 Juan Li
Juan Li Haiying Jiang
Haiying Jiang Linmiao Li
Linmiao Li Xiujuan Zhang
Xiujuan Zhang Jinping Chen
Jinping Chen- Guangdong Key Laboratory of Animal Conservation and Resource Utilization, Guangdong Public Laboratory of Wild Animal Conservation and Utilization, Guangdong Institute of Applied Biological Resources, Guangzhou, China
Increasing evidence has manifested that the gut bacterial microbiota of shrimps is closely related to the environmental factors, host developmental stage and health status like that of humans and animals does. These studies have provided an important guidance for improving shrimp culture benefits. In practice, aside from bacteria, eukaryotic microorganisms dominated by fungal microbiota (mycobiota), also play a key role in host growth, metabolism and homeostasis. However, little so far is known about the mycobiota in the digestive tract of shrimp. In this study, we used high-throughput sequencing of internal transcribed spacer 1 region to characterize the hepatopancreas and intestinal mycobiota of Pacific white shrimp and their connections with disease incidence and seasonal variation. The results showed that the hepatopancreas and intestinal mycobiota of Litopenaeus vannamei are dominated by the phyla Ascomycota and Basidiomycota, and the genera Alternaria, Tuber, Hortaea, Sarocladium, and Stagonospora. The fungal microbiota significantly varies under the influence of disease and seasonal variation. Sick shrimps had a higher level of potential pathogenic fungus, Candida in the intestine. Healthy shrimps had a higher abundance of the genera Didymella and Filobasidium in the gut, and Pyrenochaetopsis in the hepatopancreas. Of note, most of the fungi carried by Pacific white shrimps were pathogens to humans. This study has revealed the intestinal and hepatopancreas mycobiota of L. vannamei and the effects of diseases and seasonal variation to the mycobiota. Our study provides important guidance for Pacific white shrimp farming and sheds further insight on the fungal microbiota.
Introduction
The digestive system hosts a large number of microorganisms dominated by bacteria, which constitute an enormous microbial ecosystem called the microbiota (O’Hara and Shanahan, 2006; Clemente et al., 2012). The microbiota has multiple functions, such as the maintenance of the functional stability and metabolic balance in the digestive system (Xue et al., 2015; Sommer et al., 2016; Sonnenburg and Backhed, 2016) and the regulation of the host immune response (Hooper et al., 2012; Palm et al., 2015). Therefore, it is critical for the host. Although the bacterial microbiota is dominant, eukaryotic microorganisms also occur (Neville et al., 2015; Sam et al., 2017). Fungi are the paramount eukaryotic microbiota (Neville et al., 2015). Recently, many studies showed that the gut fungal population was tightly correlated to host immune- and metabolic-modulatory pathways (Mar Rodriguez et al., 2015; McAleer et al., 2016; Huseyin et al., 2017; Sokol et al., 2017) and are thus receiving an increasing amount of attention.
Crustaceans are the second most species-rich subphylum in Arthropod. Of these, shrimps have attracted extensive attention because of their commercial importance. It has been reported that shrimp farming can reach 3 million tons of production each year (Zhang et al., 2016). Pacific white shrimp, Litopenaeus vannamei, is the most popular shrimp species, contributing to their survival advantages, such as fast growth rate and strong adaptability to environment. However, high frequency of diseases has severely influenced the shrimp farming. Recent studies revealed that the intestinal bacterial microbiota of the Pacific white shrimp is closely related to host age, health conditions and immune response (Xiong et al., 2015; Cornejo-Granados et al., 2017). Stable enteric bacterial microbiota enhances the immunity of the shrimp and their resistance to external pathogens (Rungrassamee et al., 2016). Based on the results, a novel farming approach that aggregates the shrimp tolerance to diseases by maintaining or reestablishing a “healthy” gut microbiota has been developed and brought deep and positive effects on shrimp farming (Xiong, 2018). Besides, a study of pacific white shrimps has disclosed that the gut bacterial community regularly varies over shrimp disease progression and developmental stage (Xiong et al., 2017). The new findings led to that a more accurate earlier diagnosis approach that monitors microbiota has appeared and has gradually been applied to shrimp farming (Xiong et al., 2017; Xiong, 2018).
Of note, a recent study targeted the 18S rRNA gene to uncover that the gut eukaryotic microbiota of cohabitating shrimps affects the digestion and nutrition absorption and consequently alter the growth performance (Dai et al., 2017). However, the fungal community, as the most influential population in the gut eukaryotic microbiota, has not been clearly explored due to the insufficiency of sequencing depth in that study. In addition, a culture-based study found that there were various fungal pathogens in the body of the Pacific white shrimp (da Silva et al., 2011; Karthikeyan et al., 2015), which may influence the health status of the farmed shrimp. However, the number of fungi that can be cultured were limited (Huffnagle and Noverr, 2013), and the interspecies ecological interaction that existed inside the digestive tract has also barely been detected based on the conventional culture-dependent method.
In addition, differing from that of vertebrates, the shrimp digestive system is continuous and is comprised of the stomach, hepatopancreas and intestine. Of these, the hepatopancreas plays an important role in regulating host innate immunity, and it is also a vital organ, which is responsible for digestion and absorption (Cornejo-Granados et al., 2017). However, to date, few studies have focused on the bacterial microbiota in the other digestive organs apart from the intestine, not to mention the fungal microbiota. Interestingly, several recent studies of the shrimp gut bacteria found that aside from the intestine, the hepatopancreas is also a habitat of many bacteria, which can respond to the variation of environment and health condition and is closely related to the host diet (Cheung et al., 2015; Cornejo-Granados et al., 2017). However, the variation of the mycobiota in the different parts of shrimp digestive tract, such as hepatopancreas and intestine, remains unrevealed.
In this study, we used high-throughput sequencing technology to analyze the hepatopancreas and intestinal fungal microbiota of L. vannamei. The aim was to understand: (1) the composition of the hepatopancreas and enteric fungal microbiota, (2) the seasonal effects on the composition of the hepatopancreas and enteric mycobiota, and (3) the effects of disease on the composition of the hepatopancreas and enteric mycobiota of L. vannamei.
Materials and Methods
Ethics Statement
All experimental animal protocols in this study were approved by the committee on the Ethics of Animal Experiments of the Guangdong Institute of Applied Biological Resources and followed the basic principles.
Sampling
In summer, according to the disease status, three representative infected pools and a pool that cultured healthy shrimps were chosen. In winter, we randomly chose four pools for sampling as no diseased shrimps have been found. All of the culture pools chosen in this study have used the same artificial food (HAID GROUP, China). A total of 50 white shrimps from eight farms were sampled, as shown in Table 1. To avoid contamination, the live shrimp that was fished with a screen was immediately placed in 50 mL sterilized collection tubes. The collection tubes were put into ice to quickly bring back to the local lab. Each shrimp was thoroughly washed with sterile water and placed in a sterile plate. The washed samples were placed on the sterile workstation for biopsies to collect the hepatopancreas and intestinal tissue. The tissue removed was immediately placed in 2 mL EP tubes and quickly placed in dry ice. The samples were stored at −80°C in the lab until the nucleic acids were extracted.
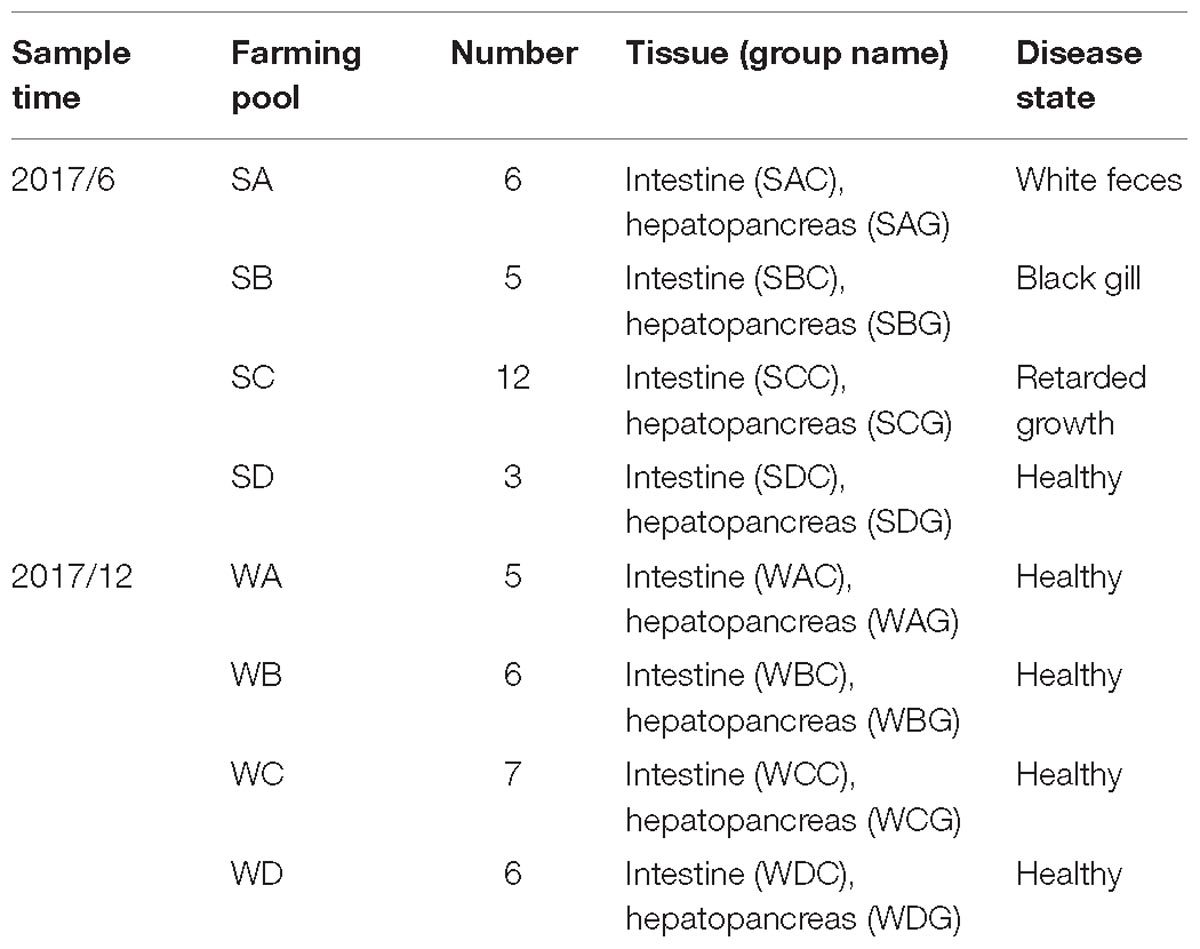
Table 1. Shrimp samples used to detect hepatopancreas and intestinal fungi.
DNA Extraction, PCR Amplification, and Sequencing
For each sample, the total genomic DNA was extracted using a PowerSoil® DNA Isolation Kit (MO BIO, United States) according to the manufacturer’s instructions. The DNA isolation was quantified using a ND-2000C spectrophotometer (NanoDrop, United States). Internal transcribed spacer 1 region (ITS1) was amplified using the universal primer set ITS1F 5′-CTTGGTCATTTAGAGGAAGTAA-3′ and ITS2 5′-GCTGCGTTCTTCATCGATGC-3′ (Mukherjee et al., 2014; Li et al., 2017). PCR reactions were performed using Phusion® Q5 High-Fidelity DNA Polymerase (New England Biolabs, United Kingdom) under the program [95°C for 5 min, 15 × (95°C for 30 s, 50°C for 30 s, 72°C for 40 s), 72°C for 7 min, hold at 4°C; 98°C for 30 min, 15 × (98°C for 30 s, 65°C for 30 s, 72°C for 30 s), 72°C for 5 min, hold at 4°C]. Sterilized water was used as the negative control. The primer set attaching barcode sequences was used during the second PCR amplification. After agarose gel electrophoresis, the PCR products was purified by using VAHTSTM DNA Clean Beads (Vazyme, China), followed by Solexa PCR, second bead purification. The final products were pooled to generate the sequencing libraries. The libraries were sequenced on an Illumina HiSeq platform (Illumina Hiseq 2500) using HiSeq Rapid Kit V2 (500 cycle) (Illumina, United States) following the manufacturer’s instructions.
Data Processing
Based on the unique barcodes, we assigned the sequence to each sample before removing the barcode and primer sequence using QIIME v1.8.0 (Caporaso et al., 2010). FLASH v1.2.7 was used to perform merging of the PE reads to obtain merged sequences (raw tags) (Magoc and Salzberg, 2011). The minimum length of the overlap was set to 10 bp, and the maximum mismatch ratio in the overlap area was set to 0.2 (Magoc and Salzberg, 2011). Trimmomatic v 0.33 was applied to conduct a comparatively stringent quality control to generate a set of high quality clean tags (Bolger et al., 2014), followed by removing the chimeric sequences using UCHIME v 4.2 (Edgar et al., 2011) and the non-fungal sequences with ITSx v 1.1.1 (Bengtsson-Palme et al., 2013). Later, the high-quality effective tags were clustered to generate operational taxonomic units (OTUs) using UCLUST v 1.2.22 at 97% similarity (Edgar, 2013). Low-abundant OTUs (the number of sequence was below three) were filtered, and normalized OTUs were generated (Bokulich et al., 2013). According to the UNITE database v 7.21 (Koljalg et al., 2013), the representative sequence of each OTU was annotated with RDP Classifier v 2.2 (Wang et al., 2007).
Statistical Analysis
The normalized OTUs were used to plot the species distribution bar charts at each taxonomic level, followed by the construction of phylogenetic trees using ClustalW22.
The abundance data were used to calculate the α diversity (Chao 1, ACE, Richness, Shannon–Wiener and Simpson diversity indices) using Mothur v.1.3.03 (Schloss et al., 2009). The differences among the groups were compared using a one-way analysis of variance (ANOVA). We plotted the rarefaction curve and the species relative abundance curve to evaluate the sufficiency of the sequencing depth (Wang et al., 2012; Koljalg et al., 2013).
The β diversity of each sample group was calculated based on the Bray–Curtis distance. Principal coordinate analysis was completed with the R package “ape,” and a clustering dendrogram was constructed using an unweighted pair-group method with arithmetic mean (UPGMA). The differences of β diversity between the groups were detected with a permutational multivariate analysis of variance (PERMANOVA).
Linear discriminative analysis effect size (LEfSe) analysis was performed at each taxonomic level to screen for the molecular markers (Biomarkers) of each group (Segata et al., 2011).
Sequence Data Accession Number
The sequencing data generated from the one hundred samples described in this study are available in a sequence read archive (SRA) at the NCBI under the accession number PRJNA495902.
Results
Assessment of Sequence Data
One hundred samples were sequenced on the Illumina Hiseq 2500 platform to generate 12.56 Gb dataset and 9,501,484 pairs of reads. Of those, 8,960,483 tags passed the strict quality control and were processed into 8,735,797 high-quality chimera-free effective tags (87,358 on average) (Supplementary Table S1). A total of 9,804 OTUs were generated at 97% similarity (Supplementary Table S2). Subsequently, the OTUs, that were below three in the sum of the sequence number, were screened to obtain 8,491 effective OTUs (Supplementary Figure S1). The diversity indices were calculated based on the OTU data. The rarefaction curves and the species relative abundance curve reached a plateau, reflecting the sufficiency the sequencing depth (Supplementary Figures S2, S3).
General Pattern of the Gut and Hepatopancreas Mycobiota of Litopenaeus vannamei
A total of 17 phyla, 53 classes, 127 orders, 325 families, 734 genera, and 990 species were identified from the classifiable sequences. As shown in Figure 1A, Ascomycota was the most dominant phylum and occupied 65.32% of the total fungal community, followed by Basidiomycota (12.45%). Other phyla in low abundance were, in descending order, Mortierellomycota (1.19%), Chytridiomycota (0.21%), and Neocallimastigomycota (0.19%).
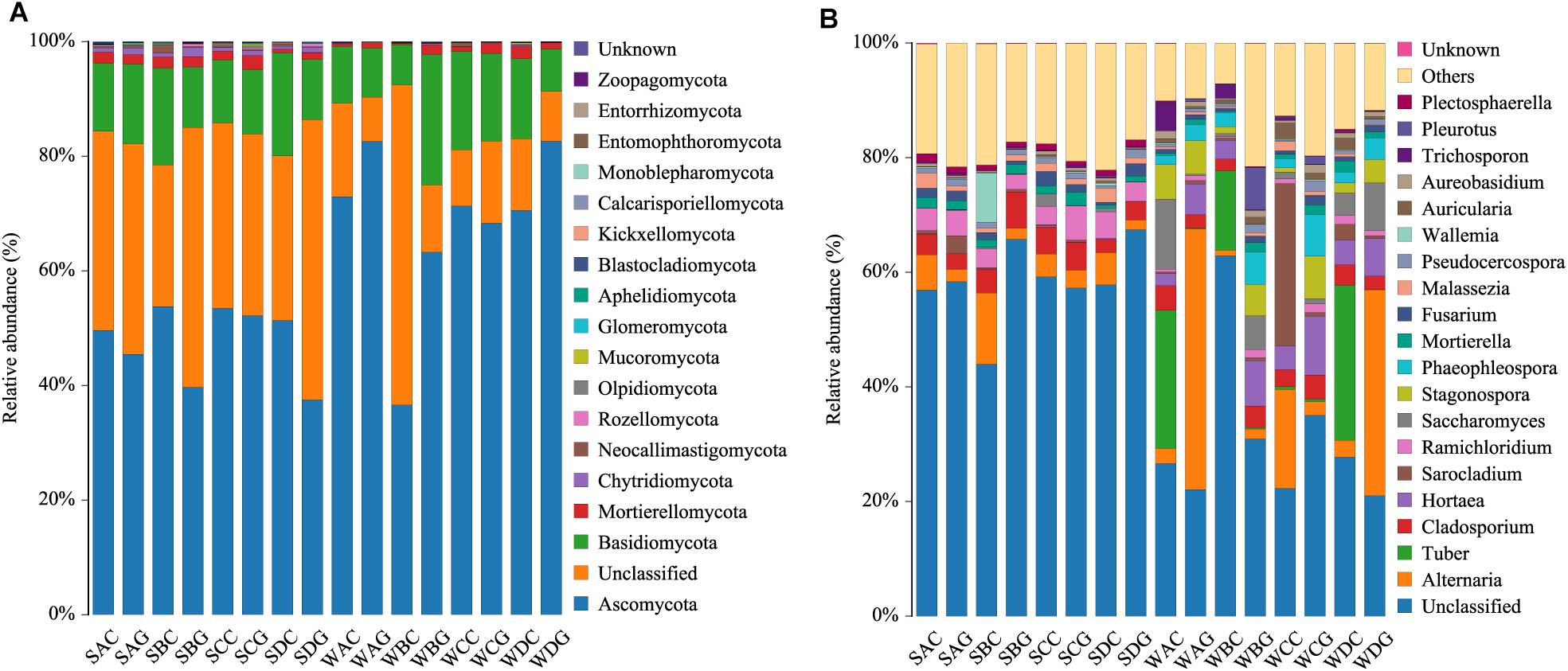
Figure 1. Species distribution of each group in 50 shrimps. Panel (A) at the phylum level; panel (B) at the genus level (top 20=). The sample ID referred to Table 1.
At the genus level, the top 20 members were affiliated with Alternaria (12.83%), Tuber (5.97%), Hortaea (4.93%), Sarocladium (3.95%), Stagonospora (3.47%), Saccharomyces (3.43%), Cladosporium (3.28%), Phaeophleospora (3.08%), Ramichloridium (1.37%), Mortierella (1.13%), Fusarium (1.05%), Auricularia (1.00%), Pleurotus (0.94%), Polyporus (0.93%), Pseudocercospora (0.86%), Trichosporon (0.84%), Aureobasidium (0.84%), Curvularia (0.74%), Malassezia (0.69%), and Candida (0.59%) (Figure 1B).
Comparison Between the Gut and Hepatopancreas Mycobiota
A comparison of the α diversity was conducted between the intestinal and hepatopancreatic fungal microbiota. The significance between the groups was analyzed using an ANOVA. To eliminate the interference of diseases, when analyzing the difference between the summer intestinal and hepatopancreas samples, we only chose the samples collected from pool SD (the intestinal group as SDC, the hepatopancreas group as SDG). The results showed that there were no detectable significant differences in the richness estimator and diversity estimator between the summer intestinal and hepatopancreas group (Supplementary Table S3A). For β diversity, although UPGMA cluster analysis seemed to show that the summer samples divided into two clades, the PCoA and PERMANOVA results both showed an overlap of the two group. This indicated the high similarity in community composition and diversity between the two groups (Figure 2 and Table 2). LEfSe analysis was used to capture the distinguished fungi between the groups. Basidiomycota, Pleosporales and Didymellacea showed a higher level in the intestinal group, and Sordariomycetes was significantly enriched in the hepatopancreas group. However, no typical representative fungi were found in genus and species level (Figure 3A).
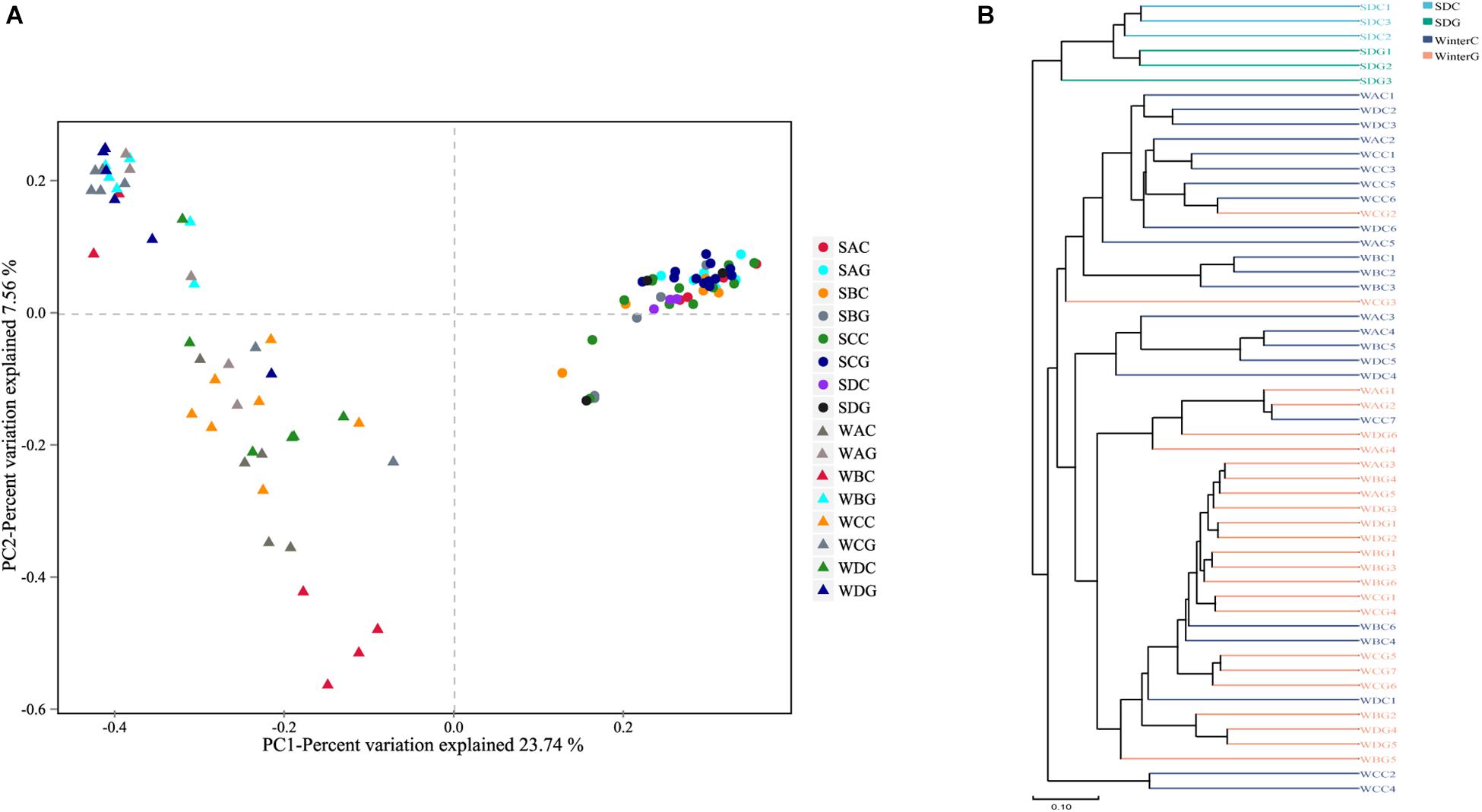
Figure 2. Boxplots depict differences of beta diversity between intestinal and hepatopancreas samples. (A) Summer shrimps; (B) winter shrimps. Beta diversity of intestinal and hepatopancreas mycobiota. (A) Principal coordinate analysis (PcoA) was based on Bray–Curtis distance. (B) A clustering dendrogram was constructed using an unweighted pair-group method with arithmetic mean (UPGMA).
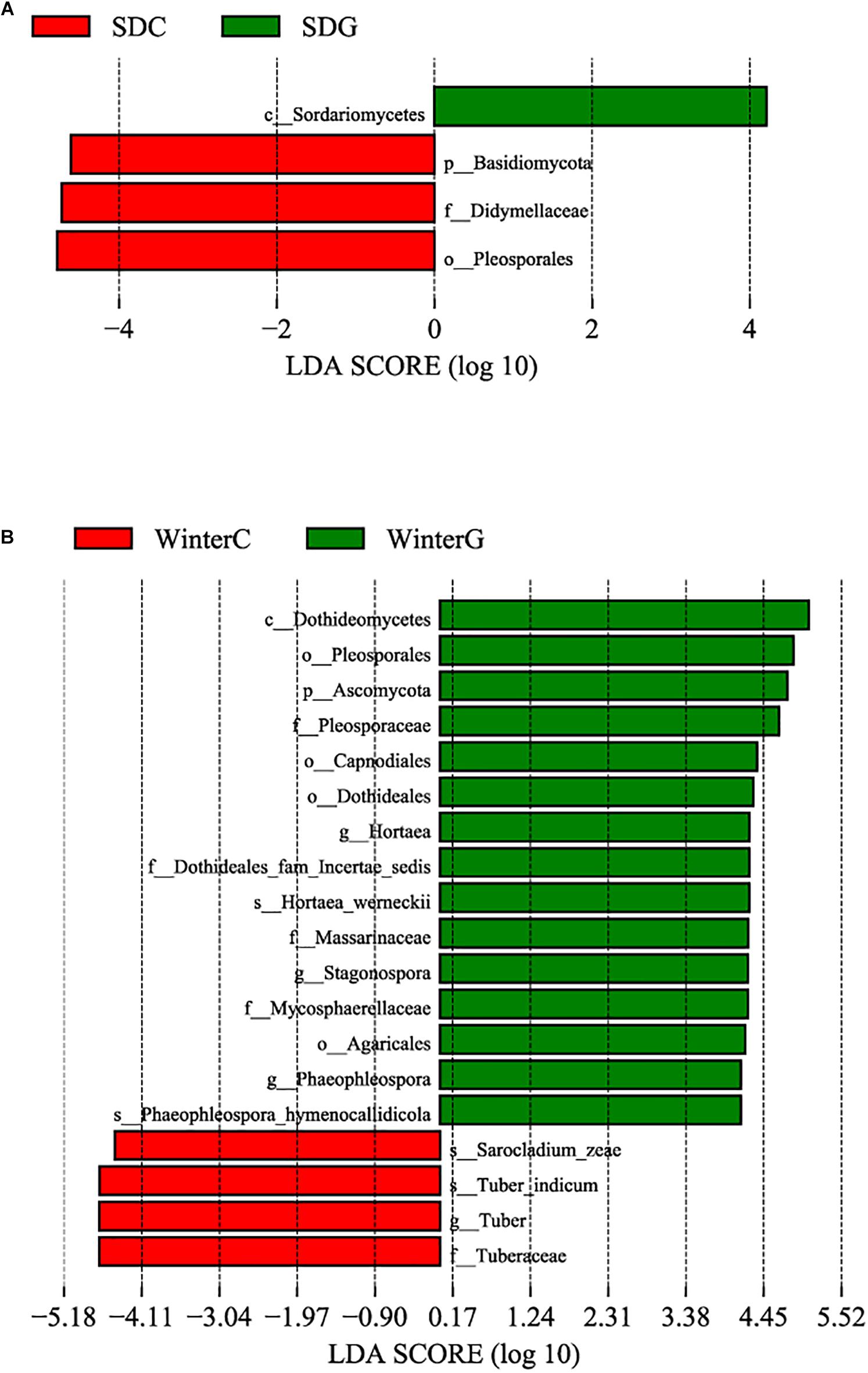
Figure 3. Differences among intestinal and hepatopancreas groups were determined by linear discriminative analysis effect size (LEfSe). The highlighted taxa were significantly enriched in the group that corresponds to each color. LDA scores can be interpreted as the degree of difference in relative abundance. (A) Comparison between the summer healthy intestinal and hepatopancreas samples; (B) comparison between the winter intestinal and hepatopancreas samples. p_, phylum, c_class, o_order, f_, family, g_, genus and s_, species; WinterC, winter intestinal group; WinterG, winter hepatopancreas.
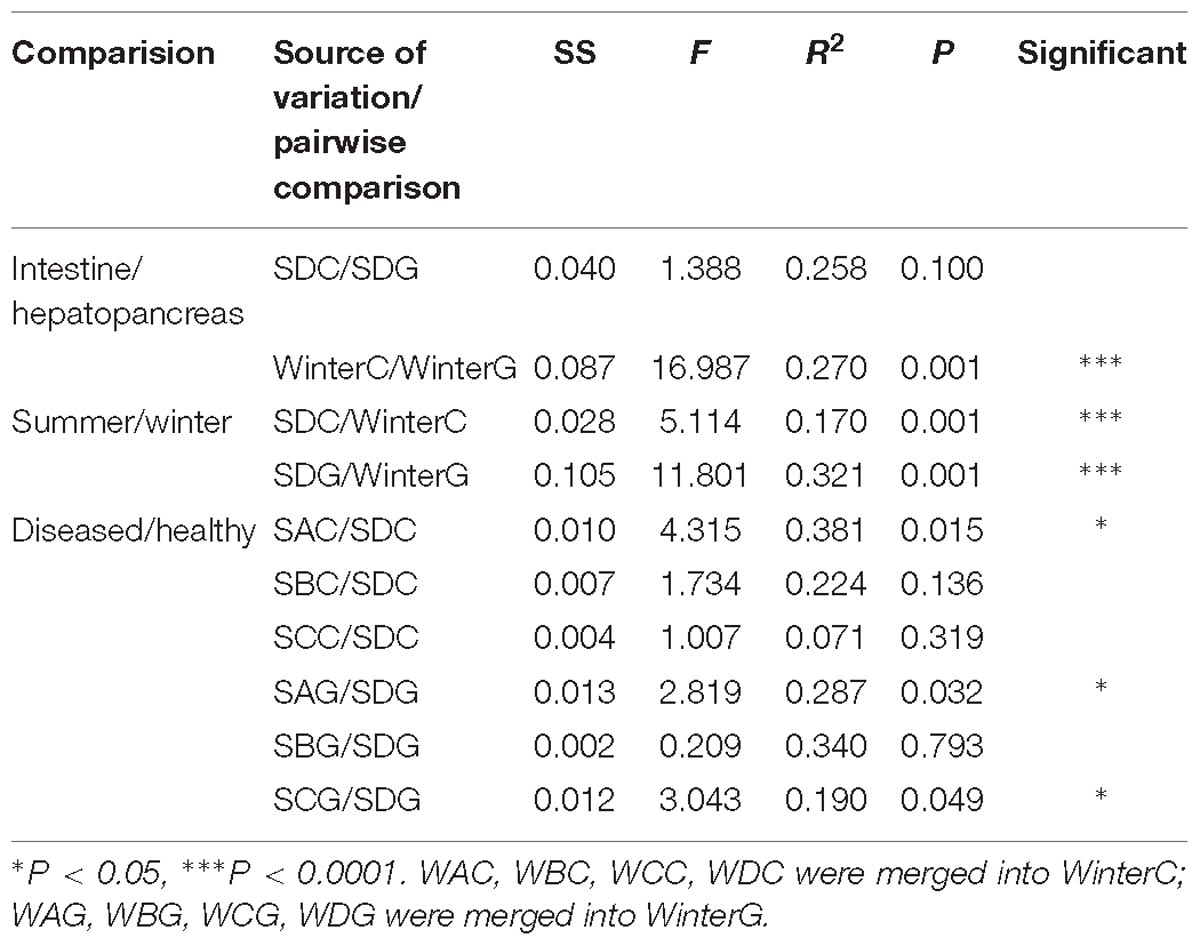
Table 2. PERMANOVA results based on Bray–Curtis distance.
When comparing the winter intestinal group with the winter hepatopancreas group, the hepatopancreas group had a higher level of OTU number, ACE, Chao, and Shannon indices than the intestinal group; the trend was reversed in Simpson index (Figure 4 and Supplementary Table S3A). The β diversity of each group was analyzed based on a Bray–Curtis distance. PCoA results reflected that the intestinal and hepatopancreas group were divided into two sets (Figure 2). The UPGMA cluster analysis showed that despite disturbing by four winter intestinal samples, the hepatopancreas samples (with the exception of two samples from pool WC) together clustered into a separate branch (Figure 2). The significant difference between the intestinal and hepatopancreas group was confirmed by a PERMANOVA, as shown in Table 2. LEfSe analysis results showed that the hepatopancreas group was significantly enriched in the genera Hortaea, Phaeophleospora, and Stagonospora, while the intestinal group had a higher abundance of Tuber and Sarocladium zeae (Figure 3B).
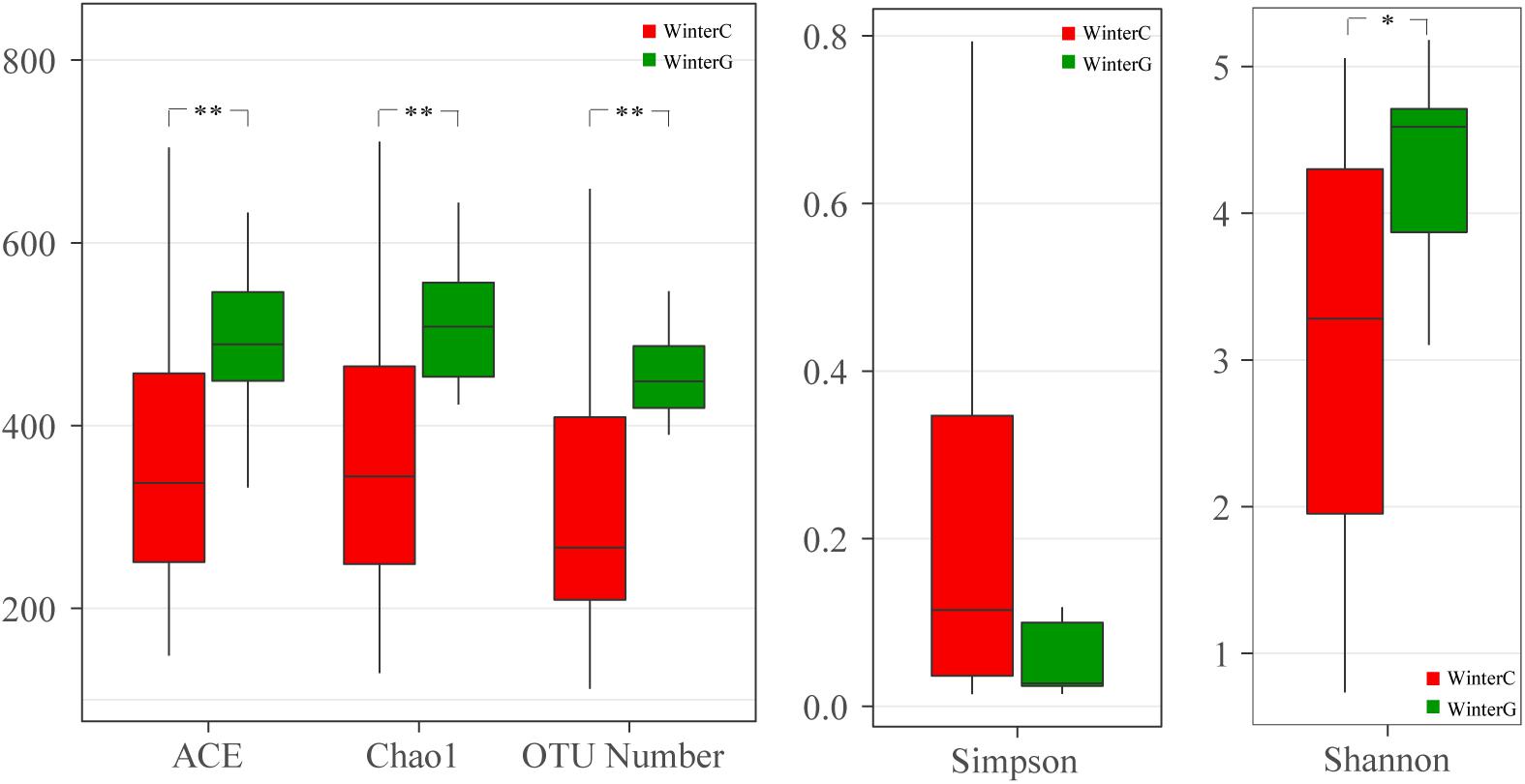
Figure 4. Boxplots depict differences of alpha diversity between winter intestinal and hepatopancreas samples. WinterC, winter intestinal samples; WinterG, winter hepatopancreas. ∗P < 0.05, ∗∗P < 0.01.
Comparison of the Mycobiota Between the Summer and Winter Samples
The influence of the season on the intestinal and hepatopancreas fungal microbiota was analyzed. To avoid the effects of diseases to the results of analysis, only SDC and SDG from summer have been used to perform the comparison analysis. The results demonstrated that there were no significant differences in the richness estimator and the community diversity estimator, as shown in Supplementary Table S3B.
To fully explore the differences in the community membership and diversity between the groups, we calculated the β diversity of each sample based on the Bray–Curtis distance. The PCoA results indicated that the summer and winter samples distinctly clustered into two sets (Figure 2A). The UPGMA cluster analysis also showed the same results (Figure 2B). The significant differences have been confirmed by a PERMANOVA as shown in Table 2.
Linear discriminative analysis effect size analysis was performed to screen the seasonally differential fungi in the intestine and hepatopancreas. For the shrimp intestine, three classes, four orders, seven families, six genera and six species presented a significant alteration in different groups (Figure 5A). The summer group had a significantly higher abundance of the genera Cryptococcus and Ramichloridium. The winter group presented a significant increase in the genera Tuber, Sarocladium, Hortaea, and Phaeophleospora. For the shrimp hepatopancreas, there were significant differences in two classes, three orders, three families, two genera and two species (Figure 5B). The winter hepatopancreas group showed an increased abundance of the genera Hortaea, Phaeophleospora, Stagonospora, but no typical representative fungi were found in the summer hepatopancreas group.
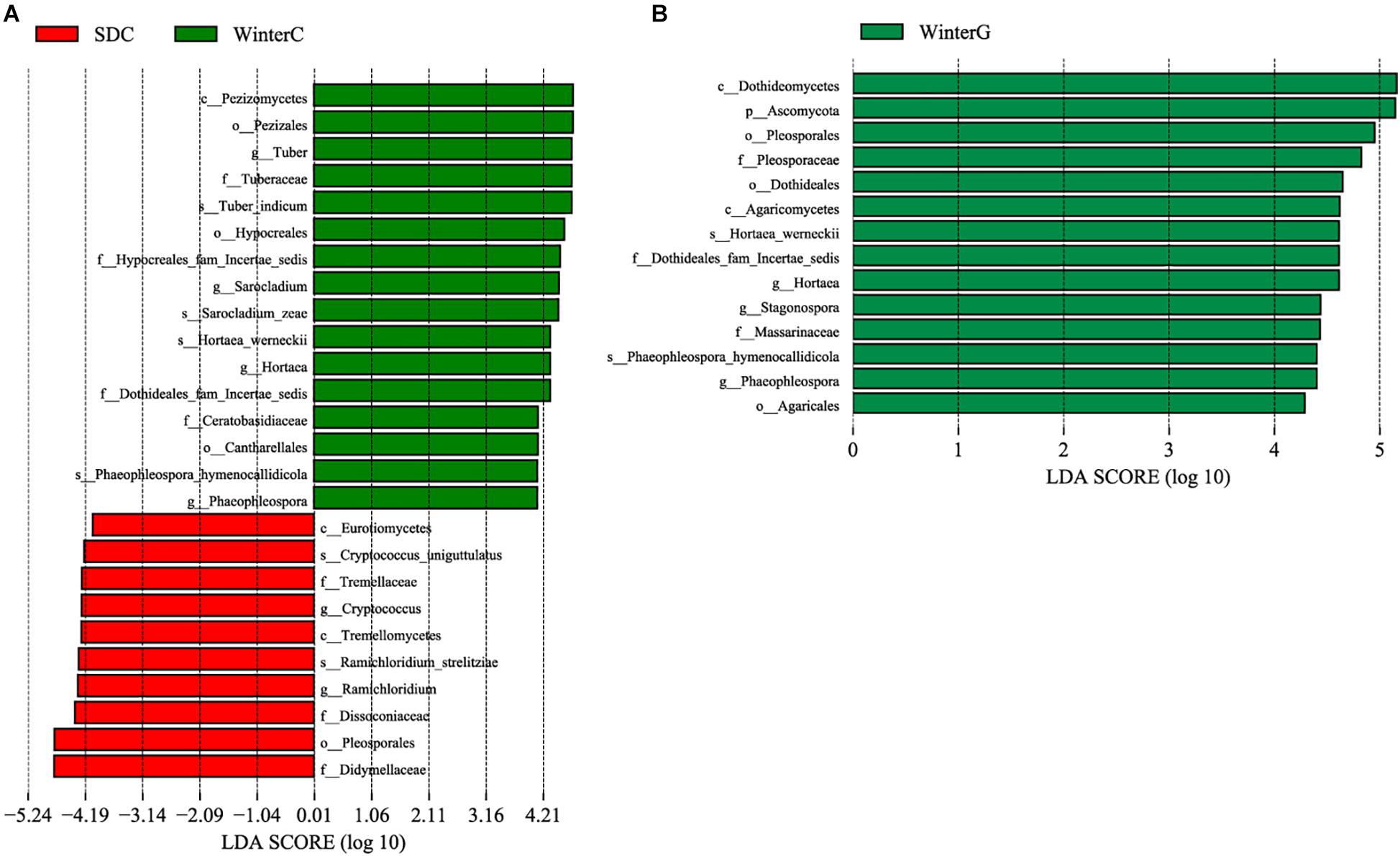
Figure 5. Differences among summer and winter groups were determined by LEfSe. The highlighted taxa were significantly enriched in the group that corresponds to each color. LDA scores can be interpreted as the degree of difference in relative abundance. (A) Comparison between summer and winter intestinal samples; (B) comparison between summer and winter hepatopancreas samples. p_, phylum, c_class, o_order, f_, family, g_, genus and s_, species; WinterC, winter intestinal group; WinterG, winter hepatopancreas.
Comparison of the Mycobiota Between Diseased and Healthy Groups
To detect the effects of disease on the fungal microbiota of L. vannamei, we compared sick intestinal groups (the intestinal samples from SA: SAC; the intestinal samples from SB: SBC, and the intestinal samples from SC: SCC) with healthy intestinal group (SDC), and sick hepatopancreas groups (the hepatopancreas samples from SA: SAG, the hepatopancreas samples from SB: SBG and the hepatopancreas samples from SC: SCG) with a healthy hepatopancreas group (SDG). The results showed that only the SAC group presented a significantly dissimilarity with the healthy intestinal group in OTU number and Shannon diversity. There were no detectable significant differences in α diversity between the rest of sick intestinal groups and healthy intestinal group, and between the sick hepatopancreas groups and the healthy hepatopancreas group (Supplementary Table S3C).
The differences in β diversity among the groups were compared using the PCoA and PERMANOVA (Figure 2 and Table 2). The PERMANOVA results showed that apart from the SAC group, there were no significant differences in community membership and community diversity between the sick and healthy intestinal groups. When the effects of disease on the hepatopancreas mycobiota were compared, the sick groups (SAG and SCG) showed a significant difference with the healthy group (SDG).
Linear discriminative analysis effect size analysis was used to screen the representative fungi in each group. First, we compared the sick SA group with the healthy SD group (Figures 6A,B) and found that in the gut, the sick group (SAC) had a significantly higher abundance in the genera Candida and Mortierella. The healthy group (SDC) had a higher abundance in the genera Didymella, Filobasidium, and Symmetrospora. In the hepatopancreas, no genera showed a significantly increased abundance in the sick group (SAG), but the genus Pyrenochaetopsis was significantly enriched in the healthy group (SDG). The comparison between the SB group and the SD group (Figure 6C) showed that in the gut, the genus Candida was enriched in the sick group (SBC). In the healthy group (SDC), there was a significant increased abundance in the genera Didymella and Filobasidium. In the hepatopancreas, no representative fungi were found in the sick (SBG) and healthy (SDG) group. Finally, we analyzed the differential fungi between the sick SC group and the healthy SD group (Figures 6D,E). The results demonstrated that in the intestine, the genera Cryptococcus, Didymella, and Symmertrospora in the healthy group (SDC) significantly outnumbered those in the sick group (SCC). In the hepatopancreas, the number of the genus Zasmidium was larger in the sick group (SCG). The healthy group (SDG) had a higher level of the genus Pyrenochaetopsis.
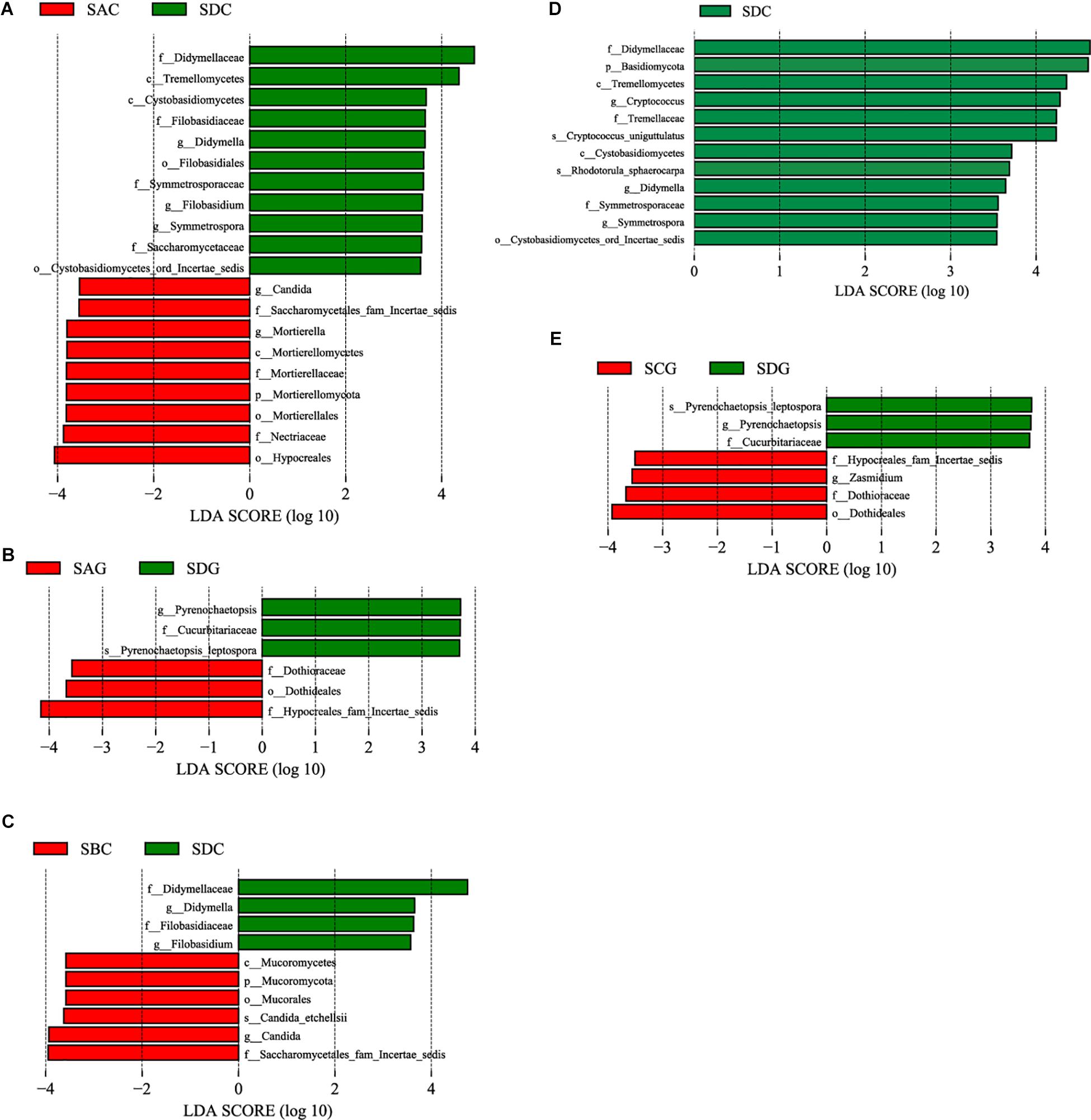
Figure 6. Differences among diseased and healthy groups were determined by LEfSe. The highlighted taxa were significantly enriched in the group that corresponds to each color. LDA scores can be interpreted as the degree of difference in relative abundance. (A) Comparison between SAC and SDC; (B) comparison between SAG and SDG; (C) comparison between SBC and SDC; (D) comparison between SCC and SDC; (E) comparison between SCG and SDG. p_, phylum, c_class, o_order, f_, family, g_, genus and s_, species; WinterC, winter intestinal group; WinterG, winter hepatopancreas.
Discussion
Composition Analysis of the Intestinal and Hepatopancreas Mycobiota of L. vannamei
Our study revealed the composition of the hepatopancreas and gut mycobiota in L. vannamei is highly diverse. Alternaria, Tuber, Hortaea, Sarocladium, and Stagonospora were identified as the most dominant five genera in the shrimp mycobiota (Figure 1B). Interestingly, Candida and Saccharomyces, which account for a large portion in humans and other animals (Neville et al., 2015; Hallen-Adams and Suhr, 2017; Schei et al., 2017; Li et al., 2018), were low-abundant in the shrimp intestinal and hepatopancreas mycobiota. It has been reported that Candida species had a higher level in hosts with carbohydrate diets (Hoffmann et al., 2013). Our recent study of the gut microbiota in bats with diverse diets has revealed that the number of Candida in the gut of insectivorous bats was significantly less than that of phytophagous bats (Li et al., 2018). In this study, the cohabitating Pacific white shrimp were fed with the same artificial food, which is high in protein and relatively low in carbohydrate. Thus, the low abundance of Candida species might partially be attributed to the specific diet. In addition, distinguishing from the bacterial microbiota, the fungal microbiota occurs primarily from the intake (De Schryver and Vadstein, 2014; Hallen-Adams and Suhr, 2017; Sam et al., 2017). L. vannamei is an aquatic anthropod. Many recent studies showed that environment factors have considerably influenced the shrimp gut bacterial microbiota by providing environment-associated microbes and disturbing the composition of the colonized microbial community (De Schryver and Vadstein, 2014; Chen et al., 2017; Cornejo-Granados et al., 2018; Huang et al., 2018; Xiong et al., 2018). Therefore, the aquatic environment may greatly affect the mycobiota of the shrimp, and thus shape the shrimp mycobiota that significantly differs from that of terrestrial organisms.
A previous culture-dependent study showed that the most primary fungal taxa in Pacific white shrimp were Aspergillus, Penicillium, and Fusarium (da Silva et al., 2011), and the presence of Penicillium was verified afterwards in another culture-based study (Laich and Andrade, 2016). Unexpectedly, in our study, Penicillium species were not found in the intestine or the hepatopancreas. This might contribute to the geographical divergence, which results in the specific aquatic microbiomes and environmental factors of each region (Seetharam et al., 2015; Chen et al., 2017).
Comparison Between the Intestinal and Hepatopancreas Mycobiota
Many studies showed that as a result of the different niches and function of each part of the digestive system, the diversity and composition of the bacterial microbiota in different parts differed from one to another (Schoster et al., 2013; Zhao et al., 2015; He et al., 2018). Recent studies of the shrimp microbiota revealed that despite the lack of significant differences among the bacterial microbiota in each part of the intestine, the bacterial microbiota in the hepatopancreas was significantly distinguished from those in the intestine (Tzuc et al., 2014; Cornejo-Granados et al., 2017). However, little attention so far was paid to the eukaryotic microbial population in the shrimp digestive system, and none on the fungal population. In this study, our results revealed that for the summer shrimp, the fungal microbiota in the hepatopancreas was extremely homologous with that in the intestine (Figure 2A, Table 2, and Supplementary Table S3A). However, when compared to the mycobiota of the winter shrimp, the hepatopancreas group showed a higher richness and Shannon diversity than the intestinal group (Figure 4) and there were significant differences in the community membership and structure between the intestine and hepatopancreas (Figure 2 and Table 2). Previous studies confirmed that external fungi were a primary source of the gastrointestinal mycobiota (De Schryver and Vadstein, 2014; Hallen-Adams and Suhr, 2017). Like most of aquatic organisms (Deus and Petrere-Junior, 2003; Guilherme and Rosa, 2014), the shrimp dietary strategies seasonally vary. Attributed to the optimal temperature and higher metabolic rate in the summer, the summer shrimp eat faster and more than the winter ones. However, in the winter, the feeding rate significantly reduces, especially when the environmental temperature is below approximately 15°C, although the shrimp is able to survive at the lowest temperature of approximately 6°C. The reduction of the shrimp feeding frequency in the winter led to the decrease in the number of food-sourced fungi. Therefore, in winter, after the digestion treatments of the hepatopancreas, the fungi that have already been low in the number would be much fewer when arriving at the intestine.
The Influence of Seasonal Variation on the Mycobiota of L. vannamei
The disease incidence of farmed aquatic animals presents a significantly seasonal alteration, which is higher in the summer but relatively lower in the winter. Recent studies showed that the gut microbiota is involved in regulating the immune system of the shrimp (Cornejo-Granados et al., 2017), and a stable microbiota can improve the host resistance to external pathogens (Xiong et al., 2015). In this study, although the comparison of alpha diversity indices showed there were no significant differences between the summer and the winter mycobiota, PCoA, UPGMA cluster analysis, and PERMANOVA demonstrated that the breeding season is a decisive factor shaping the shrimp mycobiota (Figures 2A,B and Table 2). It has been reported that the mycobiota is less stable than those of bacteria and is greatly determined by diet and fungi in the environment (Hallen-Adams and Suhr, 2017; Sam et al., 2017). Aquarium microbiome responses to the temperature variation (Van Bonn et al., 2015) and the high temperature of the summer can easily and quickly promote pathogen growth. Recent studies have demonstrated that shrimp gut microbiota is highly susceptible to environmental factors, even over short timescales (Cornejo-Granados et al., 2018; Huang et al., 2018; Xiong et al., 2018). The alteration of the number of the pathogens in the aquatic environment not only results in shrimp infections, but also directly changes the composition of the shrimp gut microbiota (Rungrassamee et al., 2016; Chen et al., 2017). Additionally, environment variation also indirectly interferes the stability of the microbiota (Huang et al., 2018; Xiong et al., 2018). Therefore, the intake of external pathogenic or opportunistically pathogenic fungi and the environmental changes might reduce the stability of the microbiome, especially the fungal population. The unstable mycobiota may negatively affect the immune function of the shrimp farmed in the summer and cause the increase of the disease incidence to some extent.
The Influence of Disease on the Mycobiota of L. vannamei
The gastrointestinal microbiota (both bacteria and fungi) is sensitive and responds to the disease state of the host. In this study, the intestinal mycobiota of the sick group SA showed significant differences in OTU number and Shannon diversity compared with those of the healthy control, but no significant differences were found between the other sick groups and the healthy group (Supplementary Table S3C). Previous studies have shown that the diversity of the gut mycobiota in sick humans has a significant alteration compared with that of healthy humans. The variation of the diversity is inconsistent in patients suffering from different types of illnesses. There was an increased fungal diversity in the gut of Crohn’s disease patients (Li et al., 2014; Liguori et al., 2016) but a decrease in the alpha diversity in ulcerative colitis (Pineton de Chambrun et al., 2012) and inflammatory bowel disease patients (Sokol et al., 2017).
When compared to the effect of disease on the composition and structure of the mycobiota, apart from SAC, no significant differences were detected between the sick intestinal groups and healthy intestinal groups as well (Table 2). However, for the hepatopancreas mycobiota, there were significant differences between the two sick groups (SAG and SCG) and the healthy group (Table 2). The results indicated that suffering white feces significantly influenced the diversity, composition and structure of the gut mycobiota of the shrimps.
Of note, we found that pathogenic Candida (Banjara et al., 2016) in the intestine of the two sick groups (SAC and SBC) significantly outnumbered that of the healthy group (Figures 6A,C). Disease might alter the homeostasis among the autochthonous microbes. The alteration resulted in that the number of the pathogens which should have been a normal commensal increased. For the healthy group, the number of plant-pathogenic Didymella (Barilli et al., 2016; Ranjbar Sistani et al., 2017) and Filobasidium, presented a significant increase in the gut compared with the sick groups (Figures 6A,C,D). The healthy hepatopancreas group had a significantly higher abundance of Pyrenochaetopsis than the sick hepatopancreas groups (Figures 6B,E). Those fungi may be fungal indicators of the healthy shrimps.
Fungal Pathogens From L. vannamei
Foodborne diseases are a hot topic around the world. Of these, food safety problems derived from microbial infections have become one of the problems of greatest concern (Liu et al., 2004; Xu and Zhang, 2012). Pacific white shrimp is a favored food commodity because of the delicious taste and rich nutrition but was also identified as a potential reservoir and disseminator of many human bacterial pathogens (Zhang et al., 2015). However, little is known about the fungal pathogens harbored by Pacific white shrimps.
Our results showed that L. vannamei harbored a variety of human fungal pathogens (Figure 1B and Supplementary Table S2). Of these, species from the three genera Aspergillus, Candida, and Cryptococcus are primarily responsible for the lethal fungal infections. Hortaea werneckii (Sharmin et al., 2002; Abliz et al., 2003) and Pseudochaetosphaeronema (Ahmed et al., 2015) are capable of causing skin infections in both humans and mammals, such as tinea nigra and subcutaneous phaeohyphomycosis. Ramichloridium, Pseudozyma, Rhodotorula, and Sporobolomyces cause hematomycosis (Arendrup et al., 2014; Etienne et al., 2016). Macrophomina (Premamalini et al., 2012), Thermomyces (Sivagnanam et al., 2013), Engyodontium alum (Augustinsky et al., 1990; Thamke et al., 2015; Wu et al., 2016) can induce different types of inflammatory diseases, such as keratitis and endocarditis. Cladophialophora, Chaetomium, Curvularia, Alternaria, and Ramichloridium are not only pathogens but also cause allergic reactions in humans (McAleer et al., 1981; Cruz et al., 1997; Paredes et al., 2013). More importantly, an increasing number of pathogenic fungi previously considered to be plant pathogens have recently been confirmed to be potential pathogens to humans (Fleming et al., 2002). Most of the newly identified human fungal pathogens are resistant to traditional antifungal drugs. Therefore, a more comprehensive risk assessment strategy for fungal pathogens derived from shrimp is required to effectively protect public health.
Conclusion
In this study, we used a high-throughput sequencing approach to explore the hepatopancreas and intestinal mycobiota of L. vannamei. The results disclosed that (1) the composition of the gut and hepatopancreas of L. vannamei is highly diverse and different from that of humans; (2) in winter, the diversity and composition of the mycobiota had a significant difference between the intestinal and hepatopancreas; (3) seasonal variation and diseases considerably affected the fungal microbiota of L. vannamei; (4) healthy shrimps was significantly enriched in the genera Didymella and Filobasidium in the gut, and Pyrenochaetopsis in the hepatopancreas. Sick groups had an increased abundance of the genera Candida in the intestine. The different fungi may be fungal indicators for health or diseases progression.
Ethics Statement
All experimental animal protocols in this study were approved by the committee on the Ethics of Animal Experiments of the Guangdong Institute of Applied Biological Resources and followed the basic principles.
Author Contributions
JC and JL designed the research. JL, HJ, LL, and XZ sampled together. JL, HJ, and LL completed the research. JL, LL, and XZ sorted and analyzed the data. JL and JC wrote and revised the manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by the Planning Funds of Science and Technology of Guangdong Province (2016B070701016), the Guangzhou Science Technology and Innovation Commission (201804020080), Funds for Technology Transfer from Guangdong Academy of Sciences and Zhongshan (2016G1FC0008), and GDAS Special of Science and Technology Development (2018GDASCX-0107).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00889/full#supplementary-material
FIGURE S1 | Operational taxonomic units (OTU) number of each group.
FIGURE S2 | Rarefaction curves. (A) OTU number rarefaction curves for summer samples; (B) OTU number rarefaction curves for winter samples; (C) Shannon index rarefaction curves for summer samples; (D) Shannon index rarefaction curves for winter samples.
FIGURE S3 | Species relative abundance curve. Red boxplots depict the sum of species; green boxplots depict the emerging probability of share species.
TABLE S1 | The sequencing data for each sample.
TABLE S2 | All the obtain OTUs and taxonomy.
TABLE S3 |(A) Differences in alpha diversity between intestine and hepatopancreas. (B) Differences in alpha diversity between summer and winter intestine; and between summer and winter hepatopancreas. (C) Differences in alpha diversity between sick and healthy group.
Footnotes
References
Abliz, P., Fukushima, K., Takizawa, K., Miyaji, M., and Nishimura, K. (2003). Specific oligonucleotide primers for identification of Hortaea werneckii, a causative agent of tinea nigra. Diagn. microbiol. Infect. Dis. 46, 89–93. doi: 10.1016/s0732-8893(03)00035-x
Ahmed, S. A., Desbois, N., Quist, D., Miossec, C., Atoche, C., Bonifaz, A., et al. (2015). Phaeohyphomycosis caused by a novel species, Pseudochaetosphaeronema martinelli. J. Clin. Microbiol. 53, 2927–2934. doi: 10.1128/JCM.01456-15
Arendrup, M. C., Boekhout, T., Akova, M., Meis, J. F., Cornely, O. A., Lortholary, O., et al. (2014). ESCMID and ECMM joint clinical guidelines for the diagnosis and management of rare invasive yeast infections. Clin Microbiol Infect 20(Suppl. 3), 76–98. doi: 10.1111/1469-0691.12360
Augustinsky, J., Kammeyer, P., Husain, A., deHoog, G. S., and Libertin, C. R. (1990). Engyodontium album endocarditis. J. Clin. Microbiol. 28, 1479–1481.
Banjara, N., Nickerson, K. W., Suhr, M. J., and Hallen-Adams, H. E. (2016). Killer toxin from several food-derived Debaryomyces hansenii strains effective against pathogenic Candida yeasts. Int. J. Food Microbiol. 222, 23–29. doi: 10.1016/j.ijfoodmicro.2016.01.016
Barilli, E., Cobos, M. J., and Rubiales, D. (2016). Clarification on host range of didymella pinodes the causal agent of pea ascochyta blight. Front. Plant Sci. 7:592. doi: 10.3389/fpls.2016.00592
Bengtsson-Palme, J., Ryberg, M., Hartmann, M., Branco, S., Wang, Z., Godhe, A., et al. (2013). Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 4, 914–919.
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336.
Chen, C. Y., Chen, P. C., Weng, F. C., Shaw, G. T., and Wang, D. (2017). Habitat and indigenous gut microbes contribute to the plasticity of gut microbiome in oriental river prawn during rapid environmental change. PLoS One 12:e0181427. doi: 10.1371/journal.pone.0181427
Cheung, M. K., Yip, H. Y., Nong, W., Law, P. T., Chu, K. H., Kwan, H. S., et al. (2015). Rapid change of microbiota diversity in the gut but not the hepatopancreas during gonadal development of the new shrimp model Neocaridina denticulata. Mar. Biotechnol. 17, 811–819. doi: 10.1007/s10126-015-9662-8
Clemente, J. C., Ursell, L. K., Parfrey, L. W., and Knight, R. (2012). The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270. doi: 10.1016/j.cell.2012.01.035
Cornejo-Granados, F., Gallardo-Becerra, L., Leonardo-Reza, M., Ochoa-Romo, J. P., and Ochoa-Leyva, A. (2018). A meta-analysis reveals the environmental and host factors shaping the structure and function of the shrimp microbiota. PeerJ 6:e5382. doi: 10.7717/peerj.5382
Cornejo-Granados, F., Lopez-Zavala, A. A., Gallardo-Becerra, L., Mendoza-Vargas, A., Sanchez, F., Vichido, R., et al. (2017). Microbiome of Pacific Whiteleg shrimp reveals differential bacterial community composition between Wild, Aquacultured and AHPND/EMS outbreak conditions. Sci. Rep. 7:11783. doi: 10.1038/s41598-017-11805-w
Cruz, A., Saenz de Santamaria, M., Martinez, J., Martinez, A., Guisantes, J., and Palacios, R. (1997). Fungal allergens from important allergenic fungi imperfecti. Allergol. Immunopathol. 25, 153–158.
da Silva, L. R. C., de Souza, O. C., Fernandes, M. J. D., Lima, D. M. M., Coelho, R. R. R., and Souza-Motta, C. M. (2011). Culturable fungal diversity of shrimp Litopenaeus vannamei boone from breeding farms in Brazil. Brazil. J. Microbiol. 42, 49–56. doi: 10.1590/S1517-83822011000100007
Dai, W., Yu, W., Zhang, J., Zhu, J., Tao, Z., and Xiong, J. (2017). The gut eukaryotic microbiota influences the growth performance among cohabitating shrimp. Appl. Microbiol. Biotechnol. 101, 6447–6457. doi: 10.1007/s00253-017-8388-0
De Schryver, P., and Vadstein, O. (2014). Ecological theory as a foundation to control pathogenic invasion in aquaculture. ISME J. 8, 2360–2368. doi: 10.1038/ismej.2014.84
Deus, C. P., and Petrere-Junior, M. (2003). Seasonal diet shifts of seven fish species in an Atlantic rainforest stream in Southeastern Brazil. Brazil. J. Biol. 63, 579–588. doi: 10.1590/s1519-69842003000400005
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Etienne, K. A., Roe, C. C., Smith, R. M., Vallabhaneni, S., Duarte, C., Escadon, P., et al. (2016). Whole-genome sequencing to determine origin of multinational outbreak of Sarocladium kiliense bloodstream infections. Emerg. Infect. Dis. 22, 476–481. doi: 10.3201/eid2203.151193
Fleming, R. V., Walsh, T. J., and Anaissie, E. J. (2002). Emerging and less common fungal pathogens. Infect. Dis. Clin. North Am. 16, 915–933. doi: 10.1016/s0891-5520(02)00041-7
Guilherme, P. D., and Rosa, L. C. (2014). Seasonal variation in body size and diet of the sea star Astropecten marginatus (Paxillosida, Astropectinidae) off coast of Parana, Southern Brazil. Rev. Biol. Trop. 62, 59–68.
Hallen-Adams, H. E., and Suhr, M. J. (2017). Fungi in the healthy human gastrointestinal tract. Virulence 8, 352–358. doi: 10.1080/21505594.2016.1247140
He, J., Yi, L., Hai, L., Ming, L., Gao, W., and Ji, R. (2018). Characterizing the bacterial microbiota in different gastrointestinal tract segments of the Bactrian camel. Sci. Rep. 8:654. doi: 10.1038/s41598-017-18298-7
Hoffmann, C., Dollive, S., Grunberg, S., Chen, J., Li, H., Wu, G. D., et al. (2013). Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One 8:e66019. doi: 10.1371/journal.pone.0066019
Hooper, L. V., Littman, D. R., and Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. doi: 10.1126/science.1223490
Huang, F., Pan, L., Song, M., Tian, C., and Gao, S. (2018). Microbiota assemblages of water, sediment, and intestine and their associations with environmental factors and shrimp physiological health. Appl. Microbiol. Biotechnol. 102,8585–8598. doi: 10.1007/s00253-018-9229-5
Huffnagle, G. B., and Noverr, M. C. (2013). The emerging world of the fungal microbiome. Trends Microbiol. 21, 334–341. doi: 10.1016/j.tim.2013.04.002
Huseyin, C. E., O’Toole, P. W., Cotter, P. D., and Scanlan, P. D. (2017). Forgotten fungi-the gut mycobiome in human health and disease. FEMS Microbiol. Rev. 41, 479–511. doi: 10.1093/femsre/fuw047
Karthikeyan, V., Selvakumar, P., and Gopalakrishnan, A. (2015). A novel report of fungal pathogen Aspergillus awamori causing black gill infection on Litopenaeus vannamei (pacific white shrimp). Aquaculture 444, 36–40. doi: 10.1016/j.aquaculture.2015.03.021
Koljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Laich, F., and Andrade, J. (2016). Penicillium pedernalense sp. nov., isolated from whiteleg shrimp heads waste compost. Int. J. Syst. Evol. Microbiol. 66, 4382–4388. doi: 10.1099/ijsem.0.001360
Li, J., Li, L., Jiang, H., Yuan, L., Zhang, L., Ma, J. E., et al. (2018). Fecal bacteriome and mycobiome in bats with diverse diets in South China. Curr. Microbiol. 75, 1352–1361. doi: 10.1007/s00284-018-1530-0
Li, Q., Huang, J., Li, Y., Zhang, Y., Luo, Y., Chen, Y., et al. (2017). Fungal community succession and major components change during manufacturing process of Fu brick tea. Sci. Rep. 7:6947. doi: 10.1038/s41598-017-07098-8
Li, Q., Wang, C., Tang, C., He, Q., Li, N., and Li, J. (2014). Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn’s disease. J. Clin. Gastroenterol. 48, 513–523. doi: 10.1097/MCG.0000000000000035
Liguori, G., Lamas, B., Richard, M. L., Brandi, G., da Costa, G., Hoffmann, T. W., et al. (2016). Fungal dysbiosis in mucosa-associated microbiota of Crohn’s Disease Patients. J. Crohns Colit. 10, 296–305. doi: 10.1093/ecco-jcc/jjv209
Liu, X., Chen, Y., Wang, X., and Ji, R. (2004). [Foodborne disease outbreaks in China from 1992 to 2001 national foodborne disease surveillance system]. J. Hyg. Res. 33:725.
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mar Rodriguez, M., Perez, D., Javier Chaves, F., Esteve, E., Marin-Garcia, P., Xifra, G., et al. (2015). Obesity changes the human gut mycobiome. Sci. Rep. 5:14600. doi: 10.1038/srep14600
McAleer, J. P., Nguyen, N. L., Chen, K., Kumar, P., Ricks, D. M., Binnie, M., et al. (2016). Pulmonary Th17 antifungal immunity is regulated by the gut microbiome. J. Immunol. 197, 97–107. doi: 10.4049/jimmunol.1502566
McAleer, R., Kroenert, D. B., Elder, J. L., and Froudist, J. H. (1981). Allergic bronchopulmonary disease caused by Curvularia lunata and Drechslera hawaiiensis. Thorax 36, 338–344. doi: 10.1136/thx.36.5.338
Mukherjee, P. K., Chandra, J., Retuerto, M., Sikaroodi, M., Brown, R. E., Jurevic, R., et al. (2014). Oral mycobiome analysis of HIV-infected patients: identification of Pichia as an antagonist of opportunistic fungi. PLoS Pathog. 10:e1003996. doi: 10.1371/journal.ppat.1003996
Neville, B. A., D’Enfert, C., and Bougnoux, M. E. (2015). Candida albicans commensalism in the gastrointestinal tract. FEMS Yeast Res. 15:fov081. doi: 10.1093/femsyr/fov081
O’Hara, A. M., and Shanahan, F. (2006). The gut flora as a forgotten organ. EMBO Rep. 7, 688–693. doi: 10.1038/sj.embor.7400731
Palm, N. W., De Zoete, M. R., and Flavell, R. A. (2015). Immune-microbiota interactions in health and disease. Clin. Immunol. 159, 122–127. doi: 10.1016/j.clim.2015.05.014
Paredes, K., Capilla, J., Sutton, D. A., Mayayo, E., Fothergill, A. W., and Guarro, J. (2013). Virulence of curvularia in a murine model. Mycoses 56, 512–515. doi: 10.1111/myc.12064
Pineton de Chambrun, G. P., Torres, J., Darfeuille-Michaud, A., and Colombel, J. F. (2012). The role of anti(myco)bacterial interventions in the management of IBD: is there evidence at all? Digest. Dis. 30, 358–367. doi: 10.1159/000338126
Premamalini, T., Ambujavalli, B. T., Vijayakumar, R., Rajyoganandh, S. V., Kalpana, S., and Kindo, A. J. (2012). Fungal keratitis caused by Macrophomina phaseolina - A case report. Med. Mycol. Case Rep. 1, 123–126. doi: 10.1016/j.mmcr.2012.10.007
Ranjbar Sistani, N., Kaul, H. P., Desalegn, G., and Wienkoop, S. (2017). Rhizobium impacts on seed productivity, quality, and protection of Pisum sativum upon disease stress caused by didymella pinodes: phenotypic, proteomic, and metabolomic traits. Front. Plant Sci. 8:1961. doi: 10.3389/fpls.2017.01961
Rungrassamee, W., Klanchui, A., Maibunkaew, S., and Karoonuthaisiri, N. (2016). Bacterial dynamics in intestines of the black tiger shrimp and the Pacific white shrimp during Vibrio harveyi exposure. J. Invertebr. Pathol. 133, 12–19. doi: 10.1016/j.jip.2015.11.004
Sam, Q. H., Chang, M. W., and Chai, L. Y. (2017). The fungal mycobiome and its interaction with gut bacteria in the host. Int. J. Mol. Sci. 18, 330. doi: 10.3390/ijms18020330
Schei, K., Avershina, E., Oien, T., Rudi, K., Follestad, T., Salamati, S., et al. (2017). Early gut mycobiota and mother-offspring transfer. Microbiome 5:107. doi: 10.1186/s40168-017-0319-x
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schoster, A., Arroyo, L. G., Staempfli, H. R., and Weese, J. S. (2013). Comparison of microbial populations in the small intestine, large intestine and feces of healthy horses using terminal restriction fragment length polymorphism. BMC Res. Notes 6:91. doi: 10.1186/1756-0500-6-91
Seetharam, A. S., Kawaler, E., Du, Z. Q., Rothschild, M. F., and Severin, A. J. (2015). Microbiome analyses of pacific white shrimp (Litopenaeus vannamei) collected from disparate geographical locations. Genom. Data 6, 67–69. doi: 10.1016/j.gdata.2015.08.009
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Sharmin, S., Haritani, K., Tanaka, R., Abliz, P., Takizawa, K., Sano, A., et al. (2002). The first isolation of Hortaea werneckii from a household guinea pig. Japan. J. Med. Mycol. 43, 175–180. doi: 10.3314/jjmm.43.175
Sivagnanam, S., Chen, S. C., Halliday, C., and Packham, D. (2013). Thermomyces lanuginosus infective endocarditis: case report and a review of endocarditis due to uncommon moulds. Med. Mycol. Case Rep. 2, 152–155. doi: 10.1016/j.mmcr.2013.09.001
Sokol, H., Leducq, V., Aschard, H., Pham, H. P., Jegou, S., Landman, C., et al. (2017). Fungal microbiota dysbiosis in IBD. Gut 66, 1039–1048. doi: 10.1136/gutjnl-2015-310746
Sommer, F., Stahlman, M., Ilkayeva, O., Arnemo, J. M., Kindberg, J., Josefsson, J., et al. (2016). The gut microbiota modulates energy metabolism in the hibernating brown bear Ursus arctos. Cell Rep. 14, 1655–1661. doi: 10.1016/j.celrep.2016.01.026
Sonnenburg, J. L., and Backhed, F. (2016). Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64. doi: 10.1038/nature18846
Thamke, D. C., Mendiratta, D. K., Dhabarde, A., and Shukla, A. K. (2015). Mycotic keratitis due to Engyodontium album: first case report from India. Ind. J. Med. Microbiol. 33, 303–304. doi: 10.4103/0255-0857.154890
Tzuc, J. T., Escalante, D. R., Rojas Herrera, R., Gaxiola Cortes, G., and Ortiz, M. L. (2014). Microbiota from Litopenaeus vannamei: digestive tract microbial community of Pacific white shrimp (Litopenaeus vannamei). SpringerPlus 3:280. doi: 10.1186/2193-1801-3-280
Van Bonn, W., LaPointe, A., Gibbons, S. M., Frazier, A., Hampton-Marcell, J., and Gilbert, J. (2015). Aquarium microbiome response to ninety-percent system water change: clues to microbiome management. Zoo. Biol. 34, 360–367. doi: 10.1002/zoo.21220
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-07
Wang, Y., Sheng, H. F., He, Y., Wu, J. Y., Jiang, Y. X., Tam, N. F., et al. (2012). Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 78, 8264–8271. doi: 10.1128/AEM.01821-12
Wu, B., Wiese, J., Wenzel-Storjohann, A., Malien, S., Schmaljohann, R., and Imhoff, J. F. (2016). Engyodontochones, antibiotic polyketides from the marine fungus Engyodontium album Strain LF069. Chemistry 22, 7452–7462. doi: 10.1002/chem.201600430
Xiong, J. (2018). Progress in the gut microbiota in exploring shrimp disease pathogenesis and incidence. Appl. Microbiol. Biotechnol. 102, 7343–7350. doi: 10.1007/s00253-018-9199-7
Xiong, J., Dai, W., Qiu, Q., Zhu, J., Yang, W., and Li, C. (2018). Response of host-bacterial colonization in shrimp to developmental stage, environment and disease. Mol. Ecol. 27, 3686–3699. doi: 10.1111/mec.14822
Xiong, J., Wang, K., Wu, J., Qiuqian, L., Yang, K., Qian, Y., et al. (2015). Changes in intestinal bacterial communities are closely associated with shrimp disease severity. Appl. Microbiol. Biotechnol. 99, 6911–6919. doi: 10.1007/s00253-015-6632-z
Xiong, J., Zhu, J., Dai, W., Dong, C., Qiu, Q., and Li, C. (2017). Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environ. Microbiol. 19, 1490–1501. doi: 10.1111/1462-2920.13701
Xu, J. F., and Zhang, J. Z. (2012). Analysis of foodborne disease outbreaks in China from 2001 to 2010. Chin. Agric. Sci. Bull. 28, 313–316.
Xue, Z., Zhang, W., Wang, L., Hou, R., Zhang, M., Fei, L., et al. (2015). The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. mBio 6:e00022-e15. doi: 10.1128/mBio.00022-15
Zhang, D., Wang, F., Dong, S., and Lu, Y. (2016). De novo assembly and transcriptome analysis of osmoregulation in Litopenaeus vannamei under three cultivated conditions with different salinities. Gene 578, 185–193. doi: 10.1016/j.gene.2015.12.026
Zhang, Z., Xiao, L., Lou, Y., Jin, M., Liao, C., Malakar, P. K., et al. (2015). Development of a multiplex real-time PCR method for simultaneous detection of Vibrio parahaemolyticus, Listeria monocytogenes and Salmonella spp. in raw shrimp. Food Control 51, 31–36. doi: 10.1016/j.foodcont.2014.11.007
Keywords: intestine, ITS amplicon, hepatopancreas, Litopenaeus vannamei, mycobiota
Citation: Li J, Jiang H, Li L, Zhang X and Chen J (2019) The Effect of Disease and Season to Hepatopancreas and Intestinal Mycobiota of Litopenaeus vannamei. Front. Microbiol. 10:889. doi: 10.3389/fmicb.2019.00889
Received: 07 December 2018; Accepted: 08 April 2019;
Published: 24 April 2019.
Edited by:
Gustavo Henrique Goldman, University of São Paulo, BrazilReviewed by:
Man Kit Cheung, The Chinese University of Hong Kong, ChinaMaryam Dadar, Razi Vaccine and Serum Research Institute, Iran
Copyright © 2019 Li, Jiang, Li, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinping Chen, Y2hlbmpwQGdpYWJyLmdkLmNu