 Yoshiki Fujii1,2
Yoshiki Fujii1,2 Yen Hai Doan1,2
Yen Hai Doan1,2 Rury Mega Wahyuni3
Rury Mega Wahyuni3 Maria Inge Lusida3
Maria Inge Lusida3 Takako Utsumi3,4
Takako Utsumi3,4 Ikuo Shoji4
Ikuo Shoji4 Kazuhiko Katayama1,2*
Kazuhiko Katayama1,2*- 1Laboratory of Viral Infection I, Department of Infection Control and Immunology, Kitasato Institute for Life Sciences and Graduate School of Infection Control Sciences, Kitasato University, Tokyo, Japan
- 2Department of Virology II, National Institute of Infectious Diseases, Tokyo, Japan
- 3Indonesia-Japan Collaborative Research Center for Emerging and Re-emerging Infectious Diseases, Institute of Tropical Disease, Airlangga University, Surabaya, Indonesia
- 4Center for Infectious Diseases, Kobe University Graduate School of Medicine, Kobe, Japan
Rotavirus A (RVA) is a major cause of gastroenteritis in infants and young children. After vaccine introduction, RVA surveillance has become more important for monitoring changes in genotype distribution, and the semi-nested multiplex-PCR is a popular method for RVA genotyping. In particular, the VP7 primer set reported by Gouvea and colleagues in 1990 is still widely used worldwide as the recommended WHO primer set in regional and national reference RVA surveillance laboratories. However, this primer set yielded some mistakes with recent epidemic strains. The newly emerged equine-like G3 strains were mistyped as G1, G8 strains were mistyped as G3, the G9 lineage 3 strains showed very weak band, and the G9 lineage 6 strains showed a G9-specific band and a non-specific band. Gouvea’s standard protocol has become relatively unreliable for identifying genotypes correctly. To overcome this limitation, we redesigned the primer set to include recent epidemic strains. Our new primer set enabled us to correctly identify the VP7 genotypes of representative epidemic strains by agarose gel electrophoresis (G1, G2, human typical G3, equine-like G3, G4, G8, G9, and G12). We believe that the multiplex-PCR method with our new primer set is a useful and valuable tool for surveillance of RVA epidemics.
Introduction
Rotavirus A (RVA), a member of the Reoviridae family, is a major cause of gastroenteritis in infants and young children worldwide. In 2016, RVA caused 128,500 deaths in children under 5 years of age globally (Troeger et al., 2018). RVA imposes a huge burden even on developed countries including Japan (Nakagomi et al., 2013). Two live attenuated RVA vaccines, Rotarix (GlaxoSmithKline, Biologicals, Belgium) and RotaTeq (Merck & Co., Inc., United States), were introduced in Japan in November 2011 and July 2012, respectively. The vaccines are very effective (Lambert et al., 2009; Leshem et al., 2014; Karafillakis et al., 2015), but the selective pressure of vaccines may induce an epidemic strain shift.
The RVA genome has 11 gene segments of double-stranded RNA that encode six structural (VPs) and six nonstructural proteins (NSPs) (Estes and Greenberg, 2013). To classify RVAs, a specific genotype is assigned to each of the 11 genome segments, according to predefined nucleotide sequence identity cutoff values (Matthijnssens et al., 2008a,b, 2011). The classification system denotes the VP7-VP4-VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5/6 genes of an RVA strain as a descriptor Gx-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx (x indicating genotype number), respectively. Human RVA strains have been mostly classified into two major and one minor genogroups, based on the genotype constellations (Nakagomi et al., 1989). The Wa, DS-1 and AU-1 genogroups are described as G1-P[8]-I1-R1-C1-M1-A1-N1-T1-E1-H1, G2-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2, and G3-P[9]-I3-R3-C3-M3-A3-N3-T3-E3-H3, respectively (Matthijnssens et al., 2008a,b, 2011). Most G1P[8] viruses are thought to possess a Wa-like genotype constellation, and most G2P[4] viruses have a DS-1-like genotype constellation (Heiman et al., 2008).
Since 2012, the unusual DS-1-like G1P[8] strains (G1-P[8]-I2-R2-C2-M2-A2-N2-T2-E2-H2) were detected worldwide, including in Japan (Fujii et al., 2014, 2019; Komoto et al., 2015; Nakagomi et al., 2017; Yamamoto et al., 2017; Jere et al., 2018). This unusual virus has remained one of the common circulating strains in Japan (Sugimoto et al., 2017). In addition, unusual equine-like G3 strains with DS-1-like genotype constellations (G3-P[8]/P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2) have been detected in many countries, including Australia (Cowley et al., 2016), Hungary (Doro et al., 2016), Spain (Arana et al., 2016), Brazil (Guerra et al., 2016), Thailand (Komoto et al., 2016), and Japan (Malasao et al., 2015). The G8 strains, which were rare in Japan, were also detected at high rates in Hokkaido in 2014 (Kondo et al., 2017). Thus, the genotype distribution in Japan is changing since vaccine introduction.
The semi-nested multiplex-PCR is the popular method for RVA genotyping. Especially, the VP7 primer set reported by Gouvea et al. (1990) is still in use worldwide. In Japan, medical doctors ask the regional institutes of public health for tests of pathogenic agents when needed. The institutes examine specimens and report results to the National Epidemiological Surveillance of Infectious Disease (NESID) system (Taniguchi et al., 2007). For RVA, about half of the reported VP7 genotypes were based on sequencing method, and remaining half were based on the multiplex-PCR and Gouvea’s primer set (calculated from NESID data between 2015 and 2017). However, the primer sequences are hardly updated over the years, and some strains have been reported to be misjudged by this primer set (Mitui et al., 2012). Iturriza-Gomara et al. (2004) improved some of the primer sequences, but the problem of the misjudged cases was not solved, and equine-like G3 and G12 strains cannot be determined by their primer set. Esona et al. (2015) reported a new primer set for VP7 genotyping, but equine-like G3 and G8 strains cannot be determined by their primer set. Nevertheless, the original Gouvea primers are still used in Japan. Therefore, in this study, we showed some misjudged cases and elucidated the mechanisms of errors caused by using that primer set for recent epidemic RVA strains. Then we updated the primer set to make genotyping more accurate.
Materials and Methods
Sample Preparation
To evaluate multiplex-PCR methods, we selected 11 representative RVA strains (SP15-09 (Wa-like G1P[8]), NT036 (Wa-like G1P[8]), SP15-06 (DS-1-like G1P[8]), To16-04 (G2P[4]), KN105 (Wa-like G3), To16-01 (equine-like, DS-1-like G3), OH279 (G4P[8]), TA15-07 (G8P[8]), To14-25 (G9 lineage 3), To16-02 (G9 lineage 6), and NS17-5 (G12) that were already genotyped by sequencing (Supplementary Table S1). RNA extraction was performed using the Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA, United States) as described (Fujii et al., 2012, 2014).
Genotyping by Semi-Nested Multiplex-PCR
To avoid false results, we manually designed a new primer set (Table 1) by comparing the VP7 sequences of more than 200 representative RVA strains. Gouvea’s primer set and our new primer set were used for RT-PCR (first PCR) and multiplex-PCR (second PCR). Extracted RNA (1 μL corresponding to 4–106 ng of RNA and 9.5 × 10-2–9.0 × 10-8 copies of the NSP3 gene) was used for RT-PCR with a TaKaRa One-Step RNA PCR Kit (AMV) (Takara, Kyoto, Japan). Before the reaction, the RNA samples were incubated at 65°C for 5 min with the first primers (10 pmol each). The initial reverse transcription reaction was carried out at 50°C for 30 min and at 94°C for 2 min, followed by 40 cycles of amplification (30 s at 94°C, 30 s at 50°C, 90s at 72°C), with a final extension of 5 min at 72°C in a GenAmp PCR System 2700 thermal cycler (Applied Biosystems, Foster, CA, United States). The first PCR products were diluted 50-fold with DNase/RNase-free water, and the 2 μL of diluted solutions were used for a second PCR. Second PCR was performed using Premix Ex TaqTM Hot Start Version (Takara) with second primers (5 pmol each). The initial denaturation step was at 94°C for 30 s, followed by 20 cycles of amplification (30 s at 94°C, 30 s at 50°C, 60 s at 72°C), with a final extension of 5 min at 72°C. The amplicons were analyzed by electrophoresis on 1.5% agarose gels with ethidium bromide. A 100 bp DNA ladder (New England BioLabs, Ipswich, MA, United States) was used as DNA size marker.
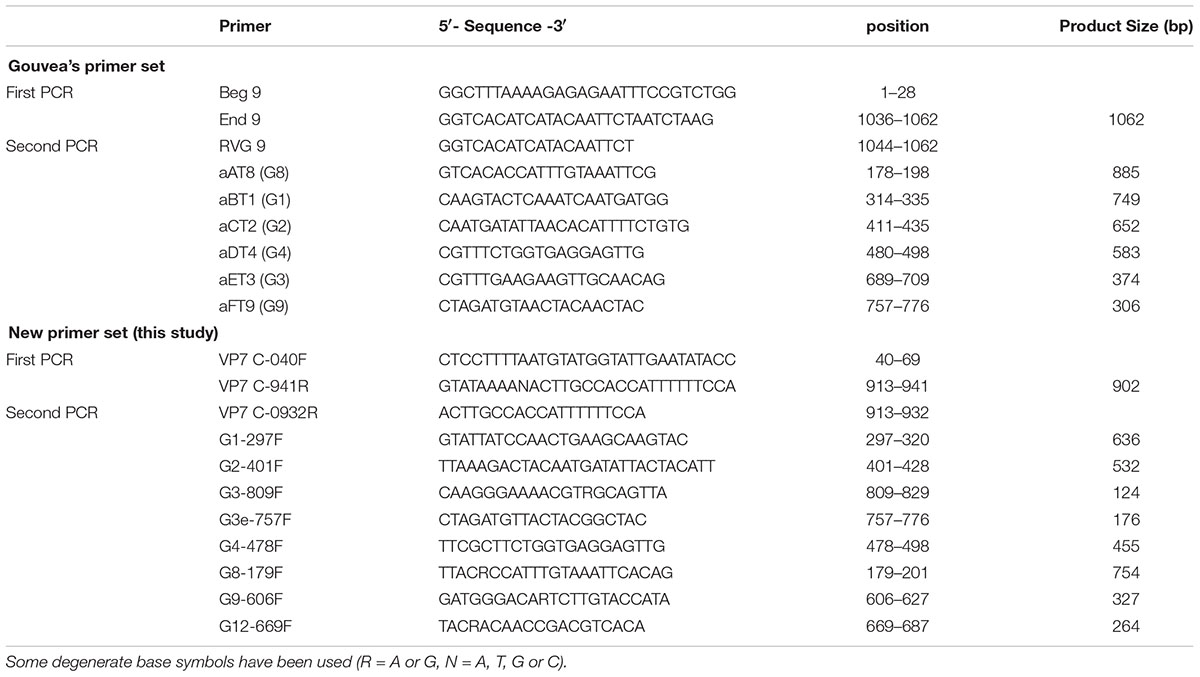
Table 1. Primer set for VP7 genotyping.>
Sequence Comparison Between Virus Strains and Primers
VP7 nucleotide sequences of representative strains were retrieved from GenBank and aligned using CLUSTAL W, which was included in the MEGA software package, version 7.0.18 and the MAFFT multiple sequence alignment software program, version 7.0 (Katoh et al., 2009). The final edit was performed with Microsoft Excel 2010 software (Microsoft Corporation, WA, United States).
Ethics Statement
The study protocols were approved by the medical research ethics committee of the National Institute of Infectious Diseases for the use of human subjects. Stool specimens were collected from patients after written informed consent was obtained from the patients or their guardians for the donation of samples.
Results
Evaluation of the Gouvea Primer Set
Ten representative RVA strains circulating in Japan (Supplementary Table S1, except for G12 strain) were selected for evaluating the Gouvea primer set. Six of 10 strains (G1 lineage 1 (Wa-like), G1 lineage 2 (Wa-like), G1 lineage 1 (DS-1-like), G2, G3 (Wa-like), and G4) were correctly genotyped by their band sizes (Figure 1A). The other four strains (equine-like G3 (DS-1-like), G8, G9 lineage 3 and G9 lineage 6) were mistyped or difficult to identify correctly (Figure 1A). Equine-like G3 strain showed an incorrect band near the size of G1. The G8 strain showed incorrect band at size of G3. The G9 lineage 3 strain showed no band or a very weak specific band that was difficult to identify correctly. In addition, the G9 lineage 6 strain showed the G9-specific band and also a weak non-specific band between sizes of G1 and G8. We confirmed the reproducibility of each failure by using more than five strains (data not shown). To better understand these errors, DNA was purified from each non-specific band and sequenced, and then the mispriming primers were determined (Supplementary Figures S1B, S2B, S3B).
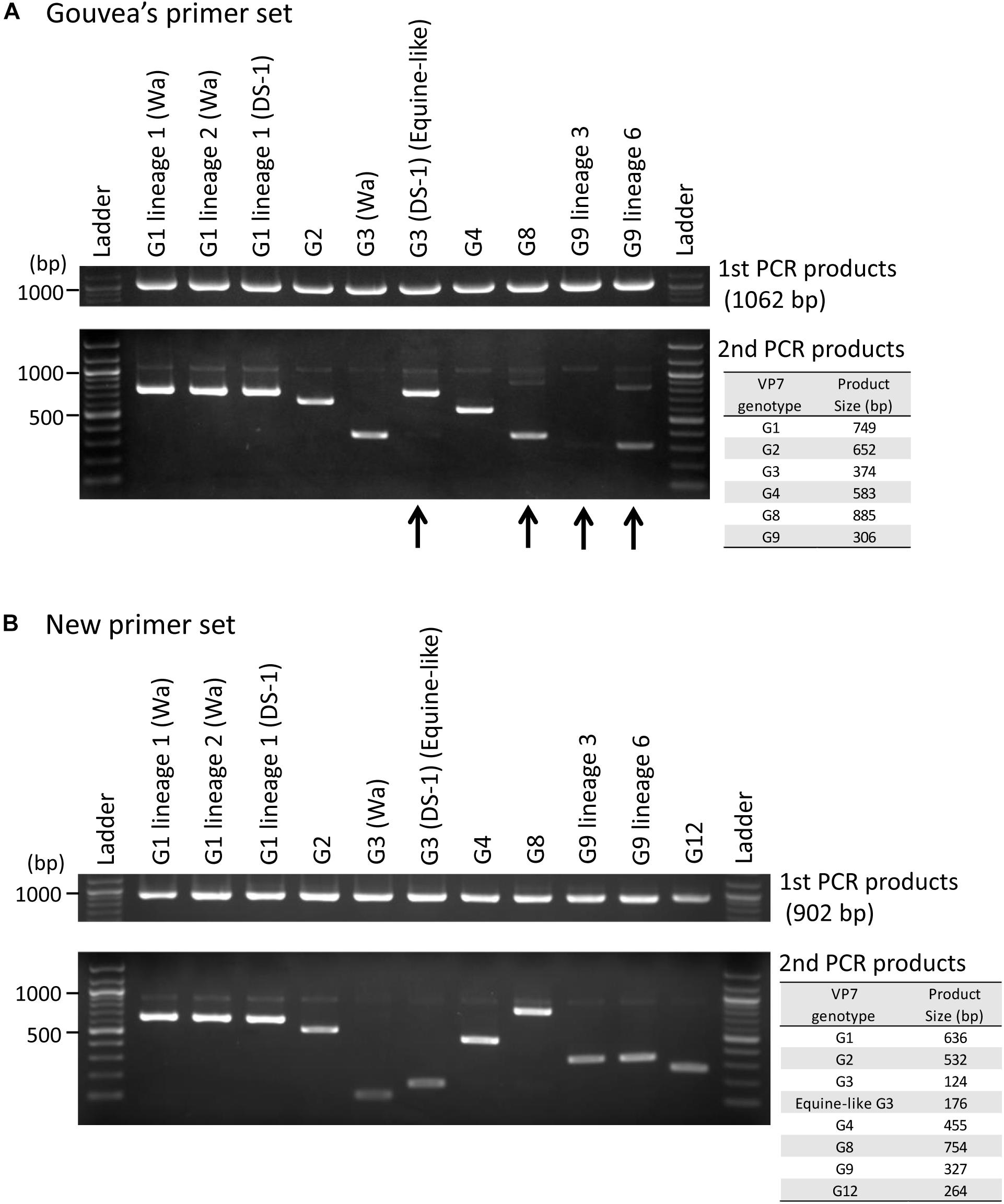
Figure 1. Evaluation of genotyping method using the Gouvea and new primer sets. Japanese representative RVA strains were evaluated by performing RT-PCR (first PCR) and multiplex-PCR (second PCR) with the Gouvea primer set (A) or our new primer set (B). The PCR products were analyzed by electrophoresis on 1.5% agarose gels. Estimated product sizes by each primer set are shown in the table to the right. Arrows indicate mistyped cases (equine-like G3 and G8), a difficult to detect case (G9 lineage 3) and a case showing a non-specific band (G9 lineage 6).
Evaluation of New Primer Set
Next, using our new primer set (Table 1), we showed that all 11 representative strains (Supplementary Table S1) could be genotyped correctly (Figure 1B). Additionally, we confirmed the reproducibility of these results by using many more strains whose VP7 genes were determined by sequencing (38 G1, 31 G2, 11 human typical G3, 48 equine-like G3, two G4, 14 G8, 32 G9, and three G12 strains, data not shown). The primer for equine-like G3 strains (DS-1-like) is different from that for human typical G3 strains (Wa-like), and these G3 strains can be distinguished by their band sizes (176 and 124 bp).
Discussion
In this study, we evaluated a semi-nested multiplex-PCR method for VP7 genotyping of RVA with the standard Gouvea primer set and our own new primer set. Of 10 representative Japanese RVA strains, four (equine-like G3 (DS-1-like), G8, G9 lineage 3 and G9 lineage 6) were mistyped or difficult to identify correctly with the Gouvea primers.
To better understand these errors, we compared the primer sequences with VP7 sequences of representative strains of each genotype. As a result, these errors were found to be caused by mispriming of genotype-specific primers (Supplementary Figures S1–S3). The Gouvea G3 primer (aET3) seems to be difficult to bind to the primer binding site (689-709) of equine-like G3 strains because of two substitutions (A705C and A708G) near the 3′ end of this site (Supplementary Figure S1A). The aET3 primer seems to prefer to bind to 292-322 region rather than 689-709 region of equine-like G3 (Supplementary Figure S1B), and induce production of a 771-bp PCR product. We designed G3-809F and G3e-757F primers as new G3 specific primers (Supplementary Figure S1C). Human typical G3 (Wa-like) strains were detected with the G3-809F primer, and equine-like G3 (DS-1-like) strains were detected with G3e-757F primer. These two primers enabled us to distinguish human typical G3 (Wa-like) strains and equine-like G3 (DS-1-like) strains by their band sizes.
For G8 primers, the Gouvea aAT8 primer was based on a 69M strain that is the prototype strain of G8 (Supplementary Figure S2A). However, most epidemic G8 strains have substitutions at the 3′ end of this primer (G198A) and are difficult to amplify with this primer (Supplementary Figure S2B). In addition, as the aET3 primer binds the nucleotides 689–709 of G8 strains, the PCR products of G3 size (374 bp) seem to be preferentially amplified with the Gouvea primer set. The new G8 primer (G8-179F) was designed by shifting three nucleotides to 3′ side and changing mismatched nucleotide (G198A) (Supplementary Figure S2C).
The Gouvea aFT9 primer was based on a WI61 strain (G9 lineage 1) that is the prototype strain of G9 (Supplementary Figure S3A). Most human G9 lineage 6 strains have two substitutions (A759T and C773A), and most G9 lineage 3 strains have three substitutions (A759T, A765G, and C773A). Thus, G9 lineage 3 strains are difficult to amplify with an aFT9 primer. Human G9 lineage 6 strains showed a non-specific band because the aAT8 primer binds the 235–258 region of G9 lineage 6 strains and generates an 825-bp amplicon (Supplementary Figure S3B). As other lineages of G9 strains have a substitution at the 3′ end of the aAT8 primer (G258A), mispriming could occur. The new G9 primer (G9-606F) enables us to detect both of lineage 3 and 6 without non-specific bands (Supplementary Figure S3C).
Our new primer set enables us to correctly identify the VP7 genotypes of most of Japanese epidemic RVA strains, including newly emerged strains (e.g., equine-like G3, G8 and G12). However, RVAs have many genotypes, and inter-species transmission and reassortment sometimes occur (Estes and Greenberg, 2013). Novel RVA strains emerge occasionally. Therefore, the primer sequences should be checked by comparing with the epidemic strains and improved as needed.
Of course, sequencing analysis is important for correct genotyping. However, a great deal of effort, time and money are required for detailed epidemiological surveillance because the genotype distributions can vary based on regions and seasons. The quantitative real-time PCR method (Liu et al., 2015) is not ideal for all institutes because of the costs. Meanwhile, the semi-nested multiplex-PCR method is low-cost and easy to introduce as a routine examination. If many regional institutes of public health can analyze RVA genotypes by multiplex-PCR method and report the results actively, it would be easy to monitor the RVA strains circulating around the country. We believe that the multiplex-PCR method using our new primer set is a useful tool for epidemiological surveillance of RVA.
Data Availability
The datasets generated for this study can be found in GenBank, LC311226, LC172271, LC311225, LC311229, LC172317, LC311227, LC311231, LC311230, LC105292, LC311228, and LC426752.
Author Contributions
YF designed new primer set, performed all experiments and was responsible for writing the manuscript. YD, RW, ML, TU, and IS gave assistance to collect samples and analyze RVA sequences. KK gave assistance for the research and helped to draft the manuscript.
Funding
This work was partially supported by the Research Program on Emerging and Re-emerging Infectious Diseases from AMED under Grant Numbers 18fk0108034j0202 and 18fk0108078j0001.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank Prof. Tsutsumi and Dr. Tsugawa, Sapporo Medical University School of Medicine, Sapporo, Japan and the staff at the hospitals for their work to collect stool specimens.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00647/full#supplementary-material
FIGURE S1 | Sequence comparison of G3 representative strains and primers. Sequences around the Gouvea aET3 primer binding site (A), non-specific binding site of aET3 primer (B), and our new primer binding sites (C) are shown. Dots indicate consensus with each primer.
FIGURE S2 | Sequence comparison of representative G8 strains and primers. Sequences around the Gouvea aAT8 primer binding site (A), non-specific binding sites of the aET3 primer (B), and our new primer binding site (C) are shown. Dots indicate consensus with each primer.
FIGURE S3 | Sequence comparison of representative G9 strains and primers. Sequences around the Gouvea aFT9 primer binding site (A), non-specific binding sites of the aAT8 primer (B), and our new primer binding sites (C) are shown. Dots indicate consensus with each primer.
TABLE S1 | The representative RVA strains used for VP7 genotyping shown in Figure 1.
References
Arana, A., Montes, M., Jere, K. C., Alkorta, M., Iturriza-Gomara, M., and Cilla, G. (2016). Emergence and spread of G3P[8] rotaviruses possessing an equine-like VP7 and a DS-1-like genetic backbone in the Basque Country (North of Spain), 2015. Infect. Genet. Evol. 44, 137–144. doi: 10.1016/j.meegid.2016.06.048
Cowley, D., Donato, C. M., Roczo-Farkas, S., and Kirkwood, C. D. (2016). Emergence of a novel equine-like G3P[8] inter-genogroup reassortant rotavirus strain associated with gastroenteritis in Australian children. J. Gen. Virol. 97, 403–410. doi: 10.1099/jgv.0.000352
Doro, R., Marton, S., Bartokne, A. H., Lengyel, G., Agocs, Z., Jakab, F., et al. (2016). Equine-like G3 rotavirus in Hungary, 2015 - Is it a novel intergenogroup reassortant pandemic strain? Acta Microbiol. Immunol. Hung. 63, 243–255. doi: 10.1556/030.63.2016.2.8
Esona, M. D., Gautam, R., Tam, K. I., Williams, A., Mijatovic-Rustempasic, S., and Bowen, M. D. (2015). Multiplexed one-step RT-PCR VP7 and VP4 genotyping assays for rotaviruses using updated primers. J. Virol. Methods 223, 96–104. doi: 10.1016/j.jviromet.2015.07.012
Estes, M. K., and Greenberg, H. B. (2013). “Rotaviruses,” in Fields Virology, eds P. M. H. David and M. Knipe (Philadelphia, PA: Lippincott Williams & Wilkins), 1347–1401.
Fujii, Y., Doan, Y. H., Suzuki, Y., Nakagomi, T., Nakagomi, O., and Katayama, K. (2019). Study of complete genome sequences of rotavirus a epidemics and evolution in japan in 2012-2014. Front. Microbiol. 10:38. doi: 10.3389/fmicb.2019.00038
Fujii, Y., Nakagomi, T., Nishimura, N., Noguchi, A., Miura, S., Ito, H., et al. (2014). Spread and predominance in Japan of novel G1P[8] double-reassortant rotavirus strains possessing a DS-1-like genotype constellation typical of G2P[4] strains. Infect. Genet. Evol. 28, 426–433. doi: 10.1016/j.meegid.2014.08.001
Fujii, Y., Shimoike, T., Takagi, H., Murakami, K., Todaka-Takai, R., Park, Y., et al. (2012). Amplification of all 11 RNA segments of group A rotaviruses based on reverse transcription polymerase chain reaction. Microbiol. Immunol. 56, 630–638. doi: 10.1111/j.1348-0421.2012.00479.x
Gouvea, V., Glass, R. I., Woods, P., Taniguchi, K., Clark, H. F., Forrester, B., et al. (1990). Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J. Clin. Microbiol. 28, 276–282.
Guerra, S. F., Soares, L. S., Lobo, P. S., Penha Junior, E. T., Sousa Junior, E. C., Bezerra, D. A., et al. (2016). Detection of a novel equine-like G3 rotavirus associated with acute gastroenteritis in Brazil. J. Gen. Virol. 97, 3131–3138. doi: 10.1099/jgv.0.000626
Heiman, E. M., McDonald, S. M., Barro, M., Taraporewala, Z. F., Bar-Magen, T., and Patton, J. T. (2008). Group A human rotavirus genomics: evidence that gene constellations are influenced by viral protein interactions. J. Virol. 82, 11106–11116. doi: 10.1128/JVI.01402-08
Iturriza-Gomara, M., Kang, G., and Gray, J. (2004). Rotavirus genotyping: keeping up with an evolving population of human rotaviruses. J. Clin. Virol. 31, 259–265. doi: 10.1016/j.jcv.2004.04.009
Jere, K. C., Chaguza, C., Bar-Zeev, N., Lowe, J., Peno, C., Kumwenda, B., et al. (2018). Emergence of double- and triple-gene reassortant G1P[8] rotaviruses possessing a DS-1-Like backbone after rotavirus vaccine introduction in malawi. J. Virol. 92, e1246–e1217. doi: 10.1128/jvi.01246-17
Karafillakis, E., Hassounah, S., and Atchison, C. (2015). Effectiveness and impact of rotavirus vaccines in Europe, 2006-2014. Vaccine 33, 2097–2107. doi: 10.1016/j.vaccine.2015.03.016
Katoh, K., Asimenos, G., and Toh, H. (2009). Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 537, 39–64. doi: 10.1007/978-1-59745-251-9-3
Komoto, S., Tacharoenmuang, R., Guntapong, R., Ide, T., Haga, K., Katayama, K., et al. (2015). Emergence and characterization of unusual DS-1-Like G1P[8] rotavirus strains in children with diarrhea in Thailand. PLoS One 10:e0141739. doi: 10.1371/journal.pone.0141739
Komoto, S., Tacharoenmuang, R., Guntapong, R., Ide, T., Tsuji, T., Yoshikawa, T., et al. (2016). Reassortment of human and animal rotavirus gene segments in emerging DS-1-Like G1P[8] rotavirus strains. PLoS One 11:e0148416. doi: 10.1371/journal.pone.0148416
Kondo, K., Tsugawa, T., Ono, M., Ohara, T., Fujibayashi, S., Tahara, Y., et al. (2017). Clinical and molecular characteristics of human rotavirus G8P[8] outbreak strain, Japan, 2014. Emerg. Infect. Dis. 23, 968–972. doi: 10.3201/eid2306.160038
Lambert, S. B., Faux, C. E., Hall, L., Birrell, F. A., Peterson, K. V., Selvey, C. E., et al. (2009). Early evidence for direct and indirect effects of the infant rotavirus vaccine program in Queensland. Med. J. Austr. 191, 157–160.
Leshem, E., Moritz, R. E., Curns, A. T., Zhou, F., Tate, J. E., Lopman, B. A., et al. (2014). Rotavirus vaccines and health care utilization for diarrhea in the United States (2007-2011). Pediatrics 134, 15–23. doi: 10.1542/peds.2013-3849
Liu, J., Lurain, K., Sobuz, S. U., Begum, S., Kumburu, H., Gratz, J., et al. (2015). Molecular genotyping and quantitation assay for rotavirus surveillance. J. Virol. Methods 213, 157–163. doi: 10.1016/j.jviromet.2014.12.001
Malasao, R., Saito, M., Suzuki, A., Imagawa, T., Nukiwa-Soma, N., Tohma, K., et al. (2015). Human G3P[4] rotavirus obtained in Japan, 2013, possibly emerged through a human-equine rotavirus reassortment event. Virus Genes 50, 129–133. doi: 10.1007/s11262-014-1135-z
Matthijnssens, J., Ciarlet, M., Heiman, E., Arijs, I., Delbeke, T., McDonald, S. M., et al. (2008a). Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 82, 3204–3219. doi: 10.1128/JVI.02257-07
Matthijnssens, J., Ciarlet, M., Rahman, M., Attoui, H., Banyai, K., Estes, M. K., et al. (2008b). Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 153, 1621–1629. doi: 10.1007/s00705-008-0155-1
Matthijnssens, J., Ciarlet, M., McDonald, S. M., Attoui, H., Banyai, K., Brister, J. R., et al. (2011). Uniformity of rotavirus strain nomenclature proposed by the rotavirus classification working group (RCWG). Arch. Virol. 156, 1397–1413. doi: 10.1007/s00705-011-1006-z
Mitui, M. T., Chandrasena, T. N., Chan, P. K., Rajindrajith, S., Nelson, E. A., Leung, T. F., et al. (2012). Inaccurate identification of rotavirus genotype G9 as genotype G3 strains due to primer mismatch. Virol. J. 9:144. doi: 10.1186/1743-422x-9-144
Nakagomi, O., Nakagomi, T., Akatani, K., and Ikegami, N. (1989). Identification of rotavirus genogroups by RNA-RNA hybridization. Mol Cell Probes 3, 251–261. doi: 10.1016/0890-8508(89)90006-6
Nakagomi, T., Kato, K., Tsutsumi, H., and Nakagomi, O. (2013). The burden of rotavirus gastroenteritis among Japanese children during its peak months: an internet survey. Jpn. J. Infect. Dis. 66, 269–275. doi: 10.7883/yoken.66.269
Nakagomi, T., Nguyen, M. Q., Gauchan, P., Agbemabiese, C. A., Kaneko, M., Do, L. P., et al. (2017). Evolution of DS-1-like G1P[8] double-gene reassortant rotavirus A strains causing gastroenteritis in children in Vietnam in 2012/2013. Arch. Virol. 162, 739–748. doi: 10.1007/s00705-016-3155-6
Sugimoto, D., Nakano, M., Inada, M., Fujitani, M., Chiba, S., and Sakai, T. (2017). Distribution of rotavirus genotypes from the 2008/2009 to 2015/2016 season in nara prefecture, Japan. Jpn. J. Infect. Dis. 70, 593–594. doi: 10.7883/yoken.JJID.2016.419
Taniguchi, K., Hashimoto, S., Kawado, M., Murakami, Y., Izumida, M., Ohta, A., et al. (2007). Overview of infectious disease surveillance system in Japan, 1999-2005. J. Epidemiol. 17, S3–S13.
Troeger, C., Khalil, I. A., Rao, P. C., Cao, S., Blacker, B. F., Ahmed, T., et al. (2018). Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 Years. JAMA Pediatr. 172, 958–965. doi: 10.1001/jamapediatrics.2018.1960
Keywords: rotavirus, genotyping, nested-PCR, multiplex-PCR, primer design, equine-like G3
Citation: Fujii Y, Doan YH, Wahyuni RM, Lusida MI, Utsumi T, Shoji I and Katayama K (2019) Improvement of Rotavirus Genotyping Method by Using the Semi-Nested Multiplex-PCR With New Primer Set. Front. Microbiol. 10:647. doi: 10.3389/fmicb.2019.00647
Received: 19 October 2018; Accepted: 14 March 2019;
Published: 29 March 2019.
Edited by:
Souvik Ghosh, Ross University School of Veterinary Medicine, Saint Kitts and NevisReviewed by:
Yashpal S. Malik, Indian Veterinary Research Institute (IVRI), IndiaMartin M. Nyaga, University of the Free State, South Africa
Copyright © 2019 Fujii, Doan, Wahyuni, Lusida, Utsumi, Shoji and Katayama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhiko Katayama, a2F0YXlhbWFAbGlzY2kua2l0YXNhdG8tdS5hYy5qcA==; a2F0YXlhbWFAbmloLmdvLmpw