 Michał Sobala
Michał Sobala Bożena Bruhn-Olszewska
Bożena Bruhn-Olszewska Michael Cashel
Michael Cashel Katarzyna Potrykus
Katarzyna Potrykus
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 24 April 2019
Sec. Microbial Physiology and Metabolism
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00859
In bacteria, the so-called stringent response is responsible for adaptation to changing environmental conditions. This response is mediated by guanosine derivatives [(p)ppGpp], synthesized by either large mono-functional RelA or bi-functional SpoT (synthesis and hydrolysis) enzymes in β- and γ-proteobacteria, such as Escherichia coli. In Firmicutes and α-, δ-, and 𝜀-proteobacteria, large bifunctional Rel-SpoT-homologs (RSH), often accompanied by small (p)ppGpp synthetases and/or hydrolases devoid of regulatory domains, are responsible for (p)ppGpp turnover. Here, we report on surprising in vitro and in vivo properties of an RSH enzyme from Methylobacterium extorquens (RSHMex). We find that this enzyme possesses some unique features, e.g., it requires cobalt cations for the most efficient (p)ppGpp synthesis, in contrast to all other known specific (p)ppGpp synthetases that require Mg2+. In addition, it can synthesize pppApp, which has not been demonstrated in vitro for any Rel/SpoT/RSH enzyme so far. In vivo, our studies also show that RSHMex is active in Escherichia coli cells, as it can complement E. coli ppGpp0 growth defects and affects rrnB P1-lacZ fusion activity in a way expected for an RSH enzyme. These studies also led us to discover pppApp synthesis in wild type E. coli cells (not carrying the RSHMex enzyme), which to our knowledge has not been demonstrated ever before. In the light of our recent discovery that pppApp directly regulates E. coli RNAP transcription in vitro in a manner opposite to (p)ppGpp, this leads to a possibility that pppApp is a new member of the nucleotide second-messenger family that is widely present in bacterial species.
In nature, bacteria are almost constantly faced with rapidly changing growth conditions that have led to evolution of complex and interconnected regulatory systems involving stress-specific sensing and responses. One of them is the stringent response, which was first characterized in Escherichia coli as a response to the onset of amino acid starvation (Cashel and Gallant, 1969; Cashel et al., 1996). This term is now generalized to include all cellular responses to virtually any environmental stress (e.g., carbon, nitrogen, phosphate, iron and lipid limitation; heat shock, and osmotic stress) that induce synthesis of specific guanosine derivatives: guanosine 5′-triphosphate-3′diphosphate (pppGpp) and guanosine 3′, 5′-bis(diphosphate) (ppGpp), collectively referred to as (p)ppGpp (Potrykus and Cashel, 2008). This response also occurs in plants (Braeken et al., 2006; Tozawa and Nomura, 2011; Field, 2018), but not in the archaea or animal kingdoms.
The hallmark of bacterial stringent response is inhibition of ribosomal RNA and tRNA synthesis with concomitant activation of stress survival gene expression, for example enhanced transcription of amino acid biosynthesis genes to cope with amino acid starvation (Cashel et al., 1996; Potrykus and Cashel, 2008). The onset of stringent response is also required for virulence of many bacterial pathogens, consistent with combatting the stress of host defense against bacterial invasion (Dalebroux et al., 2010).
In E. coli, and other γ- as well as β-proteobacteria, (p)ppGpp synthesis is catalyzed by two enzymes – RelA, activated by amino acid deprivation, and SpoT, activated by other environmental stresses (Potrykus and Cashel, 2008; Atkinson et al., 2011). SpoT also possesses another important function, i.e., it can hydrolyze (p)ppGpp, allowing bacteria to quickly adapt when environmental conditions are brought back to normal (Cashel et al., 1996; Potrykus and Cashel, 2008). These two enzymes are of similar length, however, RelA has lost the ability to hydrolyze (p)ppGpp. In other bacteria, such as α–, δ-, and 𝜀- proteobacteria, as well as Firmicutes, only one homologous enzyme exists; it is always bi-functional (i.e., has both, synthesis and hydrolysis activities) and such enzymes are often referred to as RSH (for Rel/SpoT Homolog). Bioinformatics analyses indicate that RSH is frequently accompanied by one or more shorter enzymes, i.e., SAS (small alarmone synthetase) or SAH (small alarmone hydrolase), each devoid of a large regulatory domain present in RelA, SpoT, and RSH (reviewed in: Atkinson et al., 2011; Steinchen and Bange, 2016).
Synthesis of (p)ppGpp generally involves transfer of the βγ-pyrophosphate from the donor ATP onto the ribose 3′ hydroxyl residue of either GTP or GDP as acceptor nucleotides, resulting in pppGpp or ppGpp, respectively (Cashel and Kalbacher, 1970). Upon hydrolysis, the same (p)ppGpp pyrophosphate residue is removed from the 3′ ribose, to yield GTP or GDP, respectively.
Understanding of the function and structure of SAS and E. coli RelA proteins is much advanced (Gaca et al., 2015; Steinchen et al., 2015, 2018; Beljantseva et al., 2017; Kudrin et al., 2018; Manav et al., 2018; Winther et al., 2018). In contrast, although the physiological function of full-length bi-functional RSH enzymes has been studied in several species, such as Bacillus spp., Enterococcus faecalis, Deinococcus radiodurans, and Staphylococcus aureus (Wendrich et al., 2000; Geiger et al., 2010; Gaca et al., 2013; Kim et al., 2014; Wang et al., 2016), their biochemical properties remain less well explored. Notable exceptions are Streptococcus equisimilis RelSeq (Mechold et al., 2002; Hogg et al., 2004), Mycobacterium tuberculosis RelMtb (Avarbock et al., 2005; Singal et al., 2017), and S. aureus RelSau (Gratani et al., 2018).
Interestingly, (p)ppGpp are not the only 3′pyrophosphate nucleotide derivatives found in bacteria. In the early work on the stringent response, (p)ppApp were discovered to be produced along with (p)ppGpp in Bacillus subtilis in response to addition of amino acid analogs and during sporulation, but this finding has not been pursued since (Rhaese et al., 1977; Nishino et al., 1979). So far, the only enzyme demonstrated to have such ability in vitro is a promiscuous pyrophosphotransferase secreted by Streptomyces morookaensis cells, capable of the βγ -pyrophosphate transfer from ATP or GTP onto the ribosyl-3′ hydroxyl group of any purine nucleotide (Oki et al., 1975).
We believe pppApp is worthy of further attention because recently we have demonstrated that pppApp regulates E. coli ribosomal promoter (rrnB P1) transcription in vitro, where contrary to (p)ppGpp mediated inhibition, transcriptional activation is observed (Bruhn-Olszewska et al., 2018). We had also shown that (p)ppApp binds near the catalytic center of E. coli RNA polymerase at a site distinct from (p)ppGpp site 2 (Bruhn-Olszewska et al., 2018). This suggests a regulatory role for (p)ppApp nucleotides, although their synthesis in E. coli has not been demonstrated until now.
Our immediate goal here is to rigorously substantiate and explore whether the catalytic domain of a previously uncharacterized RSH enzyme from M. extorquens (RSHMex) is capable of synthesizing (p)ppGpp and/or pppApp in vitro. This organism was chosen as a possible source for (p)ppApp synthesis because a bioinformatics search revealed a strong homology between its single SAH enzyme and eukaryotic Mesh enzymes, which we discovered to hydrolyze (p)ppApp in vitro in addition to their published cleavage activity toward (p)ppGpp (Potrykus et al., unpublished). The ensuing association described here for (p)ppApp and (p)ppGpp synthetic activities of the RSHMex catalytic domain led to observations that expression of full-length RSHMex supports accumulation of both nucleotides in E. coli cells. Controls using wild type E. coli cells lacking RSHMex, led to the surprising discovery that basal levels of pppApp are observed in wild type E. coli.
Using DELTA-BLAST (NCBI) and the RelSeq amino acid sequence, we identified a gene in the genome of M. extorquens strain AM1 (GenBank accession # ACS41145.1), termed here rshMex, encoding a potential RSHMex protein. For enhanced clarity, we decided to use the abbreviation RSHMex rather than RelMex suggested by (Atkinson et al., 2011), as we feel this better reflects its properties (see section “Discussion”). Comparison of RSHMex to RelSeq, E. coli RelA and SpoT amino acid sequences using UGENE software with MUSCLE algorithm revealed that full-length RSHMex has only 38, 32, and 39% identity with those proteins, respectively (Supplementary Figure S1). However, the conserved hydrolase and synthetase domains present in the NTD domain show a more striking homology (Figure 1). Namely, all the canonical (p)ppGpp hydrolase and synthetase motifs [reviewed in (Steinchen and Bange, 2016)] are preserved. Still, several residues previously identified as crucial for optimal activity of RelSeq are different in RSHMex (Figure 1).
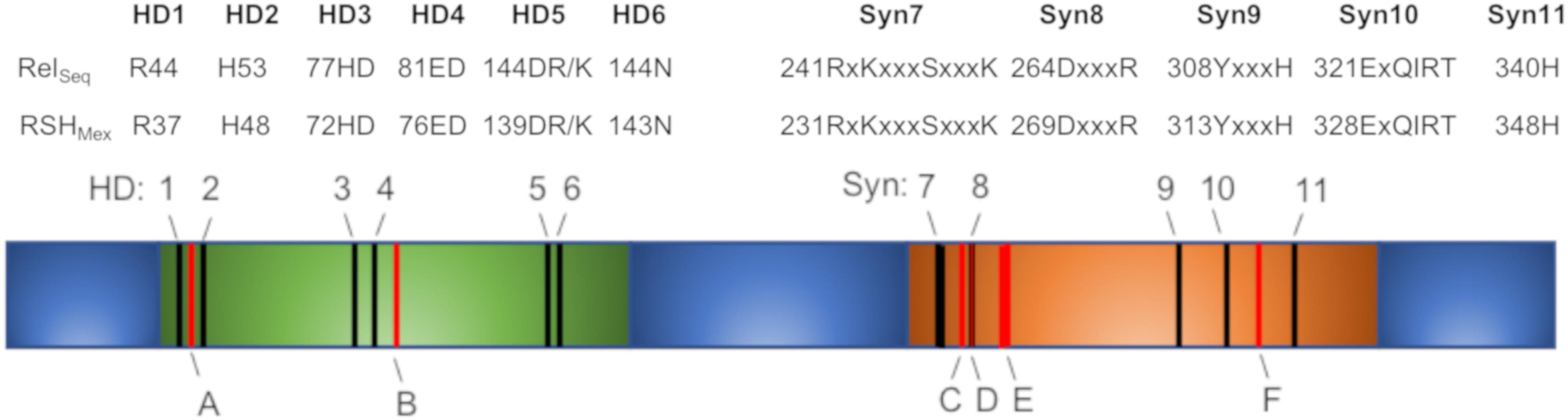
Figure 1. A schematic representation of RSHMex NTD domain, containing the synthetase, and hydrolase domains (RSHMex1-352). The CTD regulatory domain has been omitted. Hydrolase domain (green) and synthetase domain (orange) are indicated. Black lines, HD 1-6 and Syn7-11, conserved hydrolase and synthetase motifs, respectively, as reported in (Steinchen and Bange, 2016). Corresponding RelSeq and RSHMex sequences are denoted. No deviations from the conserved sequence motifs were detected in RSHMex. Red lines and A–F, previously identified in RelSeq as crucial for that enzyme’s optimal activity that are different in RSHMex. A- 50Y/F; B- 97V/I; C- 262I/L; D- 267A/G; E- 270C/V, and 272M/V; F- 333H/D (numbering based on RelSeq sequence). Supplementary Figure S1 compares full length sequences of RSHMex, RelSeq, SpoT, and RelA.
The DNA fragment encoding the N-terminal catalytic half of the RSHMex protein (containing the hydrolase and synthetase domains but lacking the regulatory domains) was cloned in the pCIOX expression vector for purification in the E. coli system. We have chosen to use the RSHMex catalytic half protein for biochemical studies and the full length protein for cellular experiments. This is because classical studies of catalytic and structural features of N-terminal half of RelSeq have provided the basis for understanding general RSH features and so far, there has not been a demonstration that substrate specificity of full length RSH enzymes differs from the catalytic half protein. The added advantage is that this experimental system is simplified because ribosomes, mRNA, and uncharged tRNA are not required for either of bifunctional (synthesis or hydrolysis) activities (Mechold et al., 2002; Hogg et al., 2004). In addition, the catalytic fragment that we used (RSHMex1-352) was designed to allow for direct comparison with the RelSeq studies just mentioned. Thus, all of the subsequent in vitro studies presented here were carried out with RSHMex1-352; for employment in several control reactions, RelSeq1-385 was cloned and purified in the same way as RSHMex1-352.
We first investigated (p)ppGpp synthesis by RSHMex1-352 since this activity is so far present in all RSH enzymes. The initial reaction conditions chosen were similar to those previously established for RelSeq1-385 (Mechold et al., 2002): 160 nM enzyme, 8 mM ATP, and 8 mM GTP were incubated at 37°C for 2 h with increasing concentrations of MgCl2; 3.3 nM [P33] γ-ATP was used as the source of label. Under these conditions, a spot corresponding to pppGpp was observed on TLC plate autoradiograms, with the highest synthesis efficiency at 16 mM Mg2+ (Figure 2A and Supplementary Figure S2). This agrees with RelSeq data, where it was observed that the reaction optimum is reached when the total nucleotide substrate concentration [(ATP) + (GTP)] equals that of Mg2+ (Mechold et al., 2002).
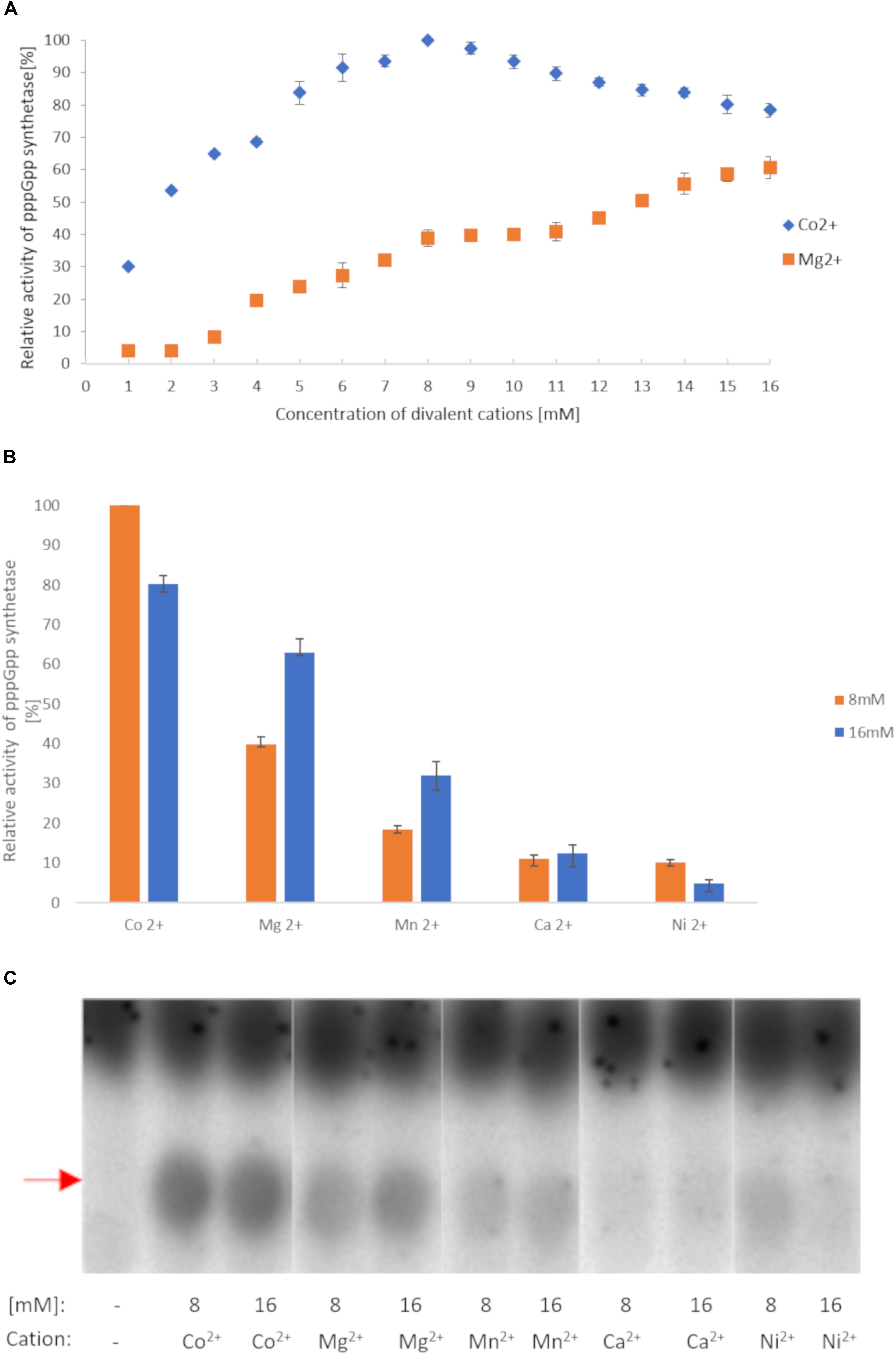
Figure 2. RSHMex pppGpp synthesis activity in the presence of different cations. (A) Mg2+ and Co2+ titration. (B) 8 and 16 mM concentrations of the indicated cations were used. In both cases, the reaction mixtures contained 3.3 nM [P33] ɣ-ATP, 8 mM unlabeled ATP, 8 mM GTP, 50 mM Tris–HCl (pH 8.9), 80 nM RSHMex1-352, and were incubated for 2 h at 37°C. The relative amount of synthesized pppGpp was determined by TLC followed by densitometry. Error bars represent SD calculated from three independent experiments. (C) TLC separation of reactions obtained in (B). Red arrow indicates pppGpp.
Next, we tested other cations at 16 mM, and found that Co2+ yields even higher RSHMex1-352 activity (1.3 fold more pppGpp), while Mn2+, Ca2+, and Ni2+ are much less efficient (2, 5, and 12.8 fold less pppGpp produced than with Mg2+, respectively; Figures 2B,C). A more detailed analysis with Co2+ revealed that contrary to Mg2+, the optimal concentration for this cation is 8 mM, yielding 1.25 fold more pppGpp than at 16 mM CoCl2 (Figure 2A). Under these conditions, RSHMex1-352 produces 2.6 fold more pppGpp than at 8 mM MgCl2, and 1.6 fold more than at 16 mM MgCl2. Mn2+, Ca2+, and Ni2+ were also tested at 8 mM concentrations, however only in the case of NiCl2 higher pppGpp synthesis was observed at 8 mM than at 16 mM concentration, but it was still very low (reaching only 10% of the pppGpp amount synthesized in the presence of 8 mM Co2+; Figures 2B,C).
In the next step, we decided to establish RSHMex1-352 kinetics for pppGpp synthesis, namely the apparent Km for ATP and GTP in this reaction. However, first we needed to establish if there would be any pppGpp hydrolysis under our synthesis reaction conditions as this would have a significant bearing on interpretation of obtained results. This was tested by incubating RSHMex1-352 with unlabeled (p)ppNpp nucleotide standards, followed by one-dimensional TLC (Supplementary Figure S3). No enzymatic hydrolysis was observed.
For Km determination, we chose two settings. In one, high GTP concentration (8 mM) was accompanied by ATP titration to determine apparent Km for ATP. In the other, ATP was kept at high concentration and GTP was being titrated. As depicted in Figure 3, RSHMex1-352 has a much higher apparent ability to bind GTP than ATP (apparent Km: 3.0 ± 0.29 mM vs. 0.39 ± 0.01 mM, a 7.8 fold difference).
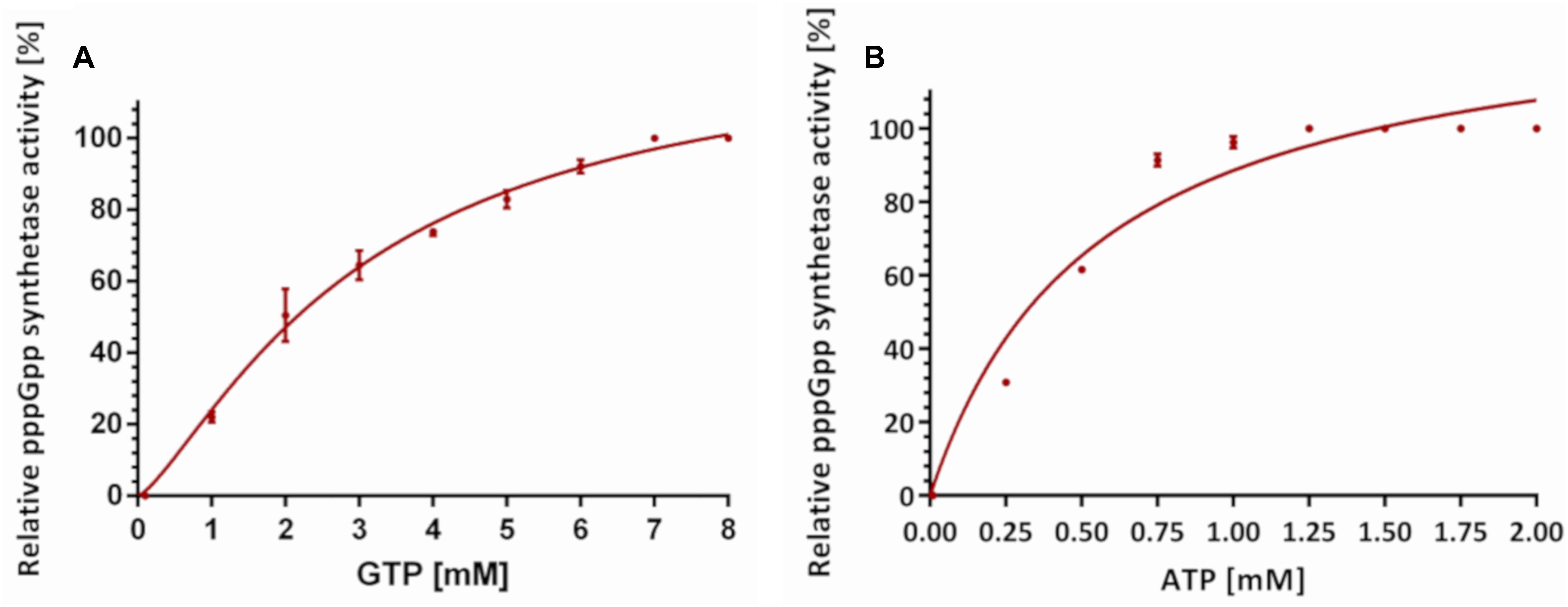
Figure 3. Effects of varying ATP and GTP concentrations on RSHMex1-352 pppGpp synthesis activities in the presence of 8 mM CoCl2. (A) Apparent Km values for ATP were determined in the presence of 8 mM GTP and varying concentrations of ATP. (B) Apparent Km values for GTP were determined in the presence of 8 mM ATP and varying concentrations of GTP. In both cases, each reaction contained 3.3 nM [P33] ɣ-ATP, 50 mM Tris–HCl (pH 8.9), and 8 mM CoCl2. Relative activity was determined by monitoring pppGpp synthesis by TLC analysis. For each curve, data was normalized to the highest amount of pppGpp obtained. Error bars indicate SD calculated from three independent experiments.
Having established optimal reaction conditions for (p)ppGpp, we tested RSHMex1-352 acceptor substrate specificity. Here, RSHMex1-352 was incubated at 37°C for 2 h with 8 mM ATP (pyrophosphate donor) and 8 mM acceptor nucleotides (either ATP, ADP, AMP, GTP, GDP, or GMP). As before, 3.3 nM [P33] γ-ATP served as the label, and 8 mM Co2+ was employed. Since S. morookaensis non-specific pyrophosphotransferase is the only known enzyme to synthesize (p)ppApp, an extract containing this enzyme was used to synthesize (p)ppNpp standards for control reactions that were resolved side by side with the RSHMex1-352 reaction products; these reactions were carried out in the presence of 16 mM MgCl2, at 37°C for 15 min.
As can be seen in Figure 4, we observed four different RSHMex1-352 reaction products, which could correspond to pppGpp, ppGpp, pppApp, and ppApp. A spot corresponding to pppApp is observed not only in reactions with ATP, but also with ADP and AMP, which is not surprising given those two reactions also contained 8 mM ATP. In reactions with GTP and GDP, both pppGpp and ppGpp were detected. In case of GTP, we could suspect a contamination of the nucleotide preparation with some minor amounts of GDP (through GTP 5′ non-enzymatic hydrolysis), however, presence of pppGpp in the reaction with GDP was puzzling. What is even more surprising, there is also a spot apparently corresponding to ppApp in those two reactions. Similar results were obtained in the presence of 16 mM Mg2+, however, in reactions containing ATP and GDP even more of the spot possibly corresponding to ppApp is produced, while the amount of pppGpp produced is roughly equal to that synthesized in a reaction containing ATP and GTP. Still, it should be noted that in this buffer system (0.85 M KH2PO4, pH 3.4), ppApp co-migrates with GTP as judged by TLC of unlabeled nucleotide standards. Co-migration with some other nucleotide derivatives could not be excluded at this point as well.
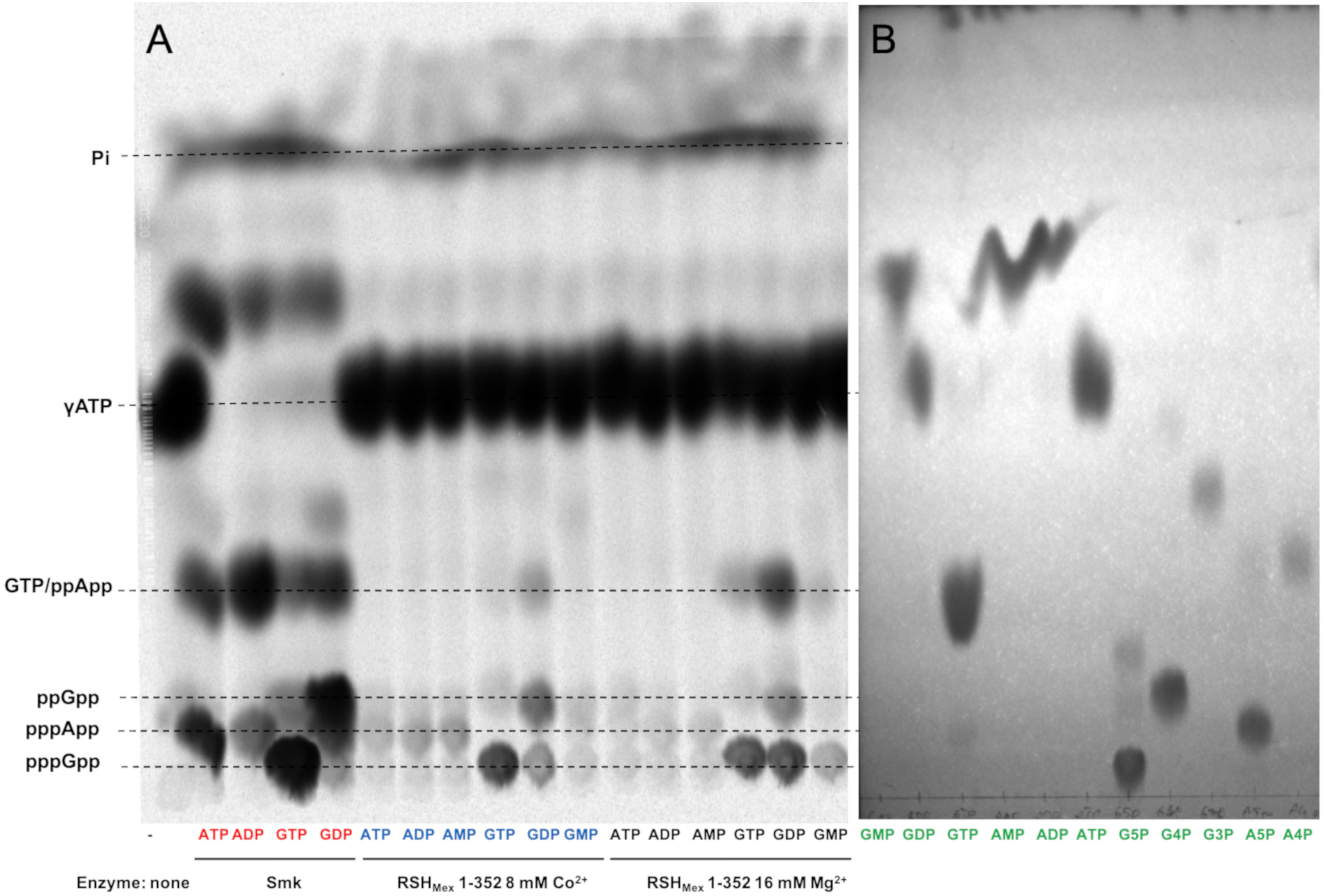
Figure 4. In vitro (p)ppNpp synthesis reactions carried out by RSHMex1-352. (A) Autoradiogram of a TLC plate with separated (p)ppNpp ’s obtained after synthesis by S. morookaensis pyrophosphokinase (A, SmK, red) and RSHMex1-352 (B, with 8 mM Co2+, blue; and C, with 16 mM Mg2+, black). Substrates used are specified under each lane. The reactions also contained 3.3 nM [P33] ɣ- ATP, 8 mM unlabeled ATP, 50 mM Tris–HCl (pH 8.9). To resolve samples on TLC plates, 0.85 M KH2PO4 (pH 3.4) was used. (B) TLC analysis of unlabeled standards (green) separated under the same conditions. G5P, pppGpp; G4P, ppGpp; G3P, pGpp; A5P, pppApp; and A4P, ppApp.
We then pursued nucleotide identification by two-dimensional TLC. The running buffers were chosen so as to distinguish between NTPs and (p)ppNpp derivatives (Supplementary Figure S4; see section “Materials and Methods” for details) (Cashel et al., 1969). The following control reactions were employed: ATP + GTP with RelSeq1-385 (to visualize pppGpp), and ATP + ADP with S. morookaensis pyrophosphotransferase (to visualize pppApp and ppApp) (Figures 5A,B). In all cases, [P33]-γATP served as the label.
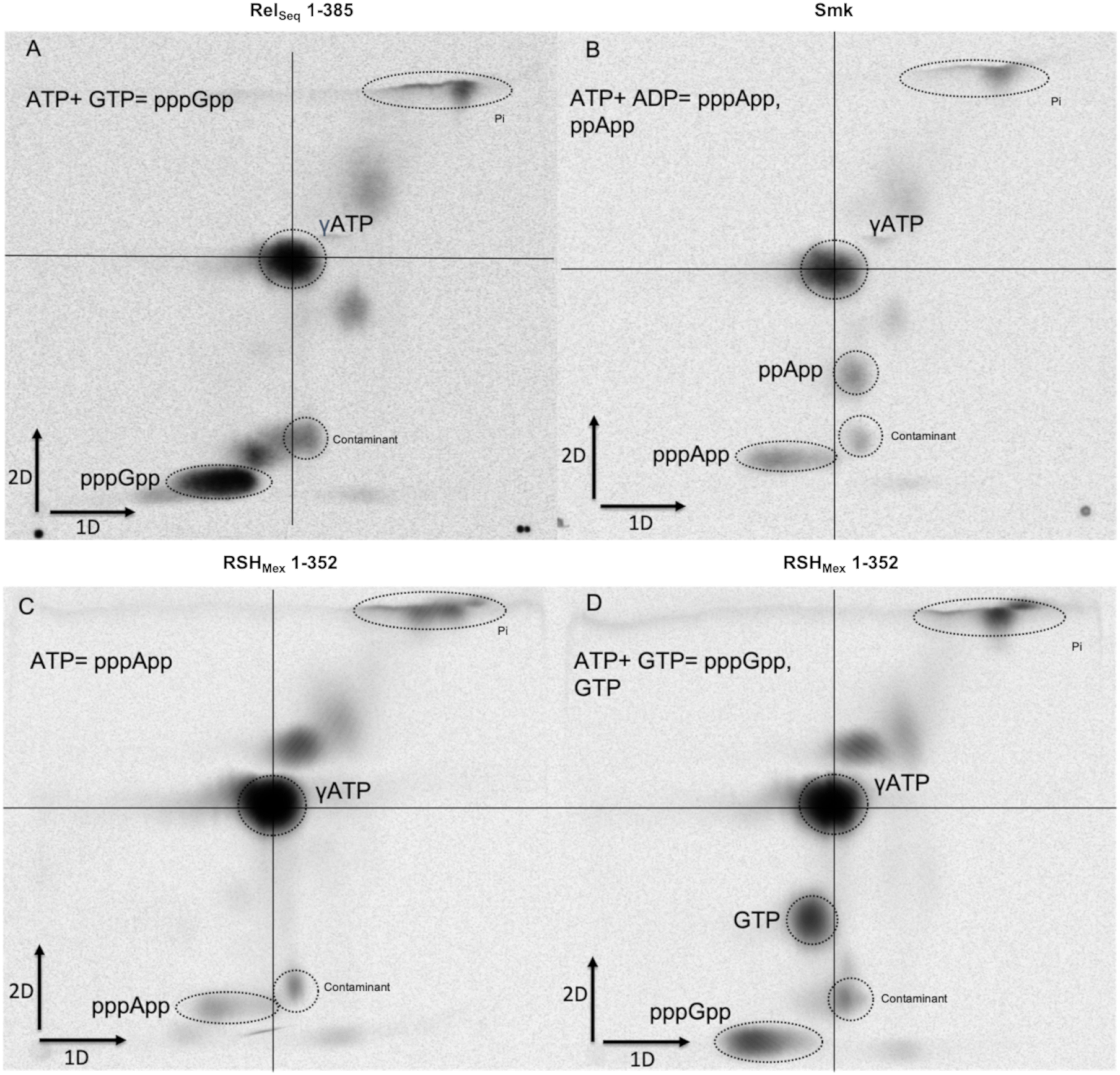
Figure 5. Two-dimensional TLC separation of (p)ppNpp’s synthetized by different enzymes. (A) RelSeq1-385 with 8 mM ATP + 8 mM GTP, (B) pyrophophokinase from S. morookaensis (Smk) with 8 mM ATP + 8 mM GDP, (C) RSHMex1-352 with 16 mM ATP, (D) RSHMex1-352 with 8 mM ATP + 8 mM GTP. The reaction mixtures contained 3.3 nM [P33] ɣ-ATP, 50 mM Tris–HCl (pH 8.9), and either 16 mM MgCl2 (RelSeq1-385 and Smk) or 8 mM CoCl2 (RSHMex1-352). 1D buffer: 3.3 M ammonium formate + 4.2% boric acid (pH 7), 2D buffer: 0.85 M KH2PO4 (pH 3.4).
We observed that in the reaction with 16 mM ATP and 8 mM Co2+, RSHMex1-352 indeed synthesized pppApp, which to our knowledge is a first documented example of an RSH enzyme synthesizing this nucleotide derivative (Figure 5C). On the other hand, in the reaction with 8 mM ATP and 8 mM GTP, we observed only pppGpp and no pppApp (Figure 5D). We did not observe ppApp (i.e., adenosine tetraphosphate) in any of the reactions catalyzed by RSHMex1-352. However, in the ATP + GTP reaction we detected a spot that corresponds to GTP, as judged by migration of unlabeled nucleotide standards (Supplementary Figure S4). In fact the spot initially ascribed by us to possibly be ppApp is GTP.
We then decided to take another approach that would also validate the observed labeling of GTP by [P33]-γATP in another way. A large scale synthesis reaction employing 8 mM ATP, 8 mM GDP, 16 mM MgCl2, and RSHMex1-352 was carried out for 24 h, using conditions that gave the highest yield of the nucleotide to be identified. The products were then separated by ion exchange chromatography on Sephadex QAE-25, using LiCl gradient for elution. This was followed by TLC, as well as by measurement of the purified compound’s absorbance in the UV-spectrum. The data obtained confirmed that GTP is indeed produced along with ppGpp, pppGpp and AMP, but not ppApp (Supplementary Figure S5).
We take the above experiments as convincing demonstration that pppApp is synthesized in vitro by the catalytic domain of RSHMex. A logical next step to establish its biological relevance would be to ask whether pppApp presence is demonstrable in vivo. We thus performed in vivo [P33] labeling of nucleotide pools in bacterial cells. In this case, a different buffer system for 2D TLC was employed than above, so as to better separate all nucleotide pools (Figure 6A) (Nishino et al., 1979). The growth medium used was not limiting for any nutrient, except that low level of inorganic phosphate had to be employed to give [P33] specific activities high enough to detect basal (p)ppNpp levels.

Figure 6. In vivo labeling of cellular nucleotide pools with [P33]. Nucleotides were labeled, extracted and resolved by TLC as described in the section “Materials and Methods.” (A) nucleotide standards, (B) M. extorquens AM1, (C) B. subtilis, (D) E. coli, (E) E. coli ppGpp0 with pUC19, and (F) E. coli ppGpp0 with pUC19-RSHMex. First dimension buffer: 1 M LiCl, 4 M formic acid; second dimension buffer: 0.85 M KH2PO4 (unadjusted pH).
As demonstrated by Figure 6B, trace amounts of pppApp and larger amounts of (p)ppGpp are detected in M. extorquens AM1 cellular nucleotide extracts. This suggests in vivo verification of our in vitro data. Importantly, similar results were obtained for B. subtilis (Figure 6C), reinforcing previous observations by others (Rhaese et al., 1977; Nishino et al., 1979).
Because of our previous interest in pppApp regulating transcription by E. coli RNA polymerase at the rrnB P1 promoter, we performed parallel experiments with wild type E. coli cells. As can be seen in Figure 6D, quite unexpectedly we were able to observe both – ppGpp and pppApp under our experimental conditions. They are not detected in the ΔrelA ΔspoT (ppGpp0) strain (Figure 6E). That deficiency is repaired when the ppGpp0 strain is transformed with a pUC19 derivative bearing full-length RSHMex, assayed by induction with 0.1 mM IPTG (Figure 6F). There, ppGpp and pppApp are again detected. We ascribe the lack of detectable pppGpp in E. coli cells due to the presence of the GppA γ-phosphate hydrolase known to convert pppGpp to ppGpp (Mechold et al., 2013).
In the final stage of this work, we tested whether expressing RSHMex in E. coli cells can provoke regulation of the sort predicted from in vitro observations with the rrnB P1 promoter for (p)ppGpp and pppApp (Bruhn-Olszewska et al., 2018). For this purpose, three different plasmid constructs were used: full length RSHMex, the catalytic domain (RSHMex1-352), and the fragment predicted to have only the synthetase domain. All plasmids were derivatives of pUC19 bearing rshMex gene fragments cloned under the plac promoter. The three different strains employed were wild type, ΔrelA and ppGpp0 and all carried a rrnB P1-lacZ fusion, whose activity is known to be inhibited by (p)ppGpp (Potrykus et al., 2006).
Growth was carried out in minimal medium supplemented with appropriate amounts of amino acids and carbon source (see section “Materials and Methods” for details) and 0.1 mM IPTG, and the rrnB P1-lacZ fusion activities were determined by β-galactosidase assays.
The results obtained are depicted in Figure 7A. In the case of strains overproducing full length RSHMex protein, we observed an increase in the rrnB P1-lacZ fusion activity in the wild type and ΔrelA strains, roughly reaching the level observed in the ppGpp0/vector control strains. There is no substantial increase in activity in case of ppGpp0/RSHMex when compared to the vector control. On the other hand RSHMex1-352 caused a slight decrease (about 25%) in the rrnB P1-lacZ fusion activity in the wild type strain background, and had no effect in the ΔrelA strain when compared to the vector control. Interestingly, we were unable to obtain transformants of the ppGpp0 strain with the RSHMex1-352 plasmid.
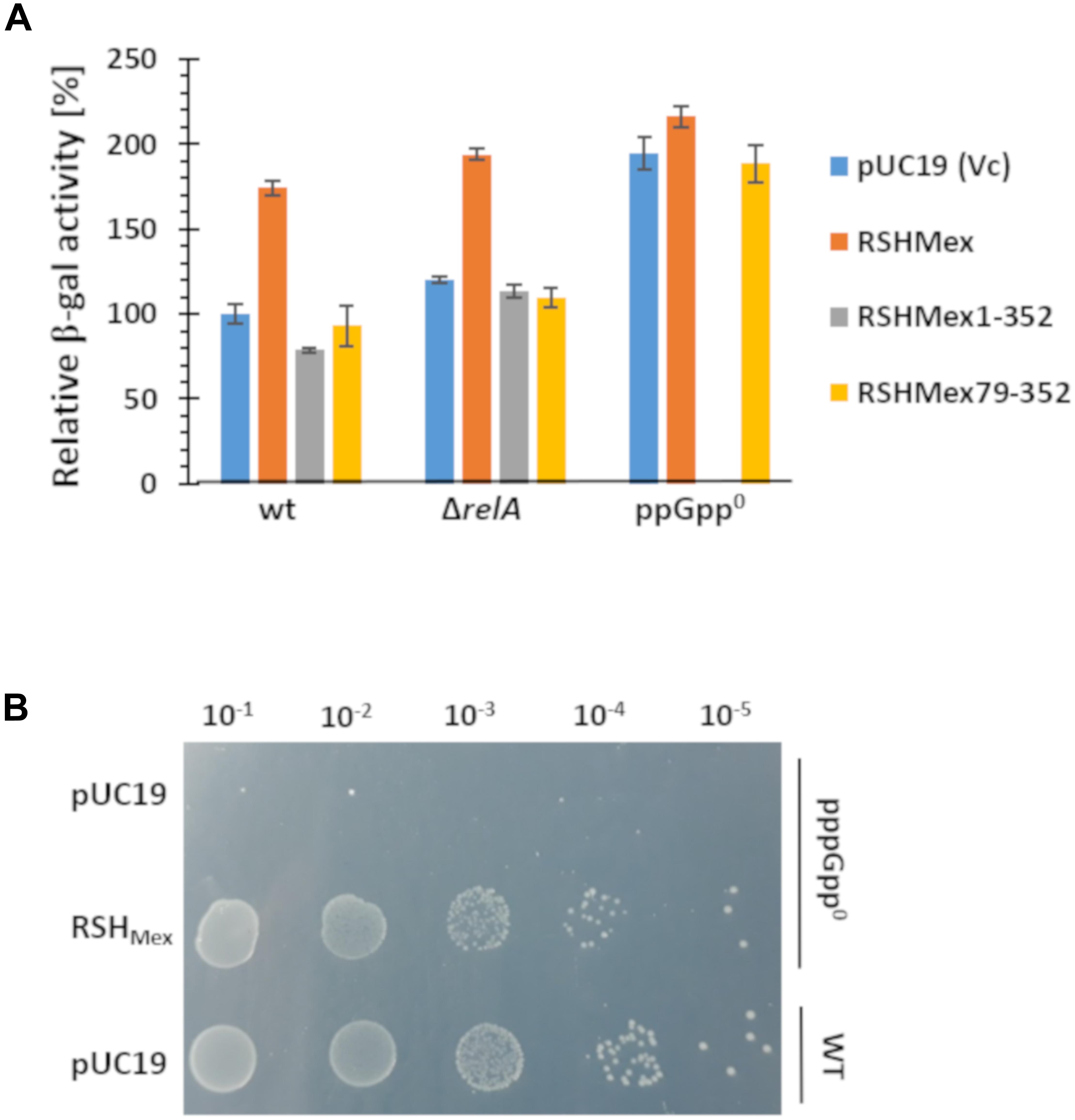
Figure 7. In vivo test of effects of (p)ppNpp synthesis. (A) RSHMex, RSHMex1-352 (NTD domain), and RSHMex79-352 (only synthetase domain) were cloned into pUC19 and transformed into the indicated host strains carrying the rrnB P1-lacZ fusion. Growth was carried out in minimal media as described in the section “Materials and Methods,” and β-galactosidase activity was assayed when the cells reached stationary phase. IPTG was added to 0.1 mM. We were unable to obtain ppGpp0/RSHMex1-352 transformants. Error bars indicate SD of the values obtained for three independent transformants and their cultures for each strain with the corresponding plasmids. (B) growth on SMG plate of the strains indicated, IPTG was added to 0.1 mM.
In addition, we observed that RSHMex79-352, expressing only the predicted synthetase domain of RSHMex, gives the same results as the vector control in all strain backgrounds, meaning this construct is inefficient in (p)ppNpp synthesis without the hydrolase domain, similarly to what was previously observed in RelSeq studies (Mechold et al., 2002).
Finally, we tested whether RSHMex could complement E. coli ppGpp0 phenotype when grown on SMG plates, i.e., minimal glucose plates containing only serine, methionine and glycine as amino acids. This is a growth test of two features. The first is for complementation of the multiple amino acid auxotrophic requirements (DEFHILSTV) of ppGpp0 strains because eight of the nine required are missing in the medium (Potrykus et al., 2011; Vinella et al., 2012). The second is for complementation of the stringent response due to an isoleucine deficiency in E. coli K-12 strains provoked in minimal medium by the presence of serine, methionine and glycine (Uzan and Danchin, 1976). Figure 7B demonstrates E. coli ppGpp0/RSHMex strain complements both, the multiple amino acid requirements of a ppGpp0 strain, as well as activation of the isoleucine synthesis (SMG resistance).
This study of the RSHMex enzyme activity is the first direct biochemical demonstration that an RSH enzyme is capable of synthesizing a nucleotide derivative other than (p)ppGpp, i.e., pppApp. This is shown in vitro with the RSHMex1-352 protein (Figures 3, 4), as well as in vivo with E. coli cells induced to express full-length RSHMex and with wild type M. extorquens AM1 cells (Figure 6). Moreover, we demonstrate pppApp accumulation in wild type E. coli cells, which points to a possible new player in bacterial stringent response.
In this work, it became evident to us that there may be a need to re-evaluate RSH nomenclature, changing it from the traditional standard of Rel(Species-name) to RSH(Species-name) and therefore in this case we use RSHMex. We suspect the problem arose when the Cashel-Mechold lab began studies on what they then thought was a relA-like monofunctional protein from a S. equisimilis strain. This led to calling it RelSeq as the RelA protein from S. equisimilis (Mechold et al., 1996). It was later discovered to be a bifunctional RSH protein but the name was not changed. The E. coli RelA is a monofunctional enzyme capable only of (p)ppGpp synthesis, but not its hydrolysis. We trust that the RSHMex full-length enzyme is bifunctional and capable of both, (p)ppNpp synthesis and hydrolysis (see below). Also, as already mentioned in the section “Results,” this enzyme has a higher homology to SpoT and RelSeq (both are bifunctional enzymes) than RelA. There are many obvious future experiments to be done, such as exploring the interaction of RSHMex with ribosomes, what sources of physiological stress activate synthesis and what responses are mediated by pppApp. This aside, classically enzymes are named for their catalytic properties and/or ortholog or paralog relatedness to other enzymes, rather than on their ability to bind to certain structures (such as ribosomes). Accordingly, RSH for RelA SpoT homologs is meaningful and clear. Thus, we propose that bifunctional homologs should be named RSH, and monofunctional large synthetases termed Rel. Again, the RSH naming does not relate to an ability to bind ribosomes. The abbreviations for small alarmone hydrolases and synthetases, SAH and SAS, respectively, remain clear.
So far, (p)ppApp synthesis has been only demonstrated in vitro by a pyrophosphotransferase from S. morookaensis, which is a non-specific enzyme transferring pyrophosphate residues from either ATP or GTP onto the ribosyl 3′hydroxyl of any purine nucleotide (Oki et al., 1975). In vivo, B. subtilis has been reported long ago to produce (p)ppApp but no biochemical evidence of synthesis was provided (Rhaese et al., 1977; Nishino et al., 1979).
In addition, we also demonstrate that (p)ppGpp synthesis by RSHMex might entail a non-canonical mechanism. For RelA and RelSeq, as well as several SAS enzymes, it has been shown that (p)ppGpp synthesis is carried out by direct transfer of the γβ-phosphate groups of ATP onto 3′ end of GTP or GDP, to yield pppGpp, or ppGpp respectively (Cashel and Kalbacher, 1970; Mechold et al., 1996, 2002; Hogg et al., 2004; Steinchen et al., 2015). Here, we found that ATP and GTP or GDP gave the expected pppGpp and ppGpp products, in addition to unexpected GTP labeled by the γ-phosphate of ATP (Figures 3, 4 and Supplementary Figure S6). Further studies will reveal whether this GTP molecule is a direct product, an intermediate or perhaps a by-product of the (p)ppGpp synthesis reaction.
Still, it must be noted here, that even though we rigorously identified that additional nucleotide as GTP, and excluded a possibility that the spot identified by us corresponds to other triphosphate guanosine nucleotide derivatives, such as ppGp and pGpp (when run on TLC in 0.85 M KH2PO4 buffer, these two nucleotides migrate much faster than GTP), it cannot be ruled out completely that this nucleotide may correspond to a yet another, previously unidentified guanosine derivative whose biochemical properties match those of GTP.
Other aspects of the RSHMex1-352 catalyzed reaction are unusual as well. First, we discovered that cobalt cations are required for the most efficient (p)ppNpp synthesis by RSHMex1-352 (Figure 2). For other RSH and SAS enzymes, including RelA, Mg2+ gives the most efficient synthesis, although we are not aware of any reports on testing Co2+ with these enzymes (for e.g., Mechold et al., 2002; Avarbock et al., 2005; Sajish et al., 2007; Steinchen et al., 2015; Ruwe et al., 2017; Manav et al., 2018). Second, Km values for ATP and GTP reveal RSHMex1-352 has a much higher affinity for ATP than GTP (Figure 5), which is in contrast with observations made with RelSeq (Mechold et al., 2002).
Still, in contrast to RSHMex we find Co2+ does not support the synthesis of (p)ppGpp by RelSeq (MS and KP, personal communication). Interestingly, M. extorquens AM1 displays cobalt requirement for methylotrophic growth, where it was found that ethylmalonyl-CoA pathway enzymes are cobalt-dependent (Kiefer et al., 2009). Intriguingly, adding CoCl2 to the liquid medium for in vivo labeling did not enhance pppApp production in our study.
It is noteworthy that under all conditions tested (even with different combinations of Mn2+, Mg2+, and Co2+) we did not observe any of the classical evidence for (p)ppNpp hydrolysis by RSHMex1-352 in vitro, namely release of an intact pyrophosphate, as documented for SpoT or RelSeq (Supplementary Figure S3, and: MS and KP, personal communication), which could mean that this activity might be regulated by the CTD domain, missing in the enzyme we studied in vitro. These results seem to be in line with our in vivo data where we were unable to obtain transformants of the ppGpp0 strain even with an uninduced multicopy plasmid carrying RSHMex1-352, however, transformation was possible with a plasmid bearing the full-length RSHMex. This observation implies that RSHMex1-352 might produce toxically high amounts of (p)ppGpp in this background (Figure 7). SpoT, present in the wild type and ΔrelA strains, is able to lower these amounts to the non-toxic levels. In line with these observations, a study was published recently on another representative of alpha-proteobacteria, namely Caulobacter crescentus RSH enzyme (called by the authors SpoT) (Ronneau et al., 2018). The authors had shown this enzymes requires the ACT domain (part of the large regulatory C-terminal domain) for ppGpp hydrolysis. Accordingly, this domain is missing in RSHMex1-352, but present in full-length RSHMex.
On the other hand, we observed an increase in the rrnB P1-lacZ fusion activity for the full-length RSHMex construct in both – the wt and ΔrelA backgrounds, while the activity was practically unchanged in the ppGpp0 strain when compared to the vector control. A possible explanation is that when SpoT is present, it can hydrolyze RSHMex – synthesized (p)ppGpp but not pppApp (or not as efficiently as (p)ppGpp). Recently, we found pppApp activates transcription initiating at the rrnB P1 promoter in vitro (Bruhn-Olszewska et al., 2018), and here the same could be true in vivo. Induction of the full length RSHMex plasmid in the wild type or relA mutant hosts does significantly elevate rrnB P1reporter activity, as if pppApp might overcome the negative regulatory effects of low levels of ppGpp to parallel pppApp activities observed in vitro (Bruhn-Olszewska et al., 2018). Lack of increase in the rrnB P1-lacZ fusion activity in the ppGpp0/RSHMex strain could be due to the maximum promoter activity already achieved in the ppGpp0 background, which simply cannot be increased any further.
Also, Figure 6F shows that in the IPTG induced ppGpp0/RSHMex strain both ppGpp and pppApp are clearly present. On the basis of ppGpp levels alone, quite strong inhibition of rrnB P1 might be predicted but is not seen. This again implies that activation by pppApp occurs, and despite its low abundance relative to ppGpp it can overcome the (p)ppGpp mediated inhibition of the rrnB P1 promoter. On the other hand, stable survival of this strain again suggests RSHMex -mediated hydrolysis, since unchecked production of (p)ppNpp would be toxic to the cell, as demonstrated by inability to transform ppGpp0 strain with RSHMex1-352. This, together with ppGpp0/RSHMex SMG plate growth (Figure 7), suggest that instead of pppApp effects, perhaps RSHMex-mediated ppGpp synthesis at low levels could be enough to induce isoleucine synthesis and high enough to complement the multiple amino acid requirements of ppGpp0 strains, as noted earlier with [P33] labeled nucleotide extracts (Figure 6).
Overall, our rrnB P1-lacZ reporter studies hint that pppApp might display positive regulatory effects in vivo. Rigorous evidence awaits devising a means of selectively manipulating incremental cellular accumulation of pppApp independently of ppGpp abundance so that competitive regulation can be assessed, much like regulatory potency for ppGpp vs. pppGpp has been investigated (Mechold et al., 2013).
Might pppApp accumulation be found among diverse bacterial species and participates in stress responses along with (p)ppGpp? It is intriguing that we observed cellular accumulations of pppApp but not ppApp, since in vitro transcription studies reveal pppApp has a much stronger effect on RNA polymerase than ppApp (Bruhn-Olszewska et al., 2018). If RNA polymerase regulatory effects of pppApp are generally present in bacteria then it is predicted that E. coli RNAP residues contacting pppApp (Bruhn-Olszewska et al., 2018) might also be found in RNA polymerases of B. subtilis and M. extorquens. Such comparisons do reveal conservation among these species with respect to residues R346, R352, A426, and Q465 of the β′ subunit, and K1242 of the β subunit. It is an especially intriguing finding for B. subtilis RNAP because in this organism, control of rRNA synthesis by (p)ppGpp is clearly known to occur indirectly through control of GTP levels rather than direct interactions of (p)ppGpp with RNAP (Krasny and Gourse, 2004).
Since pppApp co-migrates with ppGpp on TLC plates in commonly used assays employing 1 M KH2PO4 (pH 3.4) resolution buffer (Supplementary Figure S3), the question arises as to whether previous measurements of ppGpp in different bacteria were in fact distorted by unsuspected pppApp content. It is likely that instances will be found where pppApp was mistakenly included in quantitation with ppGpp by TLC resolution or dismissed as contamination. On the other hand, neither our current work nor that of others (Rhaese et al., 1977; Nishino et al., 1979) have detected cellular (p)ppApp at high levels, e.g., equal to or exceeding GTP, as commonly found for (p)ppGpp. Therefore future studies should have priority for finding conditions where (p)ppApp production levels increase and establishing the mechanism of its synthesis in vivo. In addition an effort should focus on developing better TLC methods to resolving all four nucleotides pppApp, ppGpp, ppApp, and GTP.
All strains used, plasmids and their construction are described in Supplementary Table S1.
For in vitro studies, Hisx8-SUMO-tagged proteins (RSHMex1-352 and RelSeq1-385) were purified by Ni2+-NTA affinity chromatography, followed by removal of the His-SUMO tag by digestion with Ulp1 SUMO protease. In detail, E. coli BL21 (DE3) Rosetta cells carrying a plasmid for His-SUMO-tagged protein production were grown at 30°C in 2 L of LB (0.5% NaCl), supplemented with kanamycin at 50 μg/ml. Cells were cultivated to OD600 = 0.6. Expression was induced by adding IPTG to the final concentration of 0.4 mM, and cultivation was continued overnight at 16°C. Cells were harvested by centrifugation, re-suspended in 100 ml of lysis buffer (20 mM of Tris–HCl pH 8.0, 500 mM NaCl, 20 mM imidazole, 10% glycerol, 2 mM β-mercapthoethanol), PMSF was added, and the cells were lysed by sonication. After centrifugation (37,850 × g, 30 min, 4°C), clear supernatant was loaded on a BioRad disposable column pre-loaded with 2 ml of the Ni-NTA Superflow resin (Thermo Scientific) and pre-equilibrated with 10 ml of lysis buffer but without imidazole. Following a wash with 40 ml of lysis buffer, the His-SUMO-tagged protein was eluted with 10 ml of elution buffer (lysis buffer but with 180 mM imidazole). Protein containing fractions (10 ml) were pooled and dialyzed against 2 L of dialysis buffer (20 mM of Tris–HCl pH 8.0, 250 mM NaCl, 10% glycerol) in a Slide-a-lyzer 10K cassette (Thermo Scientific). His-SUMO-tag was cleaved by an in-house purified His-tagged yeast Ulp1 SUMO protease, added to the final concentration of 10 μg/ml, and incubated at 4°C for 15 min. After cleavage, the protein sample was loaded on a new BioRad column pre-loaded with 2 ml of Ni-NTA Superflow resin, pre-equilibrated with 10 ml of dialysis buffer. Flow through fraction obtained this way contained pure, untagged proteins. Glycerol was added to the final concentration of 50% and preps were stored at -80°C. Protein concentration was determined with Qubit 2.0 (Thermo Scientific), and purity was determined by SDS-PAGE. This protocol yielded 10 ml of RSHMex1-352 at a final concentration of 0.186 mg/ml, with >99% purity (Supplementary Figure S6). For RelSeq1-385, 10 ml of 13.6 mg/ml protein were obtained (with 98% purity).
The S. morookaensis excreted ammonium sulfate precipitated protein extract containing the non-specific pyrophosphotransferase was prepared as described in (Bruhn-Olszewska et al., 2018).
All NTPs, NDPs, and NMPs used were purchased from Sigma-Aldrich. Unlabeled (p)ppNpp standards were prepared and purified as described in (Bruhn-Olszewska et al., 2018).
For RSHMex1-352, in vitro (p)ppNpp synthesis was generally carried out with either 80 or 160 nM of purified protein in a reaction containing 50 mM Tris–HCl pH 8.9, 3.3 nM [P33] γ-ATP (Perkin Elmer), and 8 mM ATP (pyrophosphate donor) and 8 mM acceptor nucleotide (ATP, ADP, AMP, GTP, GDP, or GMP). The cation concentrations used were: 8 mM Co2+ or 16 mM Mg2+ (unless indicated otherwise), provided as CoCl2 or MgCl2. MnCl2, CaCl2, or NiCl2 were also tested. The reactions were carried out at 37°C, and the reaction time varied between 1 and 16 h. Reactions were stopped by adding an equal volume of 2 M formic acid and spotted onto 20 cm × 20 cm PEI-Cellulose plates (Merck). One-dimensional thin layer chromatography (TLC) was carried out in either 0.85 or 1 M KH2PO4 (pH 3.4) buffer. For two-dimensional TLC, samples were first separated in a buffer containing 3.3 M ammonium formate and 4.2% boric acid (pH 7.0); this was followed by soaking plates for 15 min in methanol, rinsing in water, development in the second dimension in 0.85 M KH2PO4 (pH 3.4). Autoradiograms were visualized using a phosphorimager (Typhoon 9200, GE Healthcare).
For the RelSeq1-385 and S. morookaensis pyrophosphotransferase containing extract, similar reaction conditions were used as above, with 16 mM MglCl2 and 160 nM RelSeq1-385 protein or 2 μl of the S. morookaensis extract. Reactions were carried out for 30 min at 37°C.
Measurements of RSHMex1-352 enzyme kinetics were carried out as described above but with 80 nM protein. Reactions were carried out with 8 mM CoCl2 for 2 h. End-point assays were used since using continuous assays was impractical in this case due to low method sensitivity in detecting low (p)ppNpp amounts. The values obtained should be taken with caution and thus are referred to as apparent Km. The apparent Km for GTP was measured with ATP at 8 mM and titrated GTP (1–8 mM). For the apparent Km for ATP, GTP was held at 8 mM and ATP varied from 0.25 to 2 mM. The samples were processed as described above and TLC was run in 1.5 M KH2PO4 (pH 3.4) buffer. Autoradiograms were visualized using a phosphorimager (Typhoon 9200, GE Healthcare). Spots corresponding to pppGpp were quantitated using a UVP Visionworks software and the data was analyzed with GraphPad Prism 5; apparent Km values were obtained from the fit to the following equation: V = [Vmax (S)] ÷ [Km + (S)].
Methylobacterium extorquens AM1 was cultivated on LB plates supplemented with 0.1% meat extract (Sigma), 0.1% methanol and rifampicin (50 μg/ml). E. coli and B. subtilis strains were grown on LB plates. Bacteria were scraped off plates and resuspended in the Tris-glucose medium (0.1 M Tris pH 7.4, 0.1 mM KH2PO4, sodium citrate (0.42 mg/ml), MgSO4 × 7H20 (0.21 mg/ml), (NH4)2SO4 (1 mg/ml), FeCl3 (0.32 μg/ml), glucose (0.2%), and the following amino acids: lysine, proline, glycine, alanine, glutamic acid, aspartic acid, arginine, at 100 μg/ml; cysteine, methionine, tyrosine, tryptophan, and phenylalanine at 40 μg/ml) (Nishino et al., 1979). Nucleotides were labeled by the addition of [P33] – phosphoric acid to 5 μCi/ml and incubated with shaking at 30°C for 45 min. For E. coli strains carrying pUC19 derivatives, 0.1 mM IPTG was added. Reactions were stopped by the addition of an equal volume of 23.6 M formic acid, and followed by three freeze-thaw cycles in liquid nitrogen. Extracts were centrifuged (14,000 × g, 10 min, room temperature) and the supernatants were spotted onto PEI-Cellulose plates. For two-dimensional TLC separation, first dimension buffer contained 1 M LiCl and 4 M formic acid; this was followed by soaking plates for 15 min in methanol and a second dimension run in 0.85 M KH2PO4 (unadjusted pH). Autoradiograms were visualized as before.
Appropriate E. coli strains carrying rrnB P1-lacZ chromosomal fusions were transformed with pUC19-derived plasmids carrying genes of interest and streaked on minimal medium plates containing: 1 × M9 salts (BioShop), 1.5% agar, 1% vitamin B1, 0.5 μM FeSO4, 0.2% casamino acids, 0.02% glucose, and 0.1 mM IPTG. Ampicillin was added to 50 μg/ml. Plates were incubated at 30°C for 48 h. The cells were then inoculated into the same medium (but lacking agar), grown with shaking to stationary phase, and β-galactosidase activity was assayed as described in (Miller, 1972).
For E. coli ppGpp0 complementation assay, cells were collected from plates, washed three times with 0.9% NaCl, resuspended in 0.9% NaCl to adjust OD600 to 0.1, and 5 μl of appropriate dilutions were spotted on SMG plates (minimal medium plates as above but with 100 μg/ml each of serine, methionine and glycine instead of casamino acids). Growth was carried out as described above.
KP conceived this study. KP, MS, and MC designed the experiments and MS and KP performed them. BB-O participated in GTP identification. KP, MS, and MC analyzed the data. KP and MC wrote the main text. All authors discussed the results, commented on the manuscript, and approved its final version.
This work has been funded by the National Science Centre (Poland) (UMO-2013/10/E/NZ1/00657 awarded to KP), in part by the Intramural Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (MC), and the Funding for Young Researchers (538-L140-B262-16, University of Gdańsk, to MS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Mary E. Lidstrom for sharing the M. extorquens AM1 strain, Dr. Michał Miętus for providing plasmid and protocol for yeast Ulp1 SUMO-protease purification, and Dr. Llorenç Fernandez-Coll for helpful discussions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00859/full#supplementary-material
Atkinson, G. C., Tenson, T., and Hauryliuk, V. (2011). The RelA/SpoT homolog (RSH) superfamily: distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS One 6:e23479. doi: 10.1371/journal.pone.0023479
Avarbock, A., Avarbock, D., Teh, J. S., Buckstein, M., Wang, Z. M., and Rubin, H. (2005). Functional regulation of the opposing (p)ppGpp synthetase/hydrolase activities of RelMtb from Mycobacterium tuberculosis. Biochemistry 44, 9913–9923. doi: 10.1021/bi0505316
Beljantseva, J., Kudrin, P., Andresen, L., Shingler, V., Atkinson, G. C., Tenson, T., et al. (2017). Negative allosteric regulation of Enterococcus faecalis small alarmone synthetase RelQ by single-stranded RNA. Proc. Natl. Acad. Sci. U.S.A. 114, 3726–3731. doi: 10.1073/pnas.1617868114
Braeken, K., Moris, M., Daniels, R., Vanderleyden, J., and Michiels, J. (2006). New horizons for (p)ppGpp in bacterial and plant physiology. Trends Microbiol. 14, 45–54. doi: 10.1016/j.tim.2005.11.006
Bruhn-Olszewska, B., Molodtsov, V., Sobala, M., Dylewski, M., Murakami, K. S., Cashel, M., et al. (2018). Structure-function comparisons of (p)ppApp vs (p)ppGpp for Escherichia coli RNA polymerase binding sites and for rrnB P1 promoter regulatory responses in vitro. Biochim. Biophys. Acta Gene Regul. Mech. 1861, 731–742. doi: 10.1016/j.bbagrm.2018.07.005
Cashel, M., and Gallant, J. (1969). Two compounds implicated in the function of the RC gene of Escherichia coli. Nature 221, 838–841. doi: 10.1038/221838a0
Cashel, M., Gentry, D., Hernandez, V. J., and Vinella, D. (1996). “The stringent response,” in Escherichia coli and Salmonella: Cellular and Molecular Biology, ed. F. C. Neidhardt (Washington, DC: ASM Press), 1458–1496.
Cashel, M., and Kalbacher, B. (1970). The control of ribonucleic acid synthesis in Escherichia coli. V. Characterization of a nucleotide associated with the stringent response. J. Biol. Chem. 245, 2309–2318.
Cashel, M., Lazzarini, R. A., and Kalbacher, B. (1969). An improved method for thin-layer chromatography of nucleotide mixtures containing 32P-labelled orthophosphate. J. Chromatogr. 40, 103–109. doi: 10.1016/s0021-9673(01)96624-5
Dalebroux, Z. D., Svensson, S. L., Gaynor, E. C., and Swanson, M. S. (2010). ppGpp conjures bacterial virulence. Microbiol. Mol. Biol. Rev. 74, 171–199. doi: 10.1128/MMBR.00046-09
Field, B. (2018). Green magic: regulation of the chloroplast stress response by (p)ppGpp in plants and algae. J. Exp. Bot. 69, 2797–2807. doi: 10.1093/jxb/erx485
Gaca, A. O., Kajfasz, J. K., Miller, J. H., Liu, K., Wang, J. D., Abranches, J., et al. (2013). Basal levels of (p)ppGpp in Enterococcus faecalis: the magic beyond the stringent response. mBio 4:e00646-13. doi: 10.1128/mBio.00646-13
Gaca, A. O., Kudrin, P., Colomer-Winter, C., Beljantseva, J., Liu, K., Anderson, B., et al. (2015). From (p)ppGpp to (pp)pGpp: characterization of regulatory effects of pGpp synthesized by the small alarmone synthetase of Enterococcus faecalis. J. Bacteriol. 197, 2908–2919. doi: 10.1128/JB.00324-15
Geiger, T., Goerke, C., Fritz, M., Schafer, T., Ohlsen, K., Liebeke, M., et al. (2010). Role of the (p)ppGpp synthase RSH, a RelA/SpoT homolog, in stringent response and virulence of Staphylococcus aureus. Infect. Immun. 78, 1873–1883. doi: 10.1128/IAI.01439-09
Gratani, F. L., Horvatek, P., Geiger, T., Borisova, M., Mayer, C., Grin, I., et al. (2018). Regulation of the opposing (p)ppGpp synthetase and hydrolase activities in a bifunctional RelA/SpoT homologue from Staphylococcus aureus. PLoS Genet. 14:e1007514. doi: 10.1371/journal.pgen.1007514
Hogg, T., Mechold, U., Malke, H., Cashel, M., and Hilgenfeld, R. (2004). Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response. Cell 117, 57–68. doi: 10.1016/s0092-8674(04)00260-0
Kiefer, P., Buchhaupt, M., Christen, P., Kaup, B., Schrader, J., and Vorholt, J. A. (2009). Metabolite profiling uncovers plasmid-induced cobalt limitation under methylotrophic growth conditions. PLoS One 4:e7831. doi: 10.1371/journal.pone.0007831
Kim, S. K., Park, M. K., Kim, S. H., Oh, K. G., Jung, K. H., Hong, C. H., et al. (2014). Identification of stringent response-related and potential serological proteins released from Bacillus anthracis overexpressing the RelA/SpoT homolog. rsh bant. Curr. Microbiol. 69, 436–444. doi: 10.1007/s00284-014-0606-8
Krasny, L., and Gourse, R. L. (2004). An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. EMBO J. 23, 4473–4483. doi: 10.1038/sj.emboj.7600423
Kudrin, P., Dzhygyr, I., Ishiguro, K., Beljantseva, J., Maksimova, E., Oliveira, S. R. A., et al. (2018). The ribosomal A-site finger is crucial for binding and activation of the stringent factor RelA. Nucleic Acids Res. 46, 1973–1983. doi: 10.1093/nar/gky023
Manav, M. C., Beljantseva, J., Bojer, M. S., Tenson, T., Ingmer, H., Hauryliuk, V., et al. (2018). Structural basis for (p)ppGpp synthesis by the Staphylococcus aureus small alarmone synthetase RelP. J. Biol. Chem. 293, 3254–3264. doi: 10.1074/jbc.RA117.001374
Mechold, U., Cashel, M., Steiner, K., Gentry, D., and Malke, H. (1996). Functional analysis of a relA/spoT gene homolog from Streptococcus equisimilis. J. Bacteriol. 178, 1401–1411. doi: 10.1128/jb.178.5.1401-1411.1996
Mechold, U., Murphy, H., Brown, L., and Cashel, M. (2002). Intramolecular regulation of the opposing (p)ppGpp catalytic activities of rel(seq), the rel/spo enzyme from Streptococcus equisimilis. J. Bacteriol. 184, 2878–2888. doi: 10.1128/jb.184.11.2878-2888.2002
Mechold, U., Potrykus, K., Murphy, H., Murakami, K. S., and Cashel, M. (2013). Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res. 41, 6175–6189. doi: 10.1093/nar/gkt302
Miller, J. H. (1972). Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Nishino, T., Gallant, J., Shalit, P., Palmer, L., and Wehr, T. (1979). Regulatory nucleotides involved in the rel function of Bacillus subtilis. J. Bacteriol. 140, 671–679.
Oki, T., Yoshimoto, A., Sato, S., and Takamatsu, A. (1975). Purine nucleotide pyrophosphotransferase from Streptomyces morookaensis, capable of synthesizing pppApp and pppGpp. Biochim. Biophys. Acta 410, 262–272. doi: 10.1016/0005-2744(75)90228-4
Potrykus, K., and Cashel, M. (2008). (p)ppGpp: still magical? Annu. Rev. Microbiol. 62, 35–51. doi: 10.1146/annurev.micro.62.081307.162903
Potrykus, K., Murphy, H., Philippe, N., and Cashel, M. (2011). ppGpp is the major source of growth rate control in E. coli. Environ. Microbiol. 13, 563–575. doi: 10.1111/j.1462-2920.2010.02357.x
Potrykus, K., Vinella, D., Murphy, H., Szalewska-Palasz, A., D’Ari, R., and Cashel, M. (2006). Antagonistic regulation of Escherichia coli ribosomal RNA rrnB P1 promoter activity by GreA and DksA. J. Biol. Chem. 281, 15238–15248. doi: 10.1074/jbc.m601531200
Rhaese, H. J., Hoch, J. A., and Groscurth, R. (1977). Studies on the control of development: isolation of Bacillus subtilis mutants blocked early in sporulation and defective in synthesis of highly phosphorylated nucleotides. Proc. Natl. Acad. Sci. U.S.A. 74, 1125–1129. doi: 10.1073/pnas.74.3.1125
Ronneau, S., Caballero-Montes, J., Coppine, J., Mayard, A., Garcia-Pino, A., and Hallez, R. (2018). Regulation of (p)ppGpp hydrolysis by a conserved archetypal regulatory domain. Nucleic Acids Res. 47, 843–854. doi: 10.1093/nar/gky1201
Ruwe, M., Kalinowski, J., and Persicke, M. (2017). Identification and functional characterization of small alarmone synthetases in Corynebacterium glutamicum. Front. Microbiol. 8:1601. doi: 10.3389/fmicb.2017.01601
Sajish, M., Tiwari, D., Rananaware, D., Nandicoori, V. K., and Prakash, B. (2007). A charge reversal differentiates (p)ppGpp synthesis by monofunctional and bifunctional rel proteins. J. Biol. Chem. 282, 34977–34983. doi: 10.1074/jbc.m704828200
Singal, B., Balakrishna, A. M., Nartey, W., Manimekalai, M. S. S., Jeyakanthan, J., and Grüber, G. (2017). Crystallographic and solution structure of the N-terminal domain of the rel protein from Mycobacterium tuberculosis. FEBS Lett. 591, 2323–2337. doi: 10.1002/1873-3468.12739
Steinchen, W., and Bange, G. (2016). The magic dance of the alarmones (p)ppGpp. Mol. Microbiol. 101, 531–544. doi: 10.1111/mmi.13412
Steinchen, W., Schuhmacher, J. S., Altegoer, F., Fage, C. D., Srinivasan, V., Linne, U., et al. (2015). Catalytic mechanism and allosteric regulation of an oligomeric (p)ppGpp synthetase by an alarmone. Proc. Natl. Acad. Sci. U.S.A. 112, 13348–13353. doi: 10.1073/pnas.1505271112
Steinchen, W., Vogt, M. S., Altegoer, F., Giammarinaro, P. I., Horvatek, P., Wolz, C., et al. (2018). Structural and mechanistic divergence of the small (p)ppGpp synthetases RelP and RelQ. Sci. Rep. 8:2195. doi: 10.1038/s41598-018-20634-4
Tozawa, Y., and Nomura, Y. (2011). Signalling by the global regulatory molecule ppGpp in bacteria and chloroplasts of land plants. Plant Biol. 13, 699–709. doi: 10.1111/j.1438-8677.2011.00484.x
Uzan, M., and Danchin, A. (1976). A rapid test for the relA mutation in E. coli. Biochem. Biophys. Res. Commun. 69, 751–758. doi: 10.1016/0006-291x(76)90939-6
Vinella, D., Potrykus, K., Murphy, H., and Cashel, M. (2012). Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli. J. Bacteriol. 194, 261–273. doi: 10.1128/JB.06238-11
Wang, J., Tian, Y., Zhou, Z., Zhang, L., Zhang, W., Lin, M., et al. (2016). Identification and functional analysis of RelA/SpoT Homolog (RSH) genes in Deinococcus radiodurans. J. Microbiol. Biotechnol. 26, 2106–2115. doi: 10.4014/jmb.1601.01017
Wendrich, T. M., Beckering, C. L., and Marahiel, M. A. (2000). Characterization of the relA/spoT gene from Bacillus stearothermophilus. FEMS Microbiol. Lett. 190, 195–201. doi: 10.1016/s0378-1097(00)00335-9
Keywords: (p)ppGpp, pppApp, Rel-SpoT-homologs, stringent response, Methylobacterium extorquens, Escherichia coli
Citation: Sobala M, Bruhn-Olszewska B, Cashel M and Potrykus K (2019) Methylobacterium extorquens RSH Enzyme Synthesizes (p)ppGpp and pppApp in vitro and in vivo, and Leads to Discovery of pppApp Synthesis in Escherichia coli. Front. Microbiol. 10:859. doi: 10.3389/fmicb.2019.00859
Received: 03 January 2019; Accepted: 03 April 2019;
Published: 24 April 2019.
Edited by:
Marie-Joelle Virolle, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Emmanuellle Bouveret, Institut Pasteur, FranceCopyright © 2019 Sobala, Bruhn-Olszewska, Potrykus and the U.S. Government. At least a portion of this work is authored by Michael Cashel on behalf of the U.S. Government and, as regards Dr. Cashel and the US government, is not subject to copyright protection in the United States. Foreign and other copyrights may apply. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarzyna Potrykus, a2F0YXJ6eW5hLnBvdHJ5a3VzQGJpb2wudWcuZWR1LnBs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.