y., and Wang, T. (2015). Higher substitution rates and lower dN/dS for the plastid genes in Gnetales than other gymnosperms. Biochem. Syst. Ecol. 59, 278–287. doi: 10.1016/j.bse.2015.02.009
Wydrzynski, T. J., and Satoh, K. (2005). Photosystem II: the Light-Driven Water: Plastoquinone Oxidoreductase. Dordrecht: Springer.
Yamamoto, Y. (2001). Quality control of photosystem II. Plant Cell Physiol. 283, 121–128. doi: 10.1093/pcp/pce022
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yumoto, K., Kasai, F., and Kawachi, M. (2013). Taxonomic re-examination of Chlamydomonas strains maintained in the NIES-Collection. Microbiol. Cult. Collect. 29, 1–12.
Zhang, J., Nielsen, R., and Yang, Z. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479. doi: 10.1093/molbev/msi237
Keywords: higher substitution rates, Chlamydomonadales, chloroplast genome, colonial volvocine algae, unicellular species, positive selection
Citation: Hu Y, Xing W, Song H, Zhu H, Liu G and Hu Z (2019) Evolutionary Analysis of Unicellular Species in Chlamydomonadales Through Chloroplast Genome Comparison With the Colonial Volvocine Algae. Front. Microbiol. 10:1351. doi: 10.3389/fmicb.2019.01351
Received: 13 July 2018; Accepted: 31 May 2019;
Published: 18 June 2019.
Edited by:
John R. Battista, Louisiana State University, United StatesReviewed by:
Weimin Ma, Shanghai Normal University, ChinaMatthew David Herron, Georgia Institute of Technology, United States
Copyright © 2019 Hu, Xing, Song, Zhu, Liu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoxiang Liu, bGl1Z3hAaWhiLmFjLmNu