
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 26 June 2019
Sec. Virology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.01340
This article is part of the Research Topic HIV-1 Genetic Diversity View all 18 articles
 Gonzalo Bello1
Gonzalo Bello1 Ighor Arantes1
Ighor Arantes1 Vincent Lacoste2
Vincent Lacoste2 Marlene Ouka3Jacques Boncy4Raymond Césaire3Bernard Liautaud5Mathieu Nacher6
Marlene Ouka3Jacques Boncy4Raymond Césaire3Bernard Liautaud5Mathieu Nacher6 Georges Dos Santos3*
Georges Dos Santos3*The human immunodeficiency virus-type 1 (HIV-1) subtype B has probably been circulating on the island of Hispaniola since the 1960s, but information about the early viral history on this Caribbean island is scarce. In this study, we reconstruct the dissemination dynamics of early divergent non-pandemic subtype B lineages (designated BCAR) on Hispaniola by analyzing a country-balanced dataset of HIV-1 BCAR pol sequences from Haiti (n = 103) and the Dominican Republic (n = 123). Phylogenetic analyses supported that BCAR strains from Haiti and the Dominican Republic were highly intermixed between each other, although the null hypothesis of completely random mixing was rejected. Bayesian phylogeographic analyses placed the ancestral BCAR virus in Haiti and the Dominican Republic with the same posterior probability support. These analyses estimate frequent viral transmissions between Haiti and the Dominican Republic since the early 1970s onwards, and the presence of local BCAR transmission networks in both countries before first AIDS cases was officially recognized. Demographic reconstructions point that the BCAR epidemic in Hispaniola grew exponentially until the 1990s. These findings support that the HIV-1 epidemics in Haiti and the Dominican Republic have been connected by a recurrent bidirectional viral flux since the initial phase, which poses a great challenge in tracing the geographic origin of the BCAR epidemic within Hispaniola using only genetic data. These data also reinforce the notion that prevention programs have successfully reduced the rate of new HIV-1 transmissions in Hispaniola since the end of the 1990s.
The island of Hispaniola, shared by Haiti and the Dominican Republic, included in 2016 around 217,000 people living with the human immunodeficiency virus-type 1 (HIV-1), the etiologic agent of the acquired immunodeficiency syndrome (AIDS) (UNAIDS, 2013, 2017). Initially, AIDS cases recognized in Haiti (Pape et al., 1983) and the Dominican Republic (Koenig et al., 1987) mostly involved men who had sex with men, but these countries now have generalized epidemics predominantly driven by heterosexual sex (Figueroa, 2008). Despite the high HIV prevalence rates reported for the general population in Haiti (2.1%: 1.9–2.3%) and the Dominican Republic (1.0%: 0.7–1.4%) in 2016, significant declines of the HIV incidence rates were observed in both countries over the last decade (UNAIDS, 2013, 2017).
The HIV-1 epidemic in Hispaniola is dominated by subtype B (Nadai et al., 2009; Myers et al., 2012; Lopez et al., 2015). Genetic evidence suggests that the subtype B epidemic in Hispaniola, as in many other Caribbean countries, is mainly driven by the transmission of multiple early divergent non-pandemic subtype B lineages (designated “BCAR”), although the worldwide disseminated “BPANDEMIC” lineage also circulates (Cabello et al., 2014, 2015; Divino et al., 2016; Bello et al., 2018). The early dissemination dynamics of the HIV-1 subtype B epidemics between Haiti and the Dominican Republic remain largely unknown. Previous evolutionary analyses pointed that subtype B probably entered in the Americas through Haiti around the mid-1960s and then moved to other countries (Gilbert et al., 2007; Junqueira et al., 2011; Worobey et al., 2016), but no or very few (n < 15) subtype B sequences from the Dominican Republic were included in those studies. Another study with a large number of HIV-1 sequences from the Dominican Republic has shown that subtype B virus circulates in this country since the early 1960s (Lopez et al., 2015), a result comparable to the estimated age of the Haitian epidemic. This study, however, may have traced the age of the Haitian subtype B ancestor rather than of the Dominican one, given that it assumed that all subtype B infections in the Dominican Republic resulted from a single introduction and that the hypothesis of multiple independent viral introductions from Haiti was not formally tested.
The objective of this work was to reconstruct the early spatiotemporal dynamics of dissemination and demographic history of non-pandemic HIV-1 BCAR lineages on the island of Hispaniola and to estimate the current degree of geographic compartmentalization of the BCAR epidemic between Haiti and the Dominican Republic.
A total of 127 new HIV-1 subtype B pol sequences covering the complete protease (PR) and the first part of the reverse transcriptase (RT) regions (nucleotides 2253 to 3275 of reference strain HXB2) were obtained from adult patients at Port-au-Prince, Haiti, who underwent HIV genotyping tests at the Virology Laboratory of the University Hospital of Martinique (Fort-de-France, Martinique) between 2009 and 2014. All patients provided written informed consent and samples were anonymized as recommended in the study protocol approved by the Comité National d’Ethique du Ministère de la Santé Publique et de la Population de Haiti (13/07/2009). Only one sequence per subject was selected and all sites associated with major antiretroviral drug resistance were removed.
The subtype initially assigned to new Haitian sequences by the REGA HIV subtyping tool v.2 (de Oliveira et al., 2005) was confirmed by performing a maximum likelihood (ML) phylogenetic analysis with HIV-1 group M subtype reference sequences (Supplementary Table S1). We also tested for recombination using the Recombination Detection Program (RDP) v4.9 (Martin et al., 2005) with the default settings. Only statistically significant (p < 0.05) events supported by at least two methods were considered. HIV-1 subtype B pol sequences from Haiti were then aligned with subtype B sequences representative of the BPANDEMIC and the BCAR clades (Supplementary Table S2) selected from a previous study (Cabello et al., 2014) and subjected to ML phylogenetic analysis for lineage classification. The ML trees were reconstructed with the PhyML program (Guindon et al., 2010) using an online web server (Guindon et al., 2005) under the best fit nucleotide substitution model selected with the SMS tool (Lefort et al., 2017), the SPR branch-swapping algorithm of heuristic tree search, and the approximate likelihood-ratio test (aLRT) (Anisimova and Gascuel, 2006) of reliability tree topology. The ML trees were visualized using the FigTree v1.4 program (Rambaut, 2009).
The HIV-1 BCAR pol sequences from Haiti identified here were aligned with BCAR sequences from Haiti (n = 12) and the Dominican Republic (n = 123) previously characterized (Cabello et al., 2014). A Bayesian phylogenetic tree of the BCAR dataset from Hispaniola was reconstructed under the best-fit nucleotide substitution model (GTR + I + G) using the MrBayes program (Ronquist et al., 2012). Two chains were run for 25 × 106 generations, and stationarity (constant mean and variance of trace plots) and good mixing (effective sample size > 200) for all parameter estimates were assessed using TRACER v1.7 (Rambaut et al., 2018). The degree of phylogenetic mixing of BCAR sequences obtained from both countries was then quantified using the BaTS program (Parker et al., 2008), which estimates phylogeny-trait associations using the Association Index (AI) (Wang et al., 2001), the Parsimony Score (PS) (Wang et al., 2001), and the maximum clade (MC) statistics. Results were considered significant for p < 0.01.
The evolutionary rate, the age of the most recent common ancestor (TMRCA), the spatial diffusion pattern, and the rate of population growth (r, year−1) of BCAR lineages in Hispaniola were jointly estimated using a Bayesian Markov Chain Monte Carlo (MCMC) approach implemented in BEAST v1.8 (Drummond et al., 2002; Drummond and Rambaut, 2007). Regression analyses using the TempEst program (Rambaut et al., 2016) revealed that the BCAR pol dataset compiled here did not contain a sufficient temporal signal for reliable time-scale estimations [X-intercept (TMRCA) < 1910]. Thus, Bayesian MCMC analyses were performed using a relaxed uncorrelated lognormal molecular clock model (Drummond et al., 2006) with a uniform prior distribution on the substitution rate (1.7–3.0 × 10−3 subst./site/year), based on previous estimates (Hue et al., 2005; Zehender et al., 2010; Chen et al., 2011; Mendoza et al., 2014). Migration events were reconstructed using a reversible discrete phylogeographic model (Lemey et al., 2009) with a CTMC rate reference prior (Ferreira and Suchard, 2008). The number of location transitions (viral migrations between countries) throughout the evolutionary history was estimated using Markov jump counts (O’Brien et al., 2009). Changes in effective population size through time (Ne) were estimated using the non-parametric Bayesian skyline (BSKL) (Drummond et al., 2005) and Bayesian Skygrid (BSKG) (Gill et al., 2013) models. Estimates of the r were obtained under the best-fit parametric model selected using the log marginal likelihood estimation (MLE) based on the generalized stepping-stone sampling (GSS) method (Baele et al., 2016). Six MCMC chains were run for 200 million generations and then combined to ensure stationarity and good mixing as described above. The MC credibility (MCC) tree was summarized with TreeAnnotator v1.8 and visualized using the FigTree v1.4 program.
Gender and age group of Haitian individuals infected with different subtype B lineages were compared using Fisher’s exact test or χ2 implemented in Stata 13 software. Statistical significance was defined as p < 0.05.
All new HIV-1 pol sequences from Haiti obtained here (n = 127) were confirmed as non-recombinant subtype B by ML phylogenetic analysis (Supplementary Figure S1) and RDP recombination analysis (data not shown). The HIV-1 subtype B Haitian sequences were combined with viral strains representative of the BCAR diversity in different Caribbean islands (n = 200) and of the BPANDEMIC diversity in the US and France (n = 300) (Supplementary Table S2) as previously characterized (Cabello et al., 2014). The ML phylogenetic analysis revealed that most subtype B Haitian sequences (n = 91, 72%) were intermixed among basal non-pandemic BCAR lineages, whereas the remaining ones (n = 36, 28%) branched within the well-supported (aLRT = 0.89) BPANDEMIC clade (Figure 1). Analysis of the epidemiological characteristics of Haitian subjects showed that both BCAR and BPANDEMIC viral lineages circulated among males and females of different age groups (Supplementary Table S3). No significant differences were observed in the frequency of subtype B lineages according to gender (p = 0.82) or age group (p = 0.45), although subjects infected with BCAR strains had a relative younger mean age (36.0 years) as compared with those infected with the BPANDEMIC clade (40.1 years) (p = 0.03).
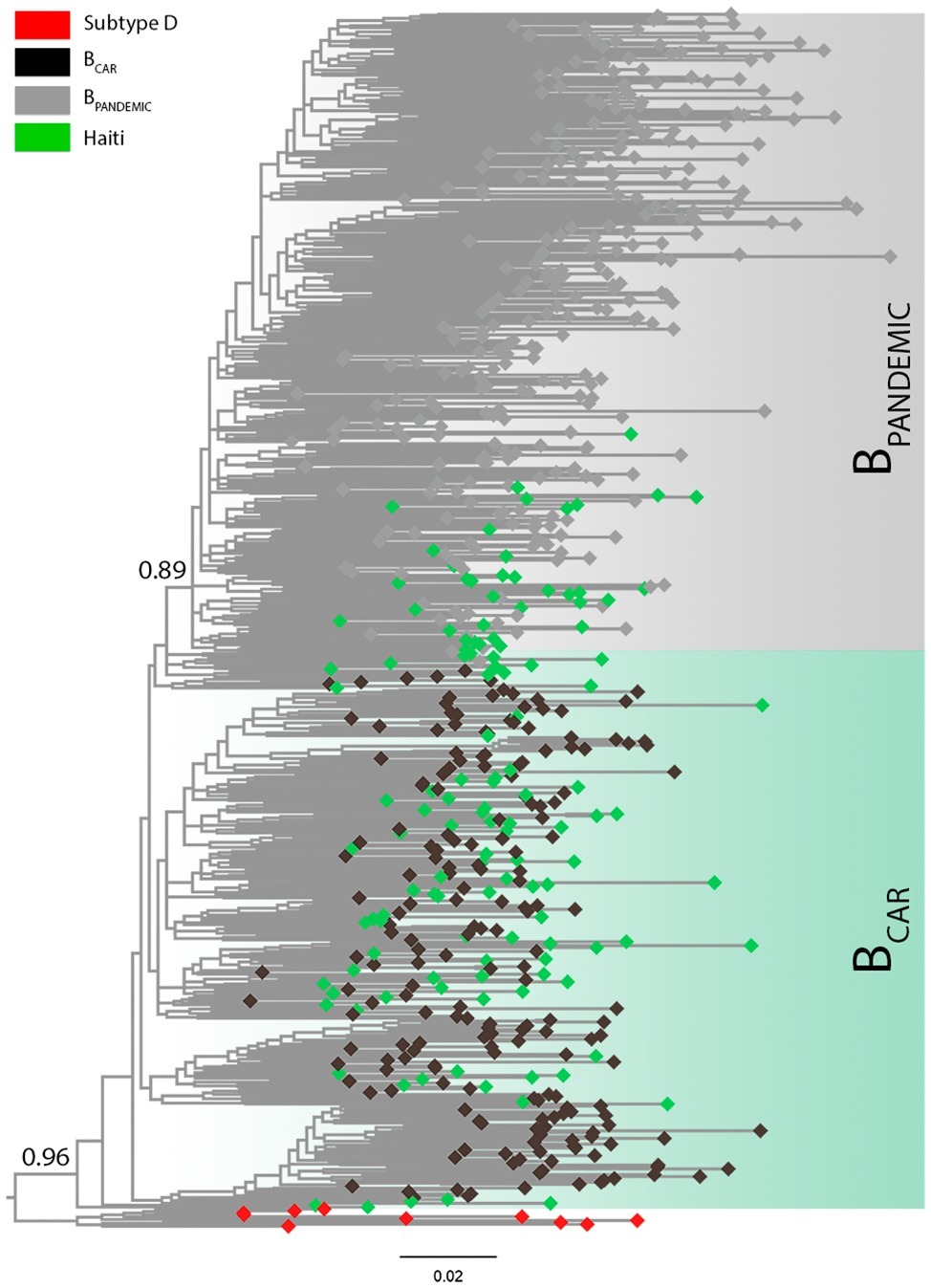
Figure 1. Classification of the HIV-1 subtype B sequences from Haiti among pandemic (BPANDEMIC) and non-pandemic (BCAR) lineages. ML phylogenetic tree of HIV-1 subtype B pol sequences (~1,000 nt) from Haiti (n = 127; green tips) together with representative sequences of the BPANDEMIC (US = 165, France = 135; gray tips) and the BCAR (Caribbean = 200; black tips) lineages. Node support (aLRT) for subtype B and BPANDEMIC monophyletic groups are indicated. Shaded boxes highlight the position of the BCAR and BPANDEMIC lineages. Tree was rooted using HIV-1 subtype D reference sequences (red tips). The branch lengths are drawn to scale with the bar at the bottom indicating nucleotide substitutions per site.
A closer inspection of the phylogenetic relationship among BCAR sequences confirm that sequences from Haiti and the Dominican Republic were highly intermixed with each other and are usually basal to sequences from other Caribbean islands (Supplementary Figure S2). Most sequences from other well-sampled islands like Jamaica and Trinidad and Tobago, by contrast, branched in country-specific subclades that were nested within the Hispaniola BCAR diversity. Very few sequences from Hispaniola (<1%) branched within Trinidadian and Jamaican clusters, confirming that most BCAR infections in Hispaniola resulted from internal viral dissemination and not from re-introductions of viral strains from other Caribbean islands. To investigate the phylogeographic structure of the HIV-1 epidemic within Hispaniola, BCAR pol sequences from Haiti here identified (n = 91) were combined with Haitian (n = 12) and Dominican (n = 123) BCAR pol sequences identified in a previous study (Cabello et al., 2014) and analyzed using BaTS. Analyses of population subdivision rejected the null hypothesis of panmixis (i.e., complete intermixing of sequences from Haiti and the Dominican Republic) (Supplementary Table S4), demonstrating that despite frequent viral intermixing between both countries, the geographic subdivision of the HIV-1 BCAR sequences from Hispaniola was greater than expected by chance.
The same dataset of BCAR pol sequences from Haiti (n = 103) and the Dominican Republic (n = 123) was then subjected to Bayesian phylogeographic analyses. Reconstruction of the spatiotemporal dissemination dynamic traced the TMRCA for the HIV-1 BCAR epidemic in Hispaniola at 1967 (95% HPD: 1961–1972), but failed to uncover its precise epicenter (Figure 2). After combining six independent Bayesian MCMC runs, the root location of the HIV-1 BCAR ancestor in Hispaniola was traced with equal probability [posterior state probability (PSP) = 0.50] to Haiti and to the Dominican Republic. The difficulty to trace the location of the BCAR root into a single country was also evidenced when the results obtained from independent Bayesian MCMC runs were visualized separately (Supplementary Table S5). Quantification of BCAR flux between countries using Markov jump counts support a mean of 18 viral migrations from Haiti to the Dominican Republic and 9 viral migration events from the Dominican Republic to Haiti. These viral migrations started in the early 1970s and were homogenously distributed between the mid-1970s and the mid-2000s (Supplementary Figure S3). The Bayesian phylogeographic analysis also revealed 20 country-specific (14 Dominican and 6 Haitian) BCAR monophyletic subclades with relative high node support [posterior probability (PP) > 0.70] (Figure 2). The Dominican BCAR subclades mostly arose between the mid-1970s and the early 1980s (Supplementary Table S6) and together comprised 55% (n = 68) of BCAR sequences from the country.
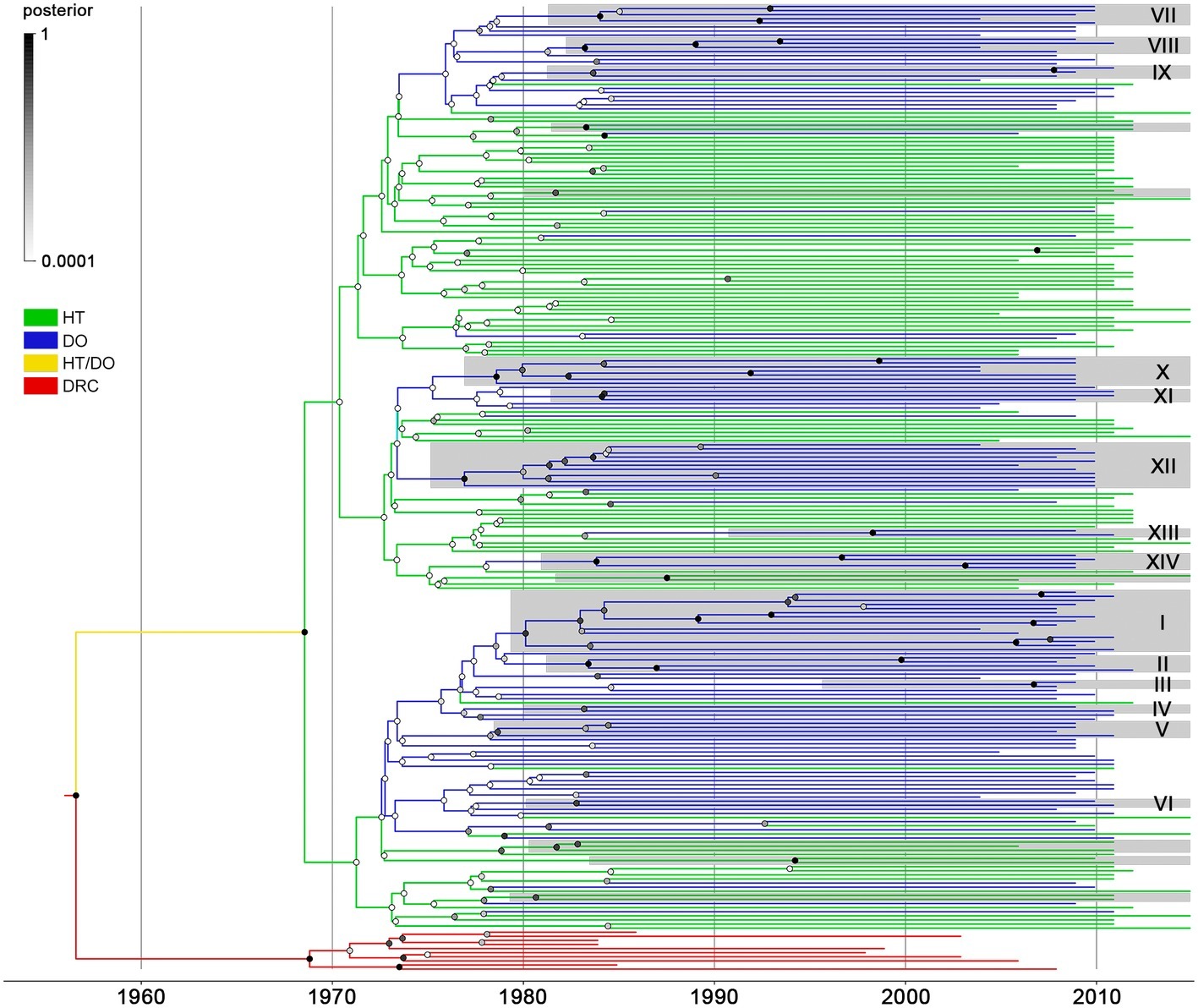
Figure 2. Spatiotemporal dissemination of HIV-1 BCAR lineages in Hispaniola. Time-scaled Bayesian MCC tree of HIV-1 BCAR pol sequences from Haiti (n = 103) and the Dominican Republic (n = 123) combined with subtype D reference sequences from the Democratic Republic of Congo (DRC; n = 10). Branches are colored according to the most probable location state of their descendent nodes as indicated in the legend on the left. Shaded boxes highlight the position of BCAR clades only composed by sequences from Haiti or the Dominican Republic (identified by numbers) and that displayed both high clade (PP ≥ 0.70) and location state (PSP ≥ 0.90) node supports. Circles at internal nodes are colored according to the corresponding PP node support as indicated in the legend on the left. Branch lengths are depicted in units of time (years). The tree was rooted under the assumption of a relaxed molecular clock.
HIV-1 BCAR sequences from Haiti (n = 103) and the Dominican Republic (n = 123) were finally used to reconstruct the demographic history of this viral epidemic in Hispaniola. Reconstruction of population dynamics with the BSKL coalescent-based model suggested that the BCAR epidemic in Hispaniola experienced an initial phase of fast exponential growth until the beginning of the 1990s, followed by a stabilization of the Ne (Figure 3A). The BSKG model, however, supported a longer exponential growth phase until the late 1990s, followed by an epidemic decline that extended until the most recent coalescent event around the mid-2000s (Figure 3B). The UNAIDS epidemiological data, in agreement with the BSKG model, indicate a growth in the number of new HIV infections in Hispaniola until the early 2000s and a subsequent phase of decline extending until the most recent date (Figure 3C). To estimate the growth rate of the BCAR epidemic at the initial phase, three parametric coalescent models (logistic, exponential, and expansion) were compared. According to the best-fit logistic growth coalescent model (Supplementary Table S7), the mean growth rate of the BCAR epidemic during the first decades of expansion in Hispaniola was 0.50 (95% HPD: 0.37–0.65).
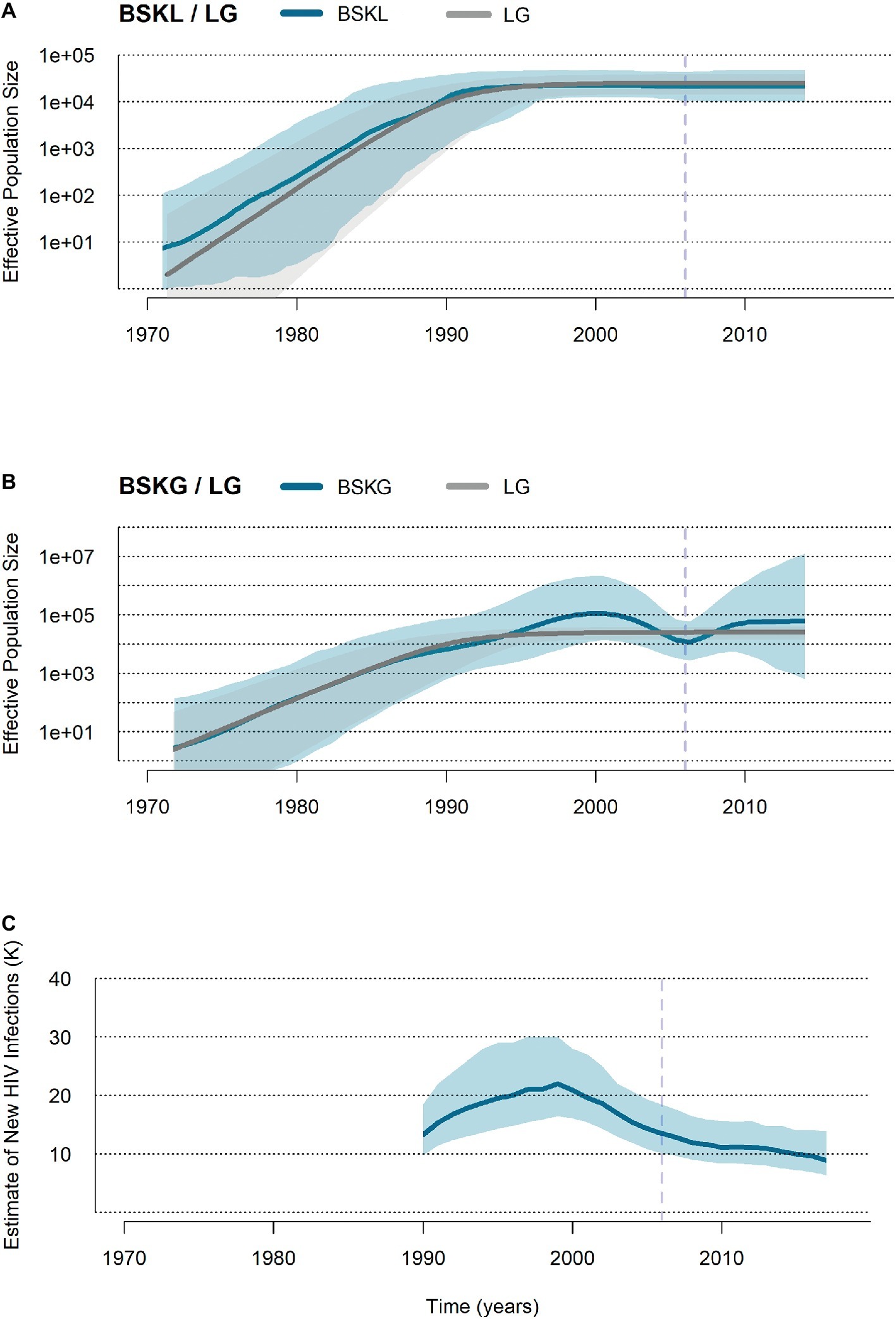
Figure 3. Population and epidemiological dynamics of the HIV-1 BCAR epidemic in Hispaniola. (A,B) Plots showing the median (solid blue lines) and the 95% HPD intervals (dashed blue areas) estimates of the effective number of HIV-1 BCAR infections (Ne, y axis) along time (years, x axis) in Hispaniola under the BSKL and BSKG models. The median Ne estimates provided by the logistic growth (LG) parametric model (dark gray line) and their 95% HPD (pale gray area) are co-plotted in both graphics. (C) Plot summarizing the number of new HIV cases in adult (>15 years old) populations from Haiti and the Dominican Republic according to the UNAIDS estimations (http://aidsinfo.unaids.org/). The dashed vertical lines indicate the time of the last coalescent event.
This study confirms that the HIV-1 subtype B epidemic in Haiti is mostly driven by dissemination of early divergent non-pandemic BCAR lineages (Cabello et al., 2014). The relative prevalence of BCAR lineages among HIV-1 subtype B infections in Haiti (72%) closely matches that estimated in the neighboring Dominican Republic (74%) (Cabello et al., 2014). Together, Haiti and the Dominican Republic host the largest population (~160,000 people) of BCAR-infected individuals in the Americas. Our results reveal a high degree of phylogenetic intermixing of the HIV-1 BCAR sequences from Haiti and the Dominican Republic, consistent with the historical intense cross-border population mobility (Cohen, 2006; Rojas et al., 2011), but also support some level of geographic structure within Hispaniola. We estimate that nearly 55% of BCAR infections in the Dominican Republic probably occurred within local transmission networks.
Previous evolutionary analyses (Gilbert et al., 2007; Junqueira et al., 2011; Worobey et al., 2016) support that the HIV-1 subtype B arrived in Haiti earlier than in any other American country. Here, we tested the hypothesis of the Haitian origin of subtype B epidemic by using for the first time a geographically balanced HIV-1 BCAR dataset of Haitian (n = 103) and Dominican (n = 123) sequences. Our Bayesian phylogeographic analysis traced the root location of the BCAR epidemic into Haiti and the Dominican Republic with the same posterior probability. This result revealed the complexity to uncover the geographic origin of the subtype B epidemic within Hispaniola by using only genetic data, probably due to the continuous cross-border viral movements between countries since the very early epidemic times. Thus, integration of non-genetic information (such as HIV incidence and prevalence data and human flows) into phylogeographic inference (Graf et al., 2015) would be probably indispensable to resolve the precise location of the HIV-1 BCAR ancestor within Hispaniola.
Although phylogeographic analyses conducted here support the Haitian or Dominican origin of subtype B with the same probability, some epidemiological and historical data favor the Haitian origin hypothesis. The first AIDS cases were recognized in Haiti (1978–1979) a few years before those of the Dominican Republic (1983) and, by the early 1980s, HIV seroprevalence among Haitians was higher than among Dominicans (Pape et al., 1983; Koenig et al., 1987). Furthermore, the estimated TMRCA of the BCAR ancestor at around the late 1960s coincides with the return of Haitian professionals from the Democratic Republic of Congo (Gilbert et al., 2007), a country with an established HIV epidemic by that time (Worobey et al., 2008). By contrast, we found no similar historical link supporting relevant human flows between the Dominican Republic and the Democratic Republic of Congo during the 1960s.
Some epidemiological studies suggested that HIV-1 transmission between populations in Haiti and the Dominican Republic was uncommon during the early years and that tourists were the most likely source of first virus transmissions to Dominicans (Pape et al., 1983; Koenig et al., 1987). Our phylogeographic analysis, however, supports that HIV-1 BCAR strains have been disseminated between Haiti and the Dominican Republic since the early 1970s and that several BCAR transmission networks were already established in the Dominican Republic by mid-1970s, nearly a decade before the first AIDS cases were officially recognized in the country. The overall time scale here obtained for the BCAR epidemic is fully consistent with that recovered in previous studies (Gilbert et al., 2007; Junqueira et al., 2011; Cabello et al., 2014; Worobey et al., 2016), supporting the reliability of our TMRCA estimates. These results clearly indicate that HIV-1 BCAR strains have been disseminated between Haitian and Dominican populations quite frequently since the early 1970s onwards.
While the BSKL model supports a stabilization of the BCAR epidemic in Hispaniola from the early 1990s onwards, the BSKG reconstruction supports a sustained expansion until the late 1990s and a subsequent decline until the most recent coalescent event. The pattern here recovered by the BSKG model is consistent with the reported decline in HIV incidence in Haiti and the Dominican Republic since the late 1990s (UNAIDS, 2013, 2017), which is likely partially driven by changes in sexual behavior since the mid-1990s (Halperin et al., 2009). This finding is in agreement with previous studies that described that the BSKG model may uncover some aspects of the population history undetected by other Bayesian models (Gill et al., 2013; Mir et al., 2018). The mean growth rate estimated here for the BCAR epidemic in Hispaniola during the first decades (0.50 year−1) is similar to those estimated for BCAR and BPANDEMIC lineages spreading in American countries with generalized heterosexual epidemics (0.35–0.45 year−1) (Cabello et al., 2014; Mendoza et al., 2014; Mir et al., 2015; Bello et al., 2018).
A drawback to consider in our study is the relative small size of our sample. According to the UNAIDS, the number of people living with HIV was estimated at around 150,000 in Haiti and 67,000 in the Dominican Republic in 2017. Assuming that 70–75% of those infections probably correspond to BCAR viruses, a very small fraction (<1%) of BCAR-infected people living in those countries was included in our study. This low sampling density does not provide adequate power to assess HIV-1 clustering in generalized epidemics and could produce misleading results (Novitsky et al., 2014). The second limitation of our study is the lack of meta-data (such as city of origin, age, sex, or potential risk behavior) for most of the HIV-infected individuals included in our analysis, avoiding the identification of trends between individuals linked within the same local cluster. Finally, it is unclear whether our sample truly represents the whole diversity of HIV-1 BCAR in Haiti because most Haitian individuals here analyzed were from the capital city (Port-au-Prince).
In summary, this study highlights that the HIV-1 epidemic in Haiti is mainly driven by dissemination of early divergent non-pandemic BCAR strains. Our findings revealed that the HIV-1 BCAR epidemics in Haiti and the Dominican Republic are highly connected by intensive bidirectional viral dispersal since the early 1970s and that local BCAR transmission was already established in both countries when the first AIDS cases were officially recognized. Despite the use of a geographically balanced BCAR dataset, probabilistic Bayesian phylogeographic models cannot uncover the root of the subtype B epidemic in Hispaniola by using only genetic sequence information. Our findings support that both national and bi-national coordinated prevention measures are necessary to further control the HIV-1 dissemination in Haiti and the Dominican Republic.
All HIV-1 sequences were deposited in the GenBank database (accession numbers MK639799–MK639925).
Comité National d’Ethique du Ministère de la Santé Publique et de la Population de Haiti (13/07/2009).
GS, GB, VL, and MN conceived and designed the study. GS, MO, JB, RC, and BL collected the samples and performed the HIV sequence amplification and genotyping. GB and IA performed the phylogenetic and phylodynamics inferences. MN performed the statistical analyses. GS, GB, IA, and VL wrote the manuscript. All authors analyzed the data and discussed and reviewed the manuscript.
IA was funded by a fellowship from “Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES-Brazil)”. GB was funded by a fellowship from “Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq”.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank T. Graf for their support with phylogeographic analyses.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01340/full#supplementary-material
Anisimova, M., and Gascuel, O. (2006). Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552. doi: 10.1080/10635150600755453
Baele, G., Lemey, P., and Suchard, M. A. (2016). Genealogical working distributions for Bayesian model testing with phylogenetic uncertainty. Syst. Biol. 65, 250–264. doi: 10.1093/sysbio/syv083
Bello, G., Nacher, M., Divino, F., Darcissac, E., Mir, D., and Lacoste, V. (2018). The HIV-1 subtype B epidemic in French Guiana and Suriname is driven by ongoing transmissions of pandemic and non-pandemic lineages. Front. Microbiol. 9:1738. doi: 10.3389/fmicb.2018.01738
Cabello, M., Junqueira, D., and Bello, G. (2015). Dissemination of nonpandemic Caribbean HIV-1 subtype B clades in Latin America. AIDS 29, 483–492. doi: 10.1097/QAD.0000000000000552
Cabello, M., Mendoza, Y., and Bello, G. (2014). Spatiotemporal dynamics of dissemination of non-pandemic HIV-1 subtype B clades in the Caribbean region. PLoS One 9:e106045. doi: 10.1371/journal.pone.0106045
Chen, J. H., Wong, K. H., Chan, K. C., To, S. W., Chen, Z., and Yam, W. C. (2011). Phylodynamics of HIV-1 subtype B among the men-having-sex-with-men (MSM) population in Hong Kong. PLoS One 6:e25286. doi: 10.1371/journal.pone.0025286
Cohen, J. (2006). HIV/AIDS: Latin America & Caribbean. Dominican Republic: a sour taste on the sugar plantations. Science 313, 473–475. doi: 10.1126/science.313.5786.473
De Oliveira, T., Deforche, K., Cassol, S., Salminen, M., Paraskevis, D., Seebregts, C., et al. (2005). An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics 21, 3797–3800. doi: 10.1093/bioinformatics/bti607
Divino, F., De Lima Guerra Corado, A., Gomes Naveca, F., Stefani, M. M., and Bello, G. (2016). High prevalence and onward transmission of non-pandemic HIV-1 Subtype B clades in northern and northeastern Brazilian regions. PLoS One 11:e0162112. doi: 10.1371/journal.pone.0162112
Drummond, A. J., Ho, S. Y., Phillips, M. J., and Rambaut, A. (2006). Relaxed phylogenetics and dating with confidence. PLoS Biol. 4:e88. doi: 10.1371/journal.pbio.0040088
Drummond, A. J., Nicholls, G. K., Rodrigo, A. G., and Solomon, W. (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161, 1307–1320.
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Drummond, A. J., Rambaut, A., Shapiro, B., and Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192. doi: 10.1093/molbev/msi103
Ferreira, M. A. R., and Suchard, M. A. (2008). Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 26, 355–368. doi: 10.1002/cjs.5550360302
Figueroa, J. P. (2008). The HIV epidemic in the Caribbean: meeting the challenges of achieving universal access to prevention, treatment and care. West Indian Med. J. 57, 195–203.
Gilbert, M. T., Rambaut, A., Wlasiuk, G., Spira, T. J., Pitchenik, A. E., and Worobey, M. (2007). The emergence of HIV/AIDS in the Americas and beyond. Proc. Natl. Acad. Sci. USA 104, 18566–18570. doi: 10.1073/pnas.0705329104
Gill, M. S., Lemey, P., Faria, N. R., Rambaut, A., Shapiro, B., and Suchard, M. A. (2013). Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol. Biol. Evol. 30, 713–724. doi: 10.1093/molbev/mss265
Graf, T., Vrancken, B., Maletich Junqueira, D., De Medeiros, R. M., Suchard, M. A., Lemey, P., et al. (2015). Contribution of epidemiological predictors in unraveling the phylogeographic history of HIV-1 subtype C in Brazil. J. Virol. 89, 12341–12348. doi: 10.1128/JVI.01681-15
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Guindon, S., Lethiec, F., Duroux, P., and Gascuel, O. (2005). PHYML Online–a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, W557–W559. doi: 10.1093/nar/gki352
Halperin, D. T., De Moya, E. A., Perez-Then, E., Pappas, G., and Garcia Calleja, J. M. (2009). Understanding the HIV epidemic in the Dominican Republic: a prevention success story in the Caribbean? J. Acquir. Immune Defic. Syndr. 51(Suppl. 1), S52–S59. doi: 10.1097/QAI.0b013e3181a267e4
Hue, S., Pillay, D., Clewley, J. P., and Pybus, O. G. (2005). Genetic analysis reveals the complex structure of HIV-1 transmission within defined risk groups. Proc. Natl. Acad. Sci. USA 102, 4425–4429. doi: 10.1073/pnas.0407534102
Junqueira, D. M., De Medeiros, R. M., Matte, M. C., Araujo, L. A., Chies, J. A., Ashton-Prolla, P., et al. (2011). Reviewing the history of HIV-1: spread of subtype B in the Americas. PLoS One 6:e27489. doi: 10.1371/journal.pone.0027489
Koenig, R. E., Pittaluga, J., Bogart, M., Castro, M., Nunez, F., Vilorio, I., et al. (1987). Prevalence of antibodies to the human immunodeficiency virus in Dominicans and Haitians in the Dominican Republic. JAMA 257, 631–634. doi: 10.1001/jama.1987.03390050057018
Lefort, V., Longueville, J. E., and Gascuel, O. (2017). SMS: smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Lemey, P., Rambaut, A., Drummond, A. J., and Suchard, M. A. (2009). Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5:e1000520. doi: 10.1371/journal.pcbi.1000520
Lopez, P., Rivera-Amill, V., Paulino-Ramirez, R., and Yamamura, Y. (2015). Short communication: HIV-1 subtype B in the dominican republic: evolution and molecular epidemiology. AIDS Res. Hum. Retrovir. 31, 679–684. doi: 10.1089/AID.2014.0304
Martin, D. P., Posada, D., Crandall, K. A., and Williamson, C. (2005). A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 21, 98–102. doi: 10.1089/aid.2005.21.98
Mendoza, Y., Martinez, A. A., Castillo Mewa, J., Gonzalez, C., Garcia-Morales, C., Avila-Rios, S., et al. (2014). Human immunodeficiency virus type 1 (HIV-1) subtype B epidemic in Panama is mainly driven by dissemination of country-specific clades. PLoS One 9:e95360. doi: 10.1371/journal.pone.0095360
Mir, D., Cabello, M., Romero, H., and Bello, G. (2015). Phylodynamics of major HIV-1 subtype B pandemic clades circulating in Latin America. AIDS 29, 1863–1869. doi: 10.1097/QAD.0000000000000770
Mir, D., Graf, T., Esteves De Matos Almeida, S., Pinto, A. R., Delatorre, E., and Bello, G. (2018). Inferring population dynamics of HIV-1 subtype C epidemics in Eastern Africa and Southern Brazil applying different Bayesian phylodynamics approaches. Sci. Rep. 8:877. doi: 10.1038/s41598-018-26824-4
Myers, J. E., Taylor, B. S., Rojas Fermin, R. A., Reyes, E. V., Vaughan, C., Jose, L., et al. (2012). Transmitted drug resistance among antiretroviral-naive patients with established HIV type 1 infection in Santo Domingo, Dominican Republic and review of the Latin American and Caribbean literature. AIDS Res. Hum. Retrovir. 28, 667–674. doi: 10.1089/aid.2010.0355
Nadai, Y., Eyzaguirre, L. M., Sill, A., Cleghorn, F., Nolte, C., Charurat, M., et al. (2009). HIV-1 epidemic in the Caribbean is dominated by subtype B. PLoS One 4:e4814. doi: 10.1371/journal.pone.0004814
Novitsky, V., Moyo, S., Lei, Q., Degruttola, V., and Essex, M. (2014). Impact of sampling density on the extent of HIV clustering. AIDS Res. Hum. Retrovir. 30, 1226–1235. doi: 10.1089/aid.2014.0173
O’brien, J. D., Minin, V. N., and Suchard, M. A. (2009). Learning to count: robust estimates for labeled distances between molecular sequences. Mol. Biol. Evol. 26, 801–814. doi: 10.1093/molbev/msp003
Pape, J. W., Liautaud, B., Thomas, F., Mathurin, J. R., St Amand, M. M., Boncy, M., et al. (1983). Characteristics of the acquired immunodeficiency syndrome (AIDS) in Haiti. N. Engl. J. Med. 309, 945–950. doi: 10.1056/NEJM198310203091603
Parker, J., Rambaut, A., and Pybus, O. G. (2008). Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect. Genet. Evol. 8, 239–246. doi: 10.1016/j.meegid.2007.08.001
Rambaut, A. (2009). FigTree v1.4: Tree Figure Drawing Tool. Available at: http://tree.bio.ed.ac.uk/software/figtree/ (accessed January 2019).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rambaut, A., Lam, T. T., Carvalho, L. M., and Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2:vew007. doi: 10.1093/ve/vew007
Rojas, P., Malow, R., Ruffin, B., Rothe, E. M., and Rosenberg, R. (2011). The HIV/AIDS epidemic in the Dominican Republic: key contributing factors. J. Int. Assoc. Physicians AIDS Care 10, 306–315. doi: 10.1177/1545109710397770
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
UNAIDS (2013). Report on the global AIDS epidemic. Available at: http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2013/gr2013/UNAIDS_Global_Report_2013_en.pdf (accessed January 2019).
UNAIDS (2017). Fact sheet—Latest global and regional statistics on the status of the AIDS epidemic. Available at: http://www.unaids.org/en/resources/documents/2017/UNAIDS_FactSheet (accessed January 2019).
Wang, T. H., Donaldson, Y. K., Brettle, R. P., Bell, J. E., and Simmonds, P. (2001). Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 75, 11686–11699. doi: 10.1128/JVI.75.23.11686-11699.2001
Worobey, M., Gemmel, M., Teuwen, D. E., Haselkorn, T., Kunstman, K., Bunce, M., et al. (2008). Direct evidence of extensive diversity of HIV-1 in Kinshasa by 1960. Nature 455, 661–664. doi: 10.1038/nature07390
Worobey, M., Watts, T. D., McKay, R. A., Suchard, M. A., Granade, T., Teuwen, D. E., et al. (2016). 1970s and ‘Patient 0’ HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 539, 98–101. doi: 10.1038/nature19827
Keywords: HIV-1, subtype B, non-pandemic, origin, phylodynamics, Haiti, Dominican Republic
Citation: Bello G, Arantes I, Lacoste V, Ouka M, Boncy J, Césaire R, Liautaud B, Nacher M and Dos Santos G (2019) Phylogeographic Analyses Reveal the Early Expansion and Frequent Bidirectional Cross-Border Transmissions of Non-pandemic HIV-1 Subtype B Strains in Hispaniola. Front. Microbiol. 10:1340. doi: 10.3389/fmicb.2019.01340
Edited by:
Joris Hemelaar, University of Oxford, United KingdomReviewed by:
Shigeyoshi Harada, National Institute of Infectious Diseases (NIID), JapanCopyright © 2019 Bello, Arantes, Lacoste, Ouka, Boncy, Césaire, Liautaud, Nacher and Dos Santos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georges Dos Santos, Z2Vvcmdlcy5kb3Mtc2FudG9zQGNodS1tYXJ0aW5pcXVlLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.