 Widad Dantoft
Widad Dantoft Kevin A. Robertson
Kevin A. Robertson W. John Watkins3
W. John Watkins3 Birgit Strobl
Birgit Strobl Peter Ghazal
Peter Ghazal
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 04 March 2019
Sec. Infectious Agents and Disease
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00355
This article is part of the Research TopicTargeting Metabolism of Innate Immune Cells as Therapeutic Strategy for Infectious DiseasesView all 5 articles
Molecular determinants underlying interferon (IFN)-macrophage biology can help delineate enzyme systems, pathways and mechanisms for enabling host-directed therapeutic approaches against infection. Notably, while the IFN antiviral response is known to be directly coupled to mevalonate-sterol biosynthesis, mechanistic insight for providing host pathway-therapeutic targets remain incomplete. Here, we show that Nampt and Sirt6 are coordinately regulated upon immune activation of macrophages and contribute to the IFN-sterol antiviral response. In silico analysis of the Nampt and Sirt6 promoter regions identified multiple core immune gene-regulatory transcription factor sites, including Stat1, implicating a molecular link to IFN control. Experimentally, we show using a range of genetically IFN-defective macrophages that the expression of Nampt is stringently regulated by the Jak/Stat-pathway while Sirt6 activation is temporally displaced in a partial IFN-dependent manner. We further show that pharmacological inhibition of Nampt and small interfering RNA (siRNA)-mediated inhibition of Nampt and Sirt6 promotes viral growth of cytomegalovirus in both fibroblasts and macrophages. Our results support the notion of pharmacologically exploiting immune regulated enzyme systems of macrophages for use as an adjuvant-based therapy for augmenting host protective pathway responses to infection.
Infection is a dynamically complex and multifaceted process requiring not only the avoidance of immune countermeasures but also the exploitation of host cellular networks and machinery by the pathogen. In many cases, parastization by pathogens and especially by viruses requires remodeling of metabolic and energy resources for the successful production of progeny. Notably, the immune system has been found to cross regulate these resources and processes as an evolutionary selected countermeasure. For example, IFNγ induced consumption of tryptophan, by the Indoleamine 2,3 Dioxygenese (IDO) pathway, has been shown to inhibit replication of several intracellular organism including hCMV (Pfefferkorn, 1984; Carlin et al., 1989; MacKenzie et al., 1998; Bodaghi et al., 1999; Heseler et al., 2013). More recently interferon regulation of the sterol biosynthesis pathway has been shown to be a central biosynthetic pathway targeted by the immune system for broad host-protection against infection.
In this scenario, Toll-like receptor activation of macrophages by pathogens leads to the production of type I interferons which coordinately regulate a marked and sustained reduction in the mevalonate-sterol biosynthetic pathway, and whereby a wide-spectrum of different human and animal viruses have been shown to be sensitive to suppression of the pathway (Adams et al., 2004; Gerbod-Giannone et al., 2006; Eguchi et al., 2008; Zhu et al., 2008; Bauman et al., 2009; Maitra et al., 2009; Haas and Mooradian, 2010; Blanc et al., 2011; Olsen et al., 2011; Chukkapalli et al., 2012; Liu et al., 2013; Mesmin et al., 2013; Reboldi et al., 2014; Singaravelu et al., 2015; Robertson et al., 2016). The currently known molecular pathways for down-regulating the sterol pathway involve the IFN induction of an hydroxylase enzyme (Ch25h) and its cognate regulatory metabolite, 25-hydroxycholesterol (25HC) that potently inhibits, at the protein level, the master transcription factor (TF) for sterol biosynthesis (SREBP2) (Blanc et al., 2013), and also the key regulated mevalonate reductase, HMGCR (Lu et al., 2015), and additionally IFN regulated microRNAs (miR342-5p), that coordinate changes in the enzymatic flux of the cholesterol pathway within the cell (Robertson et al., 2016). However, there remains yet to be identified transcriptional or epigenetic mechanisms for suppression of SREBP2 and sterol biosynthesis.
More broadly, there is increasing evidence showing connections between immune signaling, such as interferon (IFN) signaling, and the regulation of sterol, sugar, and fatty acid metabolism (Zelcer and Tontonoz, 2006; Spann and Glass, 2013; Kotzamanis et al., 2015; York et al., 2015). While the cell typically induces changes through rapid established routes such as the PI3K/AKT/mTOR signaling pathway, these changes are not sustained over a longer period time and do not support the increased needs for de novo lipogenesis. In the context of cellular stress and inflammation, Sirtuins (SIRTs) are known to play sustained roles in protecting against cellular stress through epigenetic control of metabolic pathways (Lyssiotis and Cantley, 2012; Jiang et al., 2013). This includes the regulation of glycolytic and lipid metabolism by the nicotineamide adenine dinucleotide (NAD+)-dependent deacetylases SIRT1 and SIRT6 (Liu et al., 2012a,b; Elhanati et al., 2013; Tao et al., 2013). Metabolic coupling is strictly dependent on NAD+ production through de novo biosynthesis from tryptophan or through the nicotineamide (NAM) salvage pathway, which is regulated by the rate-limiting enzyme nicotineamide phosphatidyltransferase (NAMPT). It is notable that NAD+-dependent activation of SIRT6 has been shown to repress the SREBF2 promoter (Elhanati et al., 2013), and thereby directly linking SIRT6 activity to sterol metabolism. However, whether NAMPT or SIRT6 are coordinately regulated by the IFN macrophage antiviral response is not known. Most notably this remains a central unanswered question to the notion of using macrophage interferon biology as a guiderail for identifying host-directed druggable targets as anti-infectives.
In the present report, we find that Nampt and Sirt6 are coordinately regulated upon immune activation of macrophages and contribute to the interferon antiviral response. The coupling to the IFN response is via direct transcriptional activation of NAMPT through the JAK/STAT signaling pathway. We show that pharmacological inhibition of Nampt and small interfering RNA (siRNA)-mediated inhibition of Nampt and Sirt6 enhances the viral growth of cytomegalovirus (mCMV) in both fibroblasts and macrophages. These findings support the proposition that immune regulated enzyme systems may be used as an adjuvant therapy for augmenting the host protective response to infection.
We first investgated whether murine Nampt and Sirt6 and human NAMPT and SIRT6 promoter regions contained any putative transcriptional binding sites (TFBS) for immune-regulatory TFs (Figure 1 and Supplementary Tables S1–S4). By using the sequence analysis tool PROMO (Messeguer et al., 2002; Farré et al., 2003), and manual procurement by comparing putative binding sites to published consensus binding sequences, an array of significant binding sites (restricted to 15% dissimilarity) for core immune-activated TFs, including AP-1, NFκB (defined here as DNA binding activity constituted either by p50 homodimer, a p50/p65 heterodimer, or a heterotetramer), RELA (p65 subunit of NFκB), GATA1 and GATA2, were identified within (-1 kb upstream of) the murine Nampt and Sirt6 promoter regions (Figure 1). PROMO analysis of the human NAMPT and SIRT6 promoter regions identified similar binding sites, suggesting that the overall activation mode of these genes is conserved between humans and mice. Notably, several putative Signal Transducers and Activators of Transcription 1 (STAT1) sites were identified across the Nampt promoter region, suggesting Nampt expression might be driven directly by the activation of the JAK/STAT signaling pathway (Figure 1). A putative Oct cluster [OCT1/2/(3/4)] was also identified in the distal Nampt promoter region, in close proximity to putative NFκB, RELA, and STAT1 binding sites. While the promoter region of Sirt6 did not contain any putative STAT1 binding sites, it was dominated by putative binding sites for Activator protein 1 (AP-1), c-JUN, c-FOS, and NFκB. The AP-1 structure is a heterodimer composed of proteins belonging to the c-FOS, c-JUN, Activating transcription factor (ATF), and Jun dimerization protein (JDP) families (Angel and Karin, 1991; Karin et al., 1997). Consistent with its reported activity, the putative AP-1 binding sites were found in close proximity to either c-FOS, c-JUN, or in areas containing cis-located c-FOS and c-JUN binding sites (Figure 1). AP-1, an early response TF, has been reported to regulate gene expression in response to various stimuli, including cytokine stimulation and bacterial and viral infections (Hess et al., 2004).
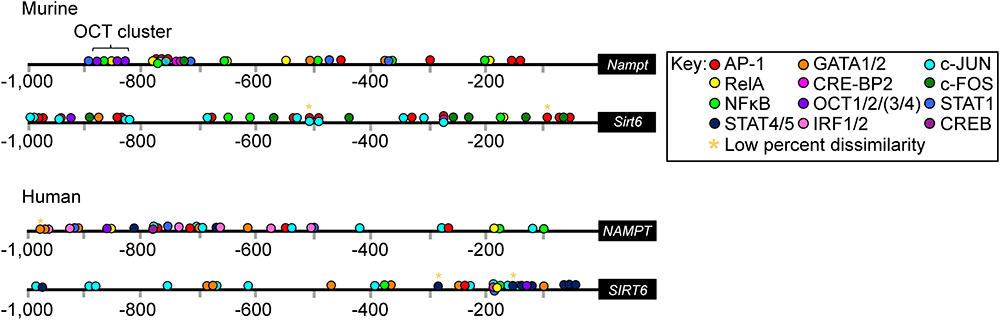
Figure 1. Putative transcription factor (TF) binding sites within the promoter regions of murine Nampt and Sirt6 and human NAMPT and SIRT6. In silico analysis using PROMO of the murine Nampt and Sirt6 and human NAMPT and SIRT6 promoter regions revealed putative binding sites for an array of different immune-regulatory TFs (Supplementary Tables S1–S4). The cut off for dissimilarity to the consensus TF binding-sequence was set to 15% (∗ marks binding sites with low percentage, 0–4%, dissimilarity) and the identified string sequences were compared to the consensus binding site sequences for each TF. Figure is not to scale.
The presence of several putative immune-gene regulatory TF binding sites, prompted us to investigate whether Nampt and Sirt6 expression was induced by infection (Figure 2). The relative expression of Nampt and Sirt6 in mCMV-infected NIH-3T3 and p53 mouse embryonic fibroblasts (p53-MEFs) was measured using Quantitative reverse-transcriptase Polymerase Chain Reaction (qRT-PCR) (Figures 2A,B). mCMV infection of NIH-3T3 and p53-MEFs resulted in significantly higher levels of Nampt expression during the first 6 h of infection (Figures 2A,B). While an early increased Sirt6 expression was not observed, a significantly higher expression was observed in NIH-3T3 after 10 h of infection, indicating a delayed response. Similarly, the temporal expression profiles (over 24 h) of Nampt and Sirt6 were investigated in mCMV infected bone marrow derived macrophages (BMDM). Following mCMV infection, cells were harvested every 2 h until 10 h post-treatment [0 (0 h after viral adsorption or poly(I:C) treatment), 2, 4, 6, 8, 10 h] and at 24 h, followed by transcriptomic profiling and modeling of their temporal expression. In these experiments, polynomial fitting of the smoothened data was used to determine whether the expression profiles of Nampt and Sirt6 changed significantly (where a R2 > 0.9 indicated significant change) with time in infected BMDM (Supplementary Tables S5, S6). In addition further statistical evaluation was performed by determining the p-value of the fitted model in relation to a horizontal flat line, where a significant p-value predicted a temporal change and a non-significant p-value is predicted of non-fluctuation in expression. Consistent with the observations in NIH-3T3s and p53-MEFs, mCMV infection of BMDM resulted in a significant early and dynamic expression of Nampt (Figure 2C and Supplementary Table S5), with a peak at 5 h, followed by a steady decline. Moreover, similarly to the observations in NIH-3T3s and p53-MEFs, temporal expression analysis revealed that mCMV infected BMDM exhibited a delayed but continuous, albeit lower than Nampt, significant Sirt6 expression (Figure 2C and Supplementary Table S6). Collectively, these results show that while Nampt and Sirt6 are both induced in response to mCMV infection, their response time differs from each other irrespective of cell type, indicative of potential differential transcriptional regulation.
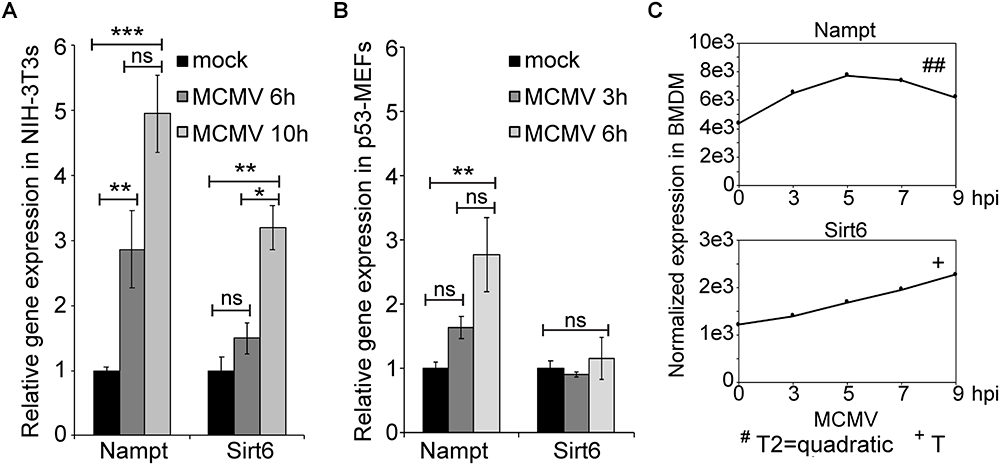
Figure 2. Expression of Nampt and Sirt6 is upregulated by MCMV infection. (A) Quantification of relative Nampt and Sirt6 mRNA expression in NIH-3T3 cells, at 6 and 10 h, using RT-qPCR following mCMV infection with mock-infected cells serving as controls (n = 3). (B) Quantification of relative Nampt and Sirt6 mRNA expression in p53-MEF cells, at 3 and 6 h, using RT-qPCR following mCMV infection (n = 3). ANOVA with Tukey post-test was used to assess statistical signficance. (C) Normalized temporal Nampt and Sirt6 expression (antilog) in mCMV infected bone marrow derived macrophages (BMDM). The expression was measured over the first 24 h of infection using microarray and compared to timepoint 0. The expression levels between 0 and 10 h were smoothened and fitted to a linear (+), quadatic (#), or cubic (∗) polynomal on time and the statistical significance (p-values) was assessed. +/#/∗p < 0.05, ++/##/∗∗p < 0.01, and +++/###/∗∗∗p < 0.001 were considered to be significant (ns, not significant). Bars represent standard error of the mean (SEM). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 were considered to be significant (ns, not significant).
The presence of the several putative STAT1 binding sites in the Nampt promoter region suggests that Nampt expression is induced in a JAK/STAT signaling pathway-dependent manner. To initially investigate this, the synthesis of Nampt mRNA was measured Tyk-2-deficient BMDM (Figure 3). Nampt mRNA synthesis was investigated in mCMV infected wild-type and Tyk2-deficient BMDM at 1–1.5 h post-infection and at 6–6.5 h post-infection (Figure 3A). The non-receptor tyrosine-protein kinase Tyk2 has been implicated in type-I IFN, IL-6, IL-10, and IL-12 signaling (Velazquez et al., 1992; Stahl et al., 1994; Heinrich et al., 1998; Shimoda et al., 2000; Shaw et al., 2006). Consistent with the identification of putative STAT1 binding sites, mCMV infection of Tyk2-deficient (Tyk2-/-) BMDM resulted in a much-reduced Nampt synthesis, compared to infected wild-type cells, suggesting that Nampt is, at least partly, induced in a JAK/STAT pathway-dependent manner (Figure 3A).
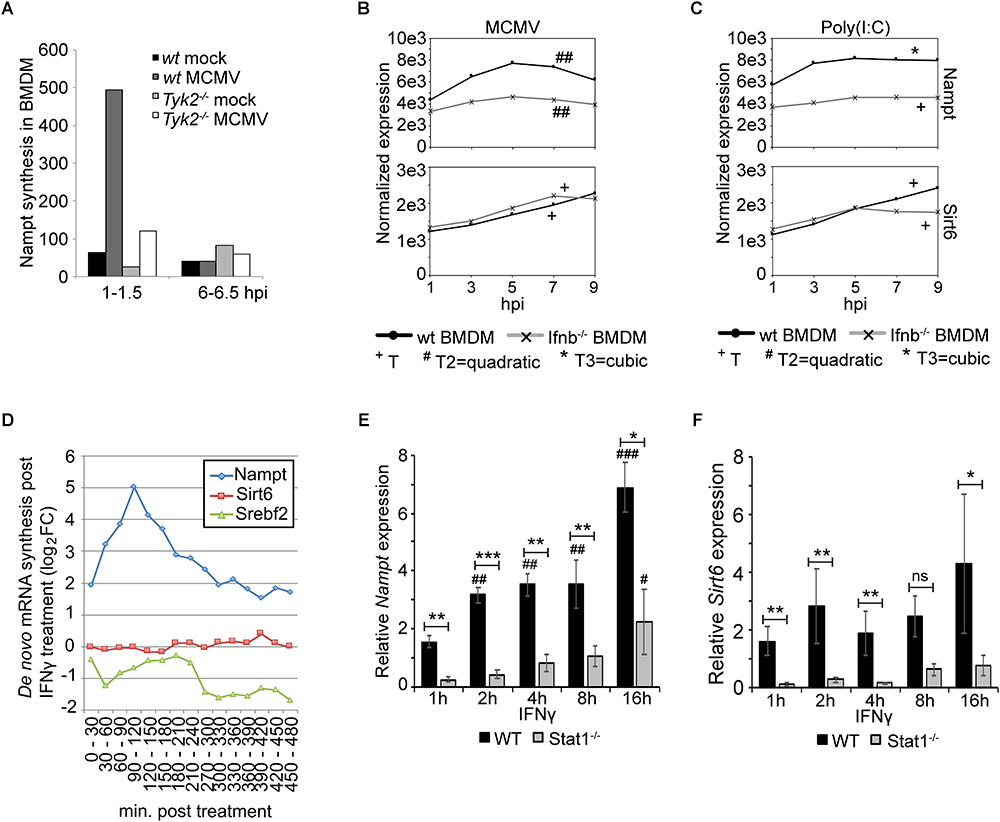
Figure 3. Expression of Nampt is induced by both type-I and type-II IFN, while respone of Sirt6 is restricted to type-I IFN response. (A) Quantification of Nampt mRNA expression, using microarray, in wild-type (wt) and Tyk2-/- BMDM following mock- (control) and mCMV-infection. (B) Normalized temporal expression (antilog) of Nampt and Sirt6 in mCMV infected wild-type and Ifnb-/- BMDM. The expression was measured over the first 24 h of infection using microarray and compared to timepoint 0. The expression levels between timepoints 0–10 h post-streatment were smoothened and fitted to a linear (+), quadatic (#), or cubic (∗) polynomal on time to assess significance (p-values). +/#/∗p < 0.05, ++/##/∗∗p < 0.01, and +++/###/∗∗∗p < 0.001 were considered to be significant (ns, not significant). (C) Normalized temporal expression (antilog) of Nampt and Sirt6 in poly(I:C) treated wild-type and Ifnb-/- BMDM. The expression was measured over the first 24 h using microarray and compared to timepoint 0. The expression levels between timepoints 0–10 h post-streatment were smoothened and fitted to a linear (+), quadatic (#), or cubic (∗) polynomal on time to assess significance (p-values). +/#/∗p < 0.05, ++/##/∗∗p < 0.01, and +++/###/∗∗∗p < 0.001 were considered to be significant (ns, not significant). (D) Quantification of de novo synthesis of Nampt, Sirt6, and Srebf2 mRNA in IFNγ-stimulated BMDMs using qRT-PCR. Expression was measured every 30 min for a total of 8 h. (E) Quantification of relative Nampt mRNA expression in wild-type and Stat1-/- p53-MEF cells, at 1, 2, 4, 8, and 16 h, using RT-qPCR following IFNγ stimulation (n = 3). Expression is relative to wild-type untreated (0 h) cells, set as 1 (not shown). One-way ANOVA with a Tukey’s post-test was used to assess statistical signficance to untreated controls. One-way ANOVA with a Sidak’s multiple comparisons test was used to assess statistical signficance between wild-type and Stat1-/- mutants. Bars represent SEM. Statisical significance between groups (wild-type and Stat1-/-) were depicted with ∗. Statistical significance in relation to untreated controls (wild-type and Stat1-/-, respectively) were depicted with #. ∗/#p < 0.05, ∗∗/##p < 0.01, ∗∗∗/###p < 0.001 were considered to be significant. (F) Quantification of relative Sirt6 mRNA expression in wild-type and Stat1-/- p53-MEF cells, at 1, 2, 4, 8, and 16 h, using RT-qPCR following IFNγ stimulation (n = 3). Expression is relative to wild-type untreated (0 h) cells, set as 1 (not shown). One-way ANOVA with a Tukey’s post-test was used to assess statistical signficance to untreated controls. One-way ANOVA with a Sidak’s multiple comparisons test was used to assess statistical signficance between wild-type and Stat1-/- mutants. Bars represent SEM. Statisical significance between groups (wild-type and Stat1-/-) were depicted with ∗. Statistical significance in relation to untreated controls (wild-type and Stat1-/-, respectively) were depicted with #. ∗/#p < 0.05, ∗∗/##p < 0.01, ∗∗∗/###p < 0.001 were considered to be significant.
The presence of putative STAT1 binding sites and the observed dependence of Nampt expression on TYK2 and on the JAK/STAT signaling pathway poses the question Whether the induced expression of Nampt and Sirt6 is dependent on type I IFN signaling? To investigate this, the expression of Nampt and Sirt6 was assessed in polyinosinic:polycytidylic acid [poly(I:C)] treated Ifnb-deficient BMDM and compared to the response in mCMV infected Ifnb1-deficient (C57BL/6J Ifnb1-/-) BMDM (Figures 3B,C). Poly(I:C), a ligand of Toll like receptor 3 (TLR3), is structurally similar to double-stranded RNA and is, thus, used to simulate viral infections. Following mCMV infection or poly(I:C) treatment, cells were, as decribed above (Figure 2), harvested every 2 h until 10 h post-treatment and at 24 h, followed by transcriptomic profiling and modeling of their temporal expression. As in Figure 2C, mCMV infection of wild-type BMDM resulted in an early dynamic expression of Nampt. Similar to Nampt, mCMV infection of wild-type BMDM significantly induced, albeit at a lower level, the expression of Sirt6 expression peaking downstream of Nampt. Poly(I:C) treatment of wild-type BMDM, resulted in a significant temporal activation of both Nampt and Sirt6, indicating that the observed expression is a host-driven response to infection (Figure 3C and Supplementary Tables S7, S8). The level of Nampt activation following mCMV infection was significantly reduced in Ifnb1-deficient cells, suggesting that a robust Nampt expression response is IFNβ-dependent (Figure 3B). While the robustness in Nampt expression was lost, a small significant temporal change in the expression profile was observed, suggesting that Nampt expression is possibly governed by other factors or pathways including by other type I Ifns but that the magnitude of expression is strongly dependent on intact IFNβ-signaling. The early expression of Sirt6 was, however, not extensively altered in mCMV infected or poly(I:C) treated Ifnb-deficient cells. The expression was similar to wild-type cells up until 7 h post-infection and up until 5 h post-poly(I:C) treatment. This was followed by a reduction in expression, indicating that early but not late activation of Sirt6 expression following mCMV infection and poly(I:C) treatment is induced independently of IFNβ (Figures 3B,C and Supplementary Tables S6, S8). Together, these results indicate that Nampt and partially Sirt6 are coupled to the type I IFN response in macrophages.
Further in our BMDM, where stimulation with physiologically relevant concentration of IFNγ has been previously determined (Kropp et al., 2011), IFNγ induced the expression of Nampt, peaking at 6 h post-treatment (Supplementary Figure S1 and Table S9). Moreover, to further investigate the dependence of Nampt and Sirt6 expression on the type-II IFN response, the level of newly transcribed Nampt and Sirt6 mRNA was measured every 30 min, over a period of 8 h, using reverse transcriptase-quantitative PCR (qRT-PCR) in BMDM stimulated with IFNγ (Figure 3D). Stimulation with IFNγ resulted, after 2 h of infection, in an eight-times increase in de novo transcribed Nampt RNA levels, followed by a rapid drop in Nampt mRNA. Sirt6 mRNA levels were on the other hand not affected by IFNγ stimulation, further suggesting that it is not a type-II IFN stimulated gene. Notably, the increase in Nampt mRNA expression was followed by a drop in Srebf2 expression, consistent with previously published data from Blanc et al. (2011).
The dependence on type-II IFN and JAK/STAT signaling was further investigated in wild-type and Stat1-deficient (Stat1-/-) p53-MEFs stimulated with IFNγ (Figures 3E,F). Steady state levels of Nampt mRNA was investigated at 1, 2, 4, 8, and 16 h post-treatment and compared to untreated (0 h) cells (Figure 3E). IFNγ activated of wild-type cells resulted, as early as 2 h post-treatment, in a significantly increased expression of Nampt compared to untreated cells (statistical significance depicted with #), suggesting that Nampt is a type-I IFN responsive gene. In the IFNγ activated Stat1-deficient (Stat1-/-) cells, a significantly increased Nampt expression, compared to the untreated Stat1-deficient control (not shown), was only observed at later time points (16 h). This expression was significantly reduced at all time points compared to activated wild-type cells, indicating that Nampt is dependent on intact Stat1 signaling, consistent with the identification of putative STAT1 binding sites within the Nampt promoter region.
Moreover, loss of Stat1 resulted in a significantly reduced expression of Sirt6 compared to the wild-type p53-MEFs (Figure 3F). Notably, unlike Nampt, Sirt6 mRNA expression in wild-type cells did not increase statistically with time (compared to untreated control), suggesting that it is not a type-II IFN stimulated gene. Nor did the expression change significantly in Stat1-deficient cells to the respective untreated control. It is possible to speculate that Stat1 signaling is required for the basal, but not induced, Sirt6 expression. The absence of identified STAT1 binding sites in combination with the absence of gene induction with time further support the notion that this activation is indirect. Collectively, these results show that Nampt and Sirt6 are both induced in response to mCMV infection and suggest that Nampt is an interferon-stimulated gene (ISG), with Nampt expression being an immediate-response gene induced by type-I and type-II IFN in a JAK/STAT dependent manner.
Infection with double-stranded DNA viruses, such as CMV, are known to trigger the common TLR signaling pathway that elicits the activation of NFκB and MAPK through the Myd88 adaptor (Compton et al., 2003), while other pathways, such as the IPS-1 and STING mediated pathways, induce type-I IFN synthesis (Seth et al., 2005; Cheng et al., 2007; Ishikawa et al., 2009) resulting in downstream target activation. To explore the gene activation of factors belonging to these pathways, the temporal gene expression profiles of Myd88, p50 (Nfκb1), p65 (Rela), Trif (Ticam1), Rig-I (Ddx58), Mda5 (Ifih1), Ips-1 (Mavs), Sting (Tmem173), and cGas (Md21d1) were investigated in mCMV infected or poly(I:C) treated wild-type and Ifnb1-deficient BMDM (Figure 4 and Supplementary Tables S10–S27). Following mCMV infection of wild-type BMDM, a significant temporal activation of Myd88, p50, p65, Trif, Rig-I, Mda5, and Sting was observed (Figure 4A). The expression profile of Ips-1 was initially suppressed up until 5 h post-infection followed by an activation, while cGas exhibited an early activation between 1 and 3 h post-infection, followed by a rapid drop in expression. Notably, the activation of Trif, which was absent until 3 h post-infection, was followed by a rapid increase in expression peaking at 7 h. In Ifnb1-deficient BMDM, mCMV infection resulted in a significant temporal change in expression of all genes but Trif. While the temporal expression of Myd88, Rig-I, Mda-5, and Sting was significantly changed over time, the level of expression was much reduced in these cells, suggesting that IFNβ-signaling is in part needed for the full induction of these genes. Notably, while the expression level of cGas was initially much higher in wild-type cells, the level expression after 5 h dropped to similar levels as those observed in the Ifnb1-deficient cells, suggesting that IFNβ is required for the early activation of this gene.
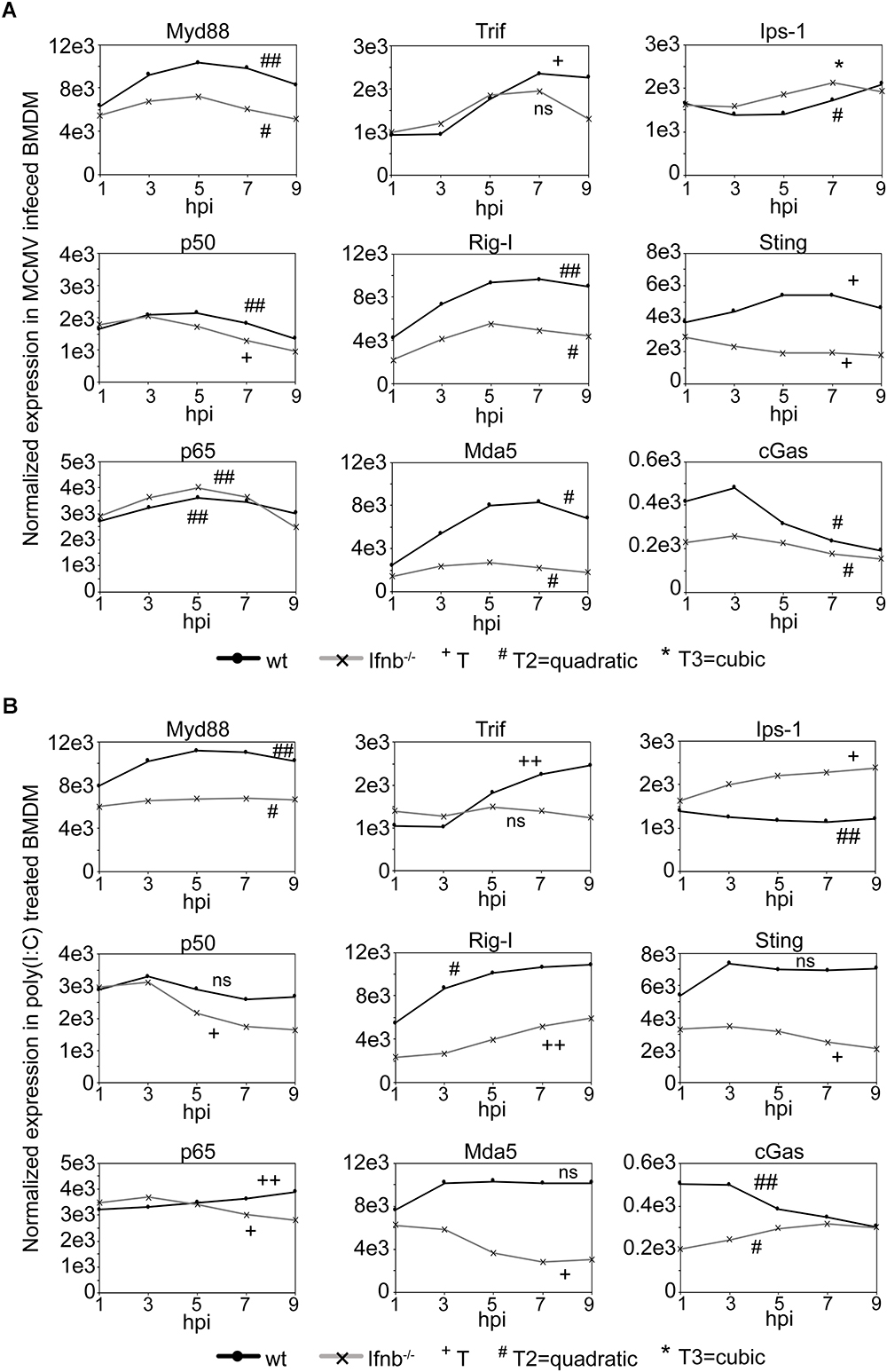
Figure 4. Expression of upstream, but not downstream, TLR signaling pathway components is dependent on IFNβ/type-I IFN signaling. (A) Normalized temporal expression (antilog) of Myd88, p50 (Nfkb1), p65 (Rela), Trif (Ticam1), Rig-I (Ddx58), Mda-5 (Ifih1), Ips-1 (Mavs), Sting (Tmem173), and cGas (Mb21d1) in mCMV infected wild-type and Ifnb-/- BMDM. The expression was measured over the first 24 h of infection using microarray and compared to timepoint 0. The expression levels between timepoints 0–10 h post-streatment were smoothened and fitted to a linear (+), quadatic (#), or cubic (∗) polynomal on time to assess significance (p-values). +/#/∗p < 0.05, ++/##/∗∗p < 0.01, and +++/###/∗∗∗p < 0.001 were considered to be significant (ns, not significant). (B) Normalized temporal expression (antilog) of Myd88, p50 (Nfkb1), p65 (Rela), Trif (Ticam1), Rig-I (Ddx58), Mda-5 (Ifih1), Ips-1 (Mavs), Sting (Tmem173), and cGas (Mb21d1) in poly(I:C) treated wild-type and Ifnb-/- BMDM. The expression was measured, as in (A), over the first 24 h using microarray and compared to timepoint 0. The expression levels between timepoints 0–10 h post-streatment were smoothened and fitted to a linear (+), quadatic (#), or cubic (∗) polynomal on time to assess significance (p-values). +/#/∗p < 0.05, ++/##/∗∗p < 0.01, and +++/###/∗∗∗p < 0.001 were considered to be significant (ns, not significant).
In poly(I:C) activated wild-type BMDM, significant temporal expression change was observed for Myd88, p65, Trif, Rig-I, Ips-1, and cGas, consistent with that observed in mCMV infected cells (Figure 4B). p50, Mda5, and Sting all exhibited an initial increase in expression between 1 and 3 h post-treatment, however, unlike Mda5 and Sting that did not significantly change, the level of p50 expression was reduced between 3 and 7 h. The modeled temporal change in the p50 expression profile was, however, not significant. In Ifnb1-deficient BMDM, a significant temporal change was observed for all genes with the exception of Trif. As in infected Ifnb1-deficient BMDM, poly(I:C) treatment resulted in a reduced temporal expression for Myd88, Rig-I, Mda-5, and Sting. Moreover, Mda5 exhibited a repressed temporal profile, as compared to its expression in poly(I:C) treated wild-type cells. Notably, Ips-1 and cGas both exhibited an increased expression over time, with the expression of Ips-1 exceeding that observed in wild-type cells (Figure 4B).
Collectively, these results suggest intact IFNβ-signaling is not required for the expression of p50 and p65 following mCMV infection, but is required for the magnitude in expression of the upstream components (Myd88, Rig-I, Mda5, and Sting) of these pathways.
The observation that Nampt and Sirt6 were coordinately induced in macrophages by immune stimulation, either by infection or the ensuing interferon response, prompted us to test whether NAMPT and SIRT6 exhibit antiviral activity. To investigate whether Sirt6 and Nampt exhibit antiviral properties, mCMV replication was measured after siRNA mediated knockdown of Sirt6 and after pharmacologic inhibition or siRNA mediated knockdown of Nampt, with the highly specific non-competitive inhibitor FK866, respectively (Hasmann and Schemainda, 2003) (Figure 5 and Supplementary Figure S2). Consistent with the reported antiviral activity of human SIRT6 (Koyuncu et al., 2014), mediated knockdown of murine Sirt6 resulted, in a siRNA concentration-dependent manner, in an increased viral replication (Figure 5A). Moreover, siRNA mediated knockdown and pharmacologic inhibition of murine Nampt also resulted in an increase in viral replication, respectively (Figures 5B,C). Together, these results indicate that Sirt6 and Nampt both display antiviral properties, providing druggable targets in bolstering interferon antiviral immunity linked to sterol metabolism.
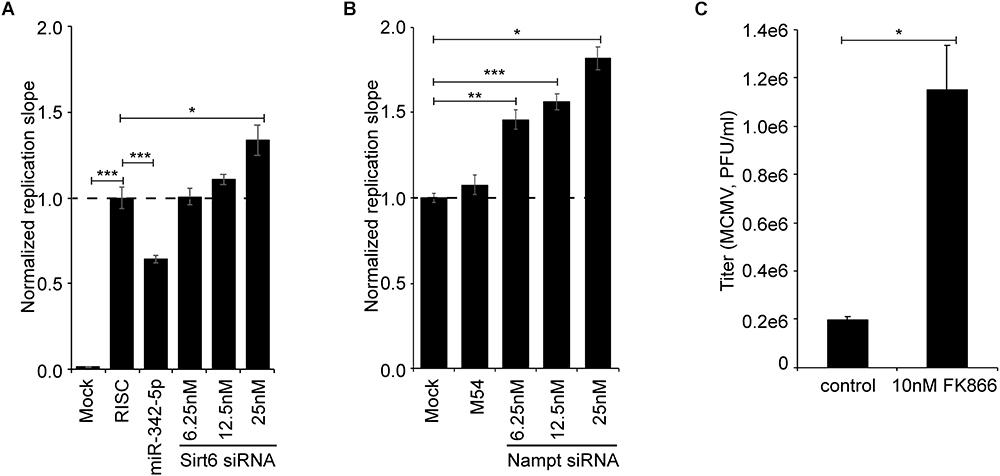
Figure 5. Inhibition of NAMPT and SIRT6 activity increases viral loads in MCMV infected cells. (A) Normalized replication slope of mCMV-GFP in Sirt6 siRNA-treated NIH-3T3. “RISC-free” siRNA served as negative control and miR342-5p siRNA served as positive control. (B) Normalized replication slope of mCMV-GFP in Nampt siRNA-treated NIH-3T3, with M54 serving as positive control. Bars represent SEM. Statistical significance was determined using One-way ANOVA with a Dunnett’s multiple comparisons test. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 were considered to be significant (ns, not significant). (C) mCMV-GFP replication (titer) in NIH-3T3 after pharmacological inhibition of NAMPT with FK866 (10 nM final concentration). mCMV-GFP was propagated in NIH-3T3 fibroblasts and quantified by plaque assay on p53-MEF monolayers in 48-well plates. Statistical significance was determined using unpaired t-test with Welch’s correction. ∗p < 0.05 was considered to be significant.
Here, we demonstrate upon infection of macrophages the serial activation of Nampt and Sirt6. The observed rapid kinetics of Nampt induction shows a strict dependency on both type I and type II IFN signal activation of transcription and, thus, represents an immediate-early class of ISG. By contrast Sirt6 shows delayed induction kinetics and is only indirectly activated downstream of viral induced type I IFN signaling. In agreement, we find the Nampt promoter region contains multiple consensus Stat1 binding sites whereas these sites are absent in the Sirt6 promoter region. Notably, pharmacological inhibition of NAMPT enzymatic activity or knock-down of Nampt or Sirt6 result in increased viral replication revealing anti-viral roles for these metabolic regulators in infection. Hence, in an apparent orchestrated and coordinated manner Nampt enzymatically drives NAD+ production that is a key rate-limiting co-factor for Sirt6 activation and thereby couples Sirt6 functions to the IFN antiviral response (Figure 6).
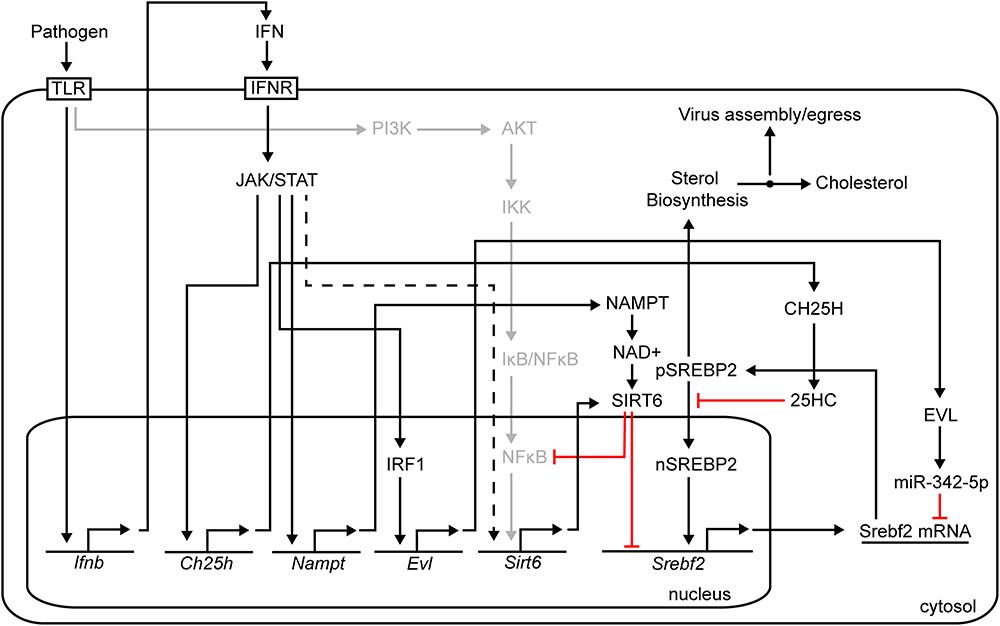
Figure 6. Summary figure.
We further find that temporal expression analysis of key pathway components of the common TLR signaling pathway, which elicits the activation of NFκB and MAPK through the Myd88 adaptor (Compton et al., 2003), and the IPS-1 and STING mediated pathways that induce type-I IFN synthesis, revealed a part dependency on intact type I IFN signaling as loss of Ifnb1 resulted in a reduced magnitude of expression (Myd88, Rig-I, Mda5, and Sting). The downstream signaling components of these pathways, p50, p65, Trif, Ips-1, and cGas were on the other hand not affected in the same way by the loss of Ifnb1. An activated expression profile was, however, observed for Ips-1 and cGas, suggesting that intact type I IFN signaling might be required for maintaining a regulated expression of these genes. As for p50 (NFκB), an initial increased expression was observed followed by a gradual (mCMV infection) or rapid (poly(I:C) treatment) declining expression. A similar expression profile was observed for p65 following mCMV infection, but not poly(I:C) treatment. Notably, in recent years, SIRT6 has been shown to inhibit NFκB expression (Kawahara et al., 2009; Santos-Barriopedro and Vaquero, 2018; Santos-Barriopedro et al., 2018) and NFκB target gene activation by interacting with p65 (Figure 6) (Li et al., 2018; Santos-Barriopedro and Vaquero, 2018). It is possible that this mode of regulation is reflected in the observed p50 and p65 expression profiles, nevertheless, further analysis would be required to confirm this. Whether Sirt6 is regulated by NFκB in this system remains to be explored, however, global profiling of p65 binding sites (by ChIP-seq) in TNFα-induced human osteosarcoma U-2 OS cells (Janus et al., 2018) and TNFα-induced or poly(I:C) stimulated Detroit 562 cells (Borghini et al., 2018) did not identify SIRT6 as a NFκB/p65 target gene. Genome-wide profiling of p65-bound sites after 3 and 6 h of LPS treatment, have on the other hand identified NAMPT as a p65-activated gene (Lim et al., 2007).
Together, these findings are consistent with observations, in other systems, that are supportive of a potential antiviral role for Nampt and Sirt6 (Schoggins et al., 2011; Van den Bergh et al., 2012; Chen et al., 2013; Koyuncu et al., 2014; Zhang et al., 2014; Li et al., 2018). In a systems-level screen for ISGs with antiviral activity, human NAMPT was identified as one of several type-I ISGs that exhibited, in infected Huh-7 cells, antiviral activity toward Venezuelan equine encephalitis virus (VEEV), a single-stranded RNA virus (Schoggins et al., 2011). NAMPT has also been reported to exhibit anti-HIV-1 activity, interfering with both early events of the life cycle (Van den Bergh et al., 2010) and Tat-induced HIV-1 long terminal repeat (LTR) transactivation (Zhang et al., 2010; Chen et al., 2013; Zhang et al., 2014). As for the role of SIRT6 in antiviral immunity this is less known. Koyuncu et al. (2014) reported in a loss-of-function study, that loss of human Sirtuin activity, including SIRT6 activity, in infected fibroblast MRC5 cells resulted, by unknown mechanism, in significant increases in viral titers (hCMV, HSV-1, Adenovirus, and Influenza A). Moreover, a recent report by Li et al. (2018) show that SIRT6 negatively regulates Dengue virus-induced inflammatory responses by targeting the DNA binding domain of NFκB p65. Notably, unlike our observations reported here those reported by Koyuncu et al. (2014), DENV replication was reduced in HEK293T cells upon silencing of SIRT6 (Li et al., 2018). It is possible that with the diverse nature of SIRT6 the mechanisms by which it exerts its antiviral function differs depending on cell type and viral strain, though the mechanism by which Nampt and Sirt6 exert antiviral effects in these studies is not known. Nevertheless, together with our findings they support the notion that NAMPT and SIRT6 constitute yet another way by which the macrophage can limit productive viral infection.
A central mechanism of action worth noting is the reciprocal increase in de novo Nampt mRNA expression, in IFNγ stimulated BMDM, is followed by a decrease in Srebf2 transcription. This is consistent with studies demonstrating IFN-antiviral suppression of transcription of multiple members of the sterol biosynthesis pathway, in part mediated by a drop in SREBP2 RNA transcription and protein levels (Blanc et al., 2011). Mechanistic studies of IFN suppression of macrophage sterol biosynthesis pathway have determined an approximately 40% contribution by Ch25h and its cognate metabolite 25HC acting at the post-translational level, and 40% by a post-transcriptional mechanism involving microRNA (miR342-5p). However, these known mechanisms fail to account for the observed transcriptional effects on Srebf2 levels. In this regard, there is good evidence to show that Sirt6 binds to and regulates Srebf2 via transcription (Lu et al., 2015; Robertson et al., 2016). Figure 6 shows a schematic of a proposed mechanism for the anti-viral activies of Nampt and Sirt6, mediated through epigentic transcriptional suppression of Srebf2, which encodes the master TF sterol biosynthesis. Figure 6 also highlights the other proposed molecular pathways for down-regulating the sterol pathway in macrophages involving the generation of 25-hydroxycholesterol (25HC) and miR342-5p microRNA, both of which contribute toward modulating the SREBP2 autoregulatory loop in response to interferon-signaling (Reboldi et al., 2014; Robertson et al., 2016).
It is noteworthy that host-directed targeting of immune modulated cellular pathways can be used as an innovative therapeutic intervention that also overcomes the antiviral drug resistance (Ghazal et al., 2000). In this regard, we note that SIRT6 inhibitors are under development as anti-cancer drugs (Hu et al., 2014; Parenti et al., 2014; Sociali et al., 2015). Some studies have investigated the efficiency of Ex527 (Selisistat), a commercially available Sirtuin inhibitor (Sigma-Aldrich) (Lugrin et al., 2013; Ekblad and Schüler, 2016) and another proposed approach in inhibiting SIRT6 activity is through administration of nicotineamide (NAM/Vitamin B3), which in addition to being a NAD+ precursor, also acts as an endogenous, non-competitive Sirtuin inhibitor (Bitterman et al., 2002). Thus, there is an opportunity for repurposing these cancer drugs for potential antiviral therapy.
C57BL/6 mice were housed in the specific pathogen-free animal facility at the University of Edinburgh. Tyk2-/- mice were maintained under specific-pathogen-free conditions at the Institute of Animal Breeding and Genetics, Department for Biomedical Sciences, University of Veterinary Medicine Vienna, Vienna, Austria. The generation or source of knockout mouse strains for Tyk2-/- has been described before (Strobl et al., 2005). All procedures were carried out under project and personal licenses approved by the Secretary of State for the Home Office, under the United Kingdom’s 1986 Animals (Scientific Procedures) Act and the Local Ethical Review Committee at Edinburgh University.
NIH-3T3 (ATCC® CRL-1658TM) immortalized cell line of embryonic mouse fibroblasts was obtained from American Type Culture Collection (ATCC) (Manassas, VA, United States) and grown in Dulbecco’s modified Eagle medium (DMEM) (Lonza, Verviers, Belgium), supplemented with 5% Calf Serum (CS) (Thermo Fisher Scientific, Waltham, MA, United States), 2 mM glutamine (Lonza) and 50 U/ml of penicillin/streptomycin (Lonza). The p53-MEF immortalized cell lines, of p53-/- embryonic mouse fibroblasts [p53-MEFs, MB355 (ATCC® CRT-2818TM)] was obtained from American Type Culture Collection (ATCC) (Manassas, VA, United States). p53-MEFs and Stat1-/- p53-MEFs were grown in DMEM (Lonza), supplemented with 5% fetal calf serum (FCS) (Thermo Fisher Scientific), 2 mM glutamine (Lonza) and 100 U/ml of penicillin/streptomycin (Lonza). BMDM were isolated and grown in DMEM/F-12 (Ham 1:1) and L-glutaMAX, supplemented with 10% Fetal Calf Serum (Lonza), 10% L929 and 100 U/ml of penicillin/streptomycin. All cells were grown in accordance to standard procedures. BMDMs were differentiated with CSF-1 derived from L929 cells for 7 days prior to further treatment.
Wild-type murine cytomegalovirus (MCMV-C3X) has been previously described (Messerle et al., 2000). The GFP-encoding MCMV (mCMV-GFP) has also been previously described (Angulo et al., 2000). For RNA expression analysis, infection was done at a multiplicity of index (MOI) of 1 unless else specified.
Small interfering RNA and “RISC-free” control siRNA were purchased from Dharmacon® RNAi Technologies (Thermo Fisher Scientific). miR-342-5p microRNA mimic were kindly gifted by Integrated DNA Technologies (WOS:000332467100005). The following siRNAs were used: “RISC-free” siRNA, SiGenomeTM Control (Cat. No. D-001220-01-05); Mouse Sirt6 siRNA (deconvoluted), ON-TARGETplus siRNA Mouse Sirt6 (Cat. No. J-061392-09, J-061392-10, J-061392-11, J-061392-12); Mouse Nampt siRNA (deconvoluted), ON-TARGETplus siRNA Mouse Nampt (Cat. No. J-040272-09, J-040272-10, J-040272-11, J-040272-12); M54 siRNA, custom made order from Dharmacon® (5′–3′ sense strand sequence is AGAAAGACGACCTGAGCTA). Mimics and siRNA were transfected into cells (NIH-3T3), in a 96 well plate, using DharmaFECT1 (Thermo Fisher Scientific) using the reverse-transfection method and in accordance to the manufacturer’s recommendations. M54 siRNA and miR-342-5p microRNA mimic were transfected at a final concentration of 25 nM and Sirt6 siRNA was transfected at a final concentration of 6.25, 12.5, and 25 nM/well. For the analysis of miR-342-5p inhibitor effects on virus replication, medium containing 3% delipidized serum [Bovine Serum, Lipid Depleted (Part number: S181L), VWR, United Kingdom] was used. After 48 h, MCMV-GFP (MOI 0.025) was used for infection. The viral growth (fluorescence in each well) was measured using a POLARstar OPTIMA plate reader (BMG Labtech, Aylesbury, United Kingdom) according to manufacturer’s recommendations. The RNAi and viral growth assay were set up as two independent experiments with 3 biological replicates per experiment (n = 6). Virus replication slopes over the linear phase were calculated, from 68 h to the end of the time course, and then normalized to control transfected wells. Statistical significance was determined using One-way ANOVA with a Dunnett’s multiple comparisons test. p-Values of <0.05, <0.01, and <0.001 were considered significant.
In silico promoter analysis of the NAMPT and SIRT6 promoter regions was done using PROMO, a virtual laboratory for identification of putative transcriptional binding sites (Messeguer et al., 2002; Farré et al., 2003). The promoter regions consisting of the 1 kb upstream regions of murine and human NAMPT and SIRT6 were retrieved from the Mouse Genome Informatics (MGI) Web Site (Bult et al., 2015; Finger et al., 2017; Smith et al., 2018) and The National Center for Biotechnology Information (NCBI) (Geer et al., 2010) resource, and imported into the online PROMO analysis tool. Species specific (Mus musculus or Homo sapiens) TFs and TF sites were chosen. For murine Nampt and Sirt6, 306, and 270 putative TF binding sites within a dissimilarity margin less or equal than 15% were identified in the promoter regions, respectively. For human NAMPT and SIRT6, 444 and 436 putative TF binding sites within a dissimilarity margin less or equal than 15% were identified in the promoter regions, respectively. All identified putative binding sites can be found in Supplementary Tables S1–S4. From these, the most probably immune-regulatory and core TF binding sites were identified via manual procurement by comparing putative binding site to publicly available/published consensus binding sequences for each TF.
Wild-type and Stat1-/- p53-MEFs were plated, in a 24-well plate, at a cell density of 3 × 105 cells/well and grown in DMEM (Lonza), supplemented with 5% FCS (Thermo Fisher Scientific), 2 mM glutamine (Lonza) and 100 U/ml of penicillin/streptomycin (Lonza), for 24 h prior to treatment with murine recombinant IFN gamma (IFNγ) and (Perbio Science). The IFNγ was diluted in complete medium and added to cells at a final concentration of 10 U/ml. Cells were harvested at 1, 2, 4, 8, and 16 h post-treatment for quantitative real-time PCR analysis using 350 μl Qiagen RTL Plus buffer (Qiagen RNeasy Plus kit) as per manufacturer’s recommendations.
Incorporation of 4-thiouridine (Sigma-Aldrich, St. Louis, MI, United States) into newly-transcribed RNA was undertaken as described by Dölken et al. (2008) and Robertson et al. (2016). In brief, at time zero medium was aspirated from all plates and 15 ml of pre-warmed medium containing IFN-γ (final concentration of 10 U/ml) or normal medium was added to the cultures. RNA labeling in BMDM during the first 30 min of the time course was undertaken by addition of 200 μM 4-Thiouridine to the medium of appropriate plates at this time. After 30 min, to end the RNA labeling period, terminate transcription and lyse the cells, medium was aspirated from the labeled BMDM and replaced with 4 ml of RLT lysis buffer (Qiagen, Hilden, DE, United States). In parallel, 10 ml of medium from the next BMDM cultures to be labeled was added to an appropriate volume of 4-thiouridine, mixed and immediately added back to the plate. BMDM cultures were then returned to the incubator. The above cycle of 4-thiouridine addition to BMDM cultures and transcriptional termination was repeated at 30-min intervals until the end of the time course. Total RNA was isolated using RNeasy Midi kit (Qiagen) according to manufacturer’s instructions, quantitated using a Nanodrop (Thermo Scientific) and integrity was confirmed using an Agilent Bioanalyser (Agilent United Kingdom). Newly transcribed RNA (ntRNA) was then isolated as described in Dölken et al. (2008) and Robertson et al. (2016) and again quantitated using a Nanodrop, followed by qRT-PCR.
Cells were harvested in 350 μl Qiagen RTL Plus buffer (Qiagen RNeasy Plus kit) as per manufacturer’s recommendations. Total RNA was extracted from cells with RNeasy Plus kit (Qiagen) according to the manufacturer’s instructions and quantitated using a Nanodrop (Thermo Scientific). All experiments were performed on three biological replicates/samples (n = 3) and expression analysis were performed with three technical replicates/sample (n = 3), unless else specified. Quantitative gene-expression analyses were performed using Taqman® Primer probe sets (Applied Biosystems, Warrington, United Kingdom). Mouse Assay ID: NAMPT: Mm00451938_m1, SIRT6: Mm01149042_m1, SREBF2: Hs01081784_m1, Actin Beta (ACTB FAM): Mm02619580_g1. Quantitative real-time PCR was performed either as one-step reactions (qRT-PCR) or two-step reactions (RT-qPCR) with an initial separate cDNA synthesis step. qRT-PCR and RT-qPCR were performed in a Stratagene MX3000P machine (Stratagene California, San Diego, CA, United States). For qRT-PCR, each sample reaction was performed in 10 μl volumes using 96-well Non-Skirted, White PCR Plates (ABgene, United Kingdom) and MicroAmp Optical Caps (Applied Biosystems, United Kingdom). For one reaction, 50 ng of diluted total RNA samples was added to 2.5 μl of 4x qScript One-Step Fast qRT-PCR (Low ROX) master-mix, 0.5 μl qScript One-step Reverse Transcriptase (Cat. No. 95081, Quanta Biosciences, United States), 0.5 μl of Taqman primer/probe set (Applied Biosystems), and RNase-free H2O to a total volume of 10 μl. cDNA synthesis by reverse-transcription was performed at 50°C for 5 min, followed by initial denaturation at 95°C for 30 s, and 40 cycles of combined denaturation at 95°C for 3 s followed by annealing/primer extension (detection) at 60°C for 30 s. For two-step analysis, 500 ng of isolated total RNA was used for cDNA synthesis with random hexamers using SuperScript® III Reverse Transcriptase (Thermo Fisher Scientific) following the manufacturer’s instructions. Following cDNA synthesis, qPCR was performed in 10 μl volumes. For one reaction, 50 ng cDNA (equivalent to 50 ng total RNA) was added to 5 μl of 2x PerfeCta® qPCR ToughMixTM (Low ROX) master-mix, 0.5 μl of Taqman primer/probe set (Applied Biosystems), and RNase-free H2O to a total volume of 10 μl. Expression of target genes was normalized to ActB. The normalized data were used to quantify the relative levels of a given mRNA according to comparative cycle threshold (2-ΔΔCT) analysis (Livak and Schmittgen, 2001; Schmittgen and Livak, 2008). Statistical significance in Nampt and Sirt6 expression was calculated using One-way ANOVA with a Tukey’s or Sidak’s multiple comparisons test (between wild-type and Stat1-/- p53-MEFs). Statistical significance for knock-down efficiency of Nampt and Sirt6 was determined using One-way ANOVA with a Dunnett’s multiple comparisons test. p-Values of < 0.05, < 0.01, and < 0.001 were considered significant.
Pharmacological inhibition of NAMPT was done using FK866 (Hasmann and Schemainda, 2003) at a final concentration of 10 mM. Briefly, NIH-3T3 cells were seeded in a 48 well plate at a cell density of 1 × 105 cells/well and infected with MCMV-GFP (MOI 0.1) the following day. After the adsorption period, the infection media was replaced with media with FK866 (10 nM) or without (control). Cells were assessed for viral growth (GFP signal) over a 4-day period at which point the cells were harvested and frozen down at -80°C. The effect of FK866 treatment on viral growth was quantified by plaque assay on p53-/- MEF monolayers in 48-well plates using standard methodology. Statistical significance was determined using unpaired t-test with Welch’s correction. p-Values of < 0.05, < 0.01 and < 0.001 were considered significant.
Temporal expression analysis for Nampt, Sirt6, and Stat1 in wild-type and IFNB-/- BMDM are based off previously generated microarray data published by Blanc et al. (2013). Briefly, wild-type, and IFNB-/- BMDM were either infected with MCMV (MOI 1) or treated with or poly(I:C) (25 μg/ml) or IFNγ (10 U/ml). Cells infected with MCMV or treated with poly(I:C) were harvested at 0, 2, 4, 6, 8, 10, and 24 h post-treatment for RNA isolation and microarray analysis (Affymetrix Mouse Gene 1.0ST microarray). Cells treated with IFNγ were harvested every 60 min for a total of 12 h post-treatment for RNA isolation and microarray analysis (Affymetrix Mouse Gene 1.0ST microarray). In brief, the arrays were normalized using the gcRMA algorithm (Wu et al., 2004) and imported into Partek Genomics Suite (Partek, United States) for downstream analysis (Blanc et al., 2013). Temporal expression data for Nampt in MCMV-infected wild-type and Tyk2-/- BMDM was generated using microarray. BMDM isolated from wild-type and Tyk2-/- mice were cultivated for 7 days. Following 7 days, cells were infected with MCMV (MOI 1) and harvested 1–1.5 and 6–6.5 h post-infection followed by RNA isolation (RNeasy Midi kit, Qiagen) and microarray analysis (Affymetrix Mouse Gene 1.0ST microarray). Time course microarray analysis data are compliant with the National Centre for Biotechnology Information Gene Expression Omnibus (GEO) (Edgar et al., 2002) under SuperSeries accession number GSE42505 (SubSeries numbers GSE42503, GSE42504) (GEO1). Macrophage microarray data of MCMV-infected wild-type and Tyk2-/- BMDM is accessible through GEO under accession number GSE126867.
Prior to statistical analysis, gene expression data [0–24 h post-mCMV infection or poly(I:C) treatment] was smoothened by taking the mean of every consecutive pair of points, i.e., mean of score at 0 and 2 h defined the score at 1 (hour) and mean of 2 and 4 h defined the score at 3 (hours), 4 and 6 h defined the score at 5 (hours), 6 and 8 h defining the score at 7 (hours), and 8 and 10 h defining the score at 9 (hours). Smoothing was not done beyond 10 h as the gap to the next point at 24 h was too large and, thus, excluded from analysis. This type of smoothing preserves the patterns in the data while removing some of the fine scale rapid changes. To investigate whether the gene expression changed over time, the mean smoothed data (or non-smoothened for the 12 h-dataset) for each gene expression was compared to a straight horizontal line. This was achieved by fitting the data to an appropriate polynomial in time t, i.e., either a linear model (y = α + βt), or a quadratic model (y = α + βt + γt2) or a cubic model (y = α + βt + γt2 + δt3). If the coefficients other than the intercept (α) were significant then the model and hence the gene expression, must vary with time. Which model was chosen depended on the model fit as given by the R2 value – the higher the better. The great majority of the fits were very strong –R2 > 0.9. To determine then if the model deviated significantly for the horizontal, the significance attached to each of the coefficients (β,γ,δ) of the time variables was investigated. If anyone of these was significant at the p < 0.05 level or if any two were significant at the 0.05 < p < 0.1 level then we considered the model and the gene expression to vary significantly with time. Refer to Supplementary Tables S5–S27 for the results of the statistical analysis.
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
WD, KR, and PG conceived and designed the experiments. WD and KR performed the experiments, and WD, KR, and PG performed the data analysis. WJ performed the statistical analysis of the time-course microarray analysis. BS contributed to the Tyk2-/- experiments. WD and PG wrote the manuscript.
This work was in part supported by the BBSRC (BB/K019112/1) and Welsh Government and EU ERDF funds to PG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00355/full#supplementary-material
FIGURE S1 | Nampt expression in macrophages is Jak/Stat-dependent and induced by IFNγ stimulation. Normalized temporal Nampt expression in IFNγ-treated BMDM. The expression was measured hourly over the first 12 h of treatment using microarray, and the expression levels were fitted to a cubic polynomal on time and assessed for statitical significance (temporal change over 12 h). ###p < 0.001 were considered to be significant.
FIGURE S2 | Knock-down efficiency of Nampt and Sirt6 in NIH-3T3 cells. (A) Quantification of relative Nampt mRNA expression in NIH-3T3 cells following using qRT-PCR following siRNA treatment (6.25, 12.5, and 25 nM siRNA) (n = 3). (B) Quantification of relative Sirt6 mRNA expression in NIH-3T3 cells following using qRT-PCR following siRNA treatment (6.25, 12.5, and 25 nM siRNA) (n = 3). One-way ANOVA with a Dunnett’s multiple comparisons test was used to determine statistical significance. Bars represent standard error of the mean (SEM). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 were considered to be significant (ns, not significant).
TABLE S1 | Putative TF binding in murine Nampt promoter region (-1 kb upstream of Nampt gene): prediction done with PROMO online based tool.
TABLE S2 | Putative TF binding in murine Sirt6 promoter region (-1 kb upstream of Sirt6 gene): prediction done with PROMO online based tool.
TABLE S3 | Putative TF binding in human NAMPT promoter region (-1 kb upstream of NAMPT gene): prediction done with PROMO online based tool.
TABLE S4 | Putative TF binding in human SIRT6 promoter region (-1 kb upstream of SIRT6 gene): prediction done with PROMO online based tool.
TABLE S5 | Coefficientsa for fit of Nampt expression over time in MCMV infected BMDM (0–10 h).
TABLE S6 | Coefficientsa for fit of Sirt6 expression over time in MCMV infected BMDM (0–10 h).
TABLE S7 | Coefficientsa for fit of Nampt expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S8 | Coefficientsa for fit of Sirt6 expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S9 | Coefficientsa for fit of Nampt expression over time in MCMV infected BMDM (0–12 h).
TABLE S10 | Coefficientsa for fit of Myd88 expression over time in MCMV infected BMDM (0–10 h).
TABLE S11 | Coefficientsa for fit of p50 (Nfkb1) expression over time in MCMV infected BMDM (0–10 h).
TABLE S12 | Coefficientsa for fit of p65 (Rela) expression over time in MCMV infected BMDM (0–10 h).
TABLE S13 | Coefficientsa for fit of Trif (Ticam1) expression over time in MCMV infected BMDM (0–10 h).
TABLE S14 | Coefficientsa for fit of Rig-I (Ddx58) expression over time in MCMV infected BMDM (0–10 h).
TABLE S15 | Coefficientsa for fit of Mda5 (Ifih1) expression over time in MCMV infected BMDM (0–10 h).
TABLE S16 | Coefficientsa for fit of Ips-1 (Mavs) expression over time in MCMV infected BMDM (0–10 h).
TABLE S17 | Coefficientsa for fit of Sting (Tmem173) expression over time in MCMV infected BMDM (0–10 h).
TABLE S18 | Coefficientsa for fit of cGas (Md21d1) expression over time in MCMV infected BMDM (0–10 h).
TABLE S19 | Coefficientsa for fit of Myd88 expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S20 | Coefficientsa for fit of p50 (Nfkb1) expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S21 | Coefficientsa for fit of p65 (Rela) expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S22 | Coefficientsa for fit of Trif (Ticam1) expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S23 | Coefficientsa for fit of Rig-I (Ddx58) expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S24 | Coefficientsa for fit of Mda5 (Ifih1) expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S25 | Coefficientsa for fit of Ips-1 (Mavs) expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S26 | Coefficientsa for fit of Sting (Tmem173) expression over time in poly(I:C) treated BMDM (0–10 h).
TABLE S27 | Coefficientsa for fit of cGas (Md21d1) expression over time in poly(I:C) treated BMDM (0–10 h).
Adams, C. M., Reitz, J., De, Brabander JK, Feramisco, J. D., Li, L., Brown, M. S., et al. (2004). Cholesterol and 25-hydroxycholesterol inhibit activation of SREBPs by different mechanisms, both involving SCAP and insigs. J. Biol. Chem. 279, 52772–52780. doi: 10.1074/jbc.M410302200
Angel, P., and Karin, M. (1991). The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072, 129–157. doi: 10.1016/0304-419X(91)90011-9
Angulo, A., Ghazal, P., and Messerle, M. (2000). The major immediate-early gene ie3 of mouse cytomegalovirus is essential for viral growth. J. Virol. 74, 11129–11136. doi: 10.1128/JVI.74.23.11129-11136.2000
Bauman, D. R., Bitmansour, A. D., McDonald, J. G., Thompson, B. M., Liang, G., and Russell, D. W. (2009). 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. U.S.A. 106, 16764–16769. doi: 10.1073/pnas.0909142106
Bitterman, K. J., Anderson, R. M., Cohen, H. Y., Latorre-Esteves, M., and Sinclair, D. A. (2002). Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 277, 45099–45107. doi: 10.1074/jbc.M205670200
Blanc, M., Hsieh, W. Y., Robertson, K. A., Kropp, K. A., Forster, T., Shui, G., et al. (2013). The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118. doi: 10.1016/j.immuni.2012.11.004
Blanc, M., Hsieh, W. Y., Robertson, K. A., Watterson, S., Shui, G., Lacaze, P., et al. (2011). Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol. 9:e1000598. doi: 10.1371/journal.pbio.1000598
Bodaghi, B., Goureau, O., Zipeto, D., Laurent, L., Virelizier, J. L., and Michelson, S. (1999). Role of IFN-gamma-induced indoleamine 2,3 dioxygenase and inducible nitric oxide synthase in the replication of human cytomegalovirus in retinal pigment epithelial cells. J. Immunol. 162, 957–964.
Borghini, L., Lu, J., Hibberd, M., and Davila, S. (2018). Variation in genome-wide NF-κB RELA binding sites upon microbial stimuli and identification of a virus response profile. J. Immunol. 201, 1295–1305. doi: 10.4049/jimmunol.1800246
Bult, C. J., Krupke, D. M., Begley, D. A., Richardson, J. E., Neuhauser, S. B., Sundberg, J. P., et al. (2015). Mouse Tumor Biology (MTB): a database of mouse models for human cancer. Nucleic Acids Res. 43, D818–D824. doi: 10.1093/nar/gku987
Carlin, J. M., Borden, E. C., and Byrne, G. I. (1989). Interferon-induced indoleamine 2,3-dioxygenase activity inhibits Chlamydia psittaci replication in human macrophages. J. Interferon Res. 9, 329–337. doi: 10.1089/jir.1989.9.329
Chen, X. Y., Zhang, H. S., Wu, T. C., Sang, W. W., and Ruan, Z. (2013). Down-regulation of NAMPT expression by miR-182 is involved in Tat-induced HIV-1 long terminal repeat (LTR) transactivation. Int. J. Biochem. Cell Biol. 45, 292–298. doi: 10.1016/j.biocel.2012.11.002
Cheng, G., Zhong, J., Chung, J., and Chisari, F. V. (2007). Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc. Natl. Acad. Sci. U.S.A. 104, 9035–9040. doi: 10.1073/pnas.0703285104
Chukkapalli, V., Heaton, N. S., and Randall, G. (2012). Lipids at the interface of virus-host interactions. Curr. Opin. Microbiol. 15, 512–518. doi: 10.1016/j.mib.2012.05.013
Compton, T., Kurt-Jones, E. A., Boehme, K. W., Belko, J., Latz, E., Golenbock, D. T., et al. (2003). Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J. Virol. 77, 4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003
Dölken, L., Ruzsics, Z., Rädle, B., Friedel, C. C., Zimmer, R., Mages, J., et al. (2008). High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14, 1959–1972. doi: 10.1261/rna.1136108
Edgar, R., Domrachev, M., and Lash, A. E. (2002). Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. doi: 10.1093/nar/30.1.207
Eguchi, J., Yan, Q. W., Schones, D. E., Kamal, M., Hsu, C. H., Zhang, M. Q., et al. (2008). Interferon regulatory factors are transcriptional regulators of adipogenesis. Cell Metab. 7, 86–94. doi: 10.1016/j.cmet.2007.11.002
Ekblad, T., and Schüler, H. (2016). Sirtuins are unaffected by parp inhibitors containing planar nicotinamide bioisosteres. Chem. Biol. Drug Des. 87, 478–482. doi: 10.1111/cbdd.12680
Elhanati, S., Kanfi, Y., Varvak, A., Roichman, A., Carmel-Gross, I., Barth, S., et al. (2013). Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep. 4, 905–912. doi: 10.1016/j.celrep.2013.08.006
Farré, D., Roset, R., Huerta, M., Adsuara, J. E., Roselló, L., Albà, M. M., et al. (2003). Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3653. doi: 10.1093/nar/gkg605
Finger, J. H., Smith, C. M., Hayamizu, T. F., McCright, I. J., Xu, J., Law, M., et al. (2017). The mouse Gene Expression Database (GXD): 2017 update. Nucleic Acids Res. 45, D730–D736. doi: 10.1093/nar/gkw1073
Geer, L. Y., Marchler-Bauer, A., Geer, R. C., Han, L., He, J., He, S., et al. (2010). The NCBI biosystems database. Nucleic Acids Res. 38, D492–D496. doi: 10.1093/nar/gkp858
Gerbod-Giannone, M., Li, C. Y., Holleboom, A., Han, S., Hsu, L. C., Tabas, I., et al. (2006). TNFalpha induces ABCA1 through NF-kappaB in macrophages and in phagocytes ingesting apoptotic cells. Proc. Natl. Acad. Sci. U.S.A. 103, 3112–3117. doi: 10.1073/pnas.0510345103
Ghazal, P., González Armas, J. C., García-Ramírez, J. J., Kurz, S., and Angulo, A. (2000). Viruses: hostages to the cell. Virology 275, 233–237. doi: 10.1006/viro.2000.0553
Haas, M. J., and Mooradian, A. D. (2010). Regulation of high-density lipoprotein by inflammatory cytokines: establishing links between immune dysfunction and cardiovascular disease. Diabetes Metab. Res. Rev. 26, 90–99. doi: 10.1002/dmrr.1057
Hasmann, M., and Schemainda, I. (2003). FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 63, 7436–7442.
Heinrich, P. C., Behrmann, I., Müller-Newen, G., Schaper, F., and Graeve, L. (1998). Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 334(Pt 2), 297–314. doi: 10.1042/bj3340297
Heseler, K., Schmidt, S. K., Spekker, K., Sinzger, C., Sorg, R. V., Quambusch, M., et al. (2013). Cytomegalovirus impairs the induction of indoleamine 2,3-dioxygenase mediated antimicrobial and immunoregulatory effects in human fibroblasts. PLoS One 8:e64442. doi: 10.1371/journal.pone.0064442
Hess, J., Angel, P., and Schorpp-Kistner, M. (2004). AP-1 subunits: quarrel and harmony among siblings. J. Cell Sci. 117(Pt 25), 5965–5973. doi: 10.1242/jcs.01589
Hu, J., Jing, H., and Lin, H. (2014). Sirtuin inhibitors as anticancer agents. Future Med. Chem. 6, 945–966. doi: 10.4155/fmc.14.44
Ishikawa, H., Ma, Z., and Barber, G. N. (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. doi: 10.1038/nature08476
Janus, P., Szołtysek, K., Zając, G., Stokowy, T., Walaszczyk, A., Widłak, W., et al. (2018). Pro-inflammatory cytokine and high doses of ionizing radiation have similar effects on the expression of NF-kappaB-dependent genes. Cell. Signal. 46, 23–31. doi: 10.1016/j.cellsig.2018.02.011
Jiang, H., Khan, S., Wang, Y., Charron, G., He, B., Sebastian, C., et al. (2013). SIRT6 regulates TNF-α secretion through hydrolysis of long-chain fatty acyl lysine. Nature 496, 110–113. doi: 10.1038/nature12038
Karin, M., Liu, Z., and Zandi, E. (1997). AP-1 function and regulation. Curr. Opin. Cell Biol. 9, 240–246. doi: 10.1016/S0955-0674(97)80068-3
Kawahara, T. L., Michishita, E., Adler, A. S., Damian, M., Berber, E., Lin, M., et al. (2009). SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 62–74. doi: 10.1016/j.cell.2008.10.052
Kotzamanis, K., Angulo, A., and Ghazal, P. (2015). Infection homeostasis: implications for therapeutic and immune programming of metabolism in controlling infection. Med. Microbiol. Immunol. 204, 395–407. doi: 10.1007/s00430-015-0402-5
Koyuncu, E., Budayeva, H. G., Miteva, Y. V., Ricci, D. P., Silhavy, T. J., Shenk, T., et al. (2014). Sirtuins are evolutionarily conserved viral restriction factors. MBio 5:e2249-14. doi: 10.1128/mBio.02249-14
Kropp, K. A., Robertson, K. A., Sing, G., Rodriguez-Martin, S., Blanc, M., Lacaze, P., et al. (2011). Reversible inhibition of murine cytomegalovirus replication by gamma interferon (IFN-γ) in primary macrophages involves a primed type I IFN-signaling subnetwork for full establishment of an immediate-early antiviral state. J. Virol. 85, 10286–10299. doi: 10.1128/JVI.00373-11
Li, P., Jin, Y., Qi, F., Wu, F., Luo, S., Cheng, Y., et al. (2018). SIRT6 acts as a negative regulator in dengue virus-induced inflammatory response by targeting the DNA binding domain of NF-κB p65. Front. Cell Infect. Microbiol. 8:113. doi: 10.3389/fcimb.2018.00113
Lim, C. A., Yao, F., Wong, J. J., George, J., Xu, H., Chiu, K. P., et al. (2007). Genome-wide mapping of RELA(p65) binding identifies E2F1 as a transcriptional activator recruited by NF-kappaB upon TLR4 activation. Mol. Cell 27, 622–635. doi: 10.1016/j.molcel.2007.06.038
Liu, S. Y., Aliyari, R., Chikere, K., Li, G., Marsden, M. D., Smith, J. K., et al. (2013). Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105. doi: 10.1016/j.immuni.2012.11.005
Liu, T. F., Brown, C. M., El Gazzar, M., McPhail, L., Millet, P., Rao, A., et al. (2012a). Fueling the flame: bioenergy couples metabolism and inflammation. J. Leukoc. Biol. 92, 499–507. doi: 10.1189/jlb.0212078
Liu, T. F., Vachharajani, V. T., Yoza, B. K., and McCall, C. E. (2012b). NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 287, 25758–25769. doi: 10.1074/jbc.M112.362343
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lu, H., Talbot, S., Robertson, K. A., Watterson, S., Forster, T., Roy, D., et al. (2015). Rapid proteasomal elimination of 3-hydroxy-3-methylglutaryl-CoA reductase by interferon-γ in primary macrophages requires endogenous 25-hydroxycholesterol synthesis. Steroids 99(Pt B), 219–229. doi: 10.1016/j.steroids.2015.02.022
Lugrin, J., Ciarlo, E., Santos, A., Grandmaison, G., dos Santos, I., Le Roy, D., et al. (2013). The sirtuin inhibitor cambinol impairs MAPK signaling, inhibits inflammatory and innate immune responses and protects from septic shock. Biochim. Biophys. Acta 1833, 1498–1510. doi: 10.1016/j.bbamcr.2013.03.004
Lyssiotis, C. A., and Cantley, L. C. (2012). SIRT6 puts cancer metabolism in the driver’s seat. Cell 151, 1155–1156. doi: 10.1016/j.cell.2012.11.020
MacKenzie, C. R., Hadding, U., and Däubener, W. (1998). Interferon-gamma-induced activation of indoleamine 2,3-dioxygenase in cord blood monocyte-derived macrophages inhibits the growth of group B streptococci. J. Infect. Dis. 178, 875–878. doi: 10.1086/515347
Maitra, U., Parks, J. S., and Li, L. (2009). An innate immunity signaling process suppresses macrophage ABCA1 expression through IRAK-1-mediated downregulation of retinoic acid receptor alpha and NFATc2. Mol. Cell Biol. 29, 5989–5997. doi: 10.1128/MCB.00541-09
Mesmin, B., Bigay, J., Moser von Filseck, J., Lacas-Gervais, S., Drin, G., and Antonny, B. (2013). A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSB. Cell 155, 830–843. doi: 10.1016/j.cell.2013.09.056
Messeguer, X., Escudero, R., Farré, D., Núñez, O., Martínez, J., and Albà, M. M. (2002). PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334. doi: 10.1093/bioinformatics/18.2.333
Messerle, M., Hahn, G., Brune, W., and Koszinowski, U. H. (2000). Cytomegalovirus bacterial artificial chromosomes: a new herpesvirus vector approach. Adv. Virus Res. 55, 463–478. doi: 10.1016/S0065-3527(00)55013-1
Olsen, B. N., Schlesinger, P. H., Ory, D. S., and Baker, N. A. (2011). 25-Hydroxycholesterol increases the availability of cholesterol in phospholipid membranes. Biophys. J. 100, 948–956. doi: 10.1016/j.bpj.2010.12.3728
Parenti, M. D., Grozio, A., Bauer, I., Galeno, L., Damonte, P., Millo, E., et al. (2014). Discovery of novel and selective SIRT6 inhibitors. J. Med. Chem. 57, 4796–4804. doi: 10.1021/jm500487d
Pfefferkorn, E. R. (1984). Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. U.S.A. 81, 908–912. doi: 10.1073/pnas.81.3.908
Reboldi, A., Dang, E. V., McDonald, J. G., Liang, G., Russell, D. W., and Cyster, J. G. (2014). Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 345, 679–684. doi: 10.1126/science.1254790
Robertson, K. A., Hsieh, W. Y., Forster, T., Blanc, M., Lu, H., Crick, P. J., et al. (2016). An interferon regulated microRNA provides broad cell-intrinsic antiviral immunity through multihit host-directed targeting of the sterol pathway. PLoS Biol. 14:e1002364. doi: 10.1371/journal.pbio.1002364
Santos-Barriopedro, I., Bosch-Presegué, L., Marazuela-Duque, A., de la Torre, C., Colomer, C., Vazquez, B. N., et al. (2018). SIRT6-dependent cysteine monoubiquitination in the PRE-SET domain of Suv39h1 regulates the NF-κB pathway. Nat. Commun. 9:101. doi: 10.1038/s41467-017-02586-x
Santos-Barriopedro, I., and Vaquero, A. (2018). Complex role of SIRT6 in NF-κB pathway regulation. Mol. Cell Oncol. 5:e1445942. doi: 10.1080/23723556.2018.1445942
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Schoggins, J. W., Wilson, S. J., Panis, M., Murphy, M. Y., Jones, C. T., Bieniasz, P., et al. (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. doi: 10.1038/nature09907
Seth, R. B., Sun, L., Ea, C. K., and Chen, Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. doi: 10.1016/j.cell.2005.08.012
Shaw, M. H., Freeman, G. J., Scott, M. F., Fox, B. A., Bzik, D. J., Belkaid, Y., et al. (2006). Tyk2 negatively regulates adaptive Th1 immunity by mediating IL-10 signaling and promoting IFN-gamma-dependent IL-10 reactivation. J. Immunol. 176, 7263–7271. doi: 10.4049/jimmunol.176.12.7263
Shimoda, K., Kato, K., Aoki, K., Matsuda, T., Miyamoto, A., Shibamori, M., et al. (2000). Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity 13, 561–571. doi: 10.1016/S1074-7613(00)00055-8
Singaravelu, R., O’Hara, S., Jones, D. M., Chen, R., Taylor, N. G., Srinivasan, P., et al. (2015). MicroRNAs regulate the immunometabolic response to viral infection in the liver. Nat. Chem. Biol. 11, 988–993. doi: 10.1038/nchembio.1940
Smith, C. L., Blake, J. A., Kadin, J. A., Richardson, J. E., Bult, C. J., and Mouse, Genome Database Group (2018). Mouse genome Database (MGD)-2018: knowledgebase for the laboratory mouse. Nucleic Acids Res. 46, D836–D842. doi: 10.1093/nar/gkx1006
Sociali, G., Galeno, L., Parenti, M. D., Grozio, A., Bauer, I., Passalacqua, M., et al. (2015). Quinazolinedione SIRT6 inhibitors sensitize cancer cells to chemotherapeutics. Eur. J. Med. Chem. 102, 530–539. doi: 10.1016/j.ejmech.2015.08.024
Spann, N. J., and Glass, C. K. (2013). Sterols and oxysterols in immune cell function. Nat. Immunol. 14, 893–900. doi: 10.1038/ni.2681
Stahl, N., Boulton, T. G., Farruggella, T., Ip, N. Y., Davis, S., Witthuhn, B. A., et al. (1994). Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 263, 92–95. doi: 10.1126/science.8272873
Strobl, B., Bubic, I., Bruns, U., Steinborn, R., Lajko, R., Kolbe, T., et al. (2005). Novel functions of tyrosine kinase 2 in the antiviral defense against murine cytomegalovirus. J. Immunol. 175, 4000–4008. doi: 10.4049/jimmunol.175.6.4000
Tao, R., Xiong, X., DePinho, R. A., Deng, C. X., and Dong, X. C. (2013). Hepatic SREBP-2 and cholesterol biosynthesis are regulated by FoxO3 and Sirt6. J. Lipid Res. 54, 2745–2753. doi: 10.1194/jlr.M039339
Van den Bergh, R., Florence, E., Vlieghe, E., Boonefaes, T., Grooten, J., Houthuys, E., et al. (2010). Transcriptome analysis of monocyte-HIV interactions. Retrovirology 7:53. doi: 10.1186/1742-4690-7-53
Van den Bergh, R., Morin, S., Sass, H. J., Grzesiek, S., Vekemans, M., Florence, E., et al. (2012). Monocytes contribute to differential immune pressure on R5 versus X4 HIV through the adipocytokine visfatin/NAMPT. PLoS One 7:e35074. doi: 10.1371/journal.pone.0035074
Velazquez, L., Fellous, M., Stark, G. R., and Pellegrini, S. (1992). A protein tyrosine kinase in the interferon alpha/beta signaling pathway. Cell 70, 313–322. doi: 10.1016/0092-8674(92)90105-L
Wu, Z., Irizarry, R. A., Gentleman, R., Martinez-Murillo, F., and Spencer, F. (2004). A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 99, 909–917. doi: 10.1198/016214504000000683
York, A. G., Williams, K. J., Argus, J. P., Zhou, Q. D., Brar, G., Vergnes, L., et al. (2015). Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling. Cell 163, 1716–1729. doi: 10.1016/j.cell.2015.11.045
Zelcer, N., and Tontonoz, P. (2006). Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Invest. 116, 607–614. doi: 10.1172/JCI27883
Zhang, H. S., Chen, X. Y., Wu, T. C., and Zhang, F. J. (2014). Tanshinone II A inhibits tat-induced HIV-1 transactivation through redox-regulated AMPK/Nampt pathway. J. Cell. Physiol. 229, 1193–1201. doi: 10.1002/jcp.24552
Zhang, H. S., Sang, W. W., Wang, Y. O., and Liu, W. (2010). Nicotinamide phosphoribosyltransferase/sirtuin 1 pathway is involved in human immunodeficiency virus type 1 Tat-mediated long terminal repeat transactivation. J. Cell. Biochem. 110, 1464–1470. doi: 10.1002/jcb.22704
Keywords: cholesterol, metabolism, lipid pathway, sterol, epigenetic, interferon, cytomegalovirus
Citation: Dantoft W, Robertson KA, Watkins WJ, Strobl B and Ghazal P (2019) Metabolic Regulators Nampt and Sirt6 Serially Participate in the Macrophage Interferon Antiviral Cascade. Front. Microbiol. 10:355. doi: 10.3389/fmicb.2019.00355
Received: 08 October 2018; Accepted: 11 February 2019;
Published: 04 March 2019.
Edited by:
Maria Laura Gennaro, Rutgers, The State University of New Jersey, United StatesReviewed by:
Sandra Marcia Muxel, University of São Paulo, BrazilCopyright © 2019 Dantoft, Robertson, Watkins, Strobl and Ghazal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Ghazal, R2hhemFsUEBjYXJkaWZmLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.