 Jia Qu
Jia Qu Yan Zhao
Yan Zhao Jun Yin
Jun Yin- School of Information and Control Engineering, China University of Mining and Technology, Xuzhou, China
Studies have shown that microbes exist widely in the human body and are closely related to human complex diseases. Predicting potential associations between microbes and diseases is conducive to understanding the mechanisms of complex diseases and can also facilitate the diagnosis and prevention of human diseases. In this paper, we put forward the Matrix Decomposition and Label Propagation for Human Microbe-Disease Association prediction (MDLPHMDA) on the basis of the dataset of known microbe-disease associations collected from the database of HMDAD and the Gaussian interaction profile kernel similarity for diseases and microbes, disease symptom similarity. Moreover, the performance of our model was evaluated by means of leave-one-out cross validation and five-fold cross validation, and the corresponding AUCs of 0.9034 and 0.8954 ± 0.0030 were gained, respectively. In case studies, 10, 9, 9, and 8 out of the top 10 predicted microbes for asthma, colorectal carcinoma, liver cirrhosis, and type 1 diabetes were confirmed by literatures, respectively. Overall, evaluation results showed that MDLPHMDA has good performance in potential microbe-diseasepositive free parameter, which associations prediction.
Introduction
Microbes are microscopic organisms that may exist in single-celled form or in a colony of cells (Madigan and Michaelt, 2015). They live in almost all the habitats from the poles to the deep sea and also make up the microbiota in all multicellular organisms (Delong and Pace, 2001). There are trillions of microbes in the human body. Lots of them are beneficial for human health, while others may cause infectious diseases (Thiele et al., 2013). Human microbiota can form an endosymbiotic relationship with their host, providing services and useful goods to humans. For example, the gut flora can contribute to gut immunity as well as digest complex carbohydrates and synthesize vitamins (O'hara and Shanahan, 2006). It is now accepted that most of the microbes are not intrinsically harmful. However, the pathogenic microorganisms and the imbalance of resident microbes are closely related to human disease.
Microorganisms are closely related to both infectious diseases and non-infectious diseases. Infectious diseases are global problems. They have induced several feared plagues in human history and new infections are still emerging today (Morse, 1995). Microorganisms are the causative pathogens for many infectious diseases. The involved organisms include pathogenic bacteria such as Mycobacterium tuberculosis and Bacillus anthracis, which can cause tuberculosis and anthrax, respectively (Hawn et al., 2014; Hendricks et al., 2014); protozoan parasites such as Plasmodium and Toxoplasma gondii, which can cause malaria and toxoplasmosis (Torgerson and Mastroiacovo, 2013; Iburg, 2015); and also fungi such as Candida albicans and Histoplasma capsulatum which can cause candidiasis or histoplasmosis (Stenn, 1960; Pappas et al., 2016). Meanwhile, most new infections appear to be caused by already discovered pathogenic microorganisms. These pathogens obtain selective advantage by changing conditions to infect new host populations or cause a new disease (Morse, 1991). On the other hand, microbiota can interact with human at multiple levels. Due to these complex microbiota-host relationships, dysbiosis can be the cause of the pathology (Forum on Microbial et al., 2014). Various factors including antibiotics, radiations, stress or nutritional changes can alter the compositions of human microbiota. This disruption of homeostasis can induce many maladies (Tamboli et al., 2004). For example, it is founded that the interactions between host immunity and gut microbiota can directly result in inflammatory bowel disease (IBD). IBD is a long-term aggravating inflammation of the intestine (Schirbel and Fiocchi, 2010). Both commensal microbiota and individual genetic susceptibility play key roles in the occurrence and development of this disease (Ferreira et al., 2014). Compared to healthy control, the composition of gut microbiota in IBD patients is distinct with decreased Firmicutes (Walker et al., 2011). The complex interplay between microbiota and human is also closely related to metabolic disease such as obesity (Ley et al., 2006). In a study about overweight and obese children, scientists found that the lower numbers of fecal Staphylococcus aureus was further linked with normal-weight development (Kalliomaki et al., 2008). Besides intestinal tract, microbial communities in respiratory tract are also closely related to various lung diseases such as sinusitis and chronic obstructive pulmonary disease (COPD) (Huang et al., 2017b). A study showed that sinusitis patients experienced an increase in Corynebacterium tuberculostearicum (Abreu et al., 2012). In COPD, increased Lactobacillus is induced by an inflammatory modulation and results in the formation of tertiary lymphoid (Sze et al., 2012). All the above studies revealed the close associations between microbes and various human diseases. Unquestionably, identifying potential microbe-disease associations is of great significance in exploring the pathogenesis, prevention, and treatment of diseases. As the traditional experimental method is time-consuming, costly, random, and blind, there is an urgent need to develop an effective calculation approach so as to help researchers in finding the regular pattern of microbe-disease associations and to provide complementary and supportive evidence for the experimental study.
Relevant research for the identification of potential microbe-disease associations are still in its infancy, and effective calculation models for the association prediction are even more scarce. Ma et al. (2017) created the first database of Human Microbe-Disease Association Database (HMDAD), which collected confirmed microbe-disease associations from published literatures. Based on the above work, several computational models were established to prioritize candidate microbes for diseases. For example, Chen et al. (2017a) introduced the network-based model of KATZ measure for Human Microbe-Disease Association prediction (KATZHMDA), the first calculation method for the identification of new microbe-disease associations through computing the number of walks of connections between microbe and disease nodes in the microbe-disease association network. Recently, the computational model of Laplacian Regularized Least Squares for Human Microbe-Disease Association (LRLSHMDA) was presented by Wang et al. (2017). It is a global measure based on a semi-supervised learning framework. In their proposed calculation model, the Laplacian regularized least squares (LapRLS) classification was adopted to prioritize candidate microbes for all interested diseases through the application of known microbe-disease associations, the Gaussian interaction profile kernel similarity for microbes and diseases. Similarly, with the same dataset of known microbe-disease associations, the Gaussian interaction profile kernel similarity for microbes and diseases mentioned above, a path-based search model of Path-Based Human Microbe-Disease Association prediction (PBHMDA) was introduced by Huang et al. (2017c). In the model, the association score of each microbe-disease pair would be computed by the integration of all paths less four between the microbe and disease with different weights. In addition, Huang et al. (2017a) put forward a Neighbor- and Graph-based combined Recommendation model for Human Microbe-Disease Association prediction (NGRHMDA). The final prediction scores of novel microbe-disease associations were attained via the integration of two prediction results predicted by neighbor-based collaborative filtering and the graph-based scoring method. Also, Peng et al. (2018) put forward a model of Adaptive Boosting for Human Microbe-Disease Association prediction (ABHMDA) by enforcing a strong classifier on the samples. Specifically, the strong classifier was constructed by the integration of 30 weak classifiers with different weights.
In this paper, by combining known microbe-disease associations collected from HMDAD, disease symptom similarity and Gaussian interaction profile kernel similarity for microbes and diseases, we introduced a computational model of Matrix Decomposition and Label Propagation for the Human Microbe-Disease Association prediction (MDLPHMDA). In our proposed algorithm, a new adjacency matrix of microbe-disease associations was first generated by employing the spare learning method (SLM) on the original association information extracted from HMDAD, and potential microbe-disease associations would be further predicted under the implementation of the label propagation algorithm (LPA). The leave-one-out cross validation (LOOCV) and five-fold cross validation were subsequently enforced for accuracy evaluation of MDLPHMDA. Assessment results showed that MDLPHMDA gained the area under the receiver operating characteristic curves (AUCs) of 0.9034 and 0.8954 ± 0.0030 in LOOCV and five-fold cross validation, respectively. In case studies, we carried out MDLPHMDA to predict potential microbes for asthma and colorectal carcinoma (CRC), respectively. Moreover, via the implementation of our developed algorithm, we prioritized microbes for liver cirrhosis and type 1 diabetes by removing their known related microbes, respectively. Finally, the results analysis of cross validations and case studies showed that MDLPHMDA is a suitable and effective model in potential microbe-disease association prediction.
Materials and Methods
Human Microbe-Disease Associations
The dataset of confirmed microbe-disease associations used in this paper were collected from HMDAD (http://www.cuilab.cn/hmdad) (Ma et al., 2017). According to the 16s RNA sequencing-based microbiome research, the database collected 483 microbe-disease associations between 39 diseases and 292 microbes from 61 previous works. Along with the deletion of the same microbe-disease associations based on different evidences in the database, we finally obtained a dataset of 450 associations between 39 diseases and 292 microbes. Moreover, the variables nd and nm were defined to represent the 39 diseases and 292 microbes, respectively. Also, adjacency matrix A of the verified microbe-disease associations was defined as follows:
Integrated Diseases Similarity
The integrated disease similarity was constructed by combining the Gaussian interaction profile kernel similarity for diseases and disease symptom similarity. First, we calculated the Gaussian interaction profile kernel similarity for diseases by adopting the calculation approach in the previous literature (Van Laarhoven et al., 2011). According to the idea that similar diseases possess similar interaction and non-interaction patterns with microbes, the Gaussian interaction profile kernel similarity for diseases was created in light of confirmed microbe-disease associations. We defined the interaction profile of each disease by using a binary vector that shows whether the disease is related to each microbe or not. For example, for disease d(i), its interaction profile IP(d(i)) is the ith row of the adjacency matrix A. Therefore, the Gaussian interaction profile kernel similarity between disease d(i) and disease d(j) can be computed as follows:
where γd indicates the normalized kernel bandwidth in light of the new bandwidth parameter . Second, according to the data of diseases and their symptoms in PubMed bibliography, disease symptom similarity DSS could be constructed (Zhou et al., 2014). Finally, in accordance with disease symptom similarity put forward by Zhou et al. (2014), taking into account of the Gaussian interaction profile kernel similarity for diseases, we constructed integrated disease similarity by using the method applied in a previous study (Chen et al., 2017a).
Gaussian Interaction Profile Kernel Similarity for Microbes
In the same way, motivated by previous literature (Van Laarhoven et al., 2011), the Gaussian interaction profile kernel similarity for microbes was established according to confirmed microbe-disease associations. For microbe m(j), its interaction profile IP(m(j)) is the jth column of the adjacency matrix A. Therefore, the Gaussian interaction profile kernel similarity between microbe m(i) and microbe m(j) can be computed as follows:
where γm indicates the normalized kernel bandwidth in light of the new bandwidth parameter .
MDLPHMDA
In this manuscript, motivated by SLM developed by Pech et al. (2017) and LPA introduced by Zhang et al. (2017), we applied the calculation model of MDLPHMDA to infer novel microbe-disease associations. Starting from the fact that redundant formation may be present in the original dataset of known microbe-disease associations, we employed matrix decomposition to eliminate the noise of known microbe-disease associations and then applied LPA for the identification of the potential microbe-disease associations (see Figure 1). It is worth mentioning that matrix decomposition has been widely used in Bioinformatics research (Chen et al., 2018b,d; Zhao et al., 2018).
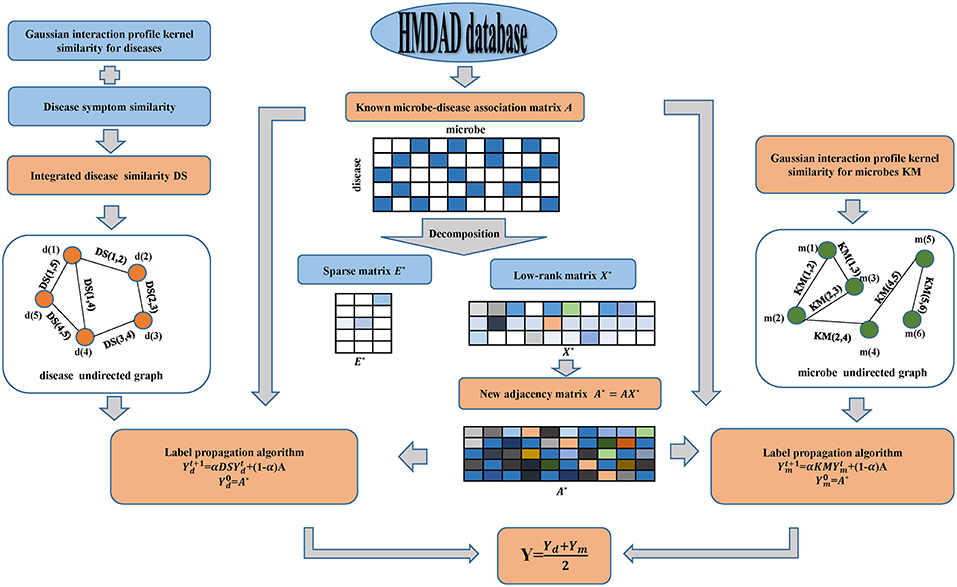
Figure 1. Flowchart of the calculation model of MDLPHMDA: We first enforced matrix decomposition to eliminate the noise of original known microbe-disease associations and gained a new adjacency matrix. Then LPA was implemented based on the created new adjacency matrix for the identification of the potential microbe-disease associations.
Since a part of microbe-disease associations in the dataset may be incorrect or redundant, we adopted SLM to remove the noise of the original data and search a lowest-rank matrix among candidates to gain a novel adjacency matrix. In our introduced model, we divided the original adjacency matrix A into two parts by using SLM. The first part is a linear combination of the original adjacency matrix A and a low-rank matrix, while the second part is a spare matrix that can be regarded as the noise of the original adjacency matrix A. Hence, the original adjacency matrix can be decomposed as follows:
In order to get a low-rank matrix X and a sparse matrix E, we could transform Equation (7) into a optimization problem by applying the nuclear norm on X and the sparse norm on E.
where
Here, α is a positive free parameter, which can balance the weight between the low-rank matrix and the sparse matrix. To transform the original optimization problem into an augmented Lagrange function, we rewrote the optimization problem into a constraint and convex optimization problem of Equation (11) and enforced an inexact augmented Lagrange multipliers (IALM) algorithm (Meng et al., 2014) to solve it (see Table 1).
where μ ≥ 0 is a penalty parameter and the detailed solution process to gain solution X* and E* of Equation (12) could be explained in previous literature (Pech et al., 2017).

Table 1. Computational procedures of the Inexact augmented Lagrange multipliers (IALM) algorithm.
As the solution of Equation (12) was solved, we gained a new adjacency matrix A* with less noise by the linear combination of the original adjacency matrix A and the low-rank matrix X* as follows:
Then, based on the Gaussian interaction profile kernel similarity for microbes and diseases, disease symptom similarity and the newly created adjacency matrix A*, we enforced LPA to infer novel microbe-disease associations. First, from the perspective of disease, we constructed an undirected graph with diseases as nodes, and similarity scores as edge weight. To combine the original microbe-disease associations information, we treated the new adjacency matrix of microbe-disease associations as the labels to propagate in the disease undirected graph and each label is updated through the absorption of its neighborhoods' label information with a rate of α and going back to its original known microbe-disease association nodes with a rate of 1 − α. Referring to previous literature (Yao et al., 2017; Zhang et al., 2017), we set α as 0.3. The label propagation process can be described as follows:
where indicates the predicted scores between microbes and diseases at step t. Specifically, refers to the newly created adjacency matrixA*. The iteration would be stable after some steps (the change in value between and measured by L1 norm is <10e-6). The final value Yd would be the predicted scores of new microbe-disease associations from the perspective of diseases.
Also, from the perspective of microbes, we can build another microbe undirected graph and employ LPA to gain another predicted scores Ym of novel microbe-disease associations. Finally, we defined the final predicted scores Y for the potential microbe-disease associations by the average of the two predicted scores mentioned above.
Results
Performance Evaluation
In order to test the prediction performance of MDLPHMDA based on the 450 confirmed microbe-disease associations collected from HMDAD (Ma et al., 2017), our model was compared with two classic algorithms (LRLSHMDA and KATZHMDA) on the basis of the evaluation method of LOOCV and five-fold cross validation. In LOOCV, each confirmed microbe-disease association was taken as test sample by turn and the rest 449 identified associations were used to train. After executing MDLPHMDA, the score of the test sample would be ranked with the scores of candidate samples that were made up of all unconfirmed microbe-disease pairs. In five-fold cross validation, we first divided the 450 microbe-disease association pairs into five equal parts and later made each part as test sample in turn and the remaining four parts of associations as training samples. In the same way, each test sample's score would be ranked with the scores of all candidate samples that were composed of unconfirmed microbe-disease pairs. As the sample divisions may cause bias, we enforced five-fold cross validation 100 times to gain an average value as the final result. If the ranking of the test sample is higher than a given threshold, our model is considered to make a successful prediction. Then, according to varying thresholds, we plotted the receiver operating characteristics (ROC) curve by computing the ratio of true positive rate (TPR, sensitivity) to false positive rate (FPR, 1-specificity). Sensitivity denotes the percentage of test samples which obtained ranks higher than the set threshold. Meanwhile, specificity denotes the percentage of negative microbe-disease pairs with ranks lower than the threshold. Finally, to assess the performance of MDLPHMDA effectively, we computed corresponding AUCs. When AUC = 1, the model possesses perfect forecast ability; when AUC = 0.5, the model possesses random forecast ability. In LOOCV, assessment results showed that MDLPHMDA, LRLSHMDA, and KATZHMDA gained the AUCs of 0.9034, 0.8909, and 0.8382, respectively (see Figure 2). In five-fold cross validation, MDLPHMDA, LRLSHMDA, and KATZHMDA gained the AUCs of 0.8954 ± 0.0030, 0.8794 ± 0.0029, and 0.8301 ± 0.0033, respectively. Stated thus, it can be seen that our model possesses good prediction ability and could be used to assist the identification of novel microbe-disease associations. Moreover, we carried out a paired t-test based on the ranking results of LOOCV to observe the statistical significance of differences among MDLPHMDA, LRLSHMDA, and KATZHMDA. As a result, the p-value of MDLPHMDA and LRLSHMDA is 0.0088, whereas the p-value of MDLPHMDA and KATZHMDA is 1.2510e-08. We can see that MDLPMDA is significantly different from LRLSHMDA and KATZHMDA on the basis of their ranking results of LOOCV (p < 0.05).
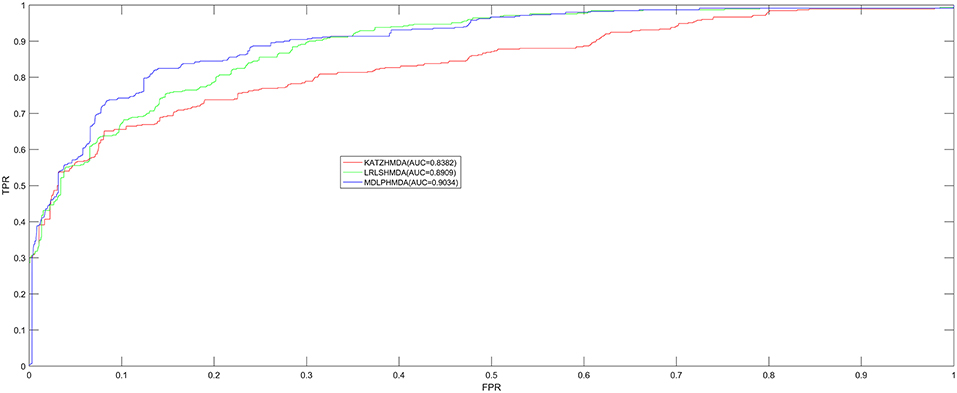
Figure 2. Performance comparison between MDLPHMDA and other two classical microbe-disease association prediction models (LRLSHMDA and KATZHMDA) by means of AUCs based on LOOCV. The results showed that MDLPHMDA gained AUCs of 0.9034 in LOOCV.
Case Study
Via two different types of case studies, we further assessed the prediction ability of MDLPHMDA based on the confirmed 450 microbe-disease associations. In the first kind, we identified potential microbes for asthma and CRC, respectively, through the implementation of MDLPHMDA. Also, we released all prediction scores for 10938 novel microbe-disease pair between 39 diseases and 292 microbes (see Supplementary Table 1). In the second kind, we enforced MDLPHMDA to identify liver cirrhosis-associated microbes by removing 62 known liver cirrhosis-associated microbes from the dataset of known microbe-disease associations and also predicted for another disease of type 1 diabetes by removing its known microbes. Based on the results of the two types of case studies, the proposed algorithm of MDLPHMDA was proven to be an effective algorithm in the identification of novel microbe-disease associations.
Asthma is a long-term inflammatory disease of the airways (Lemanske and Busse, 2010). Its common symptoms include coughing, reversible airflow obstruction, wheezing, or bronchospasm (Lemanske and Busse, 2010). Epidemiological studies indicated that microbial exposures in early life might determine microbiota composition, which can help to prevent allergy or lead to the development of asthma (Wang et al., 2003; Weber et al., 2015). A study in asthmatic children has found a low abundance of Bifidobacterium in their intestinal microbiota, which may reduce the immune function and potentially contribute to disease chronicization (Kalliomaki et al., 2001). Similarly, a probiotic strain Lactobacillus rhamnosus reduced allergic responses in the airways of neonates (Martinon et al., 2009). In this paper, via the implementation of MDLPHMDA for the inference of novel asthma-related microbes, we could see that the top 10 predicted microbes for asthma were all confirmed through literature (see Table 2). Among the top 3 confirmed associations between microbes and asthma, relevant differences in Firmicutes were found between samples from asthmatic and non-asthmatic subjects (Marri et al., 2013). Another study investigated that Clostridium difficile was associated with an increased risk for asthma (Van Nimwegen et al., 2011). Meanwhile, in a study about early intestinal colonization of infants, Clostridium coccoides was confirmed to be associated with increased risk for the development of asthma before the age of 3 years (Vael et al., 2011).
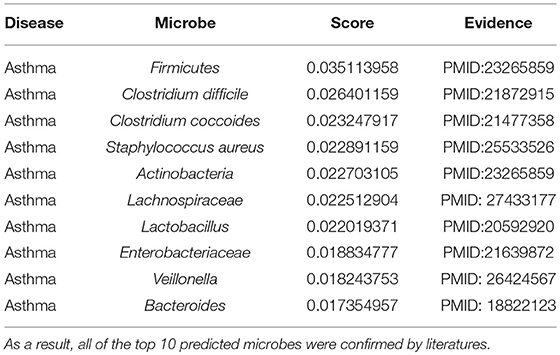
Table 2. The validation of the top 10 predicted asthma-related microbes after implementing MDLPHMDA based on the confirmed microbe-disease associations from HMDAD.
CRC is the cancer in the colon or rectum (Watson and Collins, 2011). Common symptoms include weight loss, blood in stool, and feeling tired all the time (Watson and Collins, 2011). It typically starts in the form of a polyp as a benign tumor, which becomes cancerous over time (Watson and Collins, 2011). A quantitative polymerase chain reaction (qpcr) analysis verified that Fusobacterium nucleatum, an invasive anaerobe previously linked to appendicitis and periodontitis but not to cancer, was increased in a CRC tumor vs. normal tissue (Castellarin et al., 2012). Furthermore, this overabundance is positively associated with lymph node metastasis (Castellarin et al., 2012). Another study also observed a significant difference of Bacteroides and Prevotella in a CRC group, as compared to a normal group (Sobhani et al., 2011). Moreover, we employed the proposed algorithm to predict CRC-related microbes and the outcomes displayed that all but one of the top 10 microbes for CRC were verified (see Table 3). Among the top 3 confirmed associations, according to the taxonomic results, Proteobacteria showed a higher abundance in CRC rats compared to control groups and constitute the third most abundant phyla (Zhu et al., 2014). In another analysis on CRC, the Helicobacter pylori infection was noted in 50 CRC patients. Furthermore, an infection with H. pylori CagA+ was associated with an increased risk for CRC (Shmuely et al., 2001). Moreover, a statistically significant difference in C. difficile was detected between the CRC and healthy group, suggesting a possible role of this bacteria in CRC carcinogenesis (Fukugaiti et al., 2015).
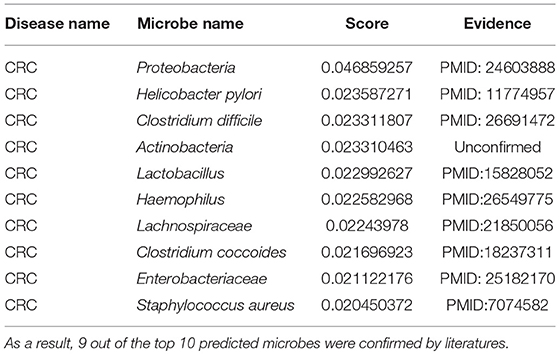
Table 3. The validation of the top 10 predicted CRC-related microbes after implementing MDLPHMDA based on the confirmed microbe-disease associations from HMDAD.
Liver cirrhosis is a disease induced by long-term damage. This damage is due to the replacement of normal tissue by scar tissue (Li et al., 1999). Typically, the disease develops slowly and there are often no significant early symptoms. As it worsens, patients may become tired, bruise easily, develop yellow skin, have fluid in the abdomen, or have swelling in the lower legs (Li et al., 1999). Liver cirrhosis is commonly caused by alcohol, non-alcoholic fatty liver disease, hepatitis B, or hepatitis C (Li et al., 1999). In a study on the alterations of the human microbiome in liver cirrhosis, quantitative metagenomics reveals 66 cognate bacterial species that differ in abundance between healthy individuals and patients, including Alistipes finegoldii, Bacteroides eggerthii, Eubacterium rectale, Faecalibacterium prausnitzii, Haemophilus parainfluenzae, and so on (Qin et al., 2014). In another study about fecal microbial communities in patients with liver cirrhosis, research has detected the prevalence of pathogenic bacteria such as Enterobacteriaceae and Streptococcaceae as well as the reduction of beneficial populations such as Lachnospiraceae (Chen et al., 2011). Here, by removing 62 known liver cirrhosis-associated microbes from the dataset of known microbe-disease associations, we enforced MDLPHMDA to identify liver cirrhosis-associated microbes on the basis of integrated disease similarity, Gaussian interaction profile kernel similarity for microbes, and the rest known microbe-disease associations. As a result, 9 out of the top 10 microbes for liver cirrhosis were confirmed by HMDAD and literature (see Table 4). Among the top 3 confirmed associations, Firmicutes was found to be highly enriched in the patients group (Chen et al., 2011). Moreover, researchers found significantly higher H. pylori prevalence in patients with previous hospital admissions (Siringo et al., 1997). This high prevalence of H. pylori is related to age and sex (Siringo et al., 1997). An analysis on the C. difficile infection in patients with liver cirrhosis showed that cirrhotic patients with the C. difficile infection have increased mortality than those without the C. difficile infection, suggesting the importance of C. difficile in the diagnosis and therapy of liver cirrhosis (Trifan et al., 2015).
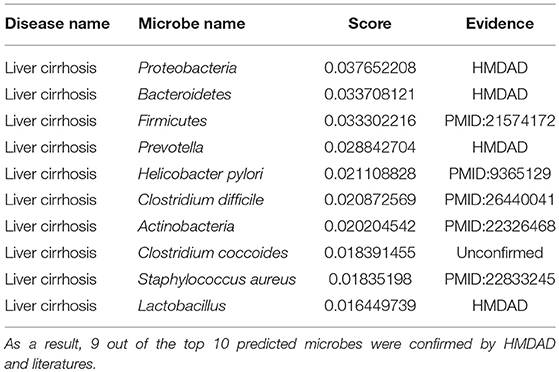
Table 4. The validation of the top 10 predicted liver cirrhosis-associated microbes after implementing MDLPHMDA by removing liver cirrhosis-related associations from the dataset of known microbe-disease associations.
Type 1 diabetes is a type of diabetes mellitus induced by very little or no insulin produced in the pancreas (Daneman, 2006). It results in high blood sugar levels in the human body. The classic symptoms include increased thirst and hunger, frequent urination and weight loss (Daneman, 2006). The cause of type 1 diabetes is still unclear. However, it is believed to involve both genetic and environmental factors (Chiang et al., 2014). One theory proposes that type 1 diabetes may be caused by an autoimmune response while the immune system attacks virus-infected insulin-producing cells in the pancreas (Knip et al., 2005). In a microbiome metagenomics analysis on type 1 diabetes, researchers identified the differences between patients and controls at the genus level. The most significant differences were noted in the genera Prevotella and Bacteroides (Brown et al., 2011). In another study defining the autoimmune microbiome for type 1 diabetes, scientists identified bacteria that correlated with the autoimmune state including Bacteroides fragilis, Clostridia, Eubacterium eligens, and so on (Giongo et al., 2011). Similarly, we employed MDLPHMDA to identify type 1 diabetes-associated microbes by removing 167 known type 1 diabetes-associated microbes from the dataset of known microbe-disease associations. The results showed that 8 out of the top 10 microbes for liver cirrhosis were confirmed (see Table 5). In a case-control study, scientists found a meaningful correlation between the H. pylori infection and the duration of diabetes in type 1 diabetic children (Bazmamoun et al., 2016). In another study, researchers found that Staphylococcus aureus is associated with the vitamin D receptor (VDR) polymorphisms in patients with type 1 diabetes (Panierakis et al., 2009).
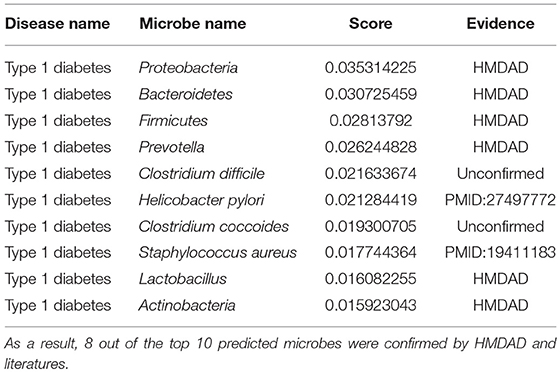
Table 5. The validation of the top 10 predicted type 1 diabetes-related microbes after implementing MDLPHMDA by removing type 1 diabetes-related associations from the dataset of known microbe-disease associations.
Discussion
Since the application of traditional experimental methods to prioritize disease-associated microbes is time consuming and expensive, the calculation approach of MDLPHMDA was put forward through the fusing of integrated disease similarity, Gaussian interaction profile kernel similarity for microbes and known microbe-disease associations. The performance of MDLPHMDA was tested using cross validations and case studies. Results on the basis of confirmed microbe-disease associations showed that the performance of our introduced algorithm is significantly improved in contrast with other two classic algorithms of LRLSHMDA and KATZHMDA. Consequently, the introduced algorithm is a suitable and effective model in the identification of novel microbe-disease associations. We further expect that the identified microbe-disease associations with high probability scores would be verified through biological experiment in the future.
The reason why MDLPHMDA could get excellent prediction performance is due to the following attractive properties. First, with the application of SLM on the original information of known microbe-disease associations, a new adjacency matrix with more accurate association information (the linear combination of low-rank matrix and the original adjacency matrix) and a noise (sparse) matrix would be gained. Obviously, in light of the new generated adjacency matrix, the forecast performance of the proposed algorithm for the identification of new microbe-disease associations could be significantly enhanced. Second, LPA was used to predict novel microbe-disease associations from the perspectives of microbe and disease, respectively, which would promote the ability of MDLPHMDA in terms of forecast accuracy. Third, in comparison with the previous calculation algorithms that only used Gaussian interaction profile kernel similarity for diseases as disease similarity, MDLPHMDA could achieve superior performance through integrating disease symptom similarity and Gaussian interaction profile kernel similarity for diseases into the final disease similarity. Moreover, the implementation of MDLPHMDA does not require negative samples and the algorithm could be applied to new diseases (microbes) without the relevant microbes (diseases).
However, the model has some main disadvantages. For instance, the amount of known microbe-disease associations used in this paper is very finite and more confirmed microbe-disease associations need to be collected. Additionally, as the computation of Gaussian interaction profile kernel similarity of microbes depended on known microbe-disease associations, other features of microbe similarity should be collected and combined to gain a more comprehensive dataset of microbe similarity such as microbe-drug associations collected by MDAD (Sun et al., 2018). For MDLPHMDA, it is difficult to find the optimum value of all the parameters to ensure that the prediction model achieves the highest accuracy. Also, the employment of SLM for creating new adjacency matrix may bring unnecessary and useless association information, which would affect the prediction result of LPA. Finally, successfully established models in the other computational fields would inspire the development of microbe-disease association prediction, such as microRNA-disease association prediction (Chen and Huang, 2017; Chen et al., 2018c), long non-coding RNA-disease association prediction (Chen and Yan, 2013; Chen et al., 2017b), drug-target interaction prediction (Chen et al., 2016b, 2018a), and synergistic drug combinations (Chen et al., 2016a).
Author Contributions
JQ developed the prediction method, implemented the experiments, analyzed the result, and wrote the paper. YZ conceived the project, designed the experiments, analyzed the result, and revised the paper. JY analyzed the result and revised the paper.
Funding
JQ, YZ, and JY was supported by the National Natural Science Foundation of China under Grant No. 61772531.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00291/full#supplementary-material
References
Abreu, N. A., Nagalingam, N. A., Song, Y., Roediger, F. C., Pletcher, S. D., Goldberg, A. N., et al. (2012). Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci. Transl. Med. 4:151ra124. doi: 10.1126/scitranslmed.3003783
Bazmamoun, H., Rafeey, M., Nikpouri, M., and Ghergherehchi, R. (2016). Helicobacter pylori infection in children with type 1 diabetes mellitus: a case-control study. J. Res. Health Sci. 16, 68–71.
Brown, C. T., Davis-Richardson, A. G., Giongo, A., Gano, K. A., Crabb, D. B., Mukherjee, N., et al. (2011). Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 6:e25792. doi: 10.1371/journal.pone.0025792
Castellarin, M., Warren, R. L., Freeman, J. D., Dreolini, L., Krzywinski, M., Strauss, J., et al. (2012). Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306. doi: 10.1101/gr.126516.111
Chen, X., Guan, N. N., Sun, Y. Z., Li, J. Q., and Qu, J. (2018a). MicroRNA-small molecule association identification: from experimental results to computational models. Brief. Bioinformatics. doi: 10.1093/bib/bby098 [Epub ahead of print].
Chen, X., and Huang, L. (2017). LRSSLMDA: laplacian regularized sparse subspace learning for miRNA-disease association prediction. PLoS Comput. Biol. 13:e1005912. doi: 10.1371/journal.pcbi.1005912
Chen, X., Huang, Y. A., You, Z. H., Yan, G. Y., and Wang, X. S. (2017a). A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 33, 733–739. doi: 10.1093/bioinformatics/btw715
Chen, X., Ren, B., Chen, M., Wang, Q., Zhang, L., and Yan, G. (2016a). NLLSS: predicting synergistic drug combinations based on semi-supervised learning. PLoS Comput. Biol. 12:e1004975. doi: 10.1371/journal.pcbi.1004975
Chen, X., Wang, L., Qu, J., Guan, N. N., and Li, J. Q. (2018b). Predicting miRNA-disease association based on inductive matrix completion. Bioinformatics 34, 4256–4265. doi: 10.1093/bioinformatics/bty503
Chen, X., Xie, D., Wang, L., Zhao, Q., You, Z. H., and Liu, H. (2018c). BNPMDA: bipartite network projection for MiRNA-Disease Association prediction. Bioinformatics. 34, 3178–3186. doi: 10.1093/bioinformatics/bty333
Chen, X., Yan, C. C., Zhang, X., and You, Z. H. (2017b). Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief. Bioinformatics 18, 558–576. doi: 10.1093/bib/bbw060
Chen, X., Yan, C. C., Zhang, X., Zhang, X., Dai, F., Yin, J., et al. (2016b). Drug-target interaction prediction: databases, web servers and computational models. Brief. Bioinformatics 17, 696–712. doi: 10.1093/bib/bbv066
Chen, X., and Yan, G. Y. (2013). Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624. doi: 10.1093/bioinformatics/btt426
Chen, X., Yin, J., Qu, J., and Huang, L. (2018d). MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Biol. 14:e1006418. doi: 10.1371/journal.pcbi.1006418
Chen, Y., Yang, F., Lu, H., Wang, B., Chen, Y., Lei, D., et al. (2011). Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54, 562–572. doi: 10.1002/hep.24423
Chiang, J. L., Kirkman, M. S., Laffel, L. M., and Peters, A. L. (2014). Type 1 diabetes through the life span: a position statement of the American Diabetes Association. Diabetes Care 37, 2034–2054. doi: 10.2337/dc14-1140
Delong, E. F., and Pace, N. R. (2001). Environmental diversity of bacteria and archaea. Syst. Biol. 50, 470–478. doi: 10.1080/10635150118513
Ferreira, C. M., Vieira, A. T., Vinolo, M. A., Oliveira, F. A., Curi, R., and Martins Fdos, S. (2014). The central role of the gut microbiota in chronic inflammatory diseases. J. Immunol. Res. 2014:689492. doi: 10.1155/2014/689492
Forum on Microbial, H., Board on Global, H., and Institute Of, M. (2014). “The National Academies Collection: Reports funded by National Institutes of Health,” in Microbial Ecology in States of Health and Disease: Workshop Summary (Washington, DC: National Academies Press).
Fukugaiti, M. H., Ignacio, A., Fernandes, M. R., Ribeiro Junior, U., Nakano, V., and Avila-Campos, M. J. (2015). High occurrence of Fusobacterium nucleatum and Clostridium difficile in the intestinal microbiota of colorectal carcinoma patients. Braz. J. Microbiol. 46, 1135–1140. doi: 10.1590/S1517-838246420140665
Giongo, A., Gano, K. A., Crabb, D. B., Mukherjee, N., Novelo, L. L., Casella, G., et al. (2011). Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 5, 82–91. doi: 10.1038/ismej.2010.92
Hawn, T. R., Day, T. A., Scriba, T. J., Hatherill, M., Hanekom, W. A., Evans, T. G., et al. (2014). Tuberculosis vaccines and prevention of infection. Microbiol. Mol. Biol. Rev. 78, 650–671. doi: 10.1128/MMBR.00021-14
Hendricks, K. A., Wright, M. E., Shadomy, S. V., Bradley, J. S., Morrow, M. G., Pavia, A. T., et al. (2014). Centers for disease control and prevention expert panel meetings on prevention and treatment of anthrax in adults. Emerging Infect. Dis. 20:e130678. doi: 10.3201/eid2002.130687
Huang, Y. A., You, Z. H., Chen, X., Huang, Z. A., Zhang, S., and Yan, G. Y. (2017a). Prediction of microbe-disease association from the integration of neighbor and graph with collaborative recommendation model. J. Transl. Med. 15:209. doi: 10.1186/s12967-017-1304-7
Huang, Y. J., Erb-Downward, J. R., Dickson, R. P., Curtis, J. L., Huffnagle, G. B., and Han, M. K. (2017b). Understanding the role of the microbiome in chronic obstructive pulmonary disease: principles, challenges, and future directions. Transl. Res. 179, 71–83. doi: 10.1016/j.trsl.2016.06.007
Huang, Z. A., Chen, X., Zhu, Z., Liu, H., Yan, G. Y., You, Z. H., et al. (2017c). PBHMDA: path-based human microbe-disease association prediction. Front. Microbiol. 8:233. doi: 10.3389/fmicb.2017.00233
Iburg, K. M. (2015). Global, regional, and national age–sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385, 117–171. doi: 10.1016/S0140-6736(14)61682-2
Kalliomaki, M., Collado, M. C., Salminen, S., and Isolauri, E. (2008). Early differences in fecal microbiota composition in children may predict overweight. Am. J. Clin. Nutr. 87, 534–538. doi: 10.1093/ajcn/87.3.534
Kalliomaki, M., Kirjavainen, P., Eerola, E., Kero, P., Salminen, S., and Isolauri, E. (2001). Distinct patterns of neonatal gut microflora in infants in whom atopy was and was not developing. J. Allergy Clin. Immunol. 107, 129–134. doi: 10.1067/mai.2001.111237
Knip, M., Veijola, R., Virtanen, S. M., Hyoty, H., Vaarala, O., and Akerblom, H. K. (2005). Environmental triggers and determinants of type 1 diabetes. Diabetes 54(Suppl. 2), S125–S136. doi: 10.2337/diabetes.54.suppl_2.S125
Lemanske, R. F. Jr., and Busse, W. W. (2010). Asthma: clinical expression and molecular mechanisms. J. Allergy Clin. Immunol. 125, S95–102. doi: 10.1016/j.jaci.2009.10.047
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Li, C. P., Lee, F. Y., Hwang, S. J., Chang, F. Y., Lin, H. C., Lu, R. H., et al. (1999). Spider angiomas in patients with liver cirrhosis: role of alcoholism and impaired liver function. Scand. J. Gastroenterol. 34, 520–523. doi: 10.1080/003655299750026272
Ma, W., Zhang, L., Zeng, P., Huang, C., Li, J., Geng, B., et al. (2017). An analysis of human microbe-disease associations. Brief. Bioinformatics 18, 85–97. doi: 10.1093/bib/bbw005
Marri, P. R., Stern, D. A., Wright, A. L., Billheimer, D., and Martinez, F. D. (2013). Asthma-associated differences in microbial composition of induced sputum. J. Allergy Clin. Immunol. 131, 346–352.e341–343. doi: 10.1016/j.jaci.2012.11.013
Martinon, F., Mayor, A., and Tschopp, J. (2009). The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27, 229–265. doi: 10.1146/annurev.immunol.021908.132715
Meng, F., Yang, X., and Zhou, C. (2014). The augmented lagrange multipliers method for matrix completion from corrupted samplings with application to mixed Gaussian-impulse noise removal. PLoS ONE 9:e108125. doi: 10.1371/journal.pone.0108125
Morse, S. S. (1991). Emerging viruses: defining the rules for viral traffic. Perspect. Biol. Med. 34, 387–409. doi: 10.1353/pbm.1991.0038
Morse, S. S. (1995). Factors in the emergence of infectious diseases. Emerg. Infect. Dis. 1, 7–15. doi: 10.3201/eid0101.950102
O'hara, A. M., and Shanahan, F. (2006). The gut flora as a forgotten organ. EMBO Rep. 7, 688–693. doi: 10.1038/sj.embor.7400731
Panierakis, C., Goulielmos, G., Mamoulakis, D., Maraki, S., Papavasiliou, E., and Galanakis, E. (2009). Staphylococcus aureus nasal carriage might be associated with vitamin D receptor polymorphisms in type 1 diabetes. Int. J. Infect. Dis. 13, e437–e443. doi: 10.1016/j.ijid.2009.02.012
Pappas, P. G., Kauffman, C. A., Andes, D. R., Clancy, C. J., Marr, K. A., Ostrosky-Zeichner, L., et al. (2016). Executive summary: clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 62, 409–417. doi: 10.1093/cid/civ1194
Pech, R., Hao, D., Po, M., and Zhou, T. (2017). Predicting drug-target interactions via sparse learning.
Peng, L. H., Yin, J., Zhou, L., Liu, M. X., and Zhao, Y. (2018). Human microbe-disease association prediction based on adaptive boosting. Front. Microbiol. 9:2440. doi: 10.3389/fmicb.2018.02440
Qin, N., Yang, F., Li, A., Prifti, E., Chen, Y., Shao, L., et al. (2014). Alterations of the human gut microbiome in liver cirrhosis. Nature 513, 59–64. doi: 10.1038/nature13568
Schirbel, A., and Fiocchi, C. (2010). Inflammatory bowel disease: established and evolving considerations on its etiopathogenesis and therapy. J. Dig. Dis. 11, 266–276. doi: 10.1111/j.1751-2980.2010.00449.x
Shmuely, H., Passaro, D., Figer, A., Niv, Y., Pitlik, S., Samra, Z., et al. (2001). Relationship between Helicobacter pylori CagA status and colorectal cancer. Am. J. Gastroenterol. 96, 3406–3410. doi: 10.1111/j.1572-0241.2001.05342.x
Siringo, S., Vaira, D., Menegatti, M., Piscaglia, F., Sofia, S., Gaetani, M., et al. (1997). High prevalence of Helicobacter pylori in liver cirrhosis: relationship with clinical and endoscopic features and the risk of peptic ulcer. Dig. Dis. Sci. 42, 2024–2030. doi: 10.1023/A:1018849930107
Sobhani, I., Tap, J., Roudot-Thoraval, F., Roperch, J. P., Letulle, S., Langella, P., et al. (2011). Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS ONE 6:e16393. doi: 10.1371/journal.pone.0016393
Stenn, F. (1960). Cave disease or speleonosis. Arch. Intern. Med. 105, 181–183. doi: 10.1001/archinte.1960.00270140003001
Sun, Y. Z., Zhang, D. H., Cai, S. B., Ming, Z., Li, J. Q., and Chen, X. (2018). MDAD: a special resource for microbe-drug associations. Front. Cell. Infect. Microbiol. 8:424. doi: 10.3389/fcimb.2018.00424
Sze, M. A., Dimitriu, P. A., Hayashi, S., Elliott, W. M., McDonough, J. E., Gosselink, J. V., et al. (2012). The lung tissue microbiome in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 185, 1073–1080. doi: 10.1164/rccm.201111-2075OC
Tamboli, C. P., Neut, C., Desreumaux, P., and Colombel, J. F. (2004). Dysbiosis in inflammatory bowel disease. Gut 53, 1–4. doi: 10.1136/gut.53.1.1
Thiele, I., Heinken, A., and Fleming, R. M. (2013). A systems biology approach to studying the role of microbes in human health. Curr. Opin. Biotechnol. 24, 4–12. doi: 10.1016/j.copbio.2012.10.001
Torgerson, P. R., and Mastroiacovo, P. (2013). The global burden of congenital toxoplasmosis: a systematic review. Bull. World Health Organ. 91, 501–508. doi: 10.2471/BLT.12.111732
Trifan, A., Stoica, O., Stanciu, C., Cojocariu, C., Singeap, A. M., Girleanu, I., et al. (2015). Clostridium difficile infection in patients with liver disease: a review. Eur. J. Clin. Microbiol. Infect. Dis. 34, 2313–2324. doi: 10.1007/s10096-015-2501-z
Vael, C., Vanheirstraeten, L., Desager, K. N., and Goossens, H. (2011). Denaturing gradient gel electrophoresis of neonatal intestinal microbiota in relation to the development of asthma. BMC Microbiol. 11:68. doi: 10.1186/1471-2180-11-68
Van Laarhoven, T., Nabuurs, S. B., and Marchiori, E. (2011). Gaussian interaction profile kernels for predicting drug-target interaction. Bioinformatics 27, 3036–3043. doi: 10.1093/bioinformatics/btr500
Van Nimwegen, F. A., Penders, J., Stobberingh, E. E., Postma, D. S., Koppelman, G. H., Kerkhof, M., et al. (2011). Mode and place of delivery, gastrointestinal microbiota, and their influence on asthma and atopy. J. Allergy Clin. Immunol. 128, 948–955.e941–943. doi: 10.1016/j.jaci.2011.07.027
Walker, A. W., Sanderson, J. D., Churcher, C., Parkes, G. C., Hudspith, B. N., Rayment, N., et al. (2011). High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 11:7. doi: 10.1186/1471-2180-11-7
Wang, F., Huang, Z. A., Chen, X., Zhu, Z., Wen, Z., Zhao, J., et al. (2017). LRLSHMDA: laplacian regularized least squares for human microbe-disease association prediction. Sci. Rep. 7:7601. doi: 10.1038/s41598-017-08127-2
Wang, X., Heazlewood, S. P., Krause, D. O., and Florin, T. H. (2003). Molecular characterization of the microbial species that colonize human ileal and colonic mucosa by using 16S rDNA sequence analysis. J. Appl. Microbiol. 95, 508–520. doi: 10.1046/j.1365-2672.2003.02005.x
Watson, A. J., and Collins, P. D. (2011). Colon cancer: a civilization disorder. Dig. Dis. 29, 222–228. doi: 10.1159/000323926
Weber, J., Illi, S., Nowak, D., Schierl, R., Holst, O., Von Mutius, E., et al. (2015). Asthma and the hygiene hypothesis. Does cleanliness matter? Am. J. Respir. Crit. Care Med. 191, 522–529. doi: 10.1164/rccm.201410-1899OC
Yao, Q., Wu, L., Li, J., Yang, L. G., Sun, Y., Li, Z., et al. (2017). Global Prioritizing disease candidate lncRNAs via a multi-level composite network. Sci. Rep. 7:39516. doi: 10.1038/srep39516
Zhang, W., Qu, Q., Zhang, Y., Wang, W., Zhang, W., Qu, Q., et al. (2017). The linear neighborhood propagation method for predicting long non-coding RNA–protein interactions. Neurocomputing 273, 526–534. doi: 10.1016/j.neucom.2017.07.065
Zhao, Y., Chen, X., and Yin, J. (2018). A novel computational method for the identification of potential miRNA-disease association based on symmetric non-negative matrix factorization and kronecker regularized least square. Front. Genet. 9:324. doi: 10.3389/fgene.2018.00324
Zhou, X., Menche, J., Barabasi, A. L., and Sharma, A. (2014). Human symptoms-disease network. Nat. Commun. 5:4212. doi: 10.1038/ncomms5212
Keywords: microbe, disease, association prediction, matrix decomposition, label propagation
Citation: Qu J, Zhao Y and Yin J (2019) Identification and Analysis of Human Microbe-Disease Associations by Matrix Decomposition and Label Propagation. Front. Microbiol. 10:291. doi: 10.3389/fmicb.2019.00291
Received: 30 November 2018; Accepted: 04 February 2019;
Published: 26 February 2019.
Edited by:
Hongsheng Liu, Liaoning University, ChinaCopyright © 2019 Qu, Zhao and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Zhao, dHMxNzA2MDA5MGEzQGN1bXQuZWR1LmNu