 Kaiyang Qu
Kaiyang Qu Fei Guo
Fei Guo Xiangrong Liu
Xiangrong Liu Yuan Lin2,3*
Yuan Lin2,3* Quan Zou
Quan Zou- 1College of Intelligence and Computing, Tianjin University, Tianjin, China
- 2School of Information Science and Technology, Xiamen University, Xiamen, China
- 3Department of System Integration, Sparebanken Vest, Bergen, Norway
- 4Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, China
- 5Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu, China
Microorganisms are ubiquitous and closely related to people’s daily lives. Since they were first discovered in the 19th century, researchers have shown great interest in microorganisms. People studied microorganisms through cultivation, but this method is expensive and time consuming. However, the cultivation method cannot keep a pace with the development of high-throughput sequencing technology. To deal with this problem, machine learning (ML) methods have been widely applied to the field of microbiology. Literature reviews have shown that ML can be used in many aspects of microbiology research, especially classification problems, and for exploring the interaction between microorganisms and the surrounding environment. In this study, we summarize the application of ML in microbiology.
Introduction
Microorganisms first appeared approximately 3.5 billion years ago, making them one of the earliest living things on Earth (Nannipieri et al., 2010). Microorganisms include bacteria, viruses, fungi, some small protozoa, and microscopic algae. These organisms, which are closely related to human beings (Ley et al., 2006a), have a wide range of beneficial and harmful uses, including in the food (Cotter et al., 2005), medicine (Petrof et al., 2012; Yu et al., 2018), agriculture (Morris et al., 1986), industrial (Souza, 2010), environmental protection and other fields (Reiff and Kelly, 2010).
Microbiology is a discipline that studies the structure and function of microbial groups, the interrelationships and mechanisms of internal communities, and the relationships between microorganisms and their environments or hosts (Alexander, 1962; Niel, 1966). The microbiome is a collection of all microbial species and their genetic information and functions in a given environment. Studies of the microbiome also include the interaction between different microorganisms (DiMucci et al., 2018), the interaction between microorganisms and other species (Xie et al., 2018), and the interaction between microorganisms and the environment (Moitinho-Silva et al., 2017). Because of their small size, the microscope is an important tool for studying microorganisms. However, microscopy analyses only allow observation and must therefore be complemented by culture techniques to study the biological, physiological, genetic, metabolic, pathogenic and other biological characteristics of microorganisms (Waldron, 2018). During cultivation, researchers can also explore the interactions between microorganisms and the environment, which reflect the breadth and diversity of microbial distribution. A variety of microorganisms living in different environments or in different hosts form microbial communities, which have extensive and complex interactions with the environment and the host and form various types of ecosystems (Srinivasan et al., 2012; Xie et al., 2018).
With the development of microbial sequencing in recent years, the microbiome has become increasingly popular in many studies. High-throughput sequencing technology has resulted in generation of an increasing amount of microbial data. Traditional methods using microscopes and biological cultures are expensive and labor intensive; therefore, machine-learning methods have been gradually applied to microbial studies (Huang Y. A. et al., 2017; Huang Z. A. et al., 2017; Wang et al., 2017; Wei et al., 2017a,b; Peng et al., 2018; Yang et al., 2018b; Zou et al., 2018a). Here, we introduce the application of machine learning (ML) in microbial analyses. Since ML is mainly applied to classification and interaction problems, we focus on these two areas. Figure 1 shows the framework of this paper.
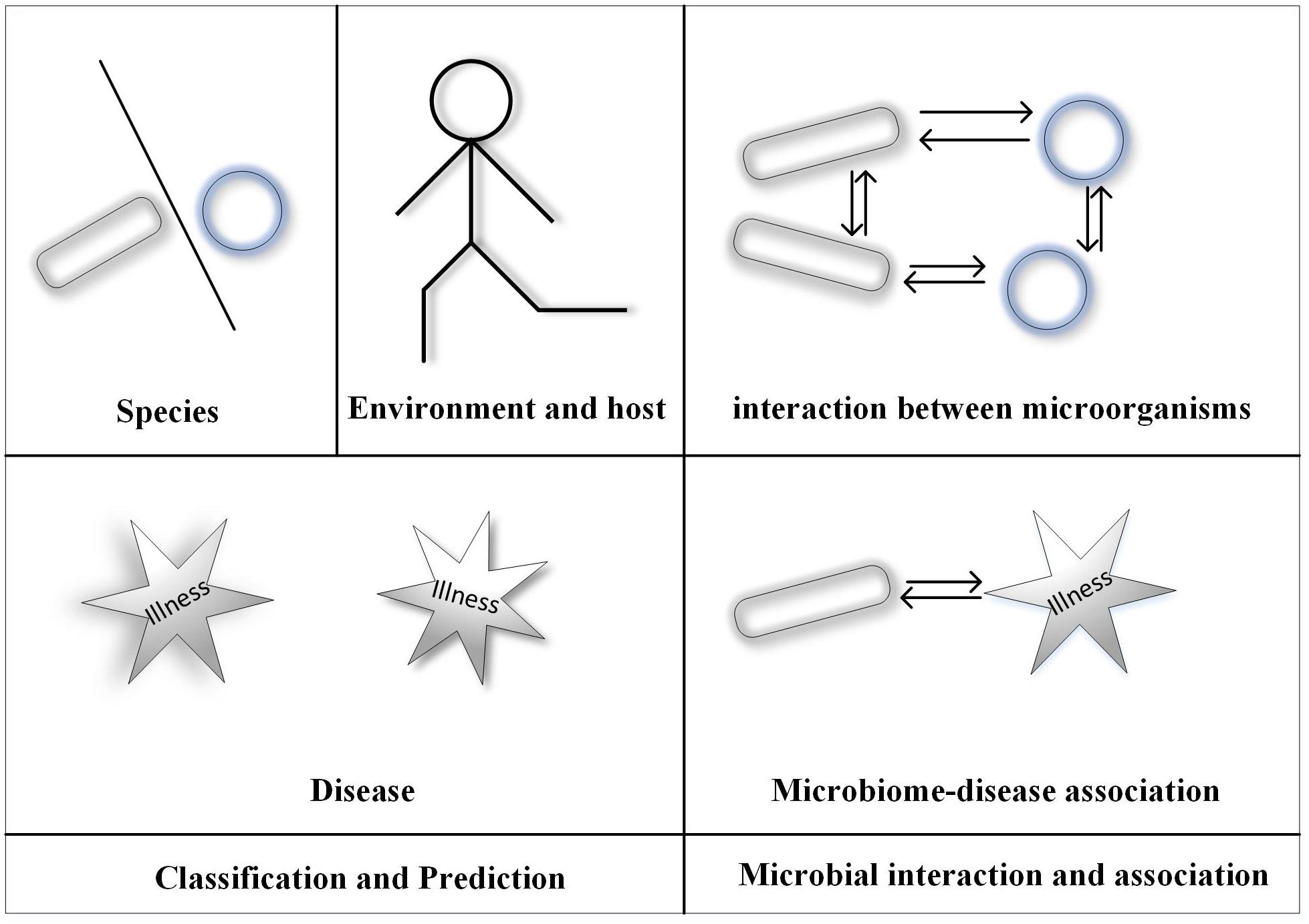
Figure 1. The framework of this paper.
Machine Learning Methods
Machine Learning is a multi-disciplinary subject involving many disciplines including probability theory, statistics, approximation theory, convex analysis, and algorithm complexity theory (Qu et al., 2017; Zou et al., 2018b). ML methods can be divided into two types (Zitnik et al., 2019), supervised learning and unsupervised learning. Supervised learning (Stoter et al., 2019) requires that the model be trained using a training set. The training sets for supervised learning include features and results. Common supervised learning algorithms include regression analysis and statistical classification. Unsupervised learning, also known as clustering, adopts k-means to establish a centriole and reduce error through iteration and descent to achieve classification. With the development of ML, more and more fields have begun to use this technique for research (Chen W. et al., 2016, Chen et al., 2017a,d, 2018a,b,e,f,g; Li et al., 2016; Zou et al., 2016, 2017; Ding et al., 2017a,b; Feng et al., 2017a; Yu et al., 2017a; Zeng et al., 2017a, 2018; Liu et al., 2018; Pan et al., 2018; Wei et al., 2018a,b; Yang et al., 2018a; Zhao et al., 2018b; He et al., 2019; Zhang et al., 2019), for example, drug repositioning (Yu et al., 2016b, 2017b), disease-related microRNA (Chen and Huang, 2017; Chen et al., 2017d, 2018b,e,g; Zhao et al., 2018a,c) identification, and disease-related long non-coding RNA identification (Chen and Yan, 2013; Chen et al., 2017e, 2018c; Hu et al., 2017, 2018). There are four main steps in developing ML algorithms (Oudah and Henschel, 2018). The first step is extraction of the features, which is critical to the ML method (Liu et al., 2015). Then, the operational classification units (OTU) table can be obtained by clustering. Next, important features that can improve the accuracy and efficiency are selected. Finally, a training dataset is used to train the model, after which a test set is used to evaluate the model. The process is summarized in Figure 2.
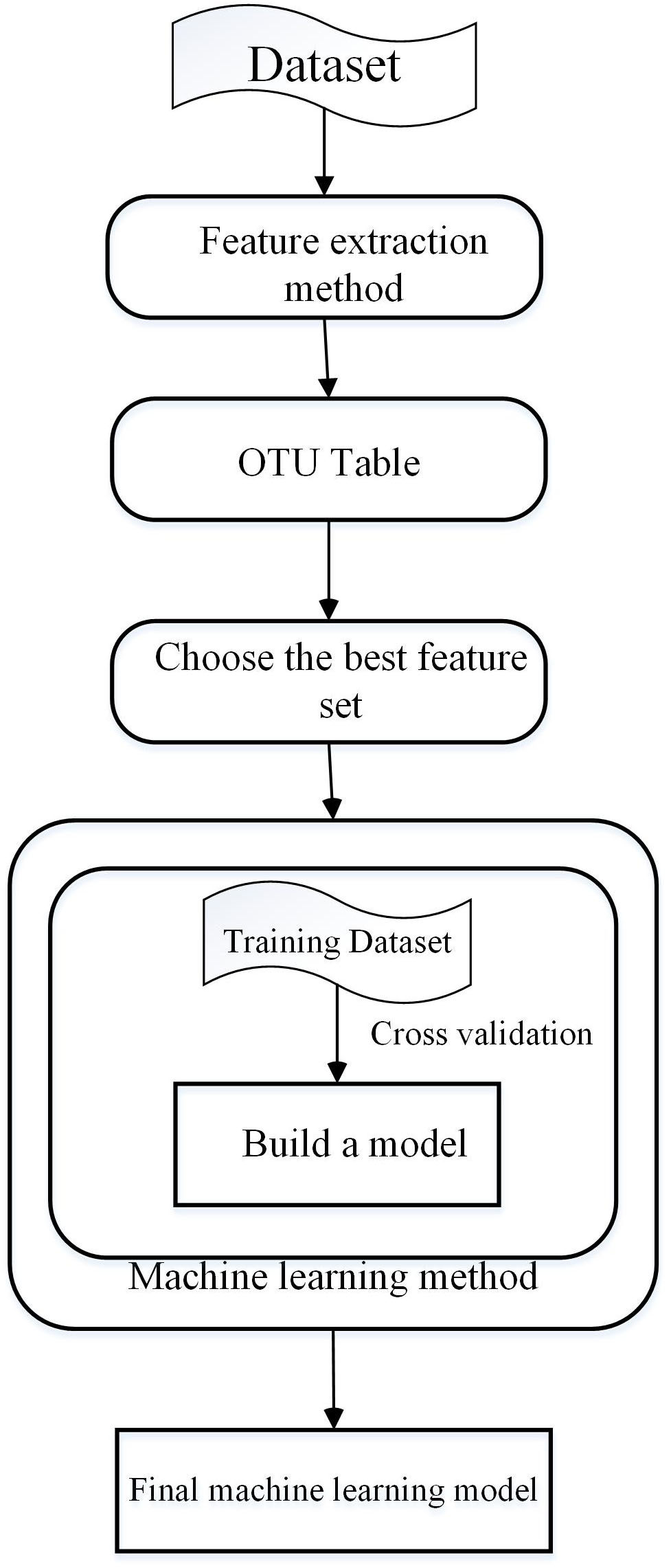
Figure 2. The main steps of machine learning in microbiology.
In microbial studies, according to the collected samples, obtaining relevant OTU is an important step in the study of microbial data. OTU is a type of similar microorganisms, which are cluster according to the similarity DNA sequences (Blaxter et al., 2005). In recent years, OTUs are always used for microbial diversity, especially when analyzing small subunit 16S or 18S rRNA datasets (Schmidt et al., 2014). Sequences can be clustered according to their similarity to one another, and the researcher sets the similarity threshold. After OTU clustering and species classification annotation for OTU, the OTU table can be obtained, which contains the OTU types and quantities for each sample, as well as species annotation information for each OTU.
As we know, some microbes have higher data dimensions, so feature dimensionality reduction is also an important part of data processing. There are some common methods for reducing the dimensionality and many studies are about how to reduce the dimensionality. For example, the principal components analysis (PCA) is a common reduction dimensionality method, which is mainly to decompose the covariance matrix to obtain the principal components and their weights (Jolliffe, 2002). PCA is often used to reduce the dimensionality of dataset while maintaining the feature that maximizes the contribution of the variance in the data set. Principal co-ordinates analysis (PCoA) is another common method. After sorting the feature values and the feature vectors, PCoA selects the features, which are in the top digits and the most significant coordinates in the distance matrix can be found (Podani and Miklós, 2002). The result is a rotation of the data matrix. It does not change the mutual positional relationship between the sample points, but only changes the coordinate system.
In microbial studies, supervised learning is always used, especially the support vector machine (SVM) (Feng et al., 2013a, 2017b; Chen X. X. et al., 2016; Yang et al., 2016), and the Naïve Bayes (NB) (Feng et al., 2013b,c), random forest (RF) (Chen et al., 2018d), and k nearest neighbor (KNN) methods (Chen et al., 2017c).
The SVM is a generalized linear classifier that can perform binary classification of data employing a decision basis, according to the maximum-margin hyperplane of the learning sample. The SVM can classify non-linear data by the kernel methods (Drucker et al., 2002). SVM is widely used in bioinformatics, such as the prediction of proteins (Xu et al., 2018a,b,c). The NB method (Meena and Chandran, 2009), which is a classification based on Bayes’ theory and the independent assumption of features that originate from classical mathematical theory (Rodríguez and Kuncheva, 2007), has a solid mathematical foundation and stable classification efficiency. The NB classifier, which requires only a few parameters, is less sensitive to missing data and simpler than other methods (Jordan, 2008). The RF is a classifier that contains multiple decision trees and its output accords to the voting on each decision tree (Svetnik et al., 2003). KNN (Cui et al., 2001) is a theoretically mature method. The method infers the sample category based on its neighbors. The main steps of the algorithm are as follows (Liao and Vemuri, 2002). First, the distance, which is between the test sample and each training sample, should be calculated. Then, the nearest k training samples are found as the nearest neighbors of the test sample. Finally, the test sample is classified according to the categories of the k nearest neighbors.
Classification and Prediction in Microbiology
Prediction of Microbial Species
There are two main types of microorganisms (Maiden et al., 1998), one of them with non-cellular morphology (Yeom and Javidi, 2006), such as viruses, and the other with cellular morphology that can divided into two types, one of them namely prokaryotes (Weinbauer, 2010), such as archaea and eubacteria, and the other namely eukaryotes (Nowrousian, 2010), such as fungi and unicellular algae. Different microorganisms have different characteristics, so it is important to identify the microorganisms properly. There are two main approaches to the identification of microorganisms. In one, the species of an unknown microorganism is determined with the goal of classifying it based on its domain, kingdom, phylum, class, order, family genus and species. In the other, the goal is to determine whether an unknown microorganism belongs to a specific species or not. For example, we can determine if an unknown microorganism is a virus or not, or more specifically, whether it is a certain virus. In this section, we will introduce recent studies that have used machine-learning methods to predict microorganisms.
In the study (Murali et al., 2018), the authors classified specific species of microorganisms using the IDTAXA, which employed the LearnTaxa and IdTaxa functions. Both of these functions are part of the R package DECIPHER, which was released under the GPLv3 license as part of the Bioconductor, which provides tools for the analysis and comprehension of high-throughput genomic data. The LearnTaxa function attempts to reclassify each training sequence into its tagged taxon using a method known as tree descent, which is similar to the decision tree, a commonly ML algorithms. IdTaxa uses the objects returned by the LearnTaxa and query sequences as input data. This system returns the classification results for each sequence in the taxonomic form and provides the relevant confidence for each level. If the confidence does not reaches the required value, which indicates that the classification cannot be accurately performed at that level. The classification of IdTaxa may lead to different conclusions in microbiological studies. Although the misclassification is small, many of the remaining misclassifications may be caused by the errors in the reference taxonomy. Fiannaca et al. (2018) presented a method for identifying the 16S short-read sequences based on k-mer and deep learning. According to their results, the method can classify both 16S shotgun (SG) and amplicon (AMP) data very well.
It is important to identify specific microbial sequences in mixed metagenomics samples. At present, gene-based similarity methods are popularly used to classify prokaryotic and host organisms from mixed samples; however, these techniques have major weakness. Therefore, many studies have been conducted to identify better methods for identification of specific microorganisms. Amgarten et al. (2018) proposed a tool known as MARVEL for predicting double-stranded DNA bacteriophage sequences in metagenomics. MARVEL uses the RF method, with a training dataset composed of 1,247 phage and 1,029 bacterial genomes and a test dataset composed of 335 bacteria and 177 phage genomes. The authors proposed six features to identify the phages, then used random forests to select features and found three features provided more information (Grazziotin et al., 2017). Ren et al. (2017) developed VirFinder, which is a ML method based on k-mer for virus overlap group identification that avoids gene-based similarity searches. VirFinder trains the ML model through known viral and non-viral (prokaryotic host) sequences to detect the specificity of viral k-mer frequencies. The model was trained with host and viral genomes prior to January 1, 2014, and the test set consisted of sequences obtained after January 1, 2014. VirSorter (Roux et al., 2015) is based on reference dependence and reference independence in different kinds of microbial sequence data to identify the viral signal. Experimental results have shown that VirSorter has good performance, especially for predicting viral sequences outside the host genome.
The above methods specifically classify microorganisms according to different needs. When we want to know the taxonomy information of microorganisms, we can use the method, which proposed by Murali et al. (2018). Moreover, MARVEL, VirSort, and VirFinder can identify specific types of microorganisms. According to the Amgarten et al. (2018), these three methods have comparable performance on specificity, but MARVEL has a better recall (sensitivity) performance. We have compiled materials for implementation of the above methods, which are shown in Table 1.

Table 1. The available data and materials for prediction of microbial species.
Prediction of Environmental and Host Phenotypes
With the development of next-generation DNA and high-throughput sequencing, a new area of microbiology has been generated. The main research in this field is to link microbial populations to phenotypes and ecological environments, which can provide favorable support for disease outbreaks and precision medicine (Atlas and Bartha, 1981). It is well known that some microorganisms are parasitic and that the surrounding environment and host cells have an important impact on the microbial population. Differences in nutrient availability and environmental conditions lead to differences in microbial communities (Moran, 2015). Because microorganisms can exchange information with the surrounding environment and host cells, we can predict the environmental and host phenotypes based on the microorganisms that are present (Xie et al., 2018). This provides a more comprehensive understanding of the environment and the host, so that we can better use the environment and protect the host. Many studies have recently been conducted to predict environmental and host phenotypes using microorganisms. In this section, we introduce these studies.
Asgari et al. (2018) used shallow subsample representation based on k-mer and deep learning, random forests, and SVMs to predict environmental and host phenotypes from 16S rRNA gene sequencing using the MicroPheno system. They found that the shallow subsample representation based on k-mer is superior to OTU in terms of body location recognition and Crohn’s disease prediction. In addition, the deep learning method is better than the RF and SVM for large datasets. This method not only can improve the performance, but also avoid overfitting. Moreover, it can reduce the time of pretreatment. Statnikov et al. (2013) used OTUs as an input feature and processed the data as follows. First, the authors sequenced the original DNA, after which they removed the human DNA sequence and defined the OTUs based on the microbial sequence. Next, they quantified the relative abundance of all sequences belonging to each OTU. The authors used SVM, kernel ridge regression, regularized logistic regression, Bayesian logistic regression, the KNN method, the RF method and probabilistic neural networks with different parameters and kernel functions. Overall, they investigated 18 ML methods. In addition, they used five feature extraction methods. The experimental results revealed that the RF, SVM, kernel-regression and Bayesian logic use Laplacian prior regression provided better performance. Based on their research, human skin microorganisms collected from objects that have been touched can be used to identify the individual from which they originated. In this work, the author used a variety of classification and dimensionality reduction methods to explore the effects of each method. It is very useful for the next work, which provides a comprehensive comparison. Schmedes et al. (2018) used the microbial community for forensic identification. In their study, they developed the hidSkinPlex, a novel targeted sequencing method using skin microbiome markers developed for human identification. In forensic science, it is important to estimate the time of death. Johnson et al. (2016) used KNN regression to predict the time interval after death using datasets from nose and ear samples. This indicates that skin microbiota can be an important tool in forensic death investigation. Traditionally, marine biological monitoring involves the classification and morphological identification of large benthic invertebrates, which requires a great deal of time and money. Cordier et al. (2017) used eDNA metabarcoding and supervised ML to build a powerful prediction model of benthic monitoring. Moitinho-Silva et al. (2017), studied the microbial flora of sponges and their HMA-LMA status demonstrated the applicability of ML to exploring host-related microbial community patterns.
Due to the specificity of microbial communities, we can better identify the environment and the host. Moreover, we can judge the existing environmental conditions and host survival status according to the existence of microbial community. We summarize the available datasets and methods, which are shown in Table 2.

Table 2. The available data and materials for prediction of environmental and host phenotypes.
Using Microbial Communities to Predict Disease
Microbiomes are important to human health and disease (Bourne et al., 2009). Indeed, there are many microbial communities in the human body. Once a microbial community is out of balance or foreign microorganisms invade, the human body is likely to get sick. For example, intestinal microbial communities are associated with obesity (Ley et al., 2006b) and pulmonary communities with pulmonary infection (Sibley et al., 2008). Because of the complexity of these communities, it is difficult to determine which kind of microbiome communities cause of the disease. Recently, many studies have investigated use of microbiome communities to predict diseases, especially bacterial vaginosis (Srinivasan et al., 2012; Deng et al., 2018) and inflammatory bowel disease (Gillevet et al., 2010). By analyzing microbial communities, we can better understand the disease and then make effective decisions regarding treatment. Therefore, in this section, we discuss current studies investigating use of microbiome communities to predict diseases.
Bacterial vaginosis (BV) is a disease associated with the vaginal microbiome. Beck and Foster (2014) used the genetic algorithm (GP), RF, and logistic regression (LR) to classify BV according to microbial communities. There are two criteria for BV, the Amsel standard, which accord to the discharge, whiff, clue cells, and pH (Amsel et al., 1983), and Nugent score, which dependents on counting gram-positive cells (Nugent et al., 1991). The dataset in Beck et al. study was from Ravel et al. (2011) and Sujatha et al. (2012). The method in the paper (Beck and Foster, 2014) first classifies BV according to vaginal microbiota and related environmental factors, then identifies the most important microbial community for predicting BV.
Hierarchical feature extraction is based on the classification of microbes from kingdoms to species. The existing stratification feature selection algorithm will lead to information loss, and the stratification information of some 16S rRNA sequences is usually incomplete, influencing the classification. Therefore, Oudah and Henschel (2018) proposed a method known as hierarchical feature engineering (HFE) to identify colorectal cancer (CRC). To accomplish this, they used RF, decision trees and the NB method to classify a dataset of Next Generation Sequencing based 16S rRNA sequences provided by metagenomics studies. This method is good for processing datasets with high dimensional features. Moreover, the available dataset and method are in https://github.com/HenschelLab/HierarchicalFeatureEngineering.
In another study (Wisittipanit, 2012), the author focused on predicting inflammatory bowel disease. In that study, patients with Crohn’s disease and ulcerative colitis were compared with healthy controls to identify differences between the mucosa and lumen in different intestinal locations. The author used the Relief algorithm (Kira and Rendell, 1992) to select features, and Metastats (White et al., 2009) to detect differential features. Finally, the author used KNN and SVM as classifiers to perform disease specificity and site specificity analysis.
In this section, we discuss using microorganisms to predict different diseases. Beck and Foster (2014) predicted BV according to the microorganisms and the diagnosis standard of BV. HFE identified the CRC according to the OTU ID and the taxonomy information. Wisittipanit proposed a method to predict Crohn’s disease, based on OTU and feature selection method. The above methods used different ideas to predict diseases by using microorganisms and obtained good results. This indicates that some diseases affect human colonies. According to these colony changes, we can not only predict the disease, but also treat the disease according to the colony condition, which is a direction for future research.
Interaction and Association in Microbiology
Interaction Between Microorganisms
The collective behavior of microbial ecosystems in biomes is the result of many interactions between community members. These interactions include metabolite exchange, signaling and quorum sensing processes, as well as growth inhibition and killing (Langille et al., 2013; DiMucci et al., 2018). Understanding the interspecific interactions within microbial communities is critical to understanding the functions of natural ecosystems and the design of synthetic consortia (Mainali et al., 2017). Therefore, in this section, we introduce the application of ML to investigation of interactions between microorganisms.
DiMucci et al. (2018) showed how the microbial interaction network can be combined with the characteristic level of individual microbes to provide an accurate inference of the missing edges in the network and a constructive mechanism of the interaction. The same authors proposed the notion of a composite vector that combined the generated trait vectors and pairwise interactions. The training set for the model is all observed interactions. The model was then used to predict the unobserved interactions. If the random forest classifier is used, feature contributions can be calculated. Microbial interactions in the soil can affect crop yields; therefore, Chang et al. (2017) used the random forest method to predict the productivity based on the microorganisms. In this study, the improved crop productivity differences were linked to the soil microbial composition.
There are cooperative and competitive relationships within the same microbial population. Moreover, there are eight relationships between the different microbial populations, which are neutralism, commensalism, synergism, mutualism, competition, amensalism, parasitism and predation. Understanding the interactions between microorganisms is important for the study of microbial species and for microbial applications. However, there are not many studies on ML in this area, which will be an important research direction.
Microbiome-Disease Association
There are many kinds of microorganisms in human bodies, and they are inseparable from human health. For example, intestinal microbial disorders can cause intestinal inflammatory diseases (Chen et al., 2017b), such as ulcerative colitis, CRC, atherosclerosis, diabetes and obesity. Accordingly, it is necessary to predict the microbial-disease association because this study not only improves the diagnosis and prognosis of human diseases, but also develops the new drugs (Yu et al., 2015, 2016a; Shi et al., 2016; Su et al., 2018; Fan et al., 2019). However, few studies have investigated predictive analysis of the microbial-disease association. Therefore, in this section, we introduce the application of ML to the study of microbial-disease association.
Fan et al. (2019) proposed a new approach to analyze the microbial-disease association by integrating multiple data sources from the human microbe-disease consortium (MDPH_HMDA) and path-based HeteSim scores. First, heterogeneity networks were constructed. Microbe-disease pair weighting was conducted according to the standardized HeteSim measurement method, after which the microbe-disease-disease pathway and microbe-microbe-disease pathway HeteSim scores were integrated. Finally, the correlation scores of potential micro genome associations were calculated. Xuezhong et al. (2014) proposed a method based on the Human Disease Network (HSDN) in which co-occurrence of disease/symptom terms based on PubMed bibliographic records was used to calculate disease similarity. KATZ (Katz, 1953) is a network based measurement method that calculates the similarly of nodes in a heterogeneous network, to solve the link prediction problem proposed by Katz. The KATZ method has been applied in many fields, including disease-gene association prediction (Xiaofei et al., 2014) and IncRNA-disease association prediction (Chen et al., 2015). Chen et al. (2017b) proposed a novel method based on KATZ to predict associations of human microbiota with non-infectious diseases (named KATZHMDA). The KATZHMDA first constructs adjacency matrix A based on known microbial-disease associations. The kernel similarity matrix KD and KM are calculated based on the disease Gaussian interaction profile and microbial Gaussian interaction profile, respectively. We can construct the integrated matrix A∗ based on KM, KD and known microbial-disease associations. Next, all walks of different lengths are integrated to obtain a single microbe-disease association measurement. Therefore, we can calculate microbe-disease association probability in a matrix form. Shi et al. (2018) proposed a prediction method based on binary matrix completion named BMCMDA. The BMCMDA assumes that the incomplete microbiome-disease association (MDA) matrix is the sum of a potential parameterization matrix and a noise matrix. Additionally, the BMCMDA assumes that the independent subscripts of the items observed in the MDA matrix follow the binomial model. Shi et al. (2018) used the same dataset, which was collected from the Human Microbe-Disease Association Database (HMDAD) and included 292 microbes and 39 human diseases, to perform comparisons. According to the study, BMCMDA is better than the KATZHMDA in AUC. BMCMDA can be integrated with other and independent microbial/disease similarities or characteristics to enhance MDA prediction. Moreover, this method can be applied to more prediction aspects. We summarize the available datasets and methods, which are shown in Table 3.

Table 3. The available data and materials for microbiome-disease association.
Conclusion
Microorganisms are involved in many life activities, and affect their surrounding environment and other organisms. Microorganisms play important roles in human heath, crop growth, livestock farming, environmental management, industrial chemical production and food production. In the 19th century, people first observed microbes using microscopes and began to study them. However, the development of high-throughput sequencing technology has led to generation of large amounts of microbial related data. As a result, machine-learning methods are now being applied to microbiological research. Here, we discuss the current application of ML in the microbiome. The results revealed that ML is widely used in microbiological research, and that it has focused on classification problems and analysis of interaction problems. However, many problems remain unresolved and will require the cooperation of researchers from different fields, such as biology, informatics and medicine, to jointly promote the development and progress of microbiological research. On the other hand, the recent developed link prediction (Liu et al., 2016; Zeng et al., 2017b) and computational intelligence methods (Cabarle et al., 2017; Song et al., 2018), can be promising in discovering the relationship between diseases and microbes.
Author Contributions
KQ drafted the manuscript. FG and XL conducted research. YL modified the manuscript. QZ conceived the idea.
Funding
The work was supported by the National Key R&D Program of China (2018YFC0910405), and the National Natural Science Foundation of China (No. 61771331).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jeremy Kamen, MSc., from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
References
Alexander, M. (1962). Introduction of soil microbiology. Soil Sci. 93:74. doi: 10.1097/00010694-196201000-00034
Amgarten, D., Braga, L. P. P., da Silva, A. M., and Setubal, J. C. (2018). MARVEL, a tool for prediction of bacteriophage sequences in metagenomic bins. Front. Genet. 9:304. doi: 10.3389/fgene.2018.00304
Amsel, R., Totten, P. A., Spiegel, C. A., Chen, K. C., Eschenbach, D., and Holmes, K. K. (1983). Nonspecific vaginitis. Diagnostic criteria and microbial and epidemiologic associations. Am. J. Med. 74, 14–22. doi: 10.1016/0002-9343(83)91112-9
Asgari, E., Garakani, K., McHardy, A. C., and Mofrad, M. R. K. (2018). MicroPheno: predicting environments and host phenotypes from 16S rRNA gene sequencing using a k-mer based representation of shallow sub-samples. Bioinformatics 34, i32–i42. doi: 10.1093/bioinformatics/bty296
Atlas, R. M., and Bartha, R. (1981). Microbial ecology:fundamentals and applications. Acta Ecol. Sin. 70:977. doi: 10.1016/j.biortech.2015.07.074
Beck, D., and Foster, J. A. (2014). Machine learning techniques accurately classify microbial communities by bacterial vaginosis characteristics. PLoS One 9:e87830. doi: 10.1371/journal.pone.0087830
Blaxter, M., Mann, J., Chapman, T., Thomas, F., Whitton, C., Floyd, R., et al. (2005). Defining operational taxonomic units using DNA barcode data. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 360, 1935–1943. doi: 10.1098/rstb.2005.1725
Bourne, D. G., Garren, M., Work, T. M., Rosenberg, E., Smith, G. W., and Harvell, C. D. (2009). Microbial disease and the coral holobiont. Trends Microbiol. 17, 554–562. doi: 10.1016/j.tim.2009.09.004
Cabarle, F. G. C., Adorna, H. N., Jiang, M., and Zeng, X. X. (2017). Spiking neural P systems with scheduled synapses. IEEE Trans. Nanobioscience 16, 792–801. doi: 10.1109/tnb.2017.2762580
Chang, H. X., Haudenshield, J. S., Bowen, C. R., and Hartman, G. L. (2017). Metagenome-wide association study and machine learning prediction of bulk soil microbiome and crop productivity. Front. Microbiol. 8:519. doi: 10.3389/fmicb.2017.00519
Chen, J., Guo, M. Y., Li, S. M., and Liu, B. (2017a). ProtDec-LTR2.0: an improved method for protein remote homology detection by combining pseudo protein and supervised Learning to Rank. Bioinformatics 33, 3473–3476. doi: 10.1093/bioinformatics/btx429
Chen, X., Huang, Y.-A., You, Z.-H., Yan, G.-Y., and Wang, X.-S. (2017b). A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 33, 733–739. doi: 10.1093/bioinformatics/btw715
Chen, X., Wu, Q. F., and Yan, G. Y. (2017c). RKNNMDA: ranking-based KNN for MiRNA-disease association prediction. RNA Biol. 14, 952–962. doi: 10.1080/15476286.2017.1312226
Chen, X., Xie, D., Zhao, Q., and You, Z.-H. (2017d). MicroRNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 20, 515–539. doi: 10.1093/bib/bbx130
Chen, X., Yan, C. C., Zhang, X., and You, Z. H. (2017e). Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 18, 558–576. doi: 10.1093/bib/bbw060
Chen, J., Guo, M. Y., Wang, X. L., and Liu, B. (2018a). A comprehensive review and comparison of different computational methods for protein remote homology detection. Brief. Bioinform. 19, 231–244. doi: 10.1093/bib/bbw108
Chen, X., Huang, L., Xie, D., and Zhao, Q. (2018b). EGBMMDA: extreme gradient boosting machine for MiRNA-disease association prediction. Cell Death Dis. 9:3. doi: 10.1038/s41419-017-0003-x
Chen, X., Sun, Y.-Z., Guan, N.-N., Qu, J., Huang, Z.-A., Zhu, Z.-X., et al. (2018c). Computational models for lncRNA function prediction and functional similarity calculation. Brief. Funct. Genomics 18, 58–82. doi: 10.1093/bfgp/ely031
Chen, X., Wang, C. C., Yin, J., and You, Z. H. (2018d). Novel human miRNA-disease association inference based on random forest. Mol. Ther. Nucleic Acids 13, 568–579. doi: 10.1016/j.omtn.2018.10.005
Chen, X., Wang, L., Qu, J., Guan, N. N., and Li, J. Q. (2018e). Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 34, 4256–4265. doi: 10.1093/bioinformatics/bty503
Chen, X., Xie, D., Wang, L., Zhao, Q., You, Z. H., and Liu, H. S. (2018f). BNPMDA: bipartite network projection for MiRNA-disease association prediction. Bioinformatics 34, 3178–3186. doi: 10.1093/bioinformatics/bty333
Chen, X., Yin, J., Qu, J., and Huang, L. (2018g). MDHGI: matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput. Biol. 14:e1006418. doi: 10.1371/journal.pcbi.1006418
Chen, W., Ding, H., Feng, P. M., Lin, H., and Chou, K. C. (2016). IACP: a sequence-based tool for identifying anticancer peptides. Oncotarget 7, 16895–16909. doi: 10.18632/oncotarget.7815
Chen, X. X., Tang, H., Li, W. C., Wu, H., Chen, W., Ding, H., et al. (2016). Identification of bacterial cell wall lyases via pseudo amino acid composition. Biomed Res. Int. 2016:1654623. doi: 10.1155/2016/1654623
Chen, X., and Huang, L. (2017). LRSSLMDA: laplacian regularized sparse subspace learning for MiRNA-disease association prediction. PLoS Comput. Biol. 13:e1005912. doi: 10.1371/journal.pcbi.1005912
Chen, X., Yan, C. C., Luo, C., Ji, W., Zhang, Y., and Dai, Q. (2015). Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci. Rep. 5:11338. doi: 10.1038/srep11338
Chen, X., and Yan, G. Y. (2013). Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624. doi: 10.1093/bioinformatics/btt426
Cordier, T., Esling, P., Lejzerowicz, F., Visco, J., Ouadahi, A., Martins, C., et al. (2017). Predicting the ecological quality status of marine environments from eDNA metabarcoding data using supervised machine learning. Environ. Sci. Technol. 51, 9118–9126. doi: 10.1021/acs.est.7b01518
Cotter, P. D., Hill, C., and Ross, R. P. (2005). Food microbiology: bacteriocins: developing innate immunity for food. Nat. Rev. Microbiol. 3, 777–788. doi: 10.1038/nrmicro1273
Cui, Y., Ooi, B. C., Tan, K. L., and Jagadish, H. V. (2001). “Indexing the distance: an efficient method to KNN processing”, in Vldb Proceedings of the 27th VLDB Conference, Rome, 421–430.
Deng, Z. L., Gottschick, C., Bhuju, S., Masur, C., Abels, C., and Wagner-Dobler, I. (2018). Metatranscriptome analysis of the vaginal microbiota reveals potential mechanisms for protection against metronidazole in bacterial vaginosis. Msphere 3:e00262-18. doi: 10.1128/mSphereDirect.00262-18
DiMucci, D., Kon, M., and Segre, D. (2018). Machine learning reveals missing edges and putative interaction mechanisms in microbial ecosystem networks. Msystems 3:e00181-18. doi: 10.1128/mSystems.00181-18
Ding, Y. J., Tang, J. J., and Guo, F. (2017a). Identification of drug-target interactions via multiple information integration. Inf. Sci. 418, 546–560. doi: 10.1016/j.ins.2017.08.045
Ding, Y. J., Tang, J. J., and Guo, F. (2017b). Identification of protein-ligand binding sites by sequence information and ensemble classifier. J. Chem. Inf. Model. 57, 3149–3161. doi: 10.1021/acs.jcim.7b00307
Drucker, H., Wu, D., and Vapnik, V. N. (2002). Support vector machines for spam categorization. IEEE Trans. Neural Netw. 10, 1048–1054. doi: 10.1109/72.788645
Fan, C. Y., Lei, X. J., Guo, L., and Zhang, A. D. (2019). Predicting the associations between microbes and diseases by integrating multiple data sources and path-based HeteSim scores. Neurocomputing 323, 76–85. doi: 10.1016/j.neucom.2018.09.054
Feng, P. M., Chen, W., Lin, H., and Chou, K. C. (2013a). iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 442, 118–125. doi: 10.1016/j.ab.2013.05.024
Feng, P. M., Ding, H., Chen, W., and Lin, H. (2013b). Naive bayes classifier with feature selection to identify phage virion proteins. Comput. Math. Methods Med. 2013:530696. doi: 10.1155/2013/530696
Feng, P. M., Lin, H., and Chen, W. (2013c). Identification of antioxidants from sequence information using naive bayes. Comput. Math. Methods Med. 2013:567529. doi: 10.1155/2013/567529
Feng, P. M., Ding, H., Lin, H., and Chen, W. (2017a). AOD: the antioxidant protein database. Sci. Rep. 7:7449. doi: 10.1038/s41598-017-08115-6
Feng, P. M., Zhang, J. D., Tang, H., Chen, W., and Lin, H. (2017b). Predicting the organelle location of noncoding RNAs using pseudo nucleotide compositions. Interdiscip. Sci. Comput. Life Sci. 9, 540–544. doi: 10.1007/s12539-016-0193-4
Fiannaca, A., Paglia, L. L., Rosa, M. L., Bosco, G. L., Renda, G., Rizzo, R., et al. (2018). Deep learning models for bacteria taxonomic classification of metagenomic data. BMC Bioinformatics 19:198. doi: 10.1186/s12859-018-2182-6
Gillevet, P., Sikaroodi, M., Keshavarzian, A., and Mutlu, E. A. (2010). Quantitative assessment of the human gut microbiome using multitag pyrosequencing. Chem. Biodivers. 7, 1065–1075. doi: 10.1002/cbdv.200900322
Grazziotin, A. L., Koonin, E. V., and Kristensen, D. M. (2017). Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 45, D491–D498. doi: 10.1093/nar/gkw975
He, W. Y., Jia, C. Z., and Zou, Q. (2019). 4mCPred: machine learning methods for DNA N-4-methylcytosine sites prediction. Bioinformatics 35, 593–601. doi: 10.1093/bioinformatics/bty668
Hu, H., Zhang, L., Ai, H. X., Zhang, H., Fan, Y. T., Zhao, Q., et al. (2018). HLPI-Ensemble: prediction of human lncRNA-protein interactions based on ensemble strategy. RNA Biol. 15, 797–806. doi: 10.1080/15476286.2018.1457935
Hu, H., Zhu, C. Y., Ai, H. X., Zhang, L., Zhao, J., Zhao, Q., et al. (2017). LPI-ETSLP: lncRNA-protein interaction prediction using eigenvalue transformation-based semi-supervised link prediction. Mol. Biosyst. 13, 1781–1787. doi: 10.1039/c7mb00290d
Huang, Y. A., You, Z. H., Chen, X., Huang, Z. A., Zhang, S. W., and Yan, G. Y. (2017). Prediction of microbe-disease association from the integration of neighbor and graph with collaborative recommendation model. J. Transl. Med. 15:209. doi: 10.1186/s12967-017-1304-7
Huang, Z. A., Chen, X., Zhu, Z. X., Liu, H. S., Yan, G. Y., You, Z. H., et al. (2017). PBHMDA: path-based human microbe-disease association prediction. Front. Microbiol. 8:233. doi: 10.3389/fmicb.2017.00233
Johnson, H. R., Trinidad, D. D., Guzman, S., Khan, Z., Parziale, J. V., DeBruyn, J. M., et al. (2016). A machine learning approach for using the postmortem skin microbiome to estimate the postmortem interval. PLoS One 11:e0167370. doi: 10.1371/journal.pone.0167370
Jordan, A. (2008). On Discriminative vs. Generative classifiers: a comparison of logistic regression and naive Bayes. Neural Process. Lett. 28:169. doi: 10.1007/s11063-008-9088-7
Katz, L. (1953). A new status index derived from sociometric analysis. Psychometrika 18, 39–43. doi: 10.1007/BF02289026
Kira, K., and Rendell, L. A. (1992). “A practical approach to feature selection,” in Proceedings of the Ninth International Workshop on Machine Learning, Aberdeen. doi: 10.1016/B978-1-55860-247-2.50037-1
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., McDonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006a). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023.
Ley, R. E., Turnbaugh, P. J., Samuel, K., and Gordon, J. I. (2006b). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023.
Li, Z., Tang, J. J., and Guo, F. (2016). Learning from real imbalanced data of 14-3-3 proteins binding specificity. Neurocomputing 217, 83–91. doi: 10.1016/j.neucom.2016.03.093
Liao, Y., and Vemuri, V. R. (2002). Use of K-Nearest Neighbor classifier for intrusion detection. Comput. Secur. 21, 439–448. doi: 10.1016/S0167-4048(02)00514-X
Liu, B., Jiang, S., and Zou, Q. (2018). HITS-PR-HHblits: protein remote homology detection by combining PageRank and Hyperlink-Induced Topic Search. Brief. Bioinform. 2018:bby104. doi: 10.1093/bib/bby104
Liu, B., Liu, F., Wang, X., Chen, J., Fang, L., and Chou, K. -C. (2015). Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 43, W65–W71. doi: 10.1093/nar/gkv458
Liu, Y., Zeng, X., He, Z., and Zou, Q. (2016). Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE/ACM Trans. Comput. Biol. Bioinform. 14, 905–915. doi: 10.1109/TCBB.2016.2550432
Maiden, M. C. J., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Mainali, K. P., Bewick, S., Thielen, P., Mehoke, T., Breitwieser, F. P., Paudel, S., et al. (2017). Statistical analysis of co-occurrence patterns in microbial presence-absence datasets. PLoS One 12:e0187132. doi: 10.1371/journal.pone.0187132
Meena, M. J., and Chandran, K. R. (2009). “Naïve Bayes text classification with positive features selected by statistical method,” in Proceedings of the First International Conference on Advanced Computing, ICAC 2009, Los Alamitos: IEEE, 28–33. doi: 10.1109/ICADVC.2009.5378273
Moitinho-Silva, L., Steinert, G., Nielsen, S., Hardoim, C. C. P., Wu, Y. C., McCormack, G. P., et al. (2017). Predicting the HMA-LMA status in marine sponges by machine learning. Front. Microbiol. 8:752. doi: 10.3389/fmicb.2017.00752
Morris, O. N., Cunningham, J. C., Finneycrawley, J. R., Jaques, R. P., and Kinoshita, G. (1986). Microbial insecticides in Canada: their registration and use in agriculture, forestry and public and animal health. Bull. Entomol. Soc. Canada 18, 1–43.
Murali, A., Bhargava, A., and Wright, E. S. (2018). IDTAXA: a novel approach for accurate taxonomic classification of microbiome sequences. Microbiome 6:140. doi: 10.1186/s40168-018-0521-5
Nannipieri, P., Ascher, J., Ceccherini, M. T., Landi, L., Pietramellara, G., and Renella, G. (2010). Microbial diversity and soil functions. Eur. J. Soil Sci. 54, 655–670. doi: 10.1046/j.1351-0754.2003.0556.x
Niel, C. B. V. (1966). Microbiology and molecular biology. Q. Rev. Biol. 41, 105–112. doi: 10.1086/404937
Nowrousian, M. (2010). Next-generation sequencing techniques for eukaryotic microorganisms: sequencing-based solutions to biological problems. Eukaryot. Cell 9, 1300–1310. doi: 10.1128/EC.00123-10
Nugent, R. P., Krohn, M. A., and Hillier, S. L. (1991). Reliability of diagnosing bacterial vaginosis is improved by a standardized method of gram stain interpretation. J. Clin. Microbiol. 29, 297–301.
Oudah, M., and Henschel, A. (2018). Taxonomy-aware feature engineering for microbiome classification. BMC Bioinformatics 19:227. doi: 10.1186/s12859-018-2205-3
Pan, G. F., Jiang, L. M., Tang, J. J., and Guo, F. (2018). A novel computational method for detecting DNA methylation sites with DNA sequence information and physicochemical properties. Int. J. Mol. Sci. 19:E511. doi: 10.3390/ijms19020511
Peng, L. H., Yin, J., Zhou, L. Q., Liu, M. X., and Zhao, Y. (2018). Human microbe-disease association prediction based on adaptive boosting. Front. Microbiol. 9:2440. doi: 10.3389/fmicb.2018.02440
Petrof, E. O., Claud, E. C., Gloor, G. B., and Allenvercoe, E. (2012). Microbial ecosystems therapeutics: a new paradigm in medicine? Benef. Microbes 4, 53–65. doi: 10.3920/BM2012.0039
Podani, J., and Miklós, I. (2002). Resemblance coefficients and the horseshoe effect in principal coordinates analysis. Ecology 83, 3331–3343. doi: 10.1890/0012-9658(2002)083[3331:RCATHE]2.0.CO;2
Qu, K. Y., Han, K., Wu, S., Wang, G. H., and Wei, L. Y. (2017). Identification of DNA-binding proteins using mixed feature representation methods. Molecules 22:E1602. doi: 10.3390/molecules22101602
Ravel, J., Gajer, P., Abdo, Z., Schneider, G. M., Koenig, S. S., and McCulle, S. L et al. (2011). Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 1), 4680–4687. doi: 10.1073/pnas.1002611107
Reiff, C., and Kelly, D. (2010). Inflammatory bowel disease, gut bacteria and probiotic therapy. Int. J. Med. Microbiol. 300, 25–33. doi: 10.1016/j.ijmm.2009.08.004
Ren, J., Ahlgren, N. A., Lu, Y. Y., Fuhrman, J. A., and Sun, F. Z. (2017). VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome 5:69. doi: 10.1186/s40168-017-0283-5
Rodríguez, J. J., and Kuncheva, L. I. (2007). “Naïve bayes ensembles with a random oracle,” in Lecture Notes in Computer Science, Vol. 4472, eds M. Haindl, J. Kittler and F. Roli (Berlin: Springer), 450–458.
Roux, S., Enault, F., Hurwitz, B. L., and Sullivan, M. B. (2015). VirSorter: mining viral signal from microbial genomic data. PeerJ 3:e985. doi: 10.7717/peerj.985
Schmedes, S. E., Woerner, A. E., Novroski, N. M. M., Wendt, F. R., King, J. L., Stephens, K. M., et al. (2018). Targeted sequencing of clade-specific markers from skin microbiomes for forensic human identification. Forensic Sci. Int. Genet. 32, 50–61. doi: 10.1016/j.fsigen.2017.10.004
Schmidt, T. S. B., Matias Rodrigues, J. F., and von Mering, C. (2014). Ecological consistency of SSU rRNA-based operational taxonomic units at a global scale. PLoS Comput. Biol. 10:e1003594. doi: 10.1371/journal.pcbi.1003594
Shi, J. Y., Huang, H., Zhang, Y. N., Cao, J. B., and Yiu, S. M. (2018). BMCMDA: a novel model for predicting human microbe-disease associations via binary matrix completion. BMC Bioinformatics 19, 169–176. doi: 10.1186/s12859-018-2274-3
Shi, J. Y., Li, J. X., and Lu, H. M. (2016). Predicting existing targets for new drugs base on strategies for missing interactions. BMC Bioinformatics 17(Suppl. 8):282. doi: 10.1186/s12859-016-1118-2
Sibley, C. D., Parkins, M. D., Rabin, H. R., Kangmin, D., Norgaard, J. C., and Surette, M. G. (2008). A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc. Natl. Acad. Sci. U.S.A. 105, 15070–15075. doi: 10.1073/pnas.0804326105
Song, T., Rodriguez-Paion, A., Zheng, P., and Zeng, X. X. (2018). Spiking neural P systems with colored spikes. IEEE Trans. Cogn. Dev. Syst. 10, 1106–1115. doi: 10.1109/tcds.2017.2785332
Souza, P. M. D. (2010). Application of microbial α-amylase in industry – A review. Braz. J. Microbiol. 41, 850–861. doi: 10.1590/S1517-83822010000400004
Srinivasan, S., Hoffman, N. G., Morgan, M. T., Matsen, F. A., Fiedler, T. L., Hall, R. W., et al. (2012). Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS One 7:e37818. doi: 10.1371/journal.pone.0037818
Statnikov, A., Henaff, M., Narendra, V., Konganti, K., Li, Z. G., Yang, L. Y., et al. (2013). A comprehensive evaluation of multicategory classification methods for microbiomic data. Microbiome 1:11. doi: 10.1186/2049-2618-1-11
Stoter, F. R., Chakrabarty, S., Edler, B., and Habetse, E. A. P. (2019). CountNet: estimating the number of concurrent speakers using supervised learning. IEEE/ACM Trans. Audio Speech Lang. Process. 27, 268–282. doi: 10.1109/taslp.2018.2877892
Su, R., Wu, H., Xu, B., Liu, X., and Wei, L. (2018). Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data. IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/tcbb.2018.2858756 [Epub ahead of print].
Sujatha, S., Hoffman, N. G., Morgan, M. T., Matsen, F. A., Fiedler, T. L., Hall, R. W., et al. (2012). Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS One 7:e37818. doi: 10.1371/journal.pone.0037818
Svetnik, V., Liaw, A., Tong, C., Culberson, J. C., Sheridan, R. P., Feuston, B. P. (2003). Random forest: a classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 43, 1947–1958. doi: 10.1021/ci034160g
Waldron, L. (2018). Data and statistical methods to analyze the human microbiome. Msystems 3:e00194-17. doi: 10.1128/mSystems.00194-17
Wang, F., Huang, Z. A., Chen, X., Zhu, Z. X., Wen, Z. K., Zhao, J. Y., et al. (2017). LRLSHMDA: laplacian regularized least squares for human microbe-disease association prediction. Sci. Rep. 7:7601. doi: 10.1038/s41598-017-08127-2
Wei, L. Y., Chen, H. R., and Su, R. (2018a). M6APred-EL: a sequence-based predictor for identifying N6-methyladenosine sites using ensemble learning. Mol. Ther. Nucleic Acids 12, 635–644. doi: 10.1016/j.omtn.2018.07.004
Wei, L. Y., Zhou, C., Chen, H. R., Song, J. N., and Su, R. (2018b). ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 34, 4007–4016. doi: 10.1093/bioinformatics/bty451
Wei, L. Y., Wan, S. X., Guo, J. S., and Wong, K. K. L. (2017a). A novel hierarchical selective ensemble classifier with bioinformatics application. Artif. Intell. Med. 83, 82–90. doi: 10.1016/j.artmed.2017.02.005
Wei, L. Y., Xing, P. W., Zeng, J. C., Chen, J. X., Su, R., and Guo, F. (2017b). Improved prediction of protein-protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Weinbauer, M. G. (2010). Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 28, 127–181. doi: 10.1016/j.femsre.2003.08.001
White, J. R., Nagarajan, N., and Pop, M. (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5:e1000352. doi: 10.1371/journal.pcbi.1000352
Wisittipanit, N. (2012). Machine Learning Approach for Profiling Human Microbiome. Ph.D. dissertation, George Mason University, Fairfax, VA. Available at: https://search.proquest.com/docview/1009703926?accountid=45721 (accessed April 8, 2019).
Xiaofei, Y., Lin, G., Xingli, G., Xinghua, S., Hao, W., Fei, S., et al. (2014). A network based method for analysis of lncRNA-disease associations and prediction of lncRNAs implicated in diseases. PLoS One 9:e87797. doi: 10.1371/journal.pone.0087797
Xie, K., Guo, L., Bai, Y., Liu, W., Yan, J., and Bucher, M. (2018). Microbiomics and plant health: an interdisciplinary and international workshop on the plant microbiome. Mol. Plant 12, 1–3. doi: 10.1016/j.molp.2018.11.004
Xu, L., Liang, G. M., Liao, C. R., Chen, G. D., and Chang, C. C. (2018a). An efficient classifier for alzheimer’s disease genes identification. Molecules 23:E3140. doi: 10.3390/molecules23123140
Xu, L., Liang, G. M., Shi, S. H., and Liao, C. R. (2018b). SeqSVM: a sequence-based support vector machine method for identifying antioxidant proteins. Int. J. Mol. Sci. 19:E1773. doi: 10.3390/ijms19061773
Xu, L., Liang, G. M., Wang, L. J., and Liao, C. R. (2018c). A novel hybrid sequence-based model for identifying anticancer peptides. Genes 9:E158. doi: 10.3390/genes9030158
Xuezhong, Z., JöRg, M., Albert-László, B., and Amitabh, S. (2014). Human symptoms-disease network. Nat. Commun. 5:4212. doi: 10.1038/ncomms5212
Yang, H., Lv, H., Ding, H., Chen, W., and Lin, H. (2018a). iRNA-2OM: a sequence-based predictor for identifying 2 ’-o-methylation sites in homo sapiens. J. Comput. Biol. 25, 1266–1277. doi: 10.1089/cmb.2018.0004
Yang, H., Qiu, W. R., Liu, G. Q., Guo, F. B., Chen, W., Chou, K. C., et al. (2018b). iRSpot-Pse6NC: identifying recombination spots in Saccharomyces cerevisiae by incorporating hexamer composition into general PseKNC. Int. J. Biol. Sci. 14, 883–891. doi: 10.7150/ijbs.24616
Yang, H., Tang, H., Chen, X. X., Zhang, C. J., Zhu, P. P., Ding, H., et al. (2016). Identification of secretory proteins in mycobacterium tuberculosis using pseudo amino acid composition. Biomed Res. Int. 2016:5413903. doi: 10.1155/2016/5413903
Yeom, S., and Javidi, B. (2006). Automatic identification of biological microorganisms using three-dimensional complex morphology. J. Biomed. Opt. 11:024017.
Yu, L., Huang, J. B., Ma, Z. X., Zhang, J., Zou, Y. P., and Gao, L. (2015). Inferring drug-disease associations based on known protein complexes. BMC Med. Genomics 8(Suppl. 2):S2. doi: 10.1186/1755-8794-8-s2-s2
Yu, L., Ma, X. K., Zhang, L., Zhang, J., and Gao, L. (2016a). Prediction of new drug indications based on clinical data and network modularity. Sci. Rep. 6:32530. doi: 10.1038/srep32530
Yu, L., Wang, B. B., Ma, X. K., and Gao, L. (2016b). The extraction of drug-disease correlations based on module distance in incomplete human interactome. BMC Syst. Biol. 10(Suppl. 4):111. doi: 10.1186/s12918-016-0364-2
Yu, L., Su, R. D., Wang, B. B., Zhang, L., Zou, Y. P., Zhang, J., et al. (2017a). Prediction of novel drugs for hepatocellular carcinoma based on multi-source random walk. IEEE/ACM Trans. Comput. Biol. Bioinform. 14, 966–977. doi: 10.1109/tcbb.2016.2550453
Yu, L., Zhao, J., and Gao, L. (2017b). Drug repositioning based on triangularly balanced structure for tissue-specific diseases in incomplete interactome. Artif. Intell. Med. 77, 53–63. doi: 10.1016/j.artmed.2017.03.009
Yu, L., Zhao, J., and Gao, L. (2018). Predicting potential drugs for breast cancer based on miRNA and tissue specificity. Int. J. Biol. Sci. 14, 971–980. doi: 10.7150/ijbs.23350
Zeng, X. X., Ding, N. X., Rodriguez-Paton, A., and Zou, Q. (2017a). Probability-based collaborative filtering model for predicting gene-disease associations. BMC Med. Genomics 10(Suppl. 5):76. doi: 10.1186/s12920-017-0313-y
Zeng, X. X., Lin, W., Guo, M. Z., and Zou, Q. (2017b). A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 13:e1005420. doi: 10.1371/journal.pcbi.1005420
Zeng, X. X., Liu, L., Lu, L. Y., and Zou, Q. (2018). Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 34, 2425–2432. doi: 10.1093/bioinformatics/bty112
Zhang, X., Zou, Q., Rodriguez-Paton, A., and Zeng, X. X. (2019). Meta-path methods for prioritizing candidate disease miRNAs. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 283–291. doi: 10.1109/tcbb.2017.2776280
Zhao, Q., Liang, D., Hu, H., Ren, G. F., and Liu, H. S. (2018a). RWLPAP: random walk for IncRNA-protein associations prediction. Protein Pept. Lett. 25, 830–837. doi: 10.2174/0929866525666180905104904
Zhao, Q., Yu, H., Ming, Z., Hu, H., Ren, G., and Liu, H. (2018b). The bipartite network projection-recommended algorithm for predicting long non-coding RNA-protein interactions. Mol. Ther. Nucleic Acids 13, 464–471. doi: 10.1016/j.omtn.2018.09.020
Zhao, Q., Zhang, Y., Hu, H., Ren, G. F., Zhang, W., and Liu, H. S. (2018c). IRWNRLPI: integrating random walk and neighborhood regularized logistic matrix factorization for lncRNA-protein interaction prediction. Front. Genet. 9:239. doi: 10.3389/fgene.2018.00239
Zitnik, M., Nguyen, F., Wang, B., Leskovec, J., Goldenberg, A., and Hoffman, M. M. (2019). Machine learning for integrating data in biology and medicine: principles, practice, and opportunities. Int. J. Inf. Fusion 50, 71–91. doi: 10.1016/j.inffus.2018.09.012
Zou, Q., Chen, L., Huang, T., Zhang, Z.G., and Xu, Y.G. (2017). Machine learning and graph analytics in computational biomedicine. Artif. Intell. Med. 83:1. doi: 10.1016/j.artmed.2017.09.003
Zou, Q., Li, J. J., Song, L., Zeng, X. X., and Wang, G. H. (2016). Similarity computation strategies in the microRNA-disease network: a survey. Brief. Funct. Genomics 15, 55–64. doi: 10.1093/bfgp/elv024
Zou, Q., Lin, G., Jiang, X., Liu, X., and Zeng, X. (2018a). Sequence clustering in bioinformatics: an empirical study. Brief. Bioinform. bby090. doi: 10.1093/bib/bby090
Keywords: microorganisms, classification, environment, species, association, diseases
Citation: Qu K, Guo F, Liu X, Lin Y and Zou Q (2019) Application of Machine Learning in Microbiology. Front. Microbiol. 10:827. doi: 10.3389/fmicb.2019.00827
Received: 31 January 2019; Accepted: 01 April 2019;
Published: 18 April 2019.
Edited by:
Hongsheng Liu, Liaoning University, ChinaReviewed by:
Yen-Wei Chu, National Chung Hsing University, TaiwanMohamed Elhoseny, Mansoura University, Egypt
Copyright © 2019 Qu, Guo, Liu, Lin and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan Lin, bGlueXVhbjE5NzlAZ21haWwuY29t Quan Zou, em91cXVhbkBuY2xhYi5uZXQ=