 Robert Czajkowski
Robert Czajkowski- Laboratory of Biologically Active Compounds, Intercollegiate Faculty of Biotechnology of University of Gdansk and Medical University of Gdansk, University of Gdansk, Gdansk, Poland
Soft Rot Pectobacteriaceae (SRP; Pectobacterium spp. and Dickeya spp., formerly known as pectinolytic Erwinia spp.) are necrotrophic bacterial pathogens infecting a large number of plant species worldwide, including agriculturally-important crops. Despite the SRP importance in agriculture, little is known about the bacteriophages infecting them, and even less about the prophages present in their genomes. Prophages are recognized as factors underlying bacterial virulence, genomic diversification and ecological fitness that contribute to the novel phenotypic properties of bacterial hosts. Likewise, they are recognized as a driving force of bacterial evolution. In this study, 57 complete genomes of Pectobacterium spp. and Dickeya spp. deposited in NCBI GenBank, were analyzed for the presence of prophage-like elements. Viral sequences were discovered in 95% of bacterial genomes analyzed with the use of PHASTER, PhiSpy, and manual curation of the candidate sequences using NCBI BLAST. In total 37 seemingly intact and 48 putatively defective prophages were found. The 37 seemingly intact prophages (27 sequences in Dickeya spp. genomes and 10 sequences in Pectobacterium spp. genomes) were annotated using RAST. Analysis of the prophage genes encoding viral structural proteins allowed classification of these prophages into different families of the order Caudovirales (tailed bacteriophages) with the SRP prophages of the Myoviridae family (81% of found prophages) being the most abundant. The phylogenetic relationships between prophages were analyzed using amino acid sequences of terminase large subunit (gene terL), integrase (gene int), holin (gene hol), and lysin (gene lys). None of these markers however proved fully useful for clear phylogenetic separation of prophages of SRP into distinct clades. Comparative analyses of prophage proteomes revealed six clusters: five present in Dickeya spp. and one within Pectobacterium spp. When screened for the presence of bacterial genes in the genomes of intact prophages, only one prophage did not contain any ORFs of bacterial origin, the other prophages contained up to 23 genes acquired from bacterial hosts. The bacterial genes present in prophages could possibly affect fitness and virulence of their hosts. The implication of prophage presence in the genomes of Pectobacterium spp. and Dickeya spp. is discussed.
Introduction
It is generally accepted that phages are the most abundant biological entities in the environment with an estimated number of 1031 particles on Earth. Consequently, they are present in virtually all habitats in which bacteria exist (Suttle, 2007). Based on their particular relationship with a host, they can be either lytic or temperate (Ackermann, 2003). Temperate bacteriophages integrate their genetic material into the host genome and persist inside bacterial cells as so-called prophages (Weinbauer, 2004). After integration, prophages are maintained in a host cell, undergoing non-lytic growth typically called a lysogenic state (Canchaya et al., 2004). During lysogeny phage DNA remains inactive, except for some regulatory and accessory genes, which are required to maintain the dormant state of the virus. This dormant bacteriophage DNA may constitute up to 20% of the host genome (Casjens, 2003).
The occurrence of prophages can contribute greatly to bacterial fitness (Bondy-Denomy and Davidson, 2014; Nanda et al., 2014). Prophages can influence host variability and evolution and may determine the adaptation of their hosts to specific ecological niches (Wang et al., 2010; Fortier and Sekulovic, 2013; Varani et al., 2013). The presence of prophages may affect bacterial genomes in several ways. For example, their integration is responsible for gene disruption or translocation which, in turn, may confer phenotypic changes in the host. Similarly, prophages may introduce new traits into the host, such as pathogenicity determinants that alter bacterial fitness. These new traits might also modulate the switch between lytic and lysogenic cycles (Brüssow et al., 2004). Consequently, prophages have been studied in a number of bacterial species including plant pathogens to understand their role in bacterial ecology (Casjens, 2003; Varani et al., 2013). To date, however, they have not been extensively studied in the Soft Rot Pectobacteriaceae (SRP).
Plant pathogenic Soft Rot Pectobacteriaceae (Adeolu et al., 2016) [consisting of Pectobacterium spp. and Dickeya spp., formerly characterized as pectinolytic Erwinia spp. (Pérombelon, 2002)] are considered to be among the top ten most important agricultural phytopathogens (Mansfield et al., 2012). They cause significant losses in crop production (up to 40%) with disease severity dependent on weather conditions, plant susceptibility and pathogen inoculum. Among the economically most important hosts worldwide are potato, carrot, tomato, onion, pineapple, maize, rice, hyacinth, chrysanthemum, and calla lily (Perombelon and Kelman, 1980; Charkowski, 2018). SRP are widespread in various ecological niches including bulk and rhizosphere soils, water, sewage, the surface of host and non-host plants, and the surfaces and interior of insects (Perombelon and Kelman, 1980; Grenier et al., 2006; Rossmann et al., 2018). Because of the diverse habitats in which they can be found, bacteria presumably also exhibit diverse lifestyles because of their transfer between these different environments; for example, from plants to soil, from plant to plant, from host plant to non-host plant, from surface and/or irrigation water to plants, from water to soil, and vice versa) (Charkowski, 2018). In all of these surroundings the SRP can encounter lytic and temperate bacteriophages and hence may become easily and repeatedly infected (Canchaya et al., 2004).
The knowledge of prophages present in SRP genomes is currently very limited as only a few temperate bacteriophages that specifically infect Pectobacterium spp. and Dickeya spp. have been characterized (for review see: Varani et al., 2013; Czajkowski, 2016). The viruses that have been characterized include temperate bacteriophage ΦEC2 infecting D. dadantii and D. solani (Resibois et al., 1984); bacteriophage ZF40 (Korol and Tovkach, 2012) infecting P. carotovorum subsp. carotovorum; bacteriophages phiTE (Blower et al., 2012), phiM1 (Blower et al., 2017), ECA29 and ECA41 (Evans et al., 2010) infecting P. atrosepticum; and bacteriophages LIMEstone 1 and LIMEstone2 infecting D. solani (Adriaenssens et al., 2012; Day et al., 2017). Likewise, the biological role of only two prophages present in SRP genomes (ECA29 and ECA41 localized in the genome of P. atrosepticum strain SCRI1043) have been elucidated to date as being involved in modulation of host swimming motility and virulence in potato (Evans et al., 2010).
The aim of this study was to identify prophage-like sequences in the complete genome sequences of Pectobacterium spp. and Dickeya spp. strains deposited in GenBank (NCBI) and to characterize these prophages using comparative genomic tools. The implications of the presence of prophage in SRP genomes and the way these genetic elements may contribute to ecological fitness of Pectobacterium spp. and Dickeya spp. are also discussed.
Materials and Methods
Data Collection and Identification of Candidate Prophage Sequences in Complete Genomes of Dickeya spp. and Pectobacterium spp.
The strategy used to identify and characterize prophages in SRP genomes is presented in Figure 1. Fifty seven complete genome sequences (17 Pectobacterium spp. genomes and 40 Dickeya spp. genomes) were accessed from NCBI (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/) (August 2018) (Table 1).

Figure 1. The workflow for identification and characterization of SRP prophages. The blue rectangles represent the tools and methods used for identification of prophages' sequences in bacterial genomes and the white rounded rectangles represent the (input) data used for the analyses.
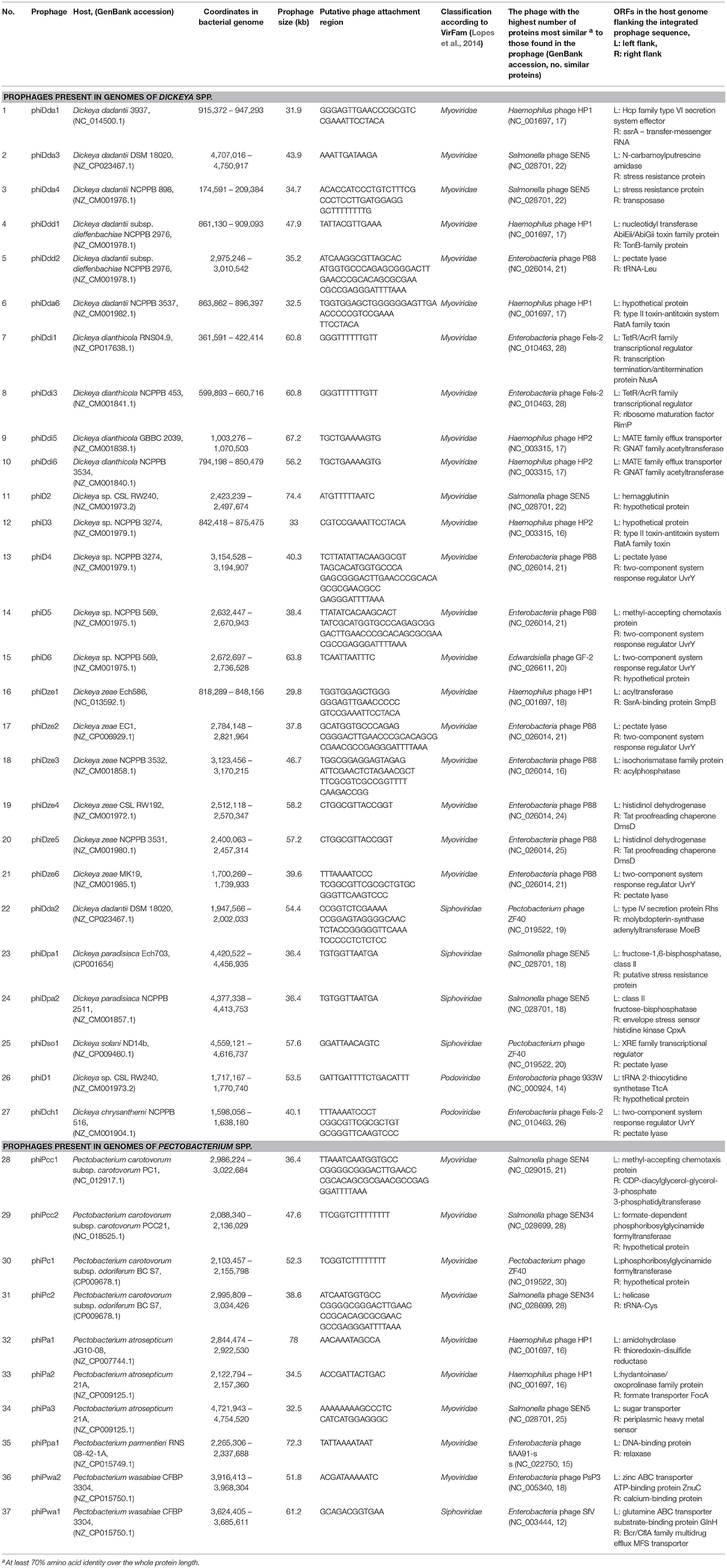
Table 1. Genomic features of intact prophages present in the complete genomes of Soft Rot Pectobacteriaceae (Dickeya spp. and Pectobacterium spp.) obtained from GenBank (NCBI).
Candidate prophage-like elements were identified with PHASTER (http://phaster.ca/) using settings described in (Arndt et al., 2016) and with PhiSpy (https://edwards.sdsu.edu/PhiSpy/index.php) using settings described in Akhter et al. (2012), followed by manual inspection of the sequences for the presence of signature genes: attachment sites (att), gene(s) encoding integrase(s), terminases(s), transposases(s), genes coding for structural viral proteins and the sequences of prophage integration sites, as suggested by others (Boyd and Brüssow, 2002). The candidate prophage-like element was defined as seemingly intact prophage when its sequence contained altogether: (i) phage attachment sites, (ii) genes encoding structural phage proteins, (iii) genes coding for proteins involved in DNA regulation, insertion to the host genome and lysis. Consequently, the candidate prophage-like element was defined as putatively defective when its sequence lacks one or more features (genes) as described above (Akhter et al., 2012; Arndt et al., 2016).
Furthermore, any two seemingly intact prophages were characterized as the same prophage if their genomes shared at least 95% nucleotide identity. Prophages were characterized on the basis of their homology with known phage sequences deposited in NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) using NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The presence of structural genes in prophage sequences was verified by the VirFam (http://biodev.cea.fr/virfam/) using settings described in Lopes et al. (2014).
Analyses of Prophage Genome Sequences and Comparative Genomics
Prophage sequences were annotated using RAST (Rapid Annotation using Subsystem Technology) (rast.nmpdr.org) as described in Aziz et al. (2008), Brettin et al. (2015) (computational settings: Classic RAST, Glimmer3 release 70, domain Viruses, genetic code:11, disable replication), and DNA Master (Lawrence, University of Pittsburgh, Pennsylvania, USA) (http://en.bio-soft.net/dna/dnamaster.html) using settings advised in Pope and Jacobs-Sera (2018).
The attL and attR attachment sites were identified using PHASTER (http://phaster.ca/) as described in Arndt et al. (2016) and manually inspected using CLC Main Workbench 7 (Qiagen) by assessing the phage localization in the host genome. Multiple sequence alignment of individual prophage genes and phylogenetic analyses were performed using Phylogenetic Pipeline of Information Génomique et Structurale, CNRS-AMU, France (http://www.phylogeny.fr/).
Because of the lack of a universal genetic marker in bacteriophages (Lawrence et al., 2002; Adriaenssens and Cowan, 2014), phylogenetic characterization of bacterial viruses and prophages may be based on comparison of different sequences e.g., encoding integrase, large subunit of terminase, holin and/or lysin (syn. endolysin, murein hydrolase). Amino acid sequences derived from int, hol, lys, terL, respectively, were used to phylogenetically analyze the 37 seemingly intact prophages in this study. For this, sequences were aligned with MUSCLE (v3.8.31) configured for highest accuracy (MUSCLE with default settings), after alignment, ambiguous regions (i.e., containing gaps and/or poorly aligned) were removed with Gblocks (v0.91b) using the following parameters: (i) minimum length of a block after gap cleaning equal to 10, (ii) no gap positions were allowed in the final alignment, (iii) all segments with contiguous non-conserved positions bigger than 8 were rejected, (iv) minimum number of sequences for a flank position equal to 85%, graphical representation and edition of the phylogenetic tree were performed with TreeDyn (v198.3).
Comparative analyses of the prophage genomes were done using EDGAR (Blom et al., 2009) accessed via (https://edgar.computational.bio.uni-giessen.de) with settings described in Blom et al. (2009), DNA Master (Lawrence, University of Pittsburgh, Pennsylvania, USA) (http://en.bio-soft.net/dna/dnamaster.html) and BLASTn (accessed via (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Pairwise comparison of sequences (BLAST) (Altschul et al., 1990) was analyzed using MAUVE as described in Darling et al. (2010) (computational settings: alignment with progressiveMauve (aligner: Muscle 3.6), default seed weight (15), full alignment (minimum island size: 50, maximum backbone gap size: 50, minimum backbone size: 50), use of seed families: yes, iterative refinement: yes, determination of LCBs: yes), and DNA Master.
The presence of genes of bacterial origin in the prophage genomes was assessed by NCBI BLAST searches. For this, the gene was classified as being of bacterial origin when altogether: (i) the gene (encoding known or hypothetical protein) is frequently present in bacterial genome(s), (ii) is unnecessary to complete bacteriophage life cycle, (iii) encodes protein with an enzymatic activity not required by the virus to interact with its hosts. The presence of putative virulence-associated genes and antibiotic resistance genes in the viral genomes was assessed using VirulenceFinder ver. 1.5 (https://cge.cbs.dtu.dk/services/VirulenceFinder/) and ResFinder ver. 3.0 (https://cge.cbs.dtu.dk/services/ResFinder/), respectively, with setting described in Kleinheinz et al. (2014).
Results
Presence of Prophage-Like Sequences in Dickeya spp. and Pectobacterium spp. Complete Genomes
The analyses of the 57 complete SRP genomes accessed from GenBank (NCBI) and interrogated with PHASTER and PhiSpy (Figure 1) resulted in discovery of the prophage-like elements in the genomes of 54 of these strains (95% of the genomes interrogated). In total, 37 seemingly intact and 48 putatively defective prophages were found among these strains (Figure 2; Table 1; Supplementary Table 1). Only three D. solani genomes, namely D. solani strain MK10 (NZ_CM001839.1), D. solani strain MK16 (NZ_CM001842.1), and D. solani strain GBC 2040 (NZ_CM001860.1) did not harbor any prophage-like elements.
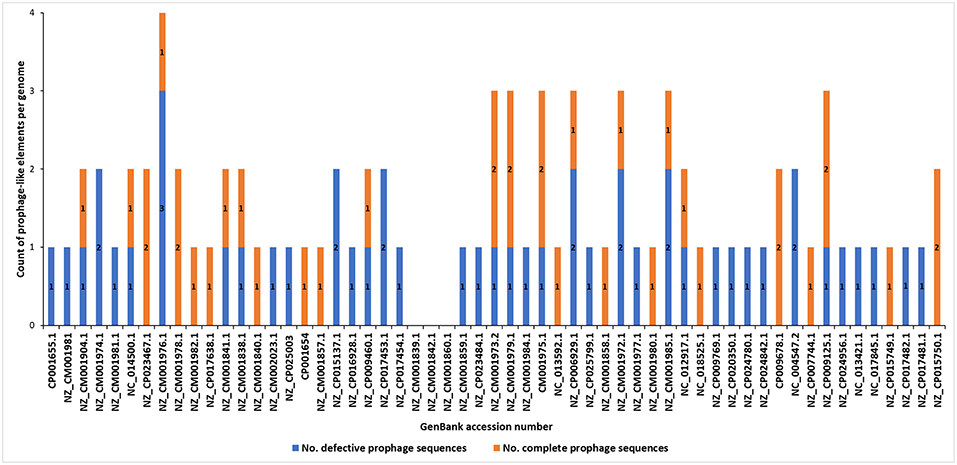
Figure 2. Distribution of putatively defective prophage-like elements and seemingly intact prophages in 57 complete genomes of Soft Rot Pectobacteriaceae acquired from GenBank (NCBI). The prophage sequences were detected using PHASTER and PhiSpy and manually curated using BLAST (NCBI) using the pipeline presented in the Figure 1.
Incomplete (putatively defective) prophage-like elements were present in the majority of the SRP genomes and ranged in size from 4.5 to 41 kb. Often more than one such an element was found in a given strain, as for example, in D. chrysanthemi strain NCPPB 402 harboring 2 putatively defective prophages, D. dadantii strain NCPPB 898 harboring 3 putatively defective prophages, and P. atrosepticum strain SCRI1043 with 2 such prophages.
Seemingly complete (intact) prophage regions were found in 27 Dickeya strains while 10 Pectobacterium spp. apparently harbored such prophage genomes (Figure 2; Table 1; Data sheets 1, 2). More than one complete prophage region was found in eight SRP (3 Pectobacterium spp. and 5 Dickeya spp.) genomes (Figure 2).
The sizes of complete prophage genomes varied from 29 to 78 kb and, on average, these viruses comprised between 0.6 to 1.8 % of the host chromosome. The integration of the prophages to the host genomes was in majority random (Table 1).
The prophages were integrated near genes coding for stress resistance proteins, transcriptional regulators, enzymes involved in the fundamental bacterial metabolism, two-component systems, transporters as well as coding for hypothetical proteins. Six prophages however viz. phiDch1, phiDdd2, phiDsol1, phiD4, phiDze2, and phiDze6 were integrated near the genes coding for pectate lyases, one of the most important virulence factors of SRP.
All of the complete prophage genomes possessed structural components that were typical of phages in the order Caudovirales (tailed bacteriophages), enabling the classification of 30 prophages (81%) to the Myoviridae family, 5 prophages (13.5%) to the Siphoviridae family, and 2 prophages (5.5%) to the Podoviridae family (Table 1).
This study did not reveal the presence of non-integrase based forms of lysogeny, such as that of transposable phages or plasmid-based replication. As the workflow included PHASTER and PhiSpy, it could be expected to detect these sorts of phages if they existed in the dataset. Furthermore, three prophages (viz. phiDda2, phiDsol1, and phiPc1) present in the genomes of D. dadantii DSM 18020, D. solani ND14b, and P. carotovorum subsp. odoriferum BC S7, respectively, share significant similarity with well-characterized temperate bacteriophage ZF40 infecting Pectobacterium carotovorum subsp. carotovorum (Table 1) (Korol and Tovkach, 2012).
Phylogenetic Relationships Between Prophages Found in SRP Genomes Based on Single Gene Analyses
The int gene encoding integrase and terL gene coding for the large subunit of terminase were present in all 37 screened prophages (Figures 3A,D), whereas genes encoding holin (hol) and lysin (lys) were found within 21 and 27 prophage sequences, respectively (Figures 3B,C). Phylogenetic analyses revealed that SRP prophages are diverse, with viruses belonging to the same viral family forming different phylogenetic clades. The phylogenetic distance between prophages calculated based on the amino acid sequences of integrase did not prove to be useful in determining a phylogenetic association with their host as no clear separation of the Pectobacterium and Dickeya prophage clades could be observed (Figure 3A).
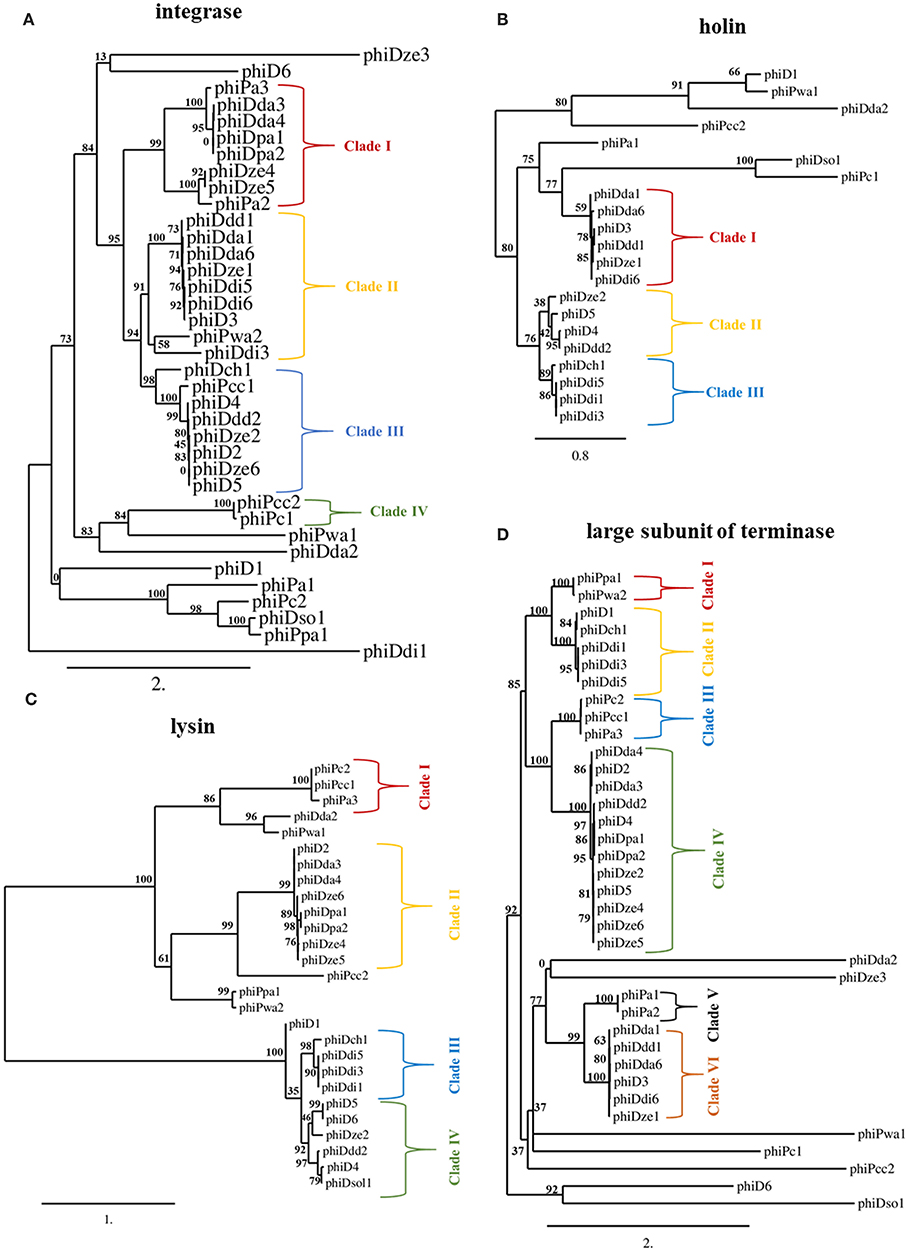
Figure 3. Maximum likelihood (ML) tree based on the aligned amino acid sequences of integrase (present in 37 prophages) (A), holin (present in 21 prophages) (B), lysin (present in 27 prophages) (C) and large subunit of terminase (present in 37 prophages) (D) genes of seemingly intact prophage sequences distributed in 57 Soft Rot Pectobacteriaceae genomes. Phylogenetic studies were performed using Phylogenetic Pipeline of Information Génomique et Structurale, CNRS-AMU, France (http://www.phylogeny.fr/) with bootstrap support for 1,000 replicates. The bar indicates the number of substitutions per sequence position. The cutoff for separating the clades was the bootstrap support for particular branch (n) of at least 70% together with the bootstrap support for particular predecessor branch (n-1) of at least 70%.
In contrast, phylogenetic analyses based on amino acid sequences of terminase large subunit, holin, and lysin revealed clades of prophages present in Dickeya spp. genomes that were distinct from those in the genomes of Pectobacterium spp. strains. This separation of clades was however only partial (Figures 3B–D). Based on terminase amino acid sequences, seven prophage clades could be distinguished each containing between two and twelve viruses. For integrase and lysin amino acid sequences, four prophage clades could be distinguished, each containing between two and nine viruses.
Phylogenetic analysis using holin sequences differentiated three prophage clades. Interestingly, phiDda1, phiDda6, phiD3, phiDdd1, phiDze1, and phiDdi6 were grouped together both in clade I of holin- and in clade II of integrase-based phylogenetic trees while phiDze2, phiD5, phiD4, and phiDdd2 were grouped both in clade III of integrase- and clade II of holin-based tries. Likewise, phiDch1, phiDdi5, phiDdi1, and phiDdi3 were present both in clade III of holin- and clade III of lysin amino acid sequence-based trees.
Comparative Genomics and Proteomics of SRP Prophages
Comparative genomics based on the RAST annotated prophage genome sequences allowed visualization of the order of ORFs present in all 37 prophage genomes (Figure 4; Supplementary Figure 1). In general, and with the few exceptions mentioned below, the organization of ORFs within the 37 SRP prophage genomes was not conserved, exhibiting a high genetic mosaicism. The genome organization of only phiDdi1 and phiDdi3 had high synteny while prophage pairs phiDze4 and phiDze5 as well as phiDpa1 and phiDpa2 exhibited somewhat lower conservation of gene order.

Figure 4. Comparative analyses of the 37 intact prophage genomes. Boxes indicate open reading frames (ORFs) in prophage genomes predicted by RAST annotation pipeline. Homological ORFs are marked with the same color. The analysis and visualization were performed with the use of DNA Master ver. 5.23.2.
The most highly syntenic prophages shared a common host bacterial species; PhiDdi1 and phiDdi3 were found in D. dianthicola strains RNS04.9 and NCPPB 453, while phiDze4 and phiDze5 were found in D. zeae isolates CSL RW192 and NCPPB 3531, and prophage phiDpa1 and phiDpa2 were found in D. paradisiaca strains Ech703 and NCPPB 2511. Only a partial conservation of the order of ORFs was present among phiD4, phiD5, and phiDdd2 (Figure 4) residing in the phylogenetically distinct hosts Dickeya sp. NCPPB 3274, Dickeya sp. NCPPB 569 and D. dadantii subsp. diffenbachiae NCPPB 2976. As noted above, bacterial genomes frequently harbored two distinct but complete prophages such as in the case of D. dadantii strain DSM 18020 (carrying phiDda2 and phiDda3), D. dadantii subsp. diffenbachiae strain NCPPB 2976 (carrying phiDdd1 and phiDdd2), Dickeya sp. CSL RW240 (carrying phiD1 and phiD2), Dickeya sp. Strain NCPPB 3274 (carrying phiD3 and phiD4), Dickeya sp. Strain NCPPB 569 (carrying phiD5 and phiD6) and P. carotovorum subsp. odoriferum strain BC S7 (carrying phiPc1 and phiPc2).
No correlation was observed between the host bacterial genome size and the aggregate prophage genome size (R2 = 0.02) (data not shown).
A dot plot matrix constructed based on average amino acid identity (AAI) of the 37 prophage proteomes revealed six visually distinctive clusters (Figure 5); two clusters (Cluster 2 and Cluster 3) (Supplementary Tables 3, 4) having a AAI > 90%, one cluster having a AAI > 85% (Cluster 1) (Supplementary Table 2), two clusters having a AAI > 80% (Cluster 4 and Cluster 6) (Supplementary Tables 5, 7), and one cluster with a AAI only > 75% (Cluster 5) (Supplementary Table 6) Five clusters (Cluster 1, 2, 3, 4, and 6) were grouping proteomes of prophages present in Dickeya spp. genomes, whereas Cluster 5 was grouping prophages hosted by Pectobacterium spp. strains as evidenced by the AAI dot plot matrix.
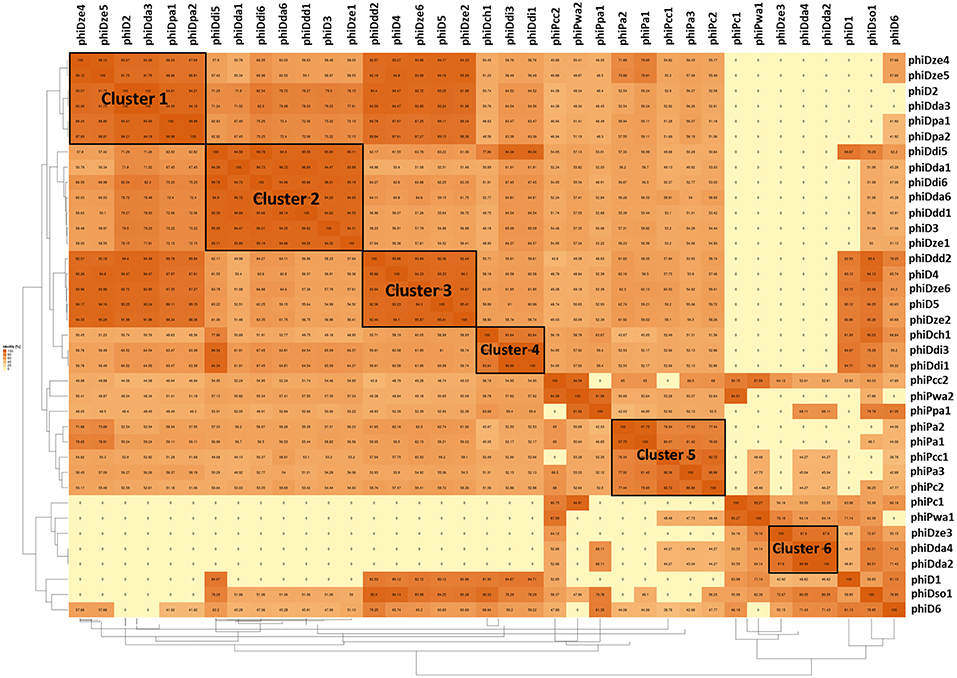
Figure 5. Pairwise average amino acid identity (AAI) heatmap among 37 intact SRP prophages. The map was generated using EDGAR—a software platform for comparative genomics (Blom et al., 2009).
Presence of Unique Genes of Bacterial Origin in the Seemingly Intact Prophage Genomes
Of 37 screened complete prophage genomes, only one, phiDze1 did not contain any ORFs of bacterial origin. The other 36 prophages contained between 1 (phiD3, phiDda1) and 23 (phiDdi1 and phiDdi3) ORFs apparently acquired from bacterial hosts (Figure 6). Most of the bacterial ORFs found in prophages encoded proteins involved in primary bacterial metabolism, proteins associated with DNA/RNA repair, energy transfer, DNA/protein regulation and modification and proteins that may be involved in niche exploitation (e.g., resistance to metal ions, nitrogen assimilation, heat shock proteins) (Supplementary Table 8).
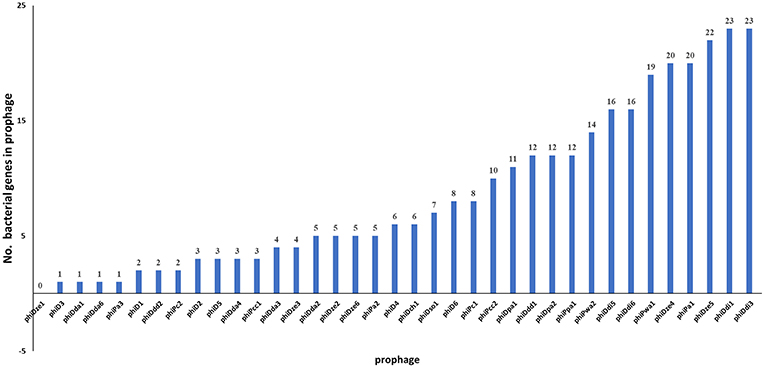
Figure 6. Distribution of genes of bacterial origin in 37 seemingly intact prophages of SRP. The particular gene found in the prophage was classified as being of bacterial origin when altogether: (i) the gene is frequently present in bacterial genome(s), (ii) is unnecessary to complete bacteriophage life cycle, (iii) encodes protein with an enzymatic activity not required by the virus to interact with its hosts.
The genes present among the large proportion of the seemingly intact prophages were those encoding: (i) methyl-directed repair DNA adenine methylase (in 21 prophage genomes), (ii) methyl-transferase (in 9 prophages), and (iii) modification methylase ScrFIA (in 6 prophages). Interestingly, similar sets of bacterial genes were found in different groups of prophages such as (1) phiD2, phiDda3, and phiDda4, (2) phiDdi1 and phiDdi3, (3) phiDdi5 and phiDdi6, (4) phiDpa1 and phiDpa2, (5) phiDze4 and phiDze5, and (6) phiPc2 and phiPcc1 (Supplementary Table 2). Prophages phiD6, phiDdi1, phiDdi1, and phiPa1 all harbored homologous genes encoding the tellurite resistance protein TerB, while prophages phiDpa1 and phiDpa2 all carried the gene for the cation-efflux pump FieF that confers resistance to cobalt, zinc and cadmium ions.
None of the prophages apparently harbored genes encoding antibiotic resistance genes and genes coding for allergens/toxins when analyzed by VirulenceFinder and ResFinder and by manual inspection with BLAST, and only phiPpa1 contained a gene potentially involved in biosynthesis of a putatively antagonistic factor (monooxygenase antibiotic).
Discussion
Despite the fact that the majority of bacterial genomes deposited in international genomic sequence databases reveal that phage DNA is commonly integrated into the host chromosome (Canchaya et al., 2003), little is known of how commonly such viruses infect SRP and the extent to which viruses might be associated with virulence or host range of this important group of bacteria (Varani et al., 2013; Czajkowski, 2016).
Initial studies of bacteriophages in this group (Erwinia chrysanthemi 3937 phage phiEC2) were reported only in 1984 (Resibois et al., 1984), and while this phage has been widely used since then for generalized transduction of Dickeya spp. even it has yet to be characterized in detail and little is known about its ecological, genomic and morphological features (for review see: Czajkowski, 2016). While other temperate SPR bacteriophages have been recently described (for review see: Czajkowski, 2016), little molecular detail is known of these viruses. This study was designed to explain to genome sequence is available for SRP to better understand the frequency of occurrence, diversity, and possible functions of viruses in this group of bacteria.
In this study all available 57 (as of August 2018) Pectobacterium spp. and Dickeya spp. complete genome sequences present in NCBI GenBank were screened for the presence of prophage-like elements. The in silico workflow used here (Figure 1) allowed the identification of prophages in 95% of SRP genomes. Although prophages are known to constitute even as much as 10 to 20% of a bacterial genome, all of the prophages analyzed comprised on average < 2% of the Pectobacterium spp. and Dickeya spp. chromosome. It is noteworthy that the related foodborne pathogen Escherichia coli O157:H7 strain Sakai that can sometimes be found in the same habitats as SRP harbors much more abundant prophages (16% of its total genome content) (Hayashi et al., 2001).
The majority of the SRP prophages (48 sequences) were putatively defective and did not apparently contain those genes essential for bacteriophage interaction with their bacterial hosts such as integrases and genes coding for viral structural proteins. Similarly, some of the screened bacterial genomes were also missing putative attachment sites for these prophage. The frequent occurrence of incomplete prophages in bacterial chromosomes has been reported for various bacteria, including human and animal pathogens as well as for saprophytic bacteria present in soil and water (Casjens, 2003; Bobay et al., 2014). It is widely accepted that bacterial hosts under natural conditions are continuously exposed to phage infections and that some of these events may result in long-term and irreversible phage-bacterial associations on a genomic level (Touchon et al., 2014). This is clearly the case for Pectobacterium spp. and Dickeya spp. since these strains have a worldwide distribution (Pérombelon, 2002). The high number of putatively defective prophage sequences reported here may further indicate an initial rapid inactivation of viable prophages in bacterial genome is followed by a slow decay of prophage genes due to the accumulations of point mutations and deletions. This so-called phage domestication has been reported for other Enterobacteriaceae as a way to cure bacterial genomes from the presence of unnecessary and/or toxic genetic material (Bobay et al., 2014).
The 37 complete prophages found in 29 genomes of Pectobacterium spp. and Dickeya spp. were characterized in detail. Bioinformatic analysis of the prophage genes encoding viral structural proteins allowed classification of these prophages into different families of the order Caudovirales (tailed bacteriophages) with the SRP prophages of the Myoviridae family being the most abundant (81% of found prophages). The order Caudovirales contains more than 97% of all described phages known to infect bacteria with at least 350 distinct phage isolates documented as members of this order to date (Ackermann, 1998; Fokine and Rossmann, 2014). The great majority of existing bioinformatic tools created to analyze bacteriophage genomes have been developed based on the known Caudovirales sequences and consequently they may not be well-suited to analyze viral genomes belonging to different orders and/or groups. Additionally, more than 99% of all SRP bacteriophages described so far also belong to the order Caudovirales and occur in three families namely Myoviridae, Podoviridae, and Siphoviridae (Czajkowski, 2016).
The genome organization and ORF arrangements was not well-conserved across the 37 seemingly intact prophages. The exceptions were the 3 prophage pairs (phiDdi1 and phiDdi3, phiDpa1 and phiiDpa2, and phiDze4 and phiDze5) that were highly conserved with respect to each other. This indicates that overall, SRP prophages are likely mobile, often being transferred between different hosts and easily undergoing rearrangements. It is well-established that prophages are often highly mosaic and that their genomes constitute modules that can be interchanged between different phages by recombination (Hendrix et al., 2000). As it is believed now, such constant recombination events and the resulting mosaicism are the major driving force both for bacteriophage and bacterial evolution (Hendrix et al., 1999; Pedulla et al., 2003).
No linkage was seen between the presence of particular seemingly intact prophages and bacterial genera, bacterial genome size, geographical location, or the environments from which the host bacteria were initially isolated. Likewise, due to the absence of universal genes in bacteriophages that can be used for phylogenetic studies (similar to 16S rDNA gene in bacteria), (pro)phage classification is difficult (Lawrence et al., 2002). In this study, contrary to the studies performed earlier in which the usefulness of integrase, holin, and lysin sequences for the phylogenetic studies of prophages were evaluated (Brüssow et al., 2004; Ventura et al., 2005, 2007), none of these genes proved useful for clear phylogenetic separation of prophages of Pectobacterium spp. and Dickeya spp. into distinct clades. Such a lack of phylogenetic association suggests an independent evolution of prophages and their SRP hosts (Colavecchio et al., 2017). This is perhaps not a surprise given that SRP are not only naturally present in many and widely different environments (e.g., soil, water, plant surface, on and inside insects) but are often dispersed from one environment to another (Perombelon, 1988; Charkowski, 2006). All these lifestyle changes would require a rapid adaptation to a new setting, a process that might not facilitate stable association of phage with a given habitat or host (Ma et al., 2007; Reverchon et al., 2016).
More than 50% of complete prophage genomes contained not only the genes encoding structural viral proteins and integrases, but also genes coding for holin and lysin. Additionally, the 13 prophages (35% of the complete prophages) contained genes encoding both proteins. Lysins and holins are viral enzymes leading to disruption of the host cells and enabling propagation of bacteriophages in the environment (Wang et al., 2000). As both holin and lysin are viewed as facilitating host infection (Young, 2014), the presence of these genes in SRP prophages may give the first assumption that those viruses may be more infective than the prophages lacking one or both genes (Feiner et al., 2015). This further indicates that in at least these 13 prophages may be possibly easily induced, thus it they may become transmittable upon encounter of particular environmental stimuli (Nanda et al., 2014). However, the point must be made that without the further experiments, the infectivity of the mentioned prophages remains rather speculative at the moment.
Likewise, the absence of holin and/or lysin or both genes in the phage genome does not necessarily characterize a bacteriophage as harmless. For example, the well-characterized infectious Dickeya spp. bacteriophage LIMEstone1 (Adriaenssens et al., 2012) and ϕD5 (Czajkowski et al., 2014) both lack the gene coding for holin, and the P. carotovorum subsp. carotovorum phage PP1 lacks the gene coding for lysin (Lee et al., 2012).
It seems likely that induction of SRP prophages will have an impact on environmental fitness and virulence of the hosts. An understanding of the conditions in which lysis is induced might make it possible to achieve some level of control of the diseases caused by these SRP by appropriately modifying the environment. Alternatively, it can be speculated that the newly found holin and lysin genes, produced at an industrial scale might be a useful tool in the biological control of such diseases as previously suggested (Fenton et al., 2010).
The high abundance of (seemingly intact) prophages in the SRP genomes may have as well a direct impact on control of Pectobacterium spp. and Dickeya spp. in agricultural applications. Prophages are known to utilize mechanism called superinfection exclusion which prevents subsequent viral infections of the same hosts (Bondy-Denomy and Davidson, 2014). It can be speculated that effectiveness of biological control of SRP with the use of lytic bacteriophages may be reduced due to the prophage-induced resistance in the target bacteria. The superinfection exclusion has been analyzed in detail in several human pathogenic bacteria including Escherichia coli, Pseudomonas aeruginosa and Salmonella spp. (for review see: Labrie et al., 2010), its ecological role has never been however assessed in SRP. Considering the increasing interest in phage therapy as a means to combat plant pathogenic bacteria, and specifically SRP, this topic undoubtedly needs further examination.
Based on average amino acid identity (AAI), six prophage clusters; five present in Dickeya spp. and one within Pectobacterium spp. could be identified in this large collection of strains. As opposed to the phylogenetic analyses based on a single given prophage gene, AAI appears to be a more powerful method to phylogenetically separate the prophages residing in the genomes of Pectobacterium spp. and Dickeya spp. While AAI has been suggested to be a better phylogenetic method for whole genome-based taxonomy of Prokaryotes (Konstantinidis and Tiedje, 2005), this method has received little usage in the phylogenetic analysis of viruses. The presented results suggest that it would prove useful in bioinformatics analyses of prophage such as in this study.
It is well-established that prophages often encode genes that are not directly involved in viral propagation and infection but which can confer a fitness benefit to their hosts (Bondy-Denomy and Davidson, 2014). These genes can enhance the virulence of the bacteria directly by prophage-encoded toxins and/or indirectly by increasing bacterial fitness which indirectly results in enhanced virulence (Hacker and Carniel, 2001). All but one of the 37 seemingly intact prophages described in this study contained at least one gene that was apparently acquired from other host bacteria (probably from Dickeya spp. and Pectobacterium spp. strains or their close relatives), as a result of infection of one or more previous hosts. Likewise, the majority of prophages analyzed in this study contained multiple genes of bacterial origin, with two prophages phiDdi1 and phiDdi3 carrying even as many as 23 bacterial genes. It remains unclear however whether the bacterial genes found in these prophage genomes are transcribed or translated. Surprisingly, several prophages present in different bacterial genomes carried homological set of bacterial genes indicating that possibly these prophages propagated in co-occurring host populations of different species at the same time. None of the 36 prophages analyzed here however acquired bacterial genes encoding well-described virulence factors exploited by Pectobacterium spp. and Dickeya spp. to infect plants (Reverchon and Nasser, 2013). Instead, the prophages carried genes that may apparently contribute to ecological fitness in complex and diverse environments; e.g., genes encoding metal ion transporters, enzymes involved in energy metabolism, heat shock proteins, nitrogen assimilation proteins as well as genes coding for DNA methylases which may be used in protecting prophage sequences in the host genome from excision by changing DNA methylation pattern (Canchaya et al., 2003). This may as well-explain the high number of prophage sequences observed in many bacterial genomes (Ohnishi et al., 2001; Matos et al., 2013) and the relatively high proportion of prophage-related genes in pathogenic strains in comparison with saprophytic, non-pathogenic bacteria (Busby et al., 2013). The most common gene present in seemingly intact prophage genomes was one encoding methyl-directed repair DNA adenine methylase (EC 2.1.1.72), being found in 21 viruses. This is a large group of enzymes that apart from being members of restriction-modification systems of many Gram-negative bacteria, plays important roles in regulation of genes encoding virulence factors in bacterial pathogens at the posttranscriptional level (Marinus and Casadesus, 2009). Unfortunately their role, if any, in pathogenicity of SRPs remains cryptic.
The biggest limitation of the in silico workflow used here is obviously that the classification of prophage element to the group of intact or defective prophages and their impact on the host fitness is based on the genome data alone. However, in general, the relatively high number of seemingly intact prophages found in the study suggest that the interaction of SRP and bacteriophages in the natural environment may be highly significant for the ecology, adaptation, and evolution of Pectobacterium spp. and Dickeya spp. Prophage induction experiments are now being conducted to further elucidate the role of prophages present in SRP strains and to better understand the molecular basis of (pro)phage-bacteria interactions.
Author Contributions
RC: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, validation, visualization, writing and original draft, writing and review, and editing.
Funding
The work was financially supported by the National Center for Research and Development, Poland (Narodowe Centrum Badań i Rozwoju, Polska) via a research grant LIDER VI (LIDER/450/L-6/14/NCBR/2015) to RC and by the University of Gdansk, Poland (Uniwersytet Gdański, Polska) grant no. DS 530-M035-D673-19.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The preprint version of this manuscript has been deposited at bioRxiv—the preprint server for biology as: Czajkowski (2018) May the phage be with you? Prophage-like elements in the genomes of Soft Rot Pectobacteriaceae (Pectobacterium spp. and Dickeya spp.) and can be accessed via https://www.biorxiv.org/search/czajkowski%252Bprophages.The author would like to express his gratitude to Prof. Steven E. Lindow (University of California-Berkeley, Berkeley, CA, USA) for his valuable comments on the manuscript and his editorial work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00138/full#supplementary-material
References
Ackermann, H. W. (1998). Tailed bacteriophages: the order Caudovirales. Adv. Virus Res. 51, 135–201. doi: 10.1016/S0065-3527(08)60785-X
Ackermann, H. W. (2003). Bacteriophage observations and evolution. Res. Microbiol. 154, 245–251. doi: 10.1016/S0923-2508(03)00067-6
Adeolu, M., Alnajar, S., Naushad, S.S, and Gupta, R. (2016). Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. System. Evol. Microbiol. 66, 5575–5599. doi: 10.1099/ijsem.0.001485
Adriaenssens, E. M., and Cowan, D. A. (2014). Using signature genes as tools to assess environmental viral ecology and diversity. Appl. Environ. Microbiol. 80, 4470–4480. doi: 10.1128/AEM.00878-14
Adriaenssens, E. M., Van Vaerenbergh, J., Vandenheuvel, D., Dunon, V., Ceyssens, P. J., De Proft, M., et al. (2012). T4-Related Bacteriophage LIMEstone isolates for the control of soft rot on potato caused by ‘Dickeya solani’. PLoS ONE 7:e33227. doi: 10.1371/journal.pone.0033227
Akhter, S., Aziz, R. K., and Edwards, R. A. (2012). PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucl. Acids Res. 40, e126–e126. doi: 10.1093/nar/gks406
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucl. Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75–75. doi: 10.1186/1471-2164-9-75
Blom, J., Albaum, S., Doppmeier, D., Pühler, A., Vorhölter, F. J., Zakrzewski, M., et al. (2009). EDGAR: A software framework for the comparative analysis of prokaryotic genomes. BMC Bioinform. 10:154. doi: 10.1186/1471-2105-10-154
Blower, T. R., Chai, R., Przybilski, R., Chindhy, S., Fang, X., Kidman, S. E., et al. (2017). Evolution of Pectobacterium bacteriophage ΦM1 to escape two bifunctional type III toxin-antitoxin and abortive infection systems through mutations in a single viral gene. Appli. Environ. Microbiol. 83, e03229–e03216. doi: 10.1128/AEM.03229-16
Blower, T. R., Evans, T. J., Przybilski, R., Fineran, P. C., and Salmond, G. P. C. (2012). Viral evasion of a bacterial suicide system by RNA–based molecular mimicry enables infectious altruism. PLoS Genetics 8:e1003023. doi: 10.1371/journal.pgen.1003023
Bobay, L. M., Touchon, M., and Rocha, E. P. C. (2014). Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. U.S.A. 111, 12127–12132. doi: 10.1073/pnas.1405336111
Bondy-Denomy, J., and Davidson, A. R. (2014). When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J. Microbiol. 52, 235–242. doi: 10.1007/s12275-014-4083-3
Boyd, E. F., and Brüssow, H. (2002). Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol. 10, 521–529. doi: 10.1016/S0966-842X(02)02459-9
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Brüssow, H., Canchaya, C., and Hardt, W. D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602. doi: 10.1128/MMBR.68.3.560-602.2004
Busby, B., Kristensen, D. M., and Koonin, E. V. (2013). Contribution of phage-derived genomic islands to the virulence of facultative bacterial pathogens. Environ. Microbiol. 15, 307–312. doi: 10.1111/j.1462-2920.2012.02886.x
Canchaya, C., Fournous, G., and Brüssow, H. (2004). The impact of prophages on bacterial chromosomes. Mol. Microbiol. 53, 9–18. doi: 10.1111/j.1365-2958.2004.04113.x
Canchaya, C., Proux, C., Fournous, G., Bruttin, A., and Brüssow, H. (2003). Prophage genomics. Microbiol. Mol. Biol. Rev. 67, 238–276. doi: 10.1128/MMBR.67.2.238-276.2003
Casjens, S. (2003). Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol. 49, 277–300. doi: 10.1046/j.1365-2958.2003.03580.x
Charkowski, A. (2018). The changing face of bacterial soft-rot diseases. Ann. Rev. Phytopathol. 56, 269–288. doi: 10.1146/annurev-phyto-080417-045906
Charkowski, A. O. (2006). The soft rot Erwinia. Plant Assoc. Bacteria Part 3, 423–505. doi: 10.1007/978-1-4020-4538-7_13
Colavecchio, A., D'souza, Y., Tompkins, E., Jeukens, J., Freschi, L., Emond-Rheault, J.-G., et al. (2017). Prophage integrase typing is a useful indicator of genomic diversity in Salmonella enterica. Front. Microbiol. 8:1283. doi: 10.3389/fmicb.2017.01283
Czajkowski, R. (2016). Bacteriophages of soft rot Enterobacteriaceae – a mini-review. FEMS Microbiol. Lett. 363:fnv230. doi: 10.1093/femsle/fnv230.
Czajkowski, R., Ozymko, Z., Zwirowski, S., and Lojkowska, E. (2014). Complete genome sequence of a broad-host-range lytic Dickeya spp. bacteriophage φD5. Arch. Virol. 159, 3153–3155. doi: 10.1007/s00705-014-2170-8
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Day, A., Ahn, J., Fang, X., and Salmond, G. P. C. (2017). Environmental bacteriophages of the emerging enterobacterial phytopathogen, Dickeya solani, show genomic conservation and capacity for horizontal gene transfer between their bacterial hosts. Front. Microbiol. 8:1654. doi: 10.3389/fmicb.2017.01654
Evans, T. J., Coulthurst, S. J., Komitopoulou, E., and Salmond, G. P. (2010). Two mobile Pectobacterium atrosepticum prophages modulate virulence. FEMS Microbiol. Lett. 304, 195–202. doi: 10.1111/j.1574-6968.2010.01901.x
Feiner, R., Argov, T., Rabinovich, L., Sigal, N., Borovok, I., and Herskovits, A. A. (2015). A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 13:641. doi: 10.1038/nrmicro3527
Fenton, M., Ross, P., Mcauliffe, O., O'mahony, J., and Coffey, A. (2010). Recombinant bacteriophage lysins as antibacterials. Bioeng. Bugs 1, 9–16. doi: 10.4161/bbug.1.1.9818
Fokine, A., and Rossmann, M. G. (2014). Molecular architecture of tailed double-stranded DNA phages. Bacteriophage 4:e28281. doi: 10.4161/bact.28281
Fortier, L.-C., and Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/viru.24498
Grenier, A. M., Duport, G., Pagès, S., Condemine, G., and Rahbé, Y. (2006). The phytopathogen Dickeya dadantii (Erwinia chrysanthemi 3937) is a pathogen of the pea aphid. Appl. Environ. Microbiol. 72, 1956–1965. doi: 10.1128/AEM.72.3.1956-1965.2006
Hacker, J., and Carniel, E. (2001). Ecological fitness, genomic islands and bacterial pathogenicity: a Darwinian view of the evolution of microbes. EMBO Rep. 2, 376–381. doi: 10.1093/embo-reports/kve097
Hayashi, T., Makino, K., Ohnishi, M., Kurokawa, K., Ishii, K., Yokoyama, K., et al. (2001). Complete genome sequence of enterohemorrhagic Eschelichia coli O157: H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8, 11–22. doi: 10.1093/dnares/8.1.11
Hendrix, R. W., Lawrence, J. G., Hatfull, G. F., and Casjens, S. (2000). The origins and ongoing evolution of viruses. Trends Microbiol. 8, 504–508. doi: 10.1016/S0966-842X(00)01863-1
Hendrix, R. W., Smith, M. C., Burns, R. N., Ford, M. E., and Hatfull, G. F. (1999). Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc. Natl. Acad. Sci. U.S.A. 96, 2192–2197.
Kleinheinz, K. A., Joensen, K. G., and Larsen, M. V. (2014). Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 4:e27943. doi: 10.4161/bact.27943
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for Prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Korol, N. A., and Tovkach, F. I. (2012). Identification of the major proteins of the virions of bacteriophage ZF40 Pectobacterium carotovorum. Microbiologia 74, 64–70.
Labrie, S. J., Samson, J. E., and Moineau, S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327. doi: 10.1038/nrmicro2315
Lawrence, J. G., Hatfull, G. F., and Hendrix, R. W. (2002). Imbroglios of viral taxonomy: genetic exchange and failings of phenetic approaches. J. Bacteriol. 184, 4891–4905. doi: 10.1128/JB.184.17.4891-4905.2002
Lee, J. H., Shin, H., Ji, S., Malhotra, S., Kumar, M., Ryu, S. H., et al. (2012). Complete genome sequence of phytopathogenic Pectobacterium carotovorum subsp. carotovorum bacteriophage PP1. J. Virol. 86, 8899–8900. doi: 10.1128/JVI.01283-12
Lopes, A., Tavares, P., Petit, M. A., Guérois, R., and Zinn-Justin, S. (2014). Automated classification of tailed bacteriophages according to their neck organization. BMC Genomics 15:1027. doi: 10.1186/1471-2164-15-1027
Ma, B., Hibbing, M. E., Kim, H. S., Reedy, R. M., Yedidia, I., Breuer, J., et al. (2007). Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya. Phytopathology 97, 1150–1163. doi: 10.1094/PHYTO-97-9-1150
Mansfield, J., Genin, S., Magori, S., Citovsky, V., Sriariyanum, M., Ronald, M., et al. (2012). Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13, 614–629. doi: 10.1111/j.1364-3703.2012.00804.x
Marinus, M. G., and Casadesus, J. (2009). Roles of DNA adenine methylation in host–pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol. Rev. 33, 488–503. doi: 10.1111/j.1574-6976.2008.00159.x
Matos, R. C., Lapaque, N., Rigottier-Gois, L., Debarbieux, L., Meylheuc, T., Gonzalez-Zorn, B., et al. (2013). Enterococcus faecalis prophage dynamics and contributions to pathogenic traits. PLoS Genet. 9:e1003539. doi: 10.1371/journal.pgen.1003539
Nanda, A. M., Thormann, K., and Frunzke, J. (2014). Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J. Bacteriol. 197, 410–419. doi: 10.1128/JB.02230-14
Ohnishi, M., Kurokawa, K., and Hayashi, T. (2001). Diversification of Escherichia coli genomes: are bacteriophages the major contributors? Trends Microbiol. 9, 481–485.
Pedulla, M. L., Ford, M. E., Houtz, J. M., Karthikeyan, T., Wadsworth, C., Lewis, J. A., et al. (2003). Origins of highly mosaic Mycobacteriophage genomes. Cell 113, 171–182. doi: 10.1016/S0092-8674(03)00233-2
Perombelon, M. C. M. (1988). “Ecology of Erwinias causing stem and tuber diseases,” in Proceedings of the Bacterial disease of Potato: Report of the Planning Conference on Bacterial Diseases of Potato [Lima: International Potato Center (CIP)].
Pérombelon, M. C. M. (2002). Potato diseases caused by soft rot Erwinias: an overview of pathogenesis. Plant Pathol. 51, 1–12. doi: 10.1046/j.0032-0862.2001.Shorttitle.doc.x
Perombelon, M. C. M., and Kelman, A. (1980). Ecology of the soft rot Erwinias. Ann. Rev. Phytopathol. 18, 361–387. doi: 10.1146/annurev.py.18.090180.002045
Pope, W. H., and Jacobs-Sera, D. (2018). “Annotation of bacteriophage genome sequences using DNA master: an overview,” in Bacteriophages: Methods and Protocols, Vol 3, eds Clokie, M. R. J, Kropinski, A. M. and Lavigne R (New York, NY: Springer New York), 217–229.
Resibois, A., Colet, M., Faelen, M., Schoonejans, E., and Toussaint, A. (1984). ϕEC2, a new generalized transducing phage of Erwinia chrysanthemi. Virology 137, 102–112. doi: 10.1016/0042-6822(84)90013-8
Reverchon, S., Muskhelisvili, G., and Nasser, W. (2016). “Chapter three - virulence program of a bacterial plant pathogen: the Dickeya model,” in Progress in Molecular Biology and Translational Science, eds M. San Francisco and B. San Francisco (San Francisco, CA: Academic Press), 51–92. Available online at: https://www.sciencedirect.com/science/article/pii/S1877117316300096?via%3Dihub
Reverchon, S., and Nasser, W. (2013). Dickeya ecology, environment sensing and regulation of virulence program. Environ. Microbiol. Rep. 5, 622–636. doi: 10.1111/1758-2229.12073
Rossmann, S., Dees, M. W., Perminow, J., Meadow, R., and Brurberg, M. B. (2018). Soft rot Enterobacteriaceae are carried by a large range of insect species in potato fields. Appl. Environ. Microbiol. 84, e00281–e00218. doi: 10.1128/AEM.00281-18
Suttle, C. A. (2007). Marine viruses — major players in the global ecosystem. Nat. Rev. Microbiol. 5:801. doi: 10.1038/nrmicro1750
Touchon, M., Bobay, L. M., and Rocha, E. P. (2014). The chromosomal accommodation and domestication of mobile genetic elements. Curr. Opin. Microbiol. 22, 22–29. doi: 10.1016/j.mib.2014.09.010
Varani, A. M., Monteiro-Vitorello, C. B., Nakaya, H. I., and Van Sluys, M.-A. (2013). The role of prophage in plant-pathogenic bacteria. Ann. Rev. Phytopathol. 51, 429–451. doi: 10.1146/annurev-phyto-081211-173010
Ventura, M., Lee, J. H., Canchaya, C., Zink, R. T., Leahy, S., Moreno-Munoz, J. A., et al. (2005). Prophage-like elements in bifidobacteria: insights from genomics, transcription, integration, distribution, and phylogenetic analysis. Appl. Environ. Microbiol. 71, 8692–8705. doi: 10.1128/AEM.71.12.8692-8705.2005
Ventura, M., Zomer, A., Canchaya, C., O'connell-Motherway, M., Kuipers, O. P., Turroni, F., et al. (2007). Comparative analyses of prophage-like elements present in two Lactococcus lactis strains. Appl. Environ. Microbiol. 73, 7771–7780. doi: 10.1128/AEM.01273-07
Wang, I. N., Smith, D. L., and Young, R. (2000). Holins: the protein clocks of bacteriophage infections. Appl. Environ. Microbiol. 54, 799–825. doi: 10.1146/annurev.micro.54.1.799
Wang, X., Kim, Y., Ma, Q., Hong, S. H., Pokusaeva, K., Sturino, J. M., et al. (2010). Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 1:147. doi: 10.1038/ncomms1146
Weinbauer, M. G. (2004). Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 28, 127–181. doi: 10.1016/j.femsre.2003.08.001
Keywords: Pectobacterium spp., Dickeya spp., integrase, attachment site, holin, lysin, bacterial gene, ecological fitness
Citation: Czajkowski R (2019) May the Phage be With You? Prophage-Like Elements in the Genomes of Soft Rot Pectobacteriaceae: Pectobacterium spp. and Dickeya spp. Front. Microbiol. 10:138. doi: 10.3389/fmicb.2019.00138
Received: 08 October 2018; Accepted: 21 January 2019;
Published: 14 February 2019.
Edited by:
Helene Sanfacon, Agriculture and Agri-Food Canada (AAFC), CanadaReviewed by:
Siân Victoria Owen, Harvard Medical School, United StatesAntonet M. Svircev, Agriculture and Agri-Food Canada (AAFC), Canada
Copyright © 2019 Czajkowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert Czajkowski, cm9iZXJ0LmN6YWprb3dza2lAYmlvdGVjaC51Zy5lZHUucGw=