
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 May 2019
Sec. Infectious Agents and Disease
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00917
This article is part of the Research Topic Pathogenomics of the genus Brucella and beyond View all 31 articles
 Xu-ming Li2†
Xu-ming Li2† Yao-xia Kang1*†
Yao-xia Kang1*† Liang Lin1En-Hou Jia1
Liang Lin1En-Hou Jia1 Dong-Ri Piao3
Dong-Ri Piao3 Hai Jiang3Cui-Cai Zhang3,4
Hai Jiang3Cui-Cai Zhang3,4 Jin He2
Jin He2 Yung-Fu Chang6*
Yung-Fu Chang6* Xiao-Kui Guo5*
Xiao-Kui Guo5* YongZhang Zhu5*
YongZhang Zhu5*As the causative agent of cattle brucellosis, Brucella abortus commonly exhibits smooth phenotype (by virtue of colony morphology) that is characteristically sensitive to specific Brucella phages, playing until recently a major role in taxonomical classification of the Brucella species by the phage typing approach. We previously reported the discrepancy between traditional phenotypic typing and MLVA results of a smooth phage-resistant (SPR) strain Bab8416 isolated from a 45-year-old custodial worker with brucellosis in a cattle farm. Here, we performed whole genome sequencing and further obtained a complete genome sequence of strain Bab8416 by a combination of multiple NGS technologies and routine PCR sequencing. The detailed genetic differences between B. abortus SPR Bab8416 and large smooth phage-sensitive (SPS) strains were investigated in a comprehensively comparative genomic study. The large indels between B. abortus SPS strains and Bab8416 showed possible divergence between two evolutionary branches at a far phylogenetic node. Compared to B. abortus SPS strain 9-941 (Bab9-941), the specific re-arrangement event in Bab8416 displaying a closer linear relationship with B. melitensis 16M than other B. abortus strains resulted in the truncation of c-di-GMP synthesis, and 3 c-di-GMP-metabolizing genes, were present in Bab8416 and B. melitensis 16M, but absent in Bab9-941 and other B. abortus strains, indicating potential SPR-associated key determinants and novel molecular mechanisms. Moreover, despite almost completely intact smooth LPS related genes, only one mutated OmpA family protein of Bab8416, functionally related to flagellar and efflux pump, was newly identified. Several point mutations were identified to be Bab8416 specific while a majority of them were verified to be B. abortus ST2 characteristic. In conclusion, our study therefore identifies new SPR-associated factors that could play a role in refining and updating Brucella taxonomic schemes and provides resources for further detailed analysis of mechanism for Brucella phage resistance.
Brucellosis is one of the most serious zoonotic infectious diseases worldwide, and is caused by pathogenic species of Brucella genus. Up to now, 12 species were defined into the genus Brucella (Godfroid et al., 2013). Six of them, including B. melitensis, B. abortus, B. suis, B. canis, B. ovis, and B. neotomae, belong to the “classical” or “traditional” Brucella species1. Generally, all Brucella species with nucleotide similarities > 90% are genetically closely related (Al Dahouk et al., 2010).
Traditional Brucella typing is primarily based on different phenotypic characteristics (Garcia et al., 1988; Jahans et al., 1997; Moreno et al., 2002; Sanogo et al., 2013), including colony morphology, CO2 requirement, H2S production, substrate utilization, growth on serum dextrose agar dye plate, agglutination with monospecific sera, Brucella phage lysis profiles at routine test dilution (RTD) and host preference (Jones et al., 1968; Morris et al., 1973; Rigby et al., 1989). The three major species in terms of disease and economic impact for man, B. melitensis, B. abortus and B. suis are further subdivided into multiple biovars (bv) based on a range of phenotypic and serological characteristics. For example, B. abortus is subdivided into bv 1–6 and 9 (Pappas et al., 2006). Furthermore, despite the close genetic relationship of several genetic loci (e.g., 16S rRNA, 98.7%) and a biochemical profile similar to Ochrobactrum spp., several non-classical Brucella species like B. microti and B. inopinata are often easily misidentified using traditional biochemical typing methods (Scholz et al., 2008a, b). Among these routine phenotypic characterizations, B. abortus with smooth Lipopolysaccharide (LPS) was identified to be sensitive to Brucella phages like Berkeley2 (BK2), Tbilisi (Tb), Weybridge (Wb), and Izatnagar (Iz) (FAO/WHO, 1986). This useful test is significant for differentiating B. abortus from other Brucella species (Jones et al., 1968; Morris et al., 1973).
Since SPR B. abortus was initially reported in Corbel and Morris (1974), there have been few studies on this distinct phenotype over the last four decades. The susceptibility of smooth B. abortus strains to lysis by Brucella phages is commonly used to type various Brucella species. We have recently reported the identification of the first SPR B. abortus strain Bab8416 from a brucellosis patient in China (Kang et al., 2015). The phage activity of Bab8416 is similar to that of B. melitensis bv 1 strain 16M and showed special biochemical characteristics distinct from that of all B. abortus biovars. It was not lysed by Tb, Iz, and Wb phage in 1 × RTD and 104 × RTD, but lysed by BK2 phage in 1 × RTD and 102 × RTD. Due to the unusual discrepancy between phenotypic profiles, Bab8416 could not be precisely classified to any of the existing B. abortus biovars. In this study, we completed the genome sequence of Bab8416 through a combination of next-generation sequencing (NGS) and common PCR-based gap closure and investigated genomic differences between Bab8416 and other Brucella strains for gene association in corresponding biochemical or physiological profiles.
This study and the protocol were carried out in accordance with the recommendations of ethics committee of the local disease control and Prevention Research Center of the Inner Mongolia Autonomous Region and Baotou City. The patient gave written informed consent for participation in this study and publication of his identifiable information, in accordance with the Declaration of Helsinki. The detailed information of strain Bab8416 referred to our previous study (Kang et al., 2015).
Using 454 GS-FLX system, a total of 190,817 reads were obtained with the average length of 566 bp. Twenty-two contigs with lengths more than 500 bp and average coverage of 33.2X were obtained by Newbler using default parameters. Using the genome of B. abortus 9-941 as a reference, the order of the contigs was sorted and gap closure using common PCR was performed with ContigsScape (Tang et al., 2013). To fix the homopolymer sequencing errors systemically caused by 454 GS-FLX sequencing system, another 180 bp Paired End (PE) library was constructed and sequenced by the Illumina Hiseq 2000 system. Genome sequencing results were refined by short reads using Pilon with default parameters (Walker et al., 2014). The coding genes were predicted by Prodigal (Hyatt et al., 2010) and these genes were annotated by BLAST against NCBI non-redundant (NR), COG, KEGG, TrEMBL, Swissprot databases with e value cutoff of 1e-5 and GO terms assigned to the annotated genes using BLAST2GO pipeline (Conesa et al., 2005). The tRNAs were detected by tRNAscan-SE (v1.23) (Schattner et al., 2005) and rRNAs were identified by blasting homologous rRNA sequences against the Bab8416 genome.
Firstly, oriC site was identified in both references and Bab8416 genome using Ori-Finder 2 and was set to be the first base of Bab8416 genome (Luo et al., 2014). Then, whole genome sequence alignments between these two genomes were processed by MUMmer 3.23 package (Kurtz et al., 2004).
Multiple-locus variable number tandem repeat analysis (MLVA) assay was employed and the markers were obtained by PCR (Jiang et al., 2013b). The MLVA markers of Bab8416 were compared to the MLVA database2. The multilocus sequence typing (MLST) schemes of Brucella species using 9 conserved housekeeping genes were performed as previously described (Whatmore et al., 2007).
All the draft genomes were linked to be two pseudo chromosomes by taking B. abortus 9–941 genome as a reference and the sequences were gaped with ‘NNNNN.’ The SNPs were firstly identified by Mauve (Darling et al., 2010) using the genome sequences in this study and after “N” removed, the remaining SNP were finally exported for further analysis.
Thirty-nine available Brucella reference genomes were utilized to perform comparative genomic and phylogenetic analyses, including all known Brucella species and all of seven biovars of B. abortus. Three strains with lower contig numbers and high coverage in each biovar of B. abortus were selected. All genes of the selected strains were ortholog clustered by PGAP (Zhao et al., 2012), a pipeline for pan-genome analysis, and genes with both coverage and identity higher than 90% were considered to be the same ortholog cluster. Hence, a total of 2,014 single copy gene families were identified and a super gene was constructed for phylogenetic analysis by combining all sequences of these genes into one ortholog cluster. A maximum likelihood phylogenetic tree was constructed by Phyml 3.0 (Guindon et al., 2010) using HKY85 nucleotide substitution model with a bootstrap value of 1000. In addition, in order to investigate the regions of differences (RD) from pan-genome analysis, we further added 200 B. melitensis genomes and 197 B. abortus genomes for detailed screening and characterization by using BLASTN program.
We downloaded all the virulence factor from Virulence Factors Database (VFDB) (Chen et al., 2005), and we aligned all the protein sequences of the strain Bab8416 to the VFDB using BLASTP program available at NCBI server (ncbi-blast-2.7.1+) with both coverage and identity higher than 80%.
The genome sequence and annotations were submitted to GenBank database with accession number CP008774–CP008775. All the reference genomes used in this paper were obtained from PATRIC (Wattam et al., 2017).
The genome size of strain Bab8416 is 3.2 Mb, and it consists of two circular chromosomes: a large chromosome of 2,116,946 bp and a smaller one of 1,156,123 bp. The average GC content of two chromosomes was 57.22% (Crasta et al., 2008; Tsolis et al., 2009). A total of 3,295 Coding DNA sequences (CDSs) have been computationally predicted. The summarized message of Bab8416 genome is showing in Figure 1. The average length of CDS was 856 bp and 2,272 CDSs (68.95%) were assigned definite biological function as well as 1,023 (31.05%) are hypothetical proteins. Figure 2 is showing GO function class of the annotated genes.
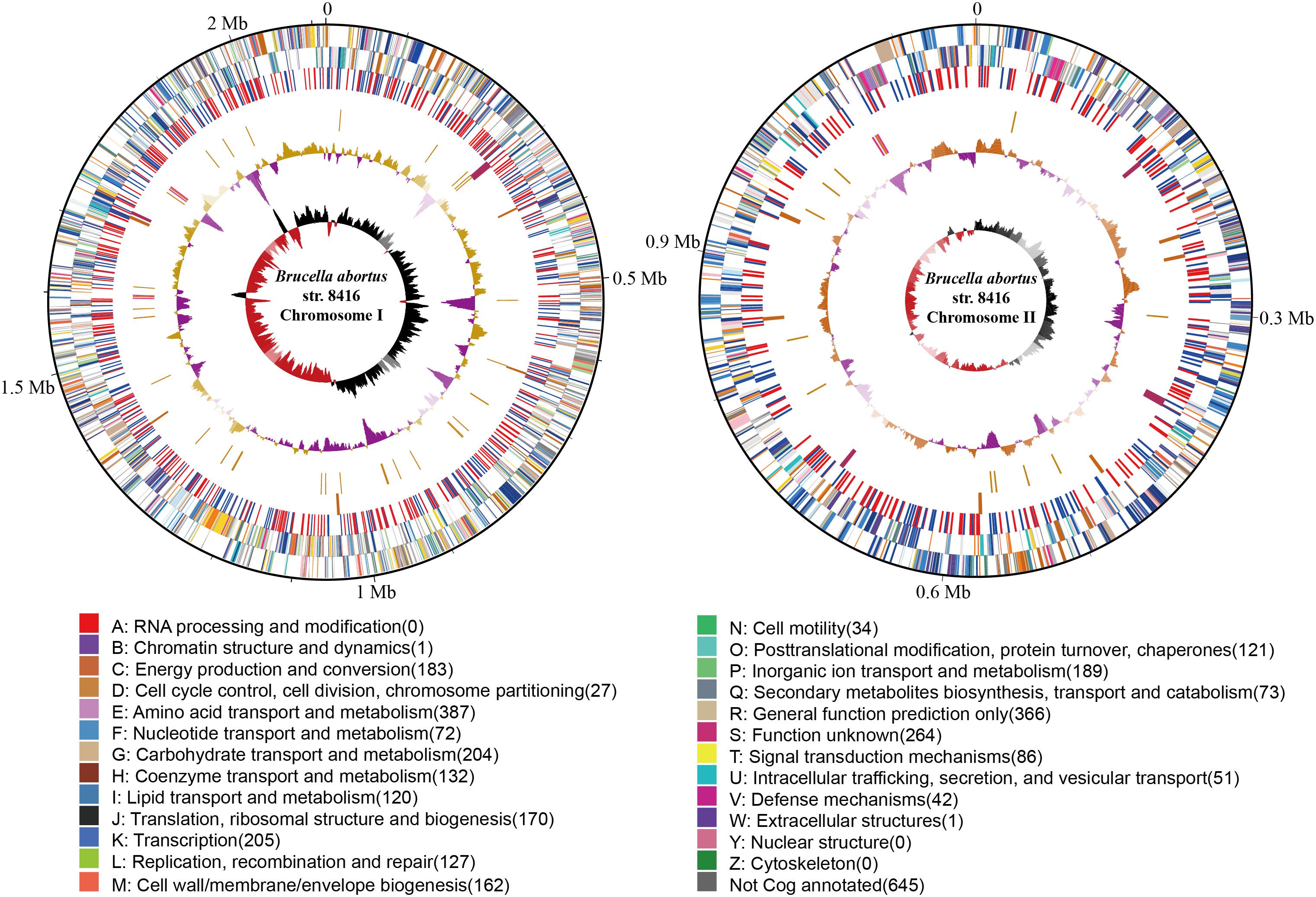
Figure 1. The atlas of Brucella abortus SPR strain Bab8416 genome. The outer black circle shows coordinates. Moving inward, the next two circles show forward and reverse strand CDS, respectively, with colors representing the functional classification, the next circle shows Bab8416 specific synonymous (red) and non-synonymous (blue) SNP, followed by the Bab8416 unique insertions (dark orange) and deletions (maroon), then is the tRNA (orange) and rRNA (deep pink). The final two are GC-content and GC-skew by using a 10-kb window and overlap at 1,000bp.
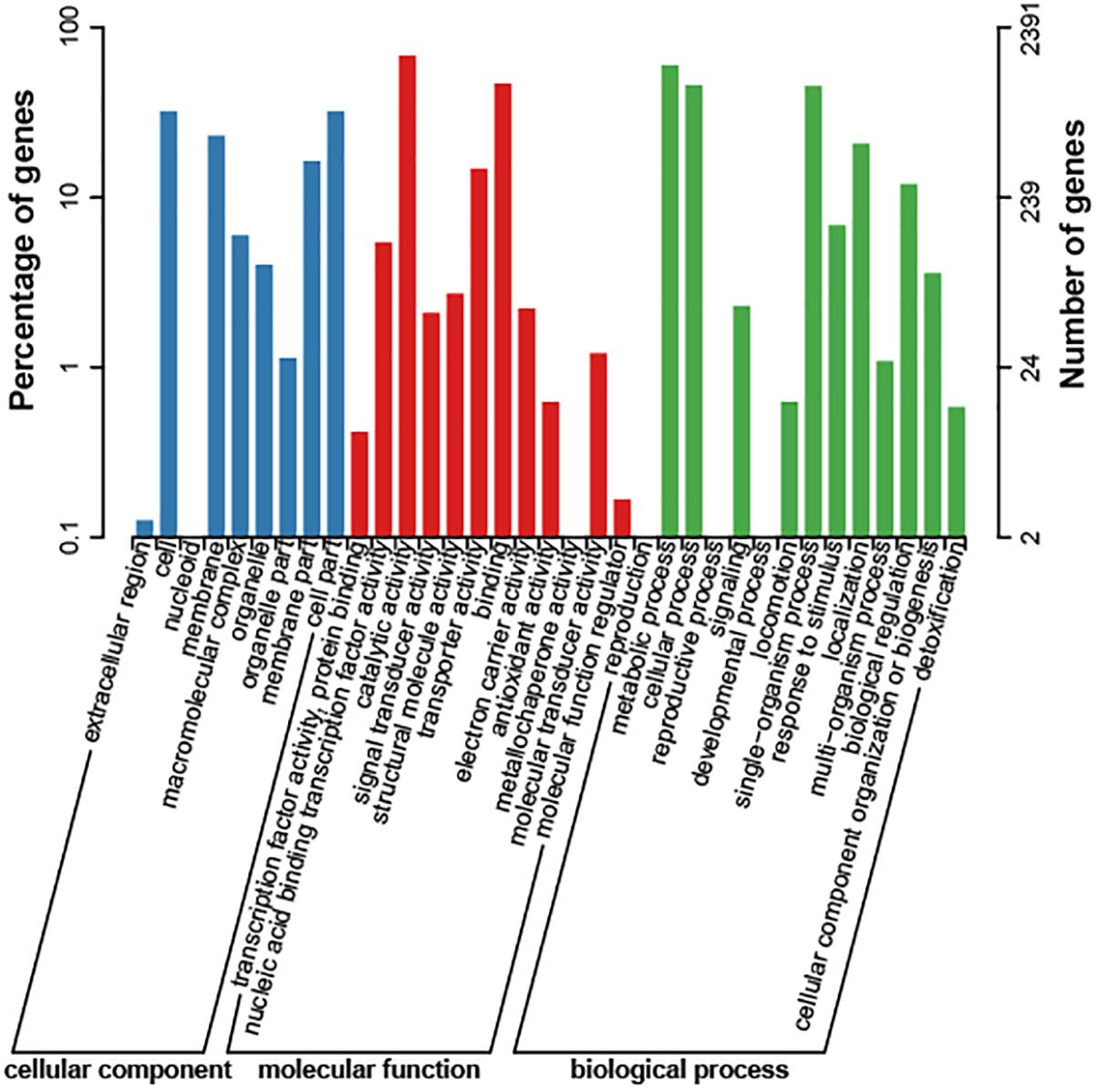
Figure 2. Cog class of Brucella abortus 8416 function genes. The result was export with e-value of 1e-5.
Except for resistance to phage Iz, Tb, and Wb shown in Table 1, the physiological and biochemical profiles of strain Bab8416 was more closely related to smooth B. abortus bv 9 (Morris et al., 1973). In addition, electron microscopy was used to investigate phage Tb/Bab8416 interaction (Figure 3); absorption but no lysis of host bacteria was observed. Here, we performed additional MLVA typing (Le Fleche et al., 2006; Al Dahouk et al., 2007; Van Belkum, 2007; Valdezate et al., 2009). While no 100% match could be found in MLVA database, the top 20 matches consistently with B. abortus bv. 3 (Figure 4).
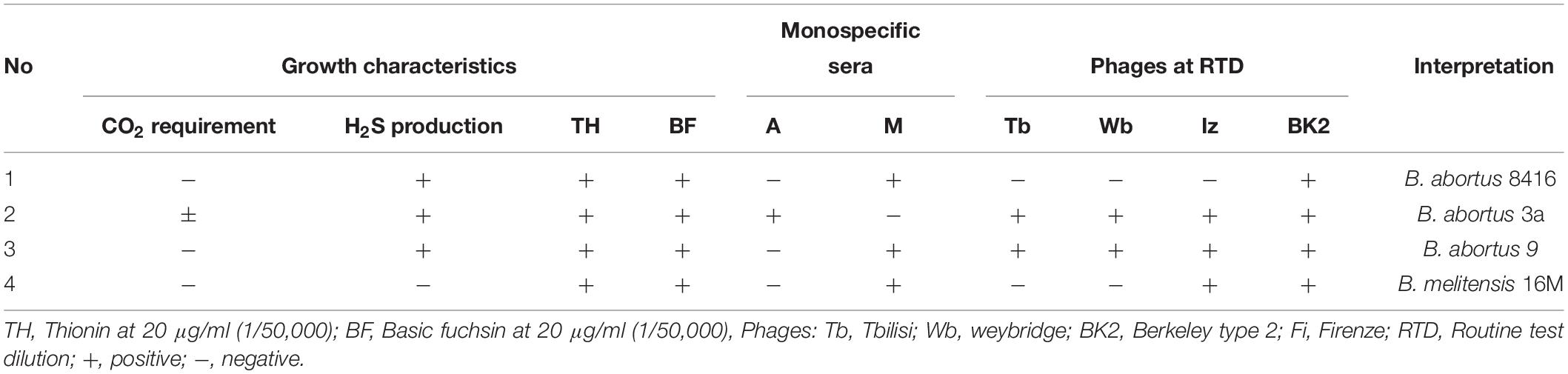
Table 1. Physiological and biochemical typing details of B. abortus Bab8416 compared with other standard strains.
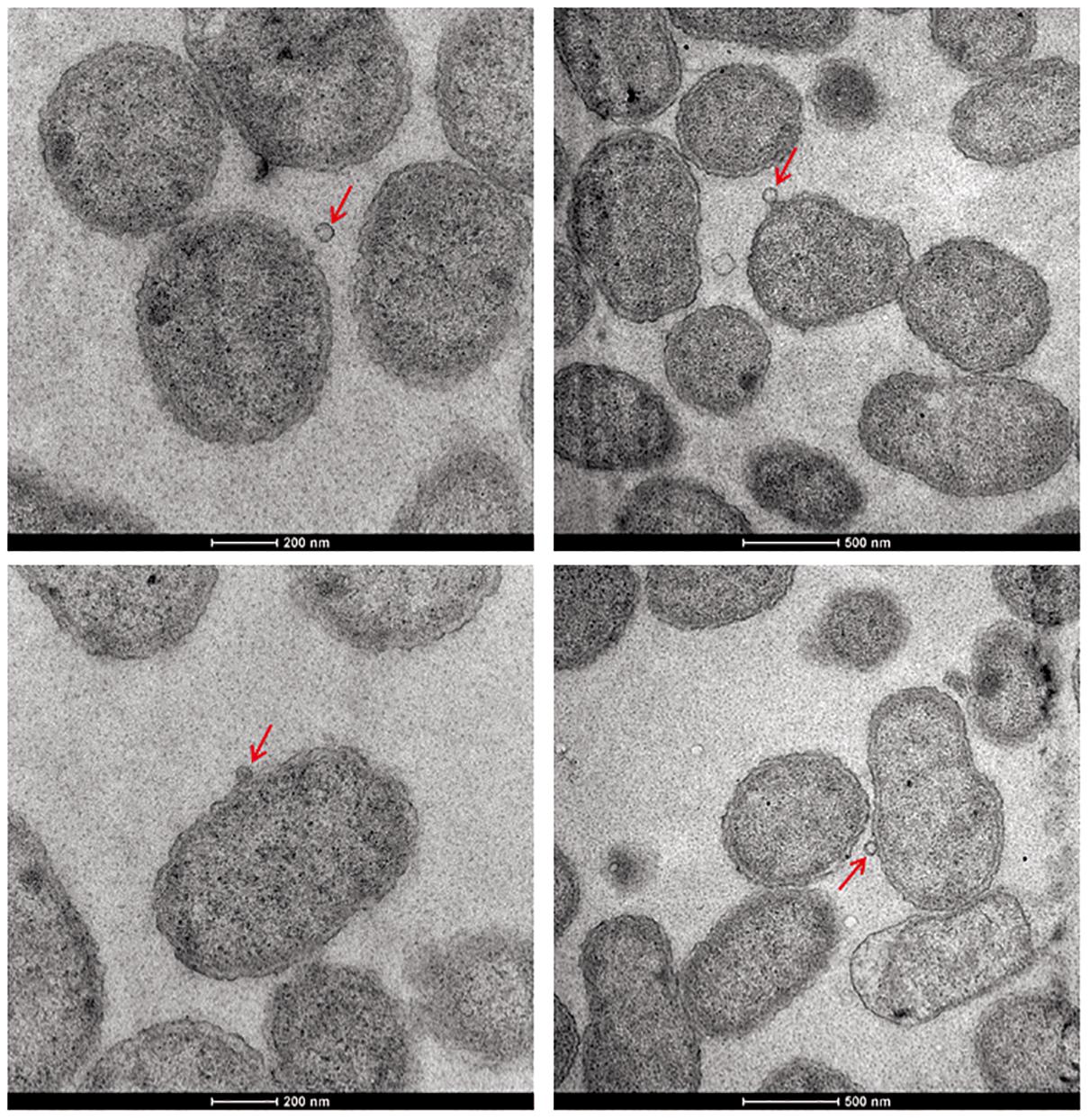
Figure 3. The electron microscope photo of interaction between Brucella phage and Brucella abortus SPR strain Bab8416. Brucella phages Tb phages were found successfully to adhere on the surface of B. abortus strain Bab8416 but failed to lyse the strain. Red arrows are showing the dissociative Tb or Tb binding to strain 8416.
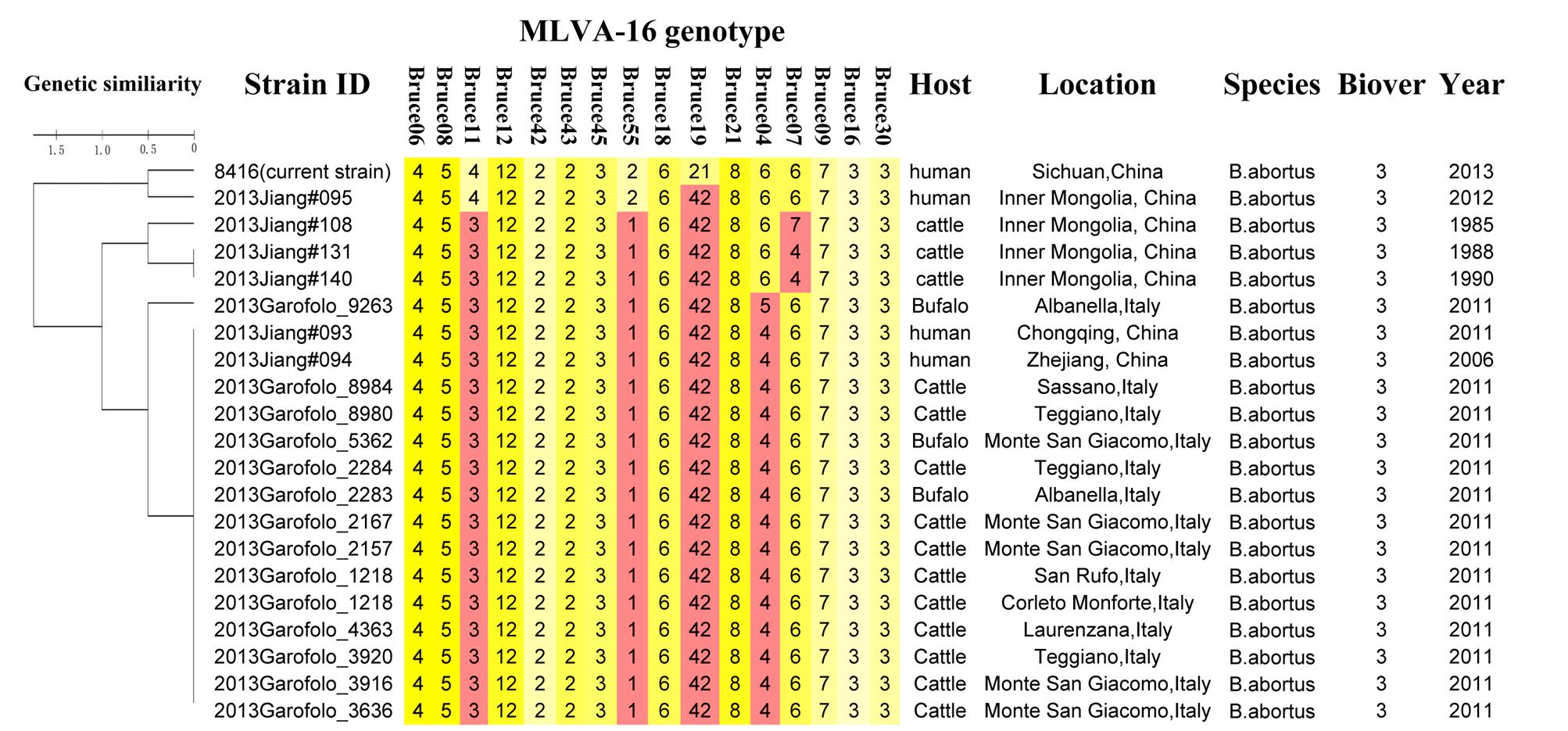
Figure 4. MLVA typing results of B. abortus strain Bab8416.
Without coincident results in both traditional phenotyping and modern MLVA genotyping, we further employed MLST method (Whatmore et al., 2007). Twenty-seven Brucella sequence types (STs) were initially identified and more STs have been found (Whatmore et al., 2016). Bab8416 was identified as an ST2 in this study.
Determining the evolutionary context of Bab8416 is essential for a detailed comparative genomic analysis and to account for the inconformity of the former two typing results from different strains and isolates of Brucella (Crasta et al., 2008). A total of 2,014 single copy genes were identified within 25 B. abortus strains with three strains in each biovar and B. melitensis str. 16M as one outgroup being used to build a maximum likelihood phylogenetic tree (Figure 5). Many strains within the same biovar are not closely genetically related; conversely, several strains in different biovars have been shown to be closely related. This finding indicates that traditional physiological and biochemical typing designations of biovars within B. abortus do not reflect genetic linkage patterns.
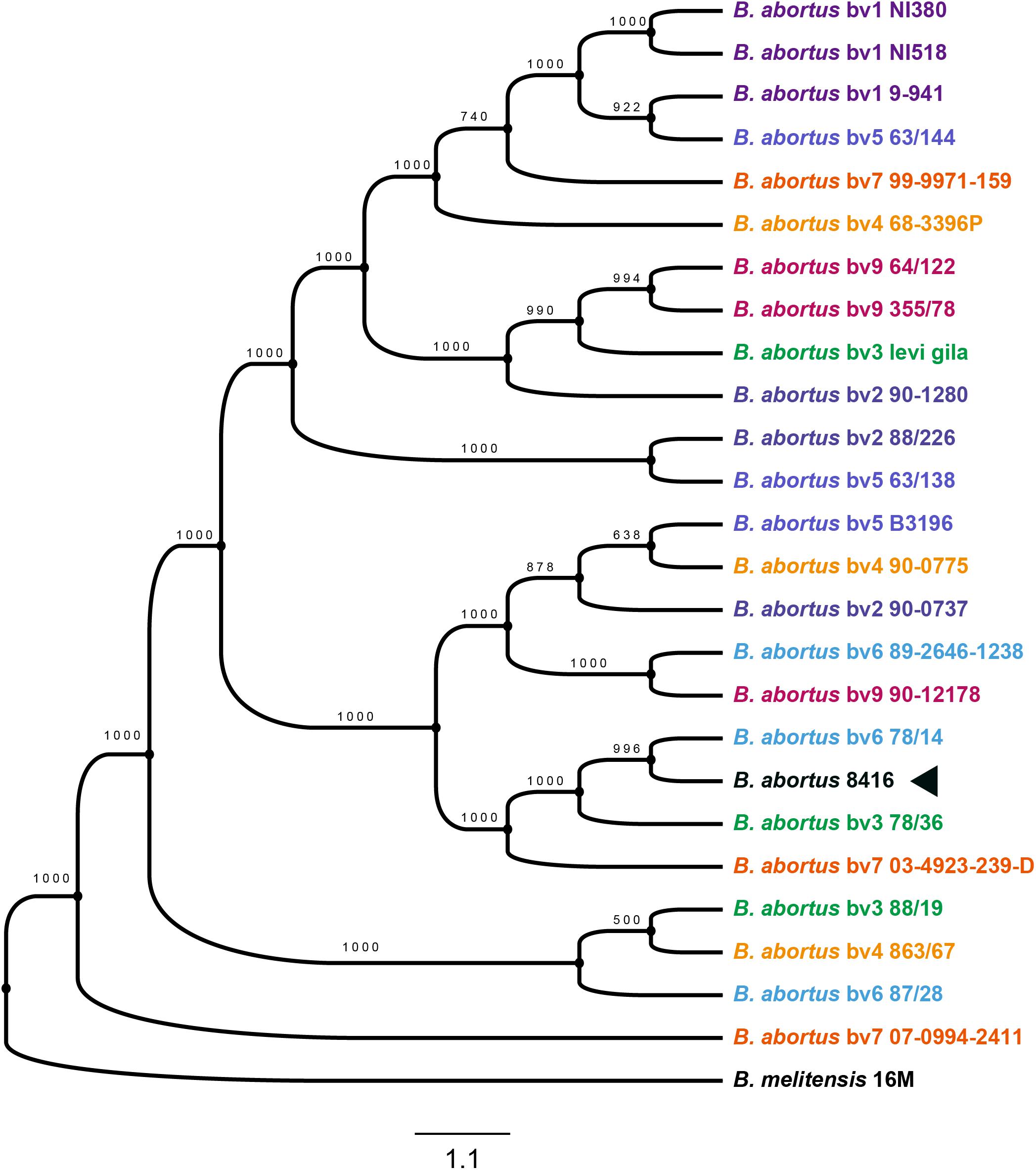
Figure 5. Phylogenetic tree of Brucellae. The phylogenetic tree was based on the 601 core genes of strains used in this analysis and it was constructed by using the maximum likelihood method with bootstrap value 1000. Black arrow is showing the phylogenetic cluster of B. abortus strain 8416.
As draft genomes often generate low resolution results in studies measuring genetic variation, we conducted a comparative genomic analysis using complete genomes as previously described (Ricker et al., 2012; Zhang et al., 2012). B. abortus strain 9-941 (Bab9-941) was a typical SPS strain with the complete genome published. Here, we chose Bab9-941 as a reference for comparative genomic analysis and the genome features of B. abortus used and were listed in Table 2.
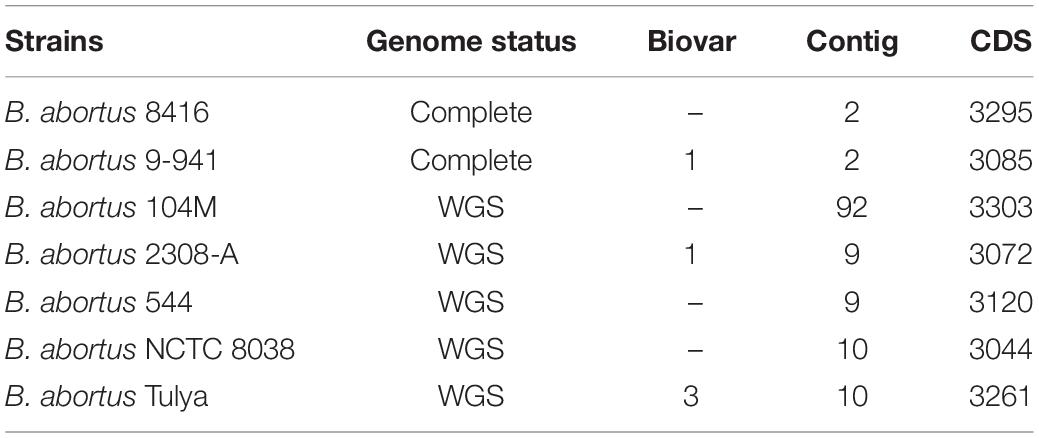
Table 2. Genome features of these strains used in comparative analysis.
In comparison with Bab9-941, a large fragment (420 kb) re-arrangement in small chromosome of Bab8416 was found by using MUMmer (Figure 6). Re-arrangements in Brucella species have been previously reported (Sieira et al., 2000; Jiang et al., 2013a), however, this one proved to be exceptional. Compared with other B. abortus genomes observed here, the re-arrangement in Bab8416 was specific and displayed a closer linear relationship with B. melitensis 16M than the other B. abortus genomes. As mentioned above, Bab8416 shared the same phage typing status with B. melitensis bv 1 strain 16M; strongly similar genomic structures were also shown to exist between these two strains.
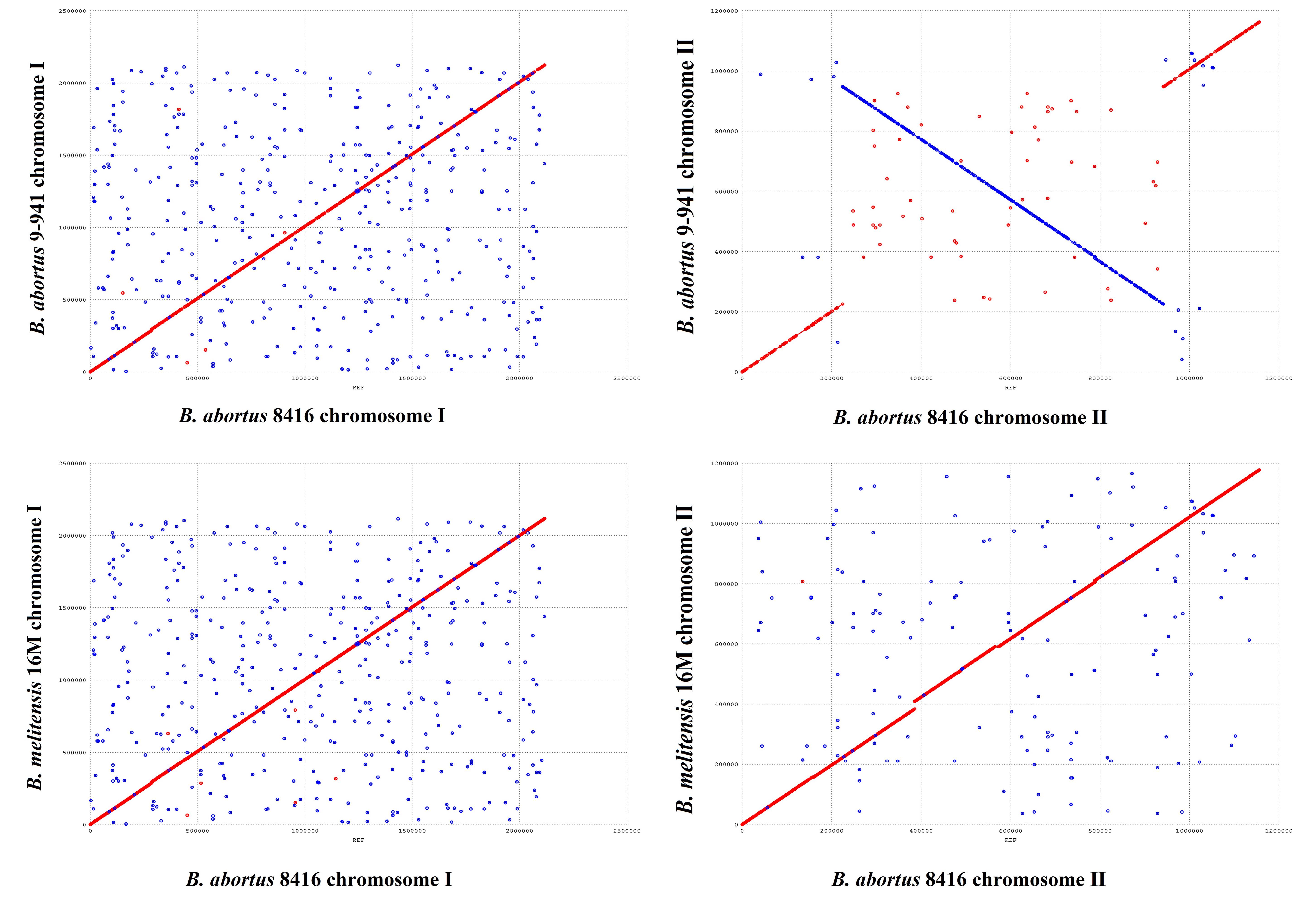
Figure 6. Genome arrangement of Brucella abortus strain Bab8416 small Chromosome. The four complete B. abortus genomes, including B. abortus Bab8416, were compared to B. abortus Bab9-941 and arrangements only appeared in small Chromosome of B. abortus Bab8416.
Nevertheless, neither IS elements nor tRNA operons usually responsible for genome re-arrangement were detected in the terminal region of the Bab8416 re-arrangement sequence. Three genes, BMEII0292, BMEII0293, and BMEII1009, were truncated or incomplete at the terminal fragment in other B. abortus strains. Both BMEII0292 and BMEII1009 contain a GGDEF domain that enables them to generate the cyclic di-GMP (c-di-GMP), a kind of secondary messenger central in regulating bacteria adaptive responses. In addition, analysis of protein-protein interactions using STRING database (Franceschini et al., 2013) indicated that BMEII0293 encodes a hypothetical protein that is tightly associated with the synthesis and degradation of c-di-GMP. In B. melitensis, 11 c-di-GMP-metabolizing proteins had been inferred to regulate c-di-GMP metabolism (Petersen et al., 2011). The structure of these 11 genes were verified to be intact in Bab8416, but BMEII0929, BMEII0292 and BMEII1009, were found absent in Bab941 and some B. abortus strains.
Compared with SPS Bab9-941, 49 indels (≥20 bps), including 25 deletions and 24 insertions, were found in the Bab8416 genome (Tables 3, 4). A 16.5 kb region is absent from Bab8416 genome and a 3.2 kb region appears to be unique. Only four large regions (>500 bp) were represented by deletions, one Region of Differences (RD) 1 in chromosome I and three (RD2–RD4) in small chromosome. Genes lost in these regions are determined by referencing the annotation of B. abortus 9–941. The details of deletions and associated ORFs are shown in Table 3.
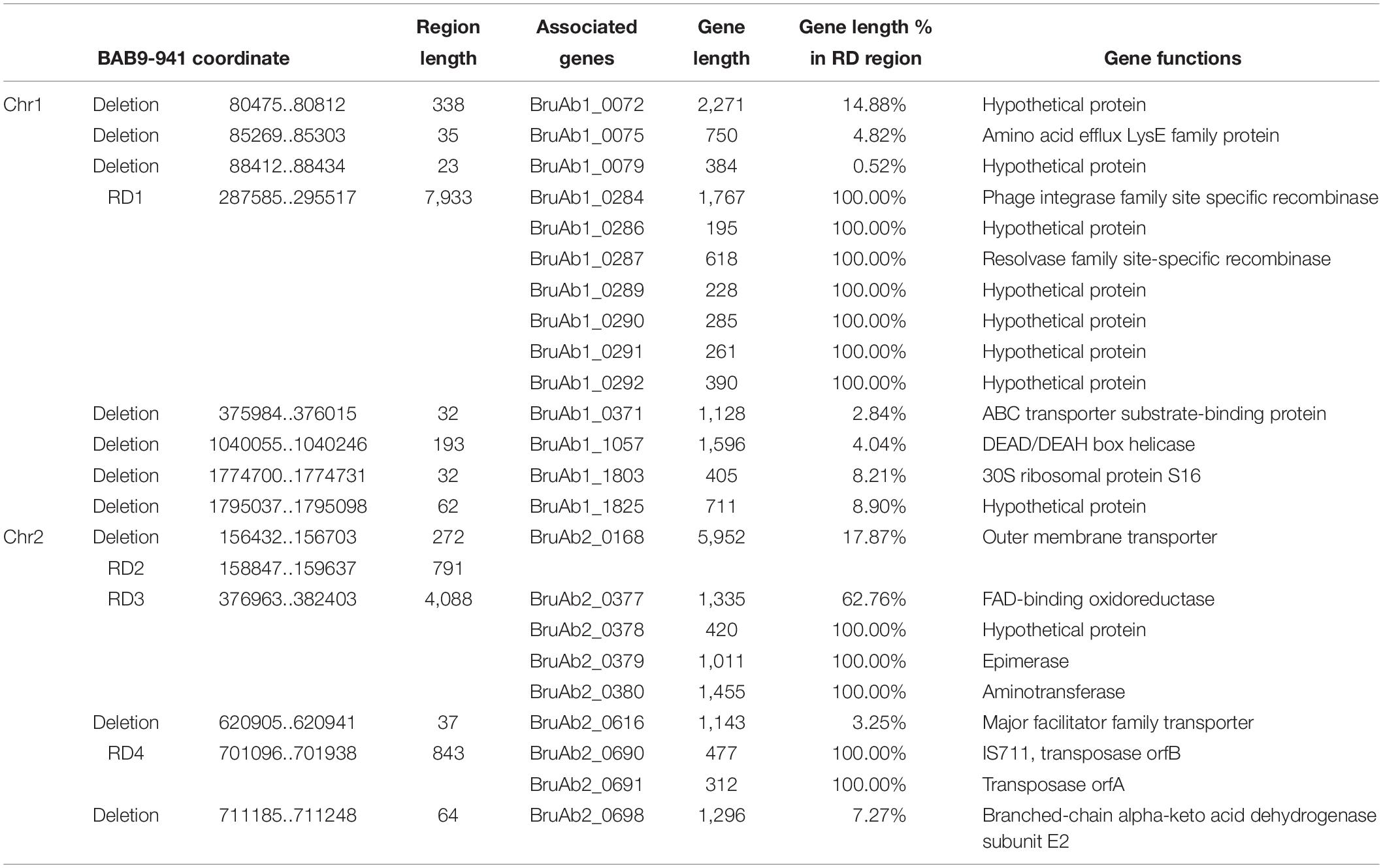
Table 3. ORFs related to deletions in B. abortus str.8416 compared to B. abortus 9–941 genome.
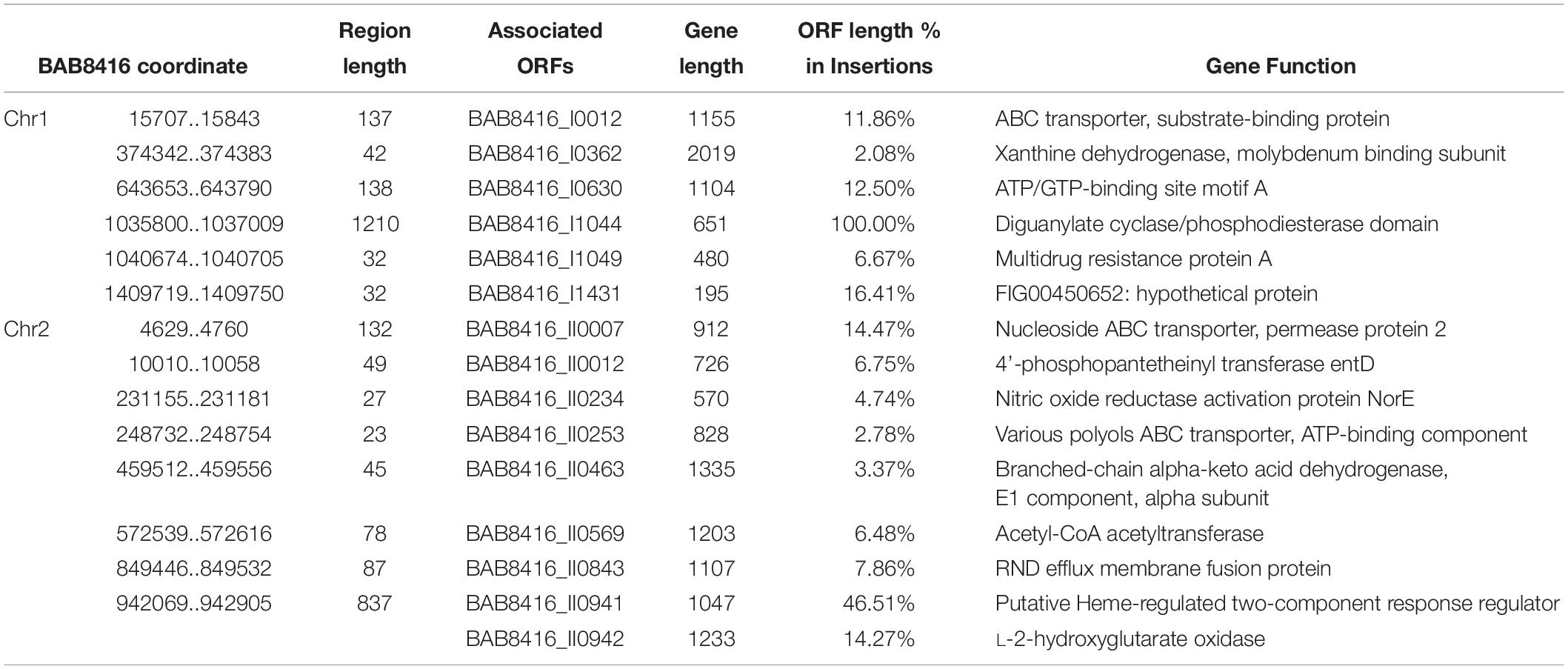
Table 4. ORFs in insertions in B. abortus 8416 compared to B. abortus 9-941 genome.
Eight genes, BruAb1_0284-0292, were located in RD1 region. BruAb1_0284 and BruAb1_0287 are specific recombinases, belonging to phage integrase and resolvase families, respectively. BruAb1_0285 and BruAb1_0288 were annotated as pseudo genes and the others were labeled hypothetical proteins. In addition, we further detected the RD1 region in 200 B. melitensis genomes and 197 B. abortus genomes by using BLASTn. In all of B. melitensis 200 strains we could not found any sequence similar with RD1. While 127 out of 197 B. abortus strains could be found the sequences with identity higher than 99% and coverage over 90% (Supplementary Table S4). These evidences above showed that RD1 was exclusively specific to B. abortus and the insert event should occur after the differentiation of the most recent common ancestor of B. abortus 9–941 and Bab8416. RD2 and another small deletion are involved in the locus of an outer membrane transporter, BruAb2_0168. An earlier study confirmed that this locus was conserved between B. abortus (Halling et al., 2005), but variation is present in Bab8416. RD3 contains four genes, BruAb2_0377 to BruAb2_0380. BruAb2_0377 encodes FAD-binding oxidoreductase. BruAb2_0378 was defined as a hypothetical protein. BruAb2_0379 encodes an epimerase that catalyzes the transformation of dTDP-glucose to dTDP-4-oxo-6-deoxy-D-glucose. BruAb2_0380 encodes an amino transferase that participates in arginine and proline metabolism, metabolic pathways and biosynthesis of secondary metabolites. Two intact genes and one partial gene are encoded by RD4. The two complete genes, BruAb2_0690 and BruAb2_069, encode transposase.
Inserted regions specific to Bab8416 are shown in Table 4. Among the 20 Bab8416 specific regions, six regions are located at intergenic spacer (IGS) and fifteen ORFs are involved in the other 14 insertions. All of these ORFs are annotated with known functions.
The variant ORFs were identified by BLASTn method. The results are shown in Table 5. In consideration of the prediction discrepancy and the restriction of software, we searched these ORFs within these five genomes. BLASTn results showed that 144 Bab9-941 ORFs were found deleted or incomplete in Bab8416 and 129 Bab8416 ORFs were found to be Bab8416 specific. These deletions may be partly responsible for the unusual Brucella phage status of Bab8416.

Table 5. Classification of B. abortus strain Bab8416 specific SNPs associated ORFS.
A total of 1,373 SNPs were identified between Bab8416 and Bab9-941. Using B. abortus 9–941 as a reference, 336 SNPs were intergenic and 1,036 SNPs were located in the ORFs. In addition, 518 genes-encoding proteins showed amino acid changes caused by 632 non-synonymous SNPs. As the SNP number was large, we inferred that these markers appeared in Bab8416 could be the characteristics of ST2. Since no other complete genomes of ST2 were available, we chose to utilize the existing draft genomes. In consideration of insuring the quality of sequencing and assembly, only the draft genomes with contig numbers less than 12 were selected. The MLST typing results of these genomes are shown in Supplementary Table S1. Fifteen out of 95 genomes were identified to be ST2. We tested the SNPs between the 16 ST2 genomes and found that overwhelming majority (95.05%) of former identified SNPs were verified to be potential markers of ST2 strains and only 68 SNPs appeared to be Bab8416 specific. The detailed SNP annotations are present in Supplementary Table S2 and the Bab8416 specific SNP involved genes are presented in Supplementary Table S3.
Lipopolysaccharide is tightly associated with the virulence of pathogens and the efficiency of corresponding vaccines. Brucella with rough lipopolysaccharide (R-LPS) was lysed by Brucella phage R/C, and is host specific (Hammerl et al., 2017). In Brucella, genes essential in synthesizing LPS and developing a smooth phenotype have been located at the Wbk region of chromosome I (Godfroid et al., 2000; Gonzalez et al., 2008; Zygmunt et al., 2009). Inactivation of formyltransferase (wbkC) gene is the significant factor that contributes to rough phenotype (Lacerda et al., 2010). BLASTn results showed that none of these genes were deleted/missing in Bab8416. Four non-synonymous mutations were identified in Bab8416 LPS genes, only one (BruAb1_1699) was found not belonging to ST2. This gene encodes an OmpA family protein, which is tightly related to flagellar protein production and also related to the efflux pump.
Bab8416 was isolated from a patient with clinical brucellosis, indicating that this strain was virulent. The presence of 23 Brucella virulence factors confirmed by VFDB was tested in the Bab8416 isolate. Bab8416 was found to have a full complement of these loci. BLASTp results showed that eleven genes were 100% identical, eight genes had point mutations, and short deletions were found in the other four genes with only one deletion being present in VFG2217. In addition, compared to BAB1_0069, a putative outer membrane protein considered to be a virulence factor, a 133 amino acid deletion is present in this locus of Bab8416. We inferred these changes might exert some influence on the virulence of Bab8416 but not that much to cause high level attenuation as it is still a pathogenic bacterium.
Combining NGS sequencing technology and comparative genomics analysis, the complete genome sequence of B. abortus SPR strain Bab8416 was obtained and specific genetic characteristics of B. abortus SPR were comprehensively investigated in this study. Study of smooth LPS related genes showed that Bab8416 does share same LPS key genes with other B. abortus SPR strains, which supported veracity of previous phenotype screening results. The gold standard for Brucella characterization is still based on specific properties of the bacteria. None of the available molecular typing methods covers all currently known species and biovars of the genus Brucella (Hammerl et al., 2017). The difference between biotyping and genotyping of some special strain need further analysis not only on genomic but protein expressive level, because the host strains co-evolute with their special phages. The importance of individual amino acids of the tail collar protein for the host range of the Brucella phages has not yet been investigated. To avoid diverging lysis patterns, examine the phage genomes by sequencing were recommended if the lysis results are inconsistent on the same indicator strains (Hammerl et al., 2017).
Bab8416 has a genetic profile different from that typically found in most B. abortus strains. The arrangement sequences in small chromosome resulted in the truncation of c-di-GMP synthesis. The indels within SPS and SPR B. abortus showed that two evolutionary branches might have diverged at a far phylogenetic node. Plentiful point mutations were identified to be Bab8416 specific while the majority of the point mutations were verified to be ST2 characteristic of B. abortus. While few Bab8416 SNPs were identified, SNPs might still exert a significant influence on phage typing status. Despite the unique genetic characteristics of Bab8416 uncovered in this study, full details of its resistance to phage have not yet been elucidated at the genomic level. Our findings established some novel molecular mechanisms underlying Brucella sensitivity to brucellapages that might contribute to improving our understanding on Brucella phenotyping.
X-ML and Y-XK performed genomic sequencing and comparative genomic analyses and wrote the manuscript. LL, E-HJ, and D-RP performed Brucella MLVA typing and phage typing. HJ, C-CZ, and JH performed Brucella MLST typing and PCR sequencing. Y-FC, X-KG, and YZ designed the whole experiments and revised the manuscript.
This study was supported by the National Natural Science Foundation of China (81460319 and 81360444).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. L. Gary Adams, for his critical reading of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00917/full#supplementary-material
Al Dahouk, S., Fleche, P. L., Nockler, K., Jacques, I., Grayon, M., Scholz, H. C., et al. (2007). Evaluation of brucella MLVA typing for human brucellosis. J. Microbiol. Methods 69, 137–145. doi: 10.1016/j.mimet.2006.12.015
Al Dahouk, S., Scholz, H. C., Tomaso, H., Bahn, P., Gollner, C., Karges, W., et al. (2010). Differential phenotyping of brucella species using a newly developed semi-automated metabolic system. BMC Microbiol. 10:269. doi: 10.1186/1471-2180-10-269
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328.
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Corbel, M. J., and Morris, J. A. (1974). Studies on a smooth phage-resistant variant of Brucella abortus. I. Immunological properties. Br. J. Exp. Pathol. 55, 78–87.
Crasta, O. R., Folkerts, O., Fei, Z., Mane, S. P., Evans, C., Martino-Catt, S., et al. (2008). Genome sequence of Brucella abortus vaccine strain S19 compared to virulent strains yields candidate virulence genes. PLoS One 3:e2193. doi: 10.1371/journal.pone.0002193
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
FAO/WHO (1986). Joint FAO/WHO expert committee on brucellosis. World Health Organ. Tech. Rep. Ser. 740, 1–132.
Franceschini, A., Szklarczyk, D., Frankild, S., Kuhn, M., Simonovic, M., Roth, A., et al. (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815. doi: 10.1093/nar/gks1094
Garcia, M. M., Brooks, B. W., Ruckerbauer, G. M., Rigby, C. E., and Forbes, L. B. (1988). Characterization of an atypical biotype of Brucella abortus. Can. J. Vet. Res. 52, 338–342.
Godfroid, F., Cloeckaert, A., Taminiau, B., Danese, I., Tibor, A., De Bolle, X., et al. (2000). Genetic organisation of the lipopolysaccharide O-antigen biosynthesis region of brucella melitensis 16M (wbk). Res. Microbiol. 151, 655–668. doi: 10.1016/s0923-2508(00)90130-x
Godfroid, J., Al Dahouk, S., Pappas, G., Roth, F., Matope, G., Muma, J., et al. (2013). A “One Health” surveillance and control of brucellosis in developing countries: moving away from improvisation. Comp. Immunol. Microbiol. Infect. Dis. 36, 241–248. doi: 10.1016/j.cimid.2012.09.001
Gonzalez, D., Grillo, M. J., De Miguel, M. J., Ali, T., Arce-Gorvel, V., Delrue, R. M., et al. (2008). Brucellosis vaccines: assessment of Brucella melitensis lipopolysaccharide rough mutants defective in core and O-polysaccharide synthesis and export. PLoS One 3:e2760. doi: 10.1371/journal.pone.0002760
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Halling, S. M., Peterson-Burch, B. D., Bricker, B. J., Zuerner, R. L., Qing, Z., Li, L. L., et al. (2005). Completion of the genome sequence of Brucella abortus and comparison to the highly similar genomes of Brucella melitensis and Brucella suis. J. Bacteriol. 187, 2715–2726. doi: 10.1128/jb.187.8.2715-2726.2005
Hammerl, J. A., Gollner, C., Jackel, C., Scholz, H. C., Nockler, K., Reetz, J., et al. (2017). Genetic diversity of brucella reference and non-reference phages and its impact on brucella-typing. Front. Microbiol. 8:408. doi: 10.3389/fmicb.2017.00408
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jahans, K. L., Foster, G., and Broughton, E. S. (1997). The characterisation of Brucella strains isolated from marine mammals. Vet. Microbiol. 57, 373–382. doi: 10.1016/s0378-1135(97)00118-1
Jiang, H., Du, P., Zhang, W., Wang, H., Zhao, H., Piao, D., et al. (2013a). Comparative genomic analysis of Brucella melitensis vaccine strain M5 provides insights into virulence attenuation. PLoS One 8:e70852. doi: 10.1371/journal.pone.0070852
Jiang, H., Wang, H., Xu, L., Hu, G., Ma, J., Xiao, P., et al. (2013b). MLVA genotyping of Brucella melitensis and Brucella abortus isolates from different animal species and humans and identification of Brucella suis vaccine strain S2 from cattle in China. PLoS One 8:e76332. doi: 10.1371/journal.pone.0076332
Jones, L. M., Merz, G., and Wilson, J. (1968). Phage typing reactions on Brucella species. Appl. Microbiol. 16, 1179–1190.
Kang, Y. X., Li, X. M., Piao, D. R., Tian, G. Z., Jiang, H., Jia, E. H., et al. (2015). Typing discrepancy between phenotypic and molecular characterization revealing an emerging biovar 9 variant of smooth phage-resistant B. abortus Strain 8416 in China. Front. Microbiol. 6:1375. doi: 10.3389/fmicb.2015.01375
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12.
Lacerda, T. L., Cardoso, P. G., Augusto De Almeida, L., Camargo, I. L., Afonso, D. A., Trant, C. C., et al. (2010). Inactivation of formyltransferase (wbkC) gene generates a Brucella abortus rough strain that is attenuated in macrophages and in mice. Vaccine 28, 5627–5634. doi: 10.1016/j.vaccine.2010.06.023
Le Fleche, P., Jacques, I., Grayon, M., Al Dahouk, S., Bouchon, P., Denoeud, F., et al. (2006). Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol. 6:9. doi: 10.1186/1471-2180-6-9
Luo, H., Zhang, C. T., and Gao, F. (2014). Ori-Finder 2, an integrated tool to predict replication origins in the archaeal genomes. Front. Microbiol. 5:482. doi: 10.3389/fmicb.2014.00482
Moreno, E., Cloeckaert, A., and Moriyon, I. (2002). Brucella evolution and taxonomy. Vet. Microbiol. 90, 209–227. doi: 10.1016/s0378-1135(02)00210-9
Morris, J. A., Corbel, M. J., and Phillip, J. I. (1973). Characterization of three phages lytic for Brucella species. J. Gen. Virol. 20, 63–73. doi: 10.1099/0022-1317-20-1-63
Pappas, G., Papadimitriou, P., Akritidis, N., Christou, L., and Tsianos, E. V. (2006). The new global map of human brucellosis. Lancet Infect. Dis. 6, 91–99. doi: 10.1016/s1473-3099(06)70382-6
Petersen, E., Chaudhuri, P., Gourley, C., Harms, J., and Splitter, G. (2011). Brucella melitensis cyclic di-GMP phosphodiesterase BpdA controls expression of flagellar genes. J. Bacteriol. 193, 5683–5691. doi: 10.1128/JB.00428-11
Ricker, N., Qian, H., and Fulthorpe, R. R. (2012). The limitations of draft assemblies for understanding prokaryotic adaptation and evolution. Genomics 100, 167–175. doi: 10.1016/j.ygeno.2012.06.009
Rigby, C. E., Cerqueira-Campos, M. L., Kelly, H. A., and Surujballi, O. P. (1989). Properties and partial genetic characterization of Nepean phage and other lytic phages of Brucella species. Can. J. Vet. Res. 53, 319–325.
Sanogo, M., Abatih, E., Thys, E., Fretin, D., Berkvens, D., and Saegerman, C. (2013). Importance of identification and typing of brucellae from west african cattle: a review. Vet. Microbiol. 164, 202–211. doi: 10.1016/j.vetmic.2013.02.009
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689.
Scholz, H. C., Al Dahouk, S., Tomaso, H., Neubauer, H., Witte, A., Schloter, M., et al. (2008a). Genetic diversity and phylogenetic relationships of bacteria belonging to the ochrobactrum-brucella group by recA and 16S rRNA gene-based comparative sequence analysis. Syst. Appl. Microbiol. 31, 1–16. doi: 10.1016/j.syapm.2007.10.004
Scholz, H. C., Pfeffer, M., Witte, A., Neubauer, H., Al Dahouk, S., Wernery, U., et al. (2008b). Specific detection and differentiation of Ochrobactrum anthropi, Ochrobactrum intermedium and Brucella spp. by a multi-primer PCR that targets the recA gene. J. Med. Microbiol. 57, 64–71. doi: 10.1099/jmm.0.47507-0
Sieira, R., Comerci, D. J., Sanchez, D. O., and Ugalde, R. A. (2000). A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence and intracellular multiplication. J. Bacteriol. 182, 4849–4855. doi: 10.1128/jb.182.17.4849-4855.2000
Tang, B., Wang, Q., Yang, M., Xie, F., Zhu, Y., Zhuo, Y., et al. (2013). ContigScape: a cytoscape plugin facilitating microbial genome gap closing. BMC Genomics 14:289. doi: 10.1186/1471-2164-14-289
Tsolis, R. M., Seshadri, R., Santos, R. L., Sangari, F. J., Lobo, J. M., De Jong, M. F., et al. (2009). Genome degradation in Brucella ovis corresponds with narrowing of its host range and tissue tropism. PLoS One 4:e5519. doi: 10.1371/journal.pone.0005519
Valdezate, S., Navarro, A., Rubio, V., Garin-Bastuji, B., Albert, D., Hernandez, P., et al. (2009). Emergence of a clonal lineage of Brucella abortus biovar 3 in clinical cases in Spain. J. Clin. Microbiol. 47, 2687–2688. doi: 10.1128/jcm.00756-09
Van Belkum, A. (2007). Tracing isolates of bacterial species by multilocus variable number of tandem repeat analysis (MLVA). FEMS Immunol. Med. Microbiol. 49, 22–27. doi: 10.1111/j.1574-695x.2006.00173.x
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wattam, A. R., Davis, J. J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., et al. (2017). Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45, D535–D542. doi: 10.1093/nar/gkw1017
Whatmore, A. M., Koylass, M. S., Muchowski, J., Edwards-Smallbone, J., Gopaul, K. K., and Perrett, L. L. (2016). Extended multilocus sequence analysis to describe the global population structure of the genus brucella: phylogeography and relationship to biovars. Front. Microbiol. 7:2049. doi: 10.3389/fmicb.2016.02049
Whatmore, A. M., Perrett, L. L., and Macmillan, A. P. (2007). Characterisation of the genetic diversity of Brucella by multilocus sequencing. BMC Microbiol. 7:34. doi: 10.1186/1471-2180-7-34
Zhang, X., Goodsell, J., and Norgren, R. B. (2012). Limitations of the rhesus macaque draft genome assembly and annotation. BMC Genomics 13:206. doi: 10.1186/1471-2164-13-206
Zhao, Y., Wu, J., Yang, J., Sun, S., Xiao, J., and Yu, J. (2012). PGAP: pan-genomes analysis pipeline. Bioinformatics 28, 416–418. doi: 10.1093/bioinformatics/btr655
Zygmunt, M. S., Blasco, J. M., Letesson, J. J., Cloeckaert, A., and Moriyon, I. (2009). DNA polymorphism analysis of Brucella lipopolysaccharide genes reveals marked differences in O-polysaccharide biosynthetic genes between smooth and rough Brucella species and novel species-specific markers. BMC Microbiol. 9:92. doi: 10.1186/1471-2180-9-92
Keywords: Brucella abortus, phage resistance, comparative genomics, genome typing, phylogenetic analysis
Citation: Li X-m, Kang Y-x, Lin L, Jia E-H, Piao D-R, Jiang H, Zhang C-C, He J, Chang Y-F, Guo X-K and Zhu Y (2019) Genomic Characterization Provides New Insights for Detailed Phage- Resistant Mechanism for Brucella abortus. Front. Microbiol. 10:917. doi: 10.3389/fmicb.2019.00917
Received: 03 December 2018; Accepted: 11 April 2019;
Published: 03 May 2019.
Edited by:
Michel Stanislas Zygmunt, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Menachem Banai, Kimron Veterinary Institute, IsraelCopyright © 2019 Li, Kang, Lin, Jia, Piao, Jiang, Zhang, He, Chang, Guo and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yao-xia Kang, a3l4YWFAMTI2LmNvbQ== Yung-Fu Chang, eWM0MkBjb3JuZWxsLmVkdQ== Xiao-Kui Guo, bWljcm9iaW9sb2d5QHNqdHUuZWR1LmNu YongZhang Zhu, eXpoemh1QGhvdG1haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.