 Xiaoqing Ru
Xiaoqing Ru Lihong Li
Lihong Li Chunyu Wang
Chunyu Wang- 1School of Information and Electrical Engineering, Hebei University of Engineering, Handan, China
- 2School of Computer Science and Technology, Harbin Institute of Technology, Harbin, China
The uniqueness of bacteriophages plays an important role in bioinformatics research. In real applications, the function of the bacteriophage virion proteins is the main area of interest. Therefore, it is very important to classify bacteriophage virion proteins and non-phage virion proteins accurately. Extracting comprehensive and effective sequence features from proteins plays a vital role in protein classification. In order to more fully represent protein information, this paper is more comprehensive and effective by combining the features extracted by the feature information representation algorithm based on sequence information (CCPA) and the feature representation algorithm based on sequence and structure information. After extracting features, the Max-Relevance-Max-Distance (MRMD) algorithm is used to select the optimal feature set with the strongest correlation between class labels and low redundancy between features. Given the randomness of the samples selected by the random forest classification algorithm and the randomness features for producing each node variable, a random forest method is employed to perform 10-fold cross-validation on the bacteriophage protein classification. The accuracy of this model is as high as 93.5% in the classification of phage proteins in this study. This study also found that, among the eight physicochemical properties considered, the charge property has the greatest impact on the classification of bacteriophage proteins These results indicate that the model discussed in this paper is an important tool in bacteriophage protein research.
Introduction
In the biological world, bacteriophages are ubiquitous, with different genomes and lifestyles. According to their morphology, they can be classified as either tail, tailless, or filamentous bacteriophages. According to morphology and nucleic acid, phages are classified as infect bacteria and infect archaea. The bacteriophage must be attached to a host cell for growth and reproduction (Seguritan et al., 2012), and directly affects the host population by lysing host cells. In addition, each bacteriophage is specific and greatly reduces the damage to host cells (Haq et al., 2012). Identification and classification of various bacteria can be performed based on the universality, diversity, dependence, and specificity of bacteriophages (Marks and Sharp, 2015). The structure of bacteriophages is simple, consisting of only a protein shell and genetic material (DNA or RNA) (Haq et al., 2012), making them important substances for simplifying experimental research in bioinformatics. As a bacteriophage can insert genes into host cells (Ding et al., 2014), it is an important tool for studying genetics (Cheng et al., 2018; Hu et al., 2018). Hershey (Hershey and Chase, 1952) performed biological experiments using the T2 bacteriophage and bacteria in 1952, and finally confirmed that DNA is the genetic material of bacteriophages and other organisms. The significance of this research in the development of biological science earned Hershey and coworkers the Nobel Prize in Physiology. Bacteriophage provide experimental systems and tools for the molecular biological science revolution. The bacteriophage rapid development has led to dection of basic principles of ecology and evolution. Besides, it is relatively easy to synthesize and has modular characteristic, which cater to the needs of synthetic biologists and carry out engineering research and implementation of biological function.
Bacteriophage proteins are classified into virion and non-viron proteins (Zhang et al., 2015), with most practical interest focusing on the function of bacteriophage virion proteins (Feng et al., 2013b). Therefore, bacteriophage proteins must be accurately classified and identified so that researchers can further study the structure and function of a particular bacteriophage. After the human genome project was officially launched in 1990, the number of bacteriophage protein sequences with unknown functions increased dramatically (Seguritan et al., 2012; Chen et al., 2018a). Faced with a large volume of data, traditional biological experimental methods could no longer keep up with the post-gene era (Chen W. et al., 2016; Cheng et al., 2019; Mrozek et al., 2016; Hu et al., 2018). For this reason, researchers introduced different machine learning algorithms into bacteriophage classification and prediction research. For example, Li et al. (2007) developed a support vector machine system called SynFPS that uses the gene–gene distance determined by k-means clustering to identify closely related genomes and perform gene function prediction. Using the protein appearance frequency of amino acids and information of isoelectric points, Seguritan et al. (2012) developed an artificial neural network method to classify viral structures. Feng et al. (2013b) used the main amino acid and dipeptide components as an encoding scheme, and modified a naive Bayes classifier to identify bacteriophage proteins. Ding et al. (2014) used g-gap dipeptide composition to represent protein sequence information, incremental feature selection to analyze the variance and identify the optimal feature set, and a support vector machine for classification. Zhang et al. (2015) obtained sequence feature vectors with various techniques, and then used the incremental feature selection algorithm to select the optimal feature subsets. Finally, the prediction results of individual classifiers trained in different feature spaces were integrated to produce the final classification effect. Machine learning algorithm (Robert, 2012; Stephenson et al., 2018) automatically analyze and obtain rules from data and use them to predict unknown data (Chen and Yan, 2013; Yu et al., 2015, 2016a; Chen and Huang, 2017; Chen et al., 2018h; Wang et al., 2018). This saves time and money, but the results from such algorithms are not as convincing as those from biological experiments. Therefore, it is especially important to choose an appropriate machine learning algorithm to ensure the most accurate classification results (Liu, 2017; Yao et al., 2017; Yu et al., 2017a). In a protein classification experiment, the classification effect depends largely on the feature set extracted (Zou et al., 2013; Bin et al., 2015; Mrozek et al., 2015; Jia et al., 2016; Yu et al., 2016b, 2018; Zhang et al., 2016; Huang et al., 2017; Qu et al., 2017; Jiang et al., 2018; Qiao et al., 2018; Xiong et al., 2018; Xu et al., 2018b). To date, feature extraction methods are divided into sequence-based and structure-based approaches (Huang et al., 2017; Qu et al., 2017) The feature set extraction part of this study is obtained by combining the features extracted by the two feature extraction methods.
In this study, we examined the final classification effect of the selected methods and the stability of the dataset when the feature dimension was reduced. First, to remove the imbalance in the reference dataset, CD-Hit was used to remove redundant data, resulting in a balanced dataset that contains comprehensive information and less redundancy. Pearson's correlation coefficient and three distance functions (Euclidean and cosine distances and the Tanimoto coefficient) (Zou et al., 2016) were then used to calculate the correlation between features and class labels and the redundancy between features. Finally, the optimal feature subset with the strongest correlation between features and class labels and low redundancy between features was selected. According to some recent studies(Wu et al., 2009; Yi et al., 2011; Chen and Lin, 2012; Yang et al., 2015; Yu et al., 2017b; Zhang and Liu, 2017; Xu et al., 2018a; Liu et al., 2019), the best algorithms for protein classification are support vector machines and random forest algorithms. However, support vector machines are more suitable for small sample sets in which the number of dimensions is greater than the number of samples. Thus, the random forest algorithm was used in this study. The random forest algorithm (Breiman, 2001; Yao et al., 2017) combines multiple weak classifiers to produce a final result that has higher accuracy and better generalization performance. It can achieve good results, mainly because of the random nature of the “forest,” which makes the algorithm resistant to overfitting and more precise. Finally, in terms of bacteriophage protein classification, the data set extracted by combining the features and the feature selection of the feature set have a positive impact on the protein classification effect. Our results also show that, among the eight physicochemical properties of amino acids, the charge property has the greatest influence on the classification of bacteriophage proteins. To evaluate the performance of the models used in this study, the results were compared with those given by the methods introduced in (Feng et al., 2013b; Ding et al., 2014; Zhang et al., 2015). Figure 1 shows the workflow of this study.
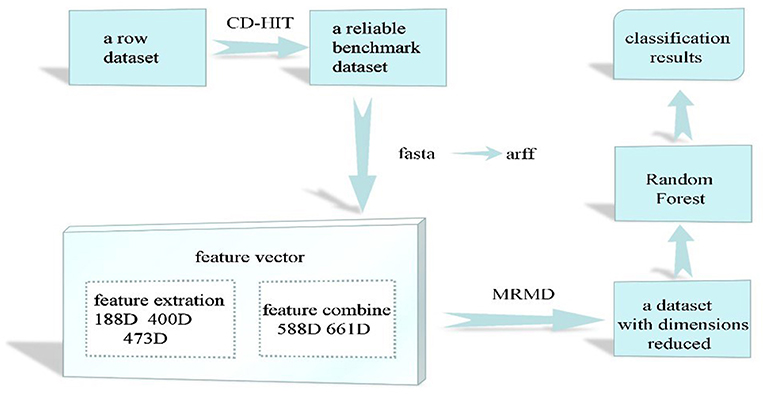
Figure 1. Outline flowchart of this study.
Methods
Dataset Processing
Source: UniProt (Rolf, 2004; Consortium, 2012) is a widely used protein sequence database that offers low protein sequence redundancy and complete protein function interpretation (Cao and Cheng, 2016a; Jiang et al., 2016). As this website is free and open, researchers can download the desired protein sequence for free. The original positive samples used in this study (a total of 15,765 data), e.g., the number of bacteriophage virion proteins, were downloaded from this database. After obtaining the bacteriophage virion protein (positive) sample set, the PFAM family of positive samples was excluded from all PFAM families, such that the remaining samples were families of non-phage virion proteins. Finally, the longest protein sequence of the remaining families was extracted to form a negative sample set. The positive and counterexample datasets obtained as described above may all contain homologous sequences. Using such sample sets would result in the classification accuracy being overestimated, which is not conducive to the establishment of prediction models. Therefore, we used the CD-Hit tool to remove redundant positive and negative samples from the datasets.
Data integration: The CD-Hit (Li et al., 2001; Li and Godzik, 2006; Huang et al., 2010; Fu et al., 2012; Chen et al., 2017) redundancy tool effectively clusters similar sequences. The basic principle is to sort protein sequences in the dataset in descending order. The longest sequence is taken as the first class, and then this is compared with the second-longest protein sequence in terms of their similarity. If the similarity between the two is greater than some threshold, they are deemed to belong to the same class. Otherwise, the second-longest sequence forms a new class. Because the bacteriophage virion protein sequences were downloaded from UniProt, which ensures relatively low redundancy, the interrupt threshold was set to 0.8. The non-phage virion proteins had a higher degree of redundancy, so their interrupt threshold was set to 0.4. Thus, 6,251 bacteriophage virion protein sequences and 9,514 non-phage virion protein sequences were obtained. The union of the resulting positive and negative sample datasets gives the total dataset, and the intersection of the two is empty.
Feature Extraction
Representation Algorithms for Amino Acid Composition and Eight Physicochemical Properties
In this study, a feature set containing 188 dimensions was extracted based on amino acid composition and eight physicochemical properties. The amino acid composition is one of the most basic features of proteins (Zhang et al., 2015; Cao and Cheng, 2016b). Eight physicochemical properties of amino acids also play a role in the functional properties of bacteriophage proteins. In 1988, Coia et al. (1988) found that amino acids having lighter side chain groups are more likely to constitute bacteriophage virion sequences. In 1994, Marvin et al. (1994) proposed that hydrophilicity, hydrophobicity, and charge have a greater impact on the function of bacteriophage virion proteins. In 2008, Shen and Chou (2008) identified the vital role that the hydrophilicity and hydrophobicity of amino acids play in the folding of proteins. In 2014, Ting et al. (2014) used logistic regression to integrate several biological features, including physicochemical properties for predicting lysine acetylation, thus demonstrating the effect of physicochemical properties on protein structure and function. Therefore, the amino acid composition and its eight physicochemical properties are used to extract features that reflect the characteristics of bacteriophage proteins.
The 20 most common amino acids are as follows:
The occurrence frequency of each amino acid in a protein sequence can be expressed as:
Where ni is the frequency with which amino acid i occurs in the protein sequence and L is the length of the protein sequence.
In addition, these 20 amino acids can be classified into three types according to their physicochemical properties (Chou and Com, 2010), as shown in Figure 2.
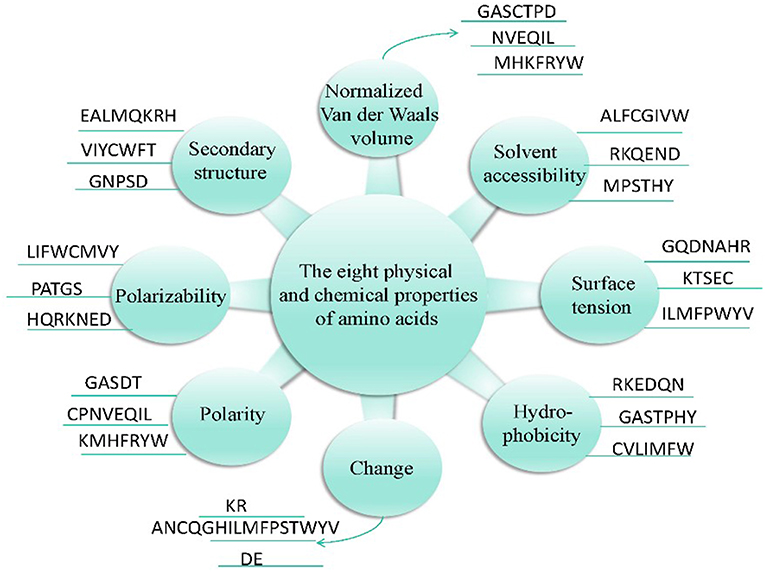
Figure 2. Eight physicochemical properties of amino acids.
The composition, transformation, and distribution of amino acids were determined by Dubchak et al. (1995) based on a global description of protein sequences. The feature extraction methods for the eight physicochemical properties of a protein sequence are as follows. Taking the electrode polarity as an example (expressed by p), the 20 amino acids are divided into high-, medium-, and low-charged polarity groups, which are expressed by ph, pp, pl, respectively. The composition, transformation, and distribution of the amino acids at this time can be represented by equations (3)–(7).
Composition features (Dubchak et al., 1995) (frequency of each charged electrode group in a sequence):
where f21, f22, f23 denote the content of the high-, medium-, and low-charged polarity groups in a sequence, respectively,L is the length of the protein sequence,n1, n2, n3 are the frequencies with which the three electrode groups appear in the sequence.
Conversion feature (Dubchak et al., 1995) (frequency of occurrence of bigeminal sequences):
Where f31, f32, f33 denote the content of the three bigeminal groups phl, php, ppl, and m1, m2, m3 are the frequencies of these three bigeminal groups appearing in sequence. There are three possible sequences of the charged polarity: phl, php, ppl In addition, in a protein sequence of length L, assuming that any two adjacent amino acids constitute a pair, the protein sequence contains L − 1 paired sequences (Zou et al., 2013).
Distribution features (Dubchak et al., 1995) (amino acid distribution of the high-, medium-, and low-charged polarity groups):
Where a1%, a25%, a50%a75%a100% represent the positions of the first, 25, 50, 75, and 100% high-charged polarity groups in a sequence, b1%, b25%, b50%, b75%, b100% represent the positions of the first, 25, 50, 75, and 100% medium-charged polarity groups in a sequence and c1%, c25%, c50%, c75%, c100% represent the positions of the first, 25, 50, 75, and 100% low-charged polarity groups in a sequence.
In summary, (3 + 3 + 3 × 5) = 21-dimensional features can be extracted from each physicochemical property, and so 8 × 21 = 168-dimensional features can be extracted from the eight physicochemical properties. The 188-dimensional features (20-dimensional + 168-dimensional) are used to express the characteristics of bacteriophage proteins, and are extracted based on the content ratio of each of the 20 amino acids in the sequence and the eight physicochemical properties.
Adaptive k-skip-n-Gram Algorithm
A feature set containing 400 dimensions is extracted based on the adaptive k-skip-n-gram method (Feng et al., 2013c; Cao et al., 2017; Wei et al., 2017a; Tang et al., 2018). In this study, the value of n was set to 2 (202 = 400).
The K value represents the separation distance between two amino acids. For example, in the protein sequence S = A1A2A3⋯AL (where L is the length of the sequence),
And Ai,Aj are the ith and jth amino acids of S.
In a bacteriophage protein dataset, the sequences have very different lengths. If the parameter K is fixed to a specific value, the sequence information cannot be properly represented, which will affect the final classification effect. Therefore, the value of k was set to be adaptive so that K could vary with the length of the sequence.
For n = 2, the combinations of the 20 most common amino acids and the number of occurrences of each combination in the sample datasets are as shown in Figure 3.
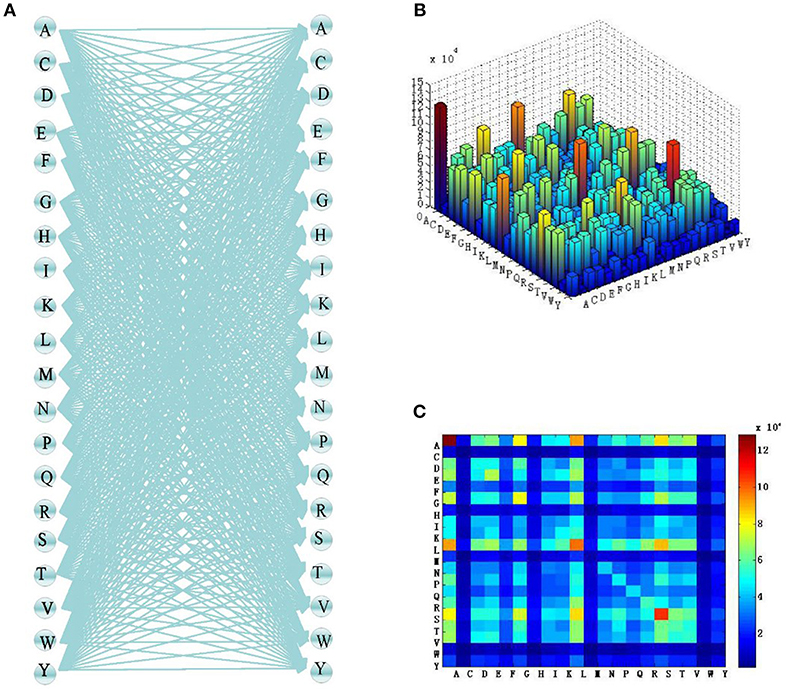
Figure 3. Two-two combination process of amino acids. (A) Two-two combination of residues. (B) Three-dimensional heat map of amino acid frequency. (C) Heat map of amino acid frequency.
This process is similar to full connection in a neural network. Among the 20 common amino acids, anyone can combine with another amino acid (or itself) in pairs, and the combination is random. In the same way as full connection, this leads to overfitting when there are too many data. Therefore, n should not be too high when using an adaptive k-skip-n-gram method. When n = 1, we have the traditional n-gram model proposed by Guthrie et al. (2006), which does not apply to shorter protein sequences. Therefore, n was set to 2 in this study.
In this feature extraction method, the combination set of two specified interval amino acids (Wei et al., 2017a) is given by:
In addition, C is used to represent a set of two amino acids that are combined at all intervals in a sequence (Wei et al., 2017a). Namely:
Finally, the feature extraction formula (Wei et al., 2017a) is:
Where N(Cskipgram) is the total number of elements in set C,am1am2⋯amn are the 20n kinds of amino acid combinations of length n, N(am1am2⋯amn) is the frequency that the two-two combination in am1am2⋯amn occurs in Cskipgram
Mixed Representation Algorithm (Seq-Str)
Some researchers have combined different feature extraction methods and achieved very good classification results (Dehzangi et al., 2013; Zou et al., 2014; Leyi et al., 2015, 2018; Chen X. et al., 2016; Ding et al., 2016, 2017a,b; Li et al., 2016; Chen et al., 2017,a,b, 2018c,d,e; Su et al., 2018 Shen et al., 2019; Wei et al., 2019; Zhu et al., 2019). Wei et al. (2015) proposed a novel feature extraction method that uses both the profile of PSI-BLAST (Altschul et al., 1997) and the profile of PSI-PRED (Jones, 1999), which contain rich evolutionary information and secondary structure information, respectively. In this way, the 473-dimensional feature can be extracted.
1) Extract 20-dimensional features based on PSI-BLAST as follows:
Sz, iindicates that during the evolution process, the residue at the “z” position in the sequence S is mutated to the fraction of the “i” species, and “i” is one of the 20 common residues. Si indicates that during the evolution, the residue in sequence S is mutated to the average score of the ith residue.
2) Extracting 420-dimensional features based on n-gram: The Adaptive k-skip-n-gram algorithm that does not consider the k value is the n-gram method. Here, take n equal to 1 and n equal to 2
3) Based on the secondary structure sequence, the following six features are extracted (Wei et al., 2015): Three feature extraction formulas for spatial arrangement
Where PHzrepresents the position index of the zth H in the secondary structure of the sequence S. nH represents the total number of occurrences of H in the secondary structure of sequence.
Two feature extraction formulas for the percentage of the maximum continuous length (Wei et al., 2015).
CH represents the length of the fragment in which H appears consecutively in the sequence of the secondary structure.
A new feature for distinguishing between two structural classes, α + β and : (Wei et al., 2015)
This formula calculates the frequency at which βαβ appears in the fragmented sequence Sseg, nβαβ represents the number of times βαβ appears in Sseg, Lsegindicates the length of Sseg.
4) Extracting 27 features based on structural probability matrices: Three features from the overall information and 24 features from local information
Feature Selection
Based on the feature extraction methods described in section Feature extraction, We extracted a 188-dimensional, 400-dimensional feature set based on sequence information, and a 473-dimensional data set based on sequence and secondary structure information representing the entire bacteriophage protein sequence dataset. Some redundant or irrelevant cases were still present in these features. The existence of invalid features wastes time and computational resources, and affects the classification accuracy of the model (Chen et al., 2018b,f,g; Dao et al., 2018; Yang et al., 2018; Zhu et al., 2018a,b). In this paper, the Max-Relevance-Max-Distance (MRMD) (Zou et al., 2016) method was used to select features and identify higher-quality feature sets, i.e., the optimal feature subset. In this method, Pearson's correlation coefficient is used to calculate the correlation between features and class labels (MR), thus enabling the selection of features with strong correlation to the target class. Three distance functions (Euclidean and cosine distances and the Tanimoto coefficient) are used to calculate the redundancy between features (MD) and identify features with low redundancy.
Taking the two eigenvectors (X,Y) as an example, Pearson's correlation coefficient (Pearson, 1909) expressed as follows:
Where σX and σY denote the standard deviation of the two vectors, cov(X, Y) is the covariance, which is used to measure the relationship between two random variables. The covariance formula is as follows:
Where and denote the mean of the respective vectors.
The formula for the Euclidean distance (Larson and Edwards, 1991; Deza and Deza, 2009) is:
Where M is the number of feature vectors,n is the total number of elements in each vector, and xq, yq are the q-th elements in X, Y, respectively.
The cosine distance formula (Tan et al., 2005) is:
Where
The Tanimoto coefficient (Rogers and Tanimoto, 1960) is given by:
Using these distance metrics, we identified the features with the strongest correlation and minimum redundancy with respect to the class labels. In different scenarios, we can increase the weights of MR and MD (max(wr × MRi + wd × MDi)) to ensure the acquired features are suitable for the classification task.
Experiments
Performance Evaluation Criteria
A 10-fold cross-validation method was employed to evaluate the models. There are four common evaluation indicators, namely the accuracy (ACC), sensitivity (SN), specificity (SP), and Matthews' correlation coefficient (MCC) (Feng et al., 2013a, 2018; Chen W. et al., 2016; Wei et al., 2017b,c; Xu et al., 2017; Jingjing et al., 2018). These are expressed as follows (Zou et al., 2013; Chen et al., 2014; Qu et al., 2017):
Where TP denotes true positive, i.e., the number of positive samples that are predicted to be positive samples, TN denotes true negative, i.e., the number of negative samples that are predicted to be negative samples, FP denotes false positive, i.e., the number of negative samples that are predicted to be positive samples, and FN denotes false negative, i.e., the number of positive samples that are predicted to be negative samples.
Classification Effects of Different Classifiers
Experiment 1: This part of the experiment is based on the feature sets of 188, 400, and 473 dimensions extracted by the method in Feature extraction. The accuracy of each classification algorithm before and after using the MRMD feature selection algorithm is presented in Table 1.
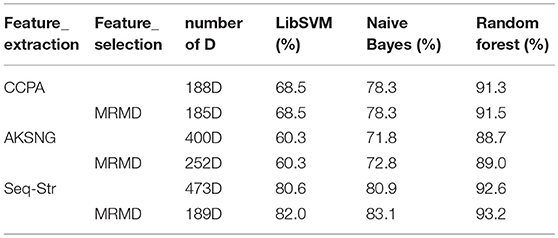
Table 1. Classification results of three data sets under different classification algorithms.
The data in Table 1 indicate that, for the classification of bacteriophage proteins, no matter which feature extraction algorithm is used, whether or not feature selection is performed, the random forest algorithm is the best classification effect.
Performance of Different Feature Extraction Methods
Experiment 2: Experiment 1 showed that the random forest algorithm produces the best classification of bacteriophage proteins. In this second experiment, the 188-dimensional and 400-dimensional datasets extracted based on sequence information (Seq Based), a 473-dimensional dataset extracted based on structure (Seq and stru Based), and two combined feature sets (Com Based) were integrated into the random forest algorithm, and the resulting performance was compared. The experimental results are presented in Table 2.
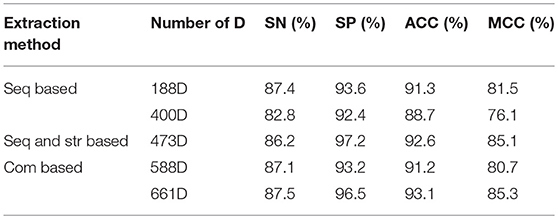
Table 2. Classification performance under different feature extraction methods.
Feature fusion can boost the recognition performance by combining the complementary information of different features (Zhu et al., 2016, 2018c). A 588-dimensional feature set was obtained by combining the features of the 188- and 400-dimensional feature sets, and a 661-dimensional feature set was obtained by combining the features of the 188- and 473-dimensional feature sets. According to the experimental results, the 188-, 473-, 588-, and 661-dimensional feature set models give better bacteriophage protein classification performance, However, based on the data of the other three evaluation indicators, the 661-dimensional feature set obtained by combining the 188-dimensional feature set extracted based on the sequence information and the features of the 473-dimensional feature set extracted based on the sequence and the secondary structure is the best. This indicates that the feature set extracted by the feature representation algorithm containing both sequence information and structural information in phage protein classification has the best influence on the classification effect, and also shows that combining some feature sets in protein classification is effective for improving classification performance.
Importance of Feature Selection
Experiment 3: This experiment used the random forest classification algorithm to classify the feature sets after MRMD. The results are given in Table 3.
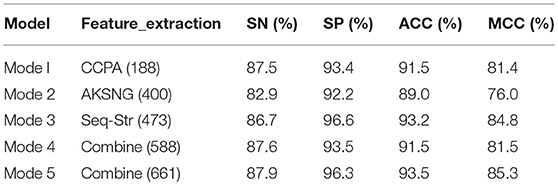
Table 3. Classification performance under each model.
The comparison of the data in Tables 2, 3 shows that after using the feature selection algorithm (MRMD), the classification effect does not change with the decrease of the dimension, and even with the decrease of the dimension, the classification effect becomes better. After removing the redundant features, the best classification performance is still the data set obtained by feature combination, that is, the 256-dimensional feature set obtained by removing redundant features from the 661-dimensional feature set.
Comparison With Recent Methods
Experiment 4: To provide an objective demonstration of the performance of the model described in this paper, this experiment compared the optimal proposed model with bacteriophage protein classification models proposed in recent years. The results are presented in Table 4.
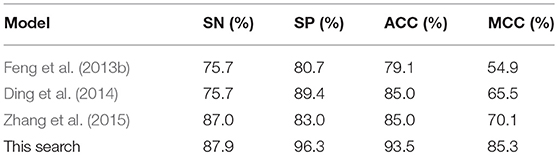
Table 4. Performance comparison against recent methods.
It is clear from Table 4 that the bacteriophage classification model proposed in this paper achieves a good classification effect, with a classification accuracy of 93.5%. Compared with Feng, it has increased by 14%, compared with Ding and Zhang by 8%. In the other three evaluation indicators, there are also different degrees of improvement, indicating that the model proposed in this paper is an effective tool for phage protein classification.
Analyzing the Impact of Eight Physicochemical Properties
This section summarizes the first eight dimensional features that have a significant impact on the classification effect of bacteriophage proteins. The top eight features are listed in Table 5 in order of their impact.
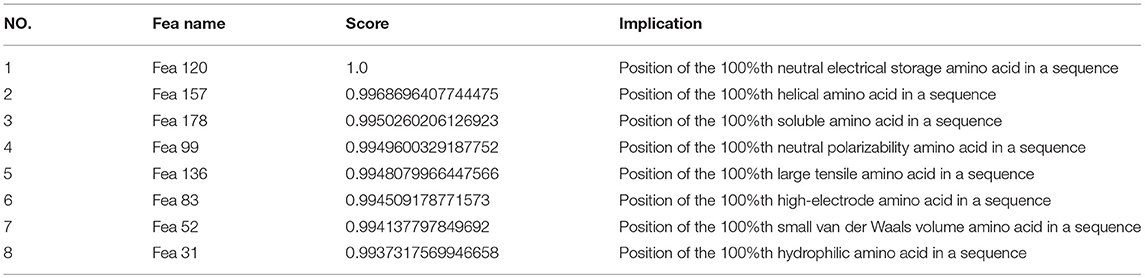
Table 5. Impact of physicochemical properties on classification.
According to the information in this table, the effects of eight physicochemical properties of amino acids on the classification of bacteriophage proteins are evenly distributed, and that which has the greatest impact on the classification is the charge property of amino acids.
Conclusion
Bacteriophage proteins are of special significance for cell typing and pathological research. It is very important to correctly classify virion and non-virion bacteriophage proteins. Therefore, this paper has proposed the following classification model: (1) higher-quality feature datasets are extracted with extraction algorithms based on feature combination; (2) the optimal feature subset is selected using the MRMD algorithm for feature selection; and (3) the random forest algorithm is applied to perform protein classification. The model can achieve accuracy of up to 93.5% for the classification of bacteriophage proteins. This demonstrates that the model developed in this paper is an important tool for the classification of bacteriophage proteins. For the future direction, link prediction paradigms, which have been successfully applied in the prediction of disease genes (Zeng et al., 2017) and miRNAs (Liu et al., 2016; Zeng et al., 2018), can be considered for identification of bacteriophage proteins. It might also be important to integrate evolutionary information using tools like evolutionary trees and networks (Yang et al., 2013, 2014). Finally, computational intelligence such as neural networks (Song et al., 2018a,b) and evolutionary algorithms (Hang et al., 2018) can be applied in this field.
Author Contributions
XR implemented the experiments and drafted the manuscript. LL and CW initiated the idea, conceived the whole process, and finalized the paper. All authors have read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported Natural Science Foundation of China (No.61872114, 91735306), and the National Key Research and Development Plan Task of China (No. 2016YFC0901902). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bin, L., Fule, L., Xiaolong, W., Junjie, C., Longyun, F., and Kuo-Chen, C. (2015). Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 43, W65–W71. doi: 10.1093/nar/gkv458
Cao, R., and Cheng, J. (2016a). Integrated protein function prediction by mining function associations, sequences, and protein-protein and gene-gene interaction networks. Methods 93, 84–91. doi: 10.1016/j.ymeth.2015.09.011
Cao, R., and Cheng, J. (2016b). Protein single-model quality assessment by feature-based probability density functions. Sci. Rep. 6:23990. doi: 10.1038/srep23990
Cao, R., Freitas, C., Chan, L., Sun, M., Jiang, H., and Chen, Z. (2017). ProLanGO: protein function prediction using neural machine translation based on a recurrent neural network. Molecules 22:1732. doi: 10.3390/molecules22101732
Chen, J., Guo, M., Wang, X., and Liu, B. (2018a). A comprehensive review and comparison of different computational methods for protein remote homology detection. Brief Bioinform. 19, 231–244. doi: 10.1093/bib/bbw108
Chen, W., Feng, P., Ding, H., and Lin, H. (2018b). Classifying included and excluded exons in exon skipping event using histone modifications. Front. Genet. 9:433. doi: 10.3389/fgene.2018.00433
Chen, W., Feng, P., Tang, H., Ding, H., and Lin, H. (2016). RAMPred: identifying the N1-methyladenosine sites in eukaryotic transcriptomes. Sci. Rep. 6:31080. doi: 10.1038/srep31080
Chen, W., Feng, P., Yang, H., Ding, H., Lin, H., and Chou, K. C. (2018c). iRNA-3typeA: identifying three types of modification at RNA's adenosine sites. Molecular therapy. Nucleic Acids 11, 468–474. doi: 10.1016/j.omtn.2018.03.012
Chen, W., Feng, P. M., Lin, H., and Chou, K. C. (2014). iSS-PseDNC: identifying splicing sites using pseudo dinucleotide composition. Biomed Res. Int. 2014, 1–12. doi: 10.1155/2014/623149
Chen, W., and Lin, H. (2012). Identification of voltage-gated potassium channel subfamilies from sequence information using support vector machine. Comput. Biol. Med. 42, 504–507. doi: 10.1016/j.compbiomed.2012.01.003
Chen, W., Yang, H., Feng, P., Ding, H., and Lin, H. (2017). iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 33, 3518–3523. doi: 10.1093/bioinformatics/btx479
Chen, X., Guan, N. N., Sun, Y. Z., Li, J. Q., and Qu, J. (2018d). MicroRNA-small molecule association identification: from experimental results to computational models. Brief. Bioinform. 2018:bby098. doi: 10.1093/bib/bby098
Chen, X., and Huang, L. (2017). LRSSLMDA: Laplacian regularized sparse subspace learning for MiRNA-disease association prediction. PLoS Comput. Biol. 13:e1005912. doi: 10.1371/journal.pcbi.1005912
Chen, X., Sun, Y. Z., Guan, N. N., Qu, J., Huang, Z. A., Zhu, Z. X., et al. (2018e). Computational models for lncRNA function prediction and functional similarity calculation. Brief Funct. Genomics 18, 58–82. doi: 10.1093/bfgp/ely031
Chen, X., Wang, L., Qu, J., Guan, N. N., and Li, J. Q. (2018f). Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 34, 4256–4265. doi: 10.1093/bioinformatics/bty503
Chen, X., Xie, D., Wang, L., Zhao, Q., You, Z. H., and Liu, H. (2018g). BNPMDA: bipartite network projection for MiRNA-disease association prediction. Bioinformatics 34, 3178–3186. doi: 10.1093/bioinformatics/bty333
Chen, X., Xie, D., Zhao, Q., and You, Z. H. (2017b). MicroRNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 2017:bbx130. doi: 10.1093/bib/bbx130
Chen, X., Yan, C. C., Zhang, X., and You, Z. H. (2017a). Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 18, 558–576. doi: 10.1093/bib/bbw060
Chen, X., Yan, C. C., Zhang, X., Zhang, X., Dai, F., Yin, J., et al. (2016). Drug-target interaction prediction: databases, web servers and computational models. Brief. Bioinform. 17, 696–712. doi: 10.1093/bib/bbv066
Chen, X., and Yan, G. Y. (2013). Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624. doi: 10.1093/bioinformatics/btt426
Chen, X., Yin, J., Qu, J., and Huang, L. (2018h). MDHGI: Matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput. Biol. 14:e1006418. doi: 10.1371/journal.pcbi.1006418
Cheng, L., Hu, Y., Sun, J., Zhou, M., and Jiang, Q. (2018). DincRNA: a comprehensive web-based bioinformatics toolkit for exploring disease associations and ncRNA function. Bioinformatics 34, 1953–1956. doi: 10.1093/bioinformatics/bty002
Cheng, L., Sun, J., Xu, W., Dong, L., Hu, Y., and Zhou, M. (2016). OAHG: an integrated resource for annotating human genes with multi-level ontologies. Sci. Rep. 6, 34820. doi: 10.1038/srep34820
Cheng, L., Yang, H., Zhao, H., Pei, X., Shi, H., Sun, J., et al. (2019). MetSigDis: a manually curated resource for the metabolic signatures of diseases. Brief. Bioinform. 20, 203–209. doi: 10.1093/bib/bbx103
Chou, K., and Com, M. P. (2010). Prediction of protein cellular attributes using pseudo-amino acid composition. Protein Struct. Funct. Bioinform. 43:246–255. doi: 10.1002/prot.1035
Coia, G., Parker, M. D., Speight, G., Byrne, M. E., and Westaway, E. G. (1988). Nucleotide and complete amino acid sequences of Kunjin virus: definitive gene order and characteristics of the virus-specified proteins. J. Gen. Virol. 69, 1–21. doi: 10.1099/0022-1317-69-1-1
Consortium, U. P. (2012). Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–D75. doi: 10.1093/nar/gkr981
Dao, F. Y., Lv, H., Wang, F., Feng, C. Q., Ding, H., Chen, W., et al. (2018). Identify origin of replication in Saccharomyces cerevisiae using two-step feature selection technique. Bioinformatics. 2018:bty943. doi: 10.1093/bioinformatics/bty943
Dehzangi, A., Paliwal, K., Sharma, A., Dehzangi, O., and Sattar, A. (2013). A combination of feature extraction methods with an ensemble of different classifiers for protein structural class prediction problem. IEEE/ACM Trans. Comput. Biol. Bioinform. 10, 564–575. doi: 10.1109/TCBB.2013.65
Deza, M. M., and Deza, E. (2009). Encyclopedia of distances. Refer. Rev. 24, 1–583. doi: 10.1007/978-3-642-00234-2
Ding, H., Feng, P. M., Chen, W., and Lin, H. (2014). Identification of bacteriophage virion proteins by the ANOVA feature selection and analysis. Mol. Biosyst. 10, 2229–2235. doi: 10.1039/C4MB00316K
Ding, Y., Tang, J., and Guo, F. (2016). Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinform. 17:398. doi: 10.1186/s12859-016-1253-9
Ding, Y., Tang, J., and Guo, F. (2017a). Identification of protein-ligand binding sites by sequence information and ensemble classifier. J. Chem. Inf. Model. 57, 3149–3161. doi: 10.1021/acs.jcim.7b00307
Ding, Y., Tang, J., and Guo, F. (2017b). Identification of drug-target interactions via multiple information integration. Inf. Sci. 418, 546–560. doi: 10.1016/j.ins.2017.08.045
Dubchak, I., Muchnik, I., Holbrook, S. R., and Kim, S. H. (1995). Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. U.S.A. 92, 8700–8704. doi: 10.1073/pnas.92.19.8700
Feng, C. Q., Zhang, Z. Y., Zhu, X. J., Lin, Y., Chen, W., Tang, H., et al. (2018). iTerm-PseKNC: a sequence-based tool for predicting bacterial transcriptional terminators. Bioinformatics. 2018:bty827. doi: 10.1093/bioinformatics/bty827
Feng, P. M., Chen, W., Lin, H., and Chou, K. C. (2013a). iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 442, 118–125. doi: 10.1016/j.ab.2013.05.024
Feng, P. M., Ding, H., Chen, W., and Lin, H. (2013b). Naïve Bayes classifier with feature selection to identify phage virion proteins. Comput. Math. Methods Med. 2013:530696. doi: 10.1155/2013/530696
Feng, P. M., Lin, H., and Chen, W. (2013c). Identification of antioxidants from sequence information using naïve Bayes. Comput. Math. Methods Med. 2013, 1–5. doi: 10.1155/2013/567529
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Guthrie, D., Allison, B., Liu, W., Guthrie, L., and Wilks, Y. (2006). “A closer look at skip-gram modelling,” in Proceedings of the 5th International Conference on Language Resources and Evaluation (Genoa: LREC), 1–4.
Hang, X., Zeng, W., Zeng, X., and Yen, G. G. (2018). An evolutionary algorithm based on minkowski distance for many-objective optimization. IEEE Trans. Cybern. 99, 1–12. doi: 10.1109/TCYB.2018.2856208
Haq, I. U., Chaudhry, W. N., Akhtar, M. N., Andleeb, S., and Qadri, I. (2012). Bacteriophages and their implications on future biotechnology: a review. Virol. J., 9:9. doi: 10.1186/1743-422X-9-9
Hershey, A. D., and Chase, M. (1952). Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol. 36, 39–56. doi: 10.1085/jgp.36.1.39
Hu, Y., Zhao, T., Zhang, N., Zang, T., Zhang, J., and Cheng, L. (2018). Identifying diseases-related metabolites using random walk. BMC Bioinform. 19(Suppl. 5):116. doi: 10.1186/s12859-018-2098-1
Huang, L., Li, X., Guo, P., Yao, Y., Liao, B., Zhang, W., et al. (2017). Matrix completion with side information and its applications in predicting the antigenicity of influenza viruses. Bioinformatics 33, 3195–3201. doi: 10.1093/bioinformatics/btx390
Huang, Y., Niu, B., Gao, Y., Fu, L., and Li, W. (2010). CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics 26, 680–682. doi: 10.1093/bioinformatics/btq003
Jia, J., Liu, Z., Xiao, X., Liu, B., and Chou, K. C. (2016). iPPBS-Opt: a sequence-based ensemble classifier for identifying protein-protein binding sites by optimizing imbalanced training datasets. Molecules 21:95. doi: 10.3390/molecules21010095
Jiang, S., Liu, B., and Zou, Q. (2018). HITS-PR-HHblits: protein remote homology detection by combining PageRank and hyperlink-induced topic search. Brief. Bioinform. 2018:bby104. doi: 10.1093/bib/bby104
Jiang, Y., Oron, T. R., Clark, W. T., Bankapur, A. R., D'Andrea, D., Lepore, R., et al. (2016). An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 17:184. doi: 10.1186/s13059-016-1037-6
Jingjing, H., Ting, F., Zizheng, Z., Bei, H., Xiaolei, Z., and Yi, X. (2018). PseUI: Pseudouridine sites identification based on RNA sequence information. BMC Bioinform. 19:306. doi: 10.1186/s12859-018-2321-0
Jones, D. T. (1999). Protein secondary structure prediction based on position-specific scoring matrices11Edited by G. Von Heijne. J. Mol. Biol. 292, 195–202. doi: 10.1006/jmbi.1999.3091
Larson, R. E., and Edwards, B. H. (1991). Elementary Linear Algebra. 2nd Edn. Lexington, MA: D.C. Heath and Company.
Leyi, W., Huangrong, C., and Ran, S. (2018). M6APred-EL: a sequence-based predictor for identifying N6-methyladenosine sites using ensemble learning. Mol. Ther. 2018, 635–644. doi: 10.1016/j.omtn.2018.07.004
Leyi, W., Minghong, L., Xing, G., and Quan, Z. (2015). An improved protein structural classes prediction method by incorporating both sequence and structure information. IEEE Trans. Nanobiosci. 14, 339–349. doi: 10.1109/TNB.2014.2352454
Li, J., Halgamuge, S. K., Kells, C. I., and Tang, S. L. (2007). Gene function prediction based on genomic context clustering and discriminative learning: an application to bacteriophages. BMC Bioinform. 8:S6. doi: 10.1186/1471-2105-8-S4-S6
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, W., Jaroszewski, L., and Godzik, A. (2001). Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 17, 282–283. doi: 10.1093/bioinformatics/17.3.282
Li, Z., Tang, J., and Guo, F. (2016). Learning from real imbalanced data of 14-3-3 proteins binding specificity. Neurocomputing 217, 83–91. doi: 10.1016/j.neucom.2016.03.093
Liu, B. (2017). BioSeq-analysis: a platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2017:bbx165. doi: 10.1093/bib/bbx165
Liu, Y., Wang, X., and Liu, B. (2019). A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief. Bioinform. 20, 330–346. doi: 10.1093/bib/bbx126
Liu, Y., Zeng, X., He, Z., and Zou, Q. (2016). Inferring microRNA-disease associations by random walk on a heterogeneous network with multiple data sources. IEEE/ACM Trans. Comput. Biol. Bioinform. 14, 905–915. doi: 10.1109/TCBB.2016.2550432
Marks, T., and Sharp, R. (2015). Bacteriophages and biotechnology: a review. J. Chem. Technol. Biotechnol. 75, 6–17. doi: 10.1002/(SICI)1097-4660(200001)75:1<6::AID-JCTB157>3.0.CO;2-A
Marvin, D. A., Hale, R. D., Nave, C., and Helmer-Citterich, M. (1994). Molecular models and structural comparisons of native and mutant class I filamentous bacteriophages Ff (fd, f1, M13), If1 and IKe. J. Mol. Biol. 235, 260–286. doi: 10.1016/S0022-2836(05)80032-4
Mrozek, D., Daniłowicz, P., and Małysiak-Mrozek, B. (2016). HDInsight4PSi: Boosting performance of 3D protein structure similarity searching with HDInsight clusters in Microsoft Azure cloud. Inf. Sci. 349, 77–101. doi: 10.1016/j.ins.2016.02.029
Mrozek, D., Socha, B., Kozielski, S., and Małysiak-Mrozek, B. (2015). An efficient and flexible scanning of databases of protein secondary structures. J. Intell. Inf. Syst. 46, 213–233. doi: 10.1007/s10844-014-0353-0
Pearson, K. (1909). Determination of the coefficient of correlation. Science 30, 23–25. doi: 10.1126/science.30.757.23
Qiao, Y., Xiong, Y., Gao, H., Zhu, X., and Chen, P. (2018). Protein-protein interface hot spots prediction based on a hybrid feature selection strategy. BMC Bioinform. 19:14. doi: 10.1186/s12859-018-2009-5
Qu, K., Han, K., Wu, S., Wang, G., and Wei, L. (2017). Identification of DNA-binding proteins using mixed feature representation methods. Molecules 22:E1602. doi: 10.3390/molecules22101602
Robert, C. (2012). Machine learning, a probabilistic perspective. Chance 27, 62–63. doi: 10.1080/09332480.2012.726570
Rogers, D. J., and Tanimoto, T. T. (1960). A computer program for classifying plants. Science 132, 1115–1118. doi: 10.1126/science.132.3434.1115
Rolf, A. (2004). UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32, D115–D119. doi: 10.1093/nar/gkh131
Seguritan, V., Alves, N. Jr., Arnoult, M., Raymond, A., Lorimer, D., Burgin, A. B. Jr., et al. (2012). Artificial neural networks trained to detect viral and phage structural proteins. PLoS Comput. Biol. 8:e1002657. doi: 10.1371/journal.pcbi.1002657
Shen, H. B., and Chou, K. C. (2008). PseAAC: A flexible web server for generating various kinds of protein pseudo amino acid composition. Anal. Biochem. 373, 386–388. doi: 10.1016/j.ab.2007.10.012
Shen, Y., Tang, J., and Guo, F. (2019). Identification of protein subcellular localization via integrating evolutionary and physicochemical information into Chou's general PseAAC. J. Theor. Biol. 462, 230–239. doi: 10.1016/j.jtbi.2018.11.012
Song, T., Rodríguez-Patón, A., Zheng, P., and Zeng, X. (2018a). Spiking neural P systems with colored spikes. IEEE Trans. Cogn. Dev. Syst. 10, 1106–1115. doi: 10.1109/TCDS.2017.2785332
Song, T., Zeng, X., Zheng, P., Jiang, M., and Rodríguez-Patón, A. (2018b). A parallel workflow pattern modeling using spiking neural p systems with colored spikes. IEEE Trans. Nanobiosci. 17, 474–484. doi: 10.1109/TNB.2018.2873221
Stephenson, N., Shane, E., Chase, J., Rowland, J., Ries, D., Justice, N., et al. (2018). Survey of machine learning techniques in drug discovery. Curr. Drug Metab. doi: 10.2174/1389200219666180820112457. [Epub ahead of print].
Su, R., Wu, H., Xu, B., Liu, X., and Wei, L. (2018). Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data IEEE/ACM Trans. Comput. Biol. Bioinform. doi: 10.1109/TCBB.2018.2858756. [Epub ahead of print].
Tan, P. N., Steinbach, M., and Kumar, V. (2005). Introduction to Data Mining, Boston, MA: Pearson Addison Wesley.
Tang, H., Zhao, Y. W., Zou, P., Zhang, C. M., Chen, R., Huang, P., et al. (2018). HBPred: a tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 14, 957–964. doi: 10.7150/ijbs.24174
Ting, H., Guangyong, Z., Pingyu, Z., Jia, J., Jing, L., Lu, X., et al. (2014). LAceP: lysine acetylation site prediction using logistic regression classifiers. PLoS ONE 9:e89575. doi: 10.1371/journal.pone.0089575
Wang, P., Zhu, W., Liao, B., Cai, L., Peng, L., and Yang, J. (2018). Predicting influenza antigenicity by matrix completion with antigen and antiserum similarity. Front. Microbiol. 9:2500. doi: 10.3389/fmicb.2018.02500
Wei, L., Liao, M., Gao, X., and Zou, Q. (2015). Enhanced protein fold prediction method through a novel feature extraction technique. IEEE Trans. Nanobiosci. 14, 649–659. doi: 10.1109/TNB.2015.2450233
Wei, L., Su, R., Wang, B., Li, X., Zou, Q., and Gao, X. (2019). Integration of deep feature representations and handcrafted features to improve the prediction of N6-methyladenosine sites. Neurocomputing 324, 3–9. doi: 10.1016/j.neucom.2018.04.082
Wei, L., Tang, J., and Zou, Q. (2017a). SkipCPP-Pred: an improved and promising sequence-based predictor for predicting cell-penetrating peptides. BMC Genomics 18:742. doi: 10.1186/s12864-017-4128-1
Wei, L., Wan, S., Guo, J., and Wong, K. K. (2017b). A novel hierarchical selective ensemble classifier with bioinformatics application. Artif. Intell. Med, 83, 82–90. doi: 10.1016/j.artmed.2017.02.005
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017c). Improved prediction of protein-protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Wu, L. C., Lee, J. X., Huang, H. D., Liu, B. J., and Horng, J. T. (2009). An expert system to predict protein thermostability using decision tree. Expert Syst. Appl. 36, 9007–9014. doi: 10.1016/j.eswa.2008.12.020
Xiong, Y., Wang, Q., Yang, J., Zhu, X., and Wei, D.-Q. (2018). PredT4SE-stack: prediction of bacterial type IV secreted effectors from protein sequences using a stacked ensemble method. Front. Microbiol. 9:2571. doi: 10.3389/fmicb.2018.02571
Xu, L., Liang, G., Shi, S., and Liao, C. (2018a). SeqSVM: a sequence-based support vector machine method for identifying antioxidant proteins. Int. J. Mol. Sci. 19:1773.
Xu, L., Liang, G., Wang, L., and Liao, C. (2018b). A novel hybrid sequence-based model for identifying anticancer peptides. Genes 9:E158. doi: 10.3390/genes9030158
Xu, Q., Xiong, Y., Dai, H., Kumari, K. M., Xu, Q., Ou, H. Y., et al. (2017). PDC-SGB: Prediction of effective drug combinations using a stochastic gradient boosting algorithm. J. Theor. Biol. 417, 1–7. doi: 10.1016/j.jtbi.2017.01.019
Yang, H., Lv, H., Ding, H., Chen, W., and Lin, H. (2018). iRNA-2OM: a sequence-based predictor for identifying 2'-O-Methylation sites in homo sapiens. J. Comput. Biol. 25, 1266–1277. doi: 10.1089/cmb.2018.0004
Yang, J., Grunewald, S., and Wan, X. F. (2013). Quartet-net: a quartet-based method to reconstruct phylogenetic networks. Mol. Biol. Evol. 30, 1206–1217. doi: 10.1093/molbev/mst040
Yang, J., Grünewald, S., Xu, Y., and Wan, X.-F. (2014). Quartet-based methods to reconstruct phylogenetic networks. BMC Syst. Biol. 8, 21–21. doi: 10.1186/1752-0509-8-21
Yang, R., Zhang, C., Gao, R., and Zhang, L. (2015). An ensemble method with hybrid features to identify extracellular matrix proteins. PLoS ONE 10:e0117804. doi: 10.1371/journal.pone.0117804
Yao, Y., Li, X., Liao, B., Huang, L., He, P., Wang, F., et al. (2017). Predicting influenza antigenicity from Hemagglutintin sequence data based on a joint random forest method. Sci. Rep. 7:1545. doi: 10.1038/s41598-017-01699-z
Yi, X., Juan, L., and Dong-Qing, W. (2011). An accurate feature-based method for identifying DNA-binding residues on protein surfaces. Proteins Struct. Funct. Bioinform. 79, 509–517. doi: 10.1002/prot.22898
Yu, L., Huang, J., Ma, Z., Zhang, J., Zou, Y., and Gao, L. (2015). Inferring drug-disease associations based on known protein complexes. BMC Med. Genomics 8:S2. doi: 10.1186/1755-8794-8-S2-S2
Yu, L., Ma, X., Zhang, L., Zhang, J., and Gao, L. (2016a). Prediction of new drug indications based on clinical data and network modularity. Sci. Rep. 6:32530. doi: 10.1038/srep32530
Yu, L., Su, R., Wang, B., Zhang, L., Zou, Y., Zhang, J., et al. (2017a). Prediction of novel drugs for hepatocellular carcinoma based on multi-source random walk. IEEE/ACM Trans. Comput. Biol. Bioinform. 14, 966–977. doi: 10.1109/TCBB.2016.2550453
Yu, L., Wang, B., Ma, X., and Gao, L. (2016b). The extraction of drug-disease correlations based on module distance in incomplete human interactome. BMC Syst. Biol. 10:111. doi: 10.1186/s12918-016-0364-2
Yu, L., Zhao, J., and Gao, L. (2017b). Drug repositioning based on triangularly balanced structure for tissue-specific diseases in incomplete interactome. Artif. Intell. Med. 77, 53–63. doi: 10.1016/j.artmed.2017.03.009
Yu, L., Zhao, J., and Gao, L. (2018). Predicting Potential Drugs for Breast Cancer based on miRNA and Tissue Specificity. Int. J. Biol. Sci. 14, 971–982. doi: 10.7150/ijbs.23350
Zeng, X., Ding, N., Rodríguez-Patón, A., and Zou, Q. (2017). Probability-based collaborative filtering model for predicting gene disease associations. BMC Med. Genomics 10:76. doi: 10.1186/s12920-017-0313-y
Zeng, X., Liu, L., Lü, L., and Zou, Q. (2018). Prediction of potential disease-associated microRNAs using structural perturbation method. Bioinformatics 34, 2425–2432. doi: 10.1093/bioinformatics/bty112
Zhang, J., Ju, Y., Lu, H., Xuan, P., and Zou, Q. (2016). Accurate Identification of cancerlectins through hybrid machine learning technology. Int. J. Genomics 2016, 1–11. doi: 10.1155/2016/7604641
Zhang, J., and Liu, B. (2017). PSFM-DBT: identifying DNA-binding proteins by combing position specific frequency matrix and distance-bigram transformation. Int. J. Mol. Sci. 18:E1856. doi: 10.3390/ijms18091856
Zhang, L., Zhang, C., Gao, R., and Yang, R. (2015). An ensemble method to distinguish bacteriophage virion from non-virion proteins based on protein sequence characteristics. Int. J. Mol. Sci. 16, 21734–21758. doi: 10.3390/ijms160921734
Zhu, P., Hu, Q., Han, Y., Zhang, C., and Du, Y. (2016). Combining neighborhood separable subspaces for classification via sparsity regularized optimization. Inf. Sci. 370, 270–287. doi: 10.1016/j.ins.2016.08.004
Zhu, P., Hu, Q., Hu, Q., Zhang, C., and Feng, Z. (2018c). Multi-view label embedding. Pattern Recognit. 84, 126–135. doi: 10.1016/j.patcog.2018.07.009
Zhu, P., Xu, Q., Hu, Q., and Zhang, C. (2018a). Co-regularized unsupervised feature selection. Neurocomputing 275, 2855–2863. doi: 10.1016/j.neucom.2017.11.061
Zhu, P., Xu, Q., Hu, Q., Zhang, C., and Zhao, H. (2018b). Multi-label feature selection with missing labels. Pattern Recognit. 74, 488–502. doi: 10.1016/j.patcog.2017.09.036
Zhu, X.-J., Feng, C.-Q., Lai, H.-Y., Chen, W., and Hao, L. (2019). Predicting protein structural classes for low-similarity sequences by evaluating different features. Knowledge Based Syst. 163, 787–793. doi: 10.1016/j.knosys.2018.10.007
Zou, Q., Li, X. B., Jiang, W. R., Lin, Z. Y., Li, G. L., and Chen, K. (2014). Survey of MapReduce frame operation in bioinformatics. Brief. Bioinform. 15, 637–647. doi: 10.1093/bib/bbs088
Zou, Q., Wang, Z., Guan, X., Liu, B., Wu, Y., and Lin, Z. (2013). An approach for identifying cytokines based on a novel ensemble classifier. Biomed Res. Int. 2013:686090. doi: 10.1155/2013/686090
Keywords: phage virion proteins, machine learning, feature extraction, feature selection, hybrid sequence features
Citation: Ru X, Li L and Wang C (2019) Identification of Phage Viral Proteins With Hybrid Sequence Features. Front. Microbiol. 10:507. doi: 10.3389/fmicb.2019.00507
Received: 24 December 2018; Accepted: 27 February 2019;
Published: 26 March 2019.
Edited by:
Hongsheng Liu, Liaoning University, ChinaReviewed by:
Zhiwei Ji, University of Texas Health Science Center, United StatesNuria Quiles Puchalt, University of Glasgow, United Kingdom
Copyright © 2019 Ru, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunyu Wang, chunyu@hit.edu.cn