 William L. King1,2
William L. King1,2 Nachshon Siboni2
Nachshon Siboni2 Nathan L. R. Williams1
Nathan L. R. Williams1 Tim Kahlke2
Tim Kahlke2 Khue Viet Nguyen1,2Cheryl Jenkins3Michael Dove4
Khue Viet Nguyen1,2Cheryl Jenkins3Michael Dove4 Wayne O’Connor4
Wayne O’Connor4 Justin R. Seymour2
Justin R. Seymour2 Maurizio Labbate1*
Maurizio Labbate1*- 1The School of Life Sciences, University of Technology Sydney, Ultimo, NSW, Australia
- 2Climate Change Cluster, University of Technology Sydney, Ultimo, NSW, Australia
- 3NSW Department of Primary Industries, Elizabeth Macarthur Agricultural Institute, Menangle, NSW, Australia
- 4NSW Department of Primary Industries, Port Stephens Fisheries Institute, Port Stephens, NSW, Australia
Oyster diseases are a major impediment to the profitability and growth of the oyster aquaculture industry. In recent years, geographically widespread outbreaks of disease caused by ostreid herpesvirus-1 microvariant (OsHV-1 μvar) have led to mass mortalities among Crassostrea gigas, the Pacific Oyster. Attempts to minimize the impact of this disease have been largely focused on breeding programs, and although these have shown some success in producing oyster families with reduced mortality, the mechanism(s) behind this protection is poorly understood. One possible factor is modification of the C. gigas microbiome. To explore how breeding for resistance to OsHV-1 μvar affects the oyster microbiome, we used 16S rRNA amplicon sequencing to characterize the bacterial communities associated with 35 C. gigas families, incorporating oysters with different levels of susceptibility to OsHV-1 μvar disease. The microbiomes of disease-susceptible families were significantly different to the microbiomes of disease-resistant families. OTUs assigned to the Photobacterium, Vibrio, Aliivibrio, Streptococcus, and Roseovarius genera were associated with low disease resistance. In partial support of this finding, qPCR identified a statistically significant increase of Vibrio-specific 16S rRNA gene copies in the low disease resistance families, possibly indicative of a reduced host immune response to these pathogens. In addition to these results, examination of the core microbiome revealed that each family possessed a small core community, with OTUs assigned to the Winogradskyella genus and the Bradyrhizobiaceae family consistent members across most disease-resistant families. This study examines patterns in the microbiome of oyster families exhibiting differing levels of OsHV-1 μvar disease resistance and reveals some key bacterial taxa that may provide a protective or detrimental role in OsHV-1 μvar disease outbreaks.
Introduction
The Pacific oyster, Crassostrea gigas is a globally cultivated oyster species, but the cultivation of this species has been increasingly impacted by disease events (Azéma et al., 2015). These disease events are largely caused by viral and bacterial etiological agents (Friedman et al., 2005; Garnier et al., 2007; Segarra et al., 2010; King et al., 2019), but in some instances no clear etiological agent is identifiable (Go et al., 2017; King et al., 2018). A major pathogen of C. gigas is the ostreid herpesvirus 1 (OsHV-1), and its micro variant form (OsHV-1 μvar) (Davison et al., 2005; Segarra et al., 2010). This virus has caused severe mortality outbreaks over the last two decades (Friedman et al., 2005; Burge et al., 2006; Segarra et al., 2010; Jenkins et al., 2013; Mortensen et al., 2016), with some outbreaks resulting in over 90% mortality and leading to the death of many millions of oysters (ASI, 2015).
To combat the impact of OsHV-1 μvar, a variety of approaches including modifying husbandry practices (e.g., increased oyster cultivation height) and breeding disease resistant oysters have been applied, with varying degrees of success (Dégremont, 2011; Paul-Pont et al., 2013; Whittington et al., 2015). Breeding programs generally involve breeding oyster genetic lines that have greater survival rates following exposure to OsHV-1 (Dégremont, 2011, 2013; Dégremont et al., 2015b; Camara et al., 2017). While these breeding programs have shown some success, resistant families still experience varying degrees of mortality (juvenile oysters 5–19%; larvae up to 86%) (Dégremont, 2011; Dégremont et al., 2013, 2016b), and the mechanism(s) underpinning resistance are often not easily distinguishable.
A number of studies have characterized the physiological and immunological factors driving OsHV-1 and OsHV-1 μvar resistance in selectively bred oysters (Sauvage et al., 2009; Dégremont, 2011, 2013; Azéma et al., 2015; Dégremont et al., 2015a, 2016a,b). Factors such as increased oyster size and weight are associated with increased resistance to infection, but why this occurs is currently unclear (Dégremont, 2013; Dégremont et al., 2015b). Other studies have determined that resistant oysters have a greater capacity to clear OsHV-1 from their tissues and supress virus replication (Dégremont, 2011; Segarra et al., 2014). When examined from an immunologic perspective, resistant oysters appear to have greater capacity to induce autophagy genes when infected by OsHV-1 compared to susceptible oysters (Moreau et al., 2015).
Another contributing factor that has received less attention is the oyster microbiome. A previous study has shown that despite being positive for OsHV-1 μvar, antibiotic-treated oysters displayed significantly reduced mortalities in comparison to untreated oysters (Petton et al., 2015). Furthermore, the total bacterial load, including the Vibrio community, is significantly elevated following OsHV-1 μvar infection and this elevation is necessary to cause mortality (de Lorgeril et al., 2018). Vibrio bacteria are commonly isolated from OsHV-1 infected oysters (Segarra et al., 2010; Garcia et al., 2011; Jenkins et al., 2013; Keeling et al., 2014), with a recent study concluding that OsHV-1 μvar infection causes immune-suppression of the oyster host, allowing opportunistic bacteria (including Vibrio species) to infect the oyster (de Lorgeril et al., 2018). In other organisms, studies have implicated the host microbiome as modulating the immune system, suggesting it is critical in host defense and overall health (reviewed by Shreiner et al., 2015) and in influencing host behavior (Shin et al., 2011).
To better understand how breeding for OsHV-1 μvar disease resistance affects the C. gigas microbiome and to elucidate whether specific taxa are associated with susceptibility and resistance, we examined the microbiome of 35 C. gigas families with varying degrees of disease-resistance. To remove the confounding effects of time and location, these oysters were deployed at a single location and sampled at the same time. In addition, the comparison of distinct C. gigas families provided the opportunity to determine whether they harbored distinct microbial community assemblages and whether persistent bacterial taxa (core microbiome) common across the families could be identified.
Materials and Methods
Sources and Sampling of C. gigas
Australian Seafood Industries (ASI) is an oyster aquaculture industry-owned company that since the first OsHV-1 μvar outbreak in 2010 has been breeding C. gigas families for OsHV-1 μvar disease resistance through field exposure. In 2016, ASI deployed 35 (n = 35) 5th generation families (5 consecutive years of biparental breeding) of juvenile C. gigas into three areas known to harbor the OsHV-1 virus, the Georges River (NSW, Australia; 34.035S, 151.145E), Pipe Clay Lagoon (TAS, Australia; 42.970S, 147.525E) and Pittwater (TAS, Australia; 42.802S, 147.509E) (Kube et al., 2018). Based on these field disease-exposure studies, expected breeding values (EBVs) were calculated by ASI. These EBVs are an estimation of how well the oysters will perform for a particular trait and the likelihood of passing those traits to their progeny. For the purposes of this study, families were classified into ‘resistance groups’ (RG) based on their OsHV-1 μvar disease resistance EBV. Families with an EBV greater than 0.6 were placed into RG1 (high disease-resistance), those with an EBV greater than 0.3 and less than 0.6 were placed into RG2 (medium disease-resistance), and families with an EBV less than 0.3 were placed into RG3 (low disease-resistance) (Table 1). The estimated heritability is the likelihood of the offspring demonstrating a particular trait, in this case OsHV-1 μvar disease resistance. Resistance is determined by the combination of many genes, since the stock used are derived from a number of genetically distinct families, each family differs in its resistance, and crosses between families differ.
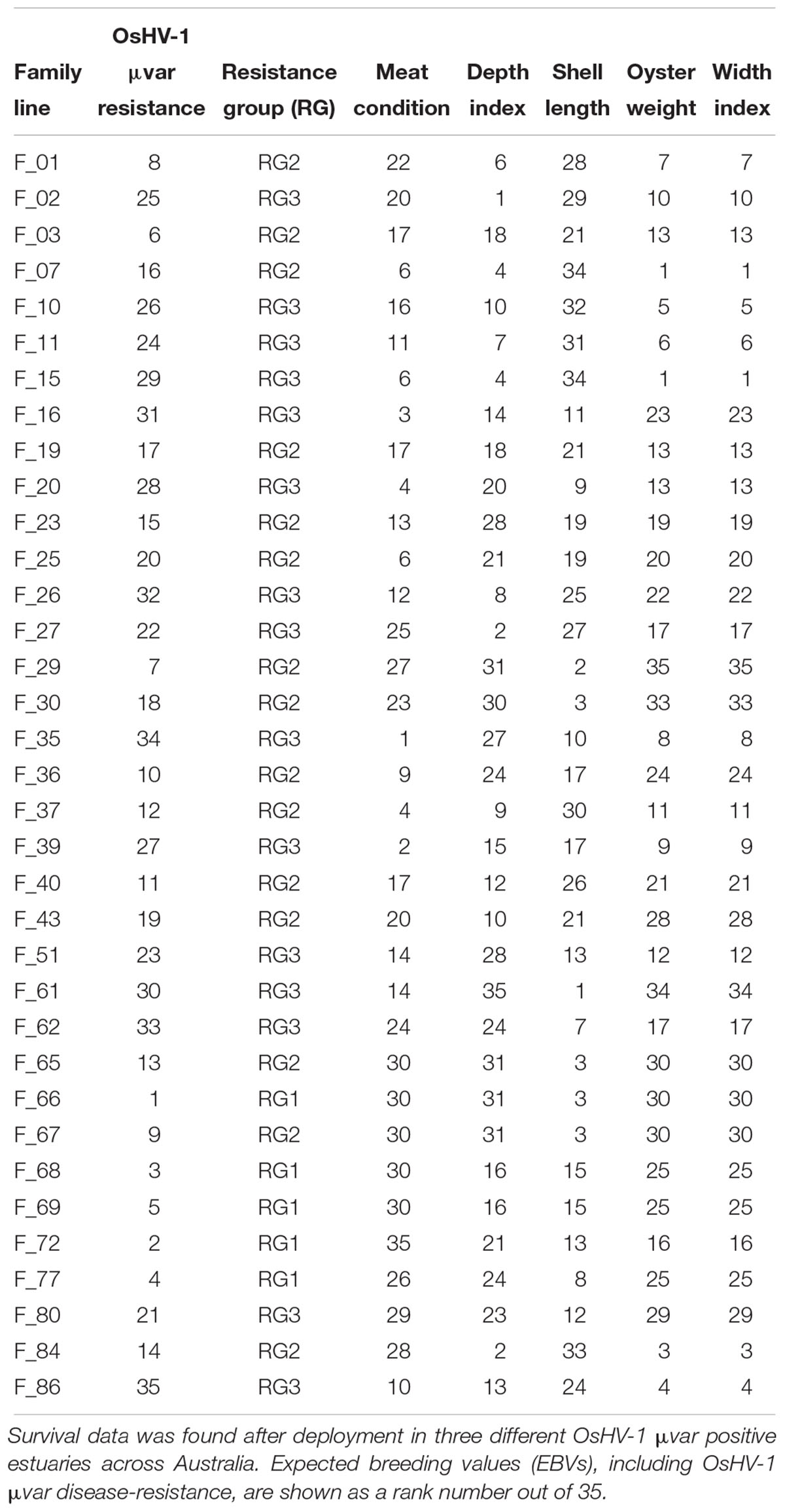
Table 1. Expected breeding value ranks for the studied oyster families including OsHV-1 μvar disease-resistance.
In addition to disease-resistance, EBVs of other oyster traits were also provided by ASI. These traits include: meat condition, the ratio of wet meat to the total weight; depth index, the ratio of shell depth to shell length; shell length; oyster weight, including the oyster shell; and width index, the ratio of shell width to shell length. As EBV’s are proprietary information, rather than providing absolute values for each index, we generated a ‘rank’ system to categorize families according to each index, with ranks of 1 being the highest (Table 1).
For this microbiome study, the families were deployed into the Georges River (34.035S, 151.145E) on the 16th of August 2016 and sampled 2 months after deployment date. The 2-month deployment time was the first opportunity to sample the deployed juvenile oysters and was sufficient time to ensure no evidence of disease or morbidity. Oysters were deployed in a resolvable incomplete block design to account for micro-geographic variation, blocks were subsections of a replicate and there were three replicates for each family, with each family stocked into a subsection of the tray (Kube et al., 2018). Five oysters from each of the 35 families (total = 175 samples) were sampled and immediately placed on ice and transported to the laboratory where they were stored at -80°C until further processing.
DNA Extraction, Sequencing, and Bioinformatics
The outer shell of the five sampled oysters was rinsed under running tap water to remove any remaining mud and debris. Defrosted oysters were then shucked with sterilized shucking knifes and approximately 25 mg of adductor muscle tissue was aseptically removed using sterile scalpel blades.
The Qiagen DNeasy blood and tissue kit (catalogue: 69506) was used to extract DNA samples, as per the manufacturer’s instructions. Microbial community composition within samples was subsequently assessed using 16S rRNA amplicon sequencing, whereby the ribosomal 16S rRNA V1–V3 region was targeted using the 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 519R (5′-GWATTACCGCGGCKGCTG-3′) primer pair. The PCR cycling conditions were as follows: 95°C for 3 min, 25 cycles of 95°C for 30 s, 55°C for 30 s and 72°C for 30 s, and a final extension at 72°C for 5 min. Amplicons were sequenced using the Illumina MiSeq platform (2 × 300 bp) using standard approaches (Ramaciotti Centre for Genomics at the University of New South Wales, Sydney, NSW, Australia). Raw data files in FASTQ format were deposited in the NCBI Sequence Read Archive (SRA) under the Bioproject number PRJNA497763.
Briefly, 16S rRNA paired-end DNA sequences were joined using Flash (Magoc and Salzberg, 2011) and subsequently trimmed using Mothur (Schloss et al., 2009) (Parameters: maxhomop = 5, maxambig = 0, minlength = 432, maxlength = 506). The resulting fragments were clustered into operational taxonomic units (OTUs) at 97% sequence identity, and chimeric sequences were identified using vsearch (Rognes et al., 2016). Taxonomy was assigned in QIIME (Caporaso et al., 2010) using the uclust algorithm (Edgar, 2010) against the Silva v128 database. Mitochondrial and chloroplast data were filtered out of the dataset and the remaining data were rarefied to allow for even coverage across all samples (Supplementary Data Sheet 2). OTUs representing less than 0.1% relative abundance in an individual sample were also filtered from the dataset (Supplementary Tables 1, 2).
Core Microbiome Analysis
To determine whether a core oyster microbiome could be characterized, we examined the microbiome of oysters at three different thresholds. First, for individual families, then for RGs, then for all samples together. A core OTU was defined as an OTU that was present in at least all but one replicate (to account for outliers) within a family. To achieve this, the panbiom.py script was used as detailed in Kahlke (2017). Briefly, the final biom file generated during the QIIME analysis was used in conjunction with a treatment file that identifies which samples are replicates within a family. The panbiom.py arguments were as follows: a replicate threshold of 1 (-r parameter) and an outlier threshold of ‘x’ (-x parameter). The -x parameter treats the replicate threshold value as an outlier threshold value, simply put, it can be absent in one replicate sample (indicated by -r = 1 and -x = x).
Quantitative PCR (qPCR)
Due to the potential role of Vibrio in OsHV-1 μvar disease dynamics (Segarra et al., 2010; Jenkins et al., 2013; Lemire et al., 2015; Petton et al., 2015; de Lorgeril et al., 2018), quantitative PCR (qPCR) was used to examine patterns in Vibrio abundance across the RGs. qPCR was performed using an epMotion 5075l Automated Liquid Handling System on a Bio-Rad CFX384 Touch Real-Time PCR Detection System with a six-point calibration curve and negative controls on every plate. The calibration curve was built from a known amount of amplicon DNA measured by Qubit, followed by a 10-fold dilution to fill out the calibration curve. All sample analyses were performed with three technical replicates, using the following reaction mixture: 2.5 μL iTaq Universal SYBR Green supermix, 0.4 μM of each forward and reverse primer, 1 μL of diluted (1:15) template DNA, and the remainder made up with water. To quantify abundance of the Vibrio community, the Vibrio-specific 16S rRNA primers Vib1-f (5′-GGCGTAAAGCGCATGCAGGT-3′) and Vib2-r (5′-GAAATTCTACCCCCCTCTACAG-3′) were used (Thompson et al., 2004; Vezzulli et al., 2011; Siboni et al., 2016). The qPCR cycling conditions were as follows: 95°C for 3 min followed by 45 cycles of 95°C for 15 s and 60°C for 1 min. The resulting data were normalized to both elution volume (200 μL) and tissue weight. A coefficient of variation (CV) was then calculated for the technical triplicates, and samples with CV > 10% were removed from the analysis. A melting curve was added to the end of every run to confirm the presence of a single PCR product.
Statistical Analysis
Comparisons of alpha diversity were performed with a one-way ANOVA followed by a Tukey’s pairwise test. Normalized [square root (x)] data were used to compare community compositions using a non-metric multidimensional scaling analysis (nMDS) with a Bray–Curtis similarity index. To determine significantly different microbial assemblage between families and RGs, and to compare qPCR data, a one-way PERMANOVA was used. To examine which OTUs contributed to differences between RGs, a SIMPER analysis with a Bray–Curtis similarity index was used. To define associations between breeding values and OTUs, breeding values were normalized (x-mean/standard deviation) and used within a canonical correspondence analysis (CCA). All analyses were performed using the PAST statistical software (Hammer et al., 2001). To determine whether an OTU was significantly elevated in a particular RG, the group_significance.py script using the default analysis (Kruskal–Wallis ANOVA) was used in QIIME. To examine correlations between EBVs, we performed a maximal information-based non-parametric exploration (MINE) analysis (Reshef et al., 2011).
Results
The C. gigas Microbiome
Following data filtering and rarefication, a total of 3294 OTUs were observed across the entire dataset, and of these, 68.5% occurred at below 1% of the total relative abundance. Conversely, across all samples and spanning all RGs, a member of the Pseudomonas genus (OTU 2034) was found to be the most relatively abundant OTU comprising 5.6% of the bacterial community. This was followed by OTUs matching an uncultured bacterium in the Psychrobacter genus (OTU 1488) and an uncultured bacterium in the Mycoplasma genus (OTU 3150), which represented 4.8 and 4% of the C. gigas microbiome across the whole dataset respectively (Figure 1).
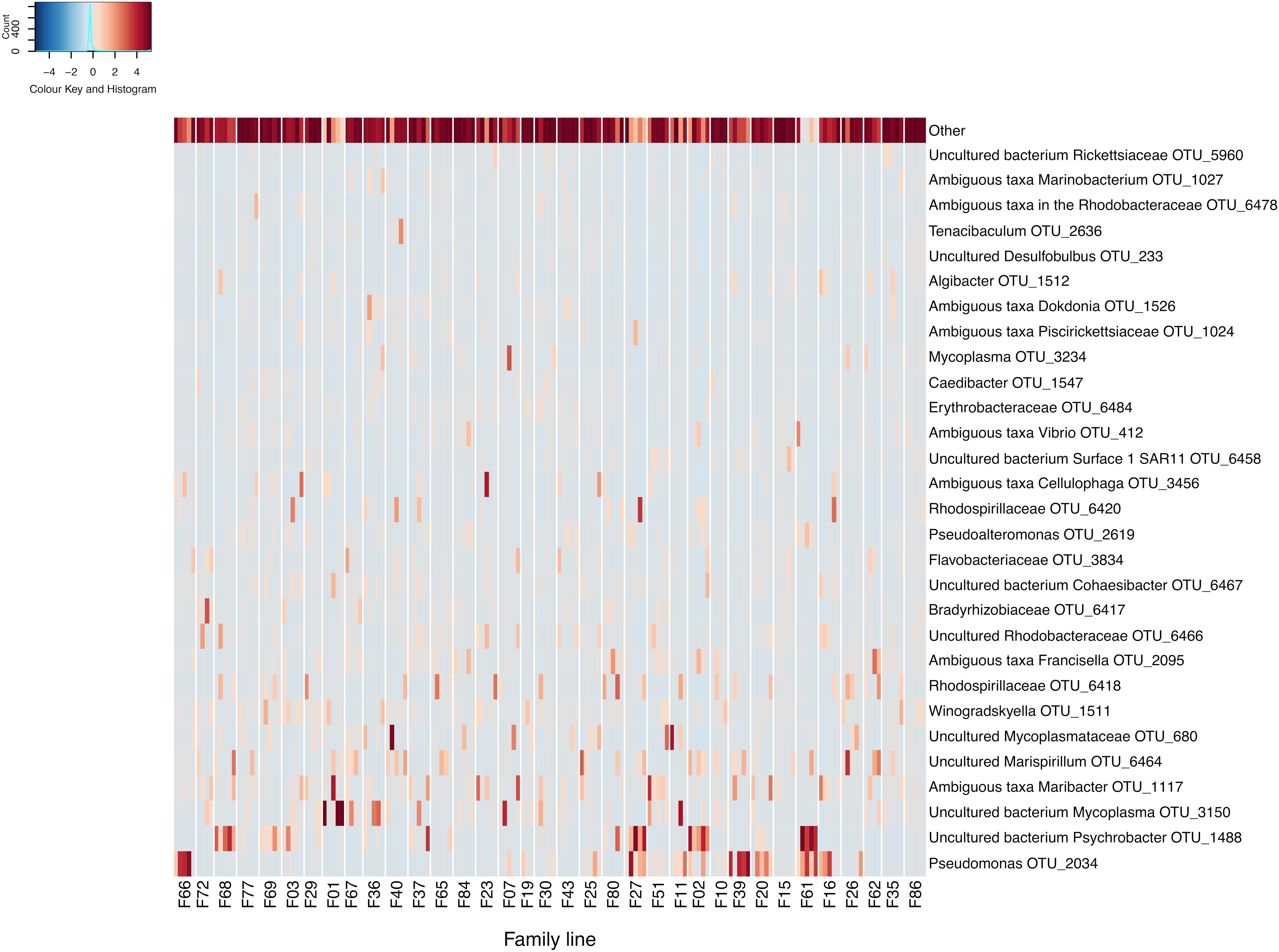
Figure 1. Heatmap of scaled OTU relative abundance for the 30 most abundant OTUs, as well as the remaining summed lowly abundant OTUs. Families ordered by OsHV-1 μvar disease-resistance. Heatmap was made using the R statistical environment using scaled data with the gplots and RColorBrewer packages (Neuwirth, 2014; Warnes et al., 2016; R Core Team, 2017).
Variability in the C. gigas Microbiome Across Different Resistance Lines
To determine whether breeding for disease resistance influences the C. gigas microbiome, the microbiome of oysters assigned to RGs were characterized and compared. Alpha diversity, quantified using Shannon’s diversity index was significantly higher in RG2 when compared to the RG3 RG (F(1,141) = 6.8, p = 0.025), but did not vary significantly when compared to RG1 (F(1,93) = 0.4, p = 0.51). Species richness (Chao1) did not differ significantly between any of the RGs (RG1 vs. RG2 – F(1,93) = 0.03, p = 0.85; RG1 vs. RG3 – F(1,94) = 1.3, p = 0.26; RG2 vs. RG3 – F(1,141) = 1.08, p = 0.30).
Comparisons of microbiome composition (beta diversity) across different RGs revealed that the microbiomes of RG1 and RG2 were both significantly different to the least disease resistant group, RG3 (p = 0.019 and p = 0.0001; F(1,94) = 1.47 and F(1,141) = 2.93 respectively). No significant difference was found between the microbiomes of RG1 and RG2 (F(1,93) = 1.29, p = 0.055). Statistical comparisons between RG2 and RG3 appeared to be stronger than those between RG1 and RG3, possibly due to more families being assigned to RG2, therefore potentially adding more microbiome variability to this group. No clear dissimilarity in the microbiome of the RGs was apparent in a 3D nMDS (Stress = 0.34), or a PCoA (Supplementary Figure 1). SIMPER comparisons showed that the composition of the microbiomes associated with RG1 and RG2 were 81.83 and 82.12% dissimilar to RG3 respectively (Supplementary Tables 3, 4).
As the RG with the lowest level of disease-resistance (RG3) was found to have a significantly different microbial assemblage to both RG2 and RG1, we examined which OTUs were responsible for driving the differences in microbiome structure between these groups (Figure 2). An OTU assigned to the Pseudomonas genus (OTU 2034; the most abundant OTU in the entire dataset) was over-represented in the RG3 microbiome relative to both RG1 (H(1,94) = 7.6, p = 0.0058) and RG2 (H(1,141) = 15, p = 0.00011). Conversely, an OTU assigned to the Tenacibaculum genus (OTU 2636) and two separate OTUs assigned to the Dokdonia genus (OTUs 2162 and 1526) were all significantly under-represented in RG3 (Tenacibaculum RG1 H(1,94) = 4.5, p = 0.033 and RG2 H(1,141) = 15.2, p = 0.0056; Dokdonia RG1 H(1,94) = 7.7, p = 0.0001 and RG2 H(1,141) = 30.3, p < 0.0001).
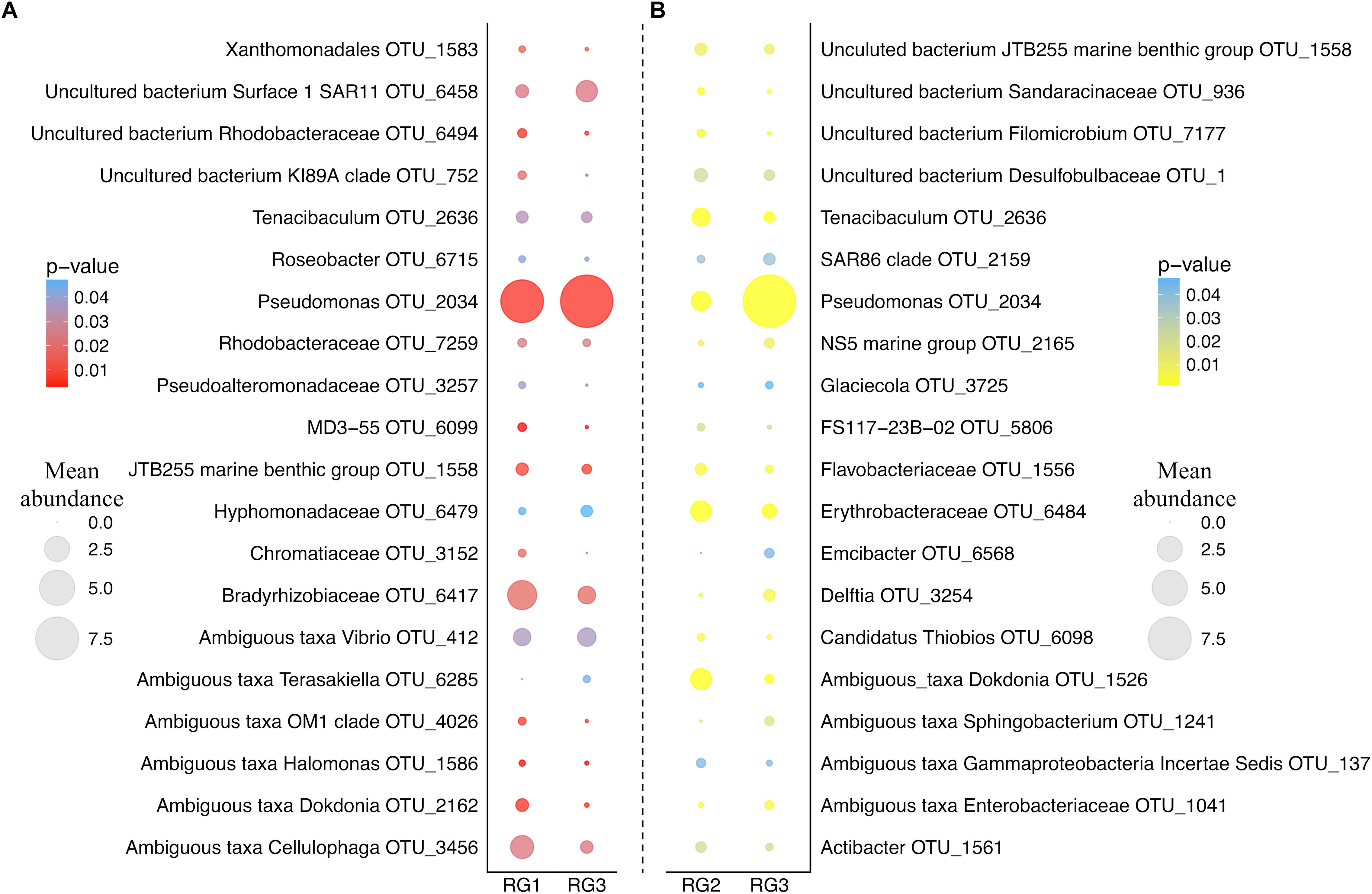
Figure 2. Bubble plot of group_significance.py analysis results using the default Kruskal–Wallis parameters. (A) Represents the comparison between RG1 and RG3. (B) Represents the comparison between RG2 and RG3. Color represents the strength of the p-value. Size represents the mean relative abundance of that OTU across the whole resistance group (RG). OTUs assigned to the genus and species level were chosen, and those 20 most abundant from each RG are displayed.
Notably, a member of the Vibrio genus (OTU 412) was found to be significantly over-represented in the least disease-resistant group (RG3) relative to the most disease-resistant group (RG1) (H(1,94) = 4.4, p = 0.036). Due to the previously demonstrated importance of Vibrio species in OsHV-1 μvar infection (Segarra et al., 2010; Jenkins et al., 2013; de Lorgeril et al., 2018), we subsequently employed a Vibrio-specific 16S rRNA qPCR assay to compare total abundances of Vibrio across RGs. A significant elevation of Vibrio 16S rRNA gene copies was observed in RG3 compared to RG1 (F(1,94) = 2.86, p = 0.027) and RG2 (F(1,141) = 3.25, p = 0.014) (average of 179, 107, and 75 gene copies mg of tissue-1 respectively; Supplementary Figure 2). Furthermore, OTUs assigned to the Vibrio genus were significantly elevated in RG3 when compared to RG1 (F(1,94) = 4.27, p = 0.011), but not RG2 (F(1,141) = 2.48, p = 0.07). To determine the extent of whether Vibrio OTUs were driving the differences between RG1 and RG3 microbiomes, OTUs assigned to the Vibrio genus were removed and the RG beta diversity comparison was reperformed. When doing this, we observed a slight weakening of the statistical comparison between RG1 and RG3, from (F(1,94) = 1.47, p = 0.019) to (F(1,94) = 1.46, p = 0.024).
A CCA was used to highlight associations between specific OTUs, OsHV-1 μvar disease-resistance and EBVs of other traits (Figure 3). OTUs matching the Cupriavidus (OTU 2182) and Psychrilyobacter (OTU 5046) genera were closely coupled with disease-resistance, followed by a member of the Tenacibaculum (OTU 2153) genus and an uncultured bacterium in the Frankiales order (OTU 5180). While OTUs assigned to members of the Photobacterium (OTU 1063; OTU 654; OTU 1053), Vibrio (OTU 651; OTU 653) and Aliivibrio (OTU 1248) genera were negatively associated with disease-resistance, but strongly associated with meat condition. Furthermore, members of the Streptococcus (OTU 814) and Roseovarius (OTU 7180) genera were closely associated with depth and width index, and also negatively associated with disease-resistance. The community composition was largely influenced by the first axis, driven by growth related EBVs. A MINE analysis identified a negative correlation between disease resistance and width index (p = 0.047; linear regression = -0.34), and a positive correlation between disease resistance and oyster weight (p = 0.038; linear regression = 0.15). Shell length and depth index had the strongest negative correlation (p = < 0.001; linear regression = -0.92), while oyster weight and shell length had the strongest positive correlation (p = 0.002; linear regression = 0.74; Supplementary Table 5).
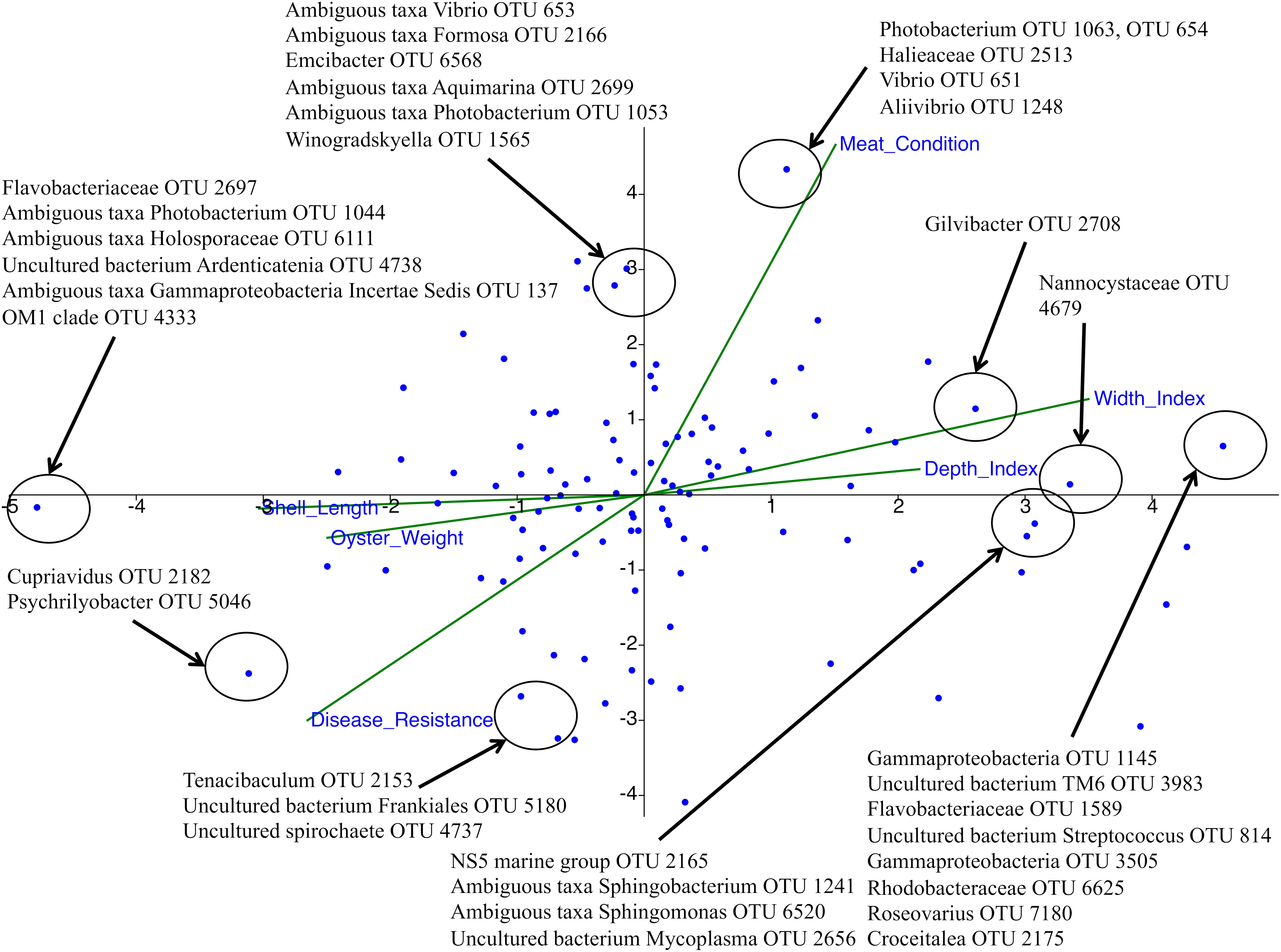
Figure 3. Canonical correspondence analysis (CCA) plot using 3% relative abundance filtered data. Cupriavidus, Psychrilyobacter, Tenacibaculum, and Frankiales were found to be strongly associated with OsHV-1 μvar disease-resistance, while OTUs assigned to the Photobacterium, Vibrio, and Aliivibrio were negatively associated with OsHV-1 μvar disease-resistance. Axis 1 and 2 were able to significantly represent 53.2% of the data (p = 0.001 for both axes with 999 permutations).
Defining the Core C. gigas Microbiome Across Different Resistance Lines
Due to the dynamic nature of oyster microbiomes, identifying a core microbiome can provide insights into which members may be driving the within-microbiome interactions and possibly shaping the community composition. While we were unable to identify a universal core microbiome across all samples, analyses of individual families revealed that each family had a small core microbiome (9–109 OTUs), with many of these OTUs shared across families. Families 30 and 84, within RG2, shared the most core OTUs (4) (Supplementary Figure 3). In contrast, family 19 of RG2 had the most unique core OTUs (27), that is those core OTUs not shared with any other family. To determine how many unique core OTUs were present in each oyster family (and therefore each RG), we compiled all of the core OTUs from the core analysis and removed duplicate bacteria. When doing this, a total of 9, 54, and 16 unique OTUs were assigned to RG1, RG2, and RG3 respectively (Table 2). When performing a separate core analysis on each RG as a whole, RG1 was comprised of two core members, a member of the Winogradskyella genus (OTU 1511) and a member of the Bradyrhizobiaceae family (OTU 6417). While, no core bacterial members were found for RG2 or RG3 microbiomes.

Table 2. Unique core bacterial members from individual oyster families collated into their respective resistance groups (RG).
Discussion
The principal goal of this study was to identify patterns in the C. gigas microbiome across 35 oyster families with differing levels of resistance to OsHV-1 μvar disease, with the objective of elucidating microbial taxa associated with disease resistance. Immunosuppression from OsHV-1 μvar infection allows opportunistic bacteria within the oyster’s microbiome to induce bacteremia, killing the host (Petton et al., 2015; de Lorgeril et al., 2018). Characterizing these interactions and gaining insights into the oyster microbiome is essential to further understand the dynamic interplay between the microbiome, OsHV-1 μvar and disease. A significant difference in the structure of the microbiome of oysters exhibiting different levels of resistance to OsHV-1 μvar disease was observed. Specifically, the microbiomes associated with the oysters showing the most resistance to OsHV-1 μvar disease (RG1) and moderately resistant oysters (RG2) were significantly different to the most disease susceptible (or least resistant) group (RG3). When considering disease resistance, we observed a strong negative association between the OsHV-1 μvar disease resistance of oyster hosts and the occurrence of OTUs assigned to the Vibrio (OTUs 651 and 653), Photobacterium (OTUs 1063, 654, and 1053), Aliivibrio (OTU 1248), Streptococcus (OTU 814) and Roseovarius (OTU 7180) genera, while on the other hand, the microbiomes of the most resistant families had an over-representation of OTUs assigned to the Cupriavidus (OTU 2182), Psychrilyobacter (OTU 5046), and Tenacibaculum (OTU 2153) genera.
The association between the occurrence of Vibrio and disease susceptibility was further supported by a significant elevation of an uncharacterized member of the Vibrio in RG3, and the results of a Vibrio-specific qPCR assay. These results are consistent with growing evidence implicating a role of the Vibrio community in oyster disease (Sugumar et al., 1998; Waechter et al., 2002; Garnier et al., 2007; Saulnier et al., 2010; Lemire et al., 2015; Petton et al., 2015; Green et al., 2018; King et al., 2018). Specifically, there is previous evidence that prior to oyster disease onset, the native Vibrio community is replaced by pathogenic Vibrio species (Lemire et al., 2015). Further, in corals, small shifts in the Vibrio community are sufficient to shift the microbiome metabolism (Thurber et al., 2009). Our data provides a new perspective on this interaction, whereby the total load of Vibrio differed between disease susceptible and resistant oyster families. This is supported by a recent study, which demonstrated that the Vibrio load following OsHV-1 μvar infection was significantly higher in disease-susceptible oysters (de Lorgeril et al., 2018). An increased Vibrio community size may provide further potential for pathogenic species to replace benign colonizers. On the other hand, a higher background load of Vibrio may become important under periods of stress, such as with OsHV-1 μvar infection, resulting in duel infection, as has recently been described (de Lorgeril et al., 2018). This is also indirectly supported by a previous study which observed reduced mortality in OsHV-1 infected oysters that were treated with antibiotics (Petton et al., 2015).
Increases in the abundance of OTUs assigned to the Photobacterium genus, as were observed here, often co-occur with an increase in the Vibrio community in oyster microbiomes (Wegner et al., 2013; Lokmer and Wegner, 2015). While members assigned to this genus have been identified as pathogens of other aquatic organisms (Pedersen et al., 2009; Liu et al., 2016), to our knowledge, no species of Photobacterium has been identified as an oyster pathogen. Members of the Streptococcus and Aliivibrio genera are known pathogens of fish and crabs (Pappalardo and Boemare, 1982; Egidius et al., 1986; Creeper and Buller, 2006; Urbanczyk et al., 2007), while a member of the Roseovarius genus is the causative agent of roseovarius oyster disease (formally juvenile oyster disease) in Crassostrea virginica (Boettcher et al., 2005; Maloy et al., 2007), yet to our knowledge these genera have not been implicated in disease of C. gigas previously, despite being over-represented in the most disease susceptible oyster families.
On the other hand, a strong positive association was observed between levels of disease resistance and the occurrence of OTUs assigned to the Cupriavidus (OTU 2182), Psychrilyobacter (OTU 5046) and Tenacibaculum (OTU 2153). Currently, little is known about the role of these genera in oysters. Cupriavidus species are commonly isolated from plants and soil (Cuadrado et al., 2010; Estrada-De Los Santos et al., 2014), but members of the Psychrilyobacter and Tenacibaculum have previously been observed in C. gigas microbiomes (Lee et al., 2009; Fernandez-Piquer et al., 2012; Wegner et al., 2013). Psychrilyobacter was observed in C. gigas microbiomes from Tasmania, Australia (Fernandez-Piquer et al., 2012), which is perhaps notable given that the oysters used in this study were initially sourced from Tasmania. In addition, we have previously identified an over-representation of a Tenacibaculum OTU in oyster microbiomes that were unaffected by a summer mortality outbreak (King et al., 2018).
As already stated, a significant elevation of OTUs belonging to the Vibrio and Photobacterium genera abundance in disease susceptible oysters has also been previously observed (de Lorgeril et al., 2018), supporting our findings. However, while we identified members of the Psychrilyobacter and Tenacibaculum genera to be associated with disease resistance, the same study (de Lorgeril et al., 2018) observed an increase in these same genera in an experimental infection experiment using disease susceptible oysters. Differences in bacterial taxa abundance and taxonomic assignment could be attributed to contrasting sequencing techniques and data analysis. For example, we used the V1–V3 hypervariable region, and clustered OTUs at the 97% identity level, compared to V3–V4 and having OTUs clustered at a three-nucleotide difference threshold (de Lorgeril et al., 2018). Furthermore, this study deployed oysters to the field, while the aforementioned study carried out their experiments in tanks. Tank based studies are known to significantly alter the oyster microbiome composition compared to oysters sourced from the environment (Lokmer et al., 2016a).
The oyster microbiome is dynamic in nature, changing in response to stressors such as disease, antibiotics, translocation, and heat (Wegner et al., 2013; Lokmer and Wegner, 2015; Lokmer et al., 2016b; de Lorgeril et al., 2018; Green et al., 2018; King et al., 2018). The microbiome assemblage can also be influenced by the oyster life stage, the genetics of the host oyster, and spatial location (Trabal et al., 2012; Wegner et al., 2013; Lokmer et al., 2016a; King et al., 2018). Because we only have one sampling point, our study would not capture the dynamic nature of the oyster microbiome, and thus the oyster microbiome could change before the onset of disease. To fully capture the importance of the taxa identified in this study, a temporal study in the field encompassing a disease outbreak would be needed. However, as disease outbreaks are often very sudden, capturing a disease outbreak in the environment can be difficult.
In addition to identifying OTUs that are over- or under-represented within the microbiomes of oysters with different levels of disease-resistance, another way to identify putatively important bacteria within the microbiome of a host organism involves the identification of “core” microbiome members (Ainsworth et al., 2015). Identifying which bacterial members are consistent and stable across microbial communities is important in unraveling the functional contribution of these core bacteria (Ainsworth et al., 2015). Notably, we could not define a universal core microbiome across all of the studied oyster families at the OTU level, suggesting significant heterogeneity in oyster microbiome structure, or possible differences in micro-geographic variation. However, we identified core microbiome members within each family microbiome, whereby a number of ‘unique’ core members often occurred exclusively in the core microbiome of a family. This is in accordance with previous observations that the composition of an oyster’s microbiome is partially governed by oyster genetics, particularly for shaping the rare specialist bacterial community (<1% abundance) (Wegner et al., 2013), although we have no information pertaining to the genetic differentiation between the studied oyster families. However, when examining the core microbiome across all of the families comprising the most highly disease-resistant group (RG1), we identified two core members, which included OTUs classified as members of the Winogradskyella genus (OTU 1511) and Bradyrhizobiaceae family (OTU 6417). OTUs assigned to the Bradyrhizobiaceae family have previously been observed in oysters (Sakowski, 2015), however, due to the coarse taxonomic assignment of this OTU, it is unclear what potential role this member of the Bradyrhizobiaceae family might have. Winogradskyella species are commonly found in numerous marine organisms, including oysters (Valdenegro-Vega et al., 2013; Park et al., 2015; Lee et al., 2017; Schellenberg et al., 2017; Franco et al., 2018), and are known for their role in amoebic-induced fish gill diseases (Embar-Gopinath et al., 2005, 2006). However, it is uncertain what function(s) Winogradskyella species play in oysters. We currently know little about the potential role, if any, of these core microbiome members in resistance, but these observations provide candidate target organisms for focused examinations of potential beneficial microbes within OsHV-1 μvar disease-resistance.
Conclusion
We have shown that the microbiome of C. gigas displays significantly different microbial assemblage structure according to oyster disease-resistance. This study provides insights into the C. gigas microbiome within the context of oysters bred for disease-resistance and highlights the potential involvement of the oyster microbiome in disease-resistance. Members of the Vibrio, Photobacterium, Aliivibrio, Streptococcus, and Roseovarius genera were over-represented features of the microbiome of oysters with high OsHV-1 μvar disease susceptibility, which is consistent with previous studies implicating Vibrio in oyster disease dynamics. Furthermore, a significant elevation of Vibrio 16S rRNA gene copies in disease-susceptible oyster families could indicate a lack of immune response against Vibrio pathogens. However, further research is required to elucidate the role of these bacteria in oyster disease dynamics. Examination of ‘core’ bacteria identified species assigned to the Winogradskyella genus and Bradyrhizobiaceae family as core members of microbiomes assigned to RG1 and may also play a role in OsHV-1 μvar disease resistance. These results deliver evidence that the C. gigas microbiome differs between oysters with different levels of susceptibility to OsHV-1 μvar disease and identifies putative microbial determinants in disease onset and resistance.
Data Availability
The datasets generated for this study can be found in NCBI SRA, PRJNA497763.
Author Contributions
WK and KN carried out the fieldwork. WK and NW processed the samples. WK and NS analyzed the data. CJ, MD, WO’C, JS, and ML conceived and designed the study. TK produced the core microbiome analysis. WK, JS, and ML wrote the manuscript.
Funding
This research was supported by an Australian Research Council Linkage Project (LP160101795) to JS and ML; a Cooperative Research Centre Project (CRC-P 2016-805; Future Oysters), led by the Australian Seafood Industry Pty Ltd. in partnership with a number of Australian Research Organisations; and Ausgem, a research partnership initiated between the University of Technology Sydney and the New South Wales Department of Primary Industries.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00473/full#supplementary-material
References
Ainsworth, T. D., Krause, L., Bridge, T., Torda, G., Raina, J.-B., Zakrzewski, M., et al. (2015). The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9, 2261–2275. doi: 10.1038/ismej.2015.39
ASI (2015). Australian Seafood Industries Oyster Breeding Programs. Available at: http://www.asioysters.com.au/breeding-program.html
Azéma, P., Travers, M. A., De Lorgeril, J., Tourbiez, D., and Dégremont, L. (2015). Can selection for resistance to OsHV-1 infection modify susceptibility to Vibrio aestuarianus infection in Crassostrea gigas? First insights from experimental challenges using primary and successive exposures. Vet. Res. 46:139. doi: 10.1186/s13567-015-0282-0
Boettcher, K. J., Geaghan, K. K., Maloy, A. P., and Barber, B. J. (2005). Roseovarius crassostreae sp. nov., a member of the Roseobacter clade and the apparent cause of juvenile oyster disease (JOD) in cultured Eastern oysters. Int. J. Syst. Evol. Microbiol. 55, 1531–1537. doi: 10.1099/ijs.0.63620-0
Burge, C. A., Griffin, F. J., and Friedman, C. S. (2006). Mortality and herpesvirus infections of the Pacific oyster Crassostrea gigas in Tomales Bay, California, USA. Dis. Aquat. Organ. 72, 31–43. doi: 10.3354/dao072031
Camara, M. D., Yen, S., Kaspar, H. F., Kesarcodi-Watson, A., King, N., Jeffs, A. G., et al. (2017). Assessment of heat shock and laboratory virus challenges to selectively breed for ostreid herpesvirus 1 (OsHV-1) resistance in the Pacific oyster, Crassostrea gigas. Aquaculture 469, 50–58. doi: 10.1016/j.aquaculture.2016.11.031
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Creeper, J. H., and Buller, N. B. (2006). An outbreak of Streptococcus iniae in barramundi (Lates calcarifera) in freshwater cage culture. Aust. Vet. J. 84, 408–411. doi: 10.1111/j.1751-0813.2006.00058.x
Cuadrado, V., Gomila, M., Merini, L., Giulietti, A. M., and Moore, E. R. B. (2010). Cupriavidus pampae sp. nov., a novel herbicide-degrading bacterium isolated from agricultural soil. Int. J. Syst. Evol. Microbiol. 60, 2606–2612. doi: 10.1099/ijs.0.018341-0
Davison, A. J., Trus, B. L., Cheng, N., Steven, A. C., Watson, M. S., Cunningham, C., et al. (2005). A novel class of herpesvirus with bivalve hosts. J. Gen. Virol. 86, 41–53. doi: 10.1099/vir.0.80382-0
de Lorgeril, J., Lucasson, A., Petton, B., Toulza, E., Montagnani, C., Clerissi, C., et al. (2018). Immune-suppression by OsHV-1 viral infection causes fatal bacteraemia in Pacific oysters. Nat. Commun. 9:4215. doi: 10.1038/s41467-018-06659-3
Dégremont, L. (2011). Evidence of herpesvirus (OsHV-1) resistance in juvenile Crassostrea gigas selected for high resistance to the summer mortality phenomenon. Aquaculture 317, 94–98. doi: 10.1016/j.aquaculture.2011.04.029
Dégremont, L. (2013). Size and genotype affect resistance to mortality caused by OsHV-1 in Crassostrea gigas. Aquaculture 416, 129–134. doi: 10.1016/j.aquaculture.2013.09.011
Dégremont, L., Lamy, J. B., Pépin, J. F., Travers, M. A., and Renault, T. (2015a). New insight for the genetic evaluation of resistance to ostreid herpesvirus infection, a worldwide disease, in Crassostrea gigas. PLoS One 10:e0127917. doi: 10.1371/journal.pone.0127917
Dégremont, L., Nourry, M., and Maurouard, E. (2015b). Mass selection for survival and resistance to OsHV-1 infection in Crassostrea gigas spat in field conditions: response to selection after four generations. Aquaculture 446, 111–121. doi: 10.1016/j.aquaculture.2015.04.029
Dégremont, L., Ledu, C., Maurouard, E., Nourry, M., and Benabdelmouna, A. (2016a). Effect of ploidy on the mortality of Crassostrea gigas spat caused by OsHV-1 in France using unselected and selected OsHV-1 resistant oysters. Aquac. Res. 47, 777–786. doi: 10.1111/are.12536
Dégremont, L., Morga, B., Trancart, S., and Pépin, J. F. (2016b). Resistance to OsHV-1 infection in crassostrea gigas larvae. Front. Mar. Sci. 3:15. doi: 10.3389/fmars.2016.00015
Dégremont, L., Tanguy, G., Delphine, T., and Jean-François, P. (2013). Is horizontal transmission of the Ostreid herpesvirus OsHV-1 in Crassostrea gigas affected by unselected or selected survival status in adults to juveniles? Aquaculture 408–409, 51–57. doi: 10.1016/j.aquaculture.2013.05.025
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Egidius, E., Wiik, R., Andersen, K., Hoff, K. A., and Hjeltnes, B. (1986). Vibrio salmonicida sp. nov., a new fish pathogen. Int. J. Syst. Evol. Microbiol. 36, 518–520. doi: 10.1099/00207713-36-4-518
Embar-Gopinath, S., Butler, R., and Nowak, B. (2005). Influence of salmonid gill bacteria on development and severity of amoebic gill disease. Dis. Aquat. Organ. 67, 55–60. doi: 10.3354/dao067055
Embar-Gopinath, S., Crosbie, P., and Nowak, B. F. (2006). Concentration effects of Winogradskyella sp. on the incidence and severity of amoebic gill disease. Dis. Aquat. Organ. 73, 43–47. doi: 10.3354/dao073043
Estrada-De Los Santos, P., Solano-Rodríguez, R., Matsumura-Paz, L. T., Vásquez-Murrieta, M. S., and Martínez-Aguilar, L. (2014). Cupriavidus plantarum sp. nov., a plant-associated species. Arch. Microbiol. 196, 811–817. doi: 10.1007/s00203-014-1018-7
Fernandez-Piquer, J., Bowman, J. P., Ross, T., and Tamplin, M. L. (2012). Molecular analysis of the bacterial communities in the live Pacific oyster (Crassostrea gigas) and the influence of postharvest temperature on its structure. J. Appl. Microbiol. 112, 1134–1143. doi: 10.1111/j.1365-2672.2012.05287.x
Franco, A., Busse, H. J., Schubert, P., Wilke, T., Kämpfer, P., and Glaeser, S. P. (2018). Winogradskyella pocilloporae sp. nov, isolated from healthy tissue of the coral Pocillopora damicornis. Int. J. Syst. Evol. Microbiol. 68, 1689–1696. doi: 10.1099/ijsem.0.002731
Friedman, C. S., Estes, R. M., Stokes, N. A., Burge, C. A., Hargove, J. S., Barber, B. J., et al. (2005). Herpes virus in juvenile Pacific oysters Crassostrea gigas from Tomales Bay, California, coincides with summer mortality episodes. Dis. Aquat. Organ. 63, 33–41. doi: 10.3354/dao063033
Garcia, C., Thebault, A., Degremont, L., Arzul, I., Miossec, L., Robert, M., et al. (2011). Ostreid herpesvirus 1 detection and relationship with Crassostrea gigas spat mortality in France between 1998 and 2006. Vet. Res. 42:73. doi: 10.1186/1297-9716-42-73
Garnier, M., Labreuche, Y., Garcia, C., Robert, M., and Nicolas, J. L. (2007). Evidence for the involvement of pathogenic bacteria in summer mortalities of the pacific oyster Crassostrea gigas. Microb. Ecol. 53, 187–196. doi: 10.1007/s00248-006-9061-9
Go, J., Deutscher, A., Spiers, Z., Dahle, K., Kirkland, P., and Jenkins, C. (2017). An investigation into mass mortalities of unknown aetiology in Pacific oysters, Crassostrea gigas, in Port Stephens, New South Wales, Australia. Dis. Aquat. Organ. 125, 227–242. doi: 10.3354/dao03146
Green, T. J., Siboni, N., King, W. L., Labbate, M., Seymour, J. R., and Raftos, D. (2018). Simulated marine heat wave alters abundance and structure of Vibrio populations associated with the Pacific Oyster resulting in a mass mortality event. Microb. Ecol. doi: 10.1007/s00248-018-1242-9 [Epub ahead of print].
Hammer,Ø., Harper, D. A. T., and Ryan, P. D. (2001). Past: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4, 1–9.
Jenkins, C., Hick, P., Gabor, M., Spiers, Z., Fell, S. A., Gu, X., et al. (2013). Identification and characterisation of an ostreid herpesvirus-1 microvariant (OsHV-1 micro-var) in Crassostrea gigas (Pacific oysters) in Australia. Dis. Aquat. Organ. 105, 109–126. doi: 10.3354/dao02623
Kahlke, T. (2017). Panbiom. Available at: https://github.com/timkahlke/panbiom
Keeling, S. E., Brosnahan, C. L., Williams, R., Gias, E., Hannah, M., Bueno, R., et al. (2014). New Zealand juvenile oyster mortality associated with ostreid herpesvirus 1-an opportunistic longitudinal study. Dis. Aquat. Organ. 109, 231–239. doi: 10.3354/dao02735
King, W. L., Jenkins, C., Go, J., Siboni, N., Seymour, J. R., and Labbate, M. (2018). Characterisation of the Pacific Oyster microbiome during a summer mortality event. Microb. Ecol. doi: 10.1007/s00248-018-1226-9 [Epub ahead of print].
King, W. L., Jenkins, C., Seymour, J. R., and Labbate, M. (2019). Oyster disease in a changing environment: decrypting the link between pathogen, microbiome and environment. Mar. Environ. Res. 143, 124–140. doi: 10.1016/j.marenvres.2018.11.007
Kube, P., Dove, M., Cunningham, M., Kirkland, P., Gu, X., Hick, P., et al. (2018). Genetic Selection for Resistance to Pacific Oyster Mortality Syndrome. Available: http://www.asioysters.com.au/uploads/2/5/9/7/ 25976125/kube_et_al__2018__genetic_selection_for_resistance_to_ poms_-_sfcrc_project_final_report.pdf [accessed December 15, 2018].
Lee, J. H., Kang, J. W., Shin, S. B., and Seong, C. N. (2017). Winogradskyella flava sp. nov., isolated from the brown alga, Sargassum fulvellum. Int. J. Syst. Evol. Microbiol. 67, 3540–3546. doi: 10.1099/ijsem.0.002161
Lee, Y. S., Baik, K. S., Park, S. Y., Kim, E. M., Lee, D. H., Kahng, H. Y., et al. (2009). Tenacibaculum crassostreae sp. nov., isolated from the Pacific oyster, Crassostrea gigas. Int. J. Syst. Evol. Microbiol. 59, 1609–1614. doi: 10.1099/ijs.0.006866-0
Lemire, A., Goudenege, D., Versigny, T., Petton, B., Calteau, A., Labreuche, Y., et al. (2015). Populations, not clones, are the unit of vibrio pathogenesis in naturally infected oysters. ISME J. 9, 1523–1531. doi: 10.1038/ismej.2014.233
Liu, F., Liu, G., and Li, F. (2016). Characterization of two pathogenic Photobacterium strains isolated from Exopalaemon carinicauda causing mortality of shrimp. Aquaculture 464, 129–135. doi: 10.1016/j.aquaculture.2016.06.019
Lokmer, A., Goedknegt, M. A., Thieltges, D. W., Fiorentino, D., Kuenzel, S., Baines, J. F., et al. (2016a). Spatial and temporal dynamics of pacific oyster hemolymph microbiota across multiple scales. Front. Microbiol. 7:1367. doi: 10.3389/fmicb.2016.01367
Lokmer, A., Kuenzel, S., Baines, J. F., and Wegner, K. M. (2016b). The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environ. Microbiol. 18, 970–987. doi: 10.1111/1462-2920.13163
Lokmer, A., and Wegner, K. M. (2015). Hemolymph microbiome of Pacific oysters in response to temperature, temperature stress and infection. ISME J. 9, 670–682. doi: 10.1038/ismej.2014.160
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Maloy, A. P., Ford, S. E., Karney, R. C., and Boettcher, K. J. (2007). Roseovarius crassostreae, the etiological agent of Juvenile Oyster Disease (now to be known as Roseovarius Oyster Disease) in Crassostrea virginica. Aquaculture 269, 71–83. doi: 10.1016/j.aquaculture.2007.04.008
Moreau, P., Moreau, K., Segarra, A., Tourbiez, D., Travers, M. A., Rubinsztein, D. C., et al. (2015). Autophagy plays an important role in protecting Pacific oysters from OsHV-1 and Vibrio aestuarianus infections. Autophagy 11, 516–526. doi: 10.1080/15548627.2015.1017188
Mortensen, S., Strand, A., Bodvin, T., Alfjorden, A., Skar, C. K., Jelmert, A., et al. (2016). Summer mortalities and detection of ostreid herpesvirus microvariant in Pacific oyster Crassostrea gigas in Sweden and Norway. Dis. Aquat. Organ. 117, 171–176. doi: 10.3354/dao02944
Neuwirth, E. (2014). RColorBrewer: ColorBrewer Palettes. Available at: https://cran.r-project.org/web/packages/RColorBrewer/index.html
Pappalardo, R., and Boemare, N. (1982). An intracellular Streptococcus, causative agent of a slowly developing disease in the Mediterranean crab, Carcinus mediterraneus. Aquaculture 28, 283–292. doi: 10.1016/0044-8486(82)90070-9
Park, S., Park, J. M., Won, S. M., and Yoon, J. H. (2015). Winogradskyella crassostreae sp. nov., isolated from an oyster (Crassostrea gigas). Int. J. Syst. Evol. Microbiol. 65, 2890–2895. doi: 10.1099/ijs.0.000350
Paul-Pont, I., Dhand, N. K., and Whittington, R. J. (2013). Influence of husbandry practices on OsHV-1 associated mortality of Pacific oysters Crassostrea gigas. Aquaculture 412-413, 202–214. doi: 10.1016/j.aquaculture.2013.07.038
Pedersen, K., Skall, H. F., Lassen-Nielsen, A. M., Bjerrum, L., and Olesen, N. J. (2009). Photobacterium damselae subsp.damselae, an emerging pathogen in Danish rainbow trout, Oncorhynchus mykiss (Walbaum), mariculture. J. Fish Dis. 32, 465–472. doi: 10.1111/j.1365-2761.2009.01041.x
Petton, B., Bruto, M., James, A., Labreuche, Y., Alunno-Bruscia, M., and Le Roux, F. (2015). Crassostrea gigas mortality in France: the usual suspect, a herpes virus, may not be the killer in this polymicrobial opportunistic disease. Front. Microbiol. 6:686. doi: 10.3389/fmicb.2015.00686
R Core Team (2017). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Reshef, D. N., Reshef, Y. A., Finucane, H. K., Grossman, S. R., Mcvean, G., Turnbaugh, P. J., et al. (2011). Detecting novel associations in large data sets. Science 334, 1518–1524. doi: 10.1126/science.1205438
Rognes, T., Flouri, T., Nichols, B., Quince, C., and Mahé, F. (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. doi: 10.7717/peerj.2584
Sakowski, E. G. (2015). The Microbiome of the Eastern Oyster, Crassostrea virginica, in Health and Disease. Ph.D. thesis, University of Delaware, Newark, DE.
Saulnier, D., De Decker, S., Haffner, P., Cobret, L., Robert, M., and Garcia, C. (2010). A large-scale epidemiological study to identify bacteria pathogenic to Pacific Oyster Crassostrea gigas and correlation between virulence and metalloprotease-like activity. Microb. Ecol. 59, 787–798. doi: 10.1007/s00248-009-9620-y
Sauvage, C., Pépin, J. F., Lapègue, S., Boudry, P., and Renault, T. (2009). Ostreid herpes virus 1 infection in families of the Pacific oyster, Crassostrea gigas, during a summer mortality outbreak: Differences in viral DNA detection and quantification using real-time PCR. Virus Res. 142, 181–187. doi: 10.1016/j.virusres.2009.02.013
Schellenberg, J., Busse, H. J., Hardt, M., Schubert, P., Wilke, T., Kämpfer, P., et al. (2017). Winogradskyella haliclonae sp. Nov., isolated from a marine sponge of the genus Haliclona. Int. J. Syst. Evol. Microbiol. 67, 4902–4910. doi: 10.1099/ijsem.0.002192
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Segarra, A., Mauduit, F., Faury, N., Trancart, S., Dégremont, L., Tourbiez, D., et al. (2014). Dual transcriptomics of virus-host interactions: comparing two Pacific oyster families presenting contrasted susceptibility to ostreid herpesvirus 1. BMC Genomics 15:580. doi: 10.1186/1471-2164-15-580
Segarra, A., Pepin, J. F., Arzul, I., Morga, B., Faury, N., and Renault, T. (2010). Detection and description of a particular Ostreid herpesvirus 1 genotype associated with massive mortality outbreaks of Pacific oysters, Crassostrea gigas, in France in 2008. Virus Res. 153, 92–99. doi: 10.1016/j.virusres.2010.07.011
Shin, S. C., Kim, S. H., You, H., Kim, B., Kim, A. C., Lee, K. A., et al. (2011). Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334, 670–674. doi: 10.1126/science.1212782
Shreiner, A. B., Kao, J. Y., and Young, V. B. (2015). The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 31, 69–75. doi: 10.1097/MOG.0000000000000139
Siboni, N., Balaraju, V., Carney, R., Labbate, M., and Seymour, J. R. (2016). Spatiotemporal dynamics of Vibrio spp. within the Sydney Harbour estuary. Front. Microbiol. 7:460. doi: 10.3389/fmicb.2016.00460
Sugumar, G., Nakai, T., Hirata, Y., Matsubara, D., and Muroga, K. (1998). Vibrio splendidus biovar II as the causative agent of bacillary necrosis of Japanese oyster Crassostrea gigas larvae. Dis. Aquat. Organ.33, 111–118. doi: 10.3354/dao033111
Thompson, J. R., Randa, M. A., Marcelino, L. A., Tomita-Mitchell, A., Lim, E., and Polz, M. F. (2004). Diversity and dynamics of a North Atlantic coastal Vibrio community. Appl. Environ. Microbiol. 70, 4103–4110. doi: 10.1128/AEM.70.7.4103-4110.2004
Thurber, R. V., Willner-Hall, D., Rodriguez-Mueller, B., Desnues, C., Edwards, R. A., Angly, F., et al. (2009). Metagenomic analysis of stressed coral holobionts. Environ. Microbiol. 11, 2148–2163. doi: 10.1111/j.1462-2920.2009.01935.x
Trabal, N., Mazon-Suastegui, J. M., Vazquez-Juarez, R., Asencio-Valle, F., Morales-Bojorquez, E., and Romero, J. (2012). Molecular analysis of bacterial microbiota associated with oysters (Crassostrea gigas and Crassostrea corteziensis) in different growth phases at two cultivation sites. Microb. Ecol. 64, 555–569. doi: 10.1007/s00248-012-0039-5
Urbanczyk, H., Ast, J. C., Higgins, M. J., Carson, J., and Dunlap, P. V. (2007). Reclassification of Vibrio fischeri, Vibrio logei, Vibrio salmonicida and Vibrio wodanis as Aliivibrio fischeri gen. nov., comb. nov., Aliivibrio logei comb. nov., Aliivibrio salmonicida comb. nov. and Aliivibrio wodanis comb. nov. Int. J. Syst. Evol. Microbiol. 57, 2823–2829. doi: 10.1099/ijs.0.65081-0
Valdenegro-Vega, V., Naeem, S., Carson, J., Bowman, J. P., Tejedor Del Real, J. L., and Nowak, B. (2013). Culturable microbiota of ranched southern bluefin tuna (Thunnus maccoyii Castelnau). J. Appl. Microbiol. 115, 923–932. doi: 10.1111/jam.12286
Vezzulli, L., Brettar, I., Pezzati, E., Reid, P. C., Colwell, R. R., Höfle, M. G., et al. (2011). Long-term effects of ocean warming on the prokaryotic community: evidence from the vibrios. ISME J. 6, 21–30. doi: 10.1038/ismej.2011.89
Waechter, M., Le Roux, F., Nicolas, J. L., Marissal, E., and Berthe, F. (2002). Characterisation of Crassostrea gigas spat pathogenic bacteria. C. R. Biol. 325, 231–238. doi: 10.1016/S1631-0691(02)01428-2
Warnes, G. R., Bolker, B., Bonebakker, L., Gentleman, R., Liaw, W. H. A., Lumley, T., et al. (2016). gplots: Various R Programming Tools for Plotting Data. R Package Version 3.0.1. Available at: https://mran.microsoft.com/snapshot/2017-04-11/web/packages/gplots/index.html
Wegner, K. M., Volkenborn, N., Peter, H., and Eiler, A. (2013). Disturbance induced decoupling between host genetics and composition of the associated microbiome. BMC Microbiol. 13:252. doi: 10.1186/1471-2180-13-252
Whittington, R. J., Dhand, N. K., Evans, O., and Paul-Pont, I. (2015). Further observations on the influence of husbandry practices on OsHV-1 μVar mortality in Pacific oysters Crassostrea gigas: age, cultivation structures and growing height. Aquaculture 438, 82–97. doi: 10.1016/j.aquaculture.2014.12.040
Keywords: Crassostrea gigas, microbiome, ostreid herpesvirus, disease resistance, Vibrio, POMS
Citation: King WL, Siboni N, Williams NLR, Kahlke T, Nguyen KV, Jenkins C, Dove M, O’Connor W, Seymour JR and Labbate M (2019) Variability in the Composition of Pacific Oyster Microbiomes Across Oyster Families Exhibiting Different Levels of Susceptibility to OsHV-1 μvar Disease. Front. Microbiol. 10:473. doi: 10.3389/fmicb.2019.00473
Received: 01 November 2018; Accepted: 22 February 2019;
Published: 11 March 2019.
Edited by:
Sebastian Fraune, University of Kiel, GermanyReviewed by:
Mathias Wegner, Alfred Wegener Institute Helmholtz Centre for Polar and Marine Research (AWI), GermanyEve Toulza, Université de Perpignan Via Domitia, France
Copyright © 2019 King, Siboni, Williams, Kahlke, Nguyen, Jenkins, Dove, O’Connor, Seymour and Labbate. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maurizio Labbate, bWF1cml6aW8ubGFiYmF0ZUB1dHMuZWR1LmF1