 I-Yin Chen1
I-Yin Chen1 Miyu Moriyama
Miyu Moriyama Ming-Fu Chang
Ming-Fu Chang Takeshi Ichinohe
Takeshi Ichinohe
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 29 January 2019
Sec. Virology
Volume 10 - 2019 | https://doi.org/10.3389/fmicb.2019.00050
Nod-like receptor family, pyrin domain-containing 3 (NLRP3) regulates the secretion of proinflammatory cytokines interleukin 1 beta (IL-1β) and IL-18. We previously showed that influenza virus M2 or encephalomyocarditis virus (EMCV) 2B proteins stimulate IL-1β secretion following activation of the NLRP3 inflammasome. However, the mechanism by which severe acute respiratory syndrome coronavirus (SARS-CoV) activates the NLRP3 inflammasome remains unknown. Here, we provide direct evidence that SARS-CoV 3a protein activates the NLRP3 inflammasome in lipopolysaccharide-primed macrophages. SARS-CoV 3a was sufficient to cause the NLRP3 inflammasome activation. The ion channel activity of the 3a protein was essential for 3a-mediated IL-1β secretion. While cells uninfected or infected with a lentivirus expressing a 3a protein defective in ion channel activity expressed NLRP3 uniformly throughout the cytoplasm, NLRP3 was redistributed to the perinuclear space in cells infected with a lentivirus expressing the 3a protein. K+ efflux and mitochondrial reactive oxygen species were important for SARS-CoV 3a-induced NLRP3 inflammasome activation. These results highlight the importance of viroporins, transmembrane pore-forming viral proteins, in virus-induced NLRP3 inflammasome activation.
Severe acute respiratory syndrome coronavirus (SARS-CoV), a member of the genus Betacoronavirus within the family Coronaviridae, is an enveloped virus with a single-stranded positive-sense RNA genome of approximately 30 kb in length. The 5′ two-thirds of the genome encodes large polyprotein precursors, open reading frame (ORF) 1 and ORF1b, which are proteolytically cleaved to generate 16 non-structural proteins (Tan et al., 2005). The 3′ one-third of the genome encodes four structural proteins, spike (S), envelope (E), matrix (M) and nucleocapsid (N), and non-structural proteins, along with a set of accessory proteins (3a, 3b, 6, 7a, 7b, 8a, 8b, and 9b) (Perlman and Dandekar, 2005; Tan et al., 2005). SARS-CoV is the etiological agent of SARS (Drosten et al., 2003; Fouchier et al., 2003; Ksiazek et al., 2003; Kuiken et al., 2003; Peiris et al., 2003). At least 8,098 laboratory-confirmed cases of human infection, with a fatality rate of 9.6%, were reported to the World Health Organization from November 2002 to July 2003. High levelsof proinflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, were detected in autopsy tissues from SARS patients (He et al., 2006). Although dysregulation of inflammatory cytokines may be involved in lung injury and the pathogenesis of SARS-CoV, the underlying molecular mechanisms are not fully understood.
The innate immune systems utilizes pattern recognition receptors (PRRs) to detect pathogen-associated molecular patterns (Medzhitov, 2001; Kawai and Akira, 2010). Recognition of virus infection plays an important role in limiting virus replication at the early stages of infection. Nod-like receptor family, pyrin domain-containing 3 (NLRP3) is activated by a wide variety of stimuli, including virus infection (Bauernfeind et al., 2011). Four models describing activation of the NLRP3 inflammasome have been proposed thus far (Hornung and Latz, 2010; Schroder et al., 2010; Tschopp and Schroder, 2010). First, the disturbances in intracellular ionic concentrations, including K+ efflux and Ca2+ influx, play an important role (Fernandes-Alnemri et al., 2007; Petrilli et al., 2007; Arlehamn et al., 2010; Ichinohe et al., 2010; Ito et al., 2012; Murakami et al., 2012; Munoz-Planillo et al., 2013). Second, cathepsin B and L, which are specific lysosomal cysteine proteases, are though to play a role after phagocytosis of cholesterol crystals (Duewell et al., 2010), fibrillar peptide amyloid-beta (Halle et al., 2008), silica crystals, and aluminum salts (Hornung et al., 2008). Third is the release of reactive oxygen species (ROS) or mitochondrial DNA from damaged mitochondria (Zhou et al., 2010, 2011; Nakahira et al., 2011; Shimada et al., 2012). Finally, viral RNA or RNA cleavage products generated by RNase L activate the NLRP3 inflammasome via the DExD/H-box helicase, DHX33 (Allen et al., 2009; Mitoma et al., 2013; Chen et al., 2014; Chakrabarti et al., 2015). Upon activation, the NLRP3 is recruited to the mitochondria via association with mitochondrial antiviral signaling (MAVS) or mitofusin 2 expressed on the outer mitochondrial membrane (Ichinohe et al., 2013; Subramanian et al., 2013); these molecules then recruit the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-caspase-1 to form the NLRP3 inflammasome. This event activates the downstream molecule, caspase-1, which catalyzes the proteolytic processing of pro-IL-1β and pro-IL-18 into their active forms and stimulates their secretion (Kayagaki et al., 2015; Shi et al., 2015).
It is increasingly evident that NLRP3 detects RNA viruses by sensing the cellular damage or distress induced by viroporins (Ichinohe et al., 2010; Ito et al., 2012; Triantafilou et al., 2013; Nieto-Torres et al., 2015), transmembrane pore-forming proteins, encoded by certain RNA viruses; these proteins alter membrane permeability to ions by forming membrane channels (Tan et al., 2005; Chen and Ichinohe, 2015). A recent study shows that the SARS-CoV E protein, which comprise only 76 amino acids, forms Ca2+-permeable ion channels and activates the NLRP3 inflammasome (Nieto-Torres et al., 2015). Although the E and 3a proteins of SARS-CoV, which comprise 274 amino acids and contain three transmembrane domains (Zeng et al., 2004; Lu et al., 2006), are thought to act as Na+/K+ and K+ channels, respectively (Wilson et al., 2004; Lu et al., 2006; Torres et al., 2007; Parthasarathy et al., 2008; Pervushin et al., 2009; Wang et al., 2011), the role of the 3a protein in activating the NLRP3 inflammasome remains unknown. Here, we examined the role of the 3a protein in activating the NLRP3 inflammasome.
Six-week-old female C57BL/6 mice were purchased from The Jackson Laboratory. All animal experiments were approved by the Animal Committees of the Institute of Medical Science (The University of Tokyo).
Bone marrow-derived macrophages (BMMs) were prepared as described previously (Ichinohe et al., 2009). In brief, bone marrow was obtained from the tibia and femur by flushing with Dulbecco’s modified Eagle’s medium (DMEM; Nacalai Tesque). Bone marrow cells were cultured for 5 days in DMEM supplemented with 30% L929 cell supernatant containing macrophage colony-stimulating factor, 10% heat-inactivated fetal bovine serum (FBS), and L-glutamine (2 mM) at 37°C/5% CO2. HEK293FT cells (a human embryonic kidney cell line) and HeLa cells (a human epithelial carcinoma cell line) were maintained in DMEM supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml) (Nacalai Tesque). MDCK cells (Madin-Darby canine kidney cells) and HT-1080 cells (a human fibrosarcoma cell line) were grown in Eagle’s minimal essential medium (E-MEM; Nacalai Tesque) supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml) (Nacalai Tesque).
Influenza A virus strain A/PR8 (H1N1) was grown at 35°C for 2 days in the allantoic cavities of 10-day-old fertile chicken eggs (Ichinohe et al., 2009). The viral titer was quantified in a standard plaque assay using MDCK cells (Pang et al., 2013).
cDNAs encoding the E and M proteins of SARS-CoV Frankfurt 1 strain (Matsuyama et al., 2005) were obtained by reverse transcription and PCR of total RNA extracted from SARS-CoV-infected Vero cells, followed by PCR amplification using specific primers. pcDNA3.1D-3a-V5His was provided by Ming-Fu Chang (National Taiwan University College of Medicine, Taipei, Taiwan). To generate the plasmids pLenti6-E-V5His, pLenti6-3a-V5His, and pLenti-M-V5His, cDNA fragments of E, 3a, and M were amplified from pcDNA3.1D-E-V5His, pcDNA3.1D-3a-V5His, and pcDNA3.1D-M-V5His using specific primer sets and then ligated into pLenti6-TOPO vectors (Invitrogen). To generate plasmids pCA7-flag-E, pCA7-flag-3a, and pCA7-flag-M, pCA7-HA-E, pCA7-HA-3a, and pCA7-HA-M, cDNA fragments of E, 3a, and M were amplified from pcDNA3.1D-E-V5His, pcDNA3.1D-3a-V5His, and pcDNA3.1D-M-V5His using specific primer sets, digested with EcoR I and Not I, and subcloned into the EcoR I-Not I sites of the pCA7-flag-ASC plasmid or pCA7-HA-M2 plasmid, respectively (Ito et al., 2012). To construct plasmids expressing the E mutant V25F, the mutated E fragments were amplified by inverse PCR with wild-type E-containing plasmids and specific primer sets. The PCR products were cleaved by Dpn I, ligated in a ligase- and T4 kinase-containing reaction and then transformed into DH5α competent cells (TOYOBO). To construct plasmids expressing the 3a mutant 3a-CS, fragments were amplified from wild-type 3a-containing plasmids using 3a-specific primer sets and transformed as described above.
HEK293FT cells were seeded in 24-well cluster plates and transfected with 1 μg pLenti6-E/3a/M-V5His, pLenti-GFP (green fluorescent protein), or pLenti-M2 using polyethylenimine (PEI) Max. At 24 h post-transfection, the cells were lysed with RIPA buffer (50 mM Tris–HCl, 1% NP-40, 0.05% sodium dodecyl sulfate (SDS), 150 mM NaCl and 1 mM EDTA). And the lysates were subjected to SDS-polyacrylamide gel electrophoresis (PAGE) followed by electroblotting onto polyvinylidene difluoride (PVDF) membranes. The membranes were incubated over night with mouse anti-V5-tag (R960-25, Invitrogen), mouse anti-influenza A virus M2 (14C2, Abcam), mouse anti-GFP (GF200, Nacalai Tesque), or rabbit anti-tubulin (DM1A, Santa Cruz) antibodies, followed by horseradish peroxide-conjugated anti-mouse IgG (Jackson Immuno Research Laboratories) or anti-rabbit IgG (Invitrogen). After washing 3 times with washing buffer (0.05% Tween-20/PBS), the membranes were exposed using Chemi-Lumi One Super (Nacalai Tesque), and the chemiluminescent signals were captured by an ImageQuant LAS-4000 mini apparatus (GE Healthcare).
To generate lentiviruses expressing V5-tagged SARS-CoV E, 3a, and M proteins, the full-length cDNA encoding each viral protein was cloned into the pLenti6.3/V5-TOPO vector (Invitrogen) using the following primers: SARS-CoV E forward, 5′-caccatgtactcattcgtttcgga-3′, and reverse, 5′-gaccagaagatcaggaactc-3′; SARS-CoV 3a forward, 5′-caccatggatttgtttatgagatt-3′, and reverse, 5′-caaaggcacgctagtagtcg-3′; SARS-CoV M forward, 5′-caccatggcagacaacggtactat-3′, and reverse, 5′-ctgtactagcaaagcaatat-3′. Sub-confluent monolayers of HEK293FT cells seeded in a collagen-coated dish (10 cm in diameter) were transfected with 3 μg of pLenti6.3/V5-TOPO vector expressing each viral protein or EGFP together with ViraPower Packaging Mix (Invitrogen) using Lipofectamine 2000 (Invitrogen). The supernatants containing lentiviruses were harvested and filtered through a 0.45 μm filter (Millipore) at 72–96 h post-transfection (Ito et al., 2012). The lentiviral titer was then quantified using HT-1080 cells as described previously (Ichinohe et al., 2013).
Bone marrow-derived macrophages were plated at a density of 8 × 105 in 24-well plate and infected with A/PR8 influenza virus or lentivirus at a multiplicity of infection (MOI) of 5 or 0.2 for 1 h, respectively. Then, BMMs were stimulated with 1 μg/ml of LPS and cultured for additional 23 h in complete media. Supernatants were collected at 24 h post-infection and centrifuged to remove cell debris. The amount of IL-1β in the supernatants was measured in an enzyme-linked immunosorbent assay (ELISA) using paired antibodies (eBioscience) (Ichinohe et al., 2010, 2013).
To clarify the cellular localization of the wild-type and mutant 3a proteins of SARS-CoV, HeLa cells were cultured on coverslips and transfected with 1 μg of pCA7-flag-3a or pCD7-flag-3a-CS together with 0.5 μg of ER-mCherry or DsRed-Golgi (Ito et al., 2012). At 24 h post-transfection, cells were fixed with 4% paraformaldehyde and permeabilized with 1% Triton X-100/PBS. After washing with PBS and blocking with 4% BSA/PBS, the cells were incubated with a mouse anti-flag antibody (M2, Sigma) followed by incubation with Alexa Fluor 488-conjugated goat anti-mouse IgG (H+L) (Life Technologies).
To observe the cellular distribution of NLRP3 in the E- or 3a-expressing cells, HeLa cells were cultured on coverslips and transfected with 1 μg of pCA7-HA-E, pCA7-HA-EV25F, pCA7-HA-3a, pCA7-HA-3a-CS, or pCA7 control vector together with 0.5 μg of pCA7-NLRP3. At 24 h post-transfection, cells were fixed and permeabilized with 4% paraformaldehyde and 1% Triton X-100/PBS. After washing and blocking, the cells were incubated with rabbit anti-HA (561, MBL) and mouse anti-NLRP3 (Cryo-2; AdipoGen) antibodies, followed by Alexa Fluor 488-conjugated goat anti-rabbit IgG (H+L) and Alexa Fluor 568-conjugated goat anti-mouse IgG (H+L) (Life Technologies). Fluorescent signals were observed by confocal microscopy (A1R+, Nikon).
Statistical significance was tested using a two-tailed Student’s t-test. P-values < 0.05 were considered statistically significant.
We previously demonstrated that the influenza virus M2 protein (a proton-selective ion channel), its H37G mutant (which has lost its proton selectivity and enables the transport of other cations such as Na+ and K+), and the EMCV 2B protein (a Ca2+ channel) stimulates NLRP3 inflammasome-mediated IL-1β secretion (Ichinohe et al., 2010; Ito et al., 2012). In addition, the SARS-CoV E protein acts as a Ca2+-permeable ion channels that activates the NLRP3 inflammasome (Nieto-Torres et al., 2015). The fact that 3a protein of SARS-CoV acts as viroporin prompted us to examine whether it also triggers inflammasome activation. Thus, we first generated lentivirus plasmids expressing V5-tagged proteins and confirmed their expression in HEK293FT cells by immunoblot analysis (Figures 1A–C). We next transduced lipopolysaccharide (LPS)-primed BMMs with the lentiviruses expressing the SARS-CoV E, 3a, M, influenza virus M2, or EMCV 2B proteins. Consistent with previous reports (Ichinohe et al., 2010; Ito et al., 2012), IL-1β was released from LPS-primed BMMs transduced with the M2- and 2B-expressing lentivirus (Figure 1D). Similarly, the lentiviruses expressing the SARS-CoV E or 3a proteins stimulated IL-1β release from LPS-primed BMMs (Figure 1D). Furthermore, IL-1β secretion from LPS-primed BMMs co-infected with E- and 3a-expressing lentiviruses was significantly higher than that from SARS-CoV E-expressing lentivirus-infected cells (Figure 1E). These data indicated that the expression of SARS-CoV viroporin 3a is sufficient to stimulate IL-1β secretion by LPS-primed BMMs.
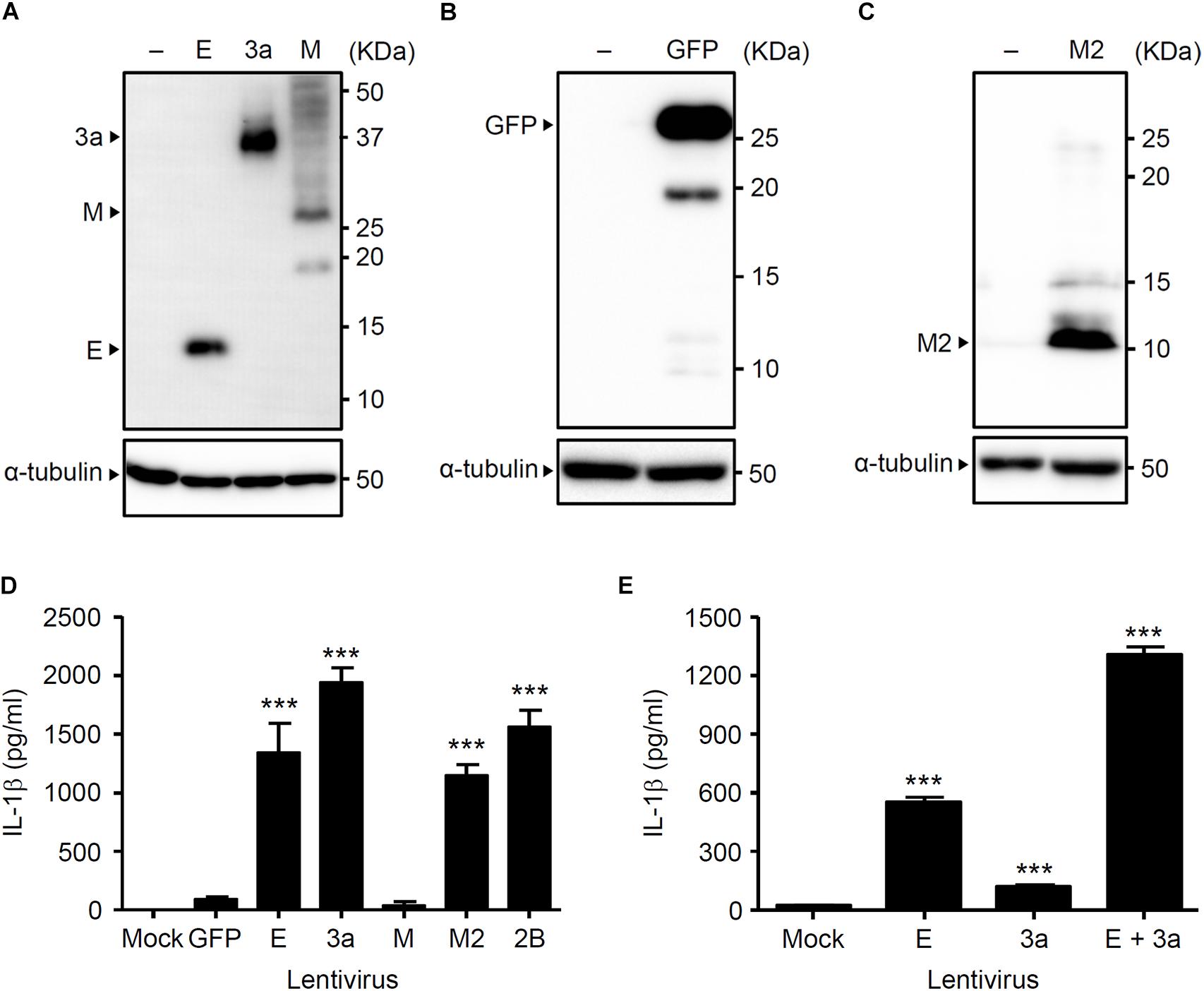
Figure 1. The 3a protein of SARS-CoV stimulates IL-1β secretion. (A–C) HEK293FT cells were transfected with pLenti6-E-V5, pLenti6-3a-V5, pLenti6-M-V5 (A), pLenti-GFP-V5 (B), or pLenti-M2-V5 plasmids (C). Samples were analyzed by immunoblot with mouse monoclonal antibodies against V5-tag (A), GFP (B), or influenza virus M2 (C). (D,E) LPS-primed BMM were infected with the lentivirus expressing SARS-CoV E, 3a, M, influenza virus M2, or EMCV 2B at MOI 0.25 (D) or 0.1 (E). Supernatants were collected at 24 h post-infection and analyzed for IL-1β by ELISA. Data are representative of at least three independent experiments, and indicate the mean ± SD (D,E); ∗∗∗P < 0.001.
Previous studies demonstrated that the N-terminal 40 amino acids of the SARS-CoV E protein are important for ion channel formation, and that mutations N15A and V25F [located in the transmembrane domain (from amino acid residues 7–38)] prevent ion conductivity (Wilson et al., 2004; Torres et al., 2007; Verdia-Baguena et al., 2012). In addition, the SARS-CoV 3a protein contains a cysteine-rich domain (amino acid residues 127–133) that is involved in the formation of a homodimer to generate the ion channel (Lu et al., 2006; Chan et al., 2009). Thus, mutation of the cysteine-rich domain blocks the ion conductivity by the 3a protein (Chan et al., 2009). To this end, we substituted amino acids Cys-127, Cys-130, and Cys-133 within the cysteine-rich domain of the SARS-CoV 3a protein with serine to generate a lentivirus expressing the ion channel activity-loss mutant, 3a-CS (Chan et al., 2009; Figure 2A). To test whether the ion channel activity of the SARS-CoV 3a protein is required to stimulate secretion of IL-1β, we transduced LPS-primed BMMs with lentiviruses expressing the SARS-CoV E, V25F, 3a, 3a-CS, or M proteins. Consistent with a previous report (Nieto-Torres et al., 2015), we found that the V25F mutant lentivirus failed to stimulate IL-1β release from BMMs (Figure 2B). Notably, the 3a-CS mutant completely abrogated IL-1β secretion (Figure 2B), suggesting that the ion channel activity of the 3a protein is required for SARS-CoV 3a-induced IL-1β secretion.
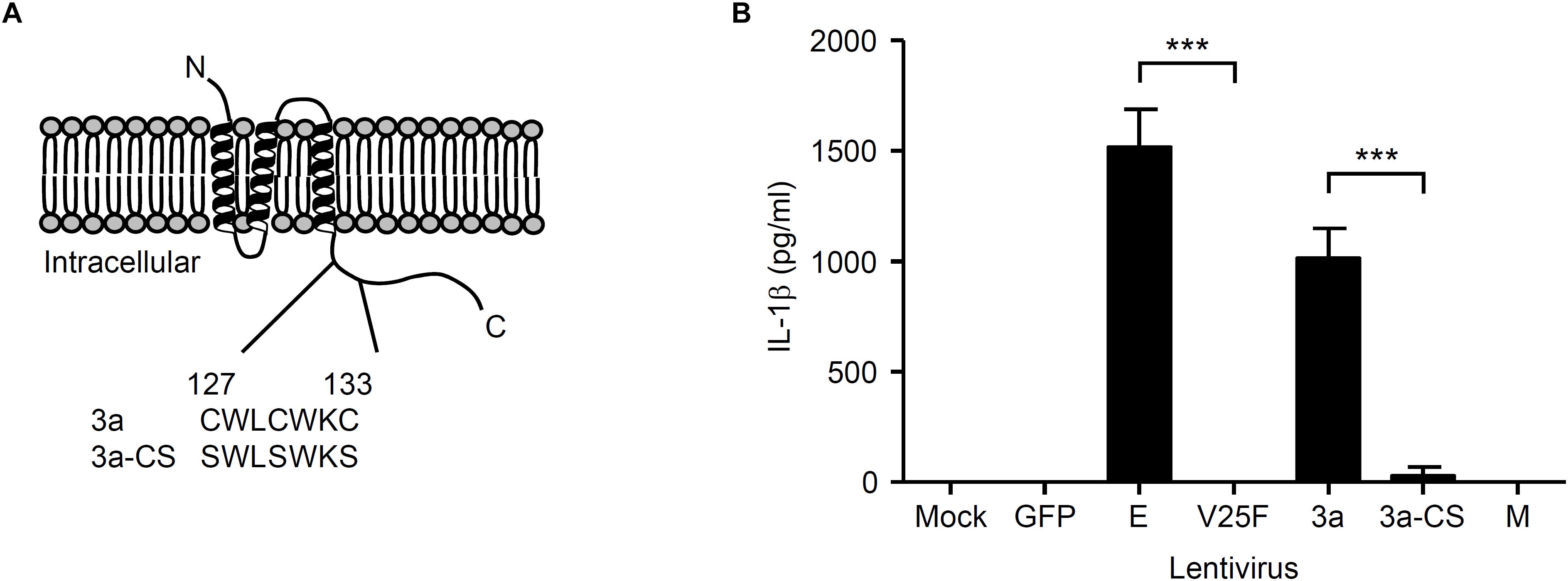
Figure 2. Ion channel activity of the 3a protein is required for IL-1β secretion. (A) SARS-CoV 3a protein; below, amino acid sequence of cysteine-rich domain (residue 127–133) of wild-type 3a and 3a-CS mutant. (B) LPS-primed BMM were infected with the lentivirus expressing SARS-CoV E, V25F, 3a, 3a-CS, or M at MOI 0.25. Supernatants were collected at 24 h post-infection and analyzed for IL-1β by ELISA. Data are representative of at least three independent experiments, and indicate the mean ± SD (B); ∗∗∗P < 0.001.
Next, we determined the subcellular localization of the SARS-CoV 3a protein using confocal microscopy. When the SARS-CoV 3a protein was expressed in HeLa cells, we observed two main distribution patterns. Consistent with previous reports (Yu et al., 2004; Yuan et al., 2005), the 3a protein localized to the Golgi apparatus (Figure 3A). In addition, the 3a proteins concentrated in spot structures, which mainly localized to the endoplasmic reticulum (ER) (Figure 3B). By contrast, the 3a-CS mutant was concentrated in the Golgi apparatus rather than in the ER and did not form spot structures (Figures 3A,B).
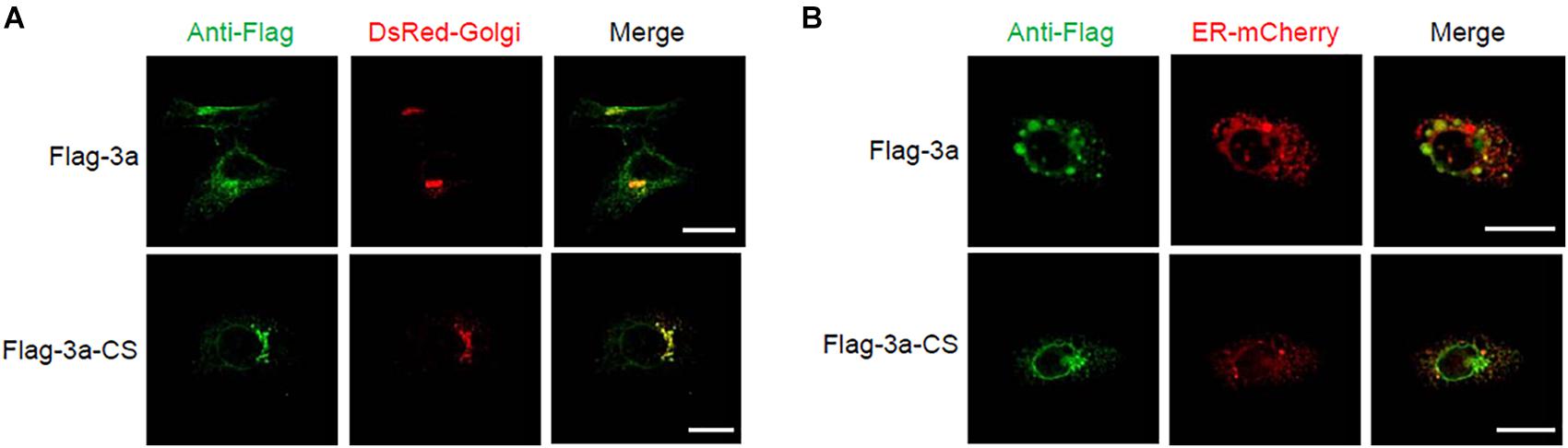
Figure 3. Subcellular localization of SARS-CoV 3a protein and 3a-CS mutant. (A,B) HeLa cells were transfected with the expression plasmid encoding flag-tagged 3a or 3a-CS and that encoding either DsRed-monomer-Golgi (A) or ER-mCherry (B), and observed with a confocal microscope at 24 h post-transfection. Scale bars, 10 μm. Data are representative of at least three independent experiments.
We next examined the intracellular localization of NLRP3. Activation of the NLRP3 inflammasome led to a redistribution from the cytosol to the perinuclear space, a process considered as a hallmark of NLRP3 activation (Zhou et al., 2011; Ito et al., 2012; Johnson et al., 2013; Moriyama et al., 2016). Although cells expressing the ion channel activity-loss mutants 3a-CS or V25F uniformly expressed NLRP3 throughout the cytoplasm, it was redistributed to the perinuclear region in SARS-CoV 3a- or E-expressing cells (Figure 4). Together, these data provide evidence that the ion channel activity of the SARS-CoV 3a protein is essential for triggering the NLRP3 inflammasome.
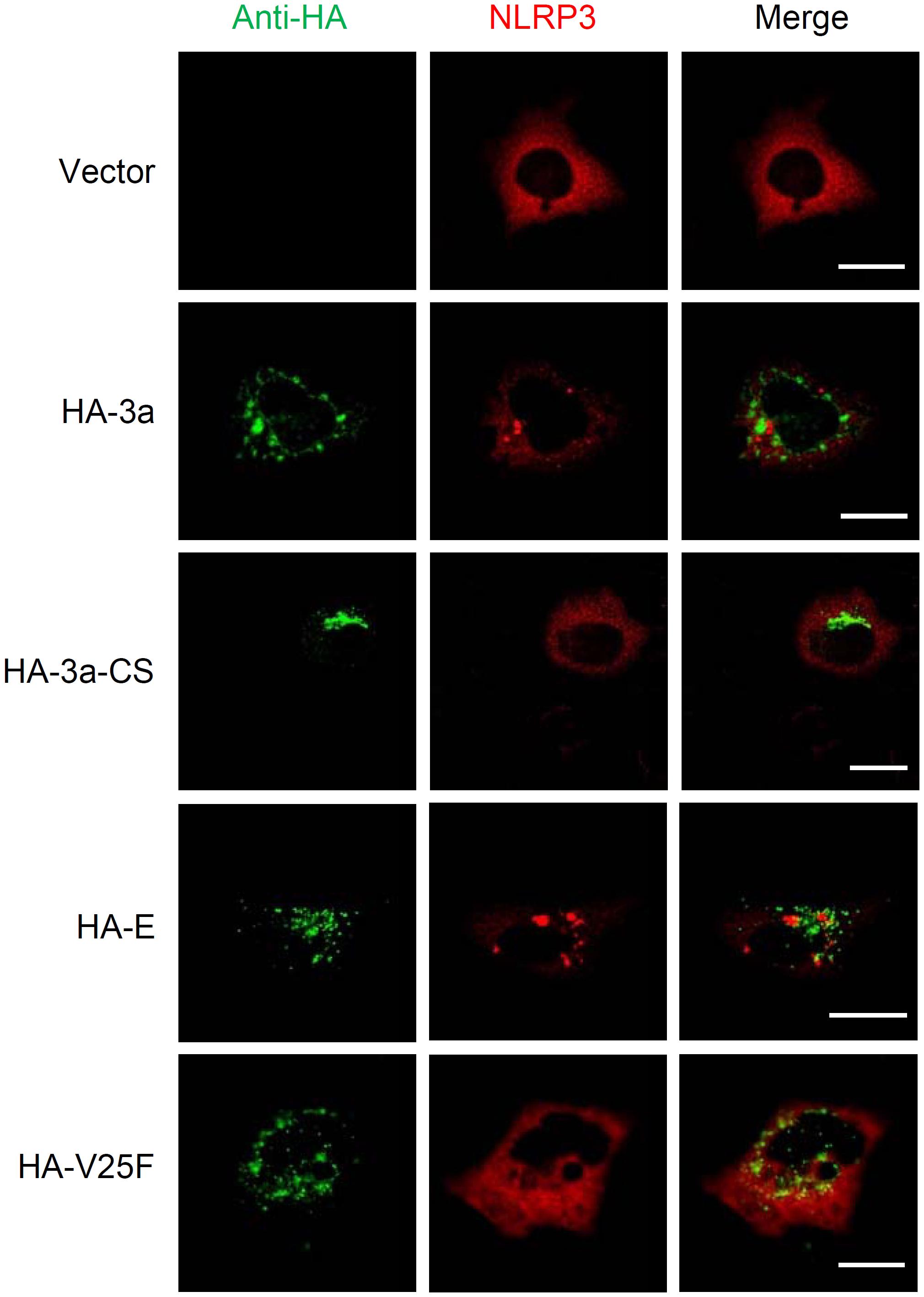
Figure 4. NLRP3 inflammasome activation by SARS-CoV 3a. HeLa cells were transfected with the expression plasmid encoding NLRP3 and that encoding HA-tagged SARS-CoV 3a, 3a-CS, E, or V25F, and by with a confocal microscope. Scale bars, 10 μm. Data are representative of at least three independent experiments.
Finally, we investigated the mechanism by which SARS-CoV 3a triggers NLRP3 inflammasome activation. A previous study showed that the 3a protein of SARS-CoV acts as a K+ channel (Lu et al., 2006). In addition, K+ efflux is a well-known activator of the NLRP3 inflammasome (Mariathasan et al., 2006; Petrilli et al., 2007). These observations prompted us to examine whether K+ efflux is required for 3a-mediated IL-1β secretion. To this end, BMMs in K+-rich medium were infected with influenza A virus or lentiviruses expressing the SARS-CoV E or 3a proteins. In agreement with a previous result (Ichinohe et al., 2010), we found that IL-1β secretion caused by influenza virus was completely blocked when the extracellular K+ concentration was increased to 130 mM (Figure 5A). The inhibitory effect of the K+-rich medium was also observed when cells were stimulated with lentiviruses expressing the SARS-CoV E or 3a proteins (Figure 5B). Since mitochondrial ROS are important for NLRP3 inflammasome activation (Nakahira et al., 2011; Zhou et al., 2011), we next stimulated BMMs with extracellular ATP or lentiviruses expressing the SARS-CoV E or 3a proteins in the presence or absence of the antioxidant, Mito-TEMPO, a scavenger that is specific for mitochondrial ROS (Jiang et al., 2009; Trnka et al., 2009). As reported previously (Nakahira et al., 2011; Ito et al., 2012), treatment of BMMs with Mito-TEMPO completely blocked IL-1β secretion in response to ATP (Figure 6A). Similarly, IL-1β release induced by the SARS-CoV E and 3a proteins was significantly inhibited by Mito-TEMPO (Figure 6B). These observations indicate that the SARS-CoV 3a protein disrupts intracellular ionic concentrations and causes mitochondrial damages, thereby activating the NLRP3 inflammasome.
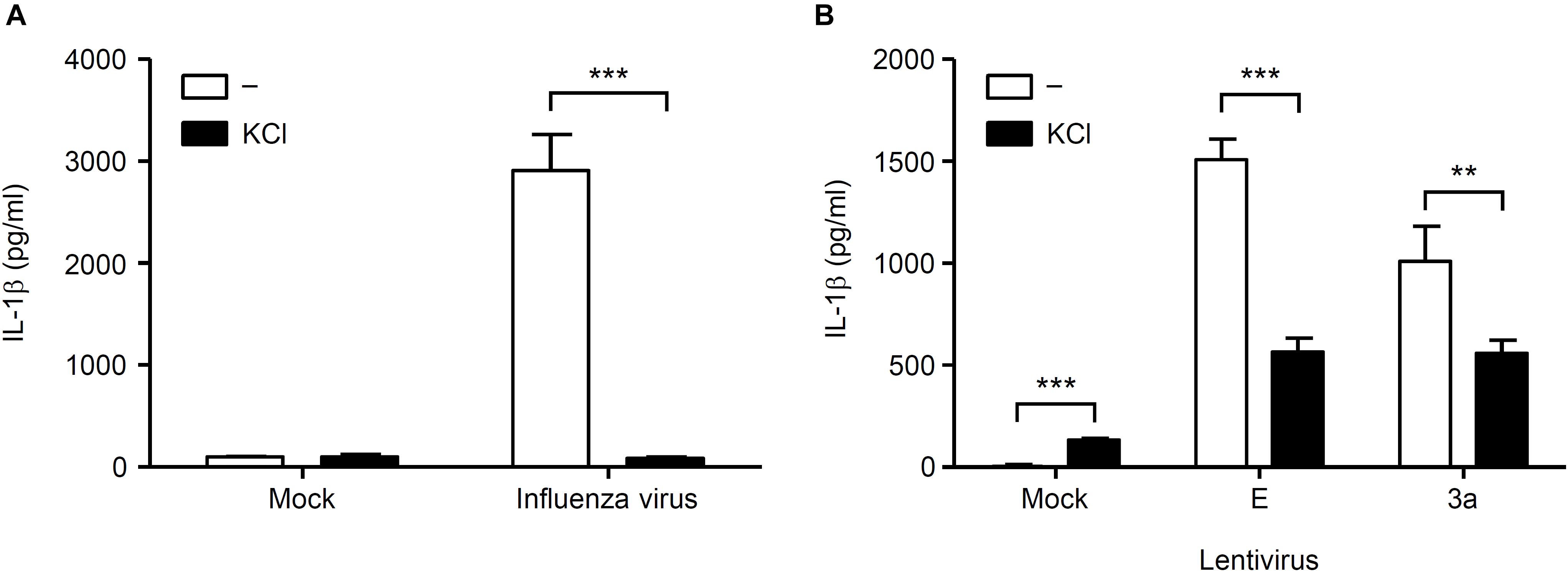
Figure 5. K+ efflux is required for activation of the NLRP3 inflammasome by SARS-CoV 3a protein. (A,B) BMMs were infected with influenza virus A/PR8 (A) or lentiviruses expressing SARS-CoV 3a or E proteins (B) and cultured in the presence or absence of KCl (130 mM). Cell-free supernatants were collected at 24 h post-infection, and analyzed for IL-1β by ELISA. Data are representative of at least three independent experiments, and indicate the mean ± SD; ∗∗P < 0.01 and ∗∗∗P < 0.001.
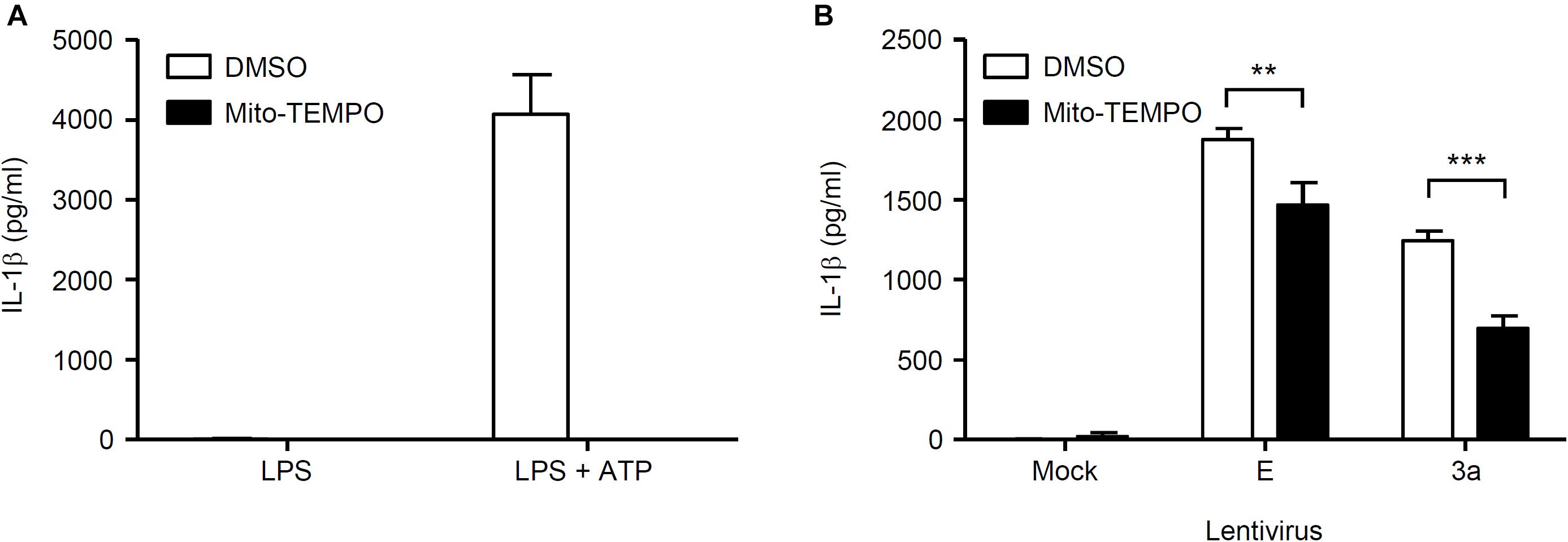
Figure 6. Mitochondrial ROS-dependent activation of the NLRP3 inflammasome by SARS-CoV 3a protein. (A,B) LPS-primed BMMs were stimulated with ATP (A) or lentiviruses expressing SARS-CoV 3a or E proteins (B) in the presence or absence of Mito-TEMPO (500 μM). Cell-free supernatants were collected at 24 h (lentiviruses) or 6 h (ATP) post-infection or stimulation, and analyzed for IL-1β by ELISA. Data are representative of at least three independent experiments, and indicate the mean ± SD; ∗∗P < 0.01 and ∗∗∗P < 0.001.
In summary, we found that the ion channel activity of SARS-CoV 3a protein is essential for activation of the NLRP3 inflammasome. In addition, both K+ efflux and mitochondrial ROS production are required for SARS-CoV 3a-mediated IL-1β secretion.
Thus far, several models have been proposed to explain NLRP3 inflammasome activation by RNA viruses. First, viral RNA or RNA cleavage products generated by RNase L activate the NLRP3 inflammasome via the DExD/H-box helicase, DHX33 (Allen et al., 2009; Mitoma et al., 2013; Chen et al., 2014; Chakrabarti et al., 2015). Second, viroporins encoded by RNA viruses activates the NLRP3 inflammasome (Ichinohe et al., 2010; Ito et al., 2012; Triantafilou et al., 2013; Nieto-Torres et al., 2015). In the case of influenza virus, the proton-selective M2 ion channel in the acidic trans-Golgi network activates the NLRP3 inflammasome (Ichinohe et al., 2010). Interestingly, an M2 mutant in which histidine was substituted with glycine at position 37 (H37G), causing loss of proton selectivity, enables transport of other cations (i.e., Na+ and K+), thereby leading to enhanced secretion of IL-1β from LPS-primed BMMs and dendritic cells when compared with the wild-type M2 protein. In addition, the 2B proteins of EMCV, poliovirus, enterovirus 71 (EV71), and human rhinovirus (a member of the Picornaviridae family) triggers NLRP3 inflammasome activation by inducing Ca2+ flux from the ER and Golgi compartments (Ito et al., 2012; Triantafilou et al., 2013). Furthermore, hepatitis C virus stimulates NLRP3 inflammasome-mediated IL-1β production though its p7 viroporin (Negash et al., 2013; Farag et al., 2017). Third, a recent study has demonstrated that the 3D protein of EV71 directly interacts with NLRP3 to facilitate the assembly of NLRP3 inflammasome complex (Wang et al., 2017).
In the case of SARS-CoV, the viroporin E forms forms Ca2+-permeable ion channels and activates the NLRP3 inflammasome (Nieto-Torres et al., 2015). In addition, another viroporin 3a was found to induce NLRP3 inflammasome activation (Yue et al., 2018). Although alanine substitution at Cys-133, which is required for dimer or tetramer formation (Lu et al., 2006), still allows activation of the NLRP3 inflammasome by interacting with caspase-1 (Yue et al., 2018), the ion channel activity-loss mutant 3a-CS (Cys-to-Ser substitution at positions Cys-127, Cys-130, and Cys-133) (Chan et al., 2009) completely abrogated IL-1β secretion from LPS-primed BMMs, suggesting that the 3a protein of SARS-CoV has the ability to induce the NLRP3 inflammasome activation by multiple mechanisms. Previous studies show that the 3a protein of SARS-CoV is localized to the plasma membrane (Minakshi and Padhan, 2014) and acts as a K+ channel (Lu et al., 2006), thereby (presumably) stimulating the K+ efflux at the plasma membrane. Indeed, we found that IL-1β secretion caused by the 3a protein was significantly inhibited when the extracellular K+ concentration increased to 130 mM. Although it remains unclear whether another viroporin 8a of SARS-CoV (Castano-Rodriguez et al., 2018) activates the NLRP3 inflammasome, these data highlights the importance of viroporins in SARS-CoV-induced NLRP3 inflammasome activation. A better understanding of the mechanism that governs the NLRP3 inflammasome will facilitate the development of more effective interventions for the treatment of infectious diseases and increase our understanding of viral pathogenesis.
I-YC and TI designed the study and wrote the manuscript. I-YC and MM performed the experiments. I-YC, MM, and TI analyzed the data. M-FC provided reagents and advice. All authors reviewed the manuscript.
This work was supported by the Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (25713018 and 15H01254), the Japanese Ministry of Health, Labour, and Welfare (H25-Shinkou-Ippan-018), the Takeda Science Foundation, the Uehara Memorial Foundation, and the SENSHIN Medical Research Foundation. I-YC was a qualified recipient of the Postdoctoral Research Abroad Program and was supported by the research grant MOST 103-2917-I-564-028 from the Ministry of Science and Technology of the Republic of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Matsuyama (National Institute of Infectious Diseases, Tokyo, Japan) for providing the total RNA extracted from SARS-CoV-infected Vero cells.
Allen, I. C., Scull, M. A., Moore, C. B., Holl, E. K., Mcelvania-Tekippe, E., Taxman, D. J., et al. (2009). The NLRP3 inflammasome mediates in vivo innate immunity to influenza a virus through recognition of viral RNA. Immunity 30, 556–565. doi: 10.1016/j.immuni.2009.02.005
Arlehamn, C. S., Petrilli, V., Gross, O., Tschopp, J., and Evans, T. J. (2010). The role of potassium in inflammasome activation by bacteria. J. Biol. Chem. 285, 10508–10518. doi: 10.1074/jbc.M109.067298
Bauernfeind, F., Ablasser, A., Bartok, E., Kim, S., Schmid-Burgk, J., Cavlar, T., et al. (2011). Inflammasomes: current understanding and open questions. Cell Mol. Life Sci. 68, 765–783. doi: 10.1007/s00018-010-0567-4
Castano-Rodriguez, C., Honrubia, J. M., Gutierrez-Alvarez, J., Dediego, M. L., Nieto-Torres, J. L., Jimenez-Guardeno, J. M., et al. (2018). Role of severe acute respiratory syndrome Coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. mBio 9:e2325-17. doi: 10.1128/mBio.02325-17
Chakrabarti, A., Banerjee, S., Franchi, L., Loo, Y. M., Gale, M. Jr., Núñez, G., et al. (2015). RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 17, 466–477. doi: 10.1016/j.chom.2015.02.010
Chan, C. M., Tsoi, H., Chan, W. M., Zhai, S., Wong, C. O., Yao, X., et al. (2009). The ion channel activity of the SARS-coronavirus 3a protein is linked to its pro-apoptotic function. Int. J. Biochem. Cell Biol. 41, 2232–2239. doi: 10.1016/j.biocel.2009.04.019
Chen, I. Y., and Ichinohe, T. (2015). Response of host inflammasomes to viral infection. Trends Microbiol. 23, 55–63. doi: 10.1016/j.tim.2014.09.007
Chen, W., Xu, Y., Li, H., Tao, W., Xiang, Y., Huang, B., et al. (2014). HCV genomic RNA activates the NLRP3 inflammasome in human myeloid cells. PLoS One 9:e84953. doi: 10.1371/journal.pone.0084953
Drosten, C., Gunther, S., Preiser, W., Van Der Werf, S., Brodt, H. R., Becker, S., et al. (2003). Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976. doi: 10.1056/NEJMoa030747
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. doi: 10.1038/nature08938
Farag, N. S., Breitinger, U., El-Azizi, M., and Breitinger, H. G. (2017). The p7 viroporin of the hepatitis C virus contributes to liver inflammation by stimulating production of Interleukin-1beta. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 712–720. doi: 10.1016/j.bbadis.2016.12.006
Fernandes-Alnemri, T., Wu, J., Yu, J. W., Datta, P., Miller, B., Jankowski, W., et al. (2007). The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604. doi: 10.1038/sj.cdd.4402194
Fouchier, R. A., Kuiken, T., Schutten, M., Van Amerongen, G., Van Doornum, G. J., Van Den Hoogen, B. G., et al. (2003). Aetiology: Koch’s postulates fulfilled for SARS virus. Nature 423:240. doi: 10.1038/423240a
Halle, A., Hornung, V., Petzold, G. C., Stewart, C. R., Monks, B. G., Reinheckel, T., et al. (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 9, 857–865. doi: 10.1038/ni.1636
He, L., Ding, Y., Zhang, Q., Che, X., He, Y., Shen, H., et al. (2006). Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 210, 288–297. doi: 10.1002/path.2067
Hornung, V., Bauernfeind, F., Halle, A., Samstad, E. O., Kono, H., Rock, K. L., et al. (2008). Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856. doi: 10.1038/ni.1631
Hornung, V., and Latz, E. (2010). Critical functions of priming and lysosomal damage for NLRP3 activation. Eur. J. Immunol. 40, 620–623. doi: 10.1002/eji.200940185
Ichinohe, T., Lee, H. K., Ogura, Y., Flavell, R., and Iwasaki, A. (2009). Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206, 79–87. doi: 10.1084/jem.20081667
Ichinohe, T., Pang, I. K., and Iwasaki, A. (2010). Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 11, 404–410. doi: 10.1038/ni.1861
Ichinohe, T., Yamazaki, T., Koshiba, T., and Yanagi, Y. (2013). Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. U.S.A. 110, 17963–17968. doi: 10.1073/pnas.1312571110
Ito, M., Yanagi, Y., and Ichinohe, T. (2012). Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome. PLoS Pathog. 8:e1002857. doi: 10.1371/journal.ppat.1002857
Jiang, J., Stoyanovsky, D. A., Belikova, N. A., Tyurina, Y. Y., Zhao, Q., Tungekar, M. A., et al. (2009). A mitochondria-targeted triphenylphosphonium-conjugated nitroxide functions as a radioprotector/mitigator. Radiat. Res. 172, 706–717. doi: 10.1667/RR1729.1
Johnson, K. E., Chikoti, L., and Chandran, B. (2013). Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 87, 5005–5018. doi: 10.1128/JVI.00082-13
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. doi: 10.1038/ni.1863
Kayagaki, N., Stowe, I. B., Lee, B. L., O’rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Ksiazek, T. G., Erdman, D., Goldsmith, C. S., Zaki, S. R., Peret, T., Emery, S., et al. (2003). A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966. doi: 10.1056/NEJMoa030781
Kuiken, T., Fouchier, R. A., Schutten, M., Rimmelzwaan, G. F., Van Amerongen, G., Van Riel, D., et al. (2003). Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362, 263–270. doi: 10.1016/S0140-6736(03)13967-0
Lu, W., Zheng, B. J., Xu, K., Schwarz, W., Du, L., Wong, C. K., et al. (2006). Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. U.S.A. 103, 12540–12545. doi: 10.1073/pnas.0605402103
Mariathasan, S., Weiss, D. S., Newton, K., Mcbride, J., O’rourke, K., Roose-Girma, M., et al. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. doi: 10.1038/nature04515
Matsuyama, S., Ujike, M., Morikawa, S., Tashiro, M., and Taguchi, F. (2005). Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. U.S.A. 102, 12543–12547. doi: 10.1073/pnas.0503203102
Medzhitov, R. (2001). Toll-like receptors and innate immunity. Nat. Rev. Immunol. 1, 135–145. doi: 10.1038/35100529
Minakshi, R., and Padhan, K. (2014). The YXXPhi motif within the severe acute respiratory syndrome coronavirus (SARS-CoV) 3a protein is crucial for its intracellular transport. Virol. J. 11:75. doi: 10.1186/1743-422X-11-75
Mitoma, H., Hanabuchi, S., Kim, T., Bao, M., Zhang, Z., Sugimoto, N., et al. (2013). The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39, 123–135. doi: 10.1016/j.immuni.2013.07.001
Moriyama, M., Chen, I. Y., Kawaguchi, A., Koshiba, T., Nagata, K., Takeyama, H., et al. (2016). The RNA- and TRIM25-binding domains of influenza virus NS1 protein are essential for suppression of NLRP3 inflammasome-mediated IL-1beta secretion. J. Virol. 90, 4105–4114. doi: 10.1128/JVI.00120-16
Munoz-Planillo, R., Kuffa, P., Martinez-Colon, G., Smith, B. L., Rajendiran, T. M., and Nunez, G. (2013). K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. doi: 10.1016/j.immuni.2013.05.016
Murakami, T., Ockinger, J., Yu, J., Byles, V., Mccoll, A., Hofer, A. M., et al. (2012). Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 109, 11282–11287. doi: 10.1073/pnas.1117765109
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230. doi: 10.1038/ni.1980
Negash, A. A., Ramos, H. J., Crochet, N., Lau, D. T., Doehle, B., Papic, N., et al. (2013). IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 9:e1003330. doi: 10.1371/journal.ppat.1003330
Nieto-Torres, J. L., Verdia-Baguena, C., Jimenez-Guardeno, J. M., Regla-Nava, J. A., Castano-Rodriguez, C., Fernandez-Delgado, R., et al. (2015). Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 485, 330–339. doi: 10.1016/j.virol.2015.08.010
Pang, I. K., Ichinohe, T., and Iwasaki, A. (2013). IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8(+) T cell responses to influenza a virus. Nat. Immunol. 14, 246–253. doi: 10.1038/ni.2514
Parthasarathy, K., Ng, L., Lin, X., Liu, D. X., Pervushin, K., Gong, X., et al. (2008). Structural flexibility of the pentameric SARS coronavirus envelope protein ion channel. Biophys. J. 95, L39–L41. doi: 10.1529/biophysj.108.133041
Peiris, J. S., Lai, S. T., Poon, L. L., Guan, Y., Yam, L. Y., Lim, W., et al. (2003). Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361, 1319–1325. doi: 10.1016/S0140-6736(03)13077-2
Perlman, S., and Dandekar, A. A. (2005). Immunopathogenesis of coronavirus infections: implications for SARS. Nat. Rev. Immunol. 5, 917–927. doi: 10.1038/nri1732
Pervushin, K., Tan, E., Parthasarathy, K., Lin, X., Jiang, F. L., Yu, D., et al. (2009). Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 5:e1000511. doi: 10.1371/journal.ppat.1000511
Petrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., and Tschopp, J. (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589. doi: 10.1038/sj.cdd.4402195
Schroder, K., Zhou, R., and Tschopp, J. (2010). The NLRP3 inflammasome: a sensor for metabolic danger? Science 327, 296–300. doi: 10.1126/science.1184003
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. doi: 10.1016/j.immuni.2012.01.009
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z., and Germain, R. N. (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. doi: 10.1016/j.cell.2013.02.054
Tan, Y. J., Lim, S. G., and Hong, W. (2005). Characterization of viral proteins encoded by the SARS-coronavirus genome. Antiviral Res. 65, 69–78. doi: 10.1016/j.antiviral.2004.10.001
Torres, J., Maheswari, U., Parthasarathy, K., Ng, L., Liu, D. X., and Gong, X. (2007). Conductance and amantadine binding of a pore formed by a lysine-flanked transmembrane domain of SARS coronavirus envelope protein. Protein Sci. 16, 2065–2071. doi: 10.1110/ps.062730007
Triantafilou, K., Kar, S., Van Kuppeveld, F. J., and Triantafilou, M. (2013). Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am. J. Respir. Cell Mol. Biol. 49, 923–934. doi: 10.1165/rcmb.2013-0032OC
Trnka, J., Blaikie, F. H., Logan, A., Smith, R. A., and Murphy, M. P. (2009). Antioxidant properties of MitoTEMPOL and its hydroxylamine. Free Radic. Res. 43, 4–12. doi: 10.1080/10715760802582183
Tschopp, J., and Schroder, K. (2010). NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 10, 210–215. doi: 10.1038/nri2725
Verdia-Baguena, C., Nieto-Torres, J. L., Alcaraz, A., Dediego, M. L., Torres, J., Aguilella, V. M., et al. (2012). Coronavirus E protein forms ion channels with functionally and structurally-involved membrane lipids. Virology 432, 485–494. doi: 10.1016/j.virol.2012.07.005
Wang, K., Xie, S., and Sun, B. (2011). Viral proteins function as ion channels. Biochim. Biophys. Acta 1808, 510–515. doi: 10.1016/j.bbamem.2010.05.006
Wang, W., Xiao, F., Wan, P., Pan, P., Zhang, Y., Liu, F., et al. (2017). EV71 3D protein binds with NLRP3 and enhances the assembly of inflammasome complex. PLoS Pathog. 13:e1006123. doi: 10.1371/journal.ppat.1006123
Wilson, L., Mckinlay, C., Gage, P., and Ewart, G. (2004). SARS coronavirus E protein forms cation-selective ion channels. Virology 330, 322–331. doi: 10.1016/j.virol.2004.09.033
Yu, C. J., Chen, Y. C., Hsiao, C. H., Kuo, T. C., Chang, S. C., Lu, C. Y., et al. (2004). Identification of a novel protein 3a from severe acute respiratory syndrome coronavirus. FEBS Lett. 565, 111–116. doi: 10.1016/j.febslet.2004.03.086
Yuan, X., Li, J., Shan, Y., Yang, Z., Zhao, Z., Chen, B., et al. (2005). Subcellular localization and membrane association of SARS-CoV 3a protein. Virus Res. 109, 191–202. doi: 10.1016/j.virusres.2005.01.001
Yue, Y., Nabar, N. R., Shi, C. S., Kamenyeva, O., Xiao, X., Hwang, I. Y., et al. (2018). SARS-Coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. 9:904. doi: 10.1038/s41419-018-0917-y
Zeng, R., Yang, R. F., Shi, M. D., Jiang, M. R., Xie, Y. H., Ruan, H. Q., et al. (2004). Characterization of the 3a protein of SARS-associated coronavirus in infected vero E6 cells and SARS patients. J. Mol. Biol. 341, 271–279. doi: 10.1016/j.jmb.2004.06.016
Zhou, R., Tardivel, A., Thorens, B., Choi, I., and Tschopp, J. (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140. doi: 10.1038/ni.1831
Keywords: SARS-CoV, viroporin, inflammasome, IL-1β, inflammation
Citation: Chen IY, Moriyama M, Chang MF and Ichinohe T (2019) Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 10:50. doi: 10.3389/fmicb.2019.00050
Received: 05 December 2018; Accepted: 14 January 2019;
Published: 29 January 2019.
Edited by:
Koichi Watashi, National Institute of Infectious Diseases (NIID), JapanReviewed by:
Heung Kyu Lee, Korea Advanced Institute of Science and Technology (KAIST), South KoreaCopyright © 2019 Chen, Moriyama, Chang and Ichinohe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takeshi Ichinohe, aWNoaW5vaGVAaW1zLnUtdG9reW8uYWMuanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.