 Danyu Hu
Danyu Hu Yang Zang
Yang Zang Yingjin Mao
Yingjin Mao Beile Gao
Beile Gao- 1CAS Key Laboratory of Tropical Marine Bio Resources and Ecology, Guangdong Key Laboratory of Marine Materia Medica, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
- 2University of Chinese Academy of Sciences, Beijing, China
The class Thermoleophilia is one of the deep-rooting lineages within the Actinobacteria phylum and metagenomic investigation of microbial diversity suggested that species associated with the class Thermoleophilia are abundant in hot spring and soil samples. However, very few species of this class have been cultivated and characterized. Our understanding of the phylogeny and taxonomy of Thermoleophilia is solely based on 16S rRNA sequence analysis of limited cultivable representatives, but no other phenotypic or genotypic characteristics are known that can clearly discriminate members of this class from the other taxonomic units within the kingdom bacteria. This study reports phylogenomic analysis for 12 sequenced members of this class and clearly resolves the interrelationship of not yet cultivated species with reconstructed genomes and known type species. Comparative genome analysis discovered 12 CSIs in different proteins and 32 CSPs that are specific to all species of this class. In addition, a large number of CSIs or CSPs were identified to be unique to certain lineages within this class. This study represents the first and most comprehensive phylogenetic analysis of the class Thermoleophilia, and the identified CSIs and CSPs provide valuable molecular markers for the identification and delineation of species belonging to this class or its subordinate taxa.
Introduction
The class Thermoleophilia is one of the deep-rooting lineages within the Actinobacteria phylum and it has only recently been recognized as independent from the class Rubrobacteria (Zhi et al., 2009; Gao and Gupta, 2012b; Ludwig et al., 2012; Suzuki and Whitman, 2012). This class encompasses two recognized orders Thermoleophilales and Solirubrobacterales according to the most updated Bergey’s Manual of Systematics of Archaea and Bacteria (Suzuki and Whitman, 2015). A deep branching order Gaiellales within the phylum Actinobacteria (Albuquerque et al., 2011) has been proposed as an order of this class based on phylogenetic position, signature nucleotides of 16S rRNA, and physicochemical characteristics (Foesel et al., 2016). However, only one type strain Gaiella occulta F2-233 from this order was included in the analyses and its position in the phylogenetic tree is between the boundary of other Thermoleophilia orders and Rubrobacteria. The order Thermoleophilales only contains one family Thermoleophilaceae with a single genus Thermoleophilum. Species of this genus are small regular rods, moderately thermophilic, and obligately aerobic (Suzuki and Whitman, 2012). Their distinct feature is growth restriction to substrate n-alkanes (Zarilla and Perry, 1986), thus these species are named as heat- and oil-loving microbes, “Thermoleophilum.” While Thermoleophilum species are generally isolated from hot springs, members of the second order Solirubrobacterales are mainly detected in soil samples, and they exhibit more species diversity and different phenotypic characteristics. According to the most updated description of the taxonomic framework of the Actinobacteria phylum (Salam et al., 2019), the order Solirubrobacterales is composed of four families including Solirubacteraceae, Conexibacteraceae, Parviterribacteraceae and Patulibacteraceae. Currently described species of this order are mostly mesophilic with some psychrotolerant (Suzuki and Whitman, 2012). For example, metagenomic surveys of microbial diversity of soil samples from Antarctica revealed the presence of Thermoleophilia organisms, which can reach 15% abundance in some samples (Ji et al., 2016; Pulschen et al., 2017). Moreover, their preferred carbon sources are more diverse, including complex proteinaceous substrates, many sugars and a few other compounds (Foesel et al., 2016).
Several microbial diversity investigations suggest that Thermoleophilia species are abundant and diverse in nature (Joseph et al., 2003; Janssen, 2006), and they play an important role in geochemical recycling (Almeida et al., 2013; Ji et al., 2017; Li et al., 2018). However, similar to other deep-rooting classes with the phylum Actinobacteria, such as Acidimicrobiia, Rubrobacteria, Nitriliruptoria, etc., the cultivated isolates of Thermoleophilia are very limited (Ludwig et al., 2012; Suzuki and Whitman, 2015). Therefore, phenotypic characteristic descriptions of higher taxonomic ranks (e.g., class, order, family, and genus) within these classes are either lacking or speculative, which may not represent other yet uncultivated members belonging to these groups. In addition, our understanding of the phylogeny or taxonomy of the class Thermoleophilia is solely based on 16S rRNA sequence analysis, including their branching patterns in the phylogenetic trees or taxon-specific 16S rRNA signature nucleotides (Foesel et al., 2016; Salam et al., 2019). Except these two standards, no other molecular, biochemical or physiological characteristics are known that can clearly distinguish Thermoleophilia species from other Actinobacteria. Consequently, the bioprospecting or utilization of this group of bacteria is limited by our lack of knowledge of them. In the recent years, efforts have been made such as the “Genomic Encyclopedia of Bacteria and Archaea” (GEBA) project to sequence a diverse collection of the underrepresented phylogenetic lineages (Mukherjee et al., 2017), or to reconstruct genomes from metagenomic data for not yet cultivated species (Parks et al., 2017; Cabello-Yeves et al., 2018; Woodcroft et al., 2018). At the time of January 2018, there are 6 complete genomes and 10 genome assemblies for the class Thermoleophilia, providing great resource to explore phenotypic and genomic features of these microbes.
Two kinds of molecular markers have been described to define or delineate different higher taxa (e.g., genus level and above) for different prokaryotic phyla (Gupta and Gao, 2010; Gao and Gupta, 2012a). One kind of these molecular markers are conserved signature indels (CSIs) that are uniquely found in the genes/proteins homologs of a certain group of organisms, but absent in species outside of this group. The other kind of molecular markers are conserved signature proteins (CSPs) that are specifically present in a monophyletic prokaryotic group. These two molecular markers represent highly reliable characteristics of specific groups of organisms, and they provide novel methods for the identification or delineation of prokaryotic taxonomic units in clear molecular terms (Gao and Gupta, 2012b; Ho et al., 2016; Zhang et al., 2016; Alnajar and Gupta, 2017). We recently identified these molecular markers for Acidimicrobiia, another deep-branch class within the phylum Actinobacteria, which proved very useful for defining the whole class or different lineages within it and also provide interesting targets for functional studies of these microbes (Hu et al., 2018).
Here, we constructed a phylogenomic tree for 12 sequenced members of the class Thermoleophilia based on concatenation of 54 widely distributed conserved proteins. This tree clearly resolved the interrelationship of not yet cultivated species with reconstructed genomes and known type species. More importantly, by analyzing the sequenced Thermoleophilia species, we discovered 12 CSIs in different proteins and 32 CSPs that are specific to all members of this class. In addition, a large number of CSIs or CSPs were identified to be unique to certain lineages within this class. This study represents the first and most comprehensive phylogenetic analysis of the class Thermoleophilia, and the identified CSIs and CSPs provide valuable molecular markers for the identification and delineation of species belonging to this class or its subordinate taxa.
Materials and Methods
Phylogenetic Analysis
A phylogenomic tree for 6 completely sequenced species and 6 metagenome-assembled genomes (MAGs) of the class Thermoleophilia (Supplementary Table 1) was constructed. These 6 MAGs were selected for phylogenomic analysis since most single copy orthologous proteins as proposed by Na et al. (2018) can be retrieved from these genomes while other MAGs lack many of these orthologs which will reduce the robustness of the phylogenetic analysis. The deep-branching order Gaiellales only has one species sequenced, Gaiella occulta F2-233, which was also added to the analyses. The final tree was based on the concatenation of 54 protein sequence alignments (Supplementary Table 2). In addition, sequences from 3 Rubrobacter species was used as outgroup to root the tree. Multiple sequence alignments for each protein were performed using the Clustal X 2.1 program (Larkin et al., 2007) and concatenated to produce a single alignment. Gblocks 0.91b program was applied to remove the poorly aligned regions (Talavera and Castresana, 2007) and the resulting alignment composed of 13,132 amino acids was used for phylogenetic analysis. A maximum-likelihood (ML) tree was constructed by MEGA 6.0 with the Whelan and Goldman substitution model based on 1000 bootstrap replicates (Tamura et al., 2013).
An ML tree based on 16S rRNA gene sequences was constructed for the representative strains of Thermoleophilia and deep-branching order Gaiellales, but no full length 16S rRNA sequences are available for the 6 MAGs. All the 16S rRNA sequences were obtained from Ribosomal Database Project (Cole et al., 2014) or NCBI GenBank, and accession number of each 16S rRNA sequences were summarized in Supplementary Table 3. Sequences from 8 Rubrobacter species were used as outgroup to root the tree. The tree was constructed by MEGA 6.0 using the General Time Reversible model with 1000 bootstrap replicates.
Identification of CSIs
CSIs were identified following the detailed method description by Gupta (Gupta, 2014). Briefly, BLASTP searches were performed on all protein sequences from the genome of Thermoleophilum album ATCC 35263 (Yakimov et al., 2003) against all sequences in the NCBI non-redundant protein sequences (nr) database, during the period from January to April, 2018. The general parameters used for BLASTP searches were default as shown in the NCBI website. Multiple sequence alignments were created for homologs of all available Thermoleophilia species and a few other bacteria by the Clustal X 2.1 program using default parameters. These sequence alignments were inspected for any conserved insertions or deletions that were restricted to Thermoleophilia species only and also flanked by at least 5–6 identical or conserved residues in the neighboring 30∼40 amino acids on each side. The indels with non-conserved flanking regions were not considered. To verify the specificity of the identified indels, another round of BLASTP searches were performed with a short indel-containing fragment (60–100 amino acids long) against the GenBank database. To further confirm that the identified signatures are restricted to Thermoleophilia homologs, the top 500 BLAST hits with the highest similarity to the query sequence were inspected for the presence or absence of these CSIs. Final alignment files were generated by two softwares Sig_Create and Sig_Style1 (Gupta, 2014). Due to page limitation, indels-containing sequence alignment in all figures and Supplementary Figures only include those that are found in all Thermoleophilia sequences and few sequences from representative strains of other bacteria.
Identification of CSPs
BLASTP searches were performed on individual proteins from the genome of T. album ATCC 35263 to identify proteins that are restricted to species of the class Thermoleophilia or the order Thermoleophilales. For CSPs that are specific to the order Solirubrobacterales or its subgroups at different taxonomic levels, the proteins from the genome of Patulibacter americanus DSM 16676 (Reddy and Garcia-Pichel, 2009) were selected as query sequences to do the BLASTP searches against all available sequences in the NCBI non-redundant protein sequences (nr) database. The parameters used for BLASTP searches were generally default except that “Max target sequences” were set to be 500. The BLAST results were manually examined for putative Thermoleophilia -specific proteins based on Expected values (E-values) (Altschul et al., 1997). Only proteins with significant hits (E-values less than 0.01) merely from Thermoleophilia genomes while no other hits or hits from non-Thermoleophilia genomes generally with E-value higher than 1.0 were considered as CSPs in this work (Gao et al., 2006; Gao and Gupta, 2012b).
Results and Discussion
Phylogenomic Analysis of the Class Thermoleophilia
Two recent comprehensive phylogenetic analyses of the Actinobacteria phylum have both applied phylogenomic methods to re-examine the evolutionary relationships or taxonomic framework of species within this phylum (Nouioui et al., 2018; Salam et al., 2019). However, both studies aimed at the entire phylogenetic structure of the phylum, only type species/strains were considered in their analyses. For the poorly represented Thermoleophilia, there are only 5∼6 species included in both studies (Nouioui et al., 2018; Salam et al., 2019). Therefore, a comprehensive phylogenomic analysis of the Thermoleophilia class is still lacking in spite of the availability of reconstructed genomes for not yet cultivated species of this class. In addition, for these assembled genomes, their exact phylogenetic relationship with type species or taxonomic assignment need to be examined although their association with this class has been suggested (Cabello-Yeves et al., 2018; Woodcroft et al., 2018). Here, we constructed a phylogenetic tree for 6 completely sequenced species and 6 MAGs of this class, for which more single-copy ortholog sequences can be retrieved for a robust phylogenomic analysis (Supplementary Table 1). Finally, 54 orthologous protein sequences that mainly belong to the functional category “translation and transcription” were extracted for the above 12 genomes (Supplementary Table 2) and ML analysis was carried out for the concatenated protein dataset. To our knowledge, this is the most comprehensive phylogenetic analysis for the class Thermoleophilia (Figure 1A). In comparison with the current taxonomic framework, we also constructed a phylogenetic tree based on 16S rRNA sequences for this class (Figure 1B). However, surprisingly no complete 16S rRNA sequence were available for the incomplete genome assemblies selected for the above phylogenomic analyses (except that genome assembly of Solirubrobacter sp. URHD0082 contained a partial 643 bp fragment of 16S rRNA).
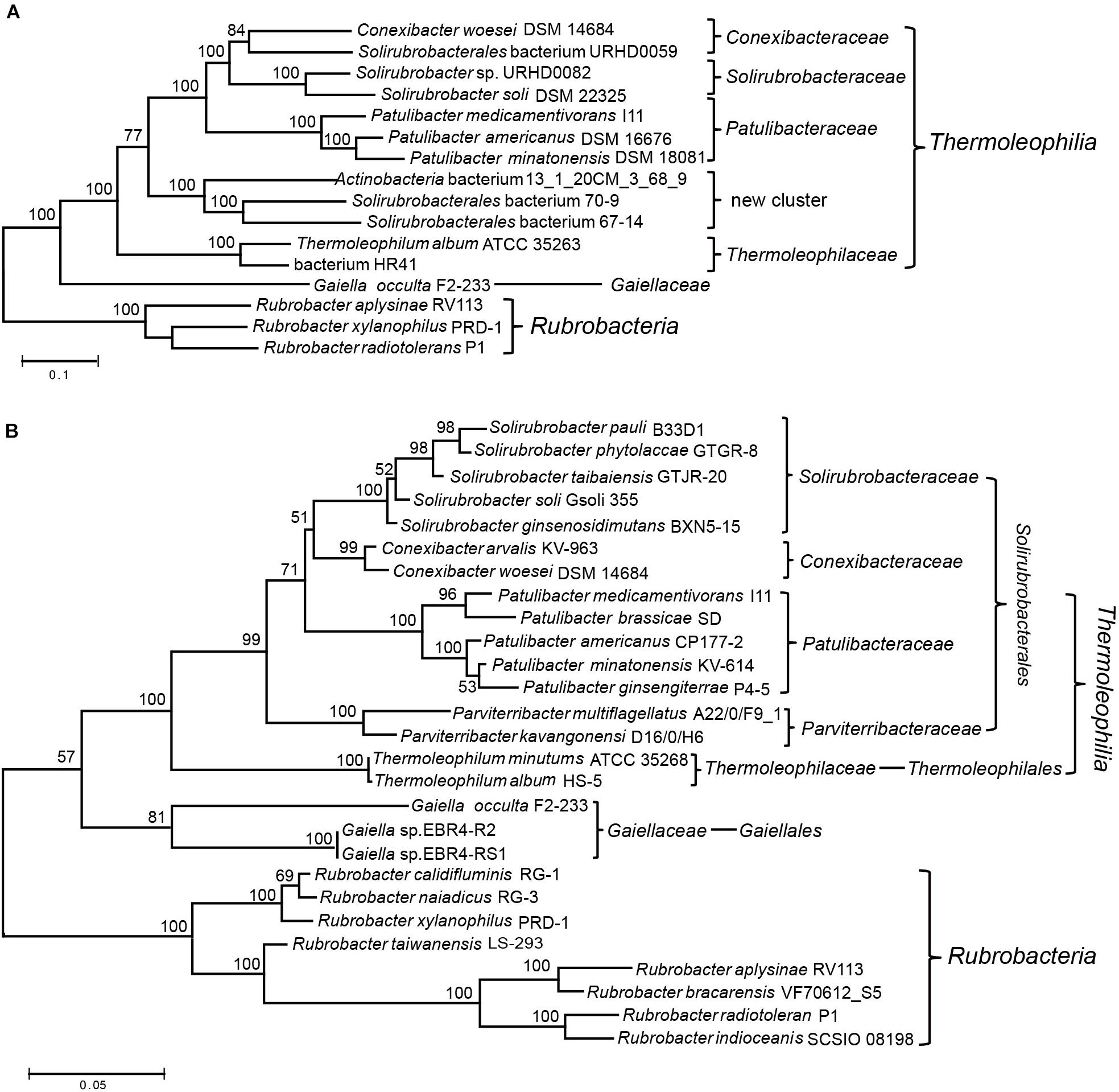
Figure 1. Phylogenetic analysis of the class Thermoleophilia. (A) Maximum-likelihood tree for Thermoleophilia species based upon concatenated sequences of 54 conserved proteins. (B) Maximum-likelihood tree based on full length 16S rRNA gene sequences of all type species within the class Thermoleophilia. Bootstrap values (%) are shown at each node and different clusters that are consistently observed in both phylogenetic trees are marked.
Overall, the combined protein tree showed a very similar branching pattern to the 16S rRNA tree. All species belonging to Thermoleophilia formed a robust cluster, separated from the class Rubrobacteria. The position of the deep branching order Gaiellales is between the boundary of other Thermoleophilia orders and the class Rubrobacteria in both trees. The single genome-sequenced species G. occulta F2-233 clusters with other Thermoleophilia orders with a very high bootstrap score 100% in the phylogenomic tree while showing a lower score 57% in the 16S rRNA tree, which is similar to the previous 16S rRNA analyses using the same G. occulta strain (Foesel et al., 2016). Within Thermoleophilia, species of the two orders Thermoleophilales and Solirubrobacterales also formed distinctive clusters in the phylogenomic tree, supporting the current order assignment based on 16S rRNA analyses (Reddy and Garcia-Pichel, 2009; Suzuki and Whitman, 2012, 2015). Compared to the diverse soil-source Solirubrobacterales, only one cultivable species T. album ATCC 35263 from the order Thermoleophilales has been genome sequenced (Yakimov et al., 2003). Our phylogenomic tree revealed that MAG “bacterium HR41” clusters together with T. album. The genome of HR41 is reconstructed from metagenomic DNA from high-temperature bioreactors, for which the initial samples were collected from an ammonia-rich geothermal groundwater stream in Japan (Kato et al., 2018). In view of their clustering pattern in the phylogenetic tree and common hot spring isolation environment, it is very likely that HR41 represents a species belonging to the family Thermoleophiliaceae or the order Thermoleophilales.
Notably, 3 MAGs- “Actinobacteria bacterium 13_1_20CM_3_68_9” from grassland (Butterfield et al., 2016), “Solirubrobacterales bacterium 67-14” and “Solirubrobacterales bacterium 70-9” from bioreactors (Kantor et al., 2015) form a distinct cluster in the phylogenomic tree, more closely related with other Solirubacteraceae families than Thermoleophilales (Figure 1A). In view of the branching pattern of these 3 MAGs, it is likely that they represent species of a novel family within the order Solirubrobacterales. Alternatively, the phylogenetic position of these MAGs is very similar to the two Parviterribacter species in the 16S rRNA tree, raising the possibility that they might be members of the Parviterribacteraceae family. However, neither the 16S rRNA of the 3 MAGs nor the genome information from the two Parviterribacter species is available at the moment, which preclude further analyses. Future new 16S rRNA or genome sequences from closely related species of either the 3 MAGs or the Parviterribacteraceae family are needed to define their relationship. In addition, assembled genomes for two monoisolates from the same study of grassland rhizosphere branched differently in our phylogenomic tree. “Solirubrobacterales bacterium URHD0059” clusters together with the type species Conexibacter woesei DSM 14684 (Pukall et al., 2010), indicating that it might be a new species belonging to the family Conexibacteraceae; while “Solirubrobacter sp. URHD0082” clusters with S. soli DSM 22325 with 100% bootstrap support, demonstrating its affiliation with the family Solirubacteraceae. The later association is also confirmed by the 16S rRNA tree based on partial sequence alignment (Supplementary Figure S1). Taken together, these phylogenomic analyses based on a concatenated protein dataset support current taxonomic structure of the class Thermoleophilia based on 16S rRNA analyses. In addition, it revealed a new cluster composed of not yet cultivated species that might be a novel family within the order Solirubrobacterales.
Molecular Markers Unique to the Class Thermoleophilia
The main purpose of this work is to identify genomic characteristics that are unique to the class Thermoleophilia or its subordinate taxa, which can be used to define their taxonomic ranks and also provide targets for functional studies. The complete genome sequences of type species and recently reported MAGs of Thermoleophilia are great resources to explore group-specific molecular markers. We focused on two molecular markers as noted earlier: CSIs and CSPs (Gao et al., 2009; Gupta and Gao, 2009; Zhang et al., 2016). Both have been identified for various prokaryotic phyla or other taxonomic ranks higher than genera in the past two decades, and proved to be very useful for phylogenetic and evolutionary studies (Gao and Gupta, 2012b; Ho et al., 2016; Alnajar and Gupta, 2017; Hu et al., 2018).
Comparative genomic analyses of species of the class Thermoleophilia and other taxonomic units within the kingdom bacteria led to the identification of 12 CSIs in various conserved universal proteins that are only found in Thermoleophilia species but not in other bacteria (Table 1). For example, a 4 amino acids (aa) insertion in a very conserved region of quinolinate synthase NadA was specifically shared by Thermoleophilia species (Figure 2). NadA is a widely distributed protein in both Archaea and Bacteria and highly conserved due to its important role in nicotinamide adenine dinucleotide (NAD) de novo biosynthesis (Ollagnier-De Choudens et al., 2005). A 4aa insertion that is located in a surface loop region of the 3D structure (Volbeda et al., 2016) is only found in homologs from Thermoleophilia but not from species outside this class. Therefore, this 4-aa insertion is a distinctive characteristic of the Thermoleophilia class. Sequence information for additional 11 CSIs that are specific to all members of this class including assembled genomes of not yet cultivated species is provided in Supplementary Figures S2–S12. In view of their specificity, these CSIs can serve as molecular markers to define and distinguish species belonging to the Thermoleophilia class. In addition, none of these 12 CSIs are found in the genome of Gaiella occulta F2-233, which is the only genome recently available from the deep-branching order Gaiellales.
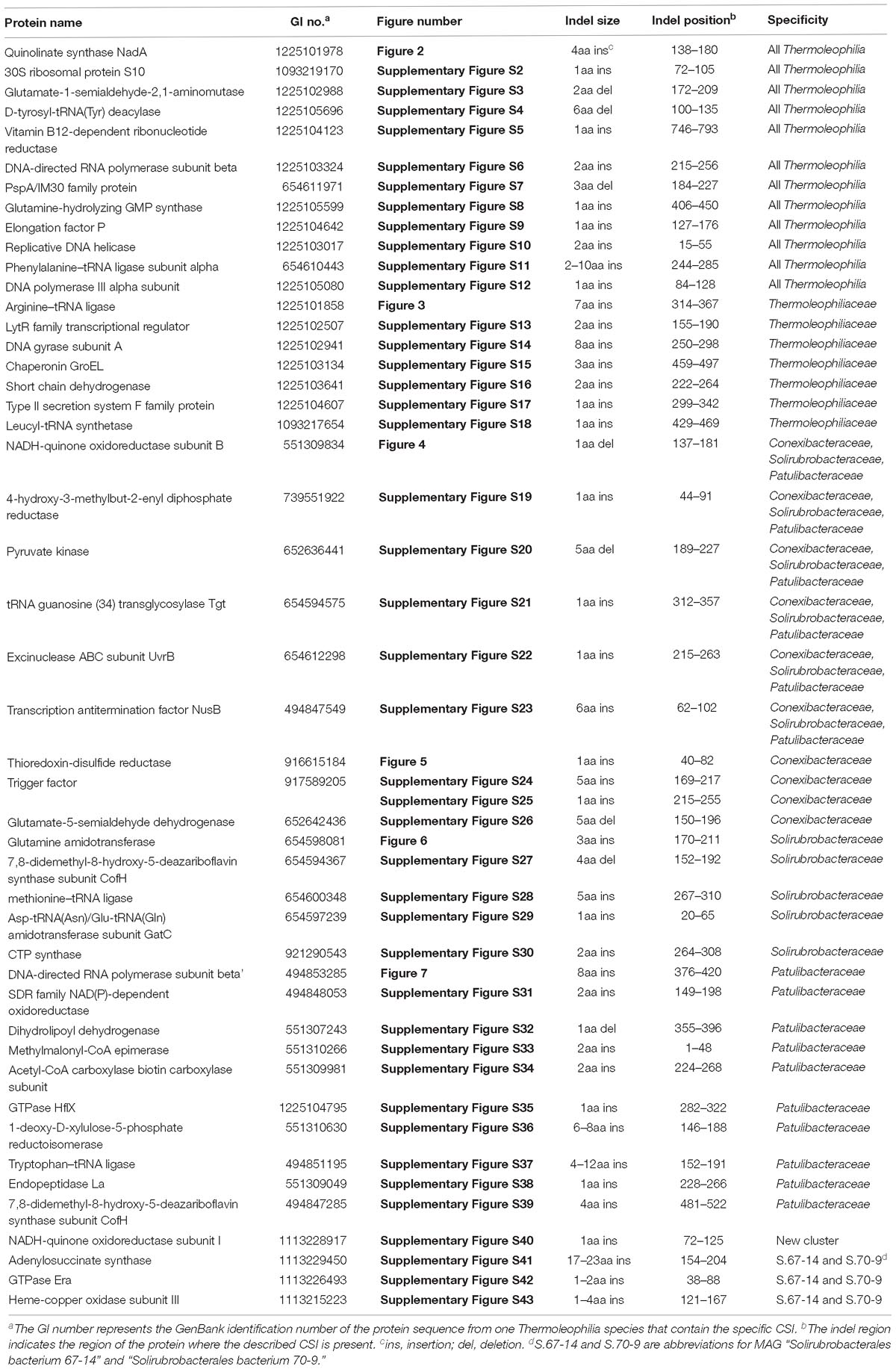
Table 1. Characteristic of Conserved Signature Indels specific to the class Thermoleophilia or its associated taxa.
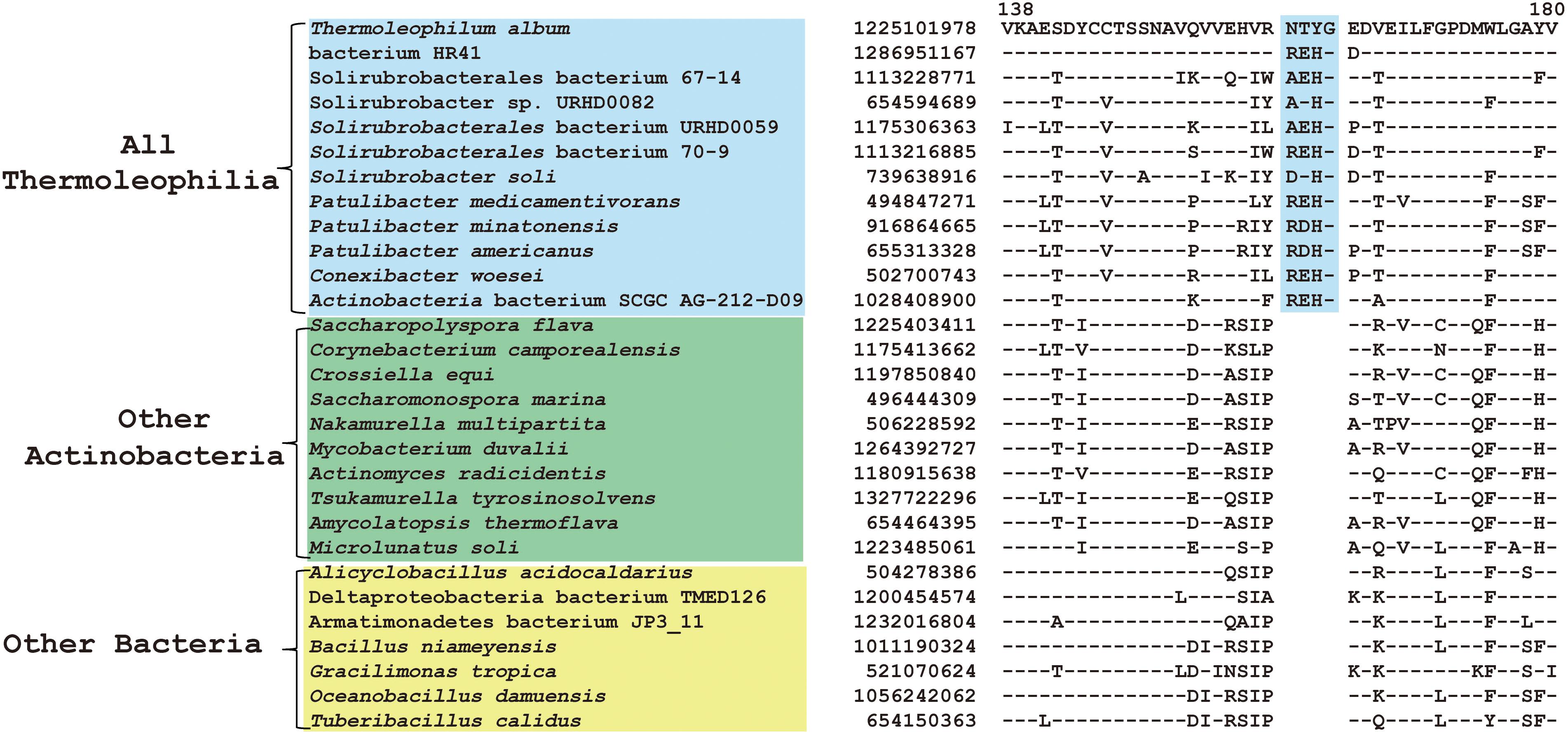
Figure 2. CSI specific to all Thermoleophilia species. Partial sequence alignment of the protein quinolinate synthase NadA showing a 4 amino acid insertion in a conserved region that is specific for members of the class Thermoleophilia. The dashes in this alignment as well as all other alignments indicate identity with the amino acid on the top line. The GenBank identification numbers of the protein sequences are shown, and the topmost numbers indicate the position of this sequence in the species shown on the top line.
Except the CSIs, we performed BLASTp searches for each protein from the type species T. album ATCC 35263 to identify CSPs that are specific to the Thermoleophilia class. In total, 32 proteins are uniquely shared by almost all sequenced Thermoleophilia genomes but not found in any other bacterial taxa except 4 present in G. occulta F2-233 (Table 2). Foesel et al. have proposed that Gaiellales is a deeply branching order within the class Thermoleophilia based on 16S rRNA analyses and some shared phenotypic features of one single strain G. occulta F2-233 and other Thermoleophilia/Rubrobacteria species (Foesel et al., 2016). The presence of 4 CSPs in the same G. occulta strain could be derived from the common ancestor of Gaiellales with the other Thermoleophilia orders or due to lateral gene transfer, which awaits confirmation from more genomes of the Gaiellales. Additionally, 3 proteins are missing in the MAGs from the newly defined potential family based on our phylogenomic analysis presented in Figure 1A but found in the other members of the class, which is possibly due to incomplete genome information. Indeed, the assembly qualities of MAGs varies as indicated by the summary of Contig-N50 statistic values in Supplementary Table 1. Therefore, it is very likely that the identified 3 CSPs are present in the species of the newly defined cluster, while the MAGs did not cover the sequence region. Together with the identified CSIs, these CSPs are additional molecular markers for Thermoleophilia. It should be mentioned that all these identified CSPs are hypothetical proteins with unknown function. Since they are restricted to species of Thermoleophilia, functional studies on them may uncover biochemical or physiological features that are unique to this class.
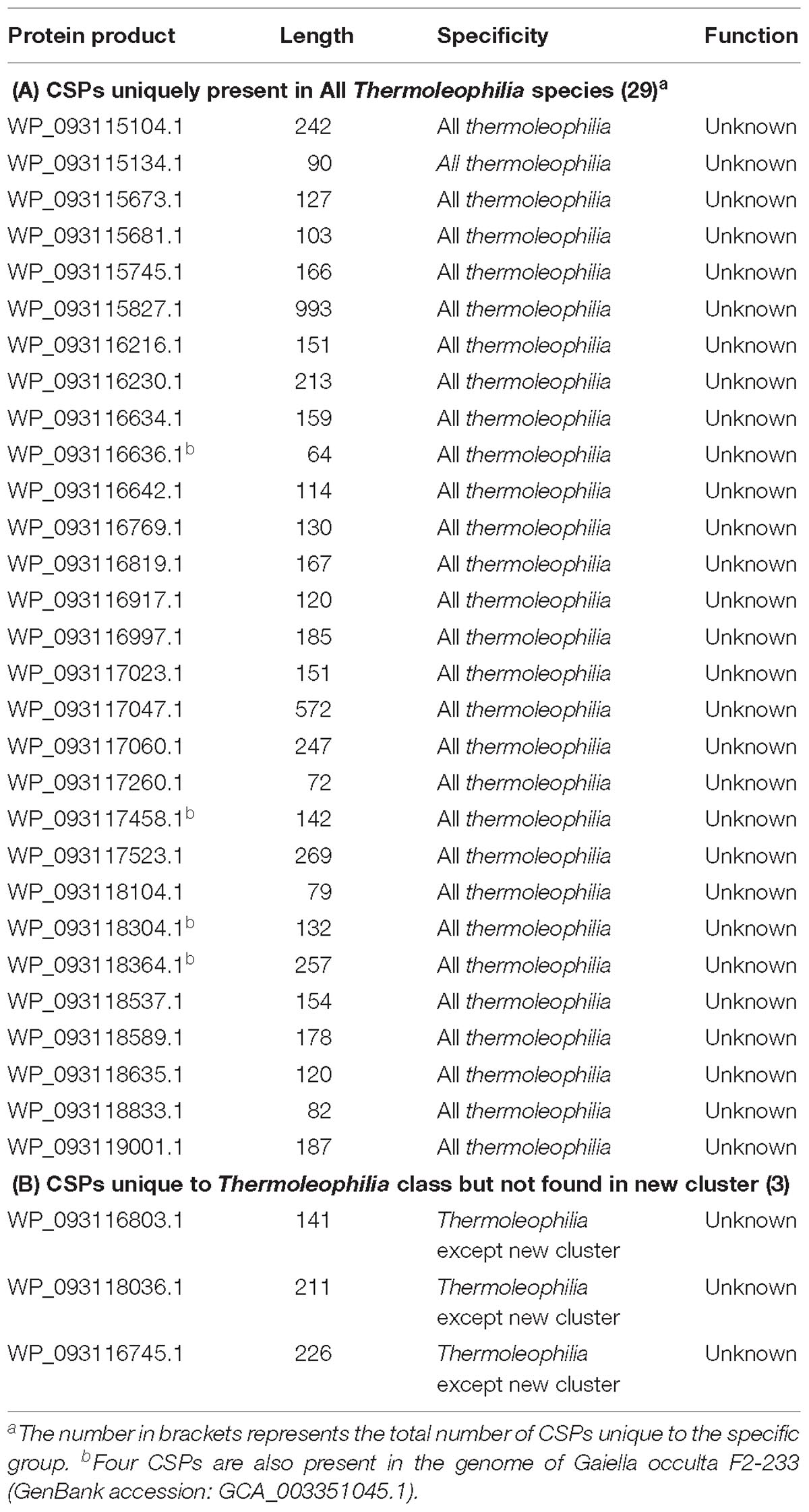
Table 2. Conserved Signature Proteins that are uniquely found in the Thermoleophilia class.
Molecular Signatures for Major Lineages Within Thermoleophilia
As described earlier, the order Thermoleophilales or its sole family Thermoleophiliaceae only have two genomes available, including T. album ATCC 35263 and MAG “bacterium HR41.” Our analyses identified 7 CSIs in different proteins (Table 1) and 29 CSPs (Table 3) that are only present in these two genomes but absent in other bacteria. Figure 3 shows one example of these CSIs. In the sequence alignment of arginine-tRNA ligase, a 7aa insertion flanked by highly conserved residues is uniquely found in homologs from both T. album and MAG “bacterium HR41.” Sequence information for further 6 CSIs with the same specificity are shown in Supplementary Figures S13–S18. Whether these identified CSIs and CSPs can constitute distinctive markers for the Thermoleophiliaceae family or even the Thermoleophilales order awaits confirmation from more sequences of other species belonging to this lineage. Nevertheless, these results provide additional evidence for the close relationship of MAG “bacterium HR41” and T. album.
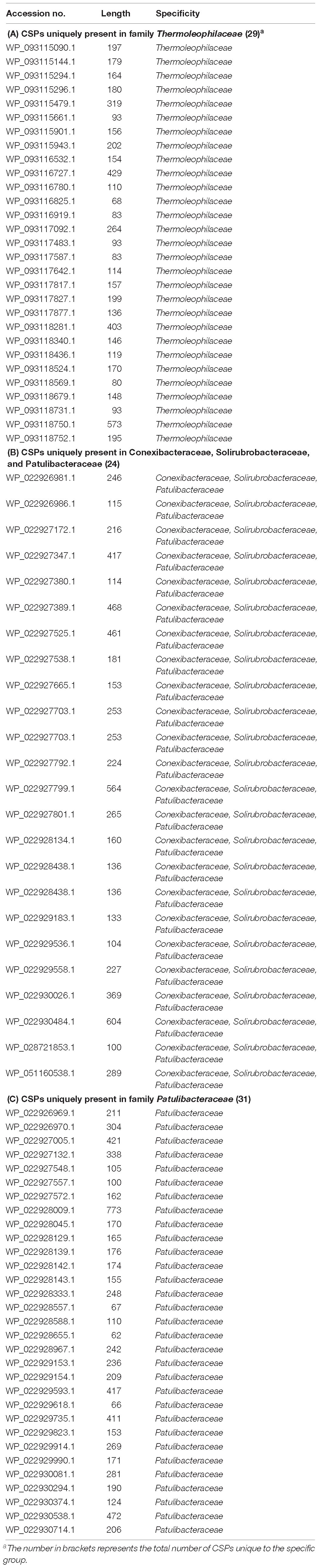
Table 3. Conserved Signature Proteins that are uniquely found in the subgroups of Thermoleophilia class.
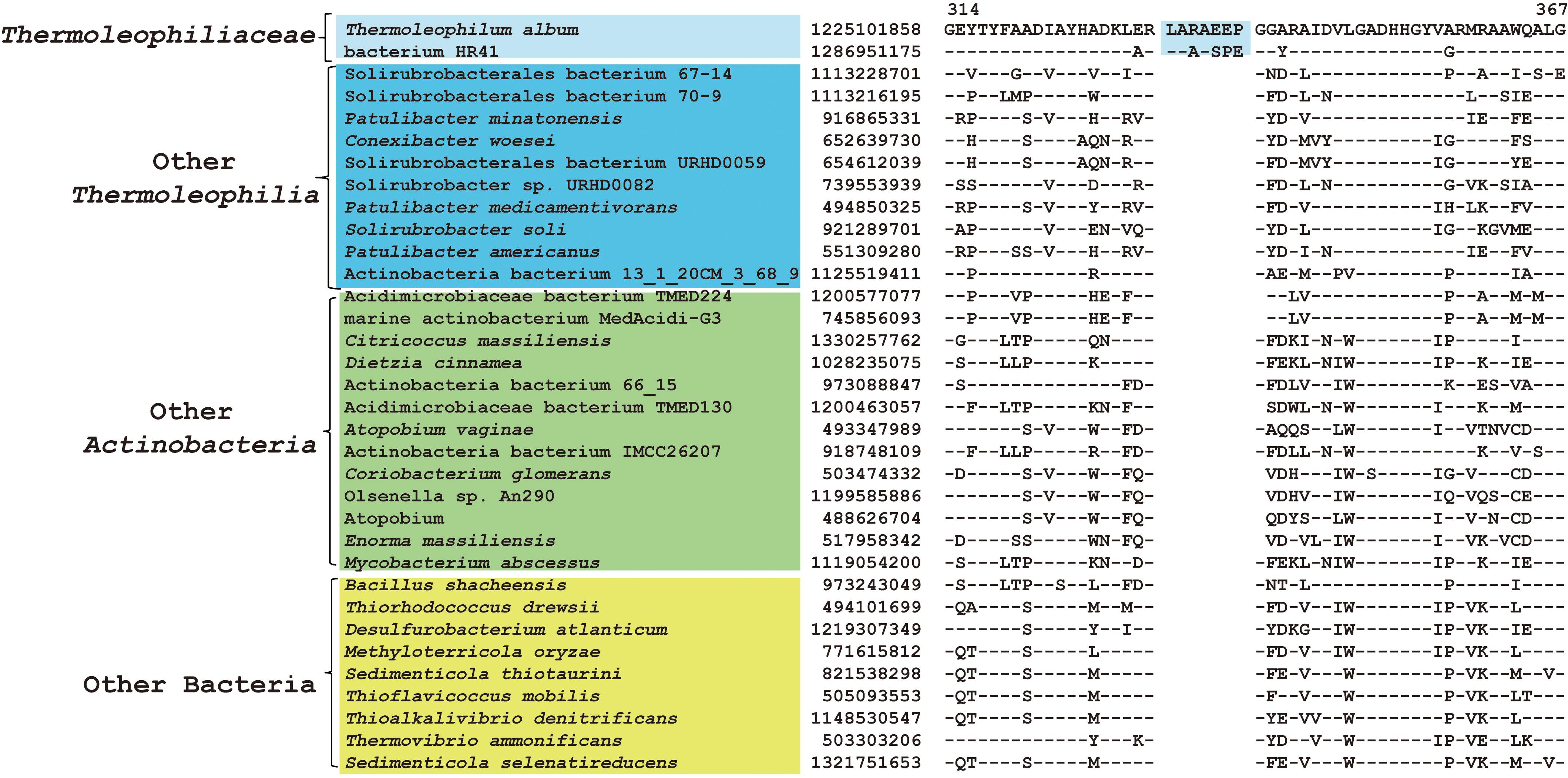
Figure 3. CSI specific to T. album and MAG HR41. Partial sequence alignment of arginine–tRNA ligase showing a 7 amino acid insertion that is uniquely shared by T. album and MAG HR41.
Within the order Solirubrobacterales, we have identified 6 CSIs that are specific to species of 3 families including Conexibacteraceae, Solirubacteraceae, and Patulibacteraceae, but no CSIs also shared by members of the new cluster (Table 1). One of these CSIs is illustrated in Figure 4, which is 1 aa deletion in a very conserved fragment of NADH-quinone oxidoreductase subunit B. Sequence information for other 5 CSIs that are uniquely shared by these 3 families are presented in Supplementary Figures S19–S23. Additionally, we discovered 24 CSPs that are only found in genomes of the named above 3 families but not in any other bacteria (Table 3). The shared presence of 6 CSIs and a number of CSPs indicate that Conexibacteraceae, Solirubacteraceae, and Patulibacteraceae are monophyletic. These two kinds of signature sequences were most likely introduced in the common ancestor of these three families and later on passed to all decedents. Moreover, if genome sequence of the fourth family Parviterribacteraceae becomes available in the future, it is worthwhile to examine whether some of these CSIs and CSPs are also shared by Parviterribacteraceae and actually constitute molecular markers of the Solirubrobacterales order.
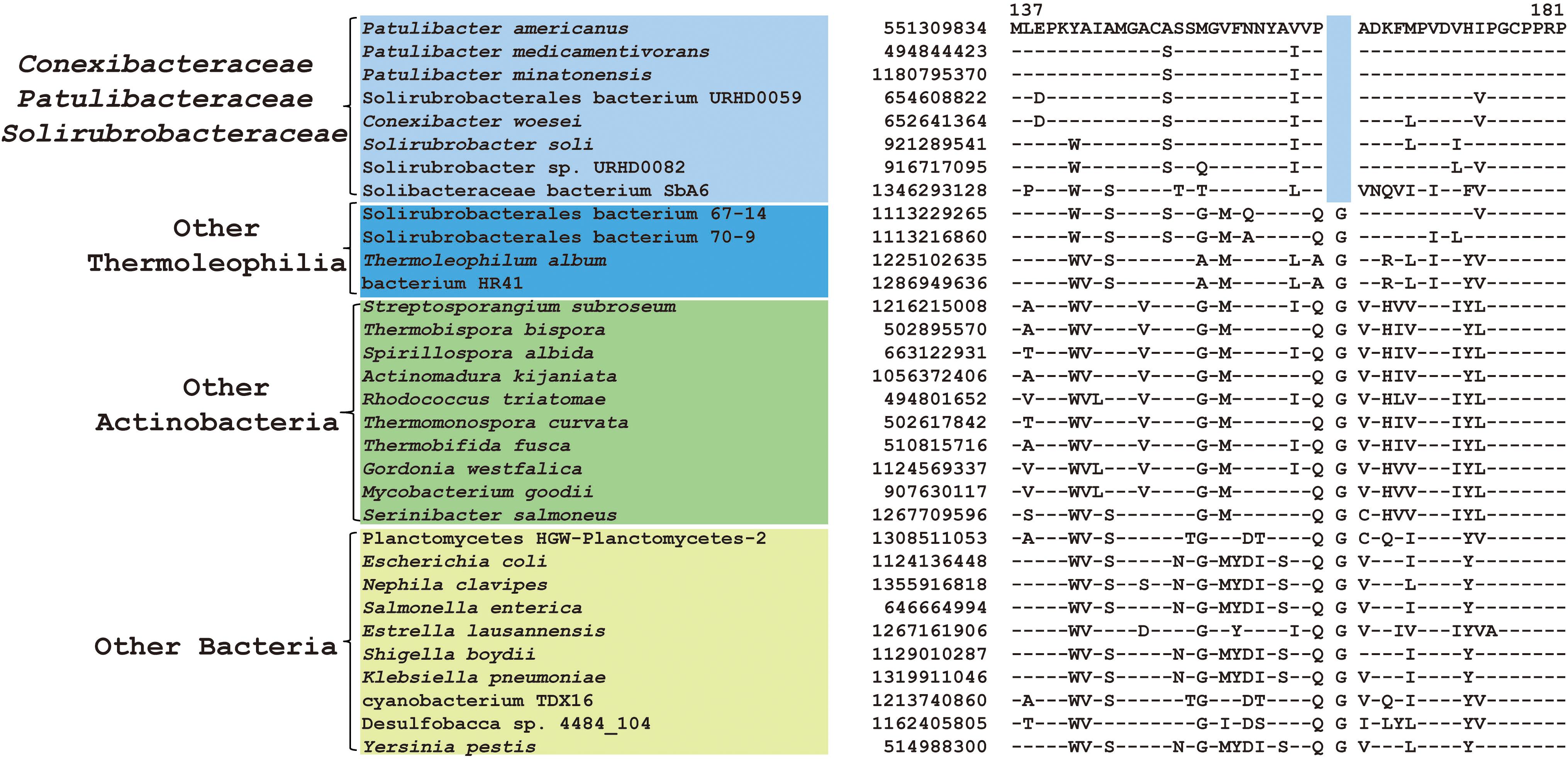
Figure 4. CSI specific to the families Conexibacteraceae, Solirubrobacteraceae and Patulibacteraceae. Partial alignment of the protein NADH-quinone oxidoreductase subunit B showing a 1 amino acid deletion that is uniquely shared by 3 families Conexibacteraceae, Solirubrobacteraceae and Patulibacteraceae.
As mentioned earlier, at family level within Thermoleophilia, only few cultivable strains are available and our current descriptions of some families such as Conexibacteraceae or Solirubacteraceae are only based on 1 or 2 strains. Here, we identified a number of CSIs that are specific to all genome-sequenced members of each family of Thermoleophilia except Parviterribacteraceae that don’t have genome sequence available (Table 1). For example, 4 CSIs were found to be unique to members of Conexibacteraceae (Figure 5 and Supplementary Figures S24–S26), 5 CSIs for Solirubacteraceae (Figure 6 and Supplementary Figures S27–S30), and totally 10 CSIs shared by 3 species of Patulibacteraceae (Figure 7 and Supplementary Figures S31–S39). We attempted to search for CSIs that are specific to the new cluster revealed by our phylogenomic analysis. Due to the incompleteness of the 3 genome assemblies, only 1 CSI is specifically shared by all three members of the new cluster (Supplementary Figure S40), while another 3 CSIs are only found in MAG “Solirubrobacterales bacterium 67-14” and “Solirubrobacterales bacterium 70-9” with two protein homologs missing in “Actinobacteria bacterium 13_1_20CM_3_68_9” (Supplementary Figures S41–S43). Furthermore, since more genomes are sequenced for Patulibacteraceae, we also identified 31 CSPs that are restricted to the genomes of this family, which provide additional markers for them (Table 3).
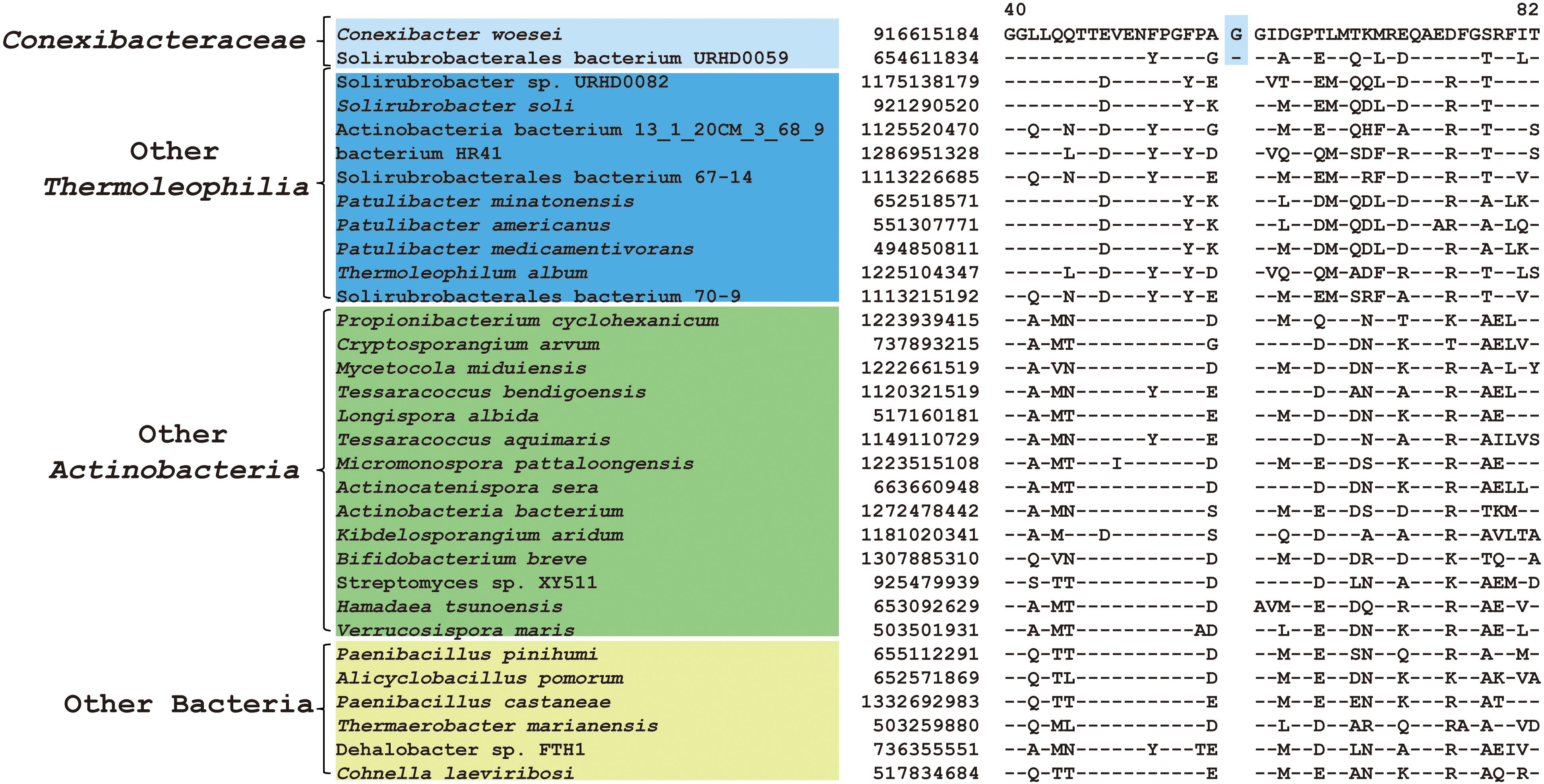
Figure 5. CSI specific to Conexibacteraceae. A 1 amino acid insertion in the protein thioredoxin-disulfide reductase that is uniquely shared by C. woesei and associated MAG.

Figure 6. CSI specific to Solirubrobacteraceae. A 3 amino acid CSI in the protein glutamine amidotransferase that is specific for S. soli and associated MAG.
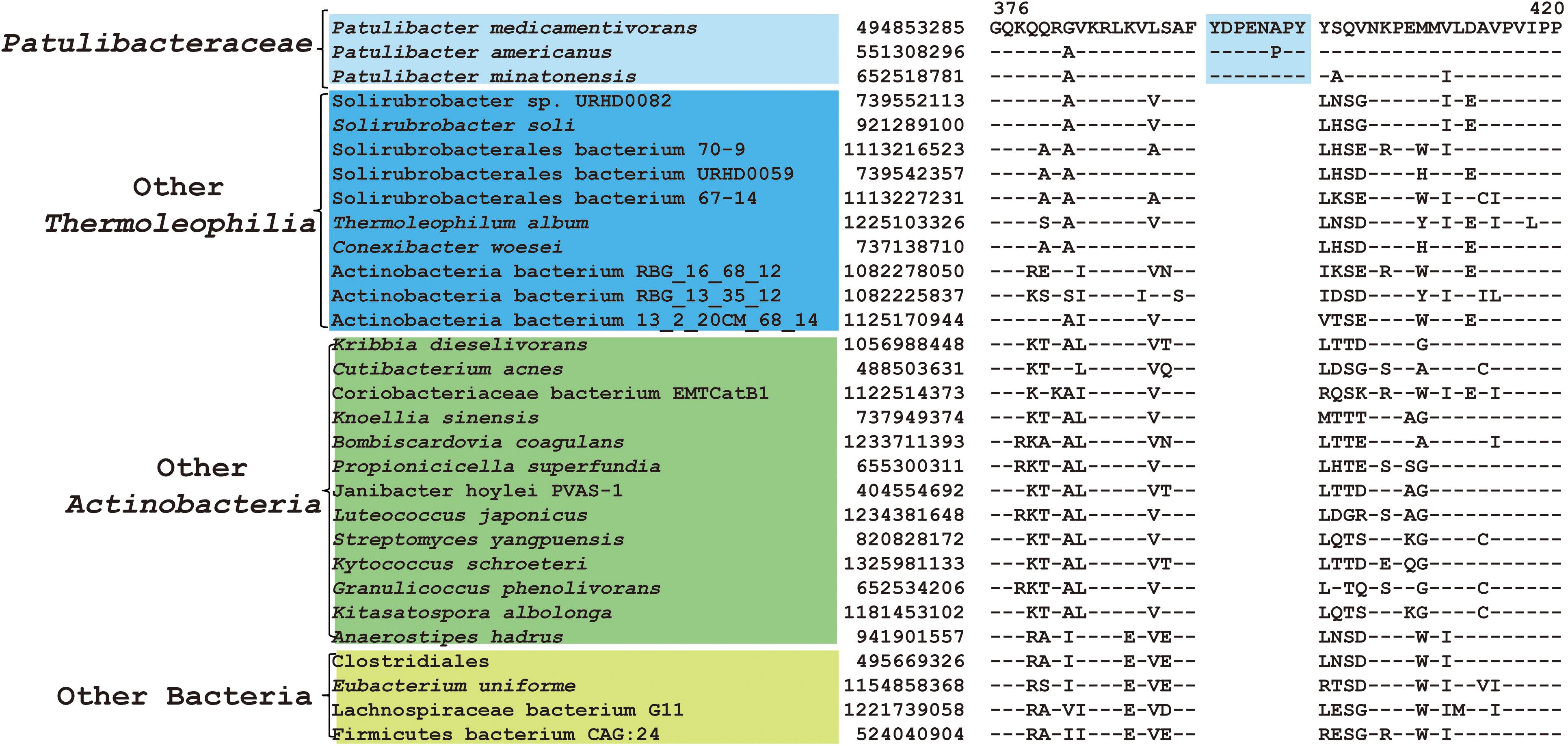
Figure 7. CSI specific to Patulibacteraceae. Partial sequence alignment of DNA-directed RNA polymerase subunit beta’ showing an 8 amino acid insertion that is specific for Patulibacteraceae.
Conclusion
Although metagenomic studies suggest that species of the class Thermoleophilia are abundant in hot spring and soil samples and they play an important role in biogeochemical cycling, very few studies have been performed on the phylogeny of this deep branch of Actinobacteria. Our current understanding of their taxonomy and phylogeny based on few cultivated species needs to be updated to better serve our exploration of this class. In this work, we have carried out detailed phylogenomic analysis of sequenced Thermoleophilia species and assembled genomes. The constructed phylogenetic tree clearly demonstrated the close affiliation of not yet cultivated MAGs with culturable type species. A new robust cluster composed of not yet cultivated MAGs is revealed within this class that might be a novel family belonging to Solirubrobacterales. Moreover, we identified a large number of CSIs and CSPs that are either specific to all species of this class or various lineages within it. These two types of signature sequences provide novel molecular markers that can be applied to define or distinguish the class Thermoleophilia or its affiliated taxa at higher taxonomic ranks, in addition to the 16S rRNA gene alone based standard.
In addition to their phylogenetic implications, these lineage-specific CSIs and CSPs can also be used to test the presence of Thermoleophilia species in different environmental samples. PCR primers could be designed for gene fragments that contain the above described CSIs or genes for CSPs, then we can detect the existence of certain lineages based on the presence or absence of the CSIs and CSPs. Furthermore, the functional significance of all CSIs and CSPs identified from this work are unknown. Due to their specificity to the Thermoleophilia class, functional studies on them might lead to identification of biochemical or physiological characteristics that are unique to this class of bacteria.
Author Contributions
DH carried out comparative analyses of the Thermoleophilia genomes to identify signatures reported here and constructed the phylogenetic trees. BG, DH, YZ and YM were responsible for the writing and editing of the manuscript. All of the work was carried out under the direction of BG.
Funding
This work was supported by National Science Foundation of China (31570011), Strategic Priority Research Program of the Chinese Academy of Sciences (XDA13020300 and XDA19060301), and Natural Science Foundation of Guangdong Province (2015A030306039). BG was also a scholar of the “100 Talents Project” of the Chinese Academy of Sciences.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We want to thank Dr. Radhey S. Gupta from McMaster University for generously providing the “SIG_CREATE” and “SIG_STYLE” programs.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01185/full#supplementary-material
Footnotes
References
Albuquerque, L., Franca, L., Rainey, F. A., Schumann, P., Nobre, M. F., and Da Costa, M. S. (2011). Gaiella occulta gen. nov., sp. nov., a novel representative of a deep branching phylogenetic lineage within the class actinobacteria and proposal of Gaiellaceae fam. nov. and Gaiellales ord. nov. Syst. Appl. Microbiol. 34, 595–599. doi: 10.1016/j.syapm.2011.07.001
Almeida, B., Kjeldal, H., Lolas, I., Knudsen, A. D., Carvalho, G., Nielsen, K. L., et al. (2013). Quantitative proteomic analysis of ibuprofen-degrading Patulibacter sp. strain I11. Biodegradation 24, 615–630. doi: 10.1007/s10532-012-9610-5
Alnajar, S., and Gupta, R. S. (2017). Phylogenomics and comparative genomic studies delineate six main clades within the family Enterobacteriaceae and support the reclassification of several polyphyletic members of the family. Infect. Genet. Evol. 54, 108–127. doi: 10.1016/j.meegid.2017.06.024
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Butterfield, C. N., Li, Z., Andeer, P. F., Spaulding, S., Thomas, B. C., Singh, A., et al. (2016). Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 4:e2687. doi: 10.7717/peerj.2687
Cabello-Yeves, P. J., Zemskaya, T. I., Rosselli, R., Coutinho, F. H., Zakharenko, A. S., Blinov, V. V., et al. (2018). Genomes of novel microbial lineages assembled from the sub-ice waters of lake baikal. Appl. Environ. Microbiol. 84:e2132-17. doi: 10.1128/AEM.02132-17
Cole, J. R., Wang, Q., Fish, J. A., Chai, B., Mcgarrell, D. M., Sun, Y., et al. (2014). Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 42, D633–D642. doi: 10.1093/nar/gkt1244
Foesel, B. U., Geppert, A., Rohde, M., and Overmann, J. (2016). Parviterribacter kavangonensis gen. nov., sp. nov. and Parviterribacter multiflagellatus sp. nov., novel members of parviterribacteraceae fam. nov. within the order solirubrobacterales, and emended descriptions of the classes thermoleophilia and rubrobacteria and their orders and families. Int. J. Syst. Evol. Microbiol. 66, 652–665. doi: 10.1099/ijsem.0.000770
Gao, B., and Gupta, R. S. (2012a). Microbial systematics in the post-genomics era. Antonie Van Leeuwenhoek 101, 45–54. doi: 10.1007/s10482-011-9663-1
Gao, B., and Gupta, R. S. (2012b). Phylogenetic framework and molecular signatures for the main clades of the phylum actinobacteria. Microbiol. Mol. Biol. Rev. 76, 66–112. doi: 10.1128/MMBR.05011-11
Gao, B., Mohan, R., and Gupta, R. S. (2009). Phylogenomics and protein signatures elucidating the evolutionary relationships among the gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 59(Pt 2), 234–247. doi: 10.1099/ijs.0.002741-0
Gao, B., Paramanathan, R., and Gupta, R. S. (2006). Signature proteins that are distinctive characteristics of actinobacteria and their subgroups. Antonie Van Leeuwenhoek 90, 69–91. doi: 10.1007/s10482-006-9061-2
Gupta, R. S. (2014). “Identification of conserved indels that are useful for classification and evolutionary studies,” in Methods in Microbiology, eds M. Goodfellow, I. Sutcliffe, and J. Chun (Oxford: Academic Press), 153–182. doi: 10.1016/bs.mim.2014.05.003
Gupta, R. S., and Gao, B. (2009). Phylogenomic analyses of clostridia and identification of novel protein signatures that are specific to the genus clostridium sensu stricto (cluster I). Int. J. Syst. Evol. Microbiol. 59, 285–294. doi: 10.1099/ijs.0.001792-0
Gupta, R. S., and Gao, B. (2010). “Recent advances in understanding microbial systematics,” in Microbial Population Genetics, ed. J. P. Xu (Norfolk: Caister Academic Press).
Ho, J., Adeolu, M., Khadka, B., and Gupta, R. S. (2016). Identification of distinctive molecular traits that are characteristic of the phylum “Deinococcus-Thermus” and distinguish its main constituent groups. Syst. Appl. Microbiol. 39, 453–463. doi: 10.1016/j.syapm.2016.07.003
Hu, D., Cha, G., and Gao, B. (2018). A phylogenomic and molecular markers based analysis of the class acidimicrobiia. Front. Microbiol. 9:987. doi: 10.3389/fmicb.2018.00987
Janssen, P. H. (2006). Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72, 1719–1728. doi: 10.1128/aem.72.3.1719-1728.2006
Ji, M., Greening, C., Vanwonterghem, I., Carere, C. R., Bay, S. K., Steen, J. A., et al. (2017). Atmospheric trace gases support primary production in Antarctic desert surface soil. Nature 552, 400–403. doi: 10.1038/nature25014
Ji, M., Van Dorst, J., Bissett, A., Brown, M. V., Palmer, A. S., Snape, I., et al. (2016). Microbial diversity at mitchell peninsula, eastern antarctica: a potential biodiversity “hotspot”. Polar Biol. 39:13.
Joseph, S. J., Hugenholtz, P., Sangwan, P., Osborne, C. A., and Janssen, P. H. (2003). Laboratory cultivation of widespread and previously uncultured soil bacteria. Appl. Environ. Microbiol. 69, 7210–7215. doi: 10.1128/aem.69.12.7210-7215.2003
Kantor, R. S., Van Zyl, A. W., Van Hille, R. P., Thomas, B. C., Harrison, S. T., and Banfield, J. F. (2015). Bioreactor microbial ecosystems for thiocyanate and cyanide degradation unravelled with genome-resolved metagenomics. Environ. Microbiol. 17, 4929–4941. doi: 10.1111/1462-2920.12936
Kato, S., Sakai, S., Hirai, M., Tasumi, E., Nishizawa, M., Suzuki, K., et al. (2018). Long-term cultivation and metagenomics reveal ecophysiology of previously uncultivated thermophiles involved in biogeochemical nitrogen cycle. Microbes Environ. 33, 107–110. doi: 10.1264/jsme2.ME17165
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., Mcwilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Li, H. Y., Wang, H., Wang, H. T., Xin, P. Y., Xu, X. H., Ma, Y., et al. (2018). The chemodiversity of paddy soil dissolved organic matter correlates with microbial community at continental scales. Microbiome 6:187. doi: 10.1186/s40168-018-0561-x
Ludwig, W., Euzeby, J., Schumann, P., Busse, H., Trujillo, M. E., Kampfer, P., et al. (2012). “Road map of the phylum actinobacteria,” in Bergey’s Manual of Systematic Bacteriology, 2 Edn, Vol. 5, eds M. Goodfellow, P. Kampfer, H. J. Busse, M. E. Trujillo, K. Suzuki, W. Ludwig, et al. (New York, NY: Springer), 1–28. doi: 10.1007/978-0-387-68233-4_1
Mukherjee, S., Seshadri, R., Varghese, N. J., Eloe-Fadrosh, E. A., Meier-Kolthoff, J. P., Goker, M., et al. (2017). 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat. Biotechnol. 35, 676–683. doi: 10.1038/nbt.3886
Na, S. I., Kim, Y. O., Yoon, S. H., Ha, S. M., Baek, I., and Chun, J. (2018). UBCG: up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 56, 280–285. doi: 10.1007/s12275-018-8014-6
Nouioui, I., Carro, L., Garcia-Lopez, M., Meier-Kolthoff, J. P., Woyke, T., Kyrpides, N. C., et al. (2018). Genome-based taxonomic classification of the phylum actinobacteria. Front. Microbiol. 9:2007. doi: 10.3389/fmicb.2018.02007
Ollagnier-De Choudens, S., Loiseau, L., Sanakis, Y., Barras, F., and Fontecave, M. (2005). Quinolinate synthetase, an iron-sulfur enzyme in NAD biosynthesis. FEBS Lett. 579, 3737–3743. doi: 10.1016/j.febslet.2005.05.065
Parks, D. H., Rinke, C., Chuvochina, M., Chaumeil, P. A., Woodcroft, B. J., Evans, P. N., et al. (2017). Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542. doi: 10.1038/s41564-017-0012-7
Pukall, R., Lapidus, A., Glavina Del Rio, T., Copeland, A., Tice, H., Cheng, J. F., et al. (2010). Complete genome sequence of conexibacter woesei type strain (ID131577). Stand. Genomic Sci. 2, 212–219. doi: 10.4056/sigs.751339
Pulschen, A. A., Bendia, A. G., Fricker, A. D., Pellizari, V. H., Galante, D., and Rodrigues, F. (2017). Isolation of uncultured bacteria from antarctica using long incubation periods and low nutritional media. Front. Microbiol. 8:1346. doi: 10.3389/fmicb.2017.01346
Reddy, G. S., and Garcia-Pichel, F. (2009). Description of Patulibacter americanus sp. nov., isolated from biological soil crusts, emended description of the genus patulibacter takahashi et al. 2006 and proposal of solirubrobacterales ord. nov. and thermoleophilales ord. nov. Int. J. Syst. Evol. Microbiol. 59, 87–94. doi: 10.1099/ijs.0.64185-0
Salam, N., Jiao, J., Zhang, X., and Li, W. (2019). Update in the classification of higher ranks in the phylum actinobacteria. Int. J. Syst. Evol. Microbiol. (in press).
Suzuki, K., and Whitman, W. B. (2012). “Class VI. thermoleophilia class. nov,” in Bergey’s Manual of Systematic Bacteriology, 2nd Edn, eds M. Goodfellow, P. Kampfer, H. J. Busse, M. E. Trujillo, K. Suzuki, W. Ludwig, et al. (New York, NY: Springer), 2010–2028.
Suzuki, K., and Whitman, W. B. (2015). “Thermoleophilia class. nov,” in Bergey’s Manual of Systematics of Archaea and Bacteria, eds W. B. Whitman, F. A. Rainey, P. Kampfer, M. Trujillo, J. Chun, P. Devos, et al. (Hoboken, NY: John Wiley & Sons, Inc.).
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Volbeda, A., Darnault, C., Renoux, O., Reichmann, D., Amara, P., Ollagnier De Choudens, S., et al. (2016). Crystal structures of quinolinate synthase in complex with a substrate analogue, the condensation intermediate, and substrate-derived product. J. Am. Chem. Soc. 138, 11802–11809. doi: 10.1021/jacs.6b05884
Woodcroft, B. J., Singleton, C. M., Boyd, J. A., Evans, P. N., Emerson, J. B., Zayed, A. A. F., et al. (2018). Genome-centric view of carbon processing in thawing permafrost. Nature 560, 49–54. doi: 10.1038/s41586-018-0338-1
Yakimov, M. M., Lunsdorf, H., and Golyshin, P. N. (2003). Thermoleophilum album and thermoleophilum minutum are culturable representatives of group 2 of the rubrobacteridae (actinobacteria). Int. J. Syst. Evol. Microbiol. 53, 377–380. doi: 10.1099/ijs.0.02425-0
Zarilla, K. A., and Perry, J. J. (1986). Deoxyribonucleic-acid homology and other comparisons among obligately thermophilic hydrocarbonoclastic bacteria, with a proposal for thermoleophilum-minutum Sp-Nov. Int. J. Syst. Evol. Microbiol. 36, 13–16. doi: 10.1099/00207713-36-1-13
Zhang, G., Gao, B., Adeolu, M., Khadka, B., and Gupta, R. S. (2016). Phylogenomic analyses and comparative studies on genomes of the bifidobacteriales: identification of molecular signatures specific for the order bifidobacteriales and its different subclades. Front. Microbiol. 7:978. doi: 10.3389/fmicb.2016.00978
Zhi, X. Y., Li, W. J., and Stackebrandt, E. (2009). An update of the structure and 16S rRNA gene sequence-based definition of higher ranks of the class actinobacteria, with the proposal of two new suborders and four new families and emended descriptions of the existing higher taxa. Int. J. Syst. Evol. Microbiol. 59, 589–608. doi: 10.1099/ijs.0.65780-0
Keywords: Thermoleophilia, phylogeny, molecular signatures, conserved signature indels, conserved signature proteins
Citation: Hu D, Zang Y, Mao Y and Gao B (2019) Identification of Molecular Markers That Are Specific to the Class Thermoleophilia. Front. Microbiol. 10:1185. doi: 10.3389/fmicb.2019.01185
Received: 25 January 2019; Accepted: 09 May 2019;
Published: 24 May 2019.
Edited by:
Haiwei Luo, The Chinese University of Hong Kong, ChinaReviewed by:
Juan Antonio Ugalde, uBiome, ChileBärbel Ulrike Fösel, Helmholtz Center Munich, Germany
Copyright © 2019 Hu, Zang, Mao and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beile Gao, Z2FvYkBzY3Npby5hYy5jbg==
†These authors have contributed equally to this work