 Yu Zheng1
Yu Zheng1 Ayana Saitou2Chiung-Mei Wang1
Ayana Saitou2Chiung-Mei Wang1 Atsushi Toyoda3
Atsushi Toyoda3 Yohei Minakuchi3Yuji Sekiguchi4Kenji Ueda5
Yohei Minakuchi3Yuji Sekiguchi4Kenji Ueda5 Hideaki Takano5Yasuteru Sakai1Keietsu Abe1
Hideaki Takano5Yasuteru Sakai1Keietsu Abe1 Akira Yokota1
Akira Yokota1 Shuhei Yabe1,6*
Shuhei Yabe1,6*- 1Graduate School of Agricultural Science, Tohoku University, Sendai, Japan
- 2Faculty of Agriculture, Tohoku University, Sendai, Japan
- 3Comparative Genomics Laboratory, National Institute of Genetics, Mishima, Japan
- 4Biomedical Research Institute, National Institute of Advanced Industrial Science and Technology, Tsukuba, Japan
- 5Life Science Research Center, College of Bioresource Sciences, Nihon University, Fujisawa, Japan
- 6Hazaka Plant Research Center, Kennan Eisei Kogyo Co., Ltd., Miyagi, Japan
The prevalence of antibiotic resistance and the decrease in novel antibiotic discovery in recent years necessitates the identification of potentially novel microbial resources to produce natural products. Ktedonobacteria, a class of deeply branched bacterial lineage in the ancient phylum Chloroflexi, are ubiquitous in terrestrial environments and characterized by their large genome size and complex life cycle. These characteristics indicate Ktedonobacteria as a potential active producer of bioactive compounds. In this study, we observed the existence of a putative “megaplasmid,” multiple copies of ribosomal RNA operons, and high ratio of hypothetical proteins with unknown functions in the class Ktedonobacteria. Furthermore, a total of 104 antiSMASH-predicted putative biosynthetic gene clusters (BGCs) for secondary metabolites with high novelty and diversity were identified in nine Ktedonobacteria genomes. Our investigation of domain composition and organization of the non-ribosomal peptide synthetase and polyketide synthase BGCs further supports the concept that class Ktedonobacteria may produce compounds structurally different from known natural products. Furthermore, screening of bioactive compounds from representative Ktedonobacteria strains resulted in the identification of broad antimicrobial activities against both Gram-positive and Gram-negative tested bacterial strains. Based on these findings, we propose the ancient, ubiquitous, and spore-forming Ktedonobacteria as a versatile and promising microbial resource for natural product discovery.
Introduction
The discovery and clinical application of antimicrobial drugs have greatly improved our well-being with regard to microbial infection over the past several decades (Demain and Sanchez, 2009). However, misuse and overuse of antibiotics has led to the accelerating development of antibiotic resistance in pathogens, which has become a major threat to modern health care and the natural environment (Pal et al., 2016; Jiang et al., 2017). Traditionally, Actinobacteria, particularly the genus Streptomyces, has served as the major producer of antimicrobial natural products, contributing 62% of the microbe-derived antibiotics from the 1950s to 1970s (Bérdy, 2012). In contrast, the ratio has decreased to 25% in the 2000s to 2010s, with the silencing of gene clusters encoding biosynthetic enzymes required for natural products under laboratory conditions further hampering the discovery of novel antibiotics via the traditional bacterial fermentation method (Rutledge and Challis, 2015). Given that, isolation and screening of rare actinomycetes and marine-derived actinomycetes have become popular. Furthermore, the application of in silico biosynthetic predictions from genome mining data and novel developed heterologous expression technologies confirm the phylum Actinobacteria as a continuing source of novel antibiotics (Bérdy, 2012; Genilloud, 2017). Nevertheless, it is of high risk to focus the discovery of novel natural products on only a single phylum as this source could become exhausted. Thus, the exploration of new microbial resources comparable to actinomycetes is extremely important.
Numerous bioactive natural products are derived from bacterial secondary metabolites, with the enzymes required for biosynthesis of these natural products being usually encoded on certain bacterial genome loci termed biosynthetic gene clusters (BGCs). Traditionally, BGCs include non-ribosomal peptide synthase (NRPS), polyketide synthase (PKS), and ribosomally synthesized and post-translationally modified peptide (RiPP) family clusters. The BGCs of NRPS and PKS encode modular enzymes that comprise functional synthesis blocks known as modules. Therefore, NRPSs and PKSs constitute a large class of structurally diverse and biomedically useful bacterial secondary metabolites and have accordingly gained considerable attention in recent years (Doroghazi and Metcalf, 2013; Bitok et al., 2017; Vila-Farres et al., 2017). Typical NRPSs contain three essential domains: an AMP-binding (A) domain responsible for the selection and activation of monomers, a peptidyl carrier protein (PCP) domain, also termed the thiolation (T) domain, required for attachment and transfer of the adenylated monomer, and a condensation (C) domain for catalyzing amide bond formation and chain elongation (Challis and Naismith, 2004). PKS pathways share some similarities with fatty acid synthase (Dutta et al., 2014) and can be further divided into three types: type I PKSs that comprise large successive modular enzymes containing multi-functional domains; type II PKSs constituting single or bifunctional enzymes; and type III PKSs that utilize co-enzyme A-tethered substrates rather than the acyl carrier protein (ACP) domain to attach and transfer monomers such as type I and type II PKSs (Shen, 2003). Similar to NRPSs, a typical type I PKS also comprises three essential domains: an acyltransferase (AT) domain, which transfers a malonyl group from malonyl-CoA onto the 40-phosphopantetheine prosthetic group of an ACP domain, and a ketosynthase (KS) domain responsible for elongation of the polyketide chain (Masschelein et al., 2017). Moreover, similarities shared by NRPSs and PKSs in their biosynthesis pathway lead to hybrid non-ribosomal peptide-polyketides, which further enlarge the diversity of bacterial natural products (Miller et al., 2002; Liu et al., 2004; Shou et al., 2016). RiPPs also represent another type of antimicrobial peptide and are being considered as promising alternative antimicrobial agents owing to their widespread bactericidal sensitivity (Lázár et al., 2018). RiPPs are divided into different subfamilies according to their biosynthetic machinery and structural features, such as lantipeptides, lasso peptides, thiopeptides, and bacteriocins (Ortega and van der Donk, 2016). However, despite the differences in classification, almost all RiPPs contain an N-terminal leader peptide and a C-terminal core peptide (Ortega and van der Donk, 2016). Additionally, unlike NRPSs and PKSs, RiPPs always follow a simple biosynthesis pathway wherein the leader peptide guides the post-translational modification of the precursor peptide and is removed by one or more peptidases in the final step of core peptide maturation (Letzel et al., 2014; Ortega and van der Donk, 2016).
The class Ktedonobacteria was first reported in 2006 and currently contains two orders, three families, four genera, and seven formally proposed species including both mesophilic (Cavaletti et al., 2006; Yabe et al., 2017b) and thermophilic strains (Yabe et al., 2010, 2011). Owing to the morphological similarities, Ktedonobacter racemifer SOSP1-21T, the first proposed Ktedonobacteria species, was initially mistakenly identified as an actinomycete (Cavaletti et al., 2006). However, phylogenetic analysis based on 16S rRNA gene sequences placed the class Ktedonobacteria in the phylum Chloroflexi (Cavaletti et al., 2006; Yabe et al., 2010). Similar to actinomycetes, isolates and environmental DNA belonging to this class were detected to be ubiquitous in various terrestrial environments from common soil (Cavaletti et al., 2006; Yabe et al., 2017b), to extreme environments, such as an acid vapor-formed spring (Jiang et al., 2016), dark oligotrophic volcanic ice cave ecosystems of Mt. Erebus in Antarctica (Tebo et al., 2015), naturally occurring CO2 gas vents (de Miera et al., 2014b), recently deglaciated high-altitude soils of the Himalaya (Stres et al., 2013), and a lava cave in a volcanic trench (Northup et al., 2011). In addition, comparable to fungi, most Streptomyces species inhabit the soil environment and live as saprophytic bacteria (Flärdh and Buttner, 2009). In contrast, unlike actinomycetes, the class Ktedonobacteria appears to preferentially predominate in oligotrophic and extreme environments, suggesting these bacteria may possess different metabolic pathways from actinomycetes.
Notably, as shown in Figure 1, some isolated Ktedonobacteria strains exhibit complex morphologies. The genus Thermosporothrix forms substrate mycelia and subsequently produces grape-like smooth exospores by budding on the aerial mycelia (Yabe et al., 2010), whereas the surfaces of exospores formed by the genus Thermogemmatispora are wrinkled (Yabe et al., 2011). In contrast, strain K. racemifer SOSP1-21T forms spherical spores with a dried plum shape on aerial mycelia (Cavaletti et al., 2006), whereas Dictyobacter aurantiacus S27T produces globose or subglobose terminal sporangia from the vegetative mycelia via short stalk cells (Yabe et al., 2019). Spore formation represents one of the numerous methods developed by bacteria to survive in conditions of nutrient depletion or harsh environment (Errington, 2003; Flärdh and Buttner, 2009). It has been suggested that bacteria with complex morphological differentiation may possess an active secondary metabolism (Flärdh and Buttner, 2009; Manivasagan et al., 2014). Previously, metabolites of new acyloins and thiazoles have been identified in the fermentation broth of Thermosporothrix hazakensis SK20-1T (Park et al., 2014, 2015), whereas the extremophilic Thermogemmatispora strain T81 produced a four (methyl) lanthionine-bridged new lanthipeptide that showed antimicrobial activity against the human pathogen Staphylococcus aureus (Xu et al., 2018). Furthermore, the released genome draft data revealed that the genome sizes of strain Thermogemmatispora onikobensis ONI-1T (Komaki et al., 2016) and Thermogemmatispora carboxidivorans PM5T were 5.56 and 5.61 Mb, respectively, whereas strain K. racemifer SOSP1-21T supported a large genome of 13.66 Mb (Chang et al., 2011), which is the second largest bacterial genome identified to date following that of Sorangium cellulosum So0157-2 (14.78 Mb) (Han et al., 2013).
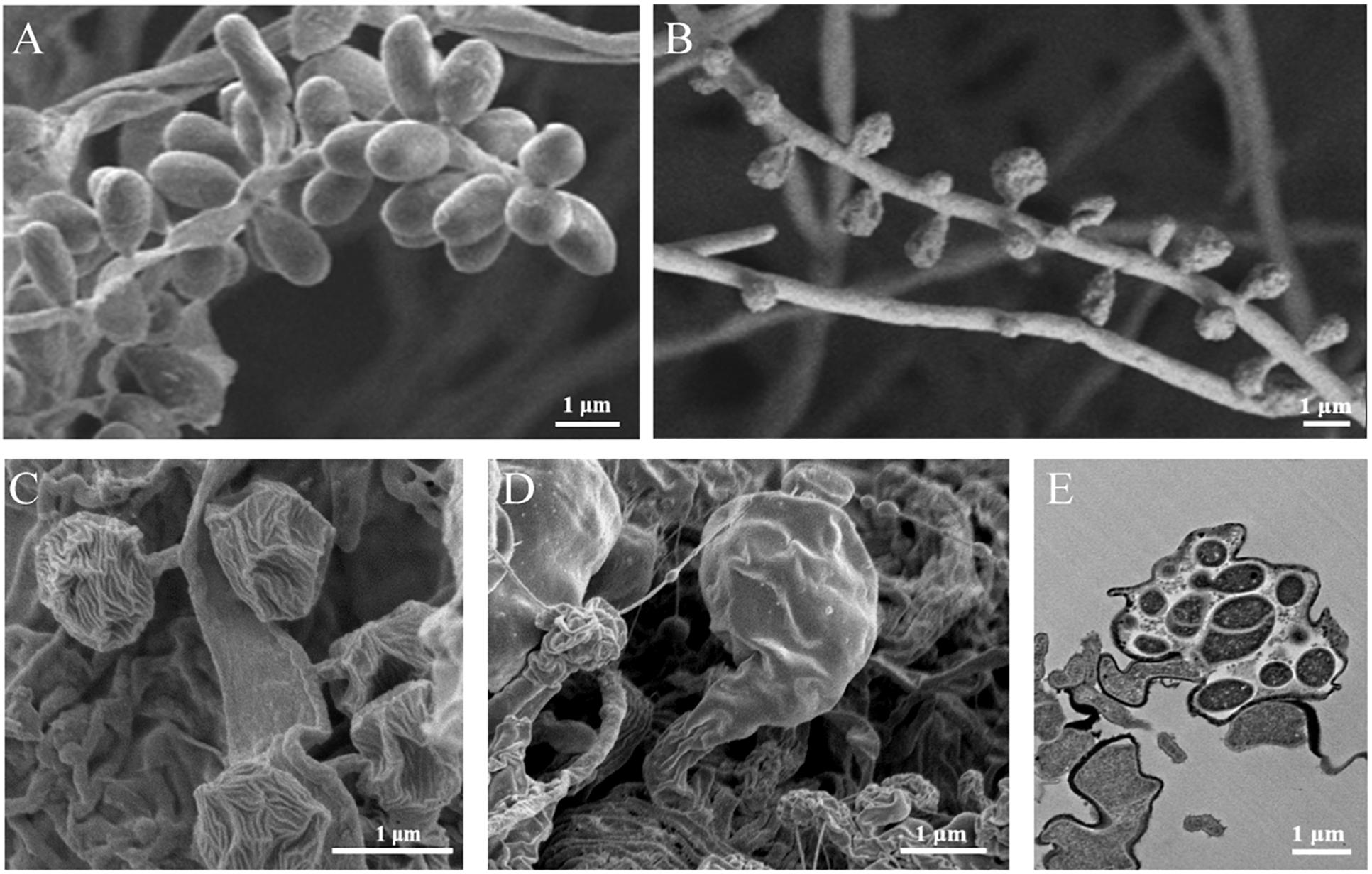
Figure 1. Morphologies of the isolated Ktedonobacteria strains. Scanning electron micrographs of strain Thermosporothrix hazakensis SK20-1T (A), Thermogemmatispora onikobensis ONI-1T (B), Ktedonobacter racemifer SOSP1-21T (C), and Dictyobacter aurantiacus S27T (D), indicating the exospores (A–C) or sporangia (D,E) formed by these strains. (E) Transmission electron micrographs of the thin-sectioned sporangium formed by strain Dictyobacter aurantiacus S27T. Bars: 1 μm.
Together, these described characteristics encouraged us to consider the class Ktedonobacteria as a Gram-positive, novel microbial resource for bioactive natural compounds discovery. However, the phylogenetic position and genome features of this class remained unclear. Moreover, although the large genome size of the Ktedonobacteria species suggest that they might encode multiple secondary metabolism gene clusters (Omura et al., 2001; Li and Walsh, 2010), few descriptions of these gene clusters or their novel natural product biosynthetic potential have been published. In this study, we therefore performed genome sequencing for another six Ktedonobacteria strains. The total nine available Ktedonobacteria genomes were then subjected to comprehensive phylogenetic, genomic, and secondary metabolic analysis, revealing the unique genome features and diversity of the predicted BGCs. Antimicrobial activity performance of the nine studied Ktedonobacteria strains against a variety of bacterial strains was also assessed.
Materials and Methods
Strain Cultural Conditions
Origins of the strains utilized in this study are described in Section “Strain Information and Whole Genome Sequencing.” Ts. hazakensis SK20-1T and Ts. hazakensis COM3 were maintained in pure culture at 50°C on ISP3 agar plates (Shirling and Gottlieb, 1966), whereas the other seven strains were maintained on 1/10 R2A gellan gum (Kanto Chemical Co., Inc., Tokyo, Japan) plates (Yabe et al., 2017b). The optimal growth temperature for K. racemifer SOSP1-21T, D. aurantiacus S27T, Ktedonobacterales bacterium Uno3, and Ktedonobacterales bacterium Uno16 was 30°C, whereas Tg. onikobensis ONI-1T, Thermogemmatispora sp. A3-2, and Tg. carboxidivorans PM5T were cultured at 60°C. For genomic DNA extraction, strains Ts. hazakensis SK20-1T and Ts. hazakensis COM3 were cultured at 50°C in ISP1 liquid medium (Shirling and Gottlieb, 1966). Strain Thermogemmatispora sp. A3-2 was cultured at 65°C, whereas strains D. aurantiacus S27T, Ktedonobacterales bacterium Uno3, and Ktedonobacterales bacterium Uno16 were cultured at 30°C in 1/10 R2A liquid medium (Yabe et al., 2017b).
The candidate testing microorganisms used for antimicrobial assays included Bacillus subtilis NBRC 3134, Pseudomonas aeruginosa NBRC 13275, Escherichia coli NBRC 3972, Salmonella enterica NBRC 100797, Micrococcus luteus NBRC 13867, Geobacillus stearothermophilus NBRC 13737, Staphylococcus aureus NBRC 13276, S. aureus NTCT8325 (MSSA) and S. aureus Mu50 (VISA) (Kuroda et al., 2001) were provided by Juntendo University. All strains were maintained in nutrient agar plates.
Genomic DNA Extraction and Whole Genome Sequencing
To extract genomic DNA from the Ktedonobacteria strains, a modified method following the protocol of the Puregene Yeast/Bact. Kit B (Qiagen, Hilden, Germany) was used. The bacterial cells were collected by centrifugation of the culture broth and an appropriate volume of purified achromopeptidase (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan) and lysozyme (MP Biomedicals, LLC, Illkirch, France) were added to the tubes to break down the cell walls. Also, 10 μL of 20 mg/mL proteinase K was added and incubated at 55°C for 1 h, then the tubes were incubated at 37°C overnight gently in a shaker to digest the residual proteins. Finally, the dried DNA pellets were dissolved in 100 μL DNA hydration solution for genome sequencing or stored at -20°C for later use.
Genome sequencing of strain Ts. hazakensis SK20-1T was conducted by TaKaRa Bio Inc., Japan on a PacBio RS II platform with long sequencing and assembled de novo using SMRT Analysis v2.2.01. The genome sequences of strain Ts. hazakensis COM3 and Thermogemmatispora sp. A3-2 were obtained from paired-end sequencing with HiSeq 2500 and long sequencing with PacBio RS II/Sequel. The genome sequencing was conducted by the National Institute of Genetics in Mishima, Shizuoka, Japan. The reads were assembled de novo using HGAP3/42, and gaps between contigs were closed with their fosmid clones. Genome data of strains K. racemifer SOSP1-21T, D. aurantiacus S27T, Ktedonobacterales bacterium Uno3, Ktedonobacterales bacterium Uno16, Tg. onikobensis ONI-1T, and Tg. carboxidivorans PM5T were recovered from NCBI and DDBJ databases for analysis. Additionally, the qualities (genome completeness and contamination) of the nine studied Ktedonobacteria genomes were estimated by CheckM, a set of tools using collocated sets of genes that are ubiquitous and single-copy within a phylogenetic lineage (Parks et al., 2015). As the class Ktedonobacteria is a relatively novel taxonomy of bacteria, both the lineage-specific workflow, taxonomic-specific workflow, and 83 single copy custom marker genes (Soo et al., 2014) were used in the CheckM estimation.
Phylogenetic Analysis Based on 16S rRNA Genes and Single-Copy Marker Genes
For phylogenetic analysis, Chloroflexi-related 16S rRNA gene sequences were obtained from the All-species Living Tree Project (LTP, release s132) (Yarza et al., 2010). The 16S rRNA gene sequence of T. hazakensis COM3 was extracted from its genome sequence (determined in this study) using CommunityM3 and aligned via the ARB software package (Ludwig et al., 2004) using the LTP database as a reference; the alignment was manually corrected using the ARB EDIT tool of the ARB package. Sequences (>1,300 nt) representing various family-level taxa in the phylum were selected in ARB and their alignments exported with lane mask filtering. Neighbor joining trees based on LogDet distance were computed using PAUP∗4 (Swofford, 2003) with 100 bootstrap resamplings. Maximum likelihood trees were built using RAxML v8.2 (GTR and Gamma models + I) with rapid 100 times bootstrapping (Stamatakis, 2006). The trees were visualized and inspected in ARB.
Phylogenetic relationships among members of the phylum Chloroflexi, including members of the class Ktedonobacteria, were assessed using the genome sequences determined in the present study along with other finished and draft genomes representing the major families in the phylum as described in Sun et al. (2016). Briefly, 38 (Darling et al., 2014) or 83 (Soo et al., 2014) single-copy marker gene products were extracted using hidden Markov model searches as described previously (Sekiguchi et al., 2015). Tree topologies were tested for robustness using the maximum likelihood methods from FastTree v2 (with default parameters, JTT model, CAT approximation) (Price et al., 2010) and RAxML v8.2 (JTT and Gamma models with rapid 100 times bootstrapping).
Genome Annotation and Comparisons
The nine Ktedonobacteria genomes and related genomes were primarily annotated using DFAST (DDBJ Fast Annotation and Submission Tool) (Tanizawa et al., 2018) to study their general features. The DFAST-annotated 16S rRNA gene sequences were extracted and blasted using NCBI Nucleotide-Nucleotide Blast4 program to compare their similarities. Meanwhile, the Kyoto Encyclopedia of Genes and Genomes (KEGG) PATHWAY database was used for functional characterization of the nine genomes (Moriya et al., 2007).
CGView Server (Grant and Stothard, 2008) and the Geneious prime 2019.0.4 software were used to generate the genome plot. To compare the genomic rearrangement among Ktedonobacteria species, the contigs were concatenated using the Geneious software. Then the concatenated genomic sequences were aligned with the ProgressiveMauve algorithm and visualized using the MAUVE plugin within Geneious suite (Darling et al., 2004). BPGA (Bacterial Pan Genome Analysis tool), an ultra-fast pan-genome analysis pipeline (Chaudhari et al., 2016), was used to compare genomes between different strains in the class Ktedonobacteria and with other classes.
Metabolites Analysis and Identification of Putative BGCs
The nine Ktedonobacteria genomes were submitted to antiSMASH version 4.2.0 (antibiotics and Secondary Metabolites Analysis Shell) (Blin et al., 2017) and the integrated ClusterFinder algorithm, an hidden Markov model based probabilistic algorithm to detect BGC-like regions in genomes of unknown types (Cimermancic et al., 2014), to identify both the characterized and unknown secondary metabolite biosynthesis gene clusters in the nine Ktedonobacteria strains. Additionally, both the KnownClusterBlast and ClusterBlast modules were selected to identify similar clusters in sequenced genomes by genome comparisons. Domain functions and genetic similarities with known BGCs in these gene clusters were further predicted and annotated using Protein-Protein BLAST and Pfam analyses (Finn et al., 2016). The AMP-binding domain amino acid substrate specificities were predicted with antiSMASH referencing three prediction algorithms, NRPSPredictor2 (Röttig et al., 2011), Stachelhaus code (Stachelhaus et al., 1999), and SANDPUMA ensemble (Chevrette et al., 2017). Only when two or all three algorithms provided consistent substrate specificity predictions was a consensus prediction called. Otherwise, the prediction results were assigned an “X” to represent an unknown amino acid residue, or all of the dubious amino acid residues were listed (Chu et al., 2016).
Phylogenetic Analysis of the PKS KS Domain, NRPS C Domain, and Lantipeptide Modification Genes
All of the PKS KS domains and NRPS C domains identified in putative BGCs from Ktedonobacteria genomes were extracted and submitted to NCBI Protein-Protein Blast (see text footnote 4). The top three reference sequences, excluding the Ktedonobacteria sequences, were selected and preserved in the FASTA file. Sequences of the Ktedonobacteria PKS KS domains, NRPS C domains, and the corresponding reference sequences were then submitted to Natural Product Domain Seeker (NaPDoS) (Ziemert et al., 2012) for functional classification. For phylogenetic tree construction of the KS and C domains, the sequences were trimmed into equal length by MEGA7.05, and the tree was constructed following the method described in Section “Phylogenetic Analysis Based on 16S rRNA Genes and Single-Copy Marker Genes” for 16S rRNA phylogenetic analysis. Additionally, the phylogenetic trees were displayed and annotated by iTOL v3, an online tool for the display, annotation, and management of phylogenetic trees (Letunic and Bork, 2016).
The lantipeptide modification genes and precursor peptides were identified via a comprehensive analysis of antiSMASH 4.2.0 and BAGEL 3.0, a web-based comprehensive mining suite to identify and characterize RiPPs in microbial genomes (van Heel et al., 2013). Reference sequences of modification genes were obtained from the NCBI database with the same method as for KS domains and C domains. The phylogenetic trees were also constructed, displayed, and annotated following the aforementioned methods. For the analysis of precursor peptides, the extracted sequences were aligned with MEGA7.0 and the figures indicating cleavage sites were visualized using WebLogo 3 (Crooks et al., 2004).
Antimicrobial Screening of the Class Ktedonobacteria
Strains Ts. hazakensis COM3 and Thermogemmatispora sp. A3-2 were selected as representatives of the genus Thermosporothrix and Thermogemmatispora, respectively. Strain Ts. hazakensis COM3 was cultured in basic medium (peptone 2 g/L, yeast extract 1 g/L, MgSO4 1 g/L, NaCl 1 g/L, and pH 7.0) at 50°C for 7 days, whereas strain Thermogemmatispora bacterium A3-2 was cultured in R2A medium at 60°C for 7 days. Strains K. racemifer SOSP1-21T, Dictyobacter aurantiacus S27T, Ktedonobacterales Uno3, and Ktedonobacterales Uno16 were cultured in Bennett’s medium (yeast extract 1 g/L, beef extract 1 g/L, N-Z amine type A 2 g/L, maltose 10 g/L, and pH 7.0) at 30°C for 14 days. Additionally, 2% Diaion® HP-20 polyaromatic adsorbent resin was added to each medium. Following 2 L culture, the cell pellets and HP-20 polyaromatic adsorbent resin were collected and extracted with the appropriate volume of acetone. Extractions from the above six strains were dried via evaporator and then redissolved in 5 mL of 80% methanol (MeOH) for antimicrobial activity screening versus a series of candidate testing microorganisms.
The antimicrobial activity screening of the class Ktedonobacteria was performed using the standard paper disc method. Paper discs (Toyobo Co., Ltd., Osaka, Japan; 6 mm) were saturated with 10 μL bacterial extracts from the above Ktedonobacteria strains and dried at room temperature. Then, the paper discs were placed on the surface of agar plates inoculated with each testing candidate microorganism and incubated for 24 h at the optimal temperature. Results of antimicrobial activity were determined by measuring diameters of zones of inhibition formed around each paper disc by the bacterial extracts. All antimicrobial tests were performed in triplicate.
Results
Strain Information and Whole Genome Sequencing
As summarized in Table 1, the nine described Ktedonobacteria isolates were all terrestrially derived, including five thermophilic strains and four mesophilic strains. Ts. hazakensis COM3 was isolated from the same compost with Ts. hazakensis SK20-1T (Yabe et al., 2010) and was identified as a homologous strain to Ts. hazakensis SK20-1T by 16S rRNA gene sequence. However, Ts. hazakensis COM3 was distinguished from Ts. hazakensis SK20-1T through the formation of red pigment in culture medium (unpublished), and thus was also studied for genome analysis in this study. Strain Thermogemmatispora sp. A3-2 was isolated from the same geothermal soils as Tg. onikobensis ONI-1T and was identified as a novel species belonging to the genus Thermogemmatispora. Thermogemmatispora sp. A3-2 is preserved in our laboratory and in the Biological Resource Center, NITE (NBRC) and China General Microbiological Culture Collection (CGMCC). Moreover, Thermogemmatispora sp. A3-2 grew at 40–78°C, the highest growth temperature in the class Ktedonobacteria identified to date. Ktedonobacterales bacterium Uno3 and Ktedonobacterales bacterium Uno16 were isolated from soil-like microbe masses termed “Tengu-no-mugimeshi” at a volcanic region located in Gunma, Japan (Okada, 1937). Optimal growth temperature of the two isolates was 30°C.
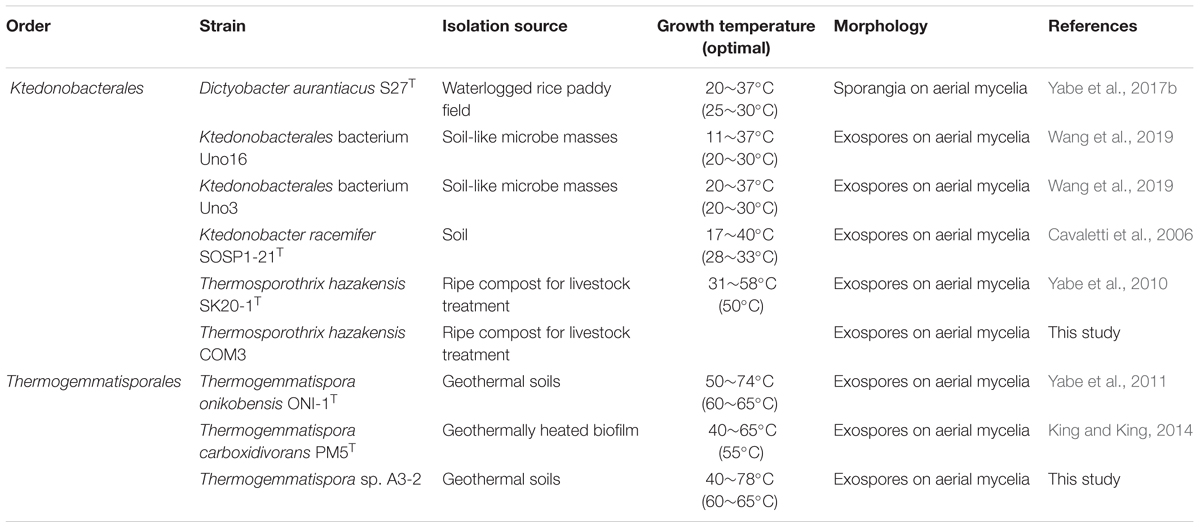
Table 1. Strain information of the nine studied Ktedonobacteria strains.
Prior to this study, the draft genome sequences of K. racemifer SOSP1-21T (Chang et al., 2011), Tg. onikobensis ONI-1T (Komaki et al., 2016), and Tg. carboxidivorans PM5T have been publically released. In addition, genome sequencing of D. aurantiacus S27T (GenBank accession: BIFQ01000001 and BIFQ01000002), Ktedonobacterales bacterium Uno3 (GenBank accession: BIFR01000001 and BIFR01000002), and Ktedonobacterales bacterium Uno16 (GenBank accession: BIFT01000001, BIFT01000002, BIFT01000003, and BIFT01000004) was performed by our group in another study (Wang et al., 2019) and the genome data were deposited in IMG or DDBJ/EMBL/GenBank databases. In the present study, we conducted genome sequencing for three strains including Ts. hazakensis SK20-1T (GenBank accession: BIFX01000001, BIFX01000002, and BIFX01000003), Ts. hazakensis COM3 (GenBank accession: AP019376), and Thermogemmatispora sp. A3-2 (GenBank accession: AP019377). Sequencing platforms, sequence coverage, assembly methods, and accession numbers of the nine Ktedonobacteria genomes are summarized in Supplementary Table S1.
As shown in Figure 2, draft genome of Ktedonobacterales bacterium Uno3 possesses two circular contigs (5.30 Mb and 2.40 Mb, respectively) whereas the complete genome of Thermogemmatispora sp. A3-2 comprises a single circular chromosome. Ts. hazakensis COM3 possesses one putative linear chromosome, whereas D. aurantiacus S27T contains two linear contigs (6.13 Mb and 2.75 Mb, respectively). Ktedonobacterales bacterium Uno16 comprises two linear contigs (5.58 Mb and 3.14 Mb) and two circular plasmids of 199,150 and 43,380 bp in size, respectively. Initially, the second circular contig of Ktedonobacterales bacterium Uno3 (2.40 Mb) and the second linear contigs of strains D. aurantiacus S27T (2.75 Mb) and Ktedonobacterales bacterium Uno16 (3.14 Mb) were thought to be part of the chromosomes because they were quite large in size compared with normal bacterial plasmids. However, the above “chromosomes” were determined to be incomplete due to the absence of most bacterial house-keeping genes in the subsequent CheckM assessment (Supplementary Table S2), suggesting that they may be “megaplasmids” (Thomas and Summers, 2008; Wu et al., 2009).
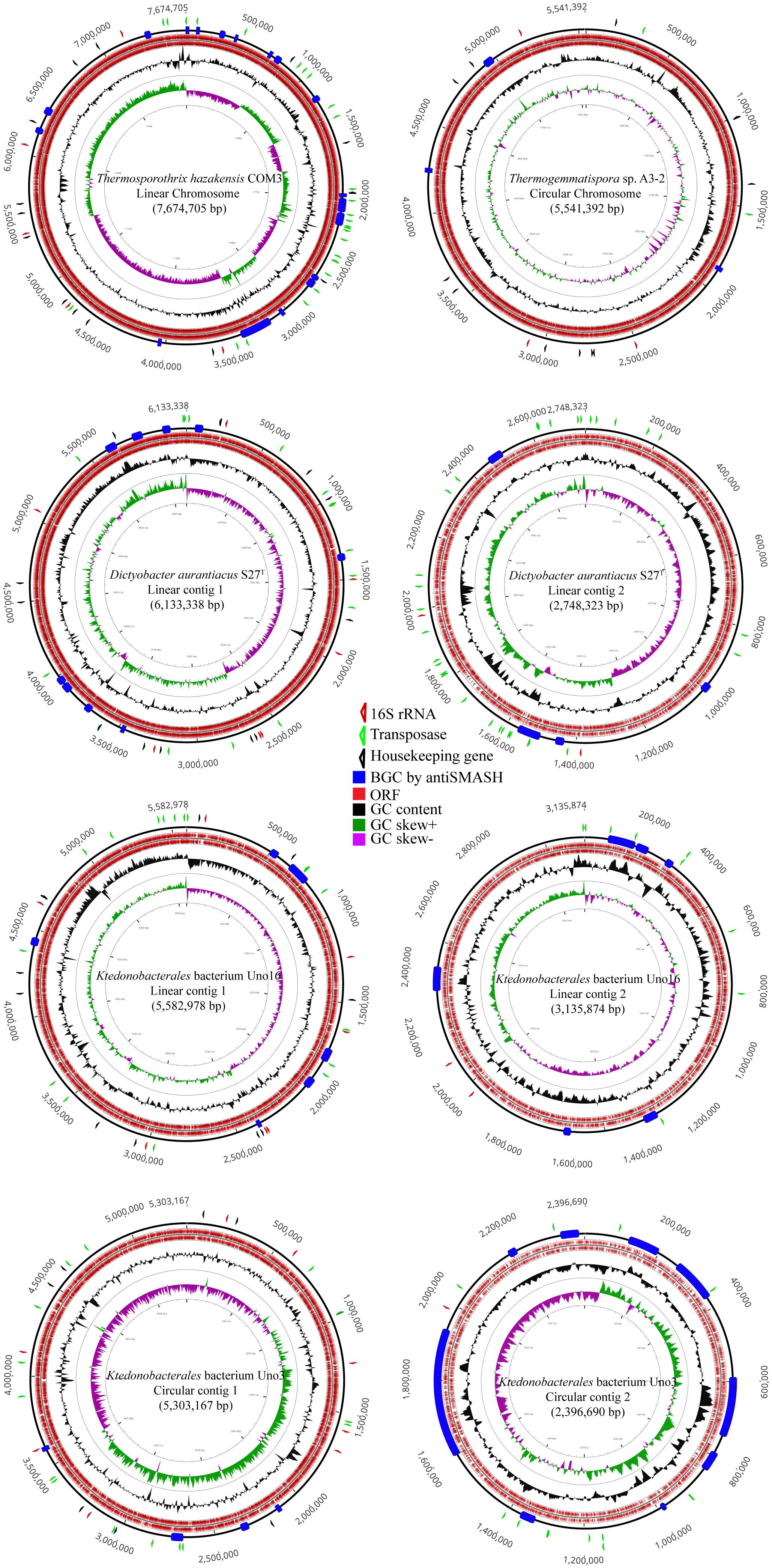
Figure 2. Genome plot of strains of Thermosporothrix hazakensis COM3, Thermogemmatispora sp. A3-2, Dictyobacter aurantiacus S27T, Ktedonobacterales bacterium Uno16, and Ktedonobacterales bacterium Uno3. The out layer triangle markers indicate the 16S rRNA gene in red, transfase in green, and part of the housekeeping genes (gmk, rpoD, recA, glyA, dnaB, gyrA, gyrB, secA, dnaK, ffh) in black. The inner layer colored boxes indicate the antiSMASH-identified BGCs in blue, ORFs in red, GC content in black, GC skew+ in green, and GC skew– in purple.
General Genome Features
As given in Table 2 and Supplementary Table S1, genome sizes of the nine Ktedonobacteria strains ranged from 5.54 to 13.66 Mb, which were larger than the other classes in the phylum Chloroflexi. The average genome size of the genus Thermosporothrix and Thermogemmatispora was 7.48 and 5.57 Mb, respectively. Excluding strain K. racemifer SOSP1-21T, the average genome size of the three mesophilic strains including strains D. aurantiacus S27T, Ktedonobacterales bacterium Uno3, and Ktedonobacterales bacterium Uno16 was 8.51 Mb. However, the nine Ktedonobacteria strains exhibited a much lower GC content (an average of 55.1%) than that of actinomycete strains, especially the genus Streptomycetes (Chater and Chandra, 2006). Additionally, the class Ktedonobacteria encoded multiple copies of ribosomal RNA operons (8∼28 copies per genome) and 16S rRNA genes (3∼9 copies per genome) on their genomes. The data of Tg. onikobensis ONI-1T was not counted due to the poor genome quality. In addition, a maximum of 1.49% 16S rRNA gene variation was observed in strain D. aurantiacus S27T (Supplementary Table S3). Moreover, 2364 (strain Tg. onikobensis ONI-1T) to 8227 (strain K. racemifer SOSP1-21T) of the detected coding sequences (CDSs) were termed as hypothetical proteins with unknown functions by DFAST annotation (Tanizawa et al., 2018), representing an average of 57.0% of the total CDSs in the nine Ktedonobacteria strains.
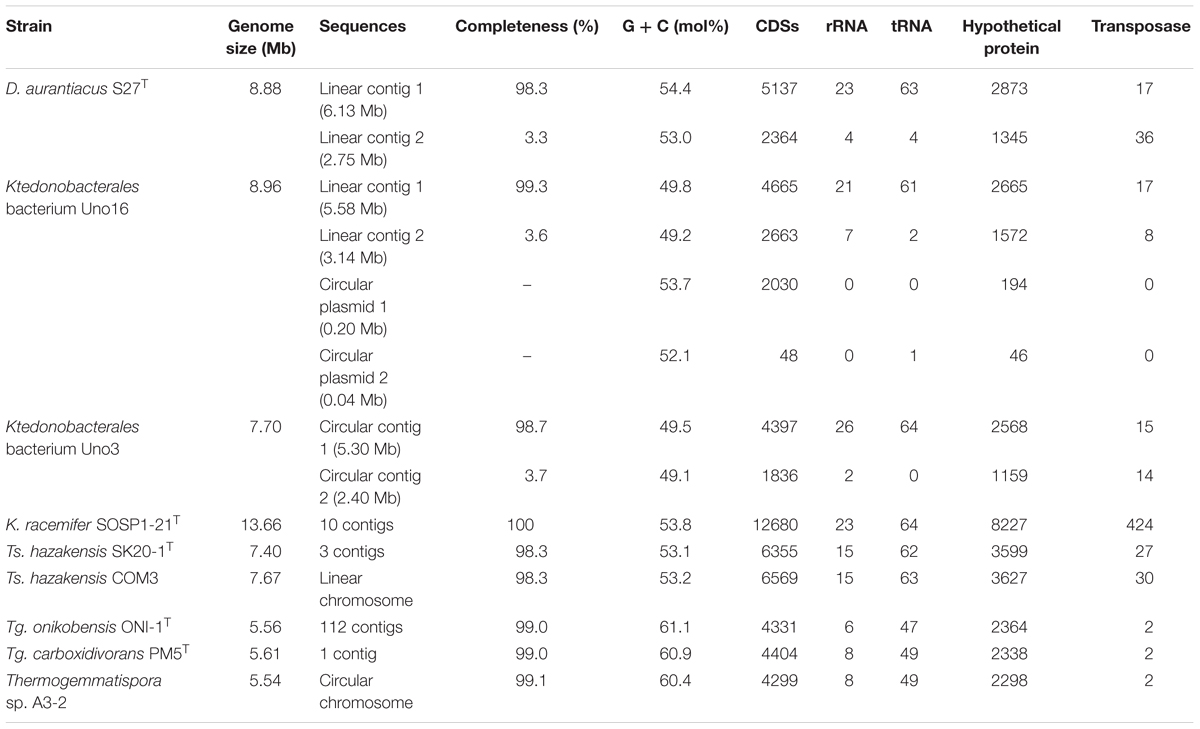
Table 2. General genome features of the nine studied Ktedonobacteria strains.
To investigate the absence/presence of genes responsible for autotrophic metabolites, the nine studied Ktedonobacteria genomes were submitted to the KEGG PATHWAY database for functional characterization. Additionally, the presence of a type I carbon monoxide dehydrogenases (cox) gene in the genome of Tg. carboxidivorans PM5T and Thermogemmatispora sp. T81 was also reported and functioned in the oxidation of carbon monoxide (CO) to carbon dioxide (CO2) (King and King, 2014; Islam et al., 2019). Herein, we aligned amino acids sequences of the cox gene with the annotated genes and their homologs in the nine studied genomes, using the NCBI Protein-Protein Blast program. As given in Supplementary Table S4A, all nine Ktedonobacteria strains possessed multiple copies of cox or their homologs in their genomes. Moreover, pyruvate synthase and α-ketoglutarate synthase, key enzymes in the reductive citric acid cycle (reductive TCA cycle) (Buchanan and Arnon, 1990), were present in the nine Ktedonobacteria strains. However, adenosine triphosphate (ATP) citrate lyase, a key enzyme responsible for the cleaving of citrate, was absent in the Ktedonobacteria genomes (Supplementary Table S4B). Additionally, comparisons between contig1 and contig2 of strains D. aurantiacus S27T and Ktedonobacterales bacterium Uno16 are also given in Table 3.
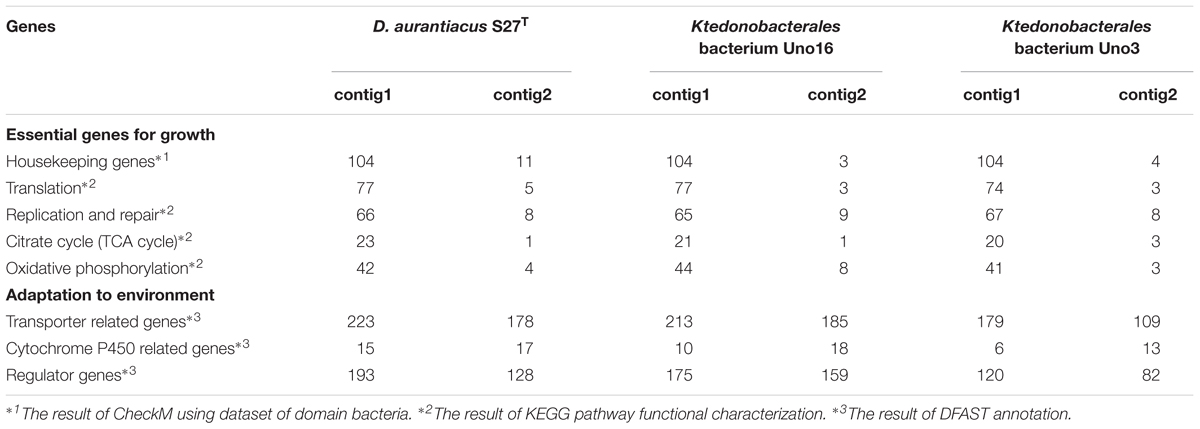
Table 3. Comparisons of contig1 and contig2 in strains D. aurantiacus S27T, Ktedonobacterales bacterium Uno16, and Ktedonobacterales bacterium Uno3.
Phylogenetic Analysis and Genome
Comparisons
Phylogenetic position of the class Ktedonobacteria was determined via a comprehensive analysis of 16S rRNA gene sequences and universally conserved protein sequences. Initially, a phylogenetic association between the class Ktedonobacteria and the phylum Chloroflexi was suspected owing to the low bootstrap values in the 16S rRNA gene sequence phylogenetic tree and the dissimilarities in morphological, physiological, and chemotaxonomic data between the two (Cavaletti et al., 2006). However, our phylogenetic analyses based on both 16S rRNA gene sequences (Figure 3A) and 38 and 83 single-copy marker genes (Figure 3B and Supplementary Figure S1) clearly inferred that the members of the class Ktedonobacteria belong to the phylum Chloroflexi. The genome-based phylogenies determined in this study added evidence supporting the idea that the members of the class Ktedonobacteria evolved from a common descendent of the phylum Chloroflexi, although they largely shared some important phenotypic traits with those of members of the phylum Actinobacteria. Notably, this is the first report of the formation of a monophyly between Ktedonobacteria and Dehalococcoidetes, another class in the phylum Chloroflexi and which is characterized with small genomes and obligate niche adaptation to reductive dehalogenation as the sole catabolic metabolism (Kaster et al., 2014).
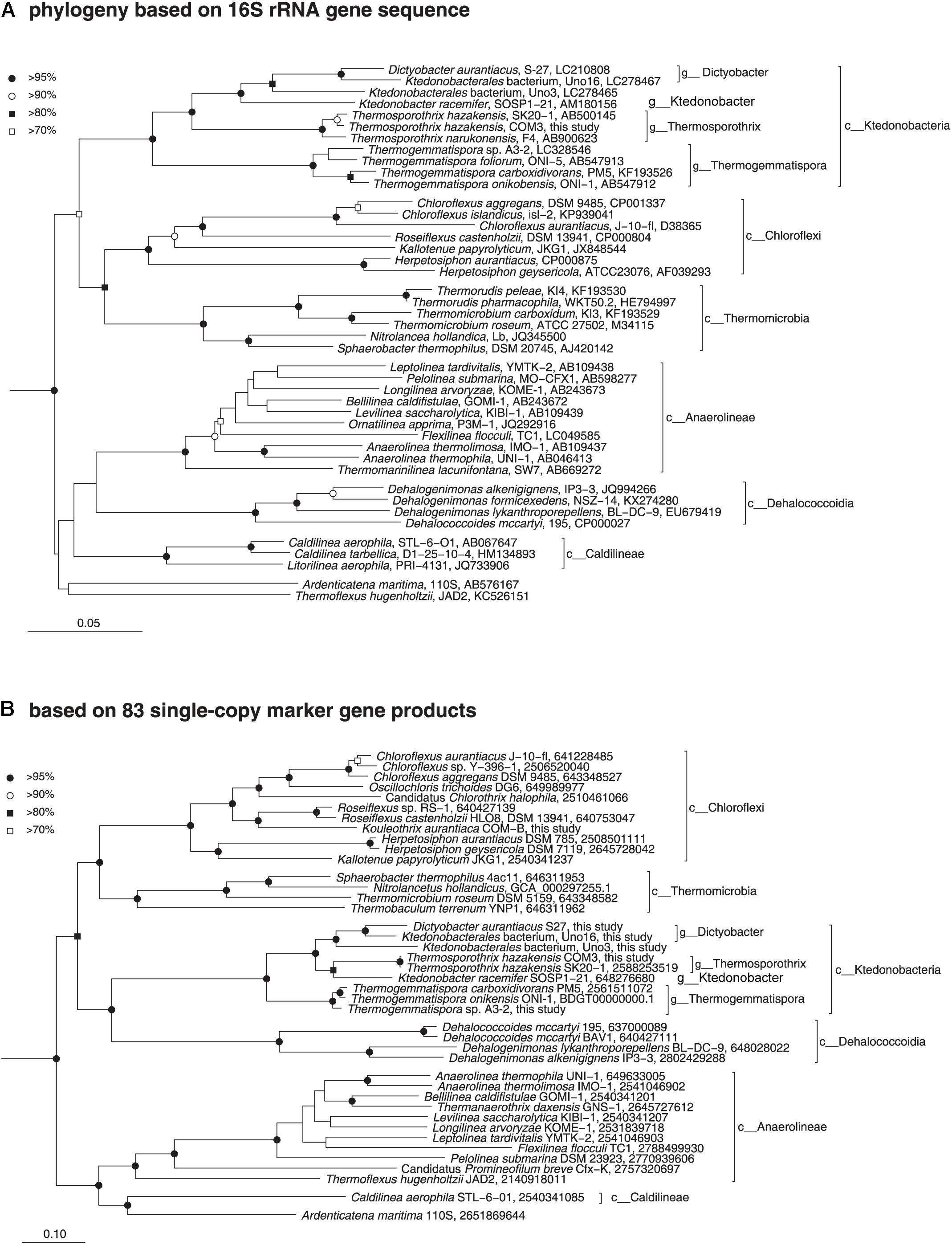
Figure 3. Phylogenetic positions of the members of the class Ktedonobacteria based on comparative analysis of 16S rRNA gene sequences (A) and conserved protein sequences (B). (A) Neighbor-joining phylogenetic tree of public data (accession numbers are shown at the end of the index of each note) and the 16S rRNA gene sequence of T. hazakensis COM3, which was extracted from the genome sequence obtained in this study. Sequences from the bacterial phylum Nitrospira were used to root the tree (not shown). Reproducible nodes are marked based on bootstrap values from neighbor-joining and maximum-likelihood inferences (closed circle, >95% for both inferences; open circle, >90%; closed square, >80%; open square, >70%). Nodes that lack symbols were not reproducible among trees. Bar: 5% estimated sequence divergence. (B) Maximum-likelihood phylogenetic inference of representative genomes of the class Chloroflexi. The tree was built using RAxML based on up to 83 universally conserved proteins. Reproducible associations based on bootstrap values (closed circle, >95% for both RAxML and FastTree inferences; open circle, >90%; closed square, >80%; open square, >70%) are indicated at interior nodes. Accession numbers of genomes (IMG or DDBJ/EMBL/GenBank databases) are shown at the end of the index of each taxon. Alignments of homologous proteins from genomes of members of the phylum Deinococci were used to root the tree (not shown). Bar: 10% estimated sequence divergence.
Meanwhile, an obvious formation of a monophyly between strain Ktedonobacterales bacterium Uno16 and D. aurantiacus S27T was observed in both 16S rRNA and genomic phylogenetic trees (Figure 3). The 16S rRNA genetic similarity and average nucleotide identity (ANI) between the two strains were 95.60% and 74.26%, respectively, suggesting strain Ktedonobacterales bacterium Uno16 could represent a novel species within the genus Dictyobacter. As for Ktedonobacterales bacterium Uno3, the most closely related strains were D. aurantiacus S27T and K. racemifer SOSP1-21T, at the 16S rRNA genetic similarities of 91.46% and 89.98%, respectively. The average nucleotide identities of Ktedonobacterales bacterium Uno3 with D. aurantiacus S27T and K. racemifer SOSP1-21T were 69.86% and 69.70%, respectively. This suggested that Ktedonobacterales bacterium Uno3 belongs to an unclassified novel genus within the order Ktedonobacterales. The full taxonomic characteristics of Uno3, Uno16, and Uno17 were conducted in another study (Wang et al., 2019).
The pan-genome concept was firstly coined by Tettelin et al. (2005) in to catalog the entire genomic genes of a given phylogenetic clade, including core genes (detected in all strains), accessory genes (detected in two or more strains), and unique genes (strain-specific genes), and was also used to estimate the genetic diversity of a studied group. In this study, we performed the pan-genome analysis to compare the nine strains within class Ktedonobacteria, and with the class Dehalococcoidetes, which formed a monophyly with the class Ktedonobacteria on the 38 and 83 single-copy marker genes based phylogenetic trees (Figure 3B and Supplementary Figure S1). As summarized in Supplementary Table S5A, 81.00∼83.73% of the total genes were shared between strains Ts. hazakensis SK20-1T and Ts. hazakensis COM3, while the numbers were 77.50∼77.80% in the genus Thermogemmatispora. By contrast, only 55.22∼54.64% of genes were shared between D. aurantiacus S27T and Ktedonobacterales bacterium Uno16. Moreover, the percentages of core genes changed to 57.45∼63.26% (Supplementary Table S5B) and 32.48∼36.59% (Supplementary Table S5C), when contig1 (chromosome) and contig2 (“megaplasmid”) were aligned separately. Additionally, we found that Ktedonobacterales bacterium Uno3 shared the highest core gene ratio (38.84%) with the genus Dictyobacter among the class Ktedonobacteria. As for K. racemifer SOSP1-21T, the highest core gene ratio was found to be with the genus Thermosporothrix, at 21.06% of its total genes. The close relationships between K. racemifer SOSP1-21T with the genus Thermosporothrix, and Ktedonobacterales bacterium Uno3 with the genus Dictyobacter were in accordance with the result of the above phylogenetic analyses based on 83 single-copy marker genes (Figure 3B). As for the class Ktedonobacteria, a total of 1181 core genes were shared among the nine strains, although they belonged to two orders and five genera. When the nine Ktedonobacteria strains were aligned with the class Dehalococcoidetes, we found the highest number of core genes (112 core genes) and highest core gene ratio (2.54∼2.59%) in the genus Thermogemmatispora. However, the entire core genes number (76 core genes) shared between the class Ktedonobacteria and the class Dehalococcoidetes was not large enough to overwhelm the number (69 core genes) shared with representative strains from other classes in the phylum Chloroflexi (Supplementary Table S5A).
To compare the genomic rearrangement between D. aurantiacus S27T and Ktedonobacterales bacterium Uno16, which were proposed to comprise putative “megaplasmids,” homologous regions in genomes were aligned between the two strains. As described above, strain Ktedonobacterales bacterium Uno16 was considered a novel species within the genus Dictyobacter by phylogenetic analyses based on both 16S rRNA gene sequences and universally conserved protein sequences. However, although genomic rearrangement in contig1 of strain Ktedonobacterales bacterium Uno16 was observed to be highly homologous to the contig1 of D. aurantiacus S27T, contig2 of the two strains were very dissimilar (Figure 4).
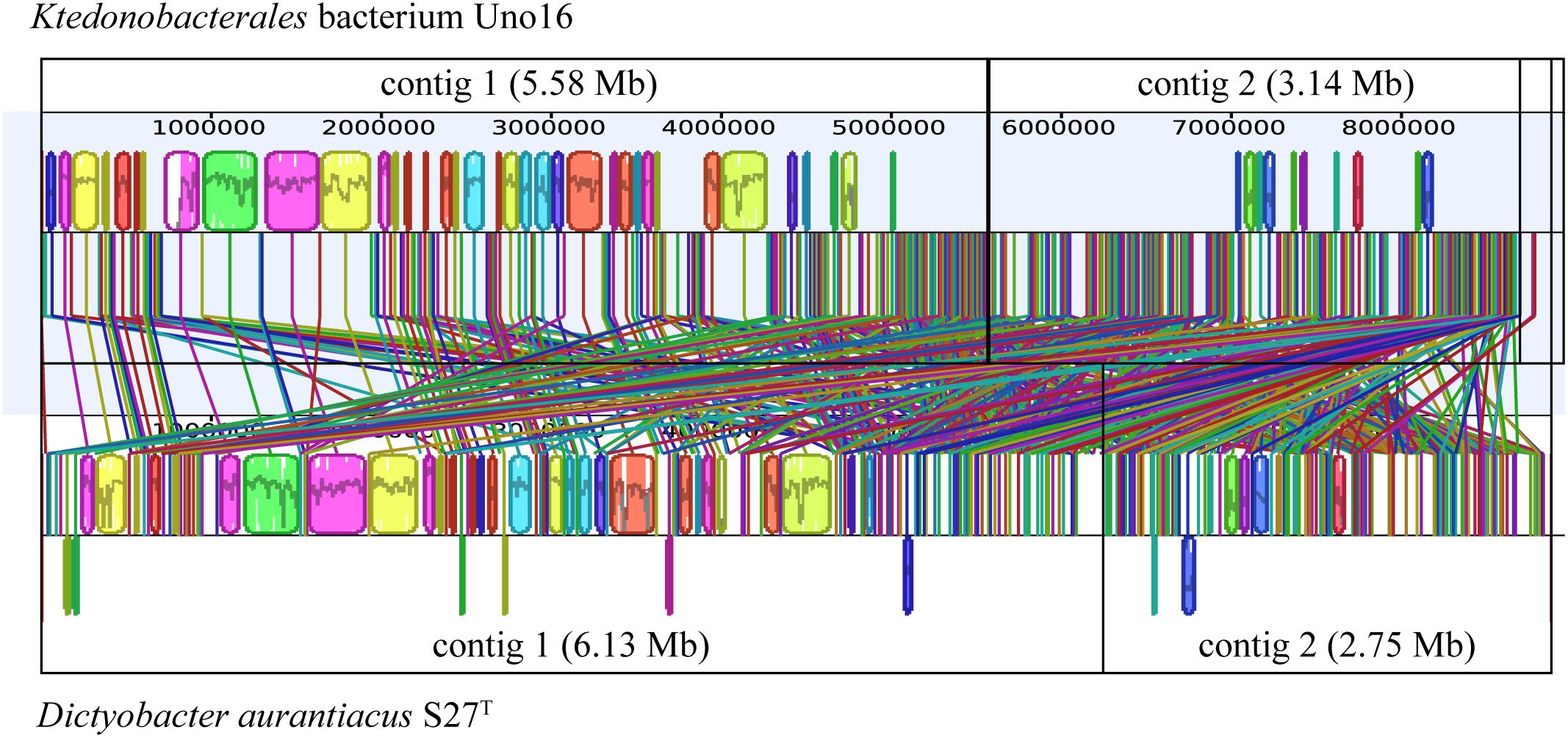
Figure 4. Comparative analyses of strains D. aurantiacus S27T and Ktedonobacterales bacterium Uno16. The colored boxes represent homologous regions with another genome. Lines between two genomes trace each homologous region between them.
Overview of the Putative Secondary Metabolites Biosynthetic Gene Clusters
To evaluate the biosynthetic potential of the class Ktedonobacteria, antiSMASH version 4.2.0 plus the integrated ClusterFinder algorithm were used to predict both characterized and unknown functioned secondary metabolite BGCs in the nine genomes. As shown in Figure 5A and Supplementary Table S6A, a total of 593 putative characterized and unknown BGCs were predicted by ClusterFinder, with 37∼117 BGCs per genome and counting for 11.75∼34.49% of the genomic sequences. To gain a deeper understanding of the Ktedonobacteria secondary metabolites, we focused on the putative BGCs with characterized functions predicted by antiSMASH. Initially, 104 antiSMASH-identified putative BGCs were found in the nine Ktedonobacteria genomes, including 14 NRPS clusters, 17 PKS clusters, 18 hybrid PKS/NRPS clusters, 27 lantipeptide clusters, and 28 other BGCs (Figure 5B and Supplementary Table S6B). However, these BGCs were distributed very unevenly in the nine strains. Strain Ts. hazakensis SK20-1T and Ts. hazakensis COM3 encoded 20 and 18 BGCs, dedicating 15.28% and 12.64% of their total genome, respectively (Supplementary Table S6B). In contrast, an average of four BGCs were identified in the genome of Thermogemmatispora strains, constituting only 1.78∼2.48% of the total genome on average. Strains D. aurantiacus S27T, Ktedonobacterales bacterium Uno16, and Ktedonobacterales bacterium Uno3 encoded 13, 12, and 13 antiSMASH-identified BGCs, dedicating 6.46%, 7.54%, and 12.89% of their total genomes, respectively. In accordance with the result of ClusterFinder, 35.27% of the genomic sequences on contig 2 of Ktedonobacterales bacterium Uno3 were predicted to encode antiSMASH-identified putative BGCs. Moreover, composition of the antiSMASH-identified putative BGC types were very diverse in the nine Ktedonobacteria strains. The main BGC types in Ts. hazakensis SK20-1T and Ts. hazakensis COM3 were NRPS/T1PKS hybrid BGCs and lantipeptide BGCs, while D. aurantiacus S27T, Ktedonobacterales bacterium Uno3, and K. racemifer SOSP1-21T were more abundant in NRPS and PKS BGCs. Unlike D. aurantiacus S27T, Ktedonobacterales bacterium Uno16 encoded more Lantipeptide and NRPS/T1PKS hybrid BGCs on its genome. Overall, it was observed that BGCs responsible for peptide compounds production including NRPS, lantipeptide, lassopeptide, and thiopeptide BGCs predominate in the nine Ktedonobacteria genomes, while PKS BGCs encoding the polyketide compounds were the second most abundant BGC types. Herein, we analyzed composition and organization of the Ktedonobacteria NRPS, PKS, NRPS/PKS hybrid, and lantipeptide BGCs in the next section. In addition, the 104 antiSMASH-identified BGCs exhibited very limited similarity with known clusters in the MIBiG database (Supplementary Table S6B).
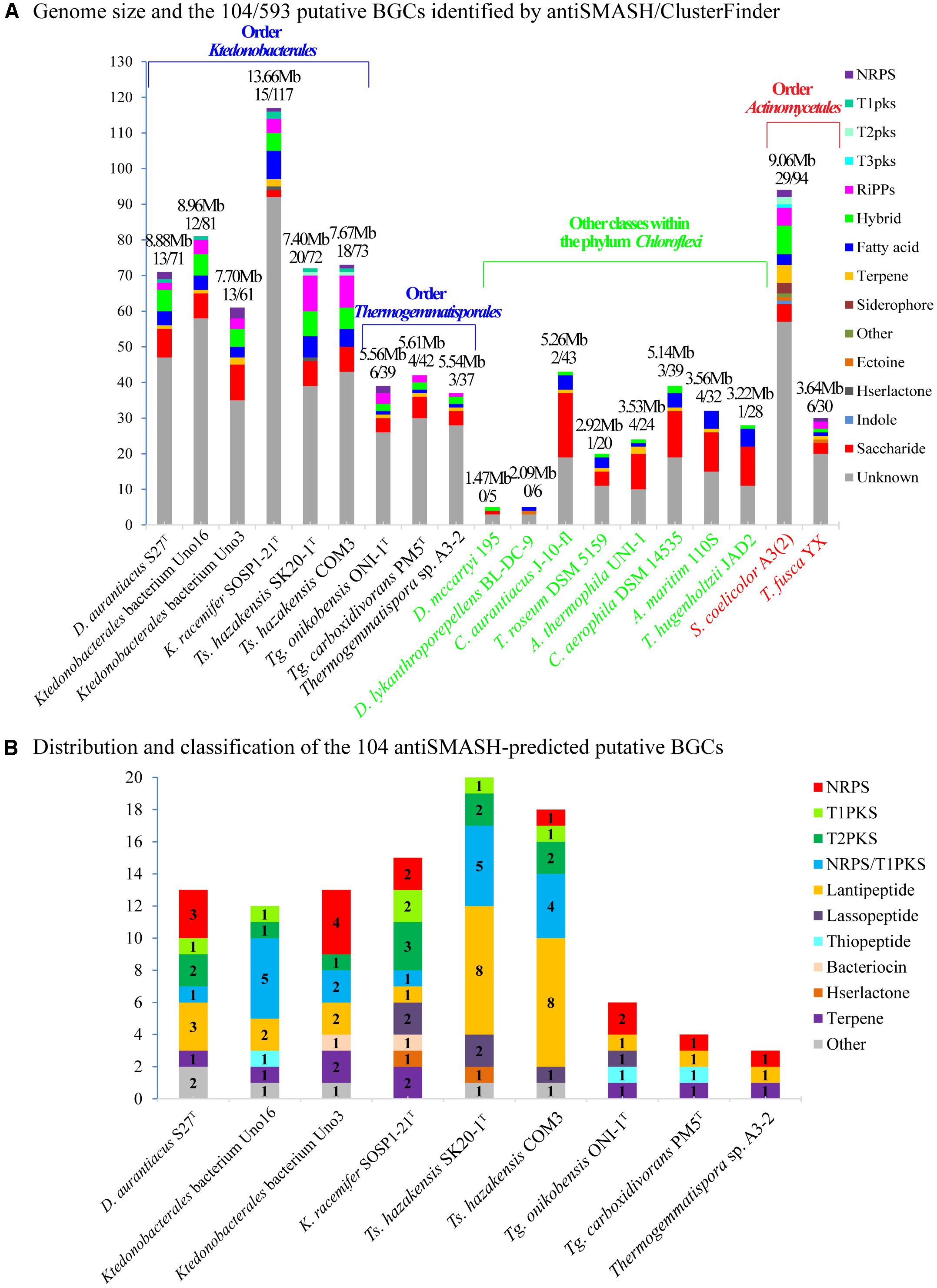
Figure 5. Composition and distribution of the ClusterFinder-identified putative BGCs (A) and antiSMASH-identified putative BGCs (B) in the nine Ktedonobacteria genomes. Strains Dehalococcoides mccartyi 195 and Dehalogenimonas lykanthroporepellens BL-DC-9 (class Dehalococcoides), Chloroflexus aurantiacus J-10-f1 (class Chloroflexi), Thermomicrobium roseum DSM 5159 (class Thermomicrobia), Anaerolinea thermophila UNI-1 (class Anaerolineae), Caldilinea aerophila DSM 14535 (class Caldilineae), Ardenticatena maritima strain 110S (class Ardenticatenia), and Thermoflexus hugenholtzii JAD2 (class Thermoflexia) of the phylum Chloroflexi and strains Streptomyces coelicolor A3(2) and Thermobifida fusca YX within order Actinomycetales, phylum Actinobacteria were used as reference strains for comparison.
NRPS, PKS, NRPS/PKS Hybrid, and Lantipeptide BGCs
A total of 14 putative antiSMASH-identified NRPS BGCs were identified albeit distributed unevenly in seven strains, ranging from 20.4 kb (cluster Toni_5 in strain Tg. onikobensis ONI-1T) to 154.4 kb (cluster Uno3_7 in strain Ktedonobacterales bacterium Uno3) in size. Domain composition and organization analysis (Supplementary Table S7A) indicated that some of the NRPS BGCs such as cluster Txc_10 and cluster Dar_12 could produce peptide products with a ring moiety, by the existence of multiple epimerization (E) domain and heterocyclization (HC) domain (Chen L.Y. et al., 2016; Bloudoff et al., 2017) in the clusters. In addition, the existence of fatty acyl-AMP ligase (FAAL) domain in cluster Uno3_5 suggests that the final product of this gene cluster may constitute a lipopeptide or its derivatives. The FAAL domain was first discovered in Mycobacterium tuberculosis and activates fatty acids such as acyl adenylates, subsequently catalyzing their transfer onto the ACPs of PKSs or non-ribosomal peptide synthetases to produce lipidic metabolites (Hayashi et al., 2011).
As for PKS BGCs, 6 T1PKS BGCs and 11 T2PKS BGCs were identified. The 6 T1PKS BGCs spanned from 34.2 kb (cluster Krac_15 in strain K. racemifer SOSP1-21T) to 46.6 kb (cluster Uno16_10 in strain Ktedonobacterales bacterium Uno16) in size whereas the 11 T2PKS BGCs spanned from 41.3 kb (cluster Uno16_1 in strain Ktedonobacterales bacterium Uno16) to 42.5 kb (cluster Krac_11 in strain K. racemifer SOSP1-21T). Domain composition and organization of the identified PKS clusters are summarized in Supplementary Tables S7B,C. Notably, cluster Uno16_10 contained two additional NRPS-A domains that were predicted to function as a long chain fatty acid CoA ligase and an acyl CoA synthetase by Protein-Protein Blast, indicating that the final products of this cluster may comprise derivatives of lipids (Hisanaga et al., 2004; Soupene and Kuypers, 2008).
Eighteen putative antiSMASH-identified BGCs were classified to be NRPS-T1PKS hybrid clusters, which were distributed mainly in strains Ts. hazakensis SK20-1T, Ts. hazakensis COM3, and Ktedonobacterales bacterium Uno16. The 18 putative hybrid NRPS-T1PKS clusters ranged in size from 54.4 kb (cluster Uno16_4 in strain Ktedonobacterales bacterium Uno16) to 333.5 kb (cluster Uno3_11 in strain Ktedonobacterales bacterium Uno3). Domain composition and organization and the AMP-binding domain amino acid substrate specificities of the hybrid clusters are summarized in Supplementary Tables S7D,E. Based on genetic similarity and domain composition and organization in these gene clusters, the 18 putative hybrid NRPS-T1PKS clusters could be divided into four groups. Moreover, the hybrid NRPSs and PKSs biosynthesis pathways obviously enlarge the diversity of bacterial natural products.
After a comprehensive analysis of antiSMASH and BAGEL3 (van Heel et al., 2013), the 27 lantipeptide BGCs were further classified into four classes according to their biosynthetic machinery (Zhang et al., 2012). Class I contains 11 lantipeptide BGCs that are synthesized by two separated modification genes, LanB and LanC. In addition, cluster Txs_13 from strain Ts. hazakensis SK20-1T comprised two LanB genes and one LanC gene. However, the two LanB genes were short in size and both lacked a lantibiotic biosynthesis dehydratase C-terminal domain, according to Pfam analysis. As the C-terminal domain is necessary for the final glutamate-elimination step in the generation of lantipeptide (Ortega et al., 2015), we propose these two LanB genes may be inactive. Class II contains 11 lantipeptide BGCs that are synthesized by a single bifunctional enzyme termed LanM. Moreover, after conducting a ClustalW alignment using MEGA7.0 and generating sequence logos with WebLogo 3 (Crooks et al., 2004), we observed F(E/D)LD (Supplementary Figure S2A) and GG (Supplementary Figure S2B) cleavage sites in class I and class II, respectively.
Based on these analyses, the antiSMASH-identified Ktedonobacteria BGCs showed very limited homology with known BGCs. The different composition and organization of tailoring genes, transport-related genes, and regulatory genes further increased the diversity of these BGCs and indicated that these clusters may encode novel natural products with novel functions.
Phylogenetic Analysis of the PKS KS Domain, NRPS C Domain, and Lantipeptide Modification Genes
Considering that class Ktedonobacteria constitutes a relatively new bacterial taxa, to date only very limited knowledge is available regarding their secondary metabolites. The domain-specific phylogenetic analysis of Ktedonobacteria-originated secondary metabolite BGCs identified in the present study may provide a better understanding of the functional and evolutionary classification of their domains. Furthermore, as C and KS domains are responsible for peptide and polyketide chain elongation in NRPS and PKS biosynthesis, respectively, the two domains represent the best candidates for domain-specific phylogenetic analysis (Rausch et al., 2007; Jenke-Kodama and Dittmann, 2009). As shown in the outlying ring of Figure 6A, the most abundant functional type among the Ktedonobacteria KS domains was assigned to hybrid KS by NaPDoS classification, owing to the high occurrence of hybrid NRPS-T1PKS clusters identified in the Ktedonobacteria genomes. Modular KS, the second most abundant KS domain functional type in the class Ktedonobacteria, was also mainly derived from hybrid NRPS-T1PKS clusters. The modular PKS is responsible for the incorporation of one building block and contains at least three domains: KS, AT, and ACP (Jenke-Kodama et al., 2005). With regard to evolutionary classification, the Ktedonobacteria-originated KS domains and reference sequences appeared to form four clades. The first clade included a sequence from strain Ktedonobacterales bacterium Uno3, a sequence from strain D. aurantiacus S27T, and two sequences derived from the phylum Cyanobacteria.
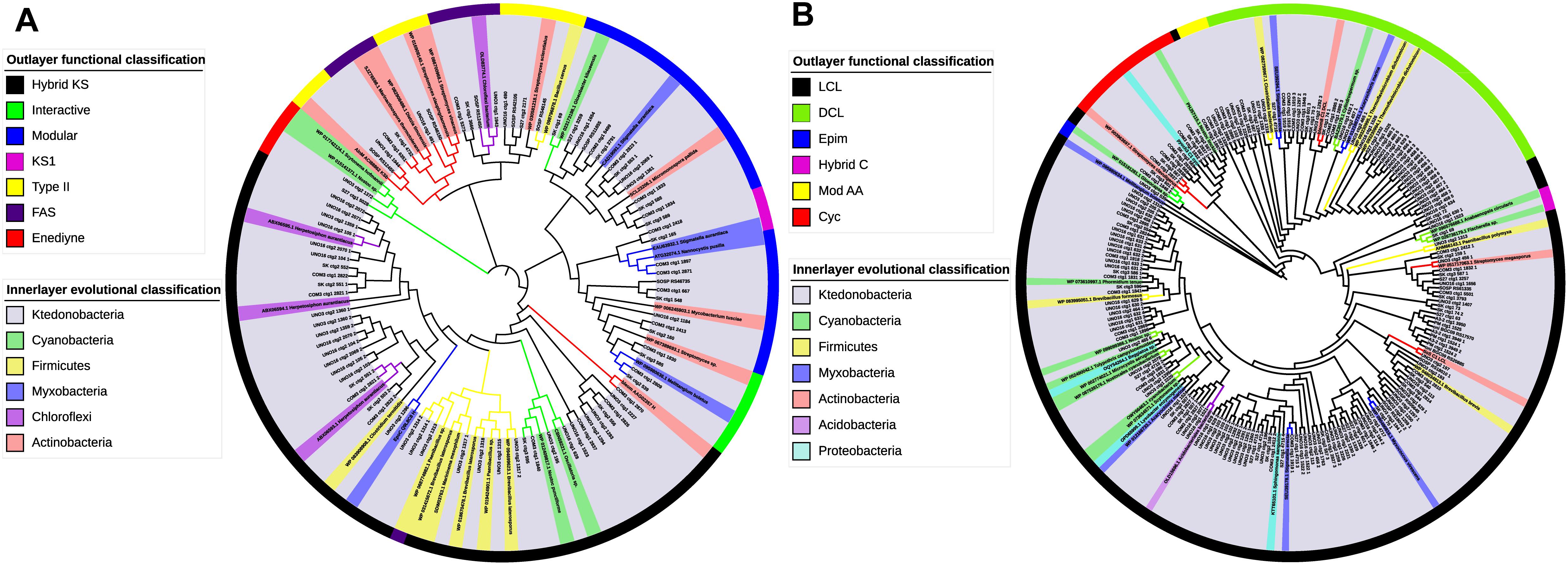
Figure 6. Functional and evolutionary analysis of Ktedonobacteria PKS KS and NRPS C domains. (A) Domain-specific phylogenetic analysis of the Ktedonobacteria amino acid sequences extracted from KS domains. The reference sequences include three top hits from Protein-Protein Blast and the NaPDoS reference database. Maximum-likelihood phylogenetic tree of the Ktedonobacteria KS domains was built in MEGA v. 7.0. The evolutionary classification was shown in the inner layer color strip whereas the outer layer color strip represents the functional classification of the KS domains. (B) Domain-specific phylogenetic analysis of the Ktedonobacteria amino acid sequences extracted from C domains. The analysis was created as for (A).
The other three clades were more diverse in representation, including sequences from the phyla Actinobacteria, Cyanobacteria, Firmicutes, Chloroflexi, and the order Myxobacteria. Moreover, part of the Ktedonobacteria KS domains clustered with Actinobacteria, Cyanobacteria, and Firmicutes reference sequences, indicating that they may be phylogenetically related to these phyla. However, the majority of the Ktedonobacteria-originated KS domains formed independent branches. Furthermore, the highest similarity between Ktedonobacteria KS domains and reference sequences was only 71%, as observed between cluster Uno3_11 and a Brevibacillus laterosporus reference sequence. According to the NaPDoS functional classification, the most abundant types of Ktedonobacteria-originated C domains are LCL and DCL. The LCL type C domain catalyzes formation of a peptide bond between two L-amino acids whereas the DCL type links an L-amino acid to a growing peptide ending with a D-amino acid in NRPS biosynthesis (Rausch et al., 2007). In agreement with our analysis of NRPS and hybrid clusters, the C domains, which are replaced by HC domains in strain SK20-1T and strain COM3, were classified as cyclization domains by NaPDoS, which catalyze both peptide bond formation and subsequent cyclization of cysteine, serine, or threonine residues. With regard to evolutionary classification, the Ktedonobacteria C domains showed more diversity compared with the KS domains. As shown in Figure 6B, only a small percentage of C domains were related to the reference sequences from Cyanobacteria, Actinobacteria, Firmicutes, Myxobacteria, Proteobacteria, and Acidobacteria. Rather, a large proportion of Ktedonobacteria C domains formed independent branches, indicating they are distinct from those that originated from other phyla in evolutionary taxonomy.
As modification genes are responsible for the maturation of lantipeptide, the phylogeny of these modification enzymes are closely correlated with the structure of final lantibiotic natural products (Willey and van der Donk, 2007; Zhang et al., 2012). In the present study, the phylogenetic analyses were also applied to Ktedonobacteria modification genes to determine their evolutionary relationship with other bacterial phyla or classes. As confirmed in the phylogenetic tree (Supplementary Figure S3), the majority of the Ktedonobacteria LanB and LanC modification genes formed independent clusters although they showed limited similarities with reference sequences from the phylum Actinobacteria. Compared with LanB and LanC modification genes, greater diversity was observed among LanM genes (Supplementary Figure S3). The two LanM genes from cluster Dar_6 and the LanMM cluster in strain S27T were related to LanMs from the phylum Cyanobacteria. The other Ktedonobacteria LanMs showed similarities with Actinobacteria and Myxobacteria sequences, whereas the majority formed independent branches, indicating that they may modify structurally different products from the known lantibiotic natural products.
Antimicrobial Activities of Ktedonobacteria
Cell pellets and HP-20 polyaromatic adsorbent resin crude extract from six representative Ktedonobacteria strains were tested for antimicrobial activities against both Gram-positive and Gram-negative bacterial strains. All six Ktedonobacteria strains demonstrated activity against at least two of the nine tested bacterial strains (Table 4). In addition, all six Ktedonobacteria strains were positive against G. stearothermophilus NBRC 13737, S. aureus NBRC 13276, and S. aureus Mu50.
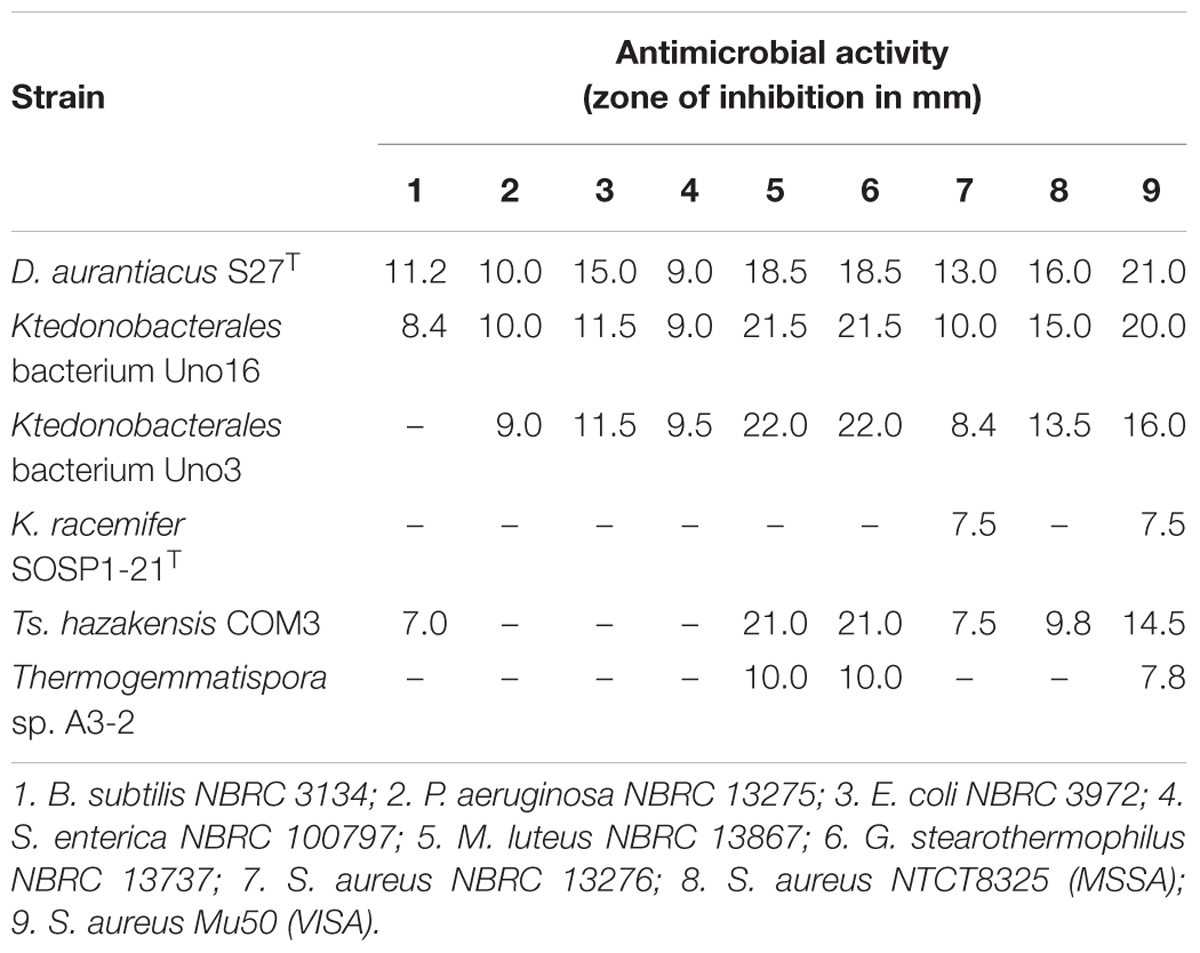
Table 4. Antimicrobial activity of the representative Ktedonobacteria strains assessed using the paper disc method.
Overall, the three mesophilic Ktedonobacteria strains including D. aurantiacus S27T, Ktedonobacterales bacterium Uno3, and Ktedonobacterales bacterium Uno16, exhibited a broader antibacterial spectra against both Gram-positive and Gram-negative (P. aeruginosa NBRC 13275, E. coli NBRC 3972, and S. enterica NBRC 100797) bacterial strains. In comparison, strain Ts. hazakensis COM3 and Thermogemmatispora sp. A3-2, the two thermophilic strains, only inhibited the Gram-positive bacterial strains (M. luteus NBRC 13867, G. stearothermophilus NBRC 13737, and S. aureus Mu50). Moreover, the three mesophilic Ktedonobacteria strains also exhibited stronger inhibitory capacities against each Gram-positive bacterial strains than that shown by the two thermophilic strains, as evidenced by the formation of larger inhibition clear zones (Table 4 and Supplementary Figure S4).
Discussion
Phylogenetic and Evolutional Analysis
Members of the phylum Chloroflexi, especially the class Chloroflexi, are generally known as filamentous anoxygenic phototrophs, also termed filamentous green non-sulfur bacteria (Björnsson et al., 2002). However, the phylum Chloroflexi also constitutes a deeply branched bacterial lineage encompassing classes of bacteria with diverse metabolic types from strictly anaerobic chlorinated hydrocarbon reducers to filamentous aerobic heterotrophs and vastly distinct genome sizes from 1.38 to 13.67 Mb (Cavaletti et al., 2006; Moe et al., 2009; Chang et al., 2011; Kaster et al., 2014). Moreover, comparative genomics and phylogenetic analyses suggest that these different classes may represent related but distinct phyla under a Chloroflexi “superphylum” (Gupta et al., 2013). Notably, maximum likelihood phylogenomic analysis based on informative amino acid sequences confirmed the class Ktedonobacteria and the SAR202 clade of dark-ocean bacterioplankton as the two deepest-branching classes related to the common ancestor of the phylum Chloroflexi (Landry et al., 2017). As the phylum Chloroflexi constitutes one of the earliest bacterial branches in the terrestrial environment (Hug et al., 2016), the class Ktedonobacteria is therefore considered to be among the earliest diverging bacterial lineages.
In the phylogenetic tree based on conserved protein sequences, class Ktedonobacteria clustered together with Dehalococcoidetes, a class of a strictly anaerobic, slow growing, and highly niche-specialized bacteria in terrestrial aquifer environments that utilize organohalide respiration as their sole source of energy (Kaster et al., 2014). However, our pan-genome analysis between class Ktedonobacteria and class Dehalococcoidetes indicated the two bacterial classes do not share a high number of conserved core genes with each other. Moreover, the two classes are clearly distinct in morphologies, genome sizes, habitats, and metabolic styles. Nonetheless, the formation of a monophyly in the phylogenetic tree based on conserved protein sequences suggest they may have evolved from a common ancestor. Herein, we hypothesize that the two bacterial classes then evolved separately to adapt to different environments and the evolutionary results reflect on their genomes. The class Dehalococcoidetes evolved a sole energy conservation mode via organohalide respiration and developed a symbiotic metabolic style, depending on co-living microbial species to acquire electron donors and cofactors (Kaster et al., 2014). As a result, species in the class Dehalococcoidetes may gradually drop unnecessary genes in their genomes, resulting in small genome sizes. On the contrary, species in the class Ktedonobacteria are very diverse in habitats, ranging from common soil to oligotrophic environments. Thus, species in the class Ktedonobacteria may tend to obtain foreign genes or foreign secondary metabolites gene clusters to adapt to these environments, resulting in relatively large genomes. Moreover, the class Ktedonobacteria developed a spore-forming morphology to survive nutrient depletion or harsh environments (Errington, 2003; Flärdh and Buttner, 2009). Of course, the currently sequenced genomes of the phylum Chloroflexi are still very limited and further studies are needed to completely characterize the evolution of class Ktedonobacteria.
Genome Features of the Class Ktedonobacteria
As described above, species in the class Ktedonobacteria are featured with low GC content and relatively large genome sizes, among which genomes of strains D. aurantiacus S27T, Ktedonobacterales bacterium Uno16, Ktedonobacterales bacterium Uno3, and K. racemifer SOSP1-21T are comparable to that of actinomycetes strains (Chen W.H. et al., 2016). As for the studies of strains in genus Thermosporothrix and genus Thermogemmatispora, their genome sizes are also quite large among thermophilic bacteria given that growth temperature and genome size in bacteria are negatively correlated and thermophilic bacteria tend to have a small genome (Sabath et al., 2013). Also, it has been reported that the Firmicutes and Gammaproteobacteria have the largest average 16S rRNA gene copy numbers (6.788 ± 2.694 and 5.141 ± 2.411, respectively) and the maximum variation (5.458 ± 5.633 and 5.298 ± 7.052, respectively) (Ibal et al., 2019). Thus, the average copy numbers of 16S rRNA genes (6.375 per genome, excluding strain Tg. onikobensis ONI-1T) in Ktedonobacteria genomes is quite high in bacterial species, although their variations are lower than Firmicutes and Gammaproteobacteria. Moreover, the relatively high percentage of functional unknown hypothetical proteins indicated that the class Ktedonobacteria may possess as yet unknown metabolites and produce some novel natural products through novel mechanisms (Galperin, 2001).
It is interesting to find that strains D. aurantiacus S27T, Ktedonobacterales bacterium Uno16, and Ktedonobacterales bacterium Uno3 may possess “megaplasmids.” As described in Section “Strain Information and Whole Genome Sequencing,” the draft genome of Ktedonobacterales bacterium Uno3 includes two circular contigs (Figure 2). As the presence of housekeeping genes are essential to define a bacterial chromosome (Thomas and Summers, 2008), herein, it is probably more appropriate to classify the second circular contig of Uno3 to be “megaplasmid” due to the absence of most housekeeping genes (Table 3 and Supplementary Table S2). The presence of megaplasmid was also observed in Thermomicrobium roseum DSM 5159, an aerobic CO oxidizing thermophile strain in the phylum Chloroflexi and composed one chromosome (2,006,217 bp) and one megaplasmid (919,596 bp) in which few standard housekeeping genes were found (Wu et al., 2009). As for the linear contig2 of strains D. aurantiacus S27T and Ktedonobacterales bacterium Uno16, the definition of “megaplasmid” could be further evidenced with the absence of most housekeeping genes, translation genes, DNA replication and repair genes, and genes involved in TCA cycle and oxidative phosphorylation (Table 3), which are essential genes for growth. Furthermore, the high dissimilarities in sequences observed between contig2 of the two strains (Figure 4) indicated they originated differently. Meanwhile, high numbers of transporter related genes, cytochrome P450 related genes, and regulator genes were identified in contig2 of the two strains (Table 3). Commonly, these genes in bacterial genomes could contribute to their adaptation or resistance to various environmental pressures such as antibiotics, heavy metals, extreme Antarctic environments, among others (Kelly and Kelly, 2013; Dziewit et al., 2015; Romaniuk et al., 2018). Thus, it could be that strains D. aurantiacus S27T and Ktedonobacterales bacterium Uno16 acquired the foreign “megaplasmids” naturally to adapt to various environments. Additionally, we found that housekeeping genes are also absent in eight contigs of SOSP1-21T (Supplementary Table S2). Nonetheless, this hypothesis of “megaplasmids” needs to be further confirmed because of their large sizes.
Unlike actinomycetes, which are saprophytic bacteria, the class Ktedonobacteria is reported to be predominate in the oligotrophic habitats (Northup et al., 2011; Stres et al., 2013; de Miera et al., 2014b; Tebo et al., 2015; Jiang et al., 2016) although they were also ubiquitous in various common terrestrial environments (Cavaletti et al., 2006; Yabe et al., 2017b). Additionally, the abundance of Ktedonobacteria was reported to increase or predominate in the bacterial communities with increasing CO2 flux in CO2 gas vents, CO2-rich hydrothermal spring and soil environments (de Miera et al., 2014a,b; Arce-Rodríguez et al., 2019). Together with our finding of the almost complete reductive TCA cycle and the presence of multiple copies of cox genes in genomes, it is highly possible that the class Ktedonobacteria may possess unknown autotrophic carbon fixation pathways.
Secondary Metabolites Biosynthetic Potential of the Class Ktedonobacteria
In this study, a total of 593 ClusterFinder-identified putative BGCs were predicted in nine Ktedonobacteria strains among which 104 putative BGCs were also predicted by antiSMASH. As the genome size of bacteria is considered to correlate with the number of gene clusters (Donadio et al., 2007), the high number of putative BGCs predicted could be a consequence of their large genomes. Moreover, we observed that the class Ktedonobacteria encode numerous NRPS, PKS, NRPS/PKS hybrid, and lantipeptide gene clusters, which may assist them in fighting against their competitors and predators in their niches (Challis and Naismith, 2004; Zhang et al., 2012; Johnston et al., 2015; Ortega and van der Donk, 2016; Naughton et al., 2017). This hypothesis could be supported by the observation that the genus Thermogemmatispora, which exhibited the lowest number of BGCs in the class Ktedonobacteria, predominates in geothermal niches in which the microbial community richness is markedly decreased (de Miera et al., 2014b; Jiang et al., 2016). Owing to a lack of competitors and predators (Yabe et al., 2017a), the genus Thermogemmatispora may tend to reduce the number of BGCs in their genomes as secondary metabolites are not necessary but are always costly for microorganisms. In addition, the majority of the 104 antiSMASH-identified putative BGCs showed no or very limited similarities with known clusters. This result could also be supported by the functional and evolutionary phylogenetic analysis of the Ktedonobacteria KS and C domains. As shown in Figure 6, the majority of Ktedonobacteria-derived KS and C domains formed independent clusters from those derived from other phyla.
Nevertheless, 38 out of the total 104 antiSMASH-identified putative Ktedonobacteria BGCs exhibited varying degrees of similarity to known gene clusters. As described in the Section “Results” and summarized in Supplementary Table S6, the 38 Ktedonobacteria BGCs were related to Actinobacteria (17 Ktedonobacteria BGCs), Cyanobacteria (14 Ktedonobacteria BGCs), Myxobacteria (4 Ktedonobacteria BGCs), Firmicutes (2 Ktedonobacteria BGCs), and Gammaproteobacteria (1 Ktedonobacteria BGCs) pathways. In addition, we observed that the 38 BGCs comprised mainly hybrid NRPS-T1PKS clusters (15 BGCs), NRPS clusters (7 BGCs), PKS clusters (7 BGCs), and RiPP clusters (7 BGCs). As aforementioned, these types of BGCs usually encode modular enzymes (Challis and Naismith, 2004; Dutta et al., 2014; Letzel et al., 2014; Ortega and van der Donk, 2016). Thus, the homolog of a single gene or single domain could result in the high similarity between two BGCs, which could be observed between the Ktedonobacteria hybrid PKS/NRPS cluster and Myxobacterium S. cellulosum pellasoren A gene cluster. The repetition of a KS-AT-cMT-KR-ACP PKS module-encoding gene in the pellasoren A gene cluster resulted in the 83% similarity between DNA sequences of the two gene clusters. Furthermore, the existence of a homologous CAL domain in cluster Uno16_7 and the puwainaphycins BGC resulted in a 20% genetic similarity between the two clusters.
However, as few descriptions regarding secondary metabolites of the class Ktedonobacteria are available, the limited similarities between BGCs from the class Ktedonobacteria and those from other bacterial phyla may contribute to our understanding of the biosynthetic pathways of secondary metabolites in Ktedonobacteria. Moreover, the majority of the Ktedonobacteria BGCs are totally novel and differ from the known BGCs with regard to composition and organization of the condensation domains in NRPS BGCs, ketosynthase domains in PKS BGCs, and leader peptide in lantipeptide clusters. In addition, the difference of transport-related genes and regulatory genes with known BGCs further increases the diversity of known Ktedonobacteria BGCs. Notably, the BGC similarities revealed by antiSMASH comparison and the results of Ktedonobacteria ketosynthase and condensation domain phylogenetic analysis (Figure 6) suggest that a portion of the secondary metabolite BGCs in the class Ktedonobacteria may have been acquired from other bacterial phyla via horizontal gene transfer (HGT). HGT is common in bacteria and may provide new functions and subsequent niche adaptations through the acquisition of valuable secondary metabolite BGCs (Tooming-Klunderud et al., 2013; Ziemert et al., 2014; Jensen, 2016; Adamek et al., 2018). BGCs encoding natural products usually include both core biosynthetic and tailoring enzymes, along with regulatory genes for biosynthesis, resistance genes to the natural products they produce, and transport genes such as efflux pumps to export these natural products from their cells to the extracellular environment (Jensen, 2016). In addition, the regulatory genes required for function may facilitate integration of the HGT-acquired genes into existing genomes (Lawrence, 1999). Thus, the existence of regulatory, resistance, and transport genes in NRPS, PKS, and their hybrid gene clusters might facilitate their HGT (Ziemert et al., 2014). However, as the class Ktedonobacteria is among the earliest diverging bacterial lineages, arising much earlier than the phyla Actinobacteria, Cyanobacteria, Firmicutes, and Proteobacteria (Hug et al., 2016; Landry et al., 2017), we cannot exclude the possibility that the class Ktedonobacteria constitutes the actual HGT origin of secondary metabolite gene clusters.
The representative Ktedonobacteria strains produced probable bioactive compounds against both Gram-positive and Gram-negative bacterial strains (Table 4 and Supplementary Figure S4), suggesting their potential utility for antimicrobial screening. Among the six screened Ktedonobacteria strains, D. aurantiacus S27T, Ktedonobacterales bacterium Uno3, and Uno16 were the most capable. Given that Gram-negative bacterial strains are becoming increasingly antibiotic resistant owing to their protective outer membranes and constitutively active efflux pumps, these mesophilic Ktedonobacteria strains may contribute to the development of novel antibiotics targeting Gram-negative bacteria (Miller, 2016; Domalaon et al., 2018). In turn, the antibacterial activities against S. aureus NBRC 13276, S. aureus NTCT8325, and S. aureus Mu50 might be explained by the production of novel lanthipeptides or other antimicrobial peptides by the Ktedonobacteria strains, as reported previously (Xu et al., 2018). Moreover, as the candidate testing microorganisms used for antimicrobial assays and activities assessed in the present study were finite and few isolated Ktedonobacteria species are available for study, this class likely harbors as-yet undisclosed bioactive functions.
Conclusion
In this study, we performed whole genome sequencing, comprehensive phylogenetic analysis, genome comparisons, and biosynthetic potential analysis of the class Ktedonobacteria, one of the deepest-branching classes related to the common ancestor of the earliest diverging bacterial lineages Chloroflexi. Our results further confirmed the classification of the spore-forming Ktedonobacteria to the phylum Chloroflexi at both the 16S rRNA and genomic level. The existence of a putative “megaplasmid” in some of the Ktedonobacteria strains was also observed in this study. Meanwhile, we found the class Ktedonobacteria were characterized with relatively large genome sizes, multiple copies of ribosomal RNA operons, and a high ratio of hypothetical proteins with unknown functions. The possibilities of reductive TCA cycle were also observed in the class Ktedonobacteria. Analysis of putative secondary metabolites BGCs in the nine Ktedonobacteria strains yielded a total of 593 ClusterFinder-identified putative BGCs (104 antiSMASH-identified putative BGCs), representing the first study to mine the biosynthetic potential of the class. The overall novelty and diversity of these BGCs provided convincing evidence that the class Ktedonobacteria possesses broad potential to produce new metabolite products with novel structures and mechanisms. Furthermore, bioactivity screening of the studied nine Ktedonobacteria strains revealed a wide spectrum of antibacterial activities. However, it should be noted that the current understanding of the Ktedonobacteria secondary metabolite pathways is still very limited, and their relationship with pathways of other bacterial phyla remains to be elucidated. Given the potential ability to produce novel and diverse bioactive natural products, we consider the ancient, ubiquitous, and mycelia-forming Ktedonobacteria to represent a versatile and promising microbial resource for pharmaceutical and biotechnological use.
Author Contributions
YZ and SY conceived and designed the study. YZ, AS, and YM performed the experiments. YZ, AT, YuS, YaS, SY, C-MW, KU, HT, KA, and AY analyzed the results and data. YZ wrote the manuscript. All authors have read and approved the manuscript, and reviewed and confirmed the manuscript for publication.
Funding
This study was supported in part by MEXT/JSPS KAKENHI (Grant Nos. 18K05406 and 16H06279), the Institute for Fermentation, Osaka (Grant No. G-2018-1-038), and the China Scholarship Council (CSC No. 201606330094).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00893/full#supplementary-material
FIGURE S1 | Maximum-likelihood phylogenetic inference of members of the class Ktedonobacteria among known bacterial phyla. The tree was constructed using RAxML or FastTree based on up to 38 marker genes (using taxon-outgroup configuration Config 3 described in Sekiguchi et al., 2015) and sequences were collapsed at the phylum level except for classes in the Proteobacteria. Ranks are indicated by prefix; p_ (phylum), c_ (class). Parameters used for phylogenetic inference using RAxML and FastTree are described in the “Materials and Methods” section. Numbers at the notes indicate bootstrap values (%) with 100 times determination. The scale bar represents 10% estimated sequence divergence.
FIGURE S2 | Sequence logos of precursor peptide conserved motifs identified in the nine Ktedonobacteria strains are underlined in red. (A) The “F(E/D)L” cleavage site for Class I lantipeptide gene clusters. (B) The “GG” cleavage site for Class II lantipeptide clusters.
FIGURE S3 | Phylogenetic analysis of the Ktedonobacteria modification genes in lantipeptide clusters. (A) Phylogenetic analysis of the Ktedonobacteria LanB genes. Amino acid sequences extracted from the Ktedonobacteria LanB and the three top hit reference sequences from Protein-Protein Blast were aligned by MEGA v. 7.0. Maximum-likelihood method was used to build the tree. The background colors represent evolutionary classification of the LanB modification genes. (B) Phylogenetic analysis of the Ktedonobacteria LanC genes. The tree was built as for (A). (C) Phylogenetic analysis of the Ktedonobacteria LanM genes. The tree was also built as for (A).
FIGURE S4 | Antimicrobial screening of six representative Ktedonobacteria strains against bacterial and fungal strains. The antimicrobial activity are indicated by formation of clear zone of S. aureus NTCT8325 (MSSA) (A), S. aureus Mu50 (VISA) (B), Escherichia coli NBRC3972 (C), Bacillus subtilis NBRC3134 (D), Staphylococcus aureus NBRC13276 (E), Geobacillus stearothermophilus NBRC13737 (F), Pseudomonas aeruginosa NBRC13275 (G), Salmonella enterica NBRC100797 (H), Micrococcus luteus NBRC13867 (I) as inhibited by Thermosporothrix hazakensis COM3 (1), Ktedonobacter racemifer SOSP1-21T (2), Dictyobacter aurantiacus S27T (3), Ktedonobacterales bacterium Uno3 (4), Ktedonobacterales bacterium Uno16 (5), Thermogemmatispora sp. A3-2 (6), and negative control (N).
TABLE S1 | Detailed sequencing information and genome features of the nine studied Ktedonobacteria strains and related taxon.
TABLE S2 | CheckM estimation of the nine studied Ktedonobacteria strains.
TABLE S3 | Similarities of multiple 16S ribosomal RNA operons within the nine studied Ktedonobacteria strains.
TABLE S4 |cox gene (A) and KEGG pathway (B) analysis of the Ktedonobacteria strains.
TABLE S5 | Pan-genome analysis of the Ktedonobacteria genomes (A), D. aurantiacus S27T and Ktedonobacterales bacterium Uno16 contig1 (B), and D. aurantiacus S27T and Ktedonobacterales bacterium Uno16 contig2 (C).
TABLE S6 | List of the putative BGCs identified by ClusterFinder (A) and antiSMASH (B) in nine Ktedonobacteria strains and their similarities to known clusters.
TABLE S7 | Domain composition and organization of the putative NRPS clusters (A), T1PKS clusters (B), T2PKS clusters (C), and NRPS-TIPKS clusters (D) identified in nine Ktedonobacteria strains, and AMP-binding domain amino acid substrate specificities in the NRPS-TIPKS clusters (E).
Footnotes
- ^https://smrt-analysis.readthedocs.io/en/latest/
- ^https://www.pacb.com/training/hgap4-de-novo-assembly-application/
- ^https://github.com/dparks1134/CommunityM
- ^https://blast.ncbi.nlm.nih.gov/Blast.cgi
- ^https://www.megasoftware.net/
References
Adamek, M., Alanjary, M., Sales-Ortells, H., Goodfellow, M., Bull, A. T., Winkler, A., et al. (2018). Comparative genomics reveals phylogenetic distribution patterns of secondary metabolites in Amycolatopsis species. BMC Genomics 19:426. doi: 10.1186/s12864-018-4809-4
Arce-Rodríguez, A., Puente-Sánchez, F., Avendaño, R., Martínez-Cruz, M., de Moor, J. M., Pieper, D. H., et al. (2019). Thermoplasmatales and sulfur-oxidizing bacteria dominate the microbial community at the surface water of a CO2-rich hydrothermal spring located in Tenorio Volcano National Park, Costa Rica. Extremophiles 23, 177–187. doi: 10.1007/s00792-018-01072-6
Bérdy, J. (2012). Thoughts and facts about antibiotics: where we are now and where we are heading. J. Antibiot. (Tokyo) 65, 385–395. doi: 10.1038/ja.2012.27
Bitok, J. K., Lemetre, C., Ternei, M. A., and Brady, S. F. (2017). Identification of biosynthetic gene clusters from metagenomic libraries using PPTase complementation in a Streptomyces host. FEMS Microbiol. Lett. 364:fnx155. doi: 10.1093/femsle/fnx155
Björnsson, L., Hugenholtz, P., Tyson, G. W., and Blackall, L. L. (2002). Filamentous Chloroflexi (green non-sulfur bacteria) are abundant in wastewater treatment processes with biological nutrient removal. Microbiology 148, 2309–2318. doi: 10.1099/00221287-148-8-2309
Blin, K., Wolf, T., Chevrette, M. G., Lu, X., Schwalen, C. J., Kautsar, S. A., et al. (2017). antiSMASH 4.0 — Improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41. doi: 10.1093/nar/gkx319
Bloudoff, K., Fage, C. D., Marahiel, M. A., and Schmeing, T. M. (2017). Structural and mutational analysis of the nonribosomal peptide synthetase heterocyclization domain provides insight into catalysis. Proc. Natl. Acad. Sci. U.S.A. 114, 95–100. doi: 10.1073/pnas.1614191114
Buchanan, B. B., and Arnon, D. I. (1990). A reverse KREBS cycle in photosynthesis: consensus at last. Photosynth. Res. 24, 47–53. doi: 10.1007/BF00032643
Cavaletti, L., Monciardini, P., Bamonte, R., Schumann, P., Rohde, M., Sosio, M., et al. (2006). New lineage of filamentous, spore-forming, gram-positive bacteria from soil. Appl. Environ. Microbiol. 72, 4360–4369. doi: 10.1128/AEM.00132-06
Challis, G. L., and Naismith, J. H. (2004). Structural aspects of non-ribosomal peptide biosynthesis. Curr. Opin. Struct. Biol. 14, 748–756. doi: 10.1016/j.sbi.2004.10.005
Chang, Y. J., Land, M., Hauser, L., Chertkov, O., Del Rio, T. G., Nolan, M., et al. (2011). Non-contiguous finished genome sequence and contextual data of the filamentous soil bacterium Ktedonobacter racemifer type strain (SOSP1-21). Stand. Genomic Sci. 5, 97–111. doi: 10.4056/sigs.2114901
Chater, K. F., and Chandra, G. (2006). The evolution of development in Streptomyces analysed by genome comparisons. FEMS Microbiol. Rev. 30, 651–672. doi: 10.1111/j.1574-6976.2006.00033.x
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chen, L. Y., Lai, Y. M., Yang, Y. L., and Zhao, X. Q. (2016). Genome mining reveals the biosynthetic potential of the marinederived strain Streptomyces marokkonensis M10. Synth. Syst. Biotechnol. 1, 56–65. doi: 10.1016/j.synbio.2016.02.005
Chen, W. H., Li, K., Guntaka, N. S., and Bruner, S. D. (2016). Interdomain and intermodule organization in epimerization domain containing nonribosomal peptide synthetases. ACS Chem. Biol. 11, 2293–2303. doi: 10.1021/acschembio.6b00332
Chevrette, M. G., Aicheler, F., Kohlbacher, O., Currie, C. R., and Medema, M. H. (2017). SANDPUMA: ensemble predictions of nonribosomal peptide chemistry reveal biosynthetic diversity across Actinobacteria. Bioinformatics 33, 3202–3210. doi: 10.1093/bioinformatics/btx400
Chu, J., Vila-Farres, X., Inoyama, D., Ternei, M., Cohen, L. J., Gordon, E. A., et al. (2016). Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat. Chem. Biol. 12, 1004–1006. doi: 10.1038/nchembio.2207
Cimermancic, P., Medema, M. H., Claesen, J., Kurita, K., Wieland, Brown, L. C., et al. (2014). Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158, 412–421. doi: 10.1016/j.cell.2014.06.034
Crooks, G. E., Hon, G., Chandonia, J. M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Darling, A. E., Jospin, G., Lowe, E., Matsen, F. A. IV, Bik, H. M., and Eisen, J. A. (2014). PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ. 2:e243. doi: 10.7717/peerj.243
Demain, A. L., and Sanchez, S. (2009). Microbial drug discovery: 80 years of progress. J. Antibiot. (Tokyo) 62, 5–16. doi: 10.1038/ja.2008.16
de Miera, L. E. S., Arroyo, P., de Luis Calabuig, E., and Ansola, G. (2014a). Effects of varying CO2 flows on bacterial communities in mesocosms created from two soils. Int. J. Greenhouse Gas Control 46, 205–214. doi: 10.1016/j.ijggc.2016.01.013
de Miera, L. E. S., Arroyo, P., de Luis Calabuig, E., Falagán, J., and Ansola, G. (2014b). High-throughput sequencing of 16S RNA genes of soil bacterial communities from a naturally occurring CO2 gas vent. Int. J. Greenhouse Gas Conrol 29, 176–184. doi: 10.1016/j.ijggc.2014.08.014
Domalaon, R., Idowu, T., Zhanel, G. G., and Schweizer, F. (2018). Antibiotic hybrids: the next generation of agents and adjuvants against gram-negative pathogens? Clin. Microbiol. Rev. 31:e00077-17. doi: 10.1128/CMR.00077-17
Donadio, S., Monciardini, P., and Sosio, M. (2007). Polyketide synthases and nonribosomal peptide synthetases: the emerging view from bacterial genomics. Nat. Prod. Rep. 24, 1073–1109. doi: 10.1039/b514050c
Doroghazi, J. R., and Metcalf, W. W. (2013). Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics 14:611. doi: 10.1186/1471-2164-14-611
Dutta, S., Whicher, J. R., Hansen, D. A., Hale, W. A., Chemler, J. A., Congdon, G. R., et al. (2014). Structure of a modular polyketide synthase. Nature 510, 512–517. doi: 10.1038/nature13423
Dziewit, L., Pyzik, A., Szuplewska, M., Matlakowska, R., Mielnicki, S., Wibberg, D., et al. (2015). Diversity and role of plasmids in adaptation of bacteria inhabiting the Lubin copper mine in Poland, an environment rich in heavy metals. Front. Microbiol. 3:152. doi: 10.3389/fmicb.2015.00152
Errington, J. (2003). Regulation of endospore formation in Bacillus subtilis. Nat. Rev. Microbiol. 1, 117–126. doi: 10.1038/nrmicro750
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Flärdh, K., and Buttner, M. J. (2009). Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat. Rev. Microbiol. 7, 36–49. doi: 10.1038/nrmicro1968
Galperin, M. Y. (2001). Conserved ‘hypothetical’ proteins: new hints and new puzzles. Comp. Funct. Genomics 2, 14–18. doi: 10.1002/cfg.66
Genilloud, O. (2017). Actinomycetes: still a source of novel antibiotics. Nat. Prod. Rep. 34, 1203–1232. doi: 10.1039/c7np00026j
Grant, J. R., and Stothard, P. (2008). The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, W181–W184. doi: 10.1093/nar/gkn179
Gupta, R. S., Chander, P., and George, S. (2013). Phylogenetic framework and molecular signatures for the class Chloroflexi and its different clades; proposal for division of the class Chloroflexia class. nov. into the suborder Chloroflexineae subord. nov., consisting of the emended family Oscillochloridaceae and the family Chloroflexaceae fam. nov., and the suborder Roseiflexineae subord. nov., containing the family Roseiflexaceae fam. nov. Antonie Van Leeuwenhoek 103, 99–119. doi: 10.1007/s10482-012-9790-3
Han, K., Li, Z. F., Peng, R., Zhu, L. P., Zhou, T., Wang, L. G., et al. (2013). Extraordinary expansion of a Sorangium cellulosum genome from an alkaline milieu. Sci. Rep. 3:2101. doi: 10.1038/srep02101
Hayashi, T., Kitamura, Y., Funa, N., Ohnishi, Y., and Horinouchi, S. (2011). Fatty acyl-AMP ligase involvement in the production of alkylresorcylic acid by a Myxococcus xanthus type III polyketide synthase. Chembiochem 12, 2166–2176. doi: 10.1002/cbic.201100344
Hisanaga, Y., Ago, H., Nakagawa, N., Hamada, K., Ida, K., Yamamoto, M., et al. (2004). Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J. Biol. Chem. 279, 31717–31726. doi: 10.1074/jbc.M400100200
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Ibal, J. C., Pham, H. Q., Park, C. E., and Shin, J. H. (2019). Information about variations in multiple copies of bacterial 16S rRNA genes may aid in species identification. PLoS One 14:e0212090. doi: 10.1371/journal.pone.0212090
Islam, Z. F., Cordero, P. F. R., Feng, J., Chen, Y.-J., Bay, S. K., Jirapanjawat, T., et al. (2019). Two Chloroflexi classes independently evolved the ability to persist on atmospheric hydrogen and carbon monoxide. ISME J. doi: 10.1038/s41396-019-0393-0
Jenke-Kodama, H., and Dittmann, E. (2009). Evolution of metabolic diversity: insights from microbial polyketide synthases. Phytochemistry 70, 1858–1866. doi: 10.1016/j.phytochem.2009.05.021
Jenke-Kodama, H., Sandmann, A., Müller, R., and Dittmann, E. (2005). Evolutionary implications of bacterial polyketide synthases. Mol. Biol. Evol. 22, 2027–2039. doi: 10.1093/molbev/msi193
Jensen, P. R. (2016). Natural products and the gene cluster revolution. Trends Microbiol. 24, 968–977. doi: 10.1016/j.tim.2016.07.006
Jiang, X., Ellabaan, M. M., Charusanti, P., Munck, C., Blin, K., Tong, Y., et al. (2017). Dissemination of antibiotic resistance genes from antibiotic producers to pathogens. Nat. Commun. 8:15784. doi: 10.1038/ncomms15784
Jiang, Z., Li, P., Jiang, D., Dai, X., Zhang, R., Wang, Y., et al. (2016). Microbial community structure and arsenic biogeochemistry in an acid vapor-formed spring in Tengchong Geothermal Area, China. PLoS One 11:e0146331. doi: 10.1371/journal.pone.0146331
Johnston, C. W., Skinnider, M. A., Wyatt, M. A., Li, X., Ranieri, M. R., Yang, L., et al. (2015). An automated genomes-to-natural products platform (GNP) for the discovery of modular natural products. Nat. Commun. 6:8421. doi: 10.1038/ncomms9421
Kaster, A. K., Mayer-Blackwell, K., Pasarelli, B., and Spormann, A. M. (2014). Single cell genomic study of Dehalococcoidetes species from deep-sea sediments of the Peruvian Margin. ISME J. 8, 1831–1842. doi: 10.1038/ismej.2014.24
Kelly, S. L., and Kelly, D. E. (2013). Microbial cytochromes P450: biodiversity and biotechnology. Where do cytochromes P450 come from, what do they do and what can they do for us? Philos. Trans. R. Soc. Lond. B Biol. Sci. 6:358. doi: 10.1098/rstb.2012.0476
King, C. E., and King, G. M. (2014). Description of Thermogemmatispora carboxidivorans sp. nov., a carbon-monoxide-oxidizing member of the class Ktedonobacteria isolated from a geothermally heated biofilm, and analysis of carbon monoxide oxidation by members of the class Ktedonobacteria. Int. J. Syst. Evol. Microbiol. 64, 1244–1251. doi: 10.1099/ijs.0.059675-0
Komaki, H., Hosoyama, A., Yabe, S., Yokota, A., Uchino, Y., and Takano, H. (2016). Draft genome sequence of Thermogemmatispora onikobensis NBRC 111776T, an aerial mycelium- and spore-forming thermophilic bacterium belonging to the class Ktedonobacteria. Genome Announc. 4:e1156-16. doi: 10.1128/genomeA.01156-16
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., and Kobayashi, I. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240. doi: 10.1016/S0140-6736(00)04403-2
Landry, Z., Swan, B. K., Herndl, G. J., Stepanauskas, R., and Giovannoni, S. J. (2017). SAR202 genomes from the dark ocean predict pathways for the oxidation of recalcitrant dissolved organic matter. MBio 8, e413–e417. doi: 10.1128/mBio.00413-17
Lawrence, J. (1999). Selfish operons: the evolutionary impact of gene clustering in prokaryotes and eukaryotes. Curr. Opin. Genet. Dev. 9, 642–648. doi: 10.1016/S0959-437X(99)00025-8
Lázár, V., Martins, A., Spohn, R., Daruka, L., Grézal, G., Fekete, G., et al. (2018). Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat. Microbiol. 3, 718–731. doi: 10.1038/s41564-018-0164-0
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Letzel, A. C., Pidot, S. J., and Hertweck, C. (2014). Genome mining for ribosomally synthesized and post-translationally modified peptides (RiPPs) in anaerobic bacteria. BMC Genomics 15:983. doi: 10.1186/1471-2164-15-983
Li, B., and Walsh, C. T. (2010). Identification of the gene cluster for the dithiolopyrrolone antibiotic holomycin in Streptomyces clavuligerus. Proc. Natl. Acad. Sci. U.S.A. 107, 19731–19735. doi: 10.1073/pnas.1014140107
Liu, F., Garneau, S., and Walsh, C. T. (2004). Hybrid nonribosomal peptide-polyketide interfaces in epothilone biosynthesis: minimal requirements at N and C termini of EpoB for elongation. Chem. Biol. 11, 1533–1542. doi: 10.1016/j.chembiol.2004.08.017
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Manivasagan, P., Venkatesan, J., Sivakumar, K., and Kim, S. K. (2014). Pharmaceutically active secondary metabolites of marine actinobacteria. Microbiol. Res. 169, 262–278. doi: 10.1016/j.micres.2013.07.014
Masschelein, J., Jenner, M., and Challis, G. L. (2017). Antibiotics from gram-negative bacteria: a comprehensive overview and selected biosynthetic highlights. Nat. Prod. Rep. 34, 712–783. doi: 10.1039/c7np00010c
Miller, D. A., Luo, L., Hillson, N., Keating, T. A., and Walsh, C. T. (2002). Yersiniabactin synthetase: a four-protein assembly line producing the nonribosomal peptide/polyketide hybrid siderophore of Yersinia pestis. Chem. Biol. 9, 333–344.
Miller, S. I. (2016). Antibiotic resistance and regulation of the gram-negative bacterial outer membrane barrier by host innate immune molecules. MBio 7, e1541–e1516. doi: 10.1128/mBio.01541-16
Moe, W. M., Yan, J., Nobre, M. F., da Costa, M. S., and Rainey, F. A. (2009). Dehalogenimonas lykanthroporepellens gen. nov., sp. nov., a reductively dehalogenating bacterium isolated from chlorinated solvent-contaminated groundwater. Int. J. Syst. Evol. Microbiol. 59, 2692–2697. doi: 10.1099/ijs.0.011502-0
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Naughton, L. M., Romano, S., O’Gara, F., and Dobson, A. D. W. (2017). Identification of secondary metabolite gene clusters in the Pseudovibrio genus reveals encouraging biosynthetic potential toward the production of novel bioactive compounds. Front. Microbiol. 8:1494. doi: 10.3389/fmicb.2017.01494
Northup, D. E., Melim, L. A., Spilde, M. N., Hathaway, J. J., Garcia, M. G., Moya, M., et al. (2011). Lava cave microbial communities within mats and secondary mineral deposits: implications for life detection on other planets. Astrobiology 11, 601–618. doi: 10.1089/ast.2010.0562
Okada, Y. (1937). Occurrence of masses of gelatinous microbes in the soil. Soil Sci. 43, 367–374. doi: 10.1097/00010694-193705000-00005
Omura, S., Ikeda, H., Ishikawa, J., Hanamoto, A., Takahashi, C., Shinose, M., et al. (2001). Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. U.S.A. 98, 12215–12220. doi: 10.1073/pnas.211433198
Ortega, M. A., Hao, Y., Zhang, Q., Walker, M. C., van der Donk, W. A., and Nair, S. K. (2015). Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB. Nature 517, 509–512. doi: 10.1038/nature13888
Ortega, M. A., and van der Donk, W. A. (2016). New insights into the biosynthetic logic of ribosomally synthesized and post-translationally modified peptide natural products. Cell Chem. Biol. 23, 31–44. doi: 10.1016/j.chembiol.2015.11.012
Pal, C., Bengtsson-Palme, J., Kristiansson, E., and Larsson, D. G. (2016). The structure and diversity of human, animal and environmental resistomes. Microbiome 4:54. doi: 10.1186/s40168-016-0199-5
Park, J. S., Kagaya, N., Hashimoto, J., Izumikawa, M., Yabe, S., Shin-ya, K., et al. (2014). Identification and biosynthesis of new acyloins from the thermophilic bacterium Thermosporothrix hazakensis SK20-1(T). Chembiochem 15, 527–532. doi: 10.1002/cbic.201300690
Park, J. S., Yabe, S., Shin-ya, K., Nishiyama, M., and Kuzuyama, T. (2015). New 2-(1’H-indole-3’-carbonyl)-thiazoles derived from the thermophilic bacterium Thermosporothrix hazakensis SK20-1(T). J. Antibiot. (Tokyo) 68, 60–62. doi: 10.1038/ja.2014.93
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/.journal.pone.0009490
Rausch, C., Hoof, I., Weber, T., Wohlleben, W., and Huson, D. H. (2007). Phylogenetic analysis of condensation domains in NRPS sheds light on their functional evolution. BMC Evol. Biol. 7:78. doi: 10.1186/1471-2148-7-78
Romaniuk, K., Golec, P., and Dziewit, L. (2018). Insight into the diversity and possible role of plasmids in the adaptation of psychrotolerant and metalotolerant Arthrobacter spp. to extreme Antarctic environments. Front. Microbiol. 9:3144. doi: 10.3389/fmicb.2018.03144
Röttig, M., Medema, M. H., Blin, K., Weber, T., Rausch, C., and Kohlbacher, O. (2011). NRPSpredictor2–a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 39, W362–W367. doi: 10.1093/nar/gkr323
Rutledge, P. J., and Challis, G. L. (2015). Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 13, 509–523. doi: 10.1038/nrmicro3496
Sabath, N., Ferrada, E., Barve, A., and Wagner, A. (2013). Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol. Evol. 5, 966–977. doi: 10.1093/gbe/evt050
Sekiguchi, Y., Ohashi, A., Parks, D. H., Yamauchi, T., Tyson, G. W., and Hugenholtz, P. (2015). First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ 3:e740. doi: 10.7717/peerj.740
Shen, B. (2003). Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 7, 285–295. doi: 10.1016/s1367-5931(03)00020-6
Shirling, E. B., and Gottlieb, D. (1966). Methods for characterization of Streptomyces species. Int. J. Syst. Bacteriol. 16, 313–340. doi: 10.1099/00207713-16-3-313
Shou, Q., Feng, L., Long, Y., Han, J., Nunnery, J. K., Powell, D. H., et al. (2016). A hybrid polyketide-nonribosomal peptide in nematodes that promotes larval survival. Nat. Chem. Biol. 12, 770–772. doi: 10.1038/nchembio.2144
Soo, R. M., Skennerton, C. T., Sekiguchi, Y., Imelfort, M., Paech, S. J., Dennis, P. G., et al. (2014). An expanded genomic representation of the phylum Cyanobacteria. Genome Biol. Evol. 6, 1031–1045. doi: 10.1093/gbe/evu073
Soupene, E., and Kuypers, F. A. (2008). Mammalian long-chain acyl-CoA synthetases. Exp. Biol. Med. (Maywood) 233, 507–521. doi: 10.3181/0710-MR-287
Stachelhaus, T., Mootz, H. D., and Marahiel, M. A. (1999). The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 6, 493–505. doi: 10.1016/S1074-5521(99)80082-9
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Stres, B., Sul, W. J., Murovec, B., and Tiedje, J. M. (2013). Recently deglaciated high-altitude soils of the Himalaya: diverse environments, heterogenous bacterial communities and long-range dust inputs from the upper troposphere. PLoS One 8:e76440. doi: 10.1371/journal.pone.0076440
Sun, L., Toyonaga, M., Ohashi, A., Tourlousse, D. M., Matsuura, N., Meng, X. Y., et al. (2016). Lentimicrobium saccharophilum gen. nov., sp. nov., a strictly anaerobic bacterium representing a new family in the phylum Bacteroidetes, and proposal of Lentimicrobiaceae fam. nov. Int. J. Syst. Evol. Microbiol. 66, 2635–2642. doi: 10.1099/ijsem.0.001103
Swofford, D. L. (2003). PAUP∗: Phylogenetic Analysis Using Parsimony, Version 4.0b10. Available at: https://paup.phylosolutions.com/ (accessed September 2018).
Tanizawa, Y., Fujisawa, T., and Nakamura, Y. (2018). DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34, 1037–1039. doi: 10.1093/bioinformatics/btx713
Tebo, B. M., Davis, R. E., Anitori, R. P., Connell, L. B., Schiffman, P., and Staudigel, H. (2015). Microbial communities in dark oligotrophic volcanic ice cave ecosystems of Mt. Erebus, Antarctica. Front. Microbiol. 6:179. doi: 10.3389/fmicb.2015.00179
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Thomas, C. M., and Summers, D. (2008). “Bacterial plasmids,” in Encyclopedia of Life Sciences (ELS), Chichester: John Wiley & Sons, Ltd. doi: 10.1002/9780470015902.a0000468.pub2
Tooming-Klunderud, A., Sogge, H., Rounge, T. B., Nederbragt, A. J., Lagesen, K., Glöckner, G., et al. (2013). From green to red: horizontal gene transfer of the phycoerythrin gene cluster between Planktothrix strains. Appl. Environ. Microbiol. 79, 6803–6812. doi: 10.1128/AE01455-13
van Heel, A. J., de Jong, A., Montalbán-López, M., Kok, J., and Kuipers, O. P. (2013). BAGEL3: automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 41, W448–W453. doi: 10.1093/nar/gkt391
Vila-Farres, X., Chu, J., Inoyama, D., Ternei, M. A., Lemetre, C., Cohen, L. J., et al. (2017). Antimicrobials inspired by nonribosomal peptide synthetase gene clusters. J. Am. Chem. Soc. 139, 1404–1407. doi: 10.1021/jacs.6b11861
Wang, C. M., Zheng, Y., Sakai, Y., Toyoda, A., Minakuchi, Y., Abe, K., et al. (2019). Tengunoibacter tsumagoiensis gen. nov., sp. nov., Dictyobacter kobayashii sp. nov., Dictyobacter alpinus sp. nov., and description of Dictyobacteraceae fam. nov. within the order Ktedonobacterales isolated from Tengu-no-mugimeshi, a soil-like granular mass of microorganisms, and emended descriptions of the genera Ktedonobacter and Dictyobacter. Int. J. Syst. Evol.Microbiol. 69 (in press).
Willey, J. M., and van der Donk, W. A. (2007). Lantibiotics: peptides of diverse structure and function. Annu. Rev. Microbiol. 61, 477–501. doi: 10.1146/annurev.micro.61.080706.093501
Wu, D., Raymond, J., Wu, M., Chatterji, S., Ren, Q., Graham, J. E., et al. (2009). Complete genome sequence of the aerobic CO-oxidizing thermophile Thermomicrobium roseum. PLoS One 4:e4207. doi: 10.1371/journal.pone.0004207
Xu, B., Aitken, E. J., Baker, B. P., Turner, C. A., Harvey, J. E., Stott, M. B., et al. (2018). Genome mining, isolation, chemical synthesis and biological evaluation of a novel lanthipeptide, tikitericin, from the extremophilic microorganism Thermogemmatispora strain T81. Chem. Sci. 9, 7311–7317. doi: 10.1039/c8sc02170h
Yabe, S., Aiba, Y., Sakai, Y., Hazaka, M., and Yokota, A. (2010). Thermosporothrix hazakensis gen. nov., sp. nov., isolated from compost, description of Thermosporotrichaceae fam. nov. within the class Ktedonobacteria Cavaletti et al. 2007 and emended description of the class Ktedonobacteria. Int. J. Syst. Evol. Microbiol. 60, 1794–1801. doi: 10.1099/ijs.0.018069-0
Yabe, S., Aiba, Y., Sakai, Y., Hazaka, M., and Yokota, A. (2011). Thermogemmatispora onikobensis gen. nov., sp. nov. and Thermogemmatispora foliorum sp. nov., isolated from fallen leaves on geothermal soils, and description of Thermogemmatisporaceae fam. nov. and Thermogemmatisporales ord. nov. within the class Ktedonobacteria. Int. J. Syst. Evol. Microbiol. 61, 903–910. doi: 10.1099/ijs.0.024877-0
Yabe, S., Sakai, Y., Abe, K., and Yokota, A. (2017a). Diversity of Ktedonobacteria with actinomycetes-like morphology in terrestrial environments. Microbes Environ. 31, 61–70. doi: 10.1264/jsme2.ME16144
Yabe, S., Sakai, Y., Abe, K., Yokota, A., Také, A., Matsumoto, A., et al. (2017b). Dictyobacter aurantiacus gen. nov., sp. nov., a member of the family Ktedonobacteraceae, isolated from soil, and emended description of the genus Thermosporothrix. Int. J. Syst. Evol. Microbiol. 67, 2615–2621. doi: 10.1099/ijsem.0.001985
Yabe, S., Wang, C. M., Zheng, Y., Sakai, Y., Abe, K., and Yokota, A. (2019). Formation of sporangiospores in Dictyobacter aurantiacus (class Ktedonobacteria in phylum Chloroflexi). J. Gen. Appl. Microbiol. 65 (in press).
Yarza, P., Ludwig, W., Euzéby, J., Amann, R., Schleifer, K. H., Glöckner, F. O., et al. (2010). Update of the all-species living tree project based on 16S and 23S rRNA sequence analyses. Syst. Appl. Microbiol. 33, 291–299. doi: 10.1016/j.syapm.2010.08.001
Zhang, Q., Yu, Y., Vélasquez, J. E., and van der Donk, W. A. (2012). Evolution of lanthipeptide synthetases. Proc. Natl. Acad. Sci. U.S.A. 109, 18361–18366. doi: 10.1073/pnas.1210393109
Ziemert, N., Lechner, A., Wietz, M., Millán-Aguiñaga, N., Chavarria, K. L., and Jensen, P. R. (2014). Diversity and evolution of secondary metabolism in the marine actinomycete genus Salinispora. Proc. Natl. Acad. Sci. U.S.A. 111, E1130–E1139. doi: 10.1073/pnas1324161111
Keywords: class Ktedonobacteria, whole genome sequencing, phylogenetic analysis, genome comparisons, NRPS, PKS, antimicrobial screening
Citation: Zheng Y, Saitou A, Wang C-M, Toyoda A, Minakuchi Y, Sekiguchi Y, Ueda K, Takano H, Sakai Y, Abe K, Yokota A and Yabe S (2019) Genome Features and Secondary Metabolites Biosynthetic Potential of the Class Ktedonobacteria. Front. Microbiol. 10:893. doi: 10.3389/fmicb.2019.00893
Received: 18 January 2019; Accepted: 08 April 2019;
Published: 26 April 2019.
Edited by:
Frank T. Robb, University of Maryland, Baltimore, United StatesReviewed by:
Xiaoying Bian, Shandong University, ChinaMarla Trindade, University of the Western Cape, South Africa
Copyright © 2019 Zheng, Saitou, Wang, Toyoda, Minakuchi, Sekiguchi, Ueda, Takano, Sakai, Abe, Yokota and Yabe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuhei Yabe, c2h1aGVpLnlhYmUuYjJAdG9ob2t1LmFjLmpw; a2VubmFuLTZAa2VubmFuLWUuY28uanA=