
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 12 March 2019
Sec. Plant Pathogen Interactions
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00407
This article is part of the Research TopicMetabolic Adjustments and Gene Expression Reprogramming for Symbiotic Nitrogen Fixation in Legume NodulesView all 12 articles
 Shintaro Hara1
Shintaro Hara1 Takashi Morikawa1
Takashi Morikawa1 Sawa Wasai1
Sawa Wasai1 Yasuhiro Kasahara2Taichi Koshiba3
Yasuhiro Kasahara2Taichi Koshiba3 Kiyoshi Yamazaki4
Kiyoshi Yamazaki4 Toru Fujiwara4Tsuyoshi Tokunaga3
Toru Fujiwara4Tsuyoshi Tokunaga3 Kiwamu Minamisawa1*
Kiwamu Minamisawa1*Sorghum (Sorghum bicolor) is cultivated worldwide for food, bioethanol, and fodder production. Although nitrogen fixation in sorghum has been studied since the 1970s, N2-fixing bacteria have not been widely examined in field-grown sorghum plants because the identification of functional diazotrophs depends on the culture method used. The aim of this study was to identify functional N2-fixing bacteria associated with field-grown sorghum by using “omics” approaches. Four lines of sorghum (KM1, KM2, KM4, and KM5) were grown in a field in Fukushima, Japan. The nitrogen-fixing activities of the roots, leaves, and stems were evaluated by acetylene reduction and 15N2-feeding assays. The highest nitrogen-fixing activities were detected in the roots of lines KM1 and KM2 at the late growth stage. Bacterial cells extracted from KM1 and KM2 roots were analyzed by metagenome, proteome, and isolation approaches and their DNA was isolated and sequenced. Nitrogenase structural gene sequences in the metagenome sequences were retrieved using two nitrogenase databases. Most sequences were assigned to nifHDK of Bradyrhizobium species, including non-nodulating Bradyrhizobium sp. S23321 and photosynthetic B. oligotrophicum S58T. Amplicon sequence and metagenome analysis revealed a relatively higher abundance (2.9–3.6%) of Bradyrhizobium in the roots. Proteome analysis indicated that three NifHDK proteins of Bradyrhizobium species were consistently detected across sample replicates. By using oligotrophic media, we purified eight bradyrhizobial isolates. Among them, two bradyrhizobial isolates possessed 16S rRNA and nif genes similar to those in S23321 and S58T which were predicted as functional diazotrophs by omics approaches. Both free-living cells of the isolates expressed N2-fixing activity in a semi-solid medium according to an acetylene reduction assay. These results suggest that major functional N2-fixing bacteria in sorghum roots are unique bradyrhizobia that resemble photosynthetic B. oligotrophicum S58T and non-nodulating Bradyrhizobium sp. S23321. Based on our findings, we discuss the N2-fixing activity level of sorghum plants, phylogenetic and genomic comparison with diazotrophic bacteria in other crops, and Bradyrhizobium diversity in N2 fixation and nodulation.
Biological N2 fixation in non-leguminous plants is required to improve agricultural sustainability by decreasing the global use of synthetic nitrogen fertilizers (Steffen et al., 2015; Yoneyama et al., 2017; Rosenblueth et al., 2018). Diazotrophic endophytes can provide fixed nitrogen to non-leguminous crops (Boddey et al., 2001; Yoneyama et al., 2017). 15N-labeling significantly contributed to N2 fixation in sugarcane (Yoneyama et al., 1997; Boddey et al., 2001). Gluconacetobacter and Herbaspirillum were isolated from sugarcane stems as candidate endophytic N2-fixing bacteria (Cavalcante and Dobereiner, 1988; James, 2000). Recent metatranscriptome analyses targeting nifH (encoding dinitrogenase reductase) suggested that Bradyrhizobium members play a role in N2 fixation in sugarcane (Thaweenut et al., 2011; Fischer et al., 2012; Rosenblueth et al., 2018). Abundant expression of Bradyrhizobium and Azorhizobium nifH was also detected in sweet potato stems and tubers (Terakado-Tonooka et al., 2008).
Sorghum [Sorghum bicolor (L.) Moench] is a C4 plant. Sorghum has little breeding history compared to sugarcane and maize but has the potential for broad agro-ecological adaptation (Khawaja et al., 2014). Sorghum provides grain for use in food and feed, sugary juice for producing syrup or bioethanol and is an excellent fodder (Khawaja et al., 2014). Omics studies of sorghum-associated microbes (Naylor et al., 2017; Xu et al., 2018) showed that drought increased the abundance and activity of monoderm bacteria including Actinobacteria in field-grown sorghum and verified that these bacteria contribute to the drought-resistance of sorghum plants. Thus, sorghum root-associated microbiomes play an important role in determining plant fitness.
For nitrogen fixation in sorghum plants, Pedersen et al. (1978) first detected the N2-fixing activities of washed root segments and soil cores of grain sorghum in NE, United States, in an acetylene reduction assay. Wani et al. (1984) observed the acetylene-reducing activity (ARA) of intact sorghum plants grown in pots. These studies suggested that sorghum-associated bacteria play a role in N2 fixation. Coelho et al. (2008) reported several diazotrophic bacteria (Paenibacillus, Azohydromonas, Ideonella, Rhizobium, and Bradyrhizobium) in the rhizosphere soils of field-grown sorghum in Brazil based on nifH PCR of soil DNA extracts. However, N2-fixing bacteria associated with sorghum plant tissues have not been fully explored.
Recent “omics” approaches have been used to identify and isolate functional diazotrophs in sugarcane (Thaweenut et al., 2011; Fischer et al., 2012), sweet potato (Terakado-Tonooka et al., 2008; Terakado-Tonooka et al., 2013), and paddy rice (Bao et al., 2014, 2016). Particularly, the combination of metagenome and metaproteome analyses based on extracted bacterial cells (EBCs) isolated from plant tissues (Ikeda et al., 2009) revealed type II methanotrophs in paddy rice roots as functional N2-fixing bacteria (Bao et al., 2014; Minamisawa et al., 2016). We adopted a similar strategy to identify diazotrophs responsible for N2 fixation in field-grown sorghum plants. We identified tissues showing significant N2-fixing activity by ARA and 15N2 fixation, identified functional diazotrophs by proteome analysis of nitrogenase proteins based on metagenomic data, and isolated bacteria with nitrogenase proteins and phylogenetic markers predicted from the omics results (Figure 1). Our results strongly suggest that bradyrhizobia fixed N2 in the roots of filed-grown sorghum plants at late growth stages. Because the N2-fixing bradyrhizobia in sorghum roots are phylogenetically close to an aquatic legume, Aeschynomene (Okubo et al., 2012a), we describe their functional roles.
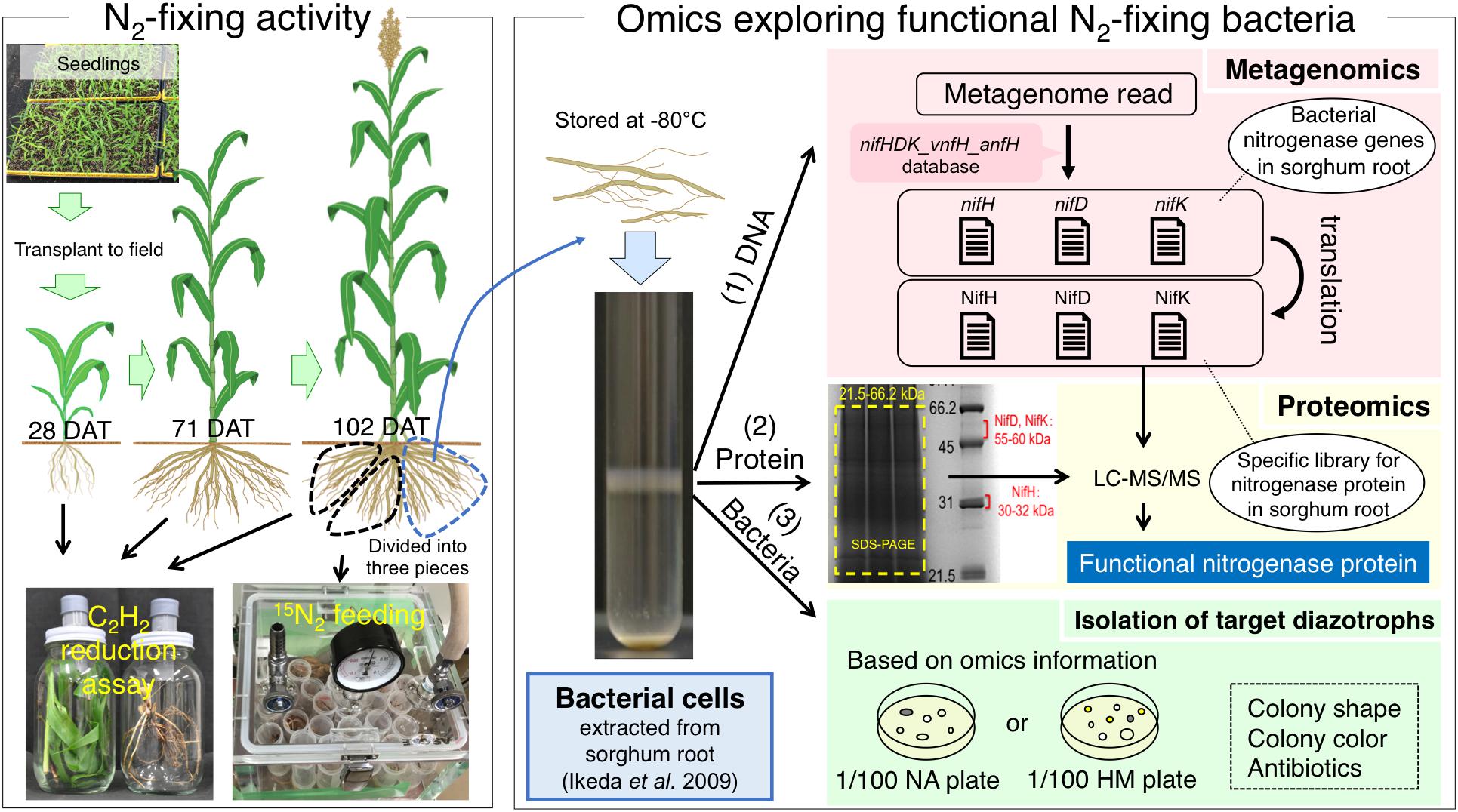
Figure 1. Outline of “omics” strategy used to explore and identify functional N2-fixing bacteria associated with sorghum plants. N2-fixing activities were monitored in tissues of sorghum at different growth stages by acetylene reduction assay and were directly confirmed in an 15N2 feeding experiment. Bacteria were extracted from sorghum root tissues with higher N2-fixing activities, and their metagenomes (1) and proteomes (2) were analyzed. Functional N2-fixing bacteria were isolated from the extracted bacteria (3). DAT = days after transplant.
We used four lines (KM1, KM2, KM4, and KM5) of sorghum developed by Earthnote Co., Ltd. (Okinawa, Japan). KM1 is a late-ripening line with vigorous leaf growth. KM2 is an early-ripening line with lodging resistance and salt tolerance. KM4 and KM5 were pre-selected because of their high (KM4) and low (KM5) N2-fixing activities as estimated by the 15N dilution method (Lee et al., unpublished).
Seeds were sown in 200-cell plug trays on May 10, 2016. The seedlings were transplanted into a field owned by Earthnote (Fukushima, Japan; 37°30′46.43′′140°34′13.7′′) on June 6, 2016. The soil had the following chemical properties: pH (H2O), 5.9; total C, 13.9 g kg-1 dry soil; total N, 0.8 g kg-1 dry soil; available phosphorus, 560.4 mg P kg-1 dry soil (Truog method). Before transplanting the seedlings, the field was treated with 85 kg N as urea, 84 kg N as controlled-release coated-urea fertilizer (LP100, JCAM Agri. Co., Ltd., Tokyo, Japan), which releases 80% of its total N over 100 days, and 85 kg K2O as potassium sulfate per hectare. This is the standard fertilization regime used for sorghum cropping under the local conditions. Each line was planted in three plots, with 50 plants per plot (0.075 m between plants, 1.00 m between rows).
Figure 1 shows the outlines of our polyphasic approach based on nitrogen-fixing activity and omics analyses. Because the experimental designs were complicated for respective analyses, we show the detailed sampling strategies used in the polyphasic approach along with the sorghum growth stages in Supplementary Figures S1, S2.
At least three plants were sampled individually from three different field plots or from the seedling trays for transplanting. When the sorghum seedlings were transplanted into the field, six seedlings were randomly selected from the plug trays [0 days after transplanting (DAT)], which were subjected to the acetylene reduction assay (Supplementary Figure S1A) as described below. At 28 DAT and 71 DAT, field-grown sorghum plants were harvested from three plots individually, and each plant tissue including the leaves, stems, and roots were subjected to the acetylene reduction assay (Supplementary Figure S1B). At 102 DAT, three plants were harvested from three plots individually, and the roots were washed with tap water, weighted, and further subdivided into three parts for the acetylene reduction assay, 15N2-feeding analysis, and omics/isolation analysis (metagenome, proteome, bacterial isolation) (Supplementary Figure S2 and Figure 1). Thus, the acetylene reduction assay, 15N2-feeding experiment, and omics analyses (metagenome and proteome) were performed based on three biological replicates.
Nitrogen-fixing activity was examined by the acetylene reduction assay (Schollhorn and Burris, 1967) at four growth stages: 0, 27, 71, and 102 DAT. Sorghum plants were maintained in the vegetative stage until 71 DAT, while the plants generally moved to the reproductive stage with ears at 102 DAT (Figure 1). One or two healthy plants were randomly sampled (Supplementary Figures S1, S2). The tissues to be used for ARA were determined according to the growth stage of the plant (Figure 1): 0 DAT, whole seedling; 28 DAT, shoot and root; 71 DAT, leaf, stem, and root; and 102 DAT, root. At 0 and 28 DAT, two plants were harvested, one of which was used for acetylene reduction with acetylene, while the others were used for control analysis without acetylene (Supplementary Figure S1). At 71 and 102 DAT, one plant was divided equally into two pieces of similar weight and structure; one piece was used to analyze acetylene reduction with acetylene and the other was used for control analysis without acetylene (Supplementary Figures S1, S2).
At 0 DAT, seedling roots were washed in tap water to remove adhering soil and placed in a 100-mL vial with a butyl rubber septum (SVG-100, Nichiden-Rika Glass Co., Ltd., Kobe, Japan) (Figure 1). At 27, 71, and 102 DAT, each tissue was placed in a 900-mL glass jar with a modified lid fitted with a butyl rubber W-plug (W-24T, Taiyo Kogyo, Tokyo, Japan) (Figure 1).
Acetylene gas (99.9999% vol/vol purity; Toho Acetylene Co., Tokyo, Japan) was injected to provide a concentration of 10% (vol/vol), and the vial or jar was incubated for 1 day at ambient temperature. We drew 20 mL of headspace gas in a sampling syringe and stored it in a vacuumed gas vial (SVG-5; Nichiden-Rika Glass Co.) until analysis. Gas samples (0.5 mL) were analyzed to determine their ethylene concentrations with a Shimadzu GC-18A gas chromatograph equipped with a flame ionization detector and Porapak R column as described previously (Elbeltagy et al., 2001).
At 102 DAT, the divided roots from the same plant used for ARA were introduced into a gas chamber with a sampling port (Figure 1 and Supplementary Figure S2). The gas phase in the chamber was replaced with 32% (v/v) 15N2 (99.4 atom % 15N; Shoko, Tokyo, Japan)/20% O2/48% Ar. The negative control included air gas containing atmospheric N2 [natural abundance of 15N (0.366 atom %)]. After static incubation in the chamber at 25°C for 23 h in the dark, the root samples were dried at 80°C for 3 days and then powdered in a Multi-Beads Shocker (Yasui Kikai, Osaka, Japan). To determine the level of 15N2 fixation in the roots, we analyzed 15N and total N contents of the powdered root tissues with an elemental analyzer/isotope ratio mass spectrometer (Flash EA1112-Delta V Advantage ConFlo IV System; Thermo Fisher Scientific, Waltham, MA, United States).
Root tissues of the same plant used for the ARA and 15N2-feeding experiments were stored at -80°C for metagenome analysis (Figure 1 and Supplementary Figure S2). EBCs, including both endophytes and epiphytes, were directly prepared from ∼100 g of root tissues as previously reported (Ikeda et al., 2009). The extraction of EBC allowed for the elimination of plant organelles and plant genomic DNA. Metagenomic DNA was prepared from the bacterial cells using an IsoPlant II kit (Nippon Gene, Tokyo, Japan) with bead beating treatment. In the first step, 0.5 g of bacterial cells were disrupted by bead beating in a Lysing Matrix B tube (MP Biomedicals, Santa Ana, CA, United States) with Solution I (Extraction Buffer) from the IsoPlant II kit. Bead beating was performed using a FastPrep FP100A (MP Biomedicals) at 5.5 m s-1 for 30 s. Other steps were conducted according to the manufacturer’s instructions, and the DNA preparation was stored at -20°C until use.
DNA libraries were prepared using the Illumina TruSeq DNA library preparation kit v. 2 (Illumina, San Diego, CA, United States). The size and quality of the libraries were assessed using a BioAnalyzer High Sensitivity Chip, and the average fragment size was around 700 base pairs (bp). Next, 250-bp paired-end libraries were sequenced on an Illumina MiSeq sequencer. The quality of the sequence data was checked by using the FastQC v. 0.10.1 software1. Reads were trimmed using Prinseq-lite 0.20.4 (Schmieder and Edwards, 2011) with the following settings: -trim_left 20 -trim_right 5 -trim_qual_right 30 -min_qual_mean 20 -min_len 70.
To extract nifH, we first assigned the metagenomic reads in each sample according to the results of BLAST analysis against the nifH database (Gaby and Buckley, 2014) with a threshold identity of 90%. The nifH database developed by Gaby and Buckley (2014) contains 32,954 nitrogenase reductase genes sequences including anfH, an alternative nitrogenase that is a paralog of nifH, and vnfH, a V–Fe alternative nitrogenase. Next, we used candidate reads in BLAST searches against the NCBI NR database with an E-value threshold of 10-10 and removed false nifH reads by manual inspection of the BLAST results. nifH genes were taxonomically assigned based on the best BLAST hit and counted. The retrieved reads were translated into amino acid sequences and incorporated into our database for proteome analysis with taxonomic information. If a best-hit was part of a genome sequence, nifD and nifK were picked up and used for database construction as described below.
To extract nifD and nifK, we modified the nifD and nifK database of Gaby and Buckley (2014), which contains only 268 and 315 sequences, respectively. To enrich the nifDK database, sequences were collected from (i) NCBI databases matched with respective keywords (“nitrogenase molybdenum-iron,” “nifD,” or “nifK”) and (ii) nifD and nifK encoded on genome sequences of nifH best-hits. The metagenomic reads in each sample were assigned according to the BLAST analysis against the nifD and nifK database with a threshold identity of 80% in-house. Next, the candidate reads of nifD and nifK from the metagenomic data were subjected to BLAST searches against the NCBI NR public database with an E-value threshold of 10-10, and false nifD or nifK reads were removed by manual inspection of the BLAST results. The resulting DNA sequences of nifD and nifK were taxonomically assigned based on the best BLAST hit, translated into amino acid sequences, and incorporated in our nitrogenase database for proteome analysis with taxonomic information.
Amplicon sequencing of 16S rRNA and metagenomics analysis targeting 31 AMPHORA genes (Wu and Eisen, 2008), which are housekeeping genes that are mostly present in single copies, were performed to analyze the bacterial community associated with the sorghum roots.
A partial bacterial 16S rRNA gene fragment was amplified by two-step PCR. First, a partial sequence of the V4 variable region was amplified with simultaneous addition of sequencing priming sites. The target region was amplified using primers 515F (5′-ACACTCTTTCCCTACACGACGCTCTTCCGATCTGTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGGACTACHVGGGTWTCTAAT-3′) (Caporaso et al., 2011). In the second round of PCR, the first-round PCR products were amplified using the above sequencing priming sites as PCR priming sites by adding dual-index tag sequences and flow cell binding sites of the Illumina adapter. The primer sequences were 5′-AATGATACGGCGACCACCGAGATCTACAC–Index2–ACACTCTTTCCCTACACGACGC-3′ (forward) and 5′-CAAGCAGAAGACGGCATACGAGAT–Index1–GTGACTGGAGTTCAGACGTGTG-3′ (reverse). Each 20-μL PCR mixture contained 0.2 μL TaKaRa ExTaq HS DNA polymerase (TaKaRa Bio, Shiga, Japan), 2.0 μL of buffer (10 × Ex buffer), 1.6 μL of 2.5 mM dNTP mix, 1.0 μL of each forward and reverse primer (10 μM), and 1 μL of template DNA. First-round PCR conditions were as follows: 94°C for 2 min, followed by 25 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 30 s. The PCR products were purified using an Agencourt AMPure XP purification system (Beckman Coulter, Brea, CA, United States) following the manufacturer’s instructions. Second-round PCR conditions were as follows: 94°C for 2 min; 12 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s; and a final 72°C for 5 min. The purified tag-indexed PCR products were quantified by using a Qubit 2.0 Fluorometer and dsDNA HS Assay Kit (Life Technologies, Carlsbad, CA, United States). Samples were then pooled in equal amounts and sequenced by using a 250-bp paired-end sequencing protocol with the MiSeq Sequencing Reagent Kit v. 2 (Illumina) at Bioengineering Lab Co., Ltd., (Kanagawa, Japan). Sequence reads of all samples were processed using QIIME2 (Caporaso et al., 2010). In summary, assembly of pair-end reads was performed with default settings and then primer removal, quality control, and chimeric sequence trimming were performed using dada2 (Callahan et al., 2016). Taxonomic classification was assigned using a SILVA 16S rRNA reference alignment [Release 132 (Quast et al., 2013)]. Each sample contained 0.3–1.7% of plant organelle reads. Therefore, all sequences classified as chloroplast or mitochondria were removed.
A universal marker set of 31 protein-encoding phylogenetic marker genes was extracted from the metagenome data of KM1 and KM2 using AMPHORA2 with default settings (Wu and Scott, 2012).
Proteins were extracted from the EBCs from the roots, and ∼50 μg of sample was separated by 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) and stained with Coomassie blue. Gel strips containing proteins ranging from 21.5 to 66.2 kDa (NifHDK protein sizes) were excised (Figure 1). Proteome analysis was performed as described previously (Kasahara et al., 2012; Bao et al., 2014). Briefly, the gel lanes were cut into strips of approximately 1 mm, and the gel strips were digested with trypsin. Nano-liquid chromatography (LC)–electrospray ionization–tandem mass spectrometry (MS/MS) analysis of the peptide mixtures was performed with an LTQ ion-trap MS (Thermo Fisher Scientific) coupled with a multidimensional high-performance LC (HPLC) Paradigm MS2 chromatograph (AMR, Inc., Tokyo, Japan) and nanospray electrospray ionization device (Michrom Bioresources, Inc., Auburn, CA, United States). The MS/MS data were searched against the protein database constructed in this study using Mascot program ver. 2.4 (Matrix Science, London, United Kingdom).
The bacterial cells extracted from the roots of lines KM1 and KM2 were pre-incubated at 28°C for 6 h in HM salt medium (Cole and Elkan, 1973) supplemented with 0.1% (w/v) arabinose and 0.025% (w/v) yeast extract to activate the bacterial cells for subsequent plating. These samples were serially diluted with sterilized water and plated on two types of agar plates, 1/100-strength nutrient medium (BD Biosciences, Franklin Lakes, NJ, United States) containing 1.5% agar and 10 mg L-1 polymixin B or 1/100-strength HM salt medium containing 1.5% agar and 10 mg L-1 polymixin B. This isolation strategy was used because Bradyrhizobium species are oligotrophic slow-growing bacteria (Okubo et al., 2012b, 2013) and resistant to polymyxin B (Hirayama et al., 2011; Piromyou et al., 2015a). After 10 days of cultivation at 28°C, slow-growing white colonies were picked up and re-streaked on the same types of agar plates for the first cultivation without polymixin B. This step was repeated one more time. Cell lysates of the resulting isolates were used to amplify the 16S rRNA gene by PCR as described previously (Itakura et al., 2009).
The 16S rRNA gene regions were amplified with Blend Taq polymerase (Toyobo, Osaka, Japan) and sequenced by direct PCR using universal forward (27f) and reverse (1492r) primers (Lane, 1991). The PCR conditions were as follows: preheating at 94°C for 2 min; 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 90 s; extension at 72°C for 60 s; a final extension at 72°C for 5 min, and cooling at 12°C. For direct PCR sequence analysis with the 27f primer, an ABI Prism 3130xl genetic analyzer was used with a BigDye Terminator v. 3.1 Cycle Sequencing Ready-Reaction Kit (Applied Biosystems, Foster City, CA, United States). Phylogenetic trees of the gene sequences were constructed in MEGA v. 7.0 software (Tamura et al., 2011) by using the neighbor-joining method (Saitou and Nei, 1987).
Genomic DNA of bradyrhizobial isolates TM122 or TM124 was extracted with an Illustra Bacteria GenomicPrep Mini Spin Kit (GE Healthcare, Little Chalfont, United Kingdom). DNA libraries were prepared with a Nextera DNA Sample Preparation Kit and sequenced on an MiSeq system using a MiSeq Reagent Nano Kit v. 2 (500 cycles) (both Illumina). Raw reads were trimmed in CLC Genomics Workbench v. 7.5.1 software (CLC Bio, Aarhus, Denmark) with the following parameters: ambiguous limit, 2; quality limit, 0.05; number of 5′ terminal nucleotides, 20; number of 3′ terminal nucleotides, 5; minimum number of nucleotides in reads, 70.
Shotgun sequence reads of the isolates were mapped to a reference genome of B. diazoefficiens USDA110T (Kaneko et al., 2002). Mapping was performed in CLC Genomics Workbench software with the following parameters: mismatch cost, 2; insertion cost, 3; deletion cost, 3; length fraction, 0.9; similarity fraction, 0.9. The shotgun sequences of TM122 and TM124 were assembled in CLC Genomics Workbench software with default parameters. The contigs containing nif genes were annotated by using the Microbial Genome Annotation Pipeline (MiGAP2).
Bradyrhizobium sp. TM122, and TM124 were pre-cultured in HM medium (Cole and Elkan, 1973) with shaking at 28°C. Five milliliters of the exponential growing culture were collected by centrifugation (13,000 × g, 3 min, 4°C) and washed twice with sterilized water. The cell density was adjusted to 107 cells/mL, and 1 mL of the cell suspension was inoculated into 7 mL of Rennie semi-solid medium (5 g of sucrose, 5 g of mannitol, 0.5 mL of sodium lactate, 0.8 g of K2HPO4, 0.2 g of KH2PO4, 0.1 g of NaCl, 28 mg of Na2FeEDTA, 25 mg of Na2MoO ⋅ 4H2O, 100 mg of yeast extract, 0.2 g of MgSO4 ⋅ 7H2O, 0.06 g of CaCl2, 5 μg of biotin, 10 μg of para-aminobenzoic acid, and 0.2 g of noble agar per L) (Rennie, 1981) in a 28-mL test tube, which was capped with a butyl rubber W-plug. After the test tubes were incubated at 28°C for 3 days, 10% (v/v) of the gas phase was replaced with acetylene (99.9999% vol/vol purity; Toho Acetylene Co., Tokyo, Japan). The samples were re-incubated at 28°C without shaking for 3 days. The gas phase (0.5 mL) was sampled to determine the concentration of ethylene by a gas chromatograph as described above.
Sorghum seeds of KM2 were shaken in 0.5% NaOCl solution for 1 min and then washed five times with sterile distilled water. Two sterilized seeds were planted in a Leonard’s jar containing sterilized vermiculite (Inaba et al., 2012) and plant nutrient solution (Mae and Ohira, 1981) with 0.05 mM of NH4NO3 under aseptic conditions (Supplementary Figure S3). A cell suspension of TM122 or TM124 was added to the sorghum seeds at a concentration of 107 cells per seed.
The plants were grown in a growth cabinet (LH300; NK System Co., Ltd., Osaka, Japan) that provided 65 mol photons m-2 s-1 of photosynthetically active radiation (400–700 nm) under a daily cycle of 16 h of light and 8 h of dark at 25°C. After 7 days, the seedlings were thinned out to one plant. At 28 days after inoculation, the roots were carefully harvested and then transferred into a 100-mL vial with a butyl rubber septum (SVG-100, Nichiden-Rika Glass Co., Ltd.). Acetylene (10 mL) was introduced into the bottles, which were incubated for 24 h at 25°C in the dark.
DNA sequences obtained by the metagenomic analysis and 16S rRNA gene amplicon, TM122 genome, and TM124 genome were deposited under the accession numbers DRA006465, DRA006466, DRA006492, and DRA006493 in the DDBJ Sequence Read Archive, respectively. The sequences of 16S rRNA genes of bacterial isolates were deposited in DDBJ/EMBL/GenBank under the accession numbers LC367220–LC367221 and LC433572–LC433596, respectively.
N2-fixing activities were monitored at different growth stages for four sorghum lines (KM1, KM2, KM4, and KM5) by an acetylene reduction assay in closed bottles (Figure 1 and Supplementary Figures S1, S2). Because sorghum tissues emit ethylene as a plant hormone, we calculated the ARA of bacterial nitrogenase as the difference in the rate of ethylene production in the presence and absence of acetylene (Supplementary Table S1).
We detected no ARA in the seedlings at 0 DAT or in the shoots and roots at 28 DAT (Table 1). However, we detected significant ARA values (KM1, 36.1 nmol plant-1 h-1, KM2, 52.6 nmol plant-1 h-1) at 71 DAT in the washed roots (Table 1). At 102 DAT, the roots of all lines showed significantly higher ARA values than at 71 DAT. Particularly, the roots of KM1 showed the highest ARA (585.8 nmol plant-1 h-1), followed by line KM2 (332.5 nmol plant-1 h-1). The ARA of KM5 roots (6.7 nmol plant-1 h-1) was significantly lower than that of KM1 (Table 1), indicating that root ARA depends on the sorghum genotype.
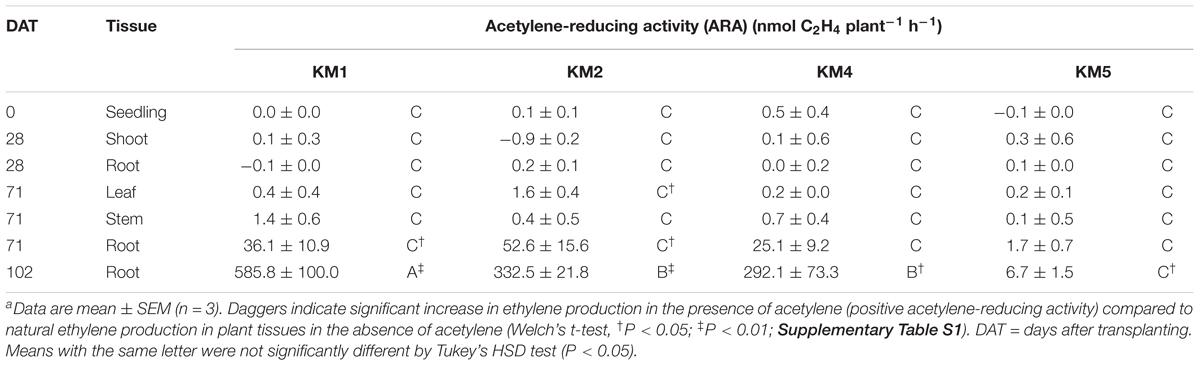
Table 1. Acetylene-reducing activity (ARA) of four sorghum lines (KM1, KM2, KM4, and KM5) at each growth stagea.
The above results suggest that N2 fixation in the roots is high in late growth stages of some sorghum lines. To confirm N2 fixation more directly, we conducted 15N2-feeding experiments using the roots at 102 DAT (Figure 1 and Supplementary Figure S2). When the roots were exposed to 15N2 for 23 h, 15N was apparently incorporated into the root tissues inhabited by bacteria including diazotrophs, indicating that the bacteria fixed N2 (Table 2). We calculated the 15N2-fixing activity from the 15N concentration (atom% excess) and total N amount (biomass × N content) (Table 2). Although the four lines showed 15N2-fixation, the highest activity was detected in the roots of KM1 (102.8 μg N plant-1 day-1), which was significantly higher than the values in the other lines (Table 2). KM2 showed the second highest activity (40.7 μg N plant-1 day-1).
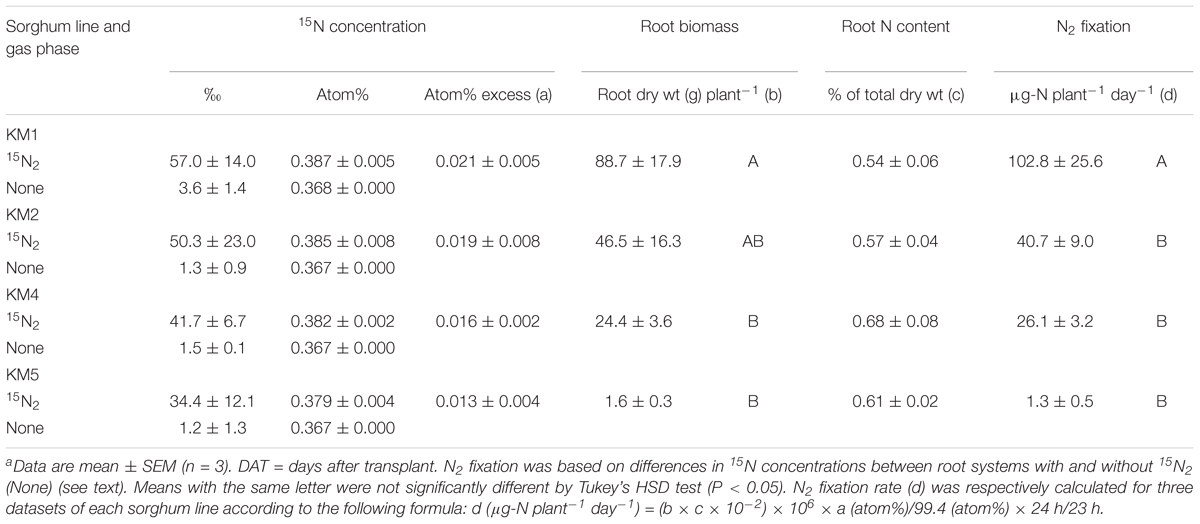
Table 2. Incorporation of 15N from 15N2 into sorghum roots at 102 days after transplanta.
ARA values were converted to gram N basis to compare the results of the ARA and 15N2-feeding experiments (Table 3). N2 fixation values in KM1 roots reached a maximum at 102 DAT and were nearly identical between the ARA and 15N2-feeding methods (98.4–102.8 μg N plant-1 day-1; Table 3). N2 fixation values in KM2 and KM4 roots were lower in the 15N2 feeding method than in ARA measurement. KM5 roots consistently showed the lowest values (∼1 μg N plant-1 day-1) in both methods. Thus, we focused on the root microbiomes of KM1 and KM2 at 102 DAT for subsequent metagenome, proteome, and isolation analyses (Figure 1) because their N2-fixing activities were the highest (Table 2).
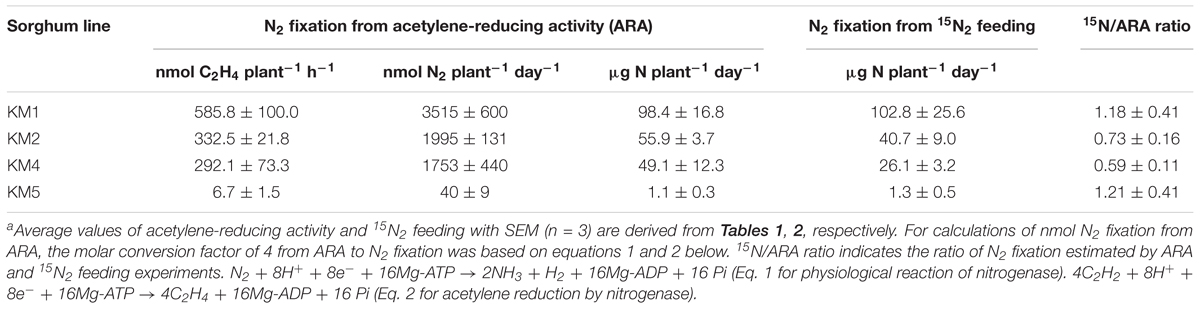
Table 3. Comparison of acetylene-reducing activity and 15N2 feeding methods of evaluating N2-fixing activity in roots of four sorghum lines at 102 days after transplanta.
We extracted bacterial cells from the roots of KM1 and KM2 with three biological replicates at 102 DAT. The metagenomes of the six DNA samples were sequenced by MiSeq. The metagenomic data comprised 4.3–5.4 × 106 reads and 196–208-bp average lengths per DNA sample (Supplementary Table S2).
By using a nif database (see section “Materials and Methods”), we retrieved genes encoding nitrogenase reductase (NifH) and dinitrogenase (NifDK) from the six datasets. Most nitrogenase genes were proteobacterial nifHDK genes (Supplementary Table S3 for KM1, Supplementary Table S4 for KM2) and did not include vnfH, anfH, or archaeal nitrogenase genes. At the genus level, Bradyrhizobium nifHDK genes were most abundant (Figure 2A, 55–86% in each nitrogenase gene of KM1 and KM2), and three nif structural genes (nifHDK) of Bradyrhizobium were consistently detected among the six datasets including sorghum lines KM1 and KM2 (Figure 2A and Supplementary Table S5). In contrast, nifHDK genes of other genera showed relatively low abundance (Figure 2A, 1–7% in each nitrogenase gene of KM1 and KM2), with parts of three nif structural genes (Supplementary Table S5). For example, nif structural genes from Azorhizobium were exclusively found in nifDK genes from the sorghum line KM1 (Supplementary Table S5).
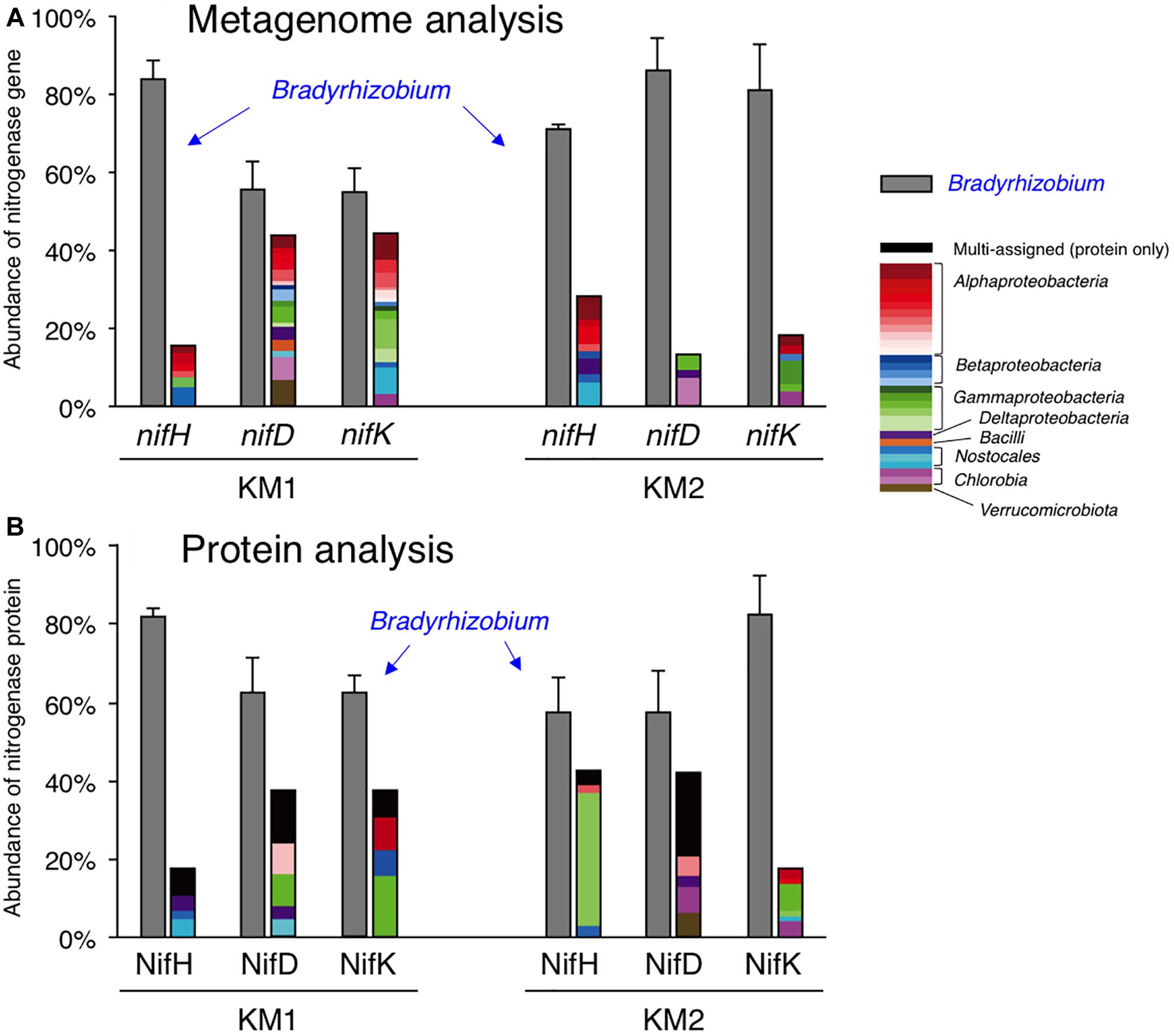
Figure 2. Relative abundance of nifHDK reads (A) and NifHDK peptides (B) of respective bacterial genera. Data represent the average of three biological replications. The color of bars indicates assigned taxonomy. Gray indicates Bradyrhizobium. Black in panel B indicates multi-assigned peptides. Bar indicates standard error of three biological replicates.
BLAST analysis indicated that most bradyrhizobial nifHDK genes belonged to the non-nodulating Bradyrhizobium sp. S23321 (AP012279) (Okubo et al., 2012a) and photosynthetic B. oligotrophicum S58T (AP012603) (Okubo et al., 2013) (Supplementary Tables S6, S7). Among the Bradyrhizobium nifHDK reads, S23321 nifH genes accounted for 40% of the nifH reads from KM1 reads and 45% of the nifH reads from KM2 reads. S58T nifH genes accounted for 17% and 21%, respectively (Supplementary Tables S6, S7). Abundant nifDK reads were also observed in S23321 (31–37%) and S58 (11–29%) among Bradyrhizobium nifDK reads of sorghum lines KM1 and KM2 (Supplementary Tables S6, S7). These results suggest that Bradyrhizobium-homologous nifHDK genes are abundant in the KM1 and KM2 root microbiomes. The expression of nitrogenase genes is regulated by oxygen and nitrogen, as microbial N2 fixation requires a large amount of energy (Yoneyama et al., 2017; Rosenblueth et al., 2018). Thus, we examined which nifHDK genes were expressed in the plant environments by determining their resulting protein levels.
We analyzed the proteomes of EBCs from the roots of KM1 and KM2 at 102 DAT (Figure 1). The proteins were separated by SDS–PAGE, and we excised gel strips with protein sizes ranging from 21.5 to 66.2 kDa because the estimated sizes of NifH as well as NifD and NifK (Boyd and Peters, 2013; Gaby and Buckley, 2014) were 30–32 and 55–60 kDa, respectively (Figure 1). First, we constructed a database of NifHDK peptides based on the DNA sequences of nifHDK genes from our metagenome data (Data Set 1). Next, amino acid sequences of the peptide generated by proteome analysis (Data Set 2) were assigned based on 100% identity (Data Set 2) in the NifHDK database (Data Set 1). The results were summarized at the genus level (Figure 2B and Table 4), which revealed that the NifHDK peptides were from 15 genera and 1 family of Alpha-, Beta-, Gamma-, and Delta-proteobacteria and cyanobacteria in the root microbiomes. Among them, Bradyrhizobium NifHDK peptides were simultaneously detected in all three sampling replicates with an apparently higher abundance than those of other genera (Table 4 and Supplementary Table S8). At the strain level, the bradyrhizobial NifHDK peptides (Table 4) were heavily assigned to NifHDK of Bradyrhizobium sp. S23321 and B. oligotrophicum S58 in sorghum lines KM1 and KM2, although the NifD peptide of B. oligotrophicum S58 was not detected in sorghum line KM2 (Supplementary Table S9). In contrast, NifH peptides of Kosakonia of Gammaproteobacteria were abundantly detected in KM2, whereas the corresponding NifDK peptides were not abundant (Table 4 and Supplementary Table S8). Three peptides (NifHDK) were not simultaneously detected in non-bradyrhizobia (Table 4).
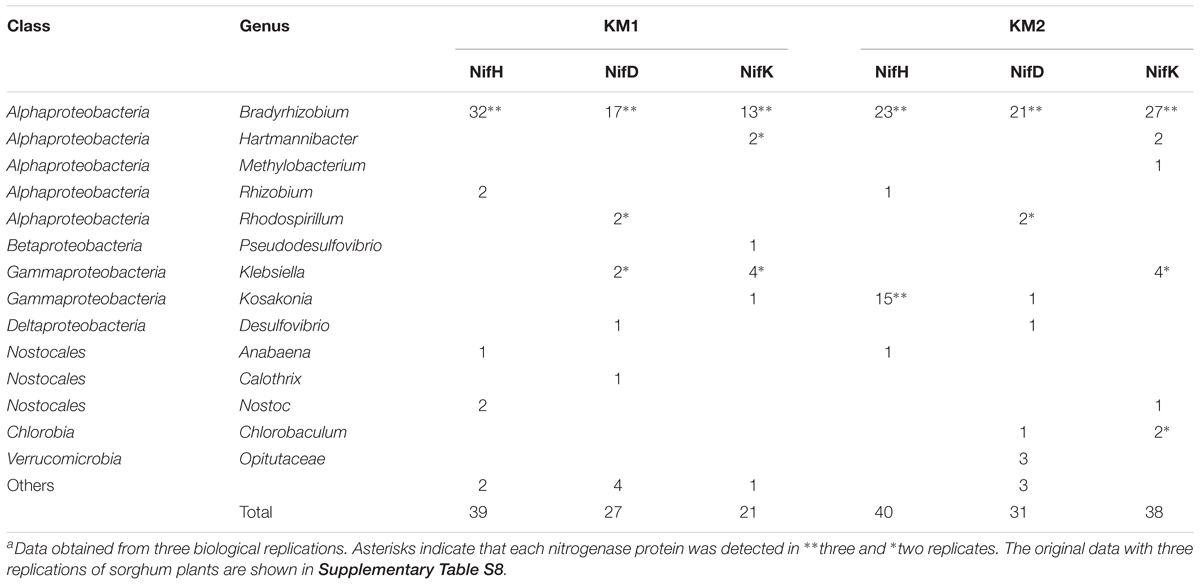
Table 4. Total number of NifHDK peptides in root microbiomes of sorghum lines KM1 and KM2a.
Therefore, our culture-independent results indicate that Bradyrhizobium species fixed N2 at the late growth stage of sorghum lines KM1 and KM2 under field conditions. Particularly, close relatives of Bradyrhizobium sp. S23321 and B. oligotrophicum S58 are likely crucial candidates as functional diazotrophs in field-grown sorghum roots according to our proteome results.
We sequenced amplicons of 16S rRNA genes to evaluate the relative abundance of Bradyrhizobium members in the root microbiomes of the four sorghum lines at 102 DAT. Taxonomic classification represented Amplicon Sequence Variants (ASVs) generated by dada2 (Callahan et al., 2016) in QIIME2 (Caporaso et al., 2010). The relative abundances of Bradyrhizobium (ASV 0004) in KM1 and KM2 root microbiomes were 2.9 and 3.0%, respectively (Figure 3). Phylogenetic analysis of our metagenomic data using 31 AMPHORA genes also showed a similar relative abundance of Bradyrhizobium of KM1 and KM2 (3.6%) (Supplementary Figure S4). These results suggest that Bradyrhizobium species are consistent members of the root microbiomes of sorghum lines KM1 and KM2.
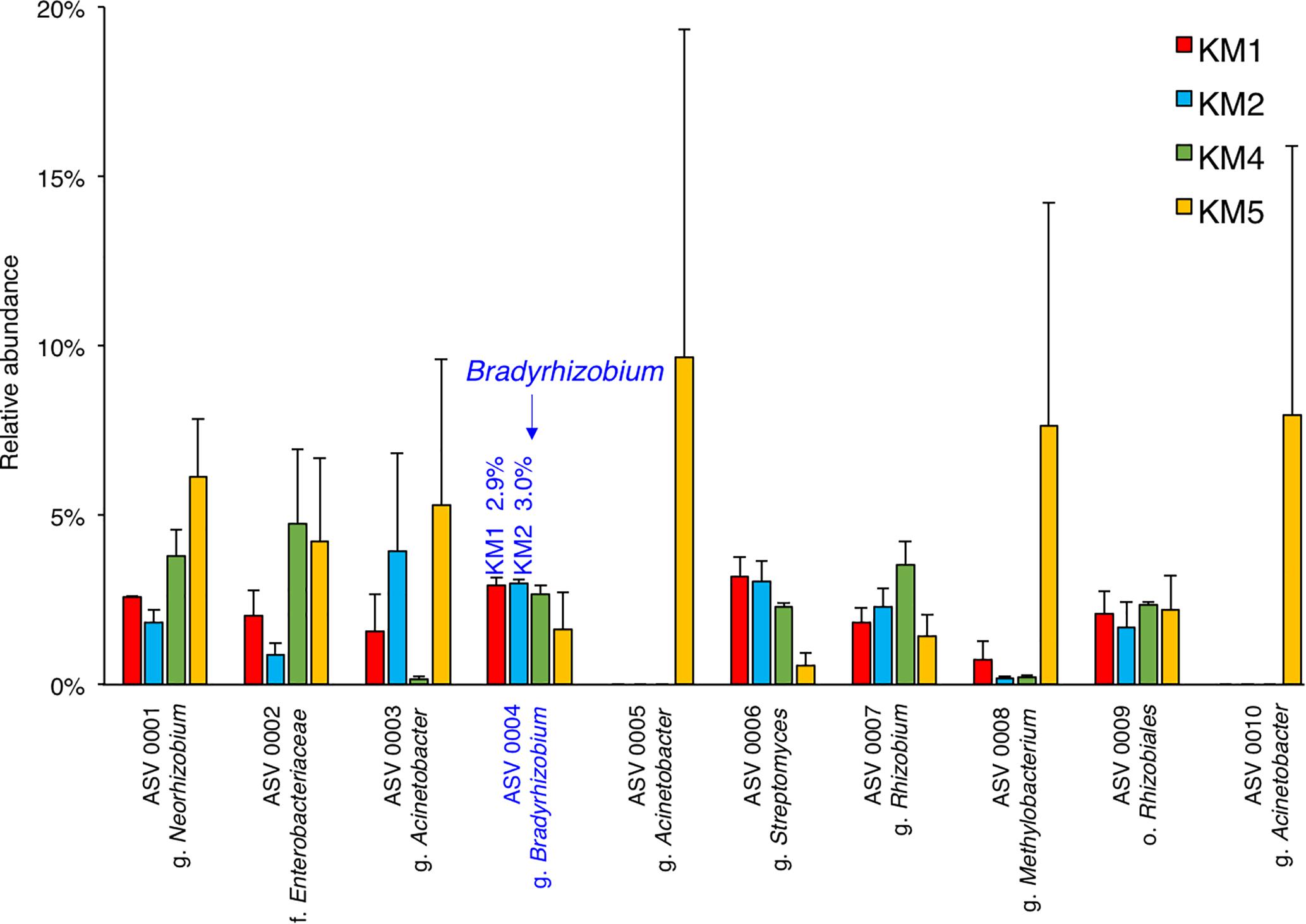
Figure 3. Relative abundance of ten most abundant ASV (amplicon sequence variants) in sorghum lines KM1, KM2, KM4, and KM5 by amplicon analysis of 16S rRNA genes. The ASVs were identified at the lowest possible classification, where o. is order, f. is family and g. is genus. Bar indicates standard error of three biological replicates.
Because horizontal transfer of nif genes among several linages of bacteria (Boyd and Peters, 2013), and even within Bradyrhizobium (Okubo et al., 2016), has been suggested, Bradyrhizobium may not always harbor bradyrhizobial nif genes. Therefore, we isolated bradyrhizobia from the sorghum roots on two types of oligotrophic media (1/100-strength nutrient agar medium or 1/100-strength HM agar medium) containing polymixin B. Twenty-eight slow-growing white colonies were isolated, eight of which were identified as Bradyrhizobium based on 16S rRNA gene sequencing (Supplementary Table S10). The other 20 isolates belonged to Ancylobacter, Boseae, Sphingobium, Mesorhizobium, Deinococcus, Mycobacterium, and Terrabacter.
Phylogenetic analysis based on 16S rRNA gene sequences indicated that bradyrhizobial isolates TM122 and TM124 were very close to photosynthetic B. oligotrophicum S58T (Okubo et al., 2013) and non-nodulating Bradyrhizobium sp. S23321 (Okubo et al., 2012a), respectively (Figure 4). The isolate TM220 apparently belonged to B. diazoefficiens, while the other bradyrhizobial isolates (TM102, TM228, TM233, TM239, and TM221) were close to B. japonicum. Both species are typical soybean endosymbionts (Kaneko et al., 2002, 2011). Metagenome and subsequent proteome analyses suggested that potential candidates of functional diazotrophs in the field-grown sorghum roots were close relatives of Bradyrhizobium sp. S23321 and B. oligotrophicum S58 (Supplementary Tables S6, S7, S9). Thus, we used the isolates TM122 and TM124 for subsequent genome analysis and N2 fixation experiments.
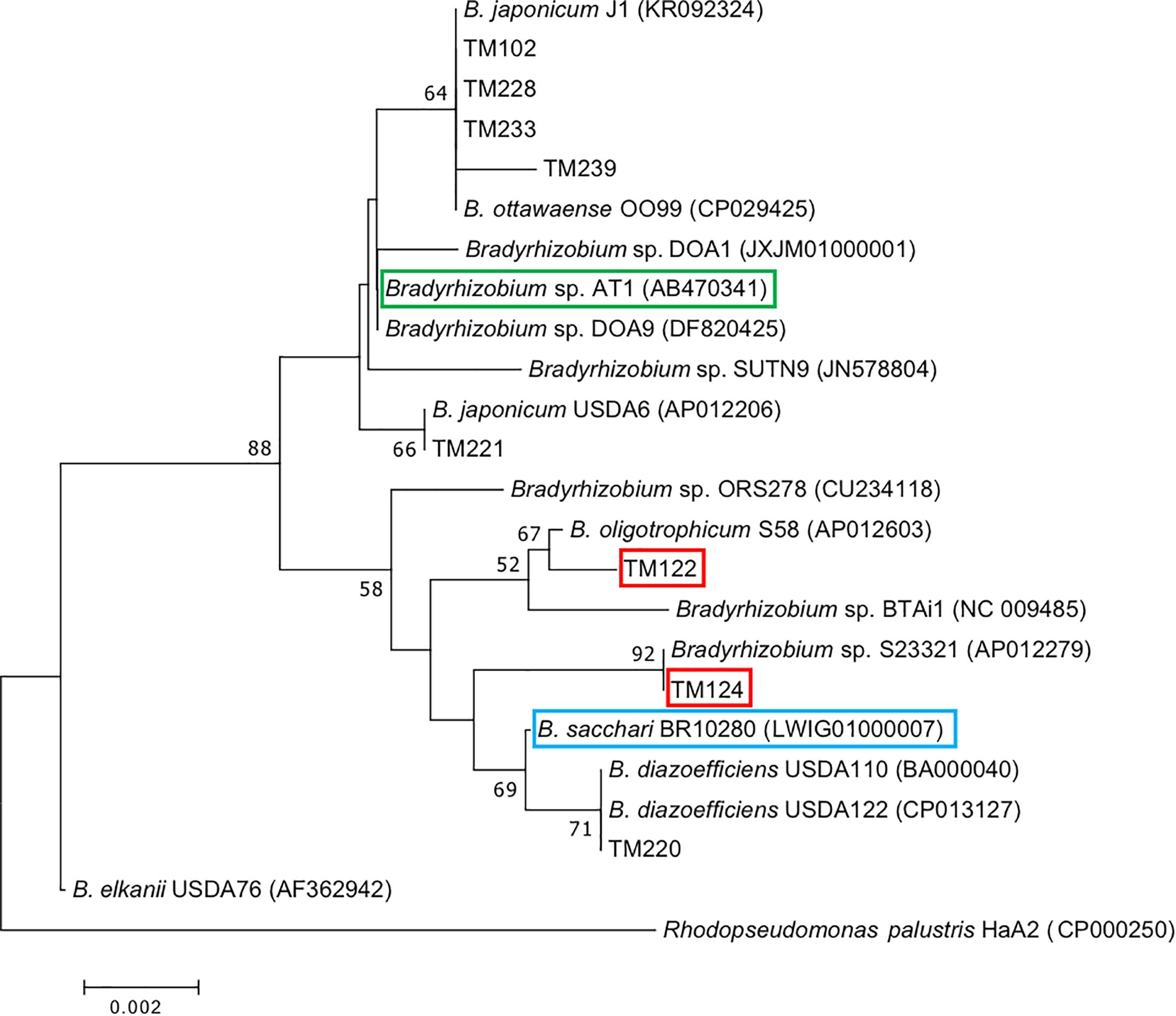
Figure 4. Phylogenetic tree of 16S rRNA genes of bradyrhizobial isolates from sorghum roots (prefix TM) constructed by the neighbor-joining method. Numbers at the nodes are percentages of 1000 bootstrap replications supporting the partition. Red boxes indicate functional diazotrophs suggested by our omics analysis derived from sorghum roots (TM122, TM124), while blue and green boxes indicate diazotrophic bradyrhizobia from sugarcane (BR10280) and sweet potato (AT1), respectively.
Mapping of the MiSeq reads of TM122 and TM124 to the soybean-nodulating B. diazoefficiens USDA110T genome showed that their genomes lacked symbiosis island structures of soybean bradyrhizobia that include nod genes (Figure 5A). A BLAST search confirmed that the genomes of both strains lacked the common nodulation genes (nodDYABC) required for legume nodulation (Andrews and Andrews, 2017). After assembling the MiSeq reads of TM122 and TM124, we retrieved the sequence contigs containing nif genes. The organization of nif genes in each genome resembled those of B. oligotrophicum S58T and Bradyrhizobium sp. S23321, but differed from those of nodulating B. diazoefficiens USDA110T, which contains symbiosis islands (Kaneko et al., 2002; Kaneko et al., 2011; Okubo et al., 2016).
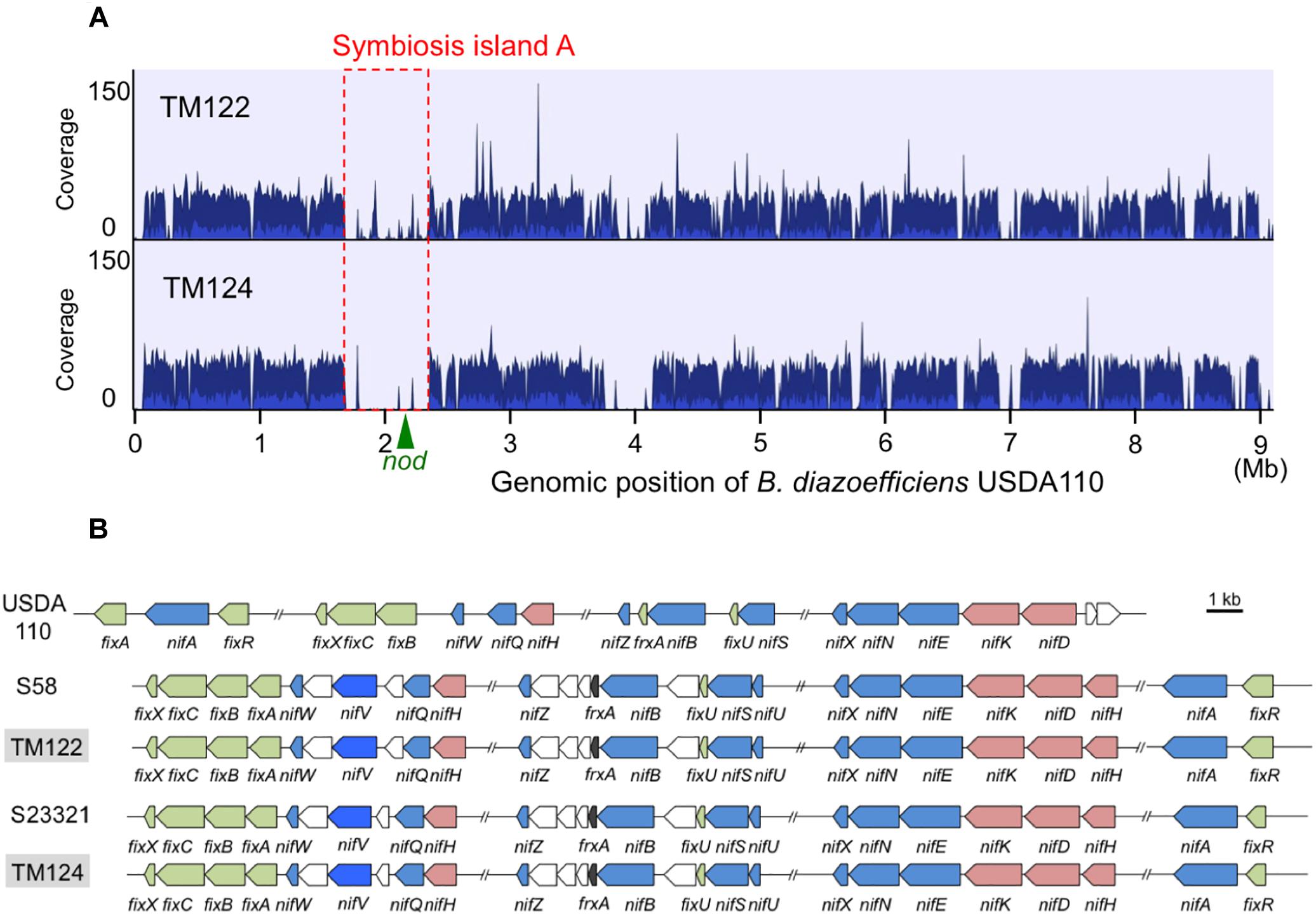
Figure 5. Genomic comparison between sorghum root isolates of TM122 and TM124 and phylogenetically close bradyrhizobia. (A) Mapping of MiSeq reads of TM122 (1,191,548 reads of 213 bp in average length) and TM124 (1,190,703 reads of 208 bp in average length) on B. diazoefficiens USDA110T genome with symbiosis island (Kaneko et al., 2002). The coverage within symbiosis island (red dotted lines) including nod genes (Kaneko et al., 2002, 2011) were apparently lower than those of other genomic regions. Cluster of nif genes of USDA110T is located on symbiosis island (Kaneko et al., 2002). (B) The organization of nif genes of TM122 and TM124 (light gray) compared to those of B. diazoefficiens USDA110T, B. oligotrophicum S58, and Bradyrhizobium sp. S23321. Colored pentagons indicate (pink) structural nifHDK, (blue) other nif, and (green) fix genes.
The potential for N2 fixation by the two bradyrhizobial isolates was evaluated in free-living and plant-associated states (Table 5). Free-living cells of TM122 and TM124 in semi-solid medium produced ethylene over time in the presence of acetylene (Supplementary Figure S5) and showed significant ARA in the semi-solid medium (Table 5). This result indicates that both isolates can fix N2 in a free-living state.
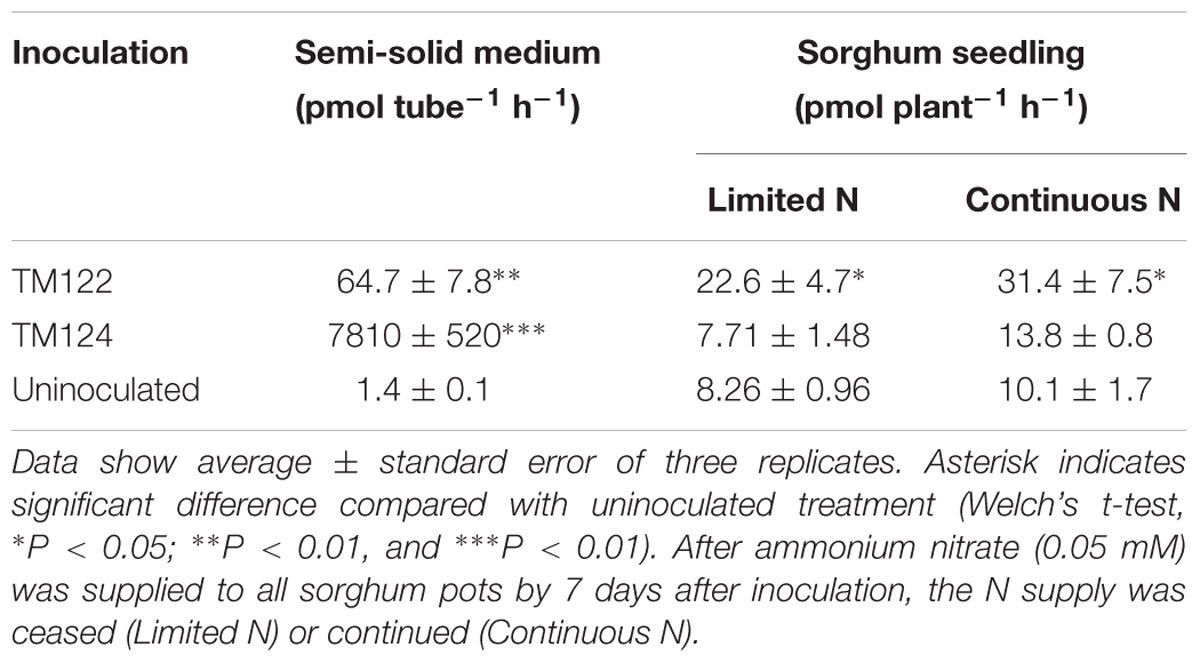
Table 5. Acetylene-reducing activity (ARA) of free-living cells of Bradyrhizobium sp. TM122 and TM124 in semi-solid medium and sorghum seedlings inoculated with TM122 and TM124.
Thereafter, seeds from the sorghum line KM2 were inoculated with isolates TM122 or TM124 and aseptically grown for 28 days after inoculation (Supplementary Figure S3). We detected weak but significant ARA (22.6–31.4 pmol plant-1 h-1) in the roots inoculated with TM122 compared to that in uninoculated roots. In contrast, TM124-inoculated roots showed no ARA under the two different treatments of N supply under our experimental conditions (Table 5 and Supplementary Figure S3), which may have been because of weak colonization or low nitrogenase expression in the sorghum seedlings following TM124 inoculation.
Our omics results revealed that functional N2-fixing bradyrhizobia associated with the roots of field-grown sorghum plants have significant N2-fixing activities in late growth stages. Experimentally, in addition to the nifH gene encoding nitrogenase reductase (NifH), the usage of nifDK genes encoding NifDK facilitated the reliable identification of functional diazotrophs in sorghum roots (Figure 2), as the nifH gene is often diversely multiplicated (Figure 5B; Okubo et al., 2016). Here, we discuss the results in terms of the level of nitrogen fixation, phylogenetic comparison with diazotrophs in other crops, and Bradyrhizobium diversity in N2 fixation and nodulation.
The N2-fixing activities of the field-grown sorghum roots (102 DAT) were determined by both the ARA and 15N2-feeding methods (Tables 1, 2). The N2-fixing activities of the roots were consistent between the methods (Table 3), indicating that the values obtained for the N2-fixing activities are reliable. Wani et al. (1984) evaluated the ARA of intact sorghum plants grown in sand, soil, and farmyard manure. Although the ARA depended on the manure content, temperature, and sorghum cultivar, the average ARA values of 15 cultivars was 625 nmol C2H4 plant-1 h-1 at 30–35°C during the day. This is similar to the value of 585.8 nmol C2H4 plant-1 h-1 in the KM1 roots (Table 1), although the experiments were performed in very different environments (Supplementary Table S11).
Active expression of the dinitrogenase reductase-encoded gene (nifH) was abundant in sugarcane stems, sweet potato stems, and pineapple leaves, which are rich in sugars and organic acids (Yoneyama et al., 2017). The leaves and stems of field-grown sorghum showed no ARA at 28 and 71 DAT except for a low ARA value for the sorghum line KM2 (Table 1 and Supplementary Table S1). In contrast, considerably higher ARA values were detected in sorghum lines KM1, KM2, and KM4 at 71 DAT (Table 1). These results suggest that the functional N2-fixing bacteria exclusively reside in the roots of field-grown sorghum.
Bradyrhizobium are versatile bacteria inhabiting soils and plants (Zhalnina et al., 2013; VanInsberghe et al., 2015; Jones et al., 2016; Szoboszlay et al., 2017). Genomic and ecological studies have focused on soybean bradyrhizobia (B. diazoefficiens and B. japonicum) because of their prominent roles in nodule formation and their agricultural significance (Masuda et al., 2016; Shiro et al., 2016; Saeki et al., 2017; Siqueira et al., 2017). However, photosynthetic diazotrophs often nodulate an aquatic legume, Aeschynomene, but sometimes lack nod genes (Giraud et al., 2007; Okubo et al., 2012a; Okazaki et al., 2016), and, thus, appear to function as an intermediate between free-living diazotrophs and classical nodulating diazotrophs (Okubo et al., 2013). Our results suggest that sorghum roots harbor functional N2-fixing bradyrhizobia that are similar to photosynthetic B. oligotrophicum S58T and Bradyrhizobium sp. S23321.
Recent metatranscriptome analyses targeting nifH genes suggested that Bradyrhizobium species play a role in sugarcane-associated N2 fixation (Thaweenut et al., 2011; Fischer et al., 2012). Representative isolates, including BR10280T, from surface-sterilized sugarcane tissues (Rouws et al., 2014) were recently classified as B. sacchari (de Matos et al., 2017). The phylogenetic position of B. sacchari BR10280T is close to that of B. diazoefficiens USDA110T, a typical soybean-nodulating bacterium (Figure 4). Bradyrhizobium sp. AT1 (Terakado-Tonooka et al., 2013; Okubo et al., 2016) from sweet potato is close to the soybean-nodulating B. japonicum USDA6T (Figure 4). Although the phylogenetic positions of eight sorghum isolates were widely distributed within Bradyrhizobium (Figure 4), our omics data suggest that the candidates responsible for in planta N2 fixation are close relatives of B. oligotrophicum S58T and Bradyrhizobium sp. S23321 such as TM122 and TM124. Recently, diverse non-diazotrophic bradyrhizobia were endophytically colonized in Arabidopsis thaliana roots (Schneijderberg et al., 2018). Therefore, functional diversity may be important for symbiosis and N2 fixation.
Two bradyrhizobial isolates from the sorghum roots based on our omics data showed potential for N2 fixation (ARA), at least in sorghum seedlings inoculated with TM122 (28 days after sowing) and in the free-living state for TM122 and TM124 (Table 5). Under field conditions, no ARA was detected in the sorghum roots at 28 DAT (53 days after sowing), but the highest ARA values were observed at 102 DAT (Table 1). There are two possibilities for explaining this growth-stage dependent phenomenon. First, N fertilization depletion may explain the highest nitrogenase activity at 102 DAT under field conditions. Second, physiological factors in plant-microbe interactions were likely involved. N2 fixation also occurs at a high rate in the late growth stages of field-grown maize, although diazotrophic bacteria were not identified (van Deynze et al., 2018). Thus, the expression of bradyrhizobial nitrogenase may require specific environments that provide appropriate carbon source supply, chemical signals, and low oxygen concentration (Terakado-Tonooka et al., 2008; Rouws et al., 2014; Yoneyama et al., 2017) along with plant stage and microbial community development in the roots. Recently, root mucilage of land maize was found to be a suitable microenvironment for N2 fixation and subsequent plant assimilation of fixed nitrogen (van Deynze et al., 2018).
Bradyrhizobium cells often endophytically colonize plant roots (Chaintreuil et al., 2000; Okubo et al., 2012a, 2013; Piromyou et al., 2015b). Bradyrhizobium sp. SUT-PR9 deeply invaded the central tissues of rice roots (Piromyou et al., 2015a,b). Cells of B. oligotrophicum S58T and Bradyrhizobium sp. ORS278 inhabited the surface tissues (epidermal and surface cortex cells) of cultivated and wild rice plants (Chaintreuil et al., 2000; Okubo et al., 2012b, 2013). Thus, bradyrhizobia may endophytically colonize sorghum root tissues. Microscopy studies should be conducted to localize bradyrhizobia in sorghum roots.
Interactions between diazotrophs and host plant genotypes have remained unclear in the use of N2-fixing bacteria in crops (Okubo et al., 2013; Yoneyama et al., 2017). Further analyses targeting bradyrhizobia and sorghum genotypes may contribute to the understanding of the interactions between N2-fixing bradyrhizobia and sorghum plants for sustainable agriculture.
The highest nitrogen-fixing activities were detected in the roots of sorghum lines KM1 and KM2 in the late growth stage (102 DAT). Nitrogenase structural genes (nifH, nifD, and nifK) in the metagenome sequences were assigned to Bradyrhizobium species by metagenome analysis. Proteome analysis indicated that three NifHDK proteins of Bradyrhizobium species were consistently detected across sample replicates. Most of these proteins were assigned to photosynthetic B. oligotrophicum S58T and non-nodulating Bradyrhizobium sp. S23321. The polyphasic approach suggested that major functional N2-fixing bacteria in the sorghum roots are unique bradyrhizobia that resemble non-nodulating Bradyrhizobium sp. S23321 and photosynthetic B. oligotrophicum S58T.
SH, TK, KY, TF, TT, and KM designed the research. SH, TM, SW, and YK analyzed the data. SH and TM analyzed the DNA sequences of PCR products and metagenome. YK performed the proteome analysis. TK, SH, and SW characterized isolated diazotrophs. SH and KM wrote the article.
This work was supported by a Grant-in-Aid for Scientific Research (A) (26252065) and (B) (18H02112) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by a grant from Earthnote Co., Ltd.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Mihoko Tokunaga, Maho Tokunaga, Hisayasu Tokunaga, Umeko Sakuma, and Junichi Yoneda (Earthnote) for their support in cultivation, sampling, acetylene reduction, and for providing information on sorghum.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00407/full#supplementary-material
Andrews, M., and Andrews, M. E. (2017). Specificity in legume-rhizobia symbioses. Int. J. Mol. Sci. 18:E705. doi: 10.3390/ijms18040705
Bao, Z., Okubo, T., Kubota, K., Kasahara, Y., Tsurumaru, H., Anda, M., et al. (2014). Metaproteomic identification of diazotrophic methanotrophs and their localization in root tissues of field-grown rice plants. Appl. Environ. Microbiol. 80, 5043–5052. doi: 10.1128/AEM.00969-14
Bao, Z., Shinoda, R., and Minamisawa, K. (2016). Draft genome sequence of Methylosinus sp. Strain 3S-1, an isolate from rice root in a low-N paddy field. Genome Announc. 4:e00932–16. doi: 10.1128/genomeA.00932-16
Boddey, R. M., Polidoro, J. C., Resende, A. S., Alves, B. J. R., and Urquiaga, S. (2001). Use of the 15N natural abundance technique for the quantification of the contribution of N2 fixation to sugar cane and other grasses. Aust. J. Plant Physiol. 28, 889–895. doi: 10.1071/PP01058
Boyd, E. S., and Peters, J. W. (2013). New insights into the evolutionary history of biological nitrogen fixation. Front. Microbiol. 4:201. doi: 10.3389/fmicb.2013.00201
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.3389/fmicb.2013.00201
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522. doi: 10.1073/pnas.1000080107
Cavalcante, V. A., and Dobereiner, J. (1988). A new acid-tolerant nitrogen-fixing bacterium associated with sugarcane. Plant Soil 108, 23–31. doi: 10.1007/BF02370096
Chaintreuil, C., Giraud, E., Prin, Y., Lorquin, J., Bâ, A., Gillis, M., et al. (2000). Photosynthetic bradyrhizobia are natural endophytes of the African wild rice Oryza breviligulata. Appl. Environ. Microbiol. 66, 5437–5447. doi: 10.1007/BF02370096
Coelho, M. R., de Vos, M., Carneiro, N. P., Marriel, I. E., Paiva, E., and Seldin, L. (2008). Diversity of nifH gene pools in the rhizosphere of two cultivars of sorghum (Sorghum bicolor) treated with contrasting levels of nitrogen fertilizer. FEMS Microbiol. Lett. 279, 15–22. doi: 10.1111/j.1574-6968.2007.00975.x
Cole, M. A., and Elkan, G. H. (1973). Transmissible resistance to penicillin G, neomycin, and chloramphenicol in Rhizobium japonicum. Antimicrob. Agents Chemother. 4, 248–253. doi: 10.1128/AAC.4.3.248
de Matos, G., Zilli, J., de Araújo, J., Parma, M., Melo, I., Radl, V., et al. (2017). Bradyrhizobium sacchari sp. nov., a legume nodulating bacterium isolated from sugarcane roots. Arch. Microbiol. 199, 1251–1258. doi: 10.1007/s00203-017-1398-6
Elbeltagy, A., Nishioka, K., Sato, T., Suzuki, H., Ye, B., Hamada, T., et al. (2001). Endophytic colonization and in planta nitogen fixation by a Herbaspirillum sp. isolated from wild rice spcies. Appl. Environ. Microbiol. 67, 5285–5293. doi: 10.1128/AEM.67.11.5285-5293.2001
Fischer, D., Pfitzner, B., Schmid, M., Simões-Araújo, J. L., Reis, V. M., Pereira, W., et al. (2012). Molecular characterisation of the diazotrophic bacterial community in uninoculated and inoculated field-grown sugarcane (Saccharum sp.). Plant Soil 356, 83–99. doi: 10.1007/s11104-011-0812-0
Gaby, J. C., and Buckley, D. H. (2014). A comprehensive aligned nifH gene database: a multipurpose tool for studies of nitrogen-fixing bacteria. Database 2014:bau001. doi: 10.1093/database/bau001
Giraud, E., Moulin, L., Vallenet, D., Barbe, V., Cytryn, E., Avarre, J. C., et al. (2007). Legumes symbioses: absence of Nod genes in photosynthetic bradyrhizobia. Science 316, 1307–1312. doi: 10.1126/science.1139548
Hirayama, J., Eda, S., Mitsui, H., and Minamisawa, K. (2011). Nitrate-dependent N2O emission from intact soybean nodules via denitrification by Bradyrhizobium japonicum bacteroids. Appl. Environ. Microbiol. 77, 8787–8790. doi: 10.1128/AEM.06262-11
Ikeda, S., Kaneko, T., Okubo, T., Rallos, L. E., Eda, S., Mitsui, H., et al. (2009). Development of a bacterial cell enrichment method and its application to the community analysis in soybean stems. Microb. Ecol. 58, 703–714. doi: 10.1007/s00248-009-9566-0
Inaba, S., Ikenishi, F., Itakura, M., Kikuchi, M., Eda, S., Chiba, N., et al. (2012). N2O emission from degraded soybean nodules depends on denitrification by Bradyrhizobium japonicum and other microbes in the rhizosphere. Microbes Environ. 27, 470–476. doi: 10.1264/jsme2.ME12100
Itakura, M., Saeki, K., Omori, H., Yokoyama, T., Kaneko, T., Tabata, S., et al. (2009). Genomic comparison of Bradyrhizobium japonicum strains with different symbiotic nitrogen-fixing capabilities and other Bradyrhizobiaceae members. ISME J. 3, 326–339. doi: 10.1038/ismej.2008.88
James, E. K. (2000). Nitrogen fixation in endophytic and associative symbiosis. Field Crops Res. 65, 197–209. doi: 10.1016/S0378-4290(99)00087-8
Jones, F. P., Clark, I. M., King, R., Shaw, L. J., Woodward, M. J., and Hirsch, P. R. (2016). Novel European free-living, non-diazotrophic Bradyrhizobium isolates from contrasting soils that lack nodulation and nitrogen fixation genes - a genome comparison. Sci. Rep. 6:25858. doi: 10.1038/srep25858
Kaneko, T., Maita, H., Hirakawa, H., Uchiike, N., Minamisawa, K., Watanabe, A., et al. (2011). Complete genome sequence of the soybean symbiont Bradyrhizobium japonicum strain USDA6T. Genes 2, 763–787. doi: 10.3390/genes2040763
Kaneko, T., Nakamura, Y., Sato, S., Minamisawa, K., Uchiumi, T., Sasamoto, S., et al. (2002). Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 9, 189–197. doi: 10.1093/dnares/9.6.189
Kasahara, Y., Morimo, H., Kuwan, M., and Kadoya, R. (2012). Genome-wide analytical approaches using semi-quantitative expression proteomics for aromatic hydrocarbon metabolism in Pseudomonas putida F1. J. Microbiol. Meth. 91, 434–442. doi: 10.1016/j.mimet.2012.09.017
Khawaja, C., Janssen, R., Rutz, D., Luquet, D., Trouche, G., Reddy, B. V. S., et al. (2014). Energy Sorghum: An alternative Energy crop A Handbook. Available at: http://oar.icrisat.org/9049/
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (Chichester: John Wiley and Sons), 115–175.
Mae, T., and Ohira, K. (1981). The remobilization of nitrogen related to leaf growth and senescence in rice plants (Oryza sativa L.). Plant Cell Physiol. 22, 1067–1074. doi: 10.1093/oxfordjournals.pcp.a076248
Masuda, S., Saito, M., Sugawara, C., Itakura, M., Eda, S., and Minamisawa, K. (2016). Identification of the hydrogen uptake gene cluster for chemolithoautotrophic growth and symbiosis hydrogen uptake in Bradyrhizobium diazoefficiens. Microbes Environ. 31, 76–78. doi: 10.1264/jsme2.ME15182
Minamisawa, K., Imaizumi-Anraku, H., Bao, Z., Shinoda, R., Okubo, T., and Ikeda, S. (2016). Are symbiotic methanotrophs key microbes for N acquisition in paddy rice root? Microbes Environ. 31, 4–10. doi: 10.1264/jsme2.ME15180
Naylor, D., DeGraaf, S., Purdom, E., and Coleman-Derr, D. (2017). Drought and host selection influence bacterial community dynamics in the grass root microbiome. ISME J. 11, 2691–2704. doi: 10.1038/ismej.2017.118
Okazaki, S., Tittabutr, P., Teulet, A., Thouin, J., Fardoux, J., Chaintreuil, C., et al. (2016). Rhizobium-legume symbiosis in the absence of Nod factors: two possible scenarios with or without the T3SS. ISME J. 10, 64–74. doi: 10.1038/ismej.2015.103
Okubo, T., Fukushima, S., Itakura, M., Oshima, K., Longtonglang, A., Teaumroong, N., et al. (2013). Genome analysis suggests that the soil oligotrophic bacterium Agromonas oligotrophica (Bradyrhizobium oligotrophicum) is a nitrogen-fixing symbiont of Aeschynomene indica. Appl. Environ. Microbiol. 79, 2542–2551. doi: 10.1128/AEM.00009-13
Okubo, T., Fukushima, S., and Minamisawa, K. (2012a). Evolution of Bradyrhizobium-Aeschynomene mutualism: living testimony of the ancient world or highly evolved state? Plant Cell Physiol. 53, 2000–2007. doi: 10.1093/pcp/pcs150
Okubo, T., Tsukui, T., Maita, H., Okamoto, S., Oshima, K., Fujisawa, T., et al. (2012b). Complete genome sequence of Bradyrhizobium sp. S23321: insights into symbiosis evolution in soil oligotrophs. Microbes Environ. 27, 306–315. doi: 10.1264/jsme2.ME11321
Okubo, T., Piromyou, P., Tittabutr, P., Teaumroong, N., and Minamisawa, K. (2016). Origin and evolution of nitrogen fixation genes on symbiosis islands and plasmid in Bradyrhizobium. Microbes Environ. 31, 260–267. doi: 10.1264/jsme2.ME15159
Pedersen, W. L., Chakrabarty, K. R. V., Klucas, R. V., and Vidaver, A. K. (1978). Nitrogen fixation (acetylene reduction) associated with roots of winter wheat and sorghum in Nebraska. Appl. Environ. Microbiol. 35, 129–135.
Piromyou, P., Greetatorn, T., Teamtisong, K., Okubo, T., Shinoda, R., Nuntakij, A., et al. (2015a). Preferential association of endophytic bradyrhizobia with different rice cultivars and its implications for rice endophyte evolution. Appl. Environ. Microbiol. 81, 3049–3061. doi: 10.1128/AEM.04253-14
Piromyou, P., Songwattana, P., Greetatorn, T., Okubo, T., Kakizaki, K. C., Prakamhang, J., et al. (2015b). The Type III secretion system (T3SS) is a determinant for rice-endophyte colonization by non-photosynthetic Bradyrhizobium. Microbes Environ. 30, 291–300. doi: 10.1264/jsme2.ME15080
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Rennie, R. J. (1981). A single medium for the isolation of acetylene-reducing (dinitrogen-fixing) bacteria from soils. Can. J. Microbiol. 27, 8–14. doi: 10.1139/m81-002
Rosenblueth, M., Ormeño-Orrillo, E., López-López, A., Rogel, M. A., Reyes-Hernández, B. J., Martínez-Romero, J. C., et al. (2018). Nitrogen fixation in cereals. Front. Microbiol. 9:1794. doi: 10.3389/fmicb.2018.01794
Rouws, L. F. M., Leite, J., de Matos, G. F., Zilli, J. E., Coelho, M. R. R., Xavier, G. R., et al. (2014). Endophytic Bradyrhizobium spp. isolates from sugarcane obtained through different culture strategies. Environ. Microbiol. Rep. 6, 354–363. doi: 10.1111/1758-2229.12122
Saeki, Y., Nakamura, M., Mason, M. L. T., Yano, T., Shiro, S., Sameshima-Saito, R., et al. (2017). Effect of flooding and the nosZ gene in bradyrhizobia on bradyrhizobial community structure in the soil. Microbes Environ. 32, 154–163. doi: 10.1264/jsme2.ME16132
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Schneijderberg, M., Schmitz, L., Cheng, X., Polman, S., Franken, C., Geurts, R., et al. (2018). A genetically and functionally diverse group of non-diazotrophic Bradyrhizobium spp. Colonizes the root endophytic compartment of Arabidopsis thaliana. BMC Plant Biol. 18:61. doi: 10.1186/s12870-018-1272-y
Schollhorn, R., and Burris, R. H. (1967). Acetylene as a competitive inhibitor of N2 fixation. Proc. Natl. Acad. Sci. U.S.A. 58, 213–216. doi: 10.1073/pnas.58.1.213
Shiro, S., Kuranaga, C., Yamamoto, A., Sameshima-Saito, R., and Saeki, Y. (2016). Temperature-dependent expression of nodC and community structure of soybean-nodulating bradyrhizobia. Microbes Environ. 31, 27–32. doi: 10.1264/jsme2.ME15114
Siqueira, A. F., Minamisawa, K., and Sánchez, C. (2017). Anaerobic reduction of nitrate to nitrous oxide is lower in Bradyrhizobium japonicum than in Bradyrhizobium diazoefficiens. Microbes Environ. 32, 398–401. doi: 10.1264/jsme2.ME17081
Steffen, W., Richardson, K., Rockström, J., Cornell, S. E., Fetzer, I., Bennett, E. M., et al. (2015). Planetary boundaries: guiding human development on a changing planet. Science 347:1259855. doi: 10.1126/science.1259855
Szoboszlay, M., Dohrmann, A. B., Poeplau, C., Don, A., and Tebbe, C. C. (2017). Impact of land-use change and soil organic carbon quality on microbial diversity in soils across Europe. FEMS Microbiol. Ecol. 93:fix146 doi: 10.1093/femsec/fix146
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Terakado-Tonooka, J., Fujihara, S., and Ohwaki, Y. (2013). Possible contribution of Bradyrhizobium on nitrogen fixation in sweet potatoes. Plant Soil 367, 639–650. doi: 10.1007/s11104-012-1495-x
Terakado-Tonooka, J., Owaki, Y., Yamakawa, H., Tanaka, F., Yoneyama, T., and Fujihara, S. (2008). Expressed nifH genes of endophytic bacteria detected in field-grown sweet potatoes (Ipomoea batatas L.). Microbes Environ. 23, 89–93. doi: 10.1264/jsme2.23.89
Thaweenut, N., Hachisuka, Y., Ando, S., Yanagisawa, S., and Yoneyama, T. (2011). Two seasons’ study on nifH gene expression and nitrogen fixation by diazotrophic endophytes in sugarcane (Saccharum spp. hybrids): expression of nifH genes similar to those of rhizobia. Plant Soil 338, 435–449. doi: 10.1007/s11104-010-0557-1
van Deynze, A., Zamora, P., Delaux, P. M., Heitmann, C., Jayaraman, D., Rajasekar, S., et al. (2018). Nitrogen fixation in a landrace of maize is supported by a mucilage-associated diazotrophic microbiota. PLoS Biol. 16:e2006352. doi: 10.1371/journal.pbio.2006352
VanInsberghe, D., Maas, K. R., Cardenas, E., Strachan, C. R., Hallam, S. J., and Mohn, W. W. (2015). Non-symbiotic Bradyrhizobium ecotypes dominate North American forest soils. ISME J. 9, 2435–2441. doi: 10.1038/ismej.2015.54
Wani, S. P., Upadhyaya, M. N., and Dart, P. J. (1984). An intact plant assay for estimating nitrogense activity (C2H2 reduction) of sorghum and millet plants grown in pots. Plant Soil 82, 15–29. doi: 10.1007/BF02220766
Wu, M., and Eisen, J. A. (2008). A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9:R151. doi: 10.1186/gb-2008-9-10-r151
Wu, M., and Scott, A. J. (2012). Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28, 1033–1034. doi: 10.1093/bioinformatics/bts079
Xu, L., Naylor, D., Dong, Z., Simmons, T., Pierroz, G., Hixson, K., et al. (2018). Drought delays development of the sorghum root microbiome and enriches for monoderm bacteria. Proc. Natl. Acad. Sci. U.S.A. 115, E4284–E4293. doi: 10.1073/pnas.1717308115
Yoneyama, T., Muraoka, T., Kim, T. H., Dacanay, E. V., and Nakanishi, Y. (1997). The natural 15N abundance of sugarcane and neighboring plants in Brazil, the Philippines and Miyako (Japan). Plant Soil 189, 239–244. doi: 10.1023/A:1004288008199
Yoneyama, T., Terakado-Tonooka, J., and Minamisawa, K. (2017). Exploration of bacterial N2-fixation systems in association with soil-grown sugarcane, sweet potato, and paddy rice: a review and synthesis. Soil Sci. Plant Nutri. 63, 578–590. doi: 10.1080/00380768.2017.1407625
Keywords: Bradyrhizobium, nitrogen fixation, sorghum, metagenome, proteome
Citation: Hara S, Morikawa T, Wasai S, Kasahara Y, Koshiba T, Yamazaki K, Fujiwara T, Tokunaga T and Minamisawa K (2019) Identification of Nitrogen-Fixing Bradyrhizobium Associated With Roots of Field-Grown Sorghum by Metagenome and Proteome Analyses. Front. Microbiol. 10:407. doi: 10.3389/fmicb.2019.00407
Received: 15 September 2018; Accepted: 15 February 2019;
Published: 12 March 2019.
Edited by:
Benjamin Gourion, Laboratoire des Interactions Plantes-Microorganismes (LIPM), FranceReviewed by:
Angela Sessitsch, Austrian Institute of Technology (AIT), AustriaCopyright © 2019 Hara, Morikawa, Wasai, Kasahara, Koshiba, Yamazaki, Fujiwara, Tokunaga and Minamisawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kiwamu Minamisawa, a2l3YW11QGlnZS50b2hva3UuYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.