 Carmen Berlanas1
Carmen Berlanas1 Mónica Berbegal2
Mónica Berbegal2 Georgina Elena2
Georgina Elena2 Meriem Laidani2José Félix Cibriain3Ana Sagües3
Meriem Laidani2José Félix Cibriain3Ana Sagües3 David Gramaje1*
David Gramaje1*- 1Instituto de Ciencias de la Vid y del Vino, Consejo Superior de Investigaciones Científicas – Universidad de la Rioja – Gobierno de La Rioja, Logroño, Spain
- 2Instituto Agroforestal Mediterráneo, Universitat Politècnica de València, Valencia, Spain
- 3EVENA, Sección de Viticultura y Enología del Gobierno de Navarra, Olite, Spain
The microbiota colonizing the rhizosphere and the endorhizosphere contribute to plant growth, productivity, carbon sequestration, and phytoremediation. Several studies suggested that different plants types and even genotypes of the same plant species harbor partially different microbiomes. Here, we characterize the rhizosphere bacterial and fungal microbiota across five grapevine rootstock genotypes cultivated in the same soil at two vineyards and sampling dates over 2 years by 16S rRNA gene and ITS high-throughput amplicon sequencing. In addition, we use quantitative PCR (qPCR) approach to measure the relative abundance and dynamic changes of fungal pathogens associated with black-foot disease. The objectives were to (1) unravel the effects of rootstock genotype on microbial communities in the rhizosphere of grapevine and (2) to compare the relative abundances of sequence reads and DNA amount of black-foot disease pathogens. Host genetic control of the microbiome was evident in the rhizosphere of the mature vineyard. Microbiome composition also shifted as year of sampling, and fungal diversity varied with sampling moments. Linear discriminant analysis identified specific bacterial (i.e., Bacillus) and fungal (i.e., Glomus) taxa associated with grapevine rootstocks. Host genotype did not predict any summary metrics of rhizosphere α- and β-diversity in the young vineyard. Regarding black-foot associated pathogens, a significant correlation between sequencing reads and qPCR was observed. In conclusion, grapevine rootstock genotypes in the mature vineyard were associated with different rhizosphere microbiomes. The latter could also have been affected by age of the vineyard, soil properties or field management practices. A more comprehensive study is needed to decipher the cause of the rootstock microbiome selection and the mechanisms by which grapevines are able to shape their associated microbial community. Understanding the vast diversity of bacteria and fungi in the rhizosphere and the interactions between microbiota and grapevine will facilitate the development of future strategies for grapevine protection.
Introduction
Plants have evolved to cope with biotic and abiotic stresses in association with soil microorganisms (Lemanceau et al., 2017). These microorganisms are known as plant microbiota and, together with the plant, they form an holobiont (Liu et al., 2018). Plant-soil microbiome interactions are complex and, until recent times, the study of these relationships has been mainly focused in the pathogenicity of some microbial agents and how they use and compete for the resources (Philippot et al., 2013; Zancarini et al., 2013; Gilbert et al., 2014; Sapkota et al., 2015). Recent investigations have shown that soil microbiota can directly and indirectly interact with the plants improving their fitness and health (Sapkota et al., 2015). For example, these interactions help plants to deal with abiotic stress and diseases, improving the exchange of substances such as nitrogen or phosphate, or by acting as biocontrol agents through competition with pathogens (Reinhold-Hurek et al., 2015; Vega-Avila et al., 2015; Gallart et al., 2018).
Roots are surrounded by a narrow zone of soil known as rhizosphere. This area, which is influenced by the roots, has a high microbial diversity and its community structure is expected to be different than the one found in the bulk soil (Reinhold-Hurek et al., 2015). The rhizosphere microbiome community composition is affected by different factors, such as ambient conditions, soil properties, and background microbial composition (Qiao et al., 2017). In addition, plants are able to shape their rhizosphere microbiome, as evidenced by the fact that different plant species host specific microbial communities when grown on the same soil (Aira et al., 2010; Berendsen et al., 2012; Bazghaleh et al., 2015).
As reviewed by Philippot et al. (2013), plant roots release a huge variety of carbon-containing compounds known as rhizodeposits (nutrients, exudates, border cells, and mucilage) which make the rhizosphere more nutritive than the bulk soil, which is mostly mesotrophic/oligotrophic, inducing therefore changes on soil microbial communities. It has been reported that the biodiversity in the rhizosphere is lower than in the corresponding bulk soil (Reinhold-Hurek et al., 2015; Lemanceau et al., 2017) since carbon availability often limits microbial growth (Dennis et al., 2010). Rhizodeposits released by the plants considerably vary according to the age and development of plants, among species and even among different genotypes of the same species (Inceoǧlu et al., 2010; Philippot et al., 2013; Gilbert et al., 2014; Bazghaleh et al., 2015; Hacquard, 2016; Wagner et al., 2016; Lemanceau et al., 2017; Qiao et al., 2017).
The rhizosphere is also the infection court where soil-borne pathogens establish a parasitic relationship with the plant. To infect root tissue, pathogens have to compete with members of the rhizosphere microbiome for available nutrients and microsites (Chapelle et al., 2016). Exploiting genetic variation in host plant species and understanding interactions between microbiota and their hosts plants will allow the rhizosphere microbiota to be incorporated into plant breeding programs to promote beneficial associations between plants and microorganisms.
Common grapevine (Vitis vinifera L.) is one of the most extensively grown and economically important woody perennial fruit crop worldwide with an annual production in 2014 exceeding 74 million tons of grapes and 30 million tons of wine (FAO, 2018). Since the late 19th century, V. vinifera cultivars have been grafted onto resistant rootstock of other Vitis species and hybrids to combat the devastating root phylloxera pest. Several major criteria have been outlined for choosing rootstocks: resistance to phylloxera and nematodes, and adaptability to drought, salinity, limestone content, and poor mineral nutrition (Reynolds and Wardle, 2001). In addition, the rootstock influence may affect scion vigor, yields, and fruit and wine qualities (Warschefsky et al., 2016).
Plant genetic control over microbial communities in the rhizosphere has been reported for different genotypes of the same species (Aira et al., 2010; Bouffaud et al., 2012; Peiffer et al., 2013; Marques et al., 2014; Jiang et al., 2017; Gallart et al., 2018). However, within grapevine species, the impact of genetic variation on the composition of the bacterial and fungal microbiota is poorly understood. In a recent study, Marasco et al. (2018) observed that five grapevine genotypes influenced the bacterial microbiome from both the root tissues and the rhizosphere fractions at a single vineyard, sampling date and year.
To better understand the players and processes that operate in the rhizosphere, a variety of molecular techniques, such as metagenomics have been applied over the past decade. Here, we characterize the rhizosphere bacterial and fungal microbiota across five grapevine rootstock genotypes cultivated in the same soil at two vineyards and sampling dates over 2 years by 16S rRNA gene and ITS high-throughput amplicon sequencing (HTAS). This design allowed us to evaluate the effect of the growing region, year, sampling date, grapevine genotype, and their interactions on the bacterial and fungal community diversity. In addition, we used quantitative Polymerase Chain Reaction (qPCR) approach to measure the relative abundance and dynamic changes of fungal pathogens associated with black-foot disease, one of the main soil-borne fungal diseases affecting grapevine production worldwide.
Materials and Methods
Sample Collection
Grapevine rhizosphere samples of five rootstocks (110 R, 140 Ru, 1103 P, 41 B, and 161-49 C) were collected at two vineyards located in Aldeanueva de Ebro (abbreviated as “Aldea”) (La Rioja, Spain) and Olite (Navarre, Spain). Features of the selected rootstocks are reported in Supplementary Table 1 (Martínez-Cutillas et al., 1990; Hidalgo, 2002; Keller, 2010). All the selected rootstocks were cultivated in the same vineyard and had been grafted onto Tempranillo cultivar. Soil physicochemical properties showed significant differences between soil types. Climate and soil management practices for fertilization, irrigation, and disease control also varied between vineyards (Supplementary Table 2). Aldea vineyard was 25-year-old vines at the moment of sampling and contained four randomized blocks of 48 vines per rootstock and block. Olite vineyard was 7-year-old vines at the moment of sampling and contained three randomized blocks of 15 vines per rootstock and block. In each vineyard, three rhizosphere samples were randomly collected per rootstock at two sampling dates (June and November) over 2 years (2016 and 2017). Sampled vines did not show any symptom of disease or nutrient deficiency. A total of 60 samples were collected per vineyard.
Rhizosphere soil samples were collected with a sterile spade close to the stem at depths of 40 to 50 cm, where the root system was denser. All samples were stored in sterile bags on dry ice at the time of sampling, and brought to the laboratory for further processing within 24 h from the time of sampling. The sampled roots with rhizosphere soil particles attached were placed in sterile tubes containing 9 mL of physiological solution (9 g/L NaCl). The tubes were vortexed for 5 min to detach the soil particles and then centrifuged at 4000 rpm for 5 min. The supernatant was discarded and the remaining soil fraction was used for DNA extraction.
DNA Extraction and Sequencing
The rhizosphere DNA was extracted from 0.5 g sample using the DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) and DNA samples were randomized across plates. The bacterial V4 region of the 16S rRNA gene was amplified using the protocol described by Lundberg et al. (2013). The universal primer pair 515F and 806R was used to generate bacterial-derived 16S rRNA amplicons. PNA PCR clamps were used to reduce host organelle contamination. The fungal ITS2 region was amplified using the universal primers ITS3/KYO2 and ITS4 (Toju et al., 2012). All primers were modified to include Illumina adapters1. Each 25 μl reaction contained 12.5 μl of HiFi HotStart Ready Mix (KAPA Biosystems, Woburn, MA, United States), 1.0 μl of each primer (10 μM), 2.5 μl of DNA template (5 ng/μl), and 8.0 μl PCR-grade water. PCR amplifications (performed in triplicate for each sample) consisted of a 3 min denaturation at 95°C; 25 cycles of 30 s at 95°C, 30 s at 55°C and 30 s at 72°C; and 5 min at 72°C. Samples were cleaned using the AMPure beads XP purification system (Beckman Coulter, United Kingdom) and sequenced on the Illumina MiSeq platform at the Fundación FISABIO (Valencia, Spain) facility using a 2 × 300 nucleotide paired reads protocol.
Data Analysis
Raw forward and reverse reads for each sample were assembled into paired-end reads considering the minimum overlapping of 50 nucleotides and a maximum of one mismatch within the region using the fastq-join tool from the ea-tools suite (Aronesty, 2011). The paired reads were then quality trimmed with a minimum of Q20. Sequences without either primer were discarded. Chimeric sequences were identified and filtered using the Usearch tool (Edgar, 2010, 2018). The UClust algorithm (Edgar, 2013) in QIIME (Caporaso et al., 2010) was used to cluster sequences at a 97% sequence similarity against UNITE dynamic database (Abarenkov et al., 2010) for ITS reads and Greengenes database (DeSantis et al., 2006) using the QIIME implementation of the RDP classifier for 16S rRNA reads (Caporaso et al., 2010). A tree was constructed from a gap-filtered alignment using FastTree (Price et al., 2009). A final OTU table was created excluding unaligned sequences and singletons. OTUs with no kingdom-level classification or matching chloroplast, mitochondrial, or Viridiplantae sequences were then removed from the data set. Good’s coverage values were calculated using the Mothur computer software (Schloss et al., 2009). The rarefied OTU table and the phylogenetic tree were used as inputs for the subsequent analyses of α- and β- diversity. The OTU table was log transformed for statistical analysis (McMurdie and Holmes, 2014). As a final filter, taxa whose total abundances were less than 1% of the mean abundance were excluded, and only the OTUs present in at least two-thirds of the replicates of each sample were selected.
Bacterial and Fungal Diversity, Taxonomy Distribution and Statistical Analysis
Biodiversity indexes and principle statistics analyses on taxonomic profiles were analyzed in R version 3.5 using the vegan (Oksanen et al., 2018) and Phyloseq packages (McMurdie and Holmes, 2014). Data in each vineyard was analyzed separately due to the differences in soil chemistry and climate (Supplementary Table 2). Technical noise (variation attributable to sequencing depth or batch effects) was controlled by including MiSeq run as a random effect.
Within sample type, α-diversity estimates were calculated by analyzing the Chao1 richness and Shannon diversity in Phyloseq package, as implemented in the tool MicrobiomeAnalyst (Dhariwal et al., 2017). The normalized OTU table was analyzed using Bray Curtis metrics (Bray and Curtis, 1957) and utilized to evaluate the β- diversity and to construct PCoA plots (Vázquez-Baeza et al., 2013) using MicrobiomeAnalyst. In order to compare bacterial and fungal communities composition and to partition of variance in different categories, Bray–Curtis distance matrices were subjected to PERMANOVA (Anderson, 2001) using the adonis function with a permutation number of 999 available in the vegan package of R. PERMANOVA was performed to investigate which OTUs significantly differed in abundance among experimental factors.
The variance-partitioning model tests for effects of year, sampling date and genotype on microbiome communities, while year-by-genotype and date-by-genotype interaction terms describe how the distinct fungal and bacterial communities at different common rootstocks respond differently to each of these factors. The linear mixed models were fit using the lme4 package (Bates et al., 2015). Statistical significance of fixed predictors (Year + Sampling Date + Genotype + Year × Genotype + Date × Genotype) was assessed using Type III ANOVA with Satterthwaite’s approximation of denominator degrees of freedom in the package InnerTest (Kuznetsova et al., 2016), and of random effects (MiSeq run) using likelihood ratio tests. This model was used to predict community descriptors that were continuous and approximately normally distributed in α-diversity metrics (Shannon entropy and Chao1 estimated richness) as described above.
The Linear Discriminant Analysis Effect Size (LEfSe) algorithm was used to identify taxa (genus level or higher) that differed in relative abundance between the rootstocks (Segata et al., 2011). The online Galaxy Version 1.0 interface (The Huttenhower Lab, 2018) was used, the threshold for the logarithmic LDA score was set at 1.0 and the Wilcoxon p-value at 0.05. The results are displayed in a cladogram and a bar graph. A Similarity Percentages (SIMPER) analysis was performed with PRIMER 6 software to explore the dissimilarities between the rootstock factor. Summarized taxa tables at the phylum and genera levels were used to investigate the phylogenetic groups that contribute to the dissimilarity. Unclassified OTUs amounting to less than 3% of the relative abundance in the rhizosphere were discarded from the analysis, according to Marasco et al. (2018). The bacterial and fungal OTUs shared among vineyards and rootstocks were defined by a Venn-diagram analysis using the software available at (Van de Peer et al., 2018).
Quantitative PCR Amplification and Quantification of Black-Foot Disease Pathogens
Quantitative PCR analyses were performed with the DNA extracted from the soil samples, as Agustí-Brisach et al. (2014) developed in previous research, using the primers YT2F and Cyl-R (Dubrovsky and Fabritius, 2007; Tewoldemedhin et al., 2011). These primers amplify the main Cylindrocarpon-like asexual morphs associated with black-foot disease, in particular those belonging to the genera Dactylonectria, Ilyonectria, Neonectria, and Thelonectria. Rotor-Gene 6000 real-time rotary analyzer (Qiagen, Hilden, Germany) was used to perform the qPCR amplifications. Each reaction contained 2 μl of DNA, 1× of SYBR Premix Ex Taq II (Tli RNase H Plus) (Takara Bio Inc., Shiga, Japan) and 0.4 μM of each primer. The reaction mix was adjusted to a final volume of 20 μl with sterile distilled water. The thermocycling profile consisted of 30 s at 95°C and 50 cycles of 10 s at 95°C, 10 s at 60°C, and 30 s at 72°C. To evaluate amplification specificity, melting curve analysis was performed at the end of the qPCR runs according to the manufacturer’s recommendations. Each analysis included three replicates of each sample, a non-template control reaction (water) and a positive control containing DNA extracted from a pure culture of the Dactylonectria torresensis isolate GTMF DT097, obtained from the collection of the Instituto Agroforestal Mediterráneo, Universitat Politècnica de Valencia, Spain. D. torresensis is the most common fungal species associated with black-foot diseased vines in Italy (Carlucci et al., 2017), Portugal (Reis et al., 2013), and Spain (Berlanas et al., 2017). For DNA extraction, fungal mycelium of this isolate grown on potato dextrose agar (PDA, Biokar-Diagnostics, Zac de Ther, France) for 2 weeks at 25°C in darkness, was scraped from the surface of the plate with a sterile scalpel. Total DNA was extracted using the E.Z.N.A. Plant Miniprep kit (Omega Bio-Tek, Doraville, United States) following the manufacturer’s instructions and mycelia was previously homogenized with 4 steel beads of 2.38 mm and 2 of 3 mm diameter (Qiagen, Hilden, Germany) using a FastPrep-24TM5G (MP Biomedicals, California, United States) at 5 m/s for 20 s twice. DNA extracted was quantified with Invitrogen Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, United States).
DNA of the Cylindrocarpon-like asexual morphs species was quantified using a standard curve constructed with the isolate GTMF DT097, consisting of a dilution series from 275 μg/μL to 0.275 fg/μL. Quantitative PCR analysis were perform as previously explained and the standard curve was generated following the MIQE guidelines (Bustin et al., 2009), by plotting quantification cycle (Cq) values obtained for each specific DNA concentration, versus the logarithm of the initial concentration of isolate DNA. The mean DNA concentration and the standard deviation were determined from three replicates per dilution. Sensitivity of the qPCR assay was assessed using the standard curve to determine the minimum DNA concentration that can be detected. The amplification efficiency (E) and the coefficient of determination (R2) of the standard curve were obtained using the Rotor-Gene 6000 Series software v. 1.7 (Qiagen, Hilden, Germany). Signal threshold levels were set automatically by the instrument software and the limit of detection (LOD) was identified by the last dilution when successful qPCR amplification of DNA occurred, accompanied by a melting curve peak temperature specific to D. torresensis.
Values from the Cylindrocarpon-like asexual morphs number of OTUs and DNA concentration were transformed by log (n/N ∗ 1000 + 1). Where n was the number of OTUs or the DNA concentration detected on each sample and N was the total number of OTUs and the total DNA concentration detected. An analysis of correlation between both transformed datasets was performed in R version 3.5 using the corrr package.
Results
High-Throughput Amplicon Sequencing
After paired-end alignments, quality filtering and deletion of chimeric, singletons, and mitochondrial and chloroplast sequences, a total of 4,337,395 bacterial 16S rRNA sequences and 6,216,366 fungal internal transcribed spacer (ITS) sequences were generated from 117 (three samples were removed from the analysis due to the low number of sequence reads) and 120 samples, respectively, and assigned to 975 bacterial and 567 fungal operational taxonomic units (OTUs) (Supplementary Table 3). Good’s coverage values indicated that on average 94.5 and 90.1% of the total species richness were accounted for in bacteria and fungal communities, respectively (Supplementary Table 4). Chao1 diversity estimator ranged from 143.6 to 549.5 in the bacterial microbiome, and from 90.5 to 254.9 in the fungal microbiome. Shannon diversity estimator ranged from 1.80 to 4.68 in the bacterial microbiome, and from 1.80 to 3.84 in the fungal microbiome (Supplementary Table 4).
Core Grapevine Phylogeny Between Vineyards
The two habitats used as vineyard sites (Aldeanueva del Ebro, abbreviated “Aldea” in the figures and tables; and Olite) were separated by 45 km, and varied in most of soil physicochemical properties (Supplementary Table 2). Bacterial communities of rhizosphere soil samples did not differ significantly between vineyards (Supplementary Table 5). However, α-diversity differed among sites when studying the fungal microbiota, and principal coordinates analysis (PCoA) of Bray Curtis data demonstrated that vineyard was the primary source of β-diversity (Supplementary Figure 1). Comparing the fungal and bacterial microbiota of the two vineyards, 82.9 and 58.7% of bacterial and fungal OTUs, respectively, were shared between vineyards, demonstrating the existence of a “core” grape phylogeny that is independent of the growing region (Figure 1).
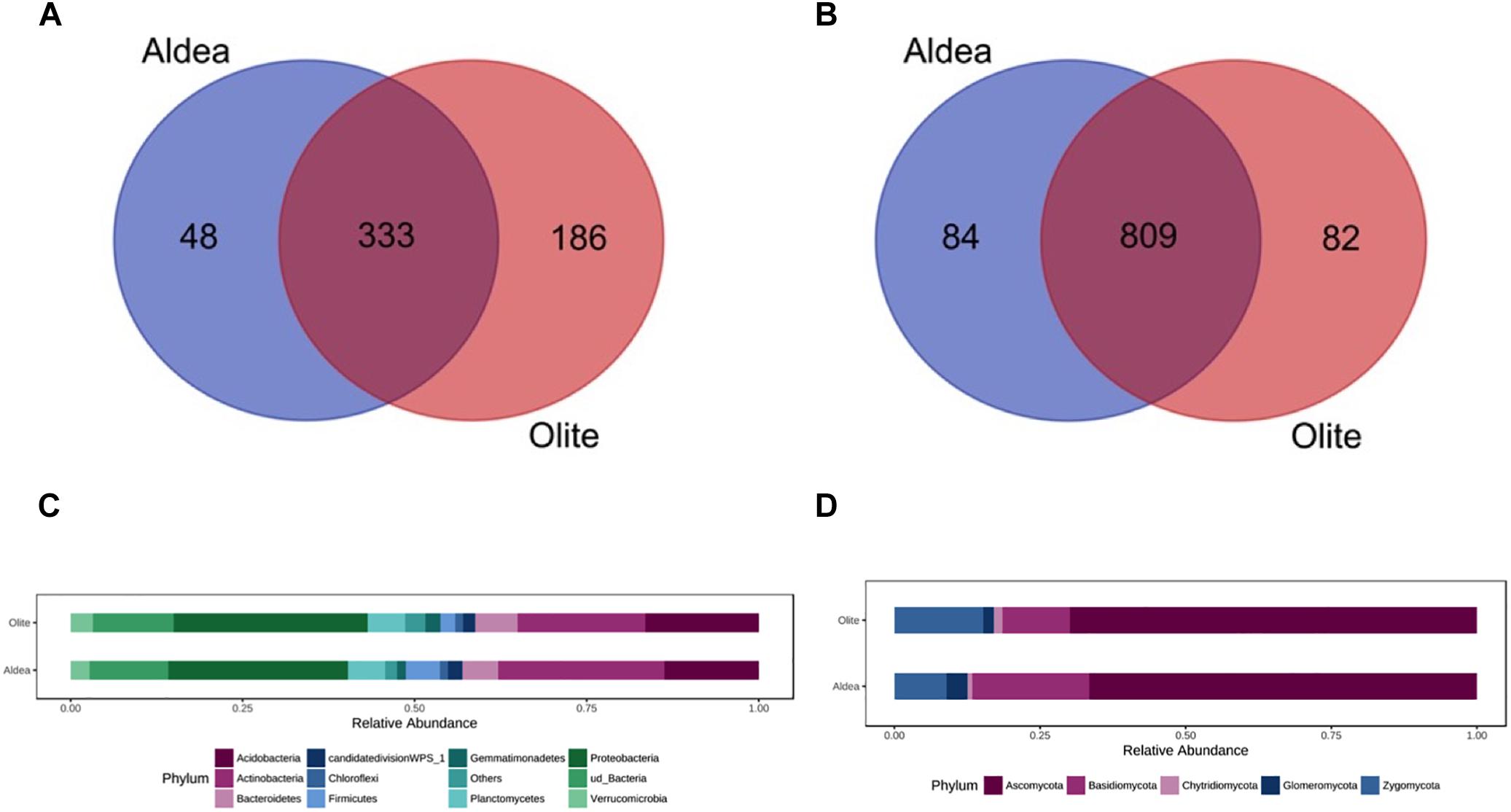
Figure 1. Venn diagram illustrating the overlap of the OTUs identified in the bacterial (A) and fungal (B) microbiota between vineyards. Relative abundance of different bacterial (C) and fungal (D) phyla in the rootstock rhizospheres in both vineyards representing OTUs showing more than 1% relative abundance of all reads and present in at least 2/3 of replicates. Phyla representing less than 1% of the total reads are grouped in “Others”.
The relative abundance of bacterial and fungal phyla detected across all samples is shown in Figure 1. In both vineyards, the bacterial phyla Proteobacteria (26.1 and 28.1% in Aldea and Olite, respectively) and Actinobacteria (24.1 and 18.5%) represented almost 50% of the total bacteria detected. These phyla were followed by Acidobacteria (13.7 and 16.4%), unidentified bacteria (11.4 and 11.7%), and Bacteroidetes (5.2 and 6.1%) (Figure 1). The most abundant families within the Proteobacteria phylum were unidentified families from the order Rhizobiales (13.0 and 10.4% in Aldea and Olite, respectively), unidentified families from the class Betaproteobacteria (9.8 and 13.0%) and Sphingomonadaceae (7.6 and 10.7%). The most abundant families within the Actinobacteria phylum were unidentified Actinobacteria (29.1 and 22.5% in Aldea and Olite, respectively), Gaiellaceae (16.0 and 15.2%) and Streptomycetaceae (6.2 and 6.7%) (Supplementary Figure 2). Regarding the fungal taxa, the most abundant fungal phylum was Ascomycota (66.6 and 69.9% in Aldea and Olite, respectively), followed by Basidiomycota (20.1 and 11.5%) and Zygomycota (8.9 and 15.2%) (Figure 1). The most abundant families within the Ascomycota phylum were Nectriaceae (15.4%), unidentified Ascomycota (8.8%), and Bionectriaceae (9.1%) in Aldea vineyard, and Nectriaceae (17.7%), unidentified Ascomycota (11.1%), Pyronemataceae (9.6%), and Trichocomaceae (8.4%) in Olite vineyard (Supplementary Figure 2).
Host Genetic Influence on the Rhizosphere Microbiota
Bacterial and fungal diversity in rhizosphere soil samples differed significantly among rootstocks in Aldea vineyard. However, plant genotype did not predict Chao1 diversity (Table 1). Host genotype was the most important factor in structuring bacterial (R2 = 0.65, P < 0.001) and fungal (R2 = 0.86, P < 0.001) communities in the entire dataset, and also when the data were split by year and date (Table 2). A PCoA further demonstrated the variation in the total dataset could be attributed to host genotype in Aldea vineyard (Figure 2). In Olite vineyard, plant genotype had a much weaker influence on rhizosphere-associated bacterial and fungal communities. Host genotype did not predict any summary metrics of rhizosphere α and β-diversities (Tables 1, 2).
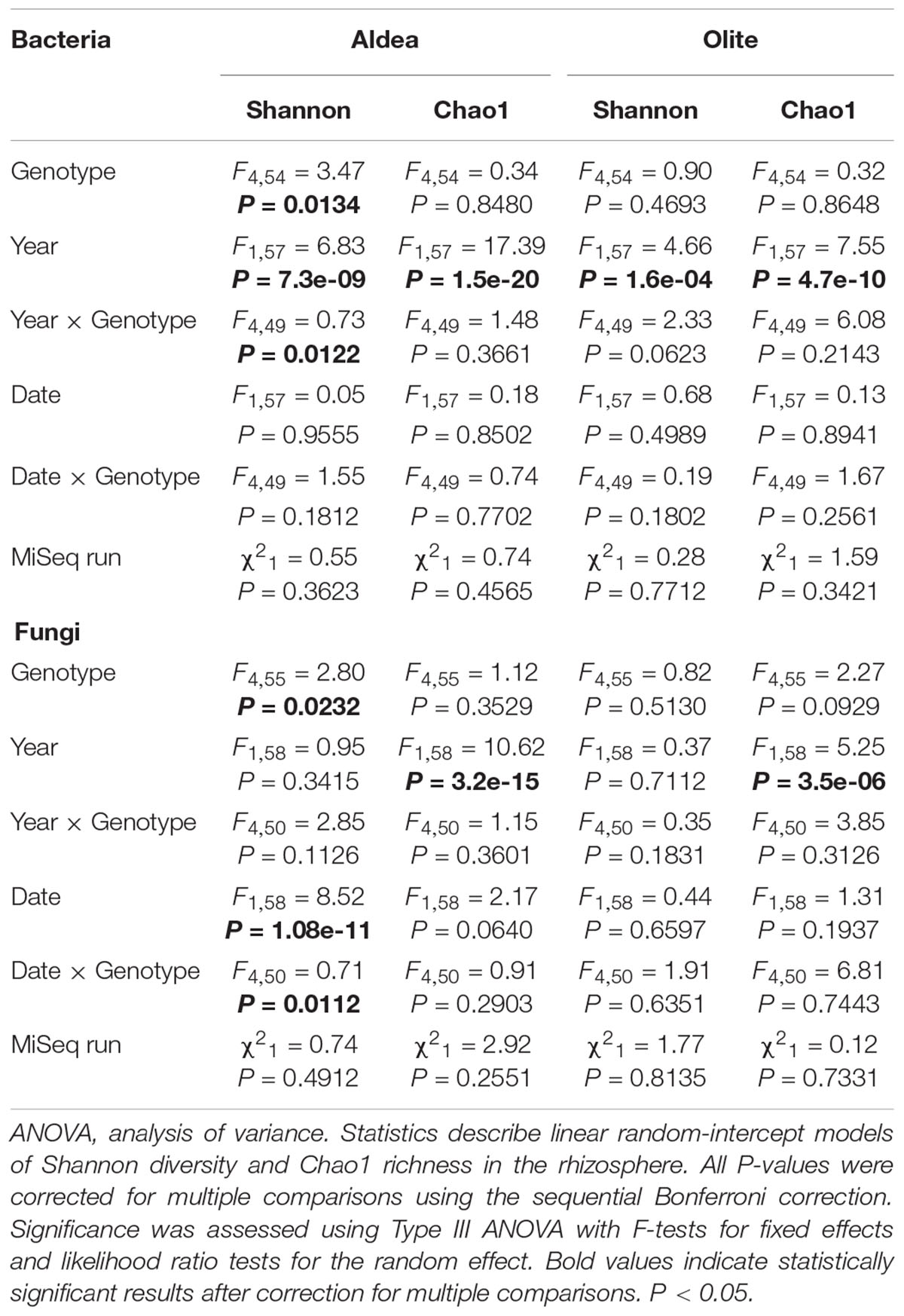
Table 1. Experimental factors predicting α-diversity of rhizosphere associated fungal and bacterial communities in Aldea and Olite vineyards.
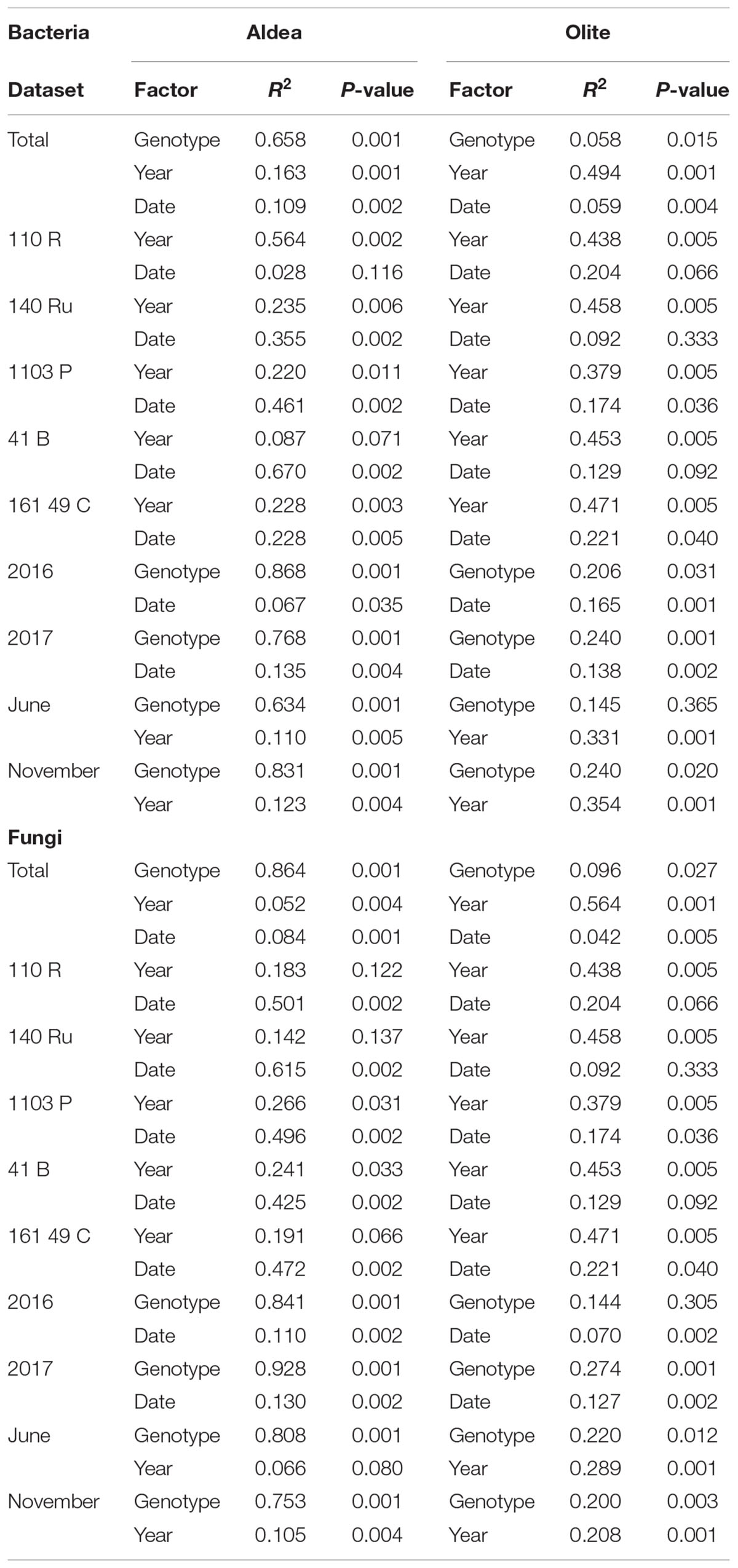
Table 2. Adonis test of category effect on bacterial and fungal Bray–Curtis distance matrix.
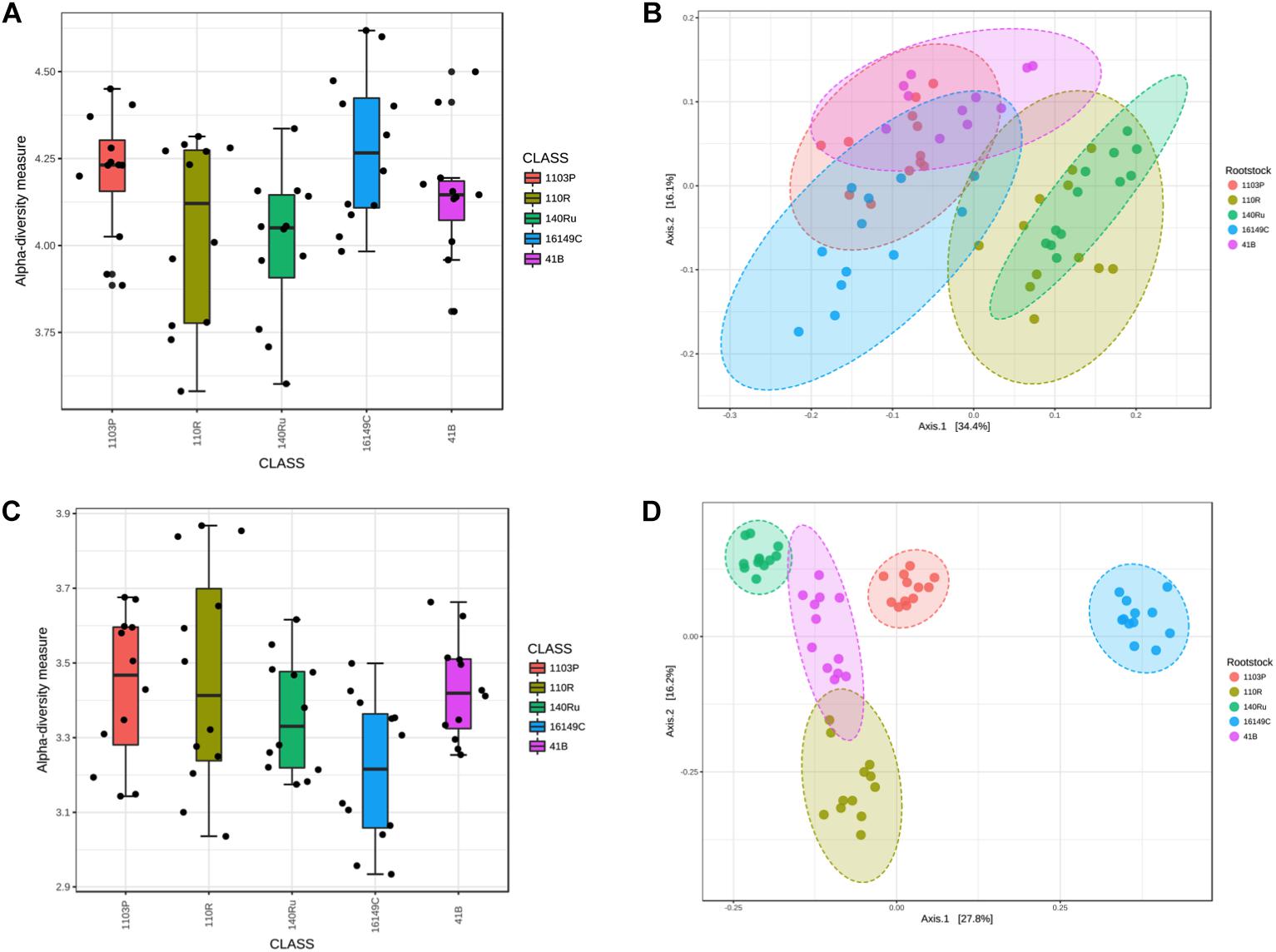
Figure 2. Box plot illustrating the differences in Shannon diversity measures of the bacterial (A) and fungal (C) communities in the grapevine rootstocks in Aldea vineyard. Principal Coordinate Analysis (PCoA) based on Bray Curtis dissimilarity metrics, showing the distance in the bacterial (B) and fungal (D) communities among grapevine rootstocks.
The linear discriminant analysis effect size (LEfSe) detected 27 bacterial and 36 fungal clades in the rhizospheres, which discriminated the microbial communities between the different rootstock genotypes in Aldea vineyard (Figures 3, 4). Both rootstocks 1103 P and 41 B showed higher number of differentially abundant bacterial clades (8 each) than the other rootstocks (5, 4, and 2 in 161-49 C, 110 R, and 140 Ru, respectively). The dominant bacterial phyla were Firmicutes (37%) in rootstock 41B, Actinobacteria and Planctomycetes (50% each) in rootstock 140 Ru, and Actinobacteria in rootstocks 161-49 C, 110 R, and 1103 P (60, 75, and 75%, respectively) (Figure 3). The dominant fungal phyla were Basidiomycota (73%) in rootstock 140 Ru, and Ascomycota in rootstocks 41 B, 161-49 C, 110 R, and 1103 P (75, 100, 36, and 71%, respectively) (Figure 4).
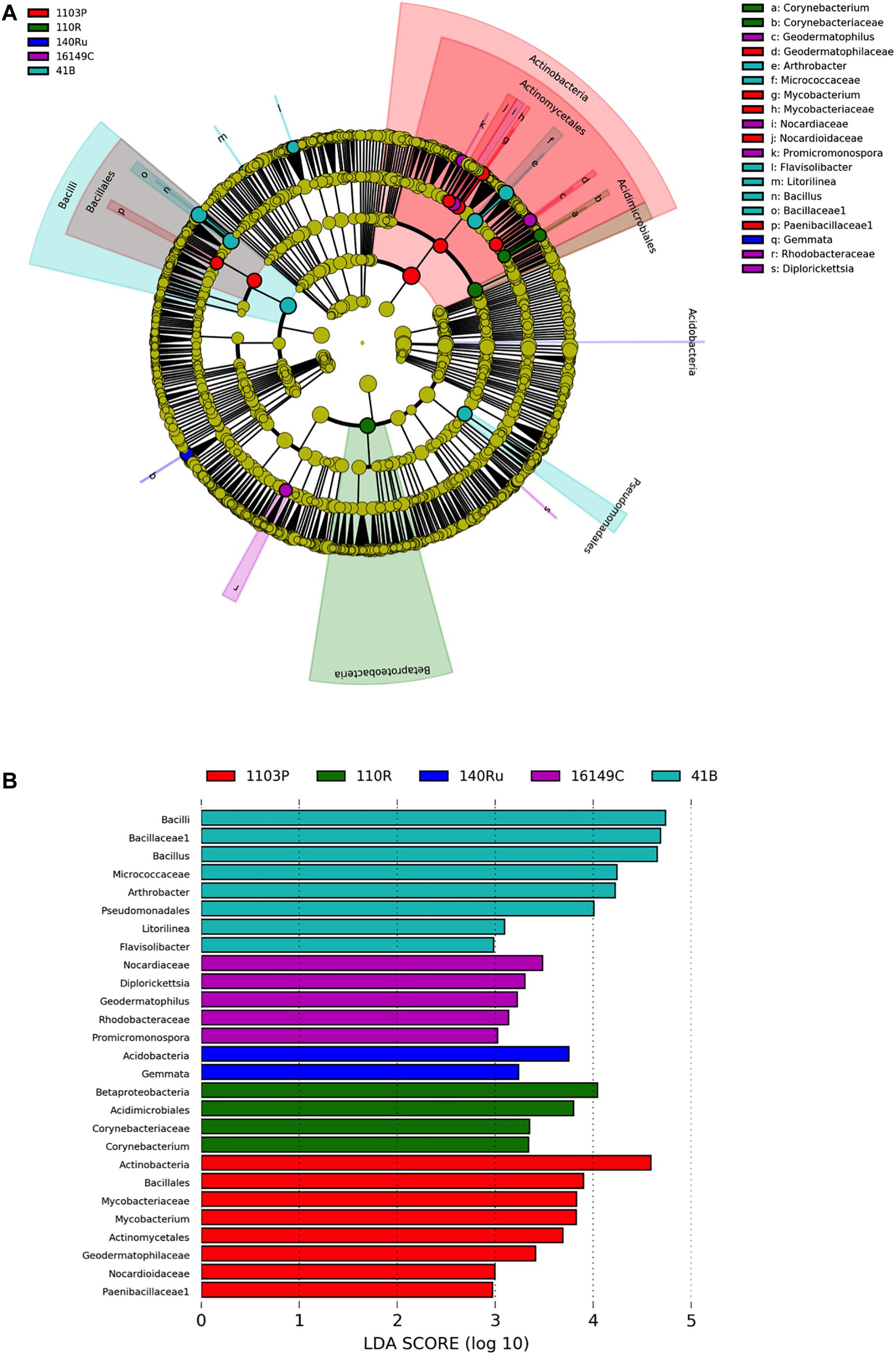
Figure 3. LEfSe was used to identify the most differentially abundant taxa among grapevine rootstocks in Aldea vineyard. Cladogram generated by LEfSe indicating differences of bacteria (A) at phylum, class, family, and genus levels between the five groups (relative abundance ≤0.5%). Each successive circle represents a phylogenetic level. Color regions indicate taxa enriched in the different rootstocks. Differing taxa are listed on the right side of the cladogram. Bar graph showing LDA scores for bacteria (B). Only taxa meeting an LDA significant threshold >2 are shown.
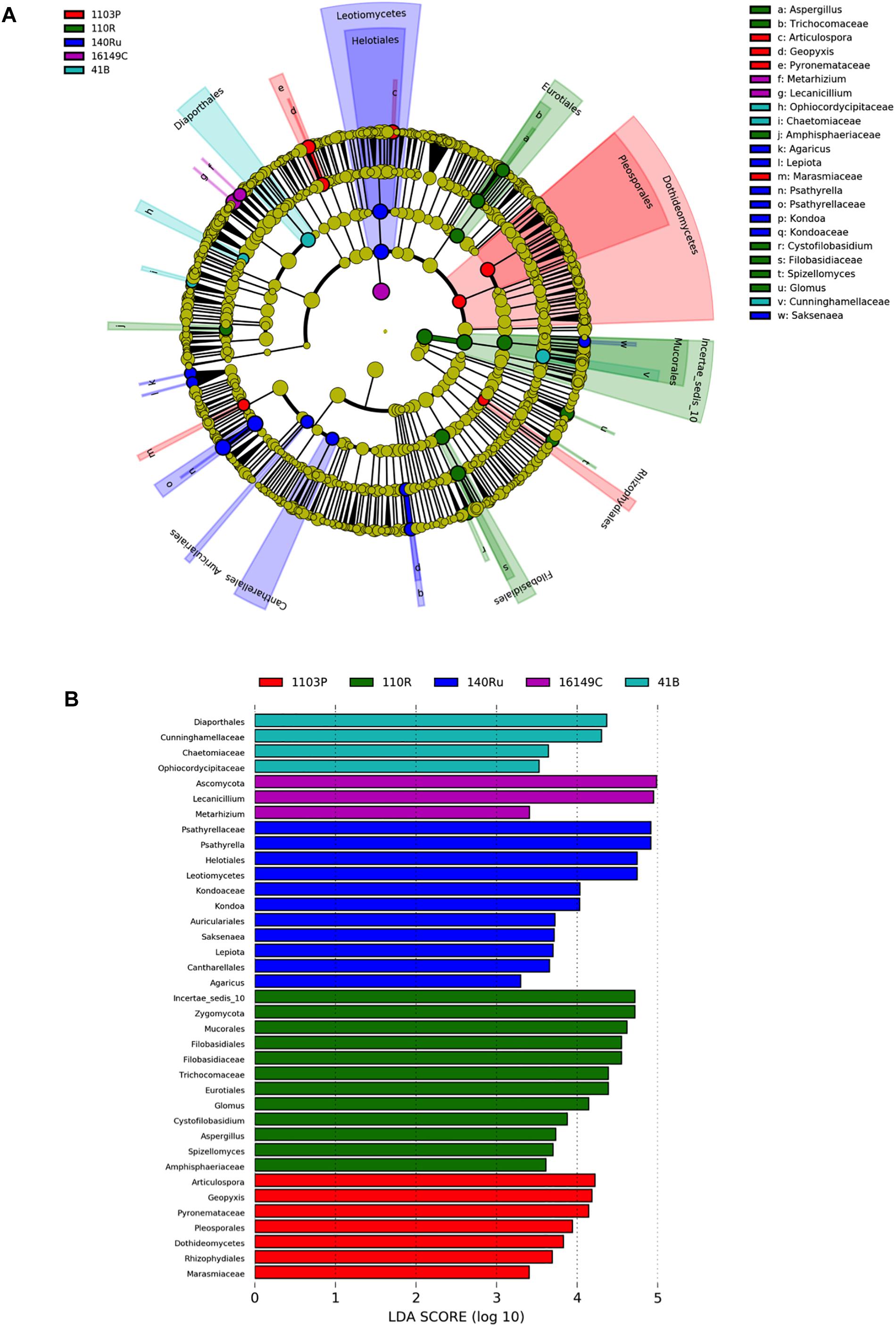
Figure 4. LEfSe was used to identify the most differentially abundant taxa among grapevine rootstocks in Aldea vineyard. Cladogram generated by LEfSe indicating differences of fungi (A) at phylum, class, family, and genus levels between the five groups (relative abundance ≤0.5%). Each successive circle represents a phylogenetic level. Color regions indicate taxa enriched in the different rootstocks. Differing taxa are listed on the right side of the cladogram. Bar graph showing LDA scores for fungi (B). Only taxa meeting an LDA significant threshold >2 are shown.
The rootstock-pairs dissimilarity, due to phyla and genera contribution in the rhizosphere was calculated by SIMPER (similarity percentages) analysis (Supplementary Table 6). Higher microbiome dissimilarity among rootstocks was revealed in Aldea vineyard compared to Olite vineyard, considering bacterial (Supplementary Table 6A) and fungal phyla (Supplementary Table 6C), and bacterial (Supplementary Table 6B) and fungal genera (Supplementary Table 6D) distribution. Firmicutes and Acidobacteria were the major phyla that contribute to differentiate the bacterial communities associated with the different rootstock types in Aldea and Olite vineyards, respectively (Supplementary Table 6A). Several genera were predominant and determined the dissimilarities among rootstocks such as Bacillus in Aldea vineyard or Aridibacter in Olite vineyard. The genus Bacillus appeared to be rhizosphere genotype biomarker of 140 Ru and 161-49 C rootstocks (Supplementary Table 6B). The fungal phyla Ascomycota and Basidiomycota contributed to the dissimilarity among rootstocks in Aldea vineyard, while only the phylum Basidiomycota contributed to differentiate fungal communities among rootstocks (Supplementary Table 6C). The fungal genera Geopyxis, Clonostachys, and Lecanicillium determined the dissimilarities among rootstocks in Aldea vineyard, being Geopyxis a rhizosphere genotype biomarker of 110 R rootstock and Clonostachys of 1103 P and 140 Ru rootstocks (Supplementary Table 6D). In Aldea vineyard, 161-49 C rootstock showed the highest dissimilarity with the other rootstocks in bacterial and fungal microbiome distribution.
Year Strongly Influenced Microbiomes
Our results demonstrate that bacterial microbiome varied profoundly between years. This pattern was consistent to community-level measure of α- diversity in both Aldea and Olite vineyards (Table 1) Richness increased between 2016 and 2017 in both vineyards (Supplementary Figure 3). However, year of sampling affected the Bray Curtis metric of β-diversity in only Olite vineyard (R2 = 0.494) (Supplementary Figure 3). Regarding the fungal microbiome, richness also varied between vineyards and increased between 2016 and 2017 in both vineyards (Table 1 and Supplementary Figure 4). However, year of sampling did not predict Shannon diversity and affected the Bray Curtis metric of β-diversity in only Olite vineyard (Table 2 and Supplementary Figure 4). Sampling date also contributed to α-diversity variation indicating temporal changes in relative abundance of fungal OTUs in Aldea vineyard. Fungal composition decreased between June and November (Table 1 and Supplementary Figure 5). Fungal community structure varied individually in each rootstock with date (R2 ranging from 0.42 to 0.61), but not in the total dataset (R2 < 0.1) (Table 2).
Rootstock-Specific and Shared Bacterial and Fungal Assemblages
The rhizosphere compartments of grapevine rootstocks showed specific fungal and bacterial OTUs for each rootstocks and a cluster of shared OTUs. In Aldea, specific OTUs associated with most of the rootstocks ranged from 4.3 to 5.8% of their bacterial communities (Figure 5). Specific OTUs associated with the rootstocks 140 Ru, 1103 P, 41 B and 110 R represented less than 9% of their fungal communities, where the 161-49C-specific OTUs enriched only 4.5% of the relative abundance (Figure 5). In Olite, specific OTUs associated with most of the rootstocks represented less than 9% of their bacterial and fungal communities, with the exception of bacterial communities associated with 140 Ru rootstock that represented 21.3% of its total (Figure 6). The OTUs that were unique in each of the grapevine rootstock are shown in Supplementary Tables 7, 8.
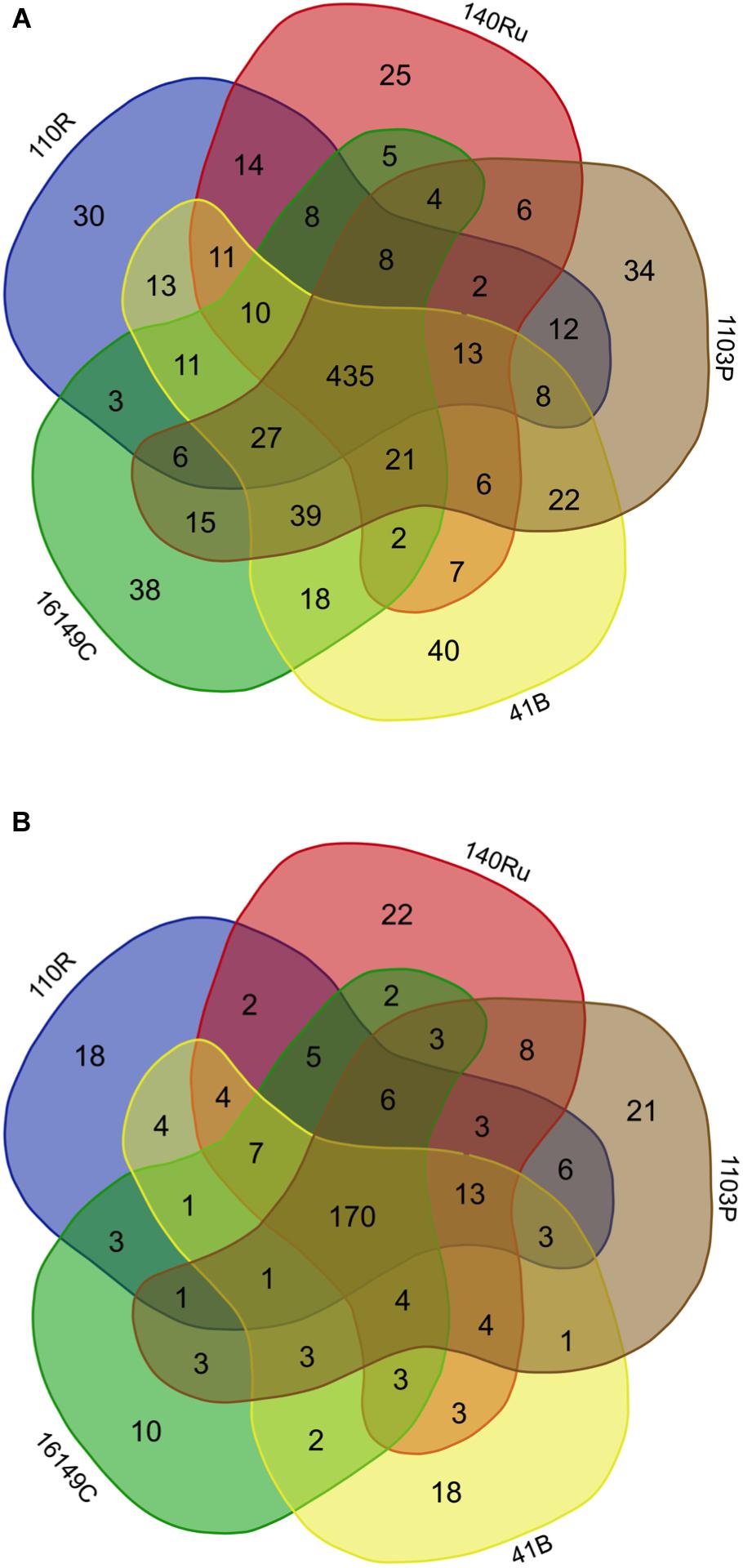
Figure 5. Venn diagrams showing the common and exclusive bacterial (A) and fungal (B) OTUs of the rhizosphere of the grapevine rootstocks in Aldea vineyard.
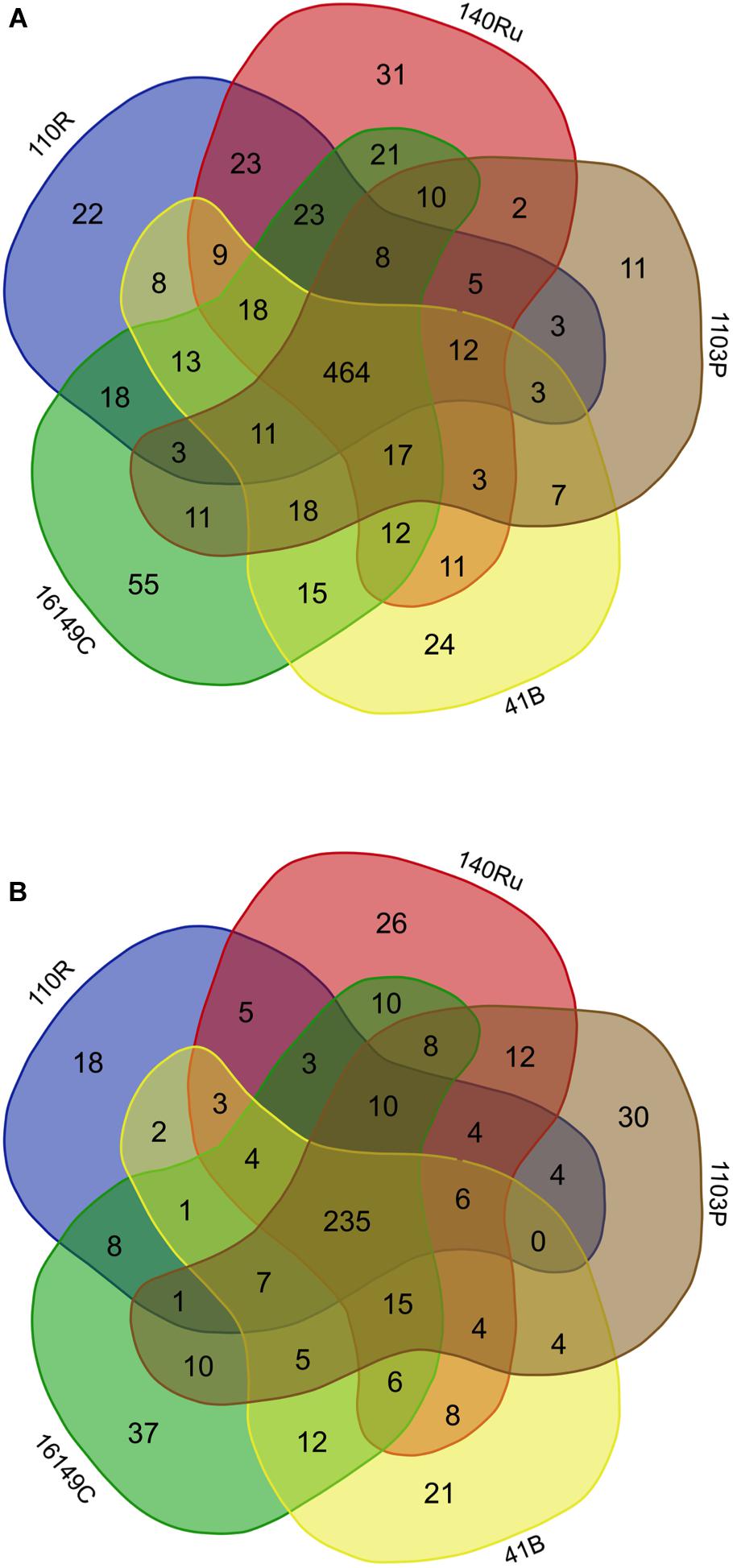
Figure 6. Venn diagrams showing the common and exclusive bacterial (A) and fungal (B) OTUs of the rhizosphere of the grapevine rootstocks in Olite vineyard.
Quantification of Black-Foot Disease Pathogens Using Quantitative PCR
The standard curve, constructed with serial dilutions of the DNA of D. torresensis isolate GTMF DT097, revealed high correlations between Cq and DNA, with R2-value of 0.99 and reaction efficiency of 0.90. The minimum DNA concentration detectable of D. torresensis was at Cq value of the dilution D7 thus, the limit of detection (LOD) was established at 2.75 fg/μL.
DNA of Cylindrocarpon-like asexual morphs was detected in all rootstock rhizosphere samples, in both vineyards and years, with concentrations ranging from 0.39 pg/μL to 4.06 pg/μL in Aldea 2016, from 3.52 pg/μL to 14.14 pg/μL in Aldea 2017, from 0.88 pg/μL to 8.45 pg/μL in Olite 2016 and from 2.65 pg/μL to 59 pg/μL in Olite 2017. The year and vineyard factors had a significant effect on Cylindrocarpon-like asexual morphs DNA concentration detected (P < 0.01). The concentration of DNA detected was significantly higher in Olite vineyard compared with Aldea vineyard, especially in year 2017. The rootstock factor had a significant effect on the DNA concentration detected in Aldea vineyard for 2017 samples (P = 0.0156). Rootstocks 161-49 C, 140 Ru, 1103 P, and 110R showed similar DNA concentrations values that were significantly lower when compared with 41 B rootstocks (Supplementary Figure 6). The analysis showed a positive significant correlation between the number of OTUs and the Cylindrocarpon-like asexual morphs DNA quantified using the real-time approach (P < 0.01, Spearman correlation coefficient = 0.72) (Supplementary Figure 7).
Discussion
In this study, we characterized the rhizosphere microbial community composition across five commercial grapevine rootstock genotypes cultivated in the same soil at two vineyards and sampling dates over 2 years. The analysis of bacterial and fungal populations in the grapevine rhizosphere targeting 16S rRNA and ITS region, respectively, have been proved effective in previous studies (Corneo et al., 2014; Holland et al., 2016; Longa et al., 2017; Manici et al., 2017; Stefanini and Cavalieri, 2018). Especially for bacterial barcoding, the choice of partial sequence regions is pivotal and can significantly affect the results because the 16S rRNA gene regions have different divergence (Youssef et al., 2009). In our study, we used the V4 region because according to recent in silico studies (Youssef et al., 2009), V4 along with V5-V6, and V6-V7 regions were considered as the most suitable regions for metagenomic purposes because they provided estimates comparable to those obtained with the complete 16S rRNA gene sequence (Youssef et al., 2009).
Our study represents the first approach to investigate the rhizosphere fungal microbiome of grapevine by HTAS. In grapevine, the ecology of fungal communities is so far largely derived from the studies using pyrosequencing approach in bulk soil (Holland et al., 2016; Castañeda and Barbosa, 2017; Longa et al., 2017) or ARISA fingerprinting (Likar et al., 2017) and PCR-DGGE (Manici et al., 2017) approaches in rhizosphere soil. Even though the ITS region was ratified by The Fungal Barcoding Consortium (Schoch et al., 2012) as the universal DNA barcode for the fungal kingdom using the same gene section proposed by White et al. (1990), some recent reports point out its limitations for specific taxa. This region does not work well with taxa having narrow or no barcode gaps in their ITS regions, such as Fusarium or Trichoderma (Schoch et al., 2012). In addition, the correct identification of morphologically similar cryptic species using the ITS regions is still problematic due to the lack of consensus in the lineage-specific cut-off value for species determination (Nilsson et al., 2008).
The bacterial microbiomes of the different rootstocks were largely composed of Proteobacteria and Actinobacteria that accounted for almost 50% of the relative abundance in both vineyards. The predominant bacterial phyla found in this work is consistent with the results obtained in other studies in vineyard soil (Opsi et al., 2014; Vega-Avila et al., 2015; Castañeda and Barbosa, 2017; Longa et al., 2017; Marasco et al., 2018). Proteobacteria and Actinobacteria are known for their role in the carbon biochemical cycle and their production of second metabolites (Jenkins et al., 2009). The major fungal phyla detected in our study were largely composed of Ascomycota and Basiodiomycota that accounted for almost 75% of the relative abundance in both vineyards. Previous studies also agree on the most common fungal phyla detected in grapevines fields (Castañeda and Barbosa, 2017; Longa et al., 2017; Manici et al., 2017). These results suggest that vineyard microbiome in Navarre and La Rioja regions is partially conserved.
The results obtained in the Aldea vineyard showed a significant fraction of variation in fungal and bacterial diversity (both the α- and β-diversity) that could be attributed to host genetics. Recent research indicated that rootstock genotypes could have a notable influence in shaping the bacteria taxa distribution in the root and rhizosphere systems of grapevine (Marasco et al., 2018). This effect of the host genotype in the rhizosphere microbiome has been reported in other woody crops, such as apple (Liu et al., 2018) and pines (Gallart et al., 2018), as well as in several annual crops, such as maize (Peiffer et al., 2013), potato (Inceoǧlu et al., 2010), and chickpea (Bazghaleh et al., 2015). This could be due to the influence of the genotype in the root metabolism, including immune response and exudate composition, which impact in the rhizosphere microbiome (Wagner et al., 2016). Rootstocks show different level of tolerance to distinct diseases; and this could be decisive in their effect in the microbiome (Sapkota et al., 2015). Moreover, as reviewed by Liu et al. (2018), several studies hint to a possible co-evolution of the holobiont. However, further research is needed to validate this hypothesis. On the other hand, the Olite vineyard showed a lower microbiome dissimilarity among rootstocks, suggesting that the effect of genotype in shaping the microbiome might be influenced by other factors.
The differences between Olite and Aldea vineyards could lie in the soil physicochemical properties, in the soil and cultivar management practices, or in the age of the plants, being vines cultivated in Olite vineyard younger than in Aldea vineyard. Environmental heterogeneity, such as the soil physicochemical properties and moisture content have been identified as major factors shaping the spatial scaling of the rhizosphere microbiome in many previous studies (Costa et al., 2006; Tan et al., 2013; Schreiter et al., 2014), including grapevine (Fernández-Calviño et al., 2010; Corneo et al., 2014; Burns et al., 2015; Zarraonaindia et al., 2015; Holland et al., 2016). Soil physicochemical properties can also influence the population structure of specific soil-borne pathogens. For instance, Berlanas et al. (2017) observed that excessive calcium carbonate in soil may increase black-foot disease inoculum density.
Field management practices have been also reported as an important driver of the microbiome diversity (Santhanam et al., 2015; Sapkota et al., 2015; Hacquard, 2016; Gallart et al., 2018), including the grapevine soil microbiome (Vega-Avila et al., 2015; Likar et al., 2017; Longa et al., 2017). Nevertheless, other studies showed a long-term effect of cultivation rather than field management on soil microbial diversity (Buckley and Schmidt, 2001; Peiffer et al., 2013). Microbiome studies should consider the high degree of temporal variability in the sample design, because sampling the same point in different times can give different results due the variability of the own microbial community through time (Redford and Fierer, 2009). The year to year variation found in our study could be explained by the different root response to distinct environmental factors, such as temperature or precipitation (Wagner et al., 2016). Further research is needed to determine if environment plays a much greater role than host genetics in determining the composition of the rhizosphere microbiome of grapevine.
Several studies have remarked the effect of the growth stage of the plant in its associated rhizosphere microbiome (Baudoin et al., 2002; Inceoǧlu et al., 2010; Li et al., 2014; Okubo et al., 2014; Yuan et al., 2015; Wagner et al., 2016; Qiao et al., 2017). Changes in the quantity and quality of root exudates as plants develop have been proposed as the main source of variation of the rhizosphere microbiome composition present during different developmental stages of maize cultivars (Baudoin et al., 2002). However, most of the published studies are focused in annual plant systems. In grapevine, Manici et al. (2017) recently investigated shifts in bacterial and fungal communities between mature and young replaced vines in Italy. At a single sampling moment, these researchers concluded that long-term growth legacy overcame plant age in shaping rhizosphere microbiome (Manici et al., 2017). Further research is therefore needed to determine the long-term effect of the grapevine age on the associated microbiome as plants develop. This could be accomplished by comparing the rhizosphere microbiome (i) in a single vineyard over time, or (ii) in two vineyards in close proximity with identical environmental conditions and soils, but with vines on different aging process.
Our results showed that the root system type is able to select specific bacterial and fungal OTUs as biomarkers for the different genotypes. Members of the bacterial genus Bacillus, which was only found in 140 Ru and 161-49 C rootstocks in Aldea vineyard, has wide diversity of physiological ability with respect to heat, pH, and salinity. Therefore, Bacillus species can be found in a wide range of habitats, being a few of them pathogenic to vertebrates or invertebrates (Holt et al., 1994). Bacillus subtilis and Bacillus amyloliquefaciens have been described as potential biocontrol agents against Aspergillus parasiticus and stem rot disease (Le et al., 2018; Siahmoshteh et al., 2018). In vitro assays of the heat stable metabolites of B. subtilis showed promising results in reducing the growth of the fungal trunk pathogens Lasiodiplodia theobromae, Phaeomoniella chlamydospora, and Phaeoacremonium minimum (Alfonzo et al., 2009). Rezgui et al. (2016) recently identified several B. subtilis strains inhabiting the wood tissues of mature grapevines in Tunisia with antagonistic traits against fungal trunk pathogens. On the other hand, some species of the arbuscular mycorrhizal (AM) fungal genus Glomus, one of the most differentially abundant taxa for 110 R rootstock in Aldea vineyard, are cataloged as biocontrol agents (Tahat et al., 2010). For instance, inoculation of grapevine roots with Rhizophagus irregularis (syn. Glomus intraradices) reduced both the disease severity and the number of root lesions caused by black-foot disease pathogens (Petit and Gubler, 2006). AM fungi form one of the most interesting beneficial plant–micro-organism associations (Smith and Read, 2008) and are known to colonize the roots of the majority of land plants, including grapevines (Schreiner and Mihara, 2009; Trouvelot et al., 2015). Several genera within the Glomeromycota phylum have been identified from the rhizosphere samples obtainted in this study, namely Claroideoglomus, Diversispora, Entrophosphora, and Rhizophagus. Trouvelot et al. (2015) reported that soil management can greatly impact the diversity of AM fungi. In fact, AM fungal communities are highly influenced by the soil characteristics but also to a smaller extent by the host plant development stage (Schreiner and Mihara, 2009; Balestrini et al., 2010).
High-throughput amplicon sequencing is a powerful method for the analysis of microbial populations. It is accomplished by sequencing specific marker genes amplified directly from environmental DNA without prior enrichment or cultivation of the target population (Franzosa et al., 2015). The advantages of this approach is the detection of rare taxa at the genus level given the availability of large and comprehensive reference databases as well as several pipelines for bioinformatics analysis (Stefanini and Cavalieri, 2018). Drawbacks of HTAS include the biased relative quantification of bacterial communities since bacterial species bear various number of copies of 16S rRNA genes, the sequencing of matrix (e.g., grape ITS, chloroplast 16S) and the low confidence for taxonomic assignment at the species level (Stefanini and Cavalieri, 2018). A step forward consists of the understanding of how changes in the composition of microbial communities impact the population’s biological functions (Ravin et al., 2015). Unfortunately, HTAS only allows inference of functional annotation while in whole-genome sequencing, functional annotation can be carried out by gene enrichment (Stefanini and Cavalieri, 2018). A further drawback of using DNA-based metagenomic data to infer the biological functions potentially exploited by microbial populations is that the detected DNA may belong to dead organisms. However, an approach based on RNA sequencing would give a direct report of the functions achievable by the viable microbial populations. In grapevine, the study of the active fungal communities of internal grapevine wood by HTAS in extracted total RNA has been recently accomplished by Eichmeier et al. (2018).
The quantitative significance of next-generation sequencing data for microorganisms is often debated (Amend et al., 2010). Fortunately, we were able to compare the relative abundance of reads with the relative abundance of DNA of black-foot disease pathogens, and we observed significant positive correlation. From the fungal soilborne pathogens affecting grapevine, Cylindrocarpon-like asexual morphs associated with black-foot disease are among the most important limiting factor of the production worldwide (Halleen et al., 2006; Agustí-Brisach and Armengol, 2013). Therefore, Cylindrocarpon-like asexual morphs can be considered model pathogens to monitor the healthy status of the grapevine planting material when analyzing the fungal microbial composition of soil/rhizosphere samples.
Grapevine rootstocks have different susceptibilities toward pathogens, including trunk disease pathogens (Eskalen et al., 2001; Alaniz et al., 2010; Gramaje et al., 2010; Brown et al., 2013; Billones-Baaijens et al., 2014), which may be an important factor in shaping not only pathogens abundance but also entire communities. Nevertheless, we did not observe a clear correlation between known disease resistances in individual genotypes and the fungal communities, although Cylindrocarpon-like asexual morphs were found in lower abundance in 161-49 C rootstock by both high-throughput amplicon sequencing and qPCR approaches. The use of 161-49 C rootstock was previously recommended within an integrated management program for other grapevine trunk diseases, such as Petri disease and esca (Gramaje et al., 2010).
Conclusion
We have studied the effects of genotype, year, sampling date, and location on bacterial and fungal communities in the grapevine rhizosphere. We found that grapevine genotype was the most important factor in shaping the microbiome in the mature vineyard. Many bacterial and fungal species were found in all rootstocks and in both locations in our study, demonstrating the existence of a “core” grape phylogeny that is independent of the growing region. Interestingly, the rhizosphere compartments of 140 Ru and 161-49 C rootstocks, the latter showing high tolerance to esca and Petri disease pathogens in previous research (Gramaje et al., 2010), harbored lower number of black-foot pathogens than the other grapevine rootstocks. Also of interest was the presence of high relative abundance of the genus Bacillus in both grapevine rootstocks, a bacterial genus recognized as biocontrol agents. A more comprehensive study is needed to decipher the cause of the rootstock microbiome selection and the mechanisms by which grapevines are able to shape their associated microbial community. Understanding the vast diversity of bacteria and fungi in the rhizosphere and the interactions between microbiota and grapevine will facilitate the development of future strategies for grapevine protection.
Author Contributions
CB, MB, and DG conceived the study. All authors contributed to the data collection. CB, MB, GE, and DG performed the data interpretation and manuscript preparation. CB, MB, DG, GE, and ML performed the experiments. CB, MB, GE, and DG contributed to the bioinformatics data analysis. All authors critically reviewed and edited the manuscript, and approved its publication.
Funding
The research was financially supported by The National Institute for Agricultural and Food Research and Technology (INIA) under the project RTA2015-00015-C02-01. DG was supported by the DOC-INIA program from the INIA, co-funded by the European Social Fund. CB was supported by the FPI-INIA program from the INIA. GE was supported by the Spanish post-doctoral grant Juan de la Cierva-Formación.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Marcos Andrés and Pilar Yécora (ICVV, Spain) for their the support in grapevine sampling.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01142/full#supplementary-material
Abbreviations
ITS, Internal transcribed spacer; OTU, Operational taxonomic unit; PCoA, Principal coordinate analysis; PCR, Polymerase chain reaction; PERMANOVA, Permutational multivariate analysis of variance; QIIME, Quantitative insights into microbial ecology; qPCR, Quantitative polymerase chain reaction; rRNA, Ribosomal RNA; SIMPER, Similarity percentages.
Footnotes
References
Abarenkov, K., Nilsson, R. H., Larsson, K. H., Alexander, I. J., Eberhardt, U., Erland, S., et al. (2010). The UNITE database for molecular identification of fungi - recent updates and future perspectives. New Phytol. 186, 281–285. doi: 10.1111/j.1469-8137.2009.03160.x
Agustí-Brisach, C., and Armengol, J. (2013). Black-foot disease of grapevine: an update on taxonomy, epidemiology and management strategies. Phytopathol. Mediterr. 52, 245–261. doi: 10.14601/Phytopathol_Mediterr-12662
Agustí-Brisach, C., Mostert, L., and Armengol, J. (2014). Detection and quantification of Ilyonectria spp. associated with black-foot disease of grapevine in nursery soils using multiplex nested PCR and quantitative PCR. Plant Pathol. 63, 316–322. doi: 10.1111/ppa.12093
Aira, M., Gómez-Brandón, M., Lazcano, C., Bååth, E., and Domínguez, J. (2010). Plant genotype strongly modifies the structure and growth of maize rhizosphere microbial communities. Soil Biol. Biochem. 42, 2276–2281. doi: 10.1016/j.soilbio.2010.08.029
Alaniz, S., García-Jiménez, J., Abad-Campos, P., and Armengol, J. (2010). Susceptibility of grapevine rootstocks to Cylindrocarpon liriodendri and C. macrodidymum. Sci. Hortic. 125, 305–308. doi: 10.1016/j.scienta.2010.04.009
Alfonzo, A., Conigliaro, G., Torta, L., Burruano, S., and Moschetti, G. (2009). Antagonism of Bacillus subtilis strain AG1 against vine wood fungal pathogens. Phytopathol. Mediterr. 48, 155–158. doi: 10.14601/phytopathol_mediterr-2886
Amend, A. S., Seifert, K. A., and Bruns, T. D. (2010). Quantifying microbial communities with 454 pyrosequencing: Does read abundance count? Mol. Ecol. 19, 5555–5565. doi: 10.1111/j.1365-294X.2010.04898.x
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46. doi: 10.1046/j.1442-9993.2001.01070.x
Aronesty, E. (2011). ea-utils: Command-Line Tools for Processing Biological Sequencing Data. Durham, NC: Expression Analysis Inc.
Balestrini, R., Magurno, F., Walker, C., Lumini, E., and Bianciotto, V. (2010). Cohorts of arbuscular mycorrhizal fungi (AMF) in Vitis vinifera, a typical Mediterranean fruit crop. Environ. Microbiol. Rep. 2, 594–604. doi: 10.1111/j.1758-2229.2010.00160.x
Bates, D., Mächler, M., Bolker, B., and Walker, S. (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48. doi: 10.18637/jss.v067.i01
Baudoin, E., Benizri, E., and Guckert, A. (2002). Impact of growth stage on the bacterial community structure along maize roots, as determined by metabolic and genetic fingerprinting. Appl. Soil Ecol. 19, 135–145. doi: 10.1016/S0929-1393(01)00185-8
Bazghaleh, N., Hamel, C., Gan, Y., Tar’an, B., and Knight, J. D. (2015). Genotype-specific variation in the structure of root fungal communities is related to chickpea plant productivity. Appl. Environ. Microbiol. 81, 2368–2377. doi: 10.1128/AEM.03692-14
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. A. H. M. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Berlanas, C., López-Manzanares, B., and Gramaje, D. (2017). Estimation of viable propagules of black-foot disease pathogens in grapevine cultivated soils and their relation to production systems and soil properties. Plant Soil 417, 467–479. doi: 10.1007/s11104-017-3272-3
Billones-Baaijens, R., Jones, E. E., Ridgway, H. J., and Jaspers, M. V. (2014). Susceptiblity of common rootstock and scion varieties of grapevines to Botryosphaeriaceae species. Australas. Plant Pathol. 43, 25–31. doi: 10.1007/s13313-013-0228-9
Bouffaud, M. L., Kyselková, M., Gouesnard, B., Grundmann, G., Muller, D., and Moënne-Loccoz, Y. (2012). Is diversification history of maize influencing selection of soil bacteria by roots? Mol. Ecol. 21, 195–206. doi: 10.1111/j.1365-294X.2011.05359.x
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 27, 325–349. doi: 10.2307/1942268
Brown, D. S., Jaspers, M. V., Ridgway, H. J., Barclay, C. J., and Jones, E. E. (2013). Susceptibility of four grapevine rootstocks to Cylindrocladiella parva. New Zeal. Plant Prot. 66, 249–253.
Buckley, D. H., and Schmidt, T. M. (2001). The structure of microbial communities in soil and the lasting impact of cultivation. Microb. Ecol. 42, 11–21. doi: 10.1007/s002480000108
Burns, K. N., Kluepfel, D. A., Strauss, S. L., Bokulich, N. A., Cantu, D., and Steenwerth, K. L. (2015). Vineyard soil bacterial diversity and composition revealed by 16S rRNA genes: differentiation by geographic features. Soil Biol. Biochem. 91, 232–247. doi: 10.1016/j.soilbio.2015.09.002
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: minimum Information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., and Costello, E. K. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Carlucci, A., Lops, F., Mostert, L., Halleen, F., and Raimondo, M. L. (2017). Occurrence fungi causing black foot on young grapevines and nursery rootstock plants in Italy. Phytopathol. Mediterr. 56, 10–39. doi: 10.14601/Phytopathol_Mediterr-18769
Castañeda, L. E., and Barbosa, O. (2017). Metagenomic analysis exploring taxonomic and functional diversity of soil microbial communities in Chilean vineyards and surrounding native forests. PeerJ 5:e3098. doi: 10.7717/peerj.3098
Chapelle, E., Mendes, R., Bakker, P. A. H., and Raaijmakers, J. M. (2016). Fungal invasion of the rhizosphere microbiome. ISME J. 10, 265–268. doi: 10.1038/ismej.2015.82
Corneo, P. E., Pellegrini, A., Cappellin, L., Gessler, C., and Pertot, I. (2014). Moderate warming in microcosm experiment does not affect microbial communities in temperate vineyard soils. Microb. Ecol. 67, 659–670. doi: 10.1007/s00248-013-0357-2
Costa, R., Salles, J. F., Berg, G., and Smalla, K. (2006). Cultivation-independent analysis of Pseudomonas species in soil and in the rhizosphere of field-grown Verticillium dahliae host plants. Environ. Microbiol. 8, 2136–2149. doi: 10.1111/j.1462-2920.2006.01096.x
Dennis, P. G., Miller, A. J., and Hirsch, P. R. (2010). Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol. Ecol. 72, 313–327. doi: 10.1111/j.1574-6941.2010.00860.x
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dhariwal, A., Chong, J., Habib, S., King, I. L., Agellon, L. B., and Xia, J. (2017). MicrobiomeAnalyst: a web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 45, W180–W188. doi: 10.1093/nar/gkx295
Dubrovsky, S., and Fabritius, A. L. (2007). Occurrence of Cylindrocarpon spp. in nursery grapevines in California. Phytopathol. Mediterr. 46, 84–86. doi: 10.14601/phytopathol_mediterr-1859
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C. (2018). USEARCH. Available at: http://drive5.com/usearch (accessed March 29, 2018).
Eichmeier, A., Pecenka, J., Penazova, E., Baranek, M., Català-García, S., León, M., et al. (2018). High-throughput amplicon sequencing-based analysis of active fungal communities inhabiting grapevine after hot-water treatments reveals unexpectedly high fungal diversity. Fungal Ecol. 36, 26–38. doi: 10.1016/j.funeco.2018.07.011
Eskalen, A., Douglas Gubler, W., and Khan, A. (2001). Rootstock susceptibility to Phaeomoniella chlamydospora and Phaeoacremonium spp. Phytopathol. Mediterr. 40, 433–438. doi: 10.14601/phytopathol_mediterr-1636
FAO (2018). FAOSTAT. Available at: http://www.fao.org/faostat/en/#data/QC (accessed May 15, 2018).
Fernández-Calviño, D., Martín, A., Arias-Estévez, M., Bååth, E., and Díaz-Raviña, M. (2010). Microbial community structure of vineyard soils with different pH and copper content. Appl. Soil Ecol. 46, 276–282. doi: 10.1016/j.apsoil.2010.08.001
Franzosa, E. A., Hsu, T., Sirota-Madi, A., Shafquat, A., Abu-Ali, G., Morgan, X. C., et al. (2015). Sequencing and beyond: integrating molecular “omics” for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372. doi: 10.1038/nrmicro3451
Gallart, M., Adair, K. L., Love, J., Meason, D. F., Clinton, P. W., Xue, J., et al. (2018). Host genotype and nitrogen form shape the root microbiome of Pinus radiata. Microb. Ecol. 75, 419–433. doi: 10.1007/s00248-017-1055-2
Gilbert, J. A., van der Lelie, D., and Zarraonaindia, I. (2014). Microbial terroir for wine grapes. Proc. Natl. Acad. Sci. U.S.A. 111, 5–6. doi: 10.1073/pnas.1320471110
Gramaje, D., García-Jiménez, J., and Armengol, J. (2010). Field evaluation of grapevine rootstocks inoculated with fungi associated with petri disease and Esca. Am. J. Enol. Vitic. 61, 512–520. doi: 10.5344/ajev.2010.10021
Hacquard, S. (2016). Disentangling the factors shaping microbiota composition across the plant holobiont. New Phytol. 209, 454–457. doi: 10.1111/nph.13760
Halleen, F., Fourie, P. H., and Crous, P. W. (2006). A review of black foot disease of grapevine. Phytopathol. Mediterr. 45, S55–S67. doi: 10.14601/PHYTOPATHOL_MEDITERR-1845
Hidalgo, L. (ed.). (2002). “Los portainjertos,” in Tratao de Viticultura General. Madrid: Ediciones Mundi-Prensa, 289–320.
Holland, T. C., Bowen, P. A., Bogdanoff, C. P., Lowery, T. D., Shaposhnikova, O., Smith, S., et al. (2016). Evaluating the diversity of soil microbial communities in vineyards relative to adjacent native ecosystems. Appl. Soil Ecol. 100, 91–103. doi: 10.1016/j.apsoil.2015.12.001
Holt, J. H., Krieg, N. R., Sneath, P. H. A., Staley, J. T., and Williams, S. T. (1994). Bergey’s Manual of Determinative Bacteriology, 9th Edn. Baltimore: Williams and Wilikins. doi: 10.1016/j.apsoil.2015.12.001
Inceoǧlu,Ö., Salles, J. F., Van Overbeek, L., and Van Elsas, J. D. (2010). Effects of plant genotype and growth stage on the betaproteobacterial communities associated with different potato cultivars in two fields. Appl. Environ. Microbiol. 76, 3675–3684. doi: 10.1128/AEM.00040-10
Jenkins, S. N., Waite, I. S., Blackburn, A., Husband, R., Rushton, S. P., Manning, D. C., et al. (2009). Actinobacterial community dynamics in long term managed grasslands. Antonie Van Leeuwenhoek 95, 319–334. doi: 10.1007/s10482-009-9317-8
Jiang, Y., Li, S., Li, R., Zhang, J., Liu, Y., Lv, L., et al. (2017). Plant cultivars imprint the rhizosphere bacterial community composition and association networks. Soil Biol. Biochem. 109, 145–155. doi: 10.1016/j.soilbio.2017.02.010
Keller, M. (ed.). (2010). “Cultivars, clones, and rootstocks,” in The Science of Grapevines: Anatomy and Physiology. Cambridge, MA: Cambridge Academic Press, 9–19.
Kuznetsova, A., Brockhoff, P. B., and Christensen, R. H. B. (2016). lmerTest: Tests in Linear Mixed Effects Models. R Package version 2.0-33. Available at: https://cran.r-project.org/package=lmerTest (accessed June 5, 2018).
Le, C. N., Hoang, T. K., Thai, T. H., Tran, T. L., Phan, T. P. N., and Raaijmakers, J. M. (2018). Isolation, characterization and comparative analysis of plant-associated bacteria for suppression of soil-borne diseases of field-grown groundnut in Vietnam. Biol. Control 121, 256–262. doi: 10.1016/J.BIOCONTROL.2018.03.014
Lemanceau, P., Barret, M., Mazurier, S., Mondy, S., Pivato, B., Fort, T., et al. (2017). “Plant communication with associated microbiota in the spermosphere, rhizosphere and phyllosphere,” in How Plants Communicate with their Biotic Environment, ed. G. Becard (Cambridge, MA: Academic Press), 101–133. doi: 10.1016/bs.abr.2016.10.007
Li, X., Rui, J., Mao, Y., Yannarell, A., and Mackie, R. (2014). Dynamics of the bacterial community structure in the rhizosphere of a maize cultivar. Soil Biol. Biochem. 68, 392–401. doi: 10.1016/j.soilbio.2013.10.017
Likar, M., Stres, B., Rusjan, D., Potisek, M., and Regvar, M. (2017). Ecological and conventional viticulture gives rise to distinct fungal and bacterial microbial communities in vineyard soils. Appl. Soil Ecol. 113, 86–95. doi: 10.1016/j.apsoil.2017.02.007
Liu, J., Abdelfattah, A., Norelli, J., Burchard, E., Schena, L., Droby, S., et al. (2018). Apple endophytic microbiota of different rootstock/scion combinations suggests a genotype-specific influence. Microbiome 6:18. doi: 10.1186/s40168-018-0403-x
Longa, C. M. O., Nicola, L., Antonielli, L., Mescalchin, E., Zanzotti, R., Turco, E., et al. (2017). Soil microbiota respond to green manure in organic vineyards. J. Appl. Microbiol. 123, 1547–1560. doi: 10.1111/jam.13606
Lundberg, D. S., Yourstone, S., Mieczkowski, P., Jones, C. D., and Dangl, J. L. (2013). Practical innovations for high-throughput amplicon sequencing. Nat. Methods 10, 999–1002. doi: 10.1038/nmeth.2634
Manici, L. M., Saccà, M. L., Caputo, F., Zanzotto, A., Gardiman, M., and Fila, G. (2017). Long- term grapevine cultivation and agro-environment affect rhizosphere microbiome rather than plant age. Appl. Soil Ecol. 119, 214–225. doi: 10.1016/j.apsoil.2017.06.027
Marasco, R., Rolli, E., Fusi, M., Michoud, G., and Daffonchio, D. (2018). Grapevine rootstocks shape underground bacterial microbiome and networking but not potential functionality. Microbiome 6:3. doi: 10.1186/s40168-017-0391-2
Marques, J. M., da Silva, T. F., Vollu, R. E., Blank, A. F., Ding, G. C., Seldin, L., et al. (2014). Plant age and genotype affect the bacterial community composition in the tuber rhizosphere of field-grown sweet potato plants. FEMS Microbiol. Ecol. 88, 424–435. doi: 10.1111/1574-6941.12313
Martínez-Cutillas, A., Erena-Arrabal, M., Carreño-Espín, J., and Fernández-Rubio, J. (1990). Patrones de Vid. Murcia: Consejería de Agricultura y Agua.
McMurdie, P. J., and Holmes, S. (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10:e1003531. doi: 10.1371/journal.pcbi.1003531
Nilsson, R. H., Kristiansson, E., Ryberg, M., Hallenberg, N., and Larsoon, K. H. (2008). Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence database and its implications for molecular species identification. Evol. Bioinform. Online 26, 193–201. doi: 10.4137/EBO.S653
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’hara, R. B., et al. (2018). Vegan: Community Ecology Package. R Package. Version 2.4-6. Available at: https://CRAN.R-project.org/package=vegan (accessed June 5, 2018).
Okubo, T., Tokida, T., Ikeda, S., Bao, Z., Tago, K., Hayatsu, M., et al. (2014). Effects of elevated carbon dioxide, elevated temperature, and rice growth stage on the community structure of rice root–associated bacteria. Microbes Environ. 29, 184–190. doi: 10.1264/jsme2.ME14011
Opsi,B., Zecca, O., Biddoccu, M., Barmaz, A., and Cavallo, E. (2014). Diversity in soil bacterial communities structure in four high-altitude vineyards cultivated using different soil management techniques. Geophys. Res. Abstr. EGU Gen. Assem. 16:14297.
Peiffer, J. A., Spor, A., Koren, O., Jin, Z., Tringe, S. G., Dangl, J. L., et al. (2013). Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 6548–6553. doi: 10.1073/pnas.1302837110
Petit, E., and Gubler, W. D. (2006). Influence of Glomus intraradices on black foot disease caused by Cylindrocarpon macrodidymum on Vitis rupestris under controlled conditions. Plant Dis. 90, 1481–1484. doi: 10.1094/PD-90-1481
Philippot, L., Raaijmakers, J. M., Lemanceau, P., and Van Der Putten, W. H. (2013). Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 11, 789–799. doi: 10.1038/nrmicro3109
Price, M. N., Dehal, P. S., and Arkin, A. P. (2009). Fasttree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650. doi: 10.1093/molbev/msp077
Qiao, Q., Wang, F., Zhang, J., Chen, Y., Zhang, C., Liu, G., et al. (2017). The variation in the rhizosphere microbiome of cotton with soil type, genotype and developmental stage. Sci. Rep. 7:3940. doi: 10.1038/s41598-017-04213-7
Ravin, N. V., Mardanov, A. V., and Skryabin, K. G. (2015). Metagenomics as a tool for the investigation of uncultured microorganisms. Russ. J. Genet. 51, 431–439. doi: 10.1134/S1022795415050063
Redford, A. J., and Fierer, N. (2009). Bacterial succession on the leaf surface: a novel system for studying successional dynamics. Microb. Ecol. 58, 189–198. doi: 10.1007/s00248-009-9495-y
Reinhold-Hurek, B., Bünger, W., Burbano, C. S., Sabale, M., and Hurek, T. (2015). Roots shaping their microbiome: global hotspots for microbial activity. Annu. Rev. Phytopathol. 53, 403–424. doi: 10.1146/annurev-phyto-082712-102342
Reis, P., Cabral, A., Nascimento, T., Oliveira, H., and Rego, C. (2013). Diversity of Ilyonectria species in a young vineyard affected by black foot disease. Phytopathol. Mediterr. 52, 335–346.
Reynolds, A. G., and Wardle, D. A. (2001). Rootstocks impact vine performance and fruit composition of grapes in British Columbia. Horttechnology 11, 419–427. doi: 10.21273/horttech.11.3.419
Rezgui, A., Ben Ghnaya-Chakroun, A., Vallance, J., Bruez, E., Hajlaoui, M. R., Sadfi-Zouaoui, N., et al. (2016). Endophytic bacteria with antagonistic traits inhabit the wood tissues of grapevines from Tunisian vineyards. Biol. Control 99, 28–37. doi: 10.1016/J.BIOCONTROL.2016.04.005
Santhanam, R., Luu, V. T., Weinhold, A., Goldberg, J., Oh, Y., and Baldwin, I. T. (2015). Native root-associated bacteria rescue a plant from a sudden-wilt disease that emerged during continuous cropping. Proc. Natl. Acad. Sci. U.S.A. 112, E5013–E5020. doi: 10.1073/pnas.1505765112
Sapkota, R., Knorr, K., Jørgensen, L. N., O’Hanlon, K. A., and Nicolaisen, M. (2015). Host genotype is an important determinant of the cereal phyllosphere mycobiome. New Phytol. 207, 1134–1144. doi: 10.1111/nph.13418
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., and Hollister, E. B. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environm. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schoch, C. L., Seifert, K. A., Huhndorf, S., Robert, V., Spouge, J. L., Levesque, C. A., et al. (2012). Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109, 6241–6246. doi: 10.1073/pnas.1117018109
Schreiner, R. P., and Mihara, K. L. (2009). The diversity of arbuscular mycorrhizal fungi amplified from grapevine roots (Vitis vinifera L.) in Oregon vineyards is seasonally stable and influenced by soil and vine age. Mycologia 101, 599–611. doi: 10.3852/08-169
Schreiter, S., Ding, G. C., Heuer, H., Neumann, G., Sandmann, M., Grosch, R., et al. (2014). Effect of the soil type on the microbiome in the rhizosphere of field-grown lettuce. Front. Microbiol. 5:144. doi: 10.3389/fmicb.2014.00144
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Siahmoshteh, F., Hamidi-Esfahani, Z., Spadaro, D., Shams-Ghahfarokhi, M., and Razzaghi-Abyaneh, M. (2018). Unraveling the mode of antifungal action of Bacillus subtilis and Bacillus amyloliquefaciens as potential biocontrol agents against aflatoxigenic Aspergillus parasiticus. Food Control 89, 300–307. doi: 10.1016/J.FOODCONT.2017.11.010
Stefanini, I., and Cavalieri, D. (2018). Metagenomic approaches to investigate the contribution of the vineyard environment to the quality of wine fermentation: potentials and difficulties. Front. Microbiol. 9:991. doi: 10.3389/fmicb.2018.00991
Tahat, M. M., Kamaruzaman, S., and Othman, R. (2010). Mycorrhizal fungi as a biocontrol agent. Plant Pathol. J. 9, 198–207. doi: 10.3923/ppj.2010.198.207
Tan, S., Yang, C., Mei, X., Shen, S., Raza, W., Shen, Q., et al. (2013). The effect of organic acids from tomato root exudates on rhizosphere colonization of Bacillus amyloliquefaciens T-5. Appl. Soil Ecol. 64, 15–22. doi: 10.1016/j.apsoil.2012.10.011
Tewoldemedhin, Y. T., Mazzola, M., Mostert, L., and McLeod, A. (2011). Cylindrocarpon species associated with apple tree roots in South Africa and their quantification using real-time PCR. Eur. J. Plant Pathol. 129, 637–651. doi: 10.1007/s10658-010-9728-4
Toju, H., Tanabe, A. S., Yamamoto, S., and Sato, H. (2012). High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. PLoS One 7:e40863. doi: 10.1371/journal.pone.0040863
Trouvelot, S., Bonneau, L., Redecker, D., van Tuinen, D., Adrian, M., and Wipf, D. (2015). Arbuscular mycorrhiza symbiosis in viticulture: a review. Agron. Sustain. Dev. 35, 1449–1467. doi: 10.1007/s13593-015-0329-7
Van de Peer, Y., Marchal, K., Maere, S., and Vandepoele, K. (2018). Bioinformatics & Systems Biology. Available at: http://bioinformatics.psb.ugent.be (accessed June 20, 2018).
Vázquez-Baeza, Y., Pirrung, M., Gonzalez, A., and Knight, R. (2013). EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2:16. doi: 10.1186/2047-217X-2-16
Vega-Avila, A. D., Gumiere, T., Andrade, P. A. M., Lima-Perim, J. E., Durrer, A., Baigori, M., et al. (2015). Bacterial communities in the rhizosphere of Vitis vinifera L. cultivated under distinct agricultural practices in Argentina. Antonie Van Leeuwenhoek 107, 575–588. doi: 10.1007/s10482-014-0353-7
Wagner, M. R., Lundberg, D. S., Del Rio, T. G., Tringe, S. G., Dangl, J. L., and Mitchell-Olds, T. (2016). Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 7:12151. doi: 10.1038/ncomms12151
Warschefsky, E. J., Klein, L. L., Frank, M. H., Chitwood, D. H., Londo, J. P., von Wettberg, E. J. B., et al. (2016). Rootstocks: diversity, domestication, and impacts on shoot phenotypes. Trends Plant Sci. 21, 418–437. doi: 10.1016/j.tplants.2015.11.008
White, T. J., Bruns, T., Lee, S. H., and Taylor, J. W. (1990). “Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics,” in PCR Protocols: A Guide to Methods and Applications, eds M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (San Diego, CA: Academic Press),315–322.
Youssef, N., Sheik, C. S., Krumholz, L. R., Najar, F. Z., Roe, B. A., Elshahed, M. S., et al. (2009). Comparison of species richness estimates obtained using nearly complete fragments and simulated pyrosequencing-generated fragments in 16S rRNA gene-based environmental surveys. Appl. Environ. Microbiol. 75, 5227–5236. doi: 10.1128/AEM.00592-09
Yuan, J., Chaparro, J. M., Manter, D. K., Zhang, R., Vivanco, J. M., and Shen, Q. (2015). Roots from distinct plant developmental stages are capable of rapidly selecting their own microbiome without the influence of environmental and soil edaphic factors. Soil Biol. Biochem. 89, 206–209. doi: 10.1016/j.soilbio.2015.07.009
Zancarini, A., Mougel, C., Terrat, S., Salon, C., and Munier-Jolain, N. (2013). Combining ecophysiological and microbial ecological approaches to study the relationship between Medicago truncatula genotypes and their associated rhizosphere bacterial communities. Plant Soil 365, 183–199. doi: 10.1007/s11104-012-1364-7
Keywords: bacterial and fungal recruitment, black-foot disease, microbial ecology, microbiome, rhizosphere, rootstock selection
Citation: Berlanas C, Berbegal M, Elena G, Laidani M, Cibriain JF, Sagües A and Gramaje D (2019) The Fungal and Bacterial Rhizosphere Microbiome Associated With Grapevine Rootstock Genotypes in Mature and Young Vineyards. Front. Microbiol. 10:1142. doi: 10.3389/fmicb.2019.01142
Received: 03 September 2018; Accepted: 06 May 2019;
Published: 22 May 2019.
Edited by:
Pierre-Emmanuel Courty, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
George Newcombe, University of Idaho, United StatesRaffaella Balestrini, Institute for Sustainable Plant Protection, Italian National Research Council (IPSP-CNR), Italy
Copyright © 2019 Berlanas, Berbegal, Elena, Laidani, Cibriain, Sagües and Gramaje. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Gramaje, ZGF2aWQuZ3JhbWFqZUBpY3Z2LmVz