
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 03 April 2019
Sec. Virology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00655
This article is part of the Research Topic HIV-1 Genetic Diversity View all 18 articles
 Elena Delgado1
Elena Delgado1 Sonia Benito1
Sonia Benito1 Vanessa Montero1
Vanessa Montero1 María Teresa Cuevas1
María Teresa Cuevas1 Aurora Fernández-García1,2
Aurora Fernández-García1,2 Mónica Sánchez-Martínez1
Mónica Sánchez-Martínez1 Elena García-Bodas1
Elena García-Bodas1 Francisco Díez-Fuertes3
Francisco Díez-Fuertes3 Horacio Gil1,4
Horacio Gil1,4 Javier Cañada1
Javier Cañada1 Cristina Carrera1Jesús Martínez-López1
Cristina Carrera1Jesús Martínez-López1 Marcos Sintes1
Marcos Sintes1 Lucía Pérez-Álvarez1
Lucía Pérez-Álvarez1 Michael M. Thomson1*the Spanish Group for the Study of New HIV Diagnoses
Michael M. Thomson1*the Spanish Group for the Study of New HIV DiagnosesIn Western Europe, the HIV-1 epidemic among men who have sex with men (MSM) is dominated by subtype B. However, recently, other genetic forms have been reported to circulate in this population, as evidenced by their grouping in clusters predominantly comprising European individuals. Here we describe four large HIV-1 non-subtype B clusters spreading among MSM in Spain. Samples were collected in 9 regions. A pol fragment was amplified from plasma RNA or blood-extracted DNA. Phylogenetic analyses were performed via maximum likelihood, including database sequences of the same genetic forms as the identified clusters. Times and locations of the most recent common ancestors (MRCA) of clusters were estimated with a Bayesian method. Five large non-subtype B clusters associated with MSM were identified. The largest one, of F1 subtype, was reported previously. The other four were of CRF02_AG (CRF02_1; n = 115) and subtypes A1 (A1_1; n = 66), F1 (F1_3; n = 36), and C (C_7; n = 17). Most individuals belonging to them had been diagnosed of HIV-1 infection in the last 10 years. Each cluster comprised viruses from 3 to 8 Spanish regions and also comprised or was related to viruses from other countries: CRF02_1 comprised a Japanese subcluster and viruses from 8 other countries from Western Europe, Asia, and South America; A1_1 comprised viruses from Portugal, United Kingom, and United States, and was related to the A1 strain circulating in Greece, Albania and Cyprus; F1_3 was related to viruses from Romania; and C_7 comprised viruses from Portugal and was related to a virus from Mozambique. A subcluster within CRF02_1 was associated with heterosexual transmission. Near full-length genomes of each cluster were of uniform genetic form. Times of MRCAs of CRF02_1, A1_1, F1_3, and C_7 were estimated around 1986, 1989, 2013, and 1983, respectively. MRCA locations for CRF02_1 and A1_1 were uncertain (however initial expansions in Spain in Madrid and Vigo, respectively, were estimated) and were most probable in Bilbao, Spain, for F1_3 and Portugal for C_7. These results show that the HIV-1 epidemic among MSM in Spain is becoming increasingly diverse through the expansion of diverse non-subtype B clusters, comprising or related to viruses circulating in other countries.
HIV-1 exhibits a characteristic high genetic variability, derived from elevated mutation and recombination rates. Through these mechanisms, the main (M) HIV-1 group, causative of the pandemic, has evolved into multiple genetic forms, designated subtypes, of which nine have been identified, subsubtypes, circulating recombinant forms (CRFs), of which 93 are currently recognized (HIV Sequence Database, 2019; Reis et al., 2019), and unique recombinant forms (URFs). The most globally prevalent HIV-1 genetic form is subtype C, estimated to represent around 47% worldwide infections, followed, in this order, by subtype B, subtype A, CRF02_AG, CRF01_AE, subtype G, and subtype D, with each of the remaining genetic forms estimated to represent less than 1% of global infections (Hemelaar et al., 2018).
In Western Europe, the predominant HIV-1 genetic form is subtype B, which was initially introduced among MSM and persons who inject drugs (PWID) (Lukashov et al., 1996; Casado et al., 2000; Kuiken et al., 2000; Hué et al., 2005; Beloukas et al., 2016). In early descriptions of HIV-1 genetic diversity in Western Europe, non-subtype B genetic forms were restricted to heterosexually infected immigrants coming from areas where those clades predominate, mainly sub-Saharan Africans, and European individuals epidemiologically linked to people from such areas (Fransen et al., 1996; Op de Coul et al., 1998; Thomson and Nájera, 2001). The first reports of HIV-1 non-subtype B genetic forms circulating in Western Europe among individuals without known epidemiological links to other geographic areas described the circulation of CRF01_AE among PWID in Finland (Liitsola et al., 2000) and of subtype G and CRF14_BG among a minority of HIV-1-infected PWID in the region of Galicia, Northwest Spain (Thomson et al., 2001; Delgado et al., 2002). Subsequent studies showed that the genetic forms circulating in Galicia derived from a subtype G variant widely circulating in Portugal, transmitted both through sexual contact and among PWID (Esteves et al., 2002, 2003; Palma et al., 2007; Carvalho et al., 2015). In recent years, an increasing prevalence of non-subtype B infections has been observed in Western Europe, reflecting both their importation from other geographical areas and their circulation among the local population (Abecasis et al., 2013; Beloukas et al., 2016; Hemelaar et al., 2018; Paraskevis et al., 2019).
In the current HIV-1 epidemic in Western Europe, the predominant propagation mode is through sexual contact among MSM (European Centre for Disease Prevention and Control, 2017 and WHO Regional Office for Europe 2017; Núñez et al., 2018), a population in which a resurgence of the HIV-1 epidemic has been observed since the 2000s, which is part of a global phenomenon (Bezemer et al., 2008; Beyrer et al., 2012). This has been accompanied by the emergence of phylogenetically identifiable transmission clusters, whose expansion is mostly driven by individuals with recent infection who are unaware of their HIV status (Lewis et al., 2008; Cuevas et al., 2009; Bezemer et al., 2010; Chalmet et al., 2010; Fisher et al., 2010; Zehender et al., 2010; Ambrosioni et al., 2012; Frange et al., 2012; Thomson et al., 2012; Audelin et al., 2013; Delgado et al., 2015; Esbjörnsson et al., 2016; Hoenigl et al., 2016; Chaillon et al., 2017; Patiño-Galindo et al., 2017; Parczewski et al., 2017; Verhofstede et al., 2018; Paraskevis et al., 2019). As expected, most clusters are of subtype B, but multiple instances of propagation of other HIV-1 genetic forms among European MSM have also been reported. These include subtypes A1 (Lai et al., 2016; Ragonnet-Cronin et al., 2016), C (de Oliveira et al., 2010; Lai et al., 2014; Ragonnet-Cronin et al., 2016), and F1 (Castro et al., 2010; Lai et al., 2012; Thomson et al., 2012; Delgado et al., 2015; Vinken et al., 2017; Verhofstede et al., 2018); CRF01_AE (von Wyl et al., 2011), CRF02_AG (Giuliani et al., 2013; Brand et al., 2014; Dauwe et al., 2015; Tamalet et al., 2015; Chaillon et al., 2017; Verhofstede et al., 2018), CRF17_BF (Fabeni et al., 2015), CRF19_cpx (Patiño-Galindo et al., 2015; González-Domenech et al., 2018; Pérez-Parra et al., 2018), CRF50_A1D (Foster et al., 2014), CRF56_cpx (Leoz et al., 2013), and CRF60_BC (Monno et al., 2012; Simonetti et al., 2014). However, the expansion of these clades has had a limited impact on the overall genetic composition of the HIV-1 epidemic among MSM in Western Europe, which is still largely dominated by subtype B. The only exception, though geographically restricted, is a large F1 subtype cluster of Brazilian ancestry, which represented a substantial proportion of new HIV-1 diagnoses among MSM in Northwest Spain (Thomson et al., 2012; Delgado et al., 2015). Here we describe four additional large non-subtype B clusters expanding among MSM in Spain, of CRF02_AG and of subtypes A1, F1 and C, each circulating in several Spanish regions and related to viruses from other countries.
Plasma or whole blood samples were collected from 1999 to 2018 from HIV-1-infected individuals attended at hospitals from 15 provinces from 9 regions of Spain (Basque Country, Galicia, Navarre, Castilla y León, La Rioja, Madrid, Castilla-La Mancha, Aragón, and Comunidad Valenciana). The regional sample representativeness is variable, being the greatest in the regions of Basque Country, where all public hospitals participated, and Galicia, where all but one public hospitals participated. The study was approved by the Bioethics and Animal Well-being Committee of Instituto de Salud Carlos III, Majadahonda, Madrid, Spain. Written informed consent was obtained from all participants in the study.
Amplification of HIV-1 fragments was done either from plasma RNA or from DNA extracted from whole blood. RNA was extracted from 1 ml plasma using Nuclisens EasyMAG kit (bioMérieux, Marcy l’Etoile, France), following the manufacturer’s instructions. DNA was extracted from 200 μl blood using QIAmp DNA Blood mini kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. An HIV-1 PR-RT fragment (approximately 1.4 kb) was amplified by RT-PCR followed by nested PCR, in the case of RNA, or by nested PCR, in the case of DNA, using previously reported primers (Delgado et al., 2015). In selected samples, NFLG amplification was done in four overlapping segments, as described (Delgado et al., 2002; Sierra et al., 2005), using RNA extracted either from plasma or from the primary isolate’s culture supernatant grown from plasma using a previously described protocol (Delgado et al., 2012). Direct sequencing of the amplified products was done using an automated capillary sequencer. Sequence electropherograms were assembled and edited with Seqman (DNASTAR, Madison, WI, United States). Newly obtained sequences are deposited in GenBank under accessions MK177651-MK177824 (PR-RT sequences) and MK177825-MK177829, KT276258, KY496622, and KY989952 (NFLG sequences).
Sequences were aligned with MAFFT v.7 (Katoh and Standley, 2013). Initial trees with all sequences were constructed with the approximate ML method implemented in FastTree v2.1.10 (Price et al., 2010) using the general time reversible (GTR)+CAT evolutionary model and assessment of node support with Shimodaira-Hasegawa (SH)-like local support values. Transmission clusters were defined as those supported by SH-like values ≥ 0.95 comprising four or more individuals, with a majority of them being native Spanish. To determine whether the identified clusters were still supported when globally sampled sequences were included and to identify viruses from other areas belonging or phylogenetically related to them, the analyses were repeated including all PR-RT sequences of 1 kb or longer of the same genetic form available at the Los Alamos HIV Sequence Database (HIV Sequence Database, 2019), downloaded with the option “one sequence per patient.” Support for clusters thus identified with FastTree was subsequently assessed with phylogenetic trees constructed via ML with PhyML v3.0 (Guindon et al., 2005), using the best-fit evolutionary model selected by Smart Model Selection (SMS) program (Lefort et al., 2017) and heuristic searches based on subtree pruning and regrafting (SPR) moves, with estimation of node support by the approximate likelihood ratio test (aLRT), SH-like procedure (Anisimova and Gascuel, 2006; Guindon et al., 2010). To keep computational times reasonable, in the analyses with PhyML only several hundred sequences (around 200–400) branching most closely to the clusters of interest in the previous FastTree analyses, together with PR-RT sequences from NFLG from databases, were included. Clusters were confirmed if in the analyses including database sequences with both FastTree and PhyML their node support values were ≥ 0.95. Trees were visualized with Dendroscope 3 (Huson and Scornavacca, 2012) or FigTree v1.4.21. Intersubtype recombination was analyzed by bootscanning with Simplot v3.5 (Lole et al., 1999), with tree construction with the neighbor-joining method, using the Kimura 2-parameter substitution model and windows of 400 or 600 nt moving in 20 nt steps.
Antiretroviral (ARV) drug resistance was analyzed with the Calibrated Population Resistance Tool (Gifford et al., 2009).
To estimate times of the most recent common ancestors (tMRCA) of clusters and their most probable geographical locations, a Bayesian MCMC coalescent method as implemented in BEAST v1.8.1 (Drummond et al., 2012) was used. Prior to these analyses, the existence of temporal signal in the datasets was assessed by an analysis of root-to-tip distances against dates of sampling using TempEst (Rambaut et al., 2016). For the BEAST analyses, we used all PR-RT sequences ≥ 1 kb from each cluster and related sequences, as determined in the ML phylogenetic analyses, excluding sequences without data on year or location of sample collection. PR-RT sequences derived from NFLG sequences of the corresponding genetic form downloaded from the HIV Sequence Database were also included in these analyses, using not more than 5 sequences per country of sampling. In the case of the CRF02_1 cluster, 40 randomly selected CRF02_AG PR-RT sequences lacking drug resistance mutations downloaded from the Los Alamos HIV Sequences database were included, since using those derived from NFLG resulted in relatively low r2 values in the TempEst analysis. We chose an HKY substitution model with gamma-distributed among-site rate heterogeneity and two partitions in codon positions (1st+2nd; 3rd) (Shapiro et al., 2006); uniform priors were used for absolute substitution rates (0-0.02 sub/site/year) and relative substitution rates in codon positions 1st+2nd and 3rd (0-10); we also used an uncorrelated lognormal relaxed clock model and a Bayesian skyline plot demographic model (Drummond et al., 2005). Each MCMC chain was run for 100 million to 200 million generations, sampling every 5,000 generations. MCMC convergence and effective sample sizes (ESS) were checked with Tracer v.1.62, ensuring that the ESS of each parameter was > 200. Results were summarized with a maximum clade credibility (MCC) tree, using TreeAnnotator v1.8.1, after removal of a 50% burn-in. The MCC trees were visualized with FigTree v1.4.2.1 Parameter uncertainty was summarized in the 95% highest posterior density (HPD) intervals.
Differences in clustering frequency between MSM and heterosexually infected individuals and changes in proportions of non-subtype B infections along time in newly diagnosed sexually infected individuals were analyzed with Fisher’s exact test. Only native Spish individuals were included in these analyses in order to focus on locally circulating strains, minimizing the confounding effect of imported HIV-1 variants acquired in other countries. Numbers of individuals used in these analyses were 7080 in the first and 2060 in the second.
Using HIV-1 PR-RT sequences from 10,506 individuals obtained by us (whose data are summarized in Supplementary Table 1), we identified 320 phylogenetic clusters comprising 4 or more individuals, 247 of subtype B and 73 of other genetic forms. Belonging to a cluster was more frequent among Spanish MSM than among heterosexually-infected Spanish individuals (68% vs. 31%; p = 7.7 × 10-6) (Supplementary Figure 1). Differences were also significant when subtype B and non-subtype B clusters were analyzed separately. An increase along time of non-subtype B infections among newly diagnosed Spanish individuals was also observed. Statistically significant increases were observed in Spanish MSM from 2005–2009 to 2010–2014 (from 10.9% to 26.7%; p = 1.4 × 10-6) and among heterosexually infected Spanish individuals from 2010–2014 to 2015–2018 (from 28.5% to 39.7%; p = 0.0097) (Supplementary Figure 2).
Of the non-subtype B clusters, 5 large ones (here defined as those comprising 10 or more individuals) were associated with MSM. The largest one, of F1 subtype (currently comprising 187 individuals), was reported previously (Thomson et al., 2012; Delgado et al., 2015). The other four were of CRF02_AG and of subtypes A1, F1, and C, henceforth designated CRF02_1, A1_1, F1_3, and C_7, respectively. All four clusters were well supported when the analyses were repeated with FastTree including all PR-RT sequences > 1 kb of the respective genetic forms available at the Los Alamos HIV Sequence database (with numbers from 1,318 of subtype F1 to 22,762 of subtype C) and with PhyML including several hundred (from 312 for subtype C to 513 for CRF02_AG) database sequences branching closer to each cluster in the FastTree analysis and derived from NFLG sequences. These analyses also allowed to identify viruses from databases belonging or closely related to the clusters. Epidemiological data of samples studied by us belonging to these clusters and to subclusters within them are shown in Table 1.
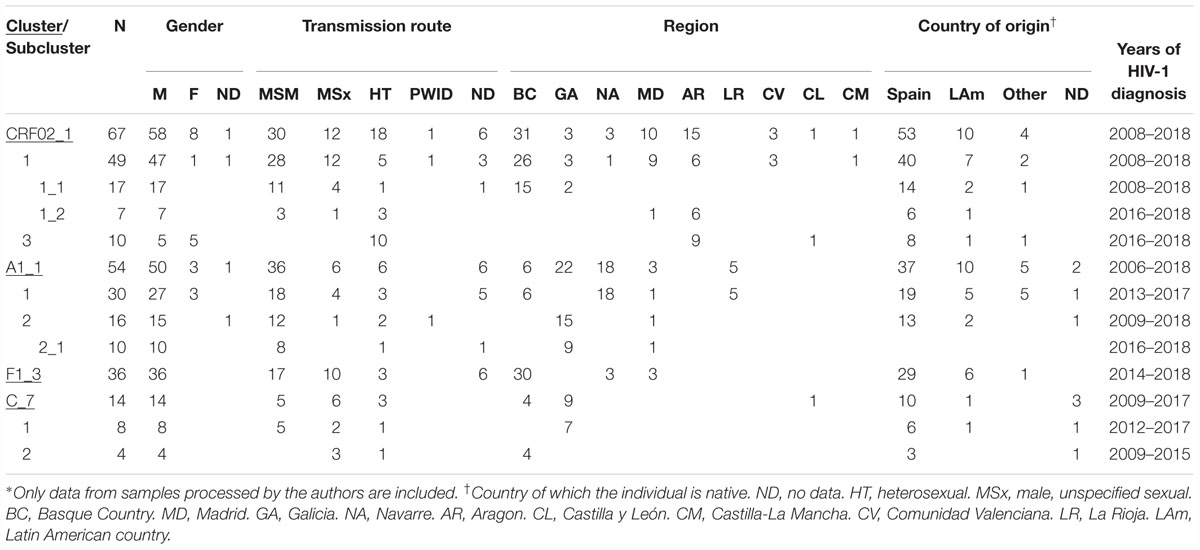
Table 1. Epidemiological data of clusters∗.
CRF02_1 comprised 115 individuals, 67 studied by us and 48 whose sequences were retrieved from databases (Figure 1). Most samples were collected in Spain, but there were also samples from Japan, Switzerland, United Kingdom, Ecuador, Netherlands, Sweden, Germany, Malaysia, and Hong Kong. Samples from Spain were from 8 regions, mainly from Basque Country, Madrid, and Aragon.
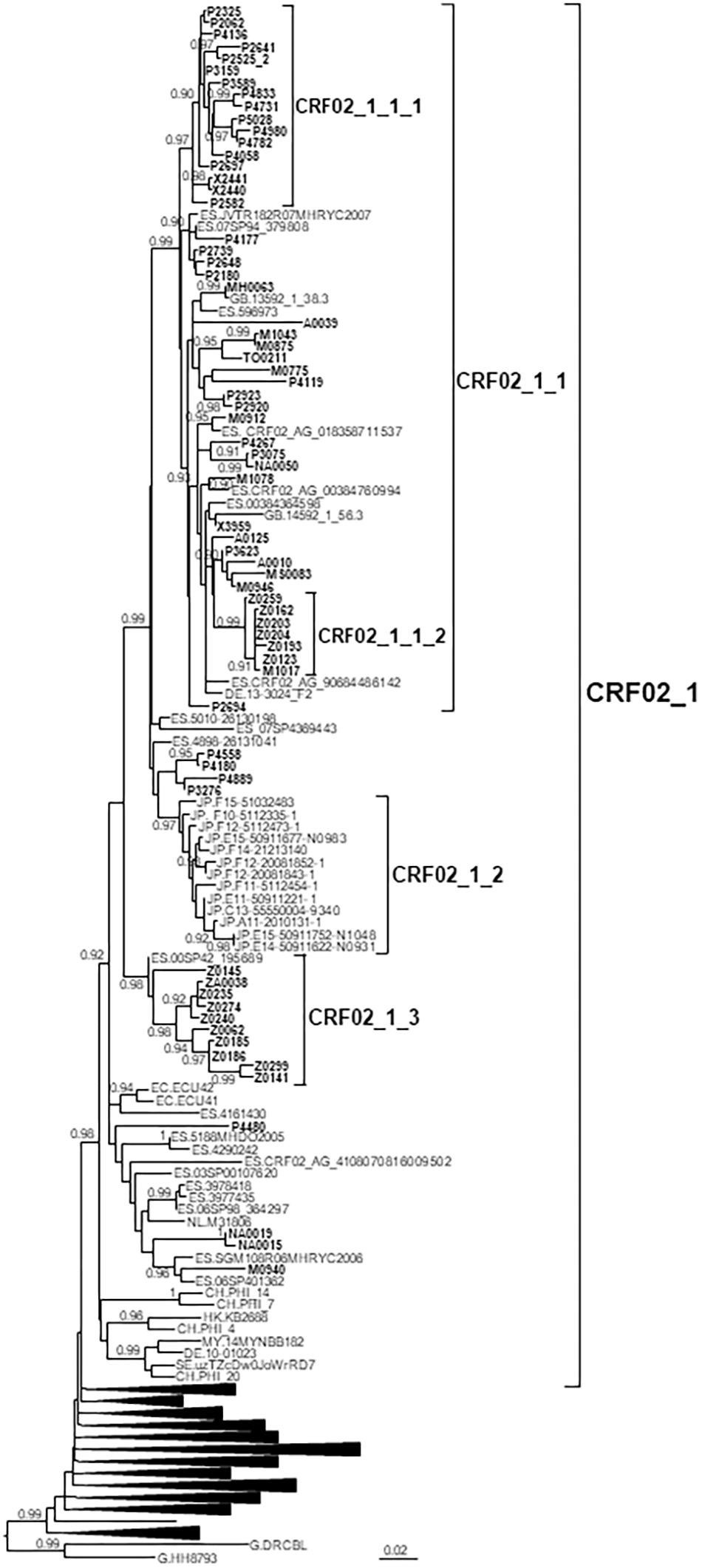
Figure 1. Maximum likelihood tree of PR-RT sequences of the CRF02_1 cluster. The tree was constructed with PhyML, with assessment of node support with the aLRT SH-like procedure. The analysis incorporates 513 CRF02_AG PR-RT sequences from databases that in preliminary analyses with FastTree branched closer to the CRF02_1 cluster and from NFLG sequences, and two subtype G sequences used to root the tree. For better viewing, clades outside of the CRF02_1 cluster are collapsed. Only aLRT SH-like node support values ≥ 0.9 are shown. Sequences obtained by us are in bold type. Sequences from databases are labeled with the two-letter ISO code of the country of sample collection followed by the sample name.
Years of HIV-1 diagnosis were 2008–2018.
CRF02_1 comprised three subclusters, designated with numerals 1-3. CRF02_1_1 comprised 60 individuals from 7 Spanish regions, two from United Kingdom and one from Germany. Within it, two subsubclusters were distinguished, associated with the Basque Country and the city of Zaragoza, Aragón, respectively; CRF02_1_2 comprised 13 individuals from Japan; and CRF02_1_3 comprised 11 individuals, 10 of them from Zaragoza.
Among samples studied by us, 88% were from men. Transmission route was sexual in the great majority, and, among those infected via sexual contact, 51% were self-declared MSM. Interestingly, all 10 individuals from subcluster CRF02_1_3 studied by us (5 women and 5 men, all from Zaragoza) were infected via heterosexual contact.
Most individuals in this and in the other clusters here described were native Spanish (Table 1).
A1_1 comprised 66 individuals, 54 studied by us and 12 with sequences deposited in databases (7 from United Kingdom, 4 from Portugal, and 1 from United States) (Figure 2). A1_1 was also related to viruses from the A1 subtype lineage circulating in Greece, Albania, and Cyprus (Ciccozzi et al., 2005; Paraskevis et al., 2007; Pineda-Peña et al., 2018). Samples from Spain were collected in 5 regions, mainly in Galicia and Navarre.

Figure 2. Maximum likelihood tree of PR-RT sequences of the A1_1 cluster. The tree was constructed with PhyML, with assessment of node support with the aLRT SH-like procedure. The analysis incorporates 338 A1 subsubtype PR-RT sequences from databases that in preliminary analyses with FastTree branched closer to the A1_1 cluster and from NFLG sequences, and two CRF01_AE sequences used to root the tree. For better viewing, clades outside of the A1_1 cluster are collapsed. Only aLRT SH-like node support values ≥ 0.90 are shown. Sequences obtained by us are in bold type. Sequences from databases are labeled with the two-letter ISO code of the country of sample collection followed by the sample name. Collapsed clades most closely related to the A1_1 cluster are labeled with the two-letter codes of the countries of sample collection, excluding countries represented by a single sequence.
Forty five of 47 individuals with available data were diagnosed in 2012–2018.
Within A1_1, there were two main subclusters: A1_1_1, comprising all individuals from Navarre, Basque Country, and La Rioja, and 1 from Madrid; and, A1_1_2 comprising individuals mostly from Galicia, with a majority grouping in a subsubcluster.
Most (93%) individuals in the cluster were men, with predominance of MSM.
F1_3 comprised 36 individuals. There were no sequences from databases belonging to F1_3, but viruses from Romania, most of them transmitted sexually (Niculescu et al., 2015), were closely related to it (Figure 3). Five viruses from Spain (4 of them sequenced by us), 3 from United Kingdom, and 1 from Poland also branched close to F1_3, interspersed among the Romanian samples. Most F1_3 samples were from Basque Country.
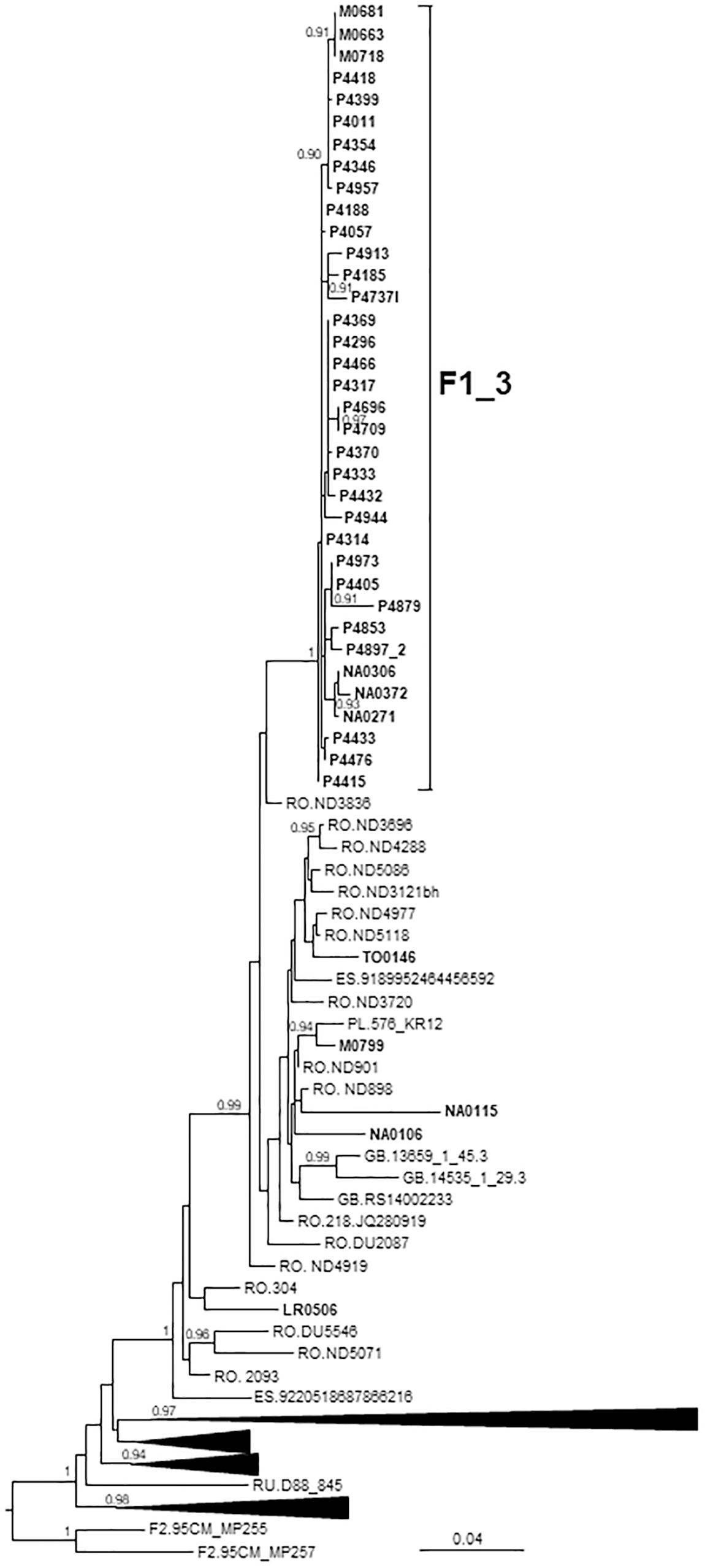
Figure 3. Maximum likelihood tree of PR-RT of viruses of the F1_3 cluster. The tree was constructed with PhyML, with assessment of node support with the aLRT SH-like procedure. The analysis incorporates 358 PR-RT F1 subsubtype sequences from databases that in preliminary analyses with FastTree branched closer to the F1_3 cluster and from NFLG sequences, and two F2 subsubtype sequences used to root the tree. For better viewing, clades outside of the F1_3 cluster, excluding those most closely related to the F1_3 cluster, are collapsed. Only aLRT SH-like node support values ≥ 0.90 are shown. Sequences obtained by us are in bold type. Sequences from databases are labeled with the two-letter ISO code of the country of sample collection followed by the sample name.
All infections were diagnosed in 2014 or later. All individuals in F1_3 were men. Transmission route was sexual in all for which data were available, with a majority of MSM.
C_7 comprised 17 individuals, including 14 studied by us and 3 from Portugal, whose sequences were retrieved from databases. One sequence from Mozambique branched basally to C_7 (Figure 4). C_7 comprised two subclusters, C_7_1, comprising all but one samples from Galicia, and C_7_2, comprising all 4 samples from Basque Country. All but one had been diagnosed in 2012–2017.
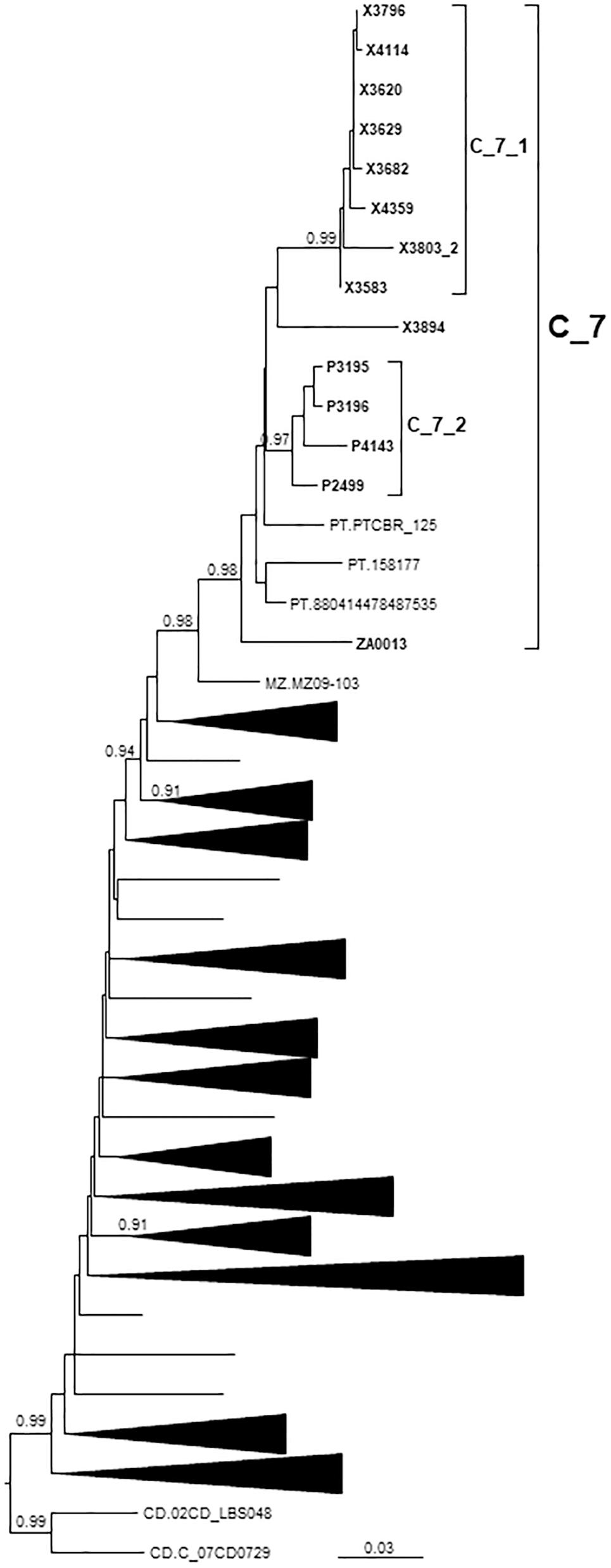
Figure 4. Maximum likelihood tree of PR-RT of viruses of the C_7 cluster. The tree was constructed with PhyML, with assessment of node support with the aLRT SH-like procedure. The analysis incorporates 312 PR-RT subtype C sequences from databases that in preliminary analyses with FastTree branched closer to the C_7 cluster and from NFLG sequences, and two subtype C sequences from the Democratic Republic of Congo used to root the tree. For better viewing, clades outside of the C_7 cluster are collapsed. Only aLRT SH-like node support values ≥ 0.90 are shown. Sequences obtained by us are in bold type. Sequences from databases are labeled with the two-letter ISO code of the country of sample collection followed by sample name.
All but one Spanish samples were from Galicia or Basque Country. All individuals in C_7 were sexually infected men, with 5 being self-declared MSM.
To estimate the temporal and geographic origins of clusters and subclusters, Bayesian coalescent analyses were performed with PR-RT sequences, summarizing the posterior distribution of trees with MCC trees. Prior to these analyses, temporal signal was analyzed, revealing a clock-like structure in all four datasets used for subsequent analyses (r2= 0.4395 in CRF02_1, r2= 0.5399 in A1_1, r2= 0.7175 in F1_3, and r2= 0.3518 in C_7), indicating that the datasets contained sufficient temporal structure for reliable estimation of divergence times.
In the Bayesian analyses, all four clusters previously defined via ML were supported by node PP values > 0.98.
The tMRCA of the entire CRF02_1 cluster was estimated around 1986, but the location of the MRCA was uncertain, since the most probable one was supported by a PP < 0.5. However, the location of the MRCA of the subcluster comprising all but the 8 most basal sequences (collected in Switzerland, Sweden, Germany, Malaysia, and Hong Kong), with tMRCA around 1988, had a strong support in Madrid, with a PP of 0.975 (Figure 5). Subcluster CRF02_1_1 emerged around 2002 in Madrid, CRF02_1_2 around 2006 in Japan, and CRF02_1_3 around 2006 in Zaragoza.
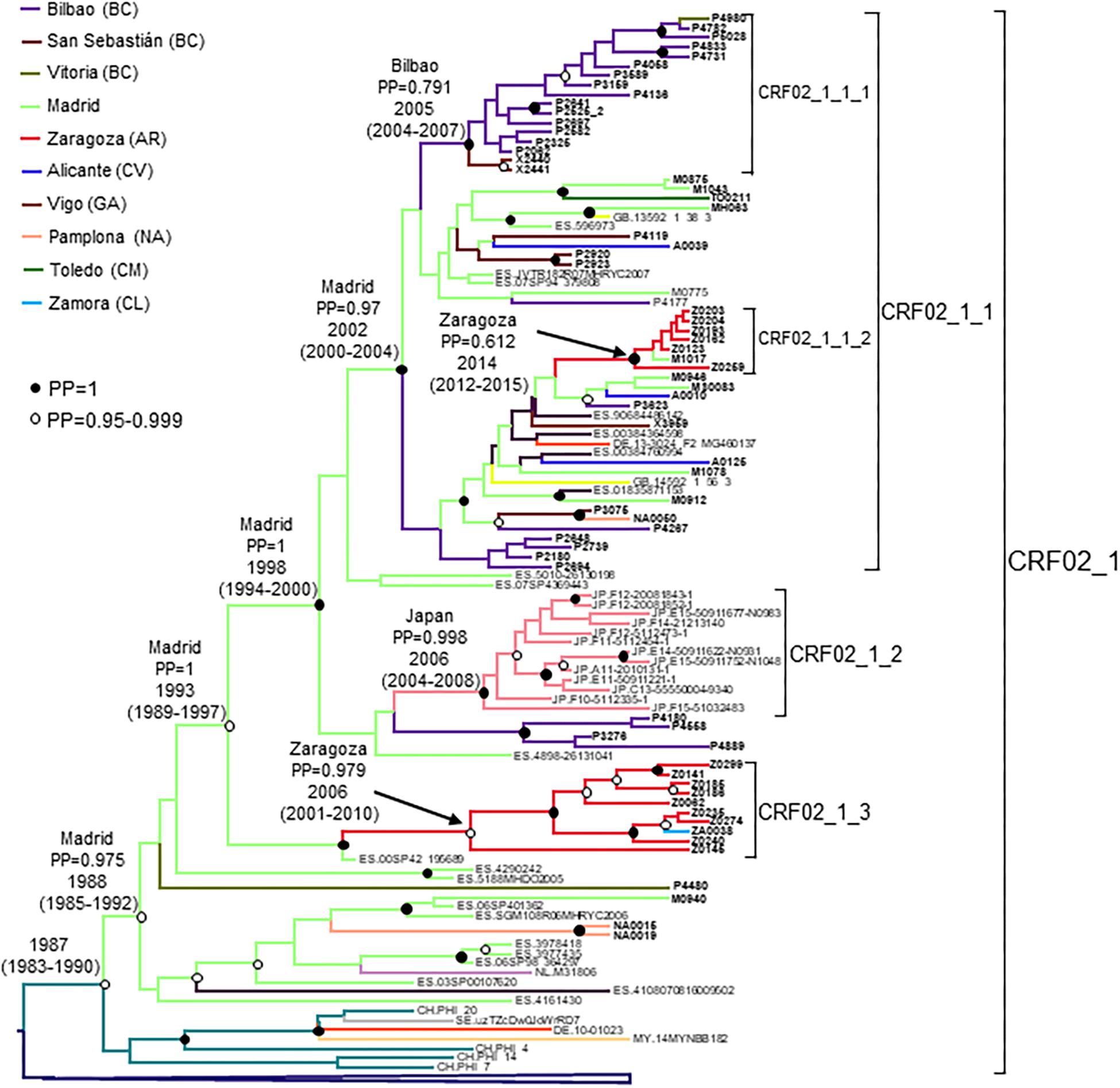
Figure 5. Maximum clade credibility tree of PR-RT sequences of the CRF02_1 cluster. The tree also includes 40 other PR-RT CRF02_AG sequences from databases. For better viewing, the clade comprising viruses branching outside of the CRF02_1 cluster is collapsed. Sequences obtained by us are in bold type. Sequences from databases are labeled with the two-letter ISO code of the country of sample collection followed by the sample name. Nodes supported by PP = 1 and PP = 0.95–0.999 are marked with filled and unfilled circles, respectively. Colors of terminal and internal branches represent sampling locations and most probable locations of the corresponding nodes, respectively, according to the legend on the left. For the nodes corresponding to CRF02_1 cluster and its major subclusters, the location with the highest PP (if > 0.5) and the tMRCAs (with 95% HPD intervals) are indicated above or close to the subtending branches.
The estimated tMRCA of the entire A1_1 cluster was around 1989, but the location of the MRCA was uncertain, since the most probable one was supported by a PP < 0.5. However, the location of the MRCA of the subcluster comprising all but the 6 most basal sequences, with tMRCA around 1994, was supported by a PP of 0.814 in the city of Vigo, Galicia. The emergence of subcluster A1_1_1 was around 2010, with highest location PP in Pamplona, and that of A1_1_2 was around 2004 in Vigo, Galicia (Figure 6). It should be pointed out that 2 samples from Portugal and 6 from United Kingdom could not be used in the Bayesian analyses, since no collection year was available for them.
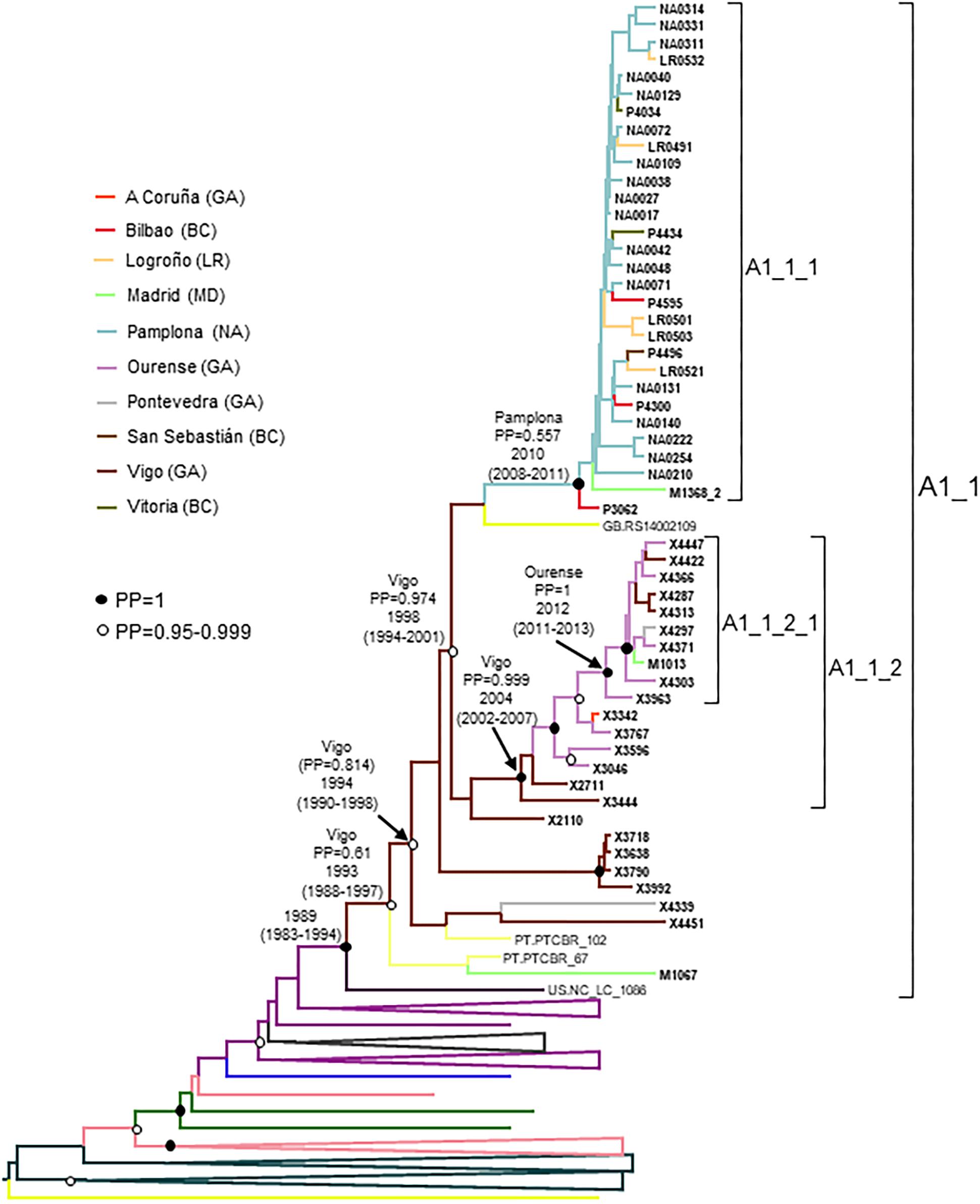
Figure 6. Maximum clade credibility tree of PR-RT sequences of the A1_1 cluster. The tree also includes a sequence from US that in ML trees branched close to the A1_1 cluster (samples from United Kingdom were excluded because no information on time of sample collection was available), and A1 subsubtype PR-RT sequences from NFLG sequences from databases. For better viewing, clades outside of the A1_1 cluster are collapsed. Sequences obtained by us are in bold type. Sequences from databases are labeled with the two-letter ISO code of country of sample collection followed by sample name. Clades most closely related to the A1_1 cluster are labeled with the two-letter ISO code of the sampling countries of viruses contained in it. Nodes supported by PP = 1 and PP = 0.95-0.999 are marked with filled and unfilled circles, respectively. Colors of terminal and internal branches represent sampling locations and most probable locations of the corresponding nodes, respectively, according to the legend on the left. For the nodes corresponding to the A1_1 cluster and its major subclusters, the location with the highest PP (if > 0.5) and the tMRCAs (with 95% HPD intervals) are indicated above or close to the subtending branches.
The estimated tMRCA of F1_3 was around 2013 in the city of Bilbao, with a strongly supported ancestry in Romania (Figure 7).
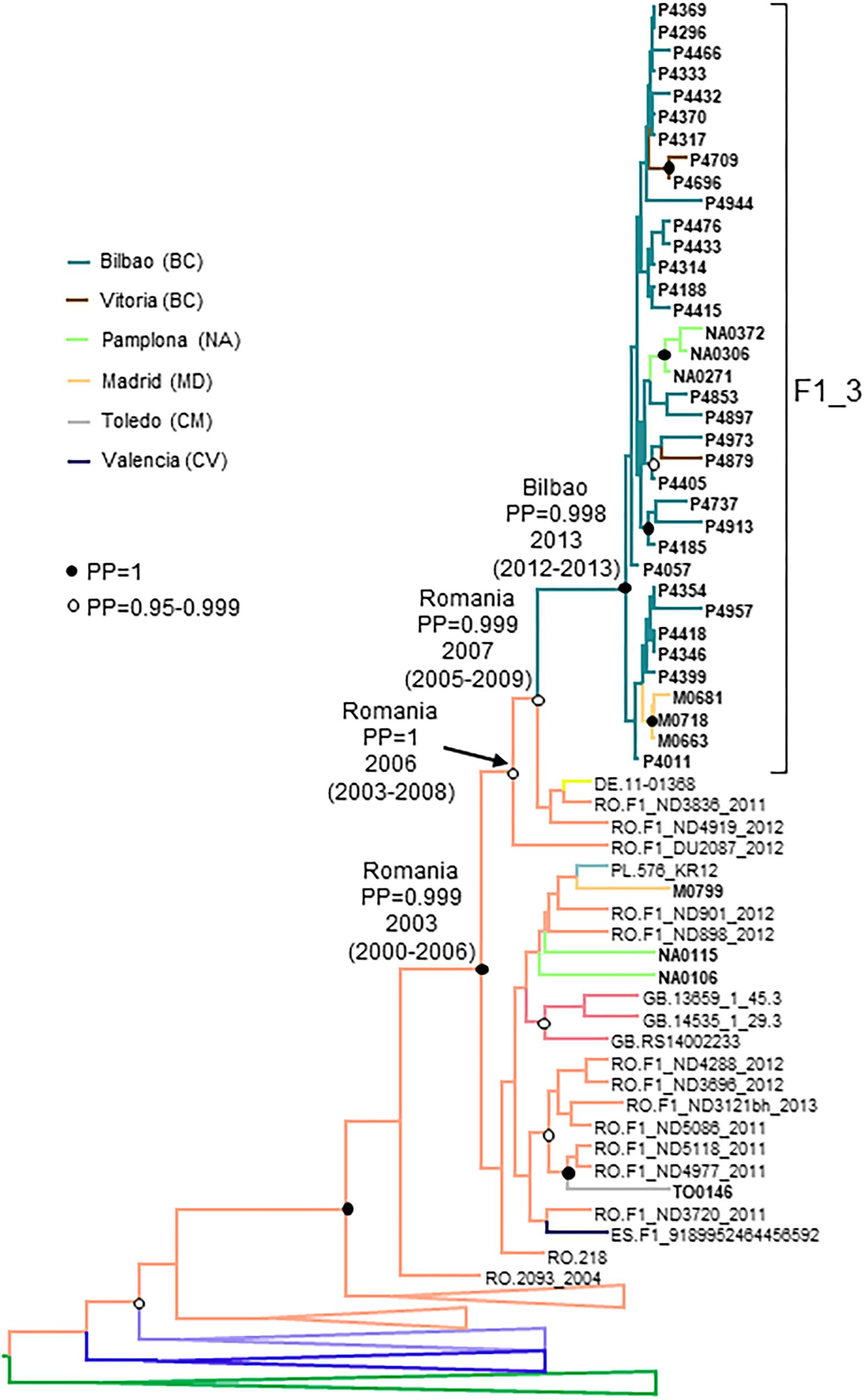
Figure 7. Maximum clade credibility tree of PR-RT sequences of the F1_3 cluster. The tree also includes F1 subsubtype sequences from databases branching close to the F1_3 cluster and F1_3 subsubtype PR-RT sequences from NFLG sequences from databases. For better viewing, clades outside of the F1 cluster are collapsed, except those most closely related to the F1_3 cluster. Sequences obtained by us are in bold type. Sequences from databases are labeled with the two-letter ISO code of country of sample collection followed by sample name. Nodes supported by PP = 1 and PP = 0.95–0.999 are marked with filled and unfilled circles, respectively. Colors of terminal and internal branches represent sampling locations and most probable locations of the corresponding nodes, respectively, according to the legend on the left. For the nodes corresponding to the F1_3 cluster and the clades within which it is contained, the location posterior probabilities and the tMRCAs (with 95% HPD intervals) are indicated above or close to the subtending branches.
Finally, the estimated tMRCA of C_7 was around 1983. Its most probable origin was Portugal, but with a PP support for the entire cluster of only 0.6. However, the location of the MRCA of the subcluster comprising all but the most basal sample had a strong support in Portugal (PP = 0.91) (Figure 8). Subcluster C_7_1 emerged around 2008 in Vigo and C_7_2 around 2004 in Vitoria, Basque Country.
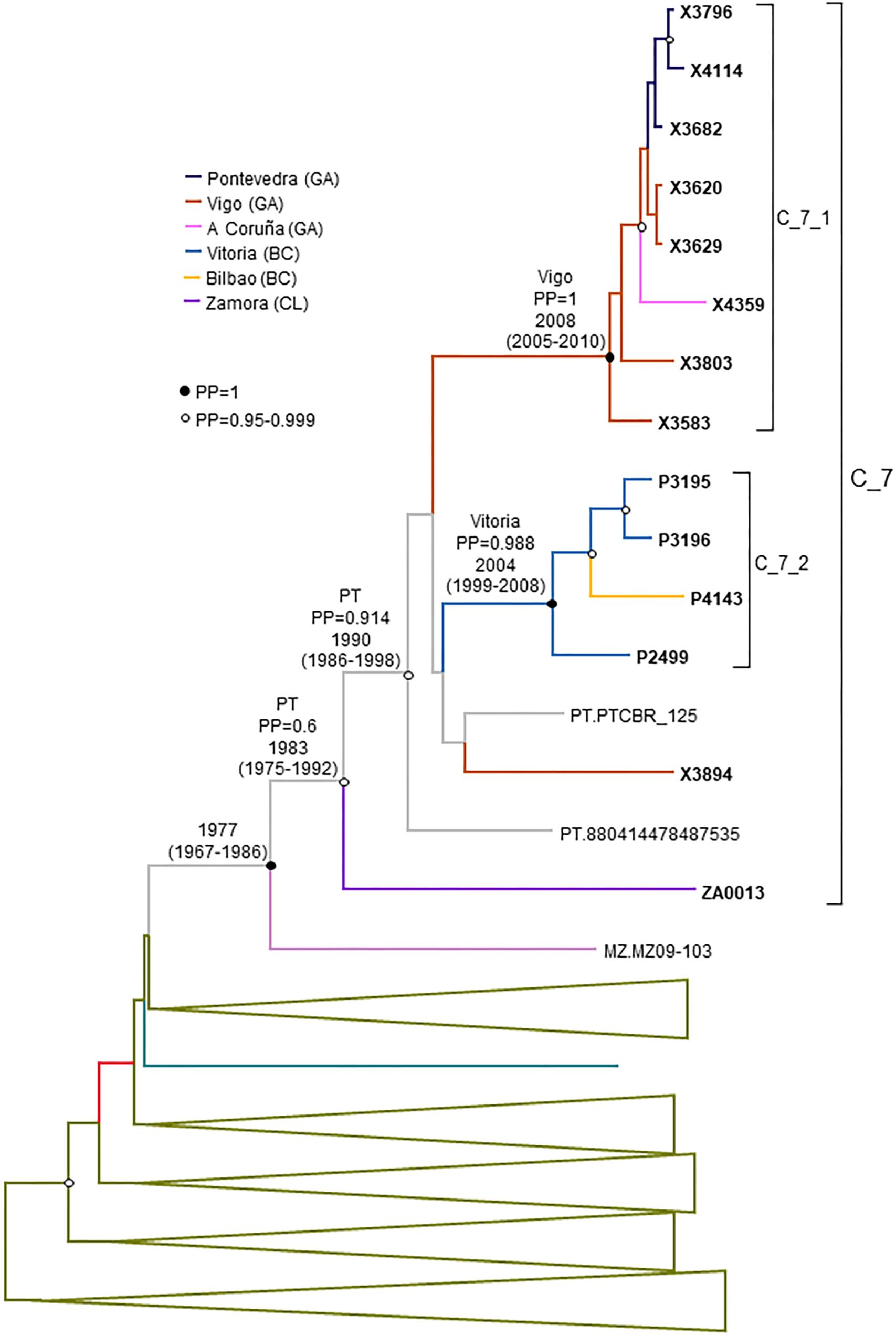
Figure 8. Maximum clade credibility tree of PR-RT sequences of the C_7 cluster. The tree also includes a subtype C sequences from Mozambique that in ML trees branched close to the C_7 cluster and subtype C PR-RT sequences from NFLG sequences from databases. For better viewing, clades outside of the C_7 cluster are collapsed. Sequences obtained by us are in bold type. Sequences from databases are labeled with the two-letter ISO code of country of sample collection followed by sample name. Nodes supported by PP = 1 and PP = 0.95–0.999 are marked with filled and unfilled circles, respectively. Colors of terminal and internal branches represent sampling locations and most probable locations of the corresponding nodes, respectively, according to the legend on the left. For the nodes corresponding to the C_7 cluster and subclusters within it, the location posterior probability and the tMRCAs (with 95% HPD intervals) are indicated above or close to the subtending branches. tMRCA is also indicated for the node corresponding to the clade including the sample from Mozambique (most probable location is omitted, since its PP is below 0.5).
In CRF02_1, 5 sequences (4 from Spain and one from Germany) had ARV drug resistance mutations. One was from a drug-experienced individual in therapeutic failure with multiple drug resistance mutations, and two, with K101E and K103N, respectively, mutations of resistance to non-nucleoside reverse transcriptase inhibitors (NNRTI), were from drug-naïve individuals. The other two, with K103N and K101E mutations, respectively, were from database sequences without data on drug treatment. In A1_1, one database sequence from United Kingdom had Y188C and G190A NNRTI resistance mutations. In C_7, all but 2 sequences had L90M mutation associated with protease inhibitor drug resistance; all 13 Spanish sequences with this mutation were from drug-naïve individuals.
To determine whether the viruses from the clusters were of uniform genetic form all along their genomes or were interclade recombinants, two NFLG sequences were obtained for each cluster, either from plasma RNA (P2648, P3075, P4496, P4346, and P4476) or from RNA extracted from culture supernatant (NA0048_2, X3303_2, and X3988). An additional NFLG from the A1_1 cluster (X2110, GenBank accession FJ670523) had been obtained previously by us (Cuevas et al., 2010). Bootscan analyses showed that all were of uniform genetic form along their genomes (Figure 9). We note that NFLG sequences of two CRF02_1 viruses from United Kingdom (Figure 1) are also available at sequence databases (Yebra et al., 2018; HIV Sequence Database, 2019) (GenBank accessions MF109381, MF109550).
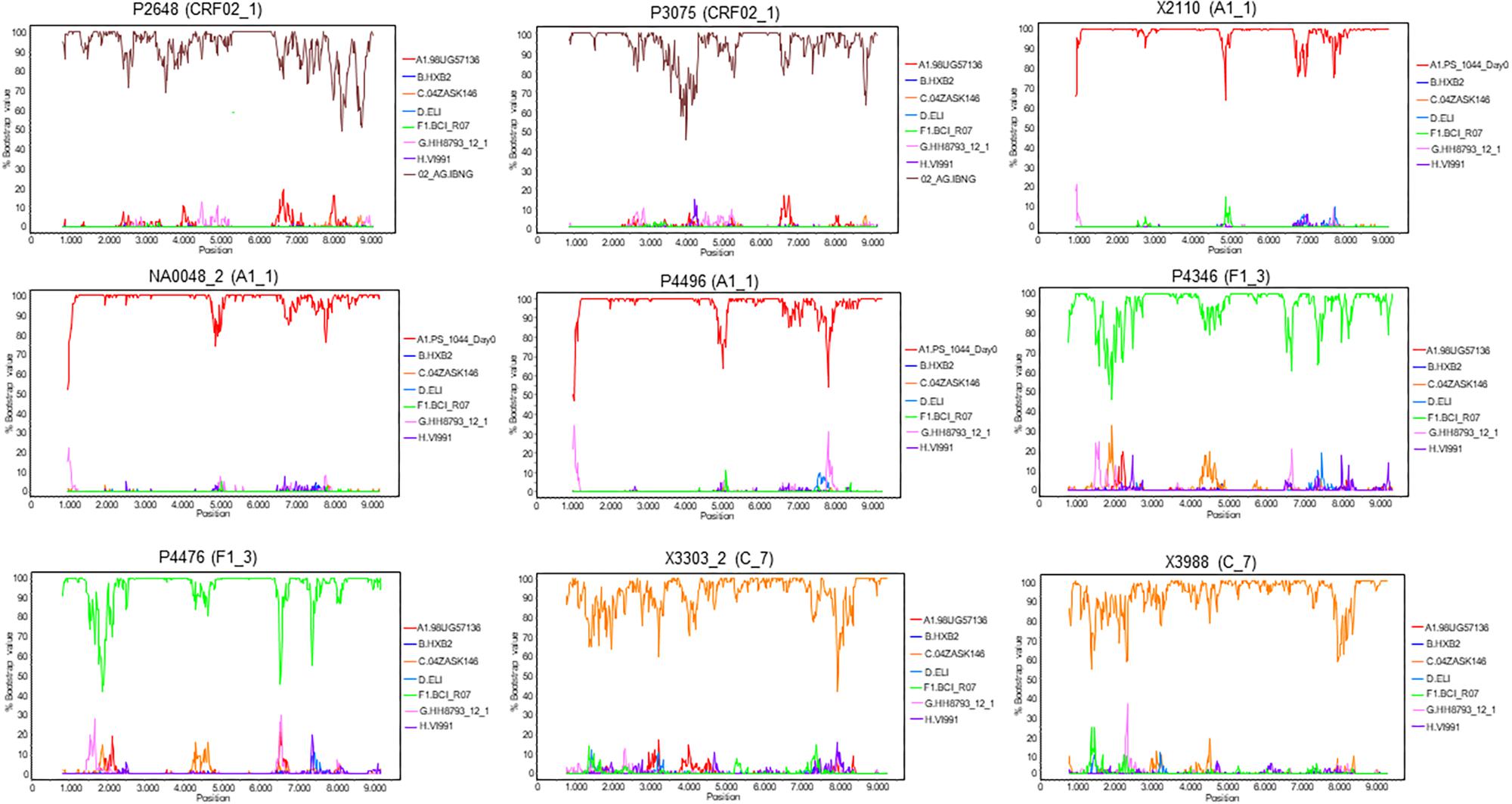
Figure 9. Bootscan analyses of NFLG sequences of viruses from the identified clusters. Virus names, with cluster in parentheses, are above each plot. A window of 400 nt was used for viruses of the A1_1, F1_3, and C_7 clusters, and of 600 nt for viruses of the CRF02_1 cluster, sliding in 20 nt increments. The horizontal axis represents the nucleotide position in the HXB2 proviral genome of the window’s midpoint. The vertical axis represents the bootstrap value supporting clustering with reference sequences, listed on the right of each plot.
In Western Europe, subtype B has been largely predominant among MSM since the early HIV-1 epidemic, but in recent years other genetic forms have been reported to be circulating in this population, as evidenced by their grouping in phylogenetic clusters comprising mostly European individuals. In this study, based on a large dataset from Spain, we found an increase in proportions of non-subtype B infections among MSM in recent years (Supplementary Figure 2) and higher clustering frequency in MSM compared to heterosexually infected individuals (Supplementary Figure 1). Among clusters associated with MSM, five large ones were of non-subtype B genetic forms, one of which, of F1 subtype, was reported previously (Thomson et al., 2012; Delgado et al., 2015). The other four were of CRF02_AG and subtypes A1, F1, and C, for which here we analyze epidemiological correlations, estimated emergence times and places, NFLGs, and drug resistance mutations.
The CRF02_AG cluster (CRF02_1) comprised 115 individuals, including 67 studied by us and 48 whose sequences were retrieved from databases, making it one of the largest non-subtype B clusters circulating among MSM reported to date in Western Europe (Delgado et al., 2015; Vinken et al., 2017). CRF02_AG is the predominant HIV-1 genetic form in most West African countries (Montavon et al., 2000; Hemelaar et al., 2018) and is common in West-Central Africa. It also propagates as a minor form in several Western European countries (Giuliani et al., 2013; Brand et al., 2014; Tamalet et al., 2015; Beloukas et al., 2016; Chaillon et al., 2017; Verhofstede et al., 2018), Tunisia (El Moussi et al., 2017), and Brazil (Delatorre et al., 2012), and in 2002 caused an outbreak among PWID in Uzbekistan (Carr et al., 2005), with subsequent dissemination to Kazakhstan (Eyzaguirre et al., 2007; Lapovok et al., 2014) and Russia (Moskaleychik et al., 2015), giving rise to CRF63_02A1 through recombination with the former Soviet Union subtype A variant (Baryshev et al., 2014; Shcherbakova et al., 2014). CRF02_AG has been reported to be one of the most common non-subtype B genetic forms in Western Europe (together with subtypes A1 and C) (Abecasis et al., 2013; Beloukas et al., 2016; Hemelaar et al., 2018) and in Spain (Yebra et al., 2012). The CRF02_AG cluster here described is not completely new, since a cluster of four individuals from the region of Valencia belonging to it was reported by other authors, who also noted that 9 sequences from databases, 7 from Spain and 2 from Ecuador, were related to the Valencian cluster (Bracho et al., 2014). However, the data here presented considerably enlarge the size and the geographic range of the cluster. CRF02_1 comprises viruses from 8 Spanish regions and from 9 other countries, from Western Europe, Asia and South America, with 13 Japanese viruses grouping in a monophyletic subcluster, indicating that it is circulating in this country. Although CRF02_1 is mainly associated with MSM, a subcluster comprising 11 individuals, 10 of them from the city of Zaragoza, propagates via heterosexual contact (Table 1). The origin of CRF02_1 is not recent, with a tMRCA estimated around 1986, with uncertain location, for the entire cluster, or 1988 in Madrid for the subcluster excluding the 8 most basal sequences, but its three major subclusters emerged in recent years, with tMRCAs in the 2000s.
The A1 subtype cluster (A1_1) is the second largest cluster here described, with 66 individuals, 54 studied by us and 12 with sequences in databases. A1 subtype circulates mainly in Eastern, Central and Western Africa (Hemelaar et al., 2018), all former Soviet Union (FSU) countries (Bobkova, 2013), Greece (Paraskevis et al., 2007), Albania (Ciccozzi et al., 2005), and Cyprus (Pineda-Peña et al., 2018), and as a minor form in India (Pandey et al., 2016), although some authors designate the variants circulating in Western Africa and FSU as distinct subsubtypes (A3 and A6, respectively) (Meloni et al., 2004; Foley et al., 2016). The lineage circulating in Greece and Albania, also detected in Cyprus, is of monophyletic origin, with estimated tMRCA around 1978 (Paraskevis et al., 2007). A1 subtype clusters have been reported in United Kingdom (Gifford et al., 2007; Hughes et al., 2009; Ragonnet-Cronin et al., 2016), Italy (Lai et al., 2016), Switzerland (von Wyl et al., 2011), and Portugal (Carvalho et al., 2015), but the one here reported is the largest reported to date in Western Europe. A1_1 comprises individuals from 5 Spanish regions and 3 other countries (United Kingdom, Portugal, and United States), and is related to the Greek-Albanian A1 lineage (Figure 2). Its origin is not recent, with estimated tMRCA around 1989, with uncertain location for the entire cluster, or around 1994 in Vigo, Galicia, excluding the 6 most basal sequences, but its two major subclusters are of recent origin, with tMRCAs around 2004 and 2010, respectively.
The F1 cluster (F1_3) comprises 36 individuals, all resident in Spain, most of them in Bilbao, Basque Country. Of the clusters here described, this is the one with the most recent origin, with estimated tMRCA around 2013 in Bilbao. It is currently increasing in size, with 6 individuals newly diagnosed in 2018. Subtype F1 is circulating in Central Africa, Brazil and Romania (Dumitrescu et al., 1994; Louwagie et al., 1994; Bandea et al., 1995; Apetrei et al., 1997; Op de Coul et al., 2000; Hemelaar et al., 2018), and F1 subtype clusters have been recently identified in Spain (Thomson et al., 2012; Delgado et al., 2015), Belgium (Vinken et al., 2017; Verhofstede et al., 2018), Switzerland (Castro et al., 2010), Italy (Lai et al., 2012), and Portugal (Carvalho et al., 2015). F1_3 is most closely related to F1 viruses from Romania (Figure 4), which are related to viruses circulating in Angola (Guimarães et al., 2009) and initially propagated among adults via sexual contact, with subsequent propagation among institutionalized children through contaminated injection equipment (Op de Coul et al., 2000; Bello et al., 2012), and more recently among PWID (Temereanca et al., 2013; Niculescu et al., 2015). The expansion of an F1 subtype cluster of Romanian ancestry in Spain has its counterpart in the recent expansion of CRF14_BG, originally described in Spain (Thomson et al., 2001; Delgado et al., 2002) and Portugal (Esteves et al., 2003; Abecasis et al., 2011; Bartolo et al., 2011), among PWID in Romania (Niculescu et al., 2015). The exchange of HIV-1 genetic forms between Romania and Spain reflects the presence of a large Romanian immigrant population in Spain (Instituto Nacional de Estadística, 2018), frequently traveling between both countries.
The subtype C cluster (C_7) comprises 17 viruses, 14 from three Spanish regions, predominantly from Galicia, and three from Portugal. It comprises a Galician and a Basque subclusters (Figure 4). This is not the first report of this cluster, as it was described by us when it comprised only 7 individuals, within the context of the description of HIV-1 clusters bearing ARV drug resistance mutations, noting that viruses belonging to it carry the L90M mutation of resistance to protease inhibitors (Vega et al., 2015). This mutation was found in 15 of 17 C_7 viruses. Subtype C is the most prevalent clade in the HIV-1 pandemic, circulating mainly in Southern and Eastern Africa, Southern Brazil, and South Asia (Hemelaar et al., 2018). In Western Europe, subtype C clusters have been reported in the United Kingdom (Hughes et al., 2009; de Oliveira et al., 2010; Ragonnet-Cronin et al., 2016), Italy (Lai et al., 2014), and Portugal (Carvalho et al., 2015).
The origin of C_7 is not recent, with a tMRCA around 1983, with a most probable origin in Portugal, but the tMRCAs of its subclusters are relatively recent, in 2008 and 2004, respectively. C_7 is related to a virus from Mozambique, a former Portuguese colony where subtype C is the predominant HIV-1 genetic form (Bellocchi et al., 2005).
It should be noted that 3 individuals in each of clusters F1_3 and C_7, composed entirely of men, declared being heterosexual, and that in clusters CRF02_1 (excluding the heterosexual-associated CRF02_1_3 subcluster) and A1_1, among sexually infected individuals the number of self-declared heterosexual men exceeds the number of women (8 vs. 3 in CRF02_1 and 6 vs. 3 in A1_1). This suggests that at least some of the self-declared heterosexual men within the clusters could in fact be MSM, who, due to social stigma and discrimination, do not declare their real sexual behaviors, as suggested by other authors who found similar discrepancies between self-reported heterosexual behavior and phylogenetic clustering with sequences from MSM (Hué et al., 2014; Hoenigl et al., 2016; Ragonnet-Cronin et al., 2018).
The descriptions of the four clusters here analyzed is in line with those of other non-subtype B clusters reported to have expanded among MSM in Western Europe. However there are some salient features of the clusters here described that should be highlighted: first, the relatively large size of CRF02_1_1 and A1_1, greater than most non-subtype B clusters reported among MSM in Western Europe; second, their wide geographic distribution among different countries, which contrasts with the predominantly within-country clustering found by other authors in Europe (Frentz et al., 2013; Paraskevis et al., 2019); and, third, the rapid expansion of F1_3, with 36 diagnoses in only 4 years.
The expansion of large clusters among MSM in recent years, as here reported, reflects the existence of high risk sexual behaviors, which should prompt public health authorities to implement public health measures aimed at preventing HIV-1 transmission in this population, including behavioral interventions to reduce risky practices, preexposure prophylaxis (Grant et al., 2010; Volk et al., 2015; McCormack et al., 2016), and early diagnosis and treatment of HIV-1 infections (European Centre for Disease Prevention and Control, 2015; United Nations Population Fund, the Global Forum on MSM and HIV, United Nations Development Programme, World Health Organization, United States Agency for International Development, and World Bank, 2015). Prevention of HIV-1 transmission among MSM could also result in a reduction of heterosexually-transmitted infections, which can have their source in MSM networks, as seen in subcluster CRF02_1_3, associated with heterosexual transmission, and as reported by other authors (Oster et al., 2015; Esbjörnsson et al., 2016).
Continued HIV-1 molecular surveillance will be necessary to gain insight in real time on the dynamics of expansion of transmission networks, which will allow to focus prophylactic efforts in populations with the highest risk of HIV-1 acquisition and ongoing transmission (Little et al., 2014; Wang et al., 2015; Poon et al., 2016; Ratmann et al., 2016; Brenner et al., 2017; Chaillon et al., 2017; German et al., 2017; Oster et al., 2018; Wertheim et al., 2018) and to monitor the efficacy of public health interventions aimed at controlling the epidemic (Wertheim et al., 2011; Magiorkinis et al., 2018). HIV-1 molecular surveillance can also provide important information for the design of vaccine immunogens adapted to the major HIV-1 variants actively propagating in different areas, considering the correlation of HIV-1 clades to susceptibility to protective immune responses (Cao et al., 2000; Thomson et al., 2002; Binley et al., 2004; Geldmacher et al., 2007; Seaman et al., 2010; Hraber et al., 2014), and with potential to induce broadly neutralizing antibody responses (Kouyos et al., 2018), and will allow to obtain reagents derived from these variants for use in vaccine-related research (Cuevas et al., 2010; Revilla et al., 2011; Hora et al., 2016). These reagents will also be useful for studies on the biological basis of increased pathogenicity (Baeten et al., 2007; Kiwanuka et al., 2010; Li et al., 2014; Pérez-Álvarez et al., 2014; Kouri et al., 2015; Venner et al., 2016) and transmissibility (Kiwanuka et al., 2009) and diminished response to ARV drugs (Pernas et al., 2014; Cid-Silva et al., 2018) exhibited by some HIV-1 variants.
Basque Country: Hospital Universitario de Basurto, Bilbao: Josefa Muñoz, María Carmen Nieto, María Zuriñe Zubero, Silvia Hernáez-Crespo; Hospital Universitario de Cruces, Bilbao: Luis Elorduy Otazua, Leyre López Soria, Koldo Agirrebengoa; Hospital de Galdakao: María José López de Goicoechea, José Mayo; Hospital Universitario Donostia, San Sebastián: Gustavo Cilla, Julio Arrizabalaga, José Antonio Iribarren, María Jesús Bustinduy, María Julia Echevarría. Hospital Universitario de Álava, Vitoria: María Jesús Lezaun, José Joaquín Portu, Carmen Gómez-González. Galicia: Complejo Hospitalario universitario de Ferrol, Ferrol, A Coruña: Ana Mariño, Patricia Ordóñez, Hortensia Álvarez, Nieves Valcarce; Complejo Hospitalario Universitario de A Coruña: Ángeles Cañizares, María Ángeles Castro. Hospital Universitario Lucus Augusti, Lugo: María Amparo Coira, María José López-Álvarez, Ramón Rabunal; Complejo Hospitalario Universitario de Ourense: Juan García-Costa, Ricardo Fernández-Rodríguez, Raúl Rodríguez-Pérez, Jorge Guitián; Complejo Hospitalario Universitario de Vigo, Vigo, Pontevedra: Antonio Ocampo, Celia Miralles, Sonia Pérez-Castro; Complejo Hospitalario de Pontevedra: Matilde Trigo, Julio Diz-Arén, María Ángeles Pallarés. Navarra: Complejo Hospitalario de Navarra, Pamplona: Carmen Ezpeleta Baquedano, Aitziber Aguinaga, María Gracia Ruiz de Alda. Madrid: Centro Sanitario Sandoval, Madrid: Jorge del Romero, Carmen Rodríguez, Mar Vera, Óscar Ayerdi; Hospital de Fuenlabrada: María Isabel García-Arata, Santiago Prieto-Menchero; Hospital Clínico Universitario San Carlos, Madrid: Esther Culebras, Iciar Rodríguez-Avial; Fundación Jiménez Díaz, Madrid: Raquel Téllez, Miguel Górgolas, Manuel Fernández-Guerrero, Olalla Calabia, Rosa García-Delgado; Hospital Severo Ochoa, Leganés: Sara María Quevedo, Lucía Puente, Manuel Álamo; Hospital Universitario Puerta de Hierro, Majadahonda: Alfonso Alfange, Sara de la Fuente. Castilla y León: Hospital Clínico Universitario de Valladolid: Carmen Hinojosa, Carlos Dueñas, Begoña Monteagudo, Edita Sánchez; Hospital Río Hortega, Valladolid: Carmen Ramos Sánchez, Pablo Bachiller, Helmuth Guillén; Hospital Virgen de la Concha, Zamora: Teresa Martínez-Domínguez, Rosa Martínez-González. La Rioja: Hospital San Pedro: José Ramón Blanco, Miriam Blasco. Aragón: Hospital Universitario Miguel Servet, Zaragoza: Ana María Martínez-Sapiña, Diego Ortega Larrea. Castilla-La Mancha: Hospital Virgen de la Salud, Toledo: César Gómez-Hernando, José Largo-Pau; Comunitat Valenciana: Hospital Universitari Sant Joan d’Alacant: Fernando Buñuel, Ana Infante.
The datasets generated for this study can be found in GenBank, MK177651–MK177824, MK177825–MK177829, KT276258, KY496622, and KY989952.
This study was carried out in accordance with the recommendations of ’name of guidelines, name of committee’ with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Bioethics and Animal Well-being Committee of Instituto de Salud Carlos III, Majadahonda, Madrid, Spain.
MT, ED, and LP-Á conceived the study and supervised the experimental work. ED, MT, MC, AF-G, FD-F, JC, JM-L, and MS processed sequences and performed phylogenetic analyses. MT and FD-F performed phylodynamic analyses. HG performed data curation and phylogenetic analyses. SB, VM, AF-G, MS-M, EG-B, and CC performed experimental work. The members of the Spanish Group for the Study of New HIV Diagnoses recruited patients and obtained epidemiological data. MT wrote the manuscript with contributions from the other authors.
This work was funded through Acción Estratégica en Salud Intramural (AESI), Instituto de Salud Carlos III, project “Estudios sobre vigilancia epidemiológica molecular del VIH-1 en España,” PI16CIII/00033; Red de Investigación en SIDA (RIS), Instituto de Salud Carlos III, Subdirección General de Evaluación y Fondo Europeo de Desarrollo Regional (FEDER), Plan Nacional I+D+I, project RD16ISCIII/0002/0004; scientific agreements with Consellería de Sanidade, Government of Galicia (MVI 1004/16) and Osakidetza-Servicio Vasco de Salud, Government of Basque Country (MVI 1001/16); European Research Infrastructures for Poverty Related Diseases (EURIPRED). Seventh Framework Program: FP7-Capacities-infrastructures-2012-1, grant agreement 312661; and Dirección General de Farmacia, Ministerio de Sanidad, Servicios Sociales e Igualdad, Government of Spain (grant EC11-272).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank José Antonio Taboada, from Consellería de Sanidade, Xunta de Galicia, and Daniel Zulaika, from Osakidetza-Servicio Vasco de Salud, for their support of this study, and the personnel at the Genomic Unit, Instituto de Salud Carlos III, for technical assistance in sequencing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00655/full#supplementary-material
MCC, maximum clade credibility; MCMC, Markov chain Monte Carlo; ML, maximum likelihood; MRCA, most recent common ancestor; MSM, men who have sex with men; NFLG, near full-length genome; PP, posterior probability; PR-RT, protease-reverse transcriptase; PWID, people who inject drugs; tMRCA, time of most recent common ancestor.
Abecasis, A. B., Martins, A., Costa, I., Carvalho, A. P., Diogo, I., Gomes, P., et al. (2011). Molecular epidemiological analysis of paired pol/env sequences from Portuguese HIV type 1 patients. AIDS Res. Hum. Retroviruses 27, 803–805. doi: 10.1089/AID.2010.0312
Abecasis, A. B., Wensing, A. M., Paraskevis, D., Vercauteren, J., Theys, K., van de Vijver, D. A., et al. (2013). HIV-1 subtype distribution and its demographic determinants in newly diagnosed patients in Europe suggest highly compartmentalized epidemics. Retrovirology 10:7. doi: 10.1186/1742-4690-10-17
Ambrosioni, J., Junier, T., Delhumeau, C., Calmy, A., Hirschel, B., Zdobnov, E., et al. (2012). Impact of highly active antiretroviral therapy on the molecular epidemiology of newly diagnosed HIV infections. AIDS 26, 2079–2086. doi: 10.1097/QAD.0b013e32835805b6
Anisimova, M., and Gascuel, O. (2006). Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst. Biol. 55, 539–552. doi: 10.1080/10635150600755453
Apetrei, C., Loussert-Ajaka, I., Collin, G., Letourneur, F., Duca, M., Saragosti, S., et al. (1997). HIV type 1 subtype F sequences in Romanian children and adults. AIDS Res. Hum. Retroviruses 13, 363–365. doi: 10.1089/aid.1997.13.363
Audelin, A. M., Cowan, S. A., Obel, N., Nielsen, C., Jorgensen, L. B., and Gerstoft, J. (2013). Phylogenetics of the Danish HIV epidemic: the role of very late presenters in sustaining the epidemic. J. Acquir. Immune Defic. Syndr. 62, 102–108. doi: 10.1097/QAI.0b013e318276becc
Baeten, J. M., Chohan, B., Lavreys, L., Chohan, V., McClelland, R. S., Certain, L., et al. (2007). HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J. Infect. Dis. 195, 1177–1180. doi: 10.1086/512682
Bandea, C. I., Ramos, A., Pieniazek, D., Pascu, R., Tanuri, A., Schochetman, G., et al. (1995). Epidemiologic and evolutionary relationships between Romanian and Brazilian HIV-subtype F strains. Emerg. Infect. Dis. 1, 91–93. doi: 10.3201/eid0103.950305
Bartolo, I., Abecasis, A. B., Borrego, P., Barroso, H., McCutchan, F., Gomes, P., et al. (2011). Origin and epidemiological history of HIV-1 CRF14_BG. PLoS One 6:e24130. doi: 10.1371/journal.pone.0024130
Baryshev, P. B., Bogachev, V. V., and Gashnikova, N. M. (2014). HIV-1 genetic diversity in Russia: CRF63_02A1, a new HIV type 1 genetic variant spreading in Siberia. AIDS Res. Hum. Retroviruses 30, 592–597. doi: 10.1089/AID.2013.0196
Bello, G., Afonso, J. M., and Morgado, M. G. (2012). Phylodynamics of HIV-1 subtype F1 in Angola, Brazil and Romania. Infect. Genet. Evol. 12, 1079–1086. doi: 10.1016/j.meegid.2012.03.014
Bellocchi, M. C., Forbici, F., Palombi, L., Gori, C., Coelho, E., Svicher, V., et al. (2005). Subtype analysis and mutations to antiviral drugs in HIV-1-infected patients from Mozambique before initiation of antiretroviral therapy: results from the DREAM programme. J. Med. Virol. 76, 452–458. doi: 10.1002/jmv.20382
Beloukas, A., Psarris, A., Giannelou, P., Kostaki, E., Hatzakis, A., and Paraskevis, D. (2016). Molecular epidemiology of HIV-1 infection in Europe: an overview. Infect. Genet. Evol. 46, 180–189. doi: 10.1016/j.meegid.2016.06.033
Beyrer, C., Baral, S. D., van Griensven, F., Goodreau, S. M., Chariyalertsak, S., Wirtz, A. L., et al. (2012). Global epidemiology of HIV infection in men who have sex with men. Lancet 380, 367–377. doi: 10.1016/S0140-6736(12)60821-6
Bezemer, D., de Wolf, F., Boerlijst, M. C., van Sighem, A., Hollingsworth, T. D., Prins, M., et al. (2008). A resurgent HIV-1 epidemic among men who have sex with men in the era of potent antiretroviral therapy. AIDS 22, 1071–1077. doi: 10.1097/QAD.0b013e3282fd167c
Bezemer, D., van Sighem, A., Lukashov, V. V., van der Hoek, L., Back, N., Schuurman, R., et al. (2010). Transmission networks of HIV-1 among men having sex with men in the Netherlands. AIDS 24, 271–282. doi: 10.1097/QAD.0b013e328333ddee
Binley, J. M., Wrin, T., Korber, B., Zwick, M. B., Wang, M., Chappey, C., et al. (2004). Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 78, 13232–13252. doi: 10.1128/JVI.78.23.13232-13252.2004
Bobkova, M. (2013). Current status of HIV-1 diversity and drug resistance monitoring in the former USSR. AIDS Rev. 15, 204–212.
Bracho, M. A., Sentandreu, V., Alastrué, I., Belda, J., Juan, A., Fernández-García, E., et al. (2014). Emerging trends in CRF02_AG variants transmission among men who have sex with men in Spain. J. Acquir. Immune Defic. Syndr. 65, e130–e133. doi: 10.1097/01.qai.0000435602.73469.56
Brand, D., Moreau, A., Cazein, F., Lot, F., Pillonel, J., Brunet, S., et al. (2014). Characteristics of patients recently infected with HIV-1 non-B subtypes in France: a nested study within the mandatory notification system for new HIV diagnoses. J. Clin. Microbiol. 52, 4010–4016. doi: 10.1128/JCM.01141-14
Brenner, B. G., Ibanescu, R. I., Hardy, I., and Roger, M. (2017). Genotypic and phylogenetic insights on prevention of the spread of HIV-1 and drug resistance in “real-world” Settings. Viruses 10:E10. doi: 10.3390/v10010010
Cao, H., Mani, I., Vincent, R., Mugerwa, R., Mugyenyi, P., Kanki, P., et al. (2000). Cellular immunity to human immunodeficiency virus type 1 (HIV-1) clades: relevance to HIV-1 vaccine trials in Uganda. J. Infect. Dis. 182, 1350–1356. doi: 10.1086/315868
Carr, J. K., Nadai, Y., Eyzaguirre, L., Saad, M. D., Khakimov, M. M., Yakubov, S. K., et al. (2005). Outbreak of a West African recombinant of HIV-1 in Tashkent, Uzbekistan. J. Acquir. Immune Defic. Syndr. 39, 570–575.
Carvalho, A., Costa, P., Triunfante, V., Branca, F., Rodrigues, F., Santos, C. L., et al. (2015). Analysis of a local HIV-1 epidemic in Portugal highlights established transmission of non-B and non-G subtypes. J. Clin. Microbiol. 53, 1506–1514. doi: 10.1128/JCM.03611-14
Casado, C., Urtasun, I., Saragosti, S., Chaix, M. L., de Rossi, A., Cattelan, A. M., et al. (2000). Different distribution of HIV type 1 genetic variants in European patients with distinct risk practices. AIDS Res. Hum. Retroviruses 16, 299–304. doi: 10.1089/088922200309403
Castro, E., Khonkarly, M., Ciuffreda, D., Burgisser, P., Cavassini, M., Yerly, S., et al. (2010). HIV-1 drug resistance transmission networks in southwest Switzerland. AIDS Res. Hum. Retroviruses 26, 1233–1238. doi: 10.1089/aid.2010.0083
Chaillon, A., Essat, A., Frange, P., Smith, D. M., Delaugerre, C., Barin, F., et al. (2017). Spatiotemporal dynamics of HIV-1 transmission in France (1999-2014) and impact of targeted prevention strategies. Retrovirology 14:15. doi: 10.1186/s12977-017-0339-4
Chalmet, K., Staelens, D., Blot, S., Dinakis, S., Pelgrom, J., Plum, J., et al. (2010). Epidemiological study of phylogenetic transmission clusters in a local HIV-1 epidemic reveals distinct differences between subtype B and non-B infections. BMC Infect. Dis. 10:262. doi: 10.1186/1471-2334-10-262
Ciccozzi, M., Gori, C., Boros, S., Ruiz-Alvarez, M. J., Harxhi, A., Dervishi, M., et al. (2005). Molecular diversity of HIV in Albania. J. Infect. Dis. 192, 475–479. doi: 10.1086/431599
Cid-Silva, P., Margusino-Framinan, L., Balboa-Barreiro, V., Martín-Herranz, I., Castro-Iglesias, A., Pernas-Souto, B., et al. (2018). Initial treatment response among HIV subtype F infected patients who started antiretroviral therapy based on integrase inhibitors. AIDS 32, 121–125. doi: 10.1097/QAD.0000000000001679
Cuevas, M. T., Fernández-García, A., Pinilla, M., García-Álvarez, V., Thomson, M., Delgado, E., et al. (2010). Short communication: biological and genetic characterization of HIV type 1 subtype B and nonsubtype B transmitted viruses: usefulness for vaccine candidate assessment. AIDS Res. Hum. Retroviruses 26, 1019–1025. doi: 10.1089/aid.2010.0018
Cuevas, M. T., Muñoz-Nieto, M., Thomson, M. M., Delgado, E., Iribarren, J. A., Cilla, G., et al. (2009). HIV-1 transmission cluster with T215D revertant mutation among newly diagnosed patients from the Basque Country, Spain. J. Acquir. Immune Defic. Syndr. 51, 99–103. doi: 10.1097/QAI.0b013e318199063e
Dauwe, K., Mortier, V., Schauvliege, M., Van Den Heuvel, A., Fransen, K., Servais, J. Y., et al. (2015). Characteristics and spread to the native population of HIV-1 non-B subtypes in two European countries with high migration rate. BMC Infect. Dis. 15:524. doi: 10.1186/s12879-015-1217-0
de Oliveira, T., Pillay, D., and Gifford, R. J. (2010). The HIV-1 subtype C epidemic in South America is linked to the United Kingdom. PLoS One 5:e9311. doi: 10.1371/journal.pone.0009311
Delatorre, E. O., Bello, G., Eyer-Silva, W. A., Chequer-Fernandez, S. L., Morgado, M. G., and Couto-Fernandez, J. C. (2012). Evidence of multiple introductions and autochthonous transmission of the HIV type 1 CRF02_AG clade in Brazil. AIDS Res. Hum. Retroviruses 28, 1369–1372. doi: 10.1089/aid.2011.0381
Delgado, E., Cuevas, M. T., Domínguez, F., Vega, Y., Cabello, M., Fernández-García, A., et al. (2015). Phylogeny and phylogeography of a recent HIV-1 subtype F outbreak among men who have sex with men in Spain deriving from a cluster with a wide geographic circulation in Western Europe. PLoS One 10:e0143325. doi: 10.1371/journal.pone.0143325
Delgado, E., Fernández-García, A., Vega, Y., Cuevas, T., Pinilla, M., García, V., et al. (2012). Evaluation of genotypic tropism prediction tests compared with in vitro co-receptor usage in HIV-1 primary isolates of diverse subtypes. J. Antimicrob. Chemother. 67, 25–31. doi: 10.1093/jac/dkr438
Delgado, E., Thomson, M. M., Villahermosa, M. L., Sierra, M., Ocampo, A., Miralles, C., et al. (2002). Identification of a newly characterized HIV-1 BG intersubtype circulating recombinant form in Galicia, Spain, which exhibits a pseudotype-like virion structure. J. Acquir. Immune Defic. Syndr. 29, 536–543. doi: 10.1097/00126334-200204150-00016
Drummond, A. J., Rambaut, A., Shapiro, B., and Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192. doi: 10.1093/molbev/msi103
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Dumitrescu, O., Kalish, M. L., Kliks, S. C., Bandea, C. I., and Levy, J. A. (1994). Characterization of human immunodeficiency virus type 1 isolates from children in Romania: identification of a new envelope subtype. J. Infect. Dis. 169, 281–288. doi: 10.1093/infdis/169.2.281
El Moussi, A., Thomson, M. M., Delgado, E., Cuevas, M. T., Nasr, M., Abid, S., et al. (2017). Genetic diversity of HIV-1 in Tunisia. AIDS Res. Hum. Retroviruses 33, 77–81. doi: 10.1089/AID.2016.0164
Esbjörnsson, J., Mild, M., Audelin, A., Fonager, J., Skar, H., Bruun, J. L., et al. (2016). HIV-1 transmission between MSM and heterosexuals, and increasing proportions of circulating recombinant forms in the Nordic Countries. Virus Evol. 2:vew010. doi: 10.1093/ve/vew010
Esteves, A., Parreira, R., Piedade, J., Venenno, T., Franco, M., Germano, et al. (2003). Spreading of HIV-1 subtype G and envB/gagG recombinant strains among injecting drug users in Lisbon, Portugal. AIDS Res. Hum. Retroviruses 19, 511–517. doi: 10.1089/088922203766774568
Esteves, A., Parreira, R., Venenno, T., Franco, M., Piedade, J., Germano, et al. (2002). Molecular epidemiology of HIV type 1 infection in Portugal: high prevalence of non-B subtypes. AIDS Res. Hum. Retroviruses 18, 313–325. doi: 10.1089/088922202753519089
European Centre for Disease Prevention and Control (2015). ECDC Guidance: HIV and STI Prevention Among Men Who Have Sex with Men. Available at: https://ecdc.europa.eu/sites/portal/files/media/en/publications/Publications/hiv-sti-prevention-among-men-who-have-sex-with-men-guidance.pdf (accessed November 18, 2018).
European Centre for Disease Prevention and Control (2017). HIV/AIDS Surveillance in Europe. Available at: https://ecdc.europa.eu/en/publications-data/hivaids-surveillance-europe-2017-2016-data (accessed November 18, 2018).
Eyzaguirre, L. M., Erasilova, I. B., Nadai, Y., Saad, M. D., Kovtunenko, N. G., Gomatos, P. J., et al. (2007). Genetic characterization of HIV-1 strains circulating in Kazakhstan. J. Acquir. Immune Defic. Syndr. 46, 19–23. doi: 10.1097/QAI.0b013e318073c620
Fabeni, L., Alteri, C., Orchi, N., Gori, C., Bertoli, A., Forbici, F., et al. (2015). Recent transmission clustering of HIV-1 C and CRF17_BF strains characterized by NNRTI-related mutations among newly diagnosed men in central Italy. PLoS One 10:e0135325. doi: 10.1371/journal.pone.0135325
Fisher, M., Pao, D., Brown, A. E., Sudarshi, D., Gill, O. N., Cane, P., et al. (2010). Determinants of HIV-1 transmission in men who have sex with men: a combined clinical, epidemiological and phylogenetic approach. AIDS 24, 1739–1747. doi: 10.1097/QAD.0b013e32833ac9e6
Foley, B. T., Leitner, T., Paraskevis, D., and Peeters, M. (2016). Primate immunodeficiency virus classification and nomenclature: review. Infect. Genet. Evol. 46, 150–158. doi: 10.1016/j.meegid.2016.10.018
Foster, G. M., Ambrose, J. C., Hué, S., Delpech, V. C., Fearnhill, E., Abecasis, A. B., et al. (2014). Novel HIV-1 recombinants spreading across multiple risk groups in the United Kingdom: the identification and phylogeography of Circulating Recombinant Form (CRF) 50_A1D. PLoS One 9:e83337. doi: 10.1371/journal.pone.0083337
Frange, P., Meyer, L., Deveau, C., Tran, L., Goujard, C., Ghosn, J., et al. (2012). Recent HIV-1 infection contributes to the viral diffusion over the French territory with a recent increasing frequency. PLoS One 7:e31695. doi: 10.1371/journal.pone.0031695
Fransen, K., Buve, A., Nkengasong, J. N., Laga, M., and van der Groen, G. (1996). Longstanding presence in Belgians of multiple non-B HIV-1 subtypes. Lancet 347:1403. doi: 10.1016/S0140-6736(96)91042-9
Frentz, D., Wensing, A. M., Albert, J., Paraskevis, D., Abecasis, A. B., Hamouda, O., et al. (2013). Limited cross-border infections in patients newly diagnosed with HIV in Europe. Retrovirology 10:36. doi: 10.1186/1742-4690-10-36
Geldmacher, C., Currier, J. R., Gerhardt, M., Haule, A., Maboko, L., Birx, D., et al. (2007). In a mixed subtype epidemic, the HIV-1 Gag-specific T-cell response is biased towards the infecting subtype. AIDS 21, 135–143. doi: 10.1097/01.aids.0000247589.77061.f7
German, D., Grabowski, M. K., and Beyrer, C. (2017). Enhanced use of phylogenetic data to inform public health approaches to HIV among men who have sex with men. Sex Health 14, 89–96. doi: 10.1071/SH16056
Gifford, R. J., de Oliveira, T., Rambaut, A., Pybus, O. G., Dunn, D., Vandamme, A. M., et al. (2007). Phylogenetic surveillance of viral genetic diversity and the evolving molecular epidemiology of human immunodeficiency virus type 1. J. Virol. 81, 13050–13056. doi: 10.1128/JVI.00889-07
Gifford, R. J., Liu, T. F., Rhee, S. Y., Kiuchi, M., Hué, S., Pillay, D., et al. (2009). The calibrated population resistance tool: standardized genotypic estimation of transmitted HIV-1 drug resistance. Bioinformatics 25, 1197–1198. doi: 10.1093/bioinformatics/btp134
Giuliani, M., Santoro, M. M., Lo, P. A., Cella, E., Scognamiglio, P., Lai, A., et al. (2013). Circulation of HIV-1 CRF02_AG among MSM population in central Italy: a molecular epidemiology-based study. Biomed. Res. Int. 2013:810617. doi: 10.1155/2013/810617
González-Domenech, C. M., Viciana, I., Delaye, L., Mayorga, M. L., Palacios, R., de la Torre, J., et al. (2018). Emergence as an outbreak of the HIV-1 CRF19_cpx variant in treatment-naive patients in southern Spain. PLoS One 13:e0190544. doi: 10.1371/journal.pone.0190544
Grant, R. M., Lama, J. R., Anderson, P. L., McMahan, V., Liu, A. Y., Vargas, L., et al. (2010). Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N. Engl. J. Med. 363, 2587–2599. doi: 10.1056/NEJMoa1011205
Guimarães, M. L., Vicente, A. C., Otsuki, K., da Silva, R. F., Francisco, M., da Silva, F. G., et al. (2009). Close phylogenetic relationship between Angolan and Romanian HIV-1 subtype F1 isolates. Retrovirology 6:39. doi: 10.1186/1742-4690-6-39
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Guindon, S., Lethiec, F., Duroux, P., and Gascuel, O. (2005). PHYML Online–a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 33, W557–W559. doi: 10.1093/nar/gki352
Hemelaar, J., Elangovan, H., Yun, J., Dickson-Tetteh, L., Fleminger, I., Kirtley, S., et al. (2018). Global and regional molecular epidemiology of HIV-1, 1990-2015: a systematic review, global survey, and trend analysis. Lancet Infect. Dis. 19, 143–155. doi: 10.1016/S1473-3099(18)30647-9
HIV Sequence Database (2019). HIV Sequence Database. Available at: https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html (accessed February 15, 2019).
Hoenigl, M., Chaillon, A., Kessler, H. H., Haas, B., Stelzl, E., Weninger, K., et al. (2016). Characterization of HIV transmission in South-East Austria. PLoS One 11:e0151478. doi: 10.1371/journal.pone.0151478
Hora, B., Keating, S. M., Chen, Y., Sanchez, A. M., Sabino, E., Hunt, G., et al. (2016). Genetic characterization of a panel of diverse HIV-1 isolates at seven international sites. PLoS One 11:e0157340. doi: 10.1371/journal.pone.0157340
Hraber, P., Korber, B. T., Lapedes, A. S., Bailer, R. T., Seaman, M. S., Gao, H., et al. (2014). Impact of clade, geography, and age of the epidemic on HIV-1 neutralization by antibodies. J. Virol. 88, 12623–12643. doi: 10.1128/JVI.01705-14
Hué, S., Brown, A. E., Ragonnet-Cronin, M., Lycett, S. J., Dunn, D. T., Fearnhill, E., et al. (2014). Phylogenetic analyses reveal HIV-1 infections between men misclassified as heterosexual transmissions. AIDS 28, 1967–1975. doi: 10.1097/QAD.0000000000000383
Hué, S., Pillay, D., Clewley, J. P., and Pybus, O. G. (2005). Genetic analysis reveals the complex structure of HIV-1 transmission within defined risk groups. Proc. Natl. Acad. Sci. U.S.A. 102, 4425–4429. doi: 10.1073/pnas.0407534102
Hughes, G. J., Fearnhill, E., Dunn, D., Lycett, S. J., Rambaut, A., and Leigh Brown, A. J. (2009). Molecular phylodynamics of the heterosexual HIV epidemic in the United Kingdom. PLoS Pathog. 5:e1000590. doi: 10.1371/journal.ppat.1000590
Huson, D. H., and Scornavacca, C. (2012). Dendroscope 3: an interactive tool for rooted phylogenetic trees and networks. Syst. Biol. 61, 1061–1067. doi: 10.1093/sysbio/sys062
Instituto Nacional de Estadística (2018). Estadística del Padrón Continuo. Datos Provisionales a 1 de Enero de. Available at: http://www.ine.es/jaxi/Datos.htm?path=/t20/e245/p04/provi/l0/&file=00000010.px (accessed November 18, 2018).
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kiwanuka, N., Laeyendecker, O., Quinn, T. C., Wawer, M. J., Shepherd, J., Robb, M., et al. (2009). HIV-1 subtypes and differences in heterosexual HIV transmission among HIV-discordant couples in Rakai, Uganda. AIDS 23, 2479–2484. doi: 10.1097/QAD.0b013e328330cc08
Kiwanuka, N., Robb, M., Laeyendecker, O., Kigozi, G., Wabwire-Mangen, F., Makumbi, F. E., et al. (2010). HIV-1 viral subtype differences in the rate of CD4+ T-cell decline among HIV seroincident antiretroviral naive persons in Rakai district, Uganda. J. Acquir. Immune Defic. Syndr. 54, 180–184. doi: 10.1097/QAI.0b013e3181c98fc0
Kouri, V., Khouri, R., Alemán, Y., Abrahantes, Y., Vercauteren, J., Pineda-Peña, A. C., et al. (2015). CRF19_cpx is an evolutionary fit HIV-1 variant strongly associated with rapid progression to AIDS in Cuba. EBioMedicine 2, 244–254. doi: 10.1016/j.ebiom.2015.01.015
Kouyos, R. D., Rusert, P., Kadelka, C., Huber, M., Marzel, A., Ebner, H., et al. (2018). Tracing HIV-1 strains that imprint broadly neutralizing antibody responses. Nature 561, 406–410. doi: 10.1038/s41586-018-0517-0
Kuiken, C., Thakallapalli, R., Esklid, A., and de Ronde, A. (2000). Genetic analysis reveals epidemiologic patterns in the spread of human immunodeficiency virus. Am. J. Epidemiol. 152, 814–822. doi: 10.1093/aje/152.9.814
Lai, A., Bozzi, G., Franzetti, M., Binda, F., Simonetti, F. R., de Luca, A., et al. (2016). HIV-1 A1 subtype epidemic in Italy originated from Africa and Eastern Europe and shows a high frequency of transmission chains involving intravenous drug users. PLoS One 11:e0146097. doi: 10.1371/journal.pone.0146097
Lai, A., Bozzi, G., Franzetti, M., Binda, F., Simonetti, F. R., Micheli, V., et al. (2014). Phylogenetic analysis provides evidence of interactions between Italian heterosexual and South American homosexual males as the main source of national HIV-1 subtype C epidemics. J. Med. Virol. 86, 729–736. doi: 10.1002/jmv.23891
Lai, A., Simonetti, F. R., Zehender, G., de Luca, A., Micheli, V., Meraviglia, P., et al. (2012). HIV-1 subtype F1 epidemiological networks among Italian heterosexual males are associated with introduction events from South America. PLoS One 7:e42223. doi: 10.1371/journal.pone.0042223
Lapovok, I., Kazennova, E., Laga, V., Vasilyev, A., Utegenova, A., Abishev, A., et al. (2014). Short communication: molecular epidemiology of HIV type 1 infection in Kazakhstan: CRF02_AG prevalence is increasing in the southeastern provinces. AIDS Res. Hum. Retroviruses 30, 769–774. doi: 10.1089/AID.2013.0291
Lefort, V., Longueville, J. E., and Gascuel, O. (2017). SMS: smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Leoz, M., Feyertag, F., Charpentier, C., Delaugerre, C., Wirden, M., Lemee, V., et al. (2013). Characterization of CRF56_cpx, a new circulating B/CRF02/G recombinant form identified in MSM in France. AIDS 27, 2309–2312. doi: 10.1097/QAD.0b013e3283632e0c
Lewis, F., Hughes, G. J., Rambaut, A., Pozniak, A., and Leigh Brown, A. J. (2008). Episodic sexual transmission of HIV revealed by molecular phylodynamics. PLoS Med. 5:e50. doi: 10.1371/journal.pmed.0050050
Li, Y., Han, Y., Xie, J., Gu, L., Li, W., Wang, H., et al. (2014). CRF01_AE subtype is associated with X4 tropism and fast HIV progression in Chinese patients infected through sexual transmission. AIDS 28, 521–530. doi: 10.1371/journal.pmed.0050050
Liitsola, K., Ristola, M., Holmstrom, P., Salminen, M., Brummer-Korvenkontio, H., Simola, S., et al. (2000). An outbreak of the circulating recombinant form AECM240 HIV-1 in the Finnish injection drug user population. AIDS 14, 2613–2615. doi: 10.1097/00002030-200011100-00028
Little, S. J., Kosakovsky Pond, S. L., Anderson, C. M., Young, J. A., Wertheim, J. O., Mehta, S. R., et al. (2014). Using HIV networks to inform real time prevention interventions. PLoS One 9:e98443. doi: 10.1371/journal.pone.0098443
Lole, K. S., Bollinger, R. C., Paranjape, R. S., Gadkari, D., Kulkarni, S. S., Novak, N. G., et al. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160.
Louwagie, J., Delwart, E. L., Mullins, J. I., McCutchan, F. E., Eddy, G., and Burke, D. S. (1994). Genetic analysis of HIV-1 isolates from Brazil reveals presence of two distinct genetic subtypes. AIDS Res. Hum. Retroviruses 10, 561–567. doi: 10.1089/aid.1994.10.561
Lukashov, V. V., Kuiken, C. L., Vlahov, D., Coutinho, R. A., and Goudsmit, J. (1996). Evidence for HIV type 1 strains of U.S. intravenous drug users as founders of AIDS epidemic among intravenous drug users in northern Europe. AIDS Res. Hum. Retroviruses 12, 1179–1183. doi: 10.1089/aid.1996.12.1179
Magiorkinis, G., Karamitros, T., Vasylyeva, T. I., Williams, L. D., Mbisa, J. L., Hatzakis, A., et al. (2018). An innovative study design to assess the community effect of interventions to mitigate HIV epidemics using transmission-chain phylodynamics. Am. J. Epidemiol. doi: 10.1093/aje/kwy160 [Epub ahead of print].
McCormack, S., Dunn, D. T., Desai, M., Dolling, D. I., Gafos, M., Gilson, R., et al. (2016). Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet 387, 53–60. doi: 10.1016/S0140-6736(15)00056-2
Meloni, S. T., Kim, B., Sankale, J. L., Hamel, D. J., Tovanabutra, S., Mboup, S., et al. (2004). Distinct human immunodeficiency virus type 1 subtype A virus circulating in West Africa: sub-subtype A3. J. Virol. 78, 12438–12445. doi: 10.1128/JVI.78.22.12438-12445.2004
Monno, L., Brindicci, G., Lai, A., Punzi, G., Altamura, M., Simonetti, F. R., et al. (2012). An outbreak of HIV-1 BC recombinants in Southern Italy. J. Clin. Virol. 55, 370–373. doi: 10.1016/j.jcv.2012.08.014
Montavon, C., Toure-Kane, C., Liegeois, F., Mpoudi, E., Bourgeois, A., Vergne, L., et al. (2000). Most env and gag subtype A HIV-1 viruses circulating in West and West Central Africa are similar to the prototype AG recombinant virus IBNG. J. Acquir. Immune Defic. Syndr. 23, 363–374. doi: 10.1097/00126334-200004150-00001
Moskaleychik, F. F., Laga, V. Y., Delgado, E., Vega, Y., Fernández-García, A., Pérez, A., et al. (2015). [Rapid spread of the HIV-1 circular recombinant CRF02-AG in Russia and neighboring countries]. Vopr. Virusol. 60, 14–19.
Niculescu, I., Paraschiv, S., Paraskevis, D., Abagiu, A., Batan, I., Banica, L., et al. (2015). Recent HIV-1 outbreak among intravenous drug users in Romania: evidence for cocirculation of CRF14_BG and subtype F1 Strains. AIDS Res. Hum. Retroviruses 31, 488–495. doi: 10.1089/aid.2014.0189
Núñez, O., Hernando, V., and Díaz, A. (2018). Estimating the number of people living with HIV and the undiagnosed fraction in Spain in 2013. AIDS 32, 2573–2581. doi: 10.1097/QAD.0000000000001989
Op de Coul, E., van den Burg, R., Asjö, B., Goudsmit, J., Cupsa, A., et al. (2000). Genetic evidence of multiple transmissions of HIV type 1 subtype F within Romania from adult blood donors to children. AIDS Res. Hum. Retroviruses 16, 327–336. doi: 10.1089/088922200309205
Op de Coul, E. L., Lukashov, V. V., van Doornum, G. J., Goudsmit, J., and Coutinho, R. A. (1998). Multiple HIV-1 subtypes present amongst heterosexuals in Amsterdam 1988-1996: no evidence for spread of non-B subtypes. AIDS 12, 1253–1255. doi: 10.1097/00002030-199810000-00024
Oster, A. M., France, A. M., Panneer, N., Bañez Ocfemia, M. C., Campbell, E., Dasgupta, S., et al. (2018). Identifying clusters of recent and rapid HIV transmission through analysis of molecular surveillance data. J. Acquir. Immune Defic. Syndr. 79, 543–550. doi: 10.1097/QAI.0000000000001856
Oster, A. M., Wertheim, J. O., Hernandez, A. L., Bañez Ocfemia, M. C., Saduvala, N., and Hall, H. I. (2015). Using molecular HIV surveillance data to understand transmission between subpopulations in the United States. J. Acquir. Immune Defic. Syndr. 70, 444–451. doi: 10.1097/QAI.0000000000000809
Palma, A. C., Araújo, F., Duque, V., Borges, F., Paixão, M. T., and Camacho, R. (2007). Molecular epidemiology and prevalence of drug resistance-associated mutations in newly diagnosed HIV-1 patients in Portugal. Infect. Genet. Evol. 7, 391–398. doi: 10.1016/j.meegid.2007.01.009
Pandey, S. S., Cherian, S., Thakar, M., and Paranjape, R. S. (2016). Short communication: phylogenetic and molecular characterization of six full-length HIV-1 genomes from India reveals a monophyletic lineage of Indian sub-subtype A1. AIDS Res. Hum. Retroviruses 32, 489–502. doi: 10.1089/AID.2015.0207
Paraskevis, D., Beloukas, A., Stasinos, K., Pantazis, N., de Mendoza, C., and Bannert, N. (2019). HIV-1 molecular transmission clusters in nine European countries and Canada: association with demographic and clinical factors. BMC Med. 17:4. doi: 10.1186/s12916-018-1241-1
Paraskevis, D., Magiorkinis, E., Magiorkinis, G., Sypsa, V., Paparizos, V., Lazanas, M., et al. (2007). Increasing prevalence of HIV-1 subtype A in Greece: estimating epidemic history and origin. J. Infect. Dis. 196, 1167–1176. doi: 10.1086/521677
Parczewski, M., Leszczyszyn-Pynka, M., Witak-Jedra, M., Szetela, B., Gasiorowski, J., Knysz, B., et al. (2017). Expanding HIV-1 subtype B transmission networks among men who have sex with men in Poland. PLoS One 12:e0172473. doi: 10.1371/journal.pone.0172473
Patiño-Galindo, J. A., Torres-Puente, M., Bracho, M. A., Alastrué, I., Juan, A., Navarro, D., et al. (2017). The molecular epidemiology of HIV-1 in the Comunidad Valenciana (Spain): analysis of transmission clusters. Sci. Rep. 7:11584. doi: 10.1038/s41598-017-10286-1
Patiño-Galindo, J. A., Torres-Puente, M., Gimeno, C., Ortega, E., Navarro, D., Galindo, M. J., et al. (2015). Expansion of the CRF19_cpx variant in Spain. J. Clin. Virol. 69, 146–149. doi: 10.1016/j.jcv.2015.06.094
Pérez-Álvarez, L., Delgado, E., Vega, Y., Montero, V., Cuevas, T., Fernández-García, A., et al. (2014). Predominance of CXCR4 tropism in HIV-1 CRF14_BG strains from newly diagnosed infections. J. Antimicrob. Chemother. 69, 246–253. doi: 10.1093/jac/dkt305
Pérez-Parra, S., Álvarez, M., Fernández-Caballero, J. A., Pérez, A. B., Santos, J., Bisbal, O., et al. (2018). Continued propagation of the CRF19_cpx variant among HIV-positive MSM patients in Spain. J. Antimicrob. Chemother. 73, 1031–1038. doi: 10.1093/jac/dkx474
Pernas, B., Grandal, M., Mena, A., Castro-Iglesias, A., Cañizares, A., Wyles, et al. (2014). High prevalence of subtype F in newly diagnosed HIV-1 persons in northwest Spain and evidence for impaired treatment response. AIDS 28, 1837–1840. doi: 10.1097/QAD.0000000000000326
Pineda-Peña, A. C., Theys, K., Stylianou, D. C., Demetriades, I., Abecasis, A. B., and Kostrikis, L. G. (2018). HIV-1 Infection in Cyprus, the Eastern Mediterranean European frontier: a densely sampled transmission dynamics analysis from 1986 to 2012. Sci. Rep. 8:1702. doi: 10.1038/s41598-017-19080-5
Poon, A. F., Gustafson, R., Daly, P., Zerr, L., Demlow, S. E., Wong, J., et al. (2016). Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: an implementation case study. Lancet HIV 3, e231–e238. doi: 10.1016/S2352-3018(16)00046-1
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Ragonnet-Cronin, M., Hué, S., Hodcroft, E. B., Tostevin, A., Dunn, D., Fawcett, T., et al. (2018). Non-disclosed men who have sex with men in UK HIV transmission networks: phylogenetic analysis of surveillance data. Lancet HIV 5, e309–e316. doi: 10.1016/S2352-3018(18)30062-6
Ragonnet-Cronin, M., Lycett, S. J., Hodcroft, E. B., Hué, S., Fearnhill, E., Brown, A. E., et al. (2016). Transmission of non-B HIV subtypes in the United Kingdom is increasingly driven by large non-heterosexual transmission clusters. J. Infect. Dis. 213, 1410–1418. doi: 10.1093/infdis/jiv758
Rambaut, A., Lam, T. T., Max, C. L., and Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2:vew007. doi: 10.1093/ve/vew007
Ratmann, O., van Sighem, A., Bezemer, D., Gavryushkina, A., Jurriaans, S., Wensing, A., et al. (2016). Sources of HIV infection among men having sex with men and implications for prevention. Sci. Transl. Med. 8:320ra2. doi: 10.1126/scitranslmed.aad1863
Reis, M. N. G, Guimarñes, M. L., Bello, G., and Stefani, M. M. A. (2019). Identification of new HIV-1 circulating recombinant forms CRF81_cpx and CRF99_BF1 in central western brazil and of unique BF1 recombinant forms. Front. Microbiol. 10:97. doi: 10.3389/fmicb.2019.00097
Revilla, A., Delgado, E., Christian, E. C., Dalrymple, J., Vega, Y., Carrera, C., et al. (2011). Construction and phenotypic characterization of HIV type 1 functional envelope clones of subtypes G and F. AIDS Res. Hum. Retroviruses 27, 889–901. doi: 10.1089/AID.2010.0177
Seaman, M. S., Janes, H., Hawkins, N., Grandpre, L. E., Devoy, C., Giri, A., et al. (2010). Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J. Virol. 84, 1439–1452. doi: 10.1128/JVI.02108-09
Shapiro, B., Rambaut, A., and Drummond, A. J. (2006). Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 23, 7–9. doi: 10.1093/molbev/msj021
Shcherbakova, N. S., Shalamova, L. A., Delgado, E., Fernández-García, A., Vega, Y., Karpenko, L. I., et al. (2014). Short communication: molecular epidemiology, phylogeny, and phylodynamics of CRF63_02A1, a recently originated HIV-1 circulating recombinant form spreading in Siberia. AIDS Res. Hum. Retroviruses 30, 912–919. doi: 10.1089/AID.2014.0075
Sierra, M., Thomson, M. M., Ríos, M., Casado, G., de Castro, R. O., Delgado, E., et al. (2005). The analysis of near full-length genome sequences of human immunodeficiency virus type 1 BF intersubtype recombinant viruses from Chile, Venezuela and Spain reveals their relationship to diverse lineages of recombinant viruses related to CRF12_BF. Infect. Genet. Evol. 5, 209–217. doi: 10.1016/j.meegid.2004.07.010
Simonetti, F. R., Lai, A., Monno, L., Binda, F., Brindicci, G., Punzi, G., et al. (2014). Identification of a new HIV-1 BC circulating recombinant form (CRF60_BC) in Italian young men having sex with men. Infect. Genet. Evol. 23, 176–181. doi: 10.1016/j.meegid.2014.02.007
Tamalet, C., Ravaux, I., Moreau, J., Bregigeon, S., Tourres, C., Richet, H., et al. (2015). Emergence of clusters of CRF02_AG and B human immunodeficiency viral strains among men having sex with men exhibiting HIV primary infection in southeastern France. J. Med. Virol. 87, 1327–1333. doi: 10.1002/jmv.24184
Temereanca, A., Ene, L., Mehta, S., Manolescu, L., Duiculescu, D., and Ruta, S. (2013). Transmitted HIV drug resistance in treatment-naive Romanian patients. J. Med. Virol. 85, 1139–1147. doi: 10.1002/jmv.23572
Thomson, M. M., Delgado, E., Manjón, N., Ocampo, A., Villahermosa, M. L., Mariño, A., et al. (2001). HIV-1 genetic diversity in Galicia Spain: BG intersubtype recombinant viruses circulating among injecting drug users. AIDS 15, 509–516. doi: 10.1097/00002030-200103090-00010
Thomson, M. M., Fernández-García, A., Delgado, E., Vega, Y., Díez-Fuertes, F., Sánchez-Martínez, M., et al. (2012). Rapid expansion of a HIV-1 subtype F cluster of recent origin among men who have sex with men in Galicia, Spain. J. Acquir. Immune Defic. Syndr. 59, e49–e51. doi: 10.1097/QAI.0b013e3182400fc4
Thomson, M. M., and Nájera, R. (2001). Travel and the introduction of human immunodeficiency virus type 1 non-B subtype genetic forms into Western countries. Clin. Infect. Dis. 32, 1732–1737. doi: 10.1086/320764
Thomson, M. M., Pérez-Álvarez, L., and Nájera, R. (2002). Molecular epidemiology of HIV-1 genetic forms and its significance for vaccine development and therapy. Lancet Infect. Dis. 2, 461–471. doi: 10.1016/S1473-3099(02)00343-2
United Nations Population Fund, the Global Forum on MSM and HIV, United Nations Development Programme, World Health Organization, United States Agency for International Development, and World Bank (2015). Implementing Comprehensive HIV and STI Programmes with Men Who Have Sex with Men. Available at: https://www.unfpa.org/es/node/13155
Vega, Y., Delgado, E., Fernández-García, A., Cuevas, M. T., Thomson, M. M., Montero, V., et al. (2015). Epidemiological surveillance of HIV-1 transmitted drug resistance in Spain in 2004-2012: relevance of transmission clusters in the propagation of resistance mutations. PLoS One 10:e0125699. doi: 10.1371/journal.pone.0125699
Venner, C. M., Nankya, I., Kyeyune, F., Demers, K., Kwok, C., Chen, P. L., et al. (2016). Infecting HIV-1 subtype predicts disease progression in women of sub-Saharan Africa. EBioMedicine 13, 305–314. doi: 10.1016/j.ebiom.2016.10.014
Verhofstede, C., Dauwe, K., Fransen, K., Van Laethem, K., Van den Wijngaert, S., Ruelle, J., et al. (2018). Phylogenetic analysis of the Belgian HIV-1 epidemic reveals that local transmission is almost exclusively driven by men having sex with men despite presence of large African migrant communities. Infect. Genet. Evol. 61, 36–44. doi: 10.1016/j.meegid.2018.03.002
Vinken, L., Fransen, K., Pineda-Peña, A. C., Alexiev, I., Balotta, C., Debaisieux, L., et al. (2017). HIV-1 sub-subtype F1 outbreak among MSM in Belgium. Virus Evol. 3(Suppl. 1):vew036.020. doi: 10.1093/ve/vew036.020
Volk, J. E., Marcus, J. L., Phengrasamy, T., Blechinger, D., Nguyen, D. P., Follansbee, S., et al. (2015). No new HIV infections with increasing use of HIV preexposure prophylaxis in a clinical practice setting. Clin. Infect. Dis. 61, 1601–1603. doi: 10.1093/cid/civ778
von Wyl, V., Kouyos, R. D., Yerly, S., Boni, J., Shah, C., Burgisser, P., et al. (2011). The role of migration and domestic transmission in the spread of HIV-1 non-B subtypes in Switzerland. J. Infect. Dis. 204, 1095–1103. doi: 10.1093/infdis/jir491
Wang, X., Wu, Y., Mao, L., Xia, W., Zhang, W., Dai, L., et al. (2015). Targeting HIV prevention based on molecular epidemiology among deeply sampled subnetworks of men who have sex with men. Clin. Infect. Dis. 61, 1462–1468. doi: 10.1093/cid/civ526
Wertheim, J. O., Kosakovsky Pond, S. L., Little, S. J., and De Gruttola, V. (2011). Using HIV transmission networks to investigate community effects in HIV prevention trials. PLoS One 6:e27775. doi: 10.1371/journal.pone.0027775
Wertheim, J. O., Murrell, B., Mehta, S. R., Forgione, L. A., Kosakovsky Pond, S. L., Smith, D. M., et al. (2018). Growth of HIV-1 molecular transmission clusters in New York City. J. Infect. Dis. 218, 1943–1953. doi: 10.1093/infdis/jiy431
Yebra, G., de Mulder, M., Martín, L., Rodríguez, C., Labarga, P., Viciana, I., et al. (2012). Most HIV type 1 non-B infections in the Spanish cohort of antiretroviral treatment-naive HIV-infected patients (CoRIS) are due to recombinant viruses. J. Clin. Microbiol. 50, 407–413. doi: 10.1128/JCM.05798-11
Yebra, G., Frampton, D., Gallo Cassarino, T., Raffle, J., Hubb, J., Ferns, R. B., et al. (2018). A high HIV-1 strain variability in London, UK, revealed by full-genome analysis: results from the ICONIC project. PLoS One 13:e0192081. doi: 10.1371/journal.pone.0192081
Keywords: HIV-1, molecular epidemiology, phylogeny, phylodynamics, men who have sex with men, subtypes, circulating recombinant forms, clusters
Citation: Delgado E, Benito S, Montero V, Cuevas MT, Fernández-García A, Sánchez-Martínez M, García-Bodas E, Díez-Fuertes F, Gil H, Cañada J, Carrera C, Martínez-López J, Sintes M, Pérez-Álvarez L, Thomson MM and the Spanish Group for the Study of New HIV Diagnoses (2019) Diverse Large HIV-1 Non-subtype B Clusters Are Spreading Among Men Who Have Sex With Men in Spain. Front. Microbiol. 10:655. doi: 10.3389/fmicb.2019.00655
Received: 25 November 2018; Accepted: 15 March 2019;
Published: 03 April 2019.
Edited by:
Gkikas Magiorkinis, National and Kapodistrian University of Athens, GreeceReviewed by:
Apostolos Beloukas, University of West Attica, GreeceCopyright © 2019 Delgado, Benito, Montero, Cuevas, Fernández-García, Sánchez-Martínez, García-Bodas, Díez-Fuertes, Gil, Cañada, Carrera, Martínez-López, Sintes, Pérez-Álvarez, Thomson and the Spanish Group for the Study of New HIV Diagnoses. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael M. Thomson, bXRob21zb25AaXNjaWlpLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.