 Yichao Wu
Yichao Wu Norazean Zaiden
Norazean Zaiden Bin Cao
Bin Cao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 12 December 2018
Sec. Evolutionary and Genomic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.03096
Comamonas is often reported to be one of the major members of microbial communities in various natural and engineered environments. Versatile catabolic capabilities of Comamonas have been studied extensively in the last decade. In contrast, little is known about the ecological roles and adaptation of Comamonas to different environments as well as the virulence of potentially pathogenic Comamonas strains. In this study, we provide genomic insights into the potential ecological roles and virulence of Comamonas by analysing the entire gene set (pangenome) and the genes present in all genomes (core genome) using 34 genomes of 11 different Comamonas species. The analyses revealed that the metabolic pathways enabling Comamonas to acquire energy from various nutrient sources are well conserved. Genes for denitrification and ammonification are abundant in Comamonas, suggesting that Comamonas plays an important role in the nitrogen biogeochemical cycle. They also encode sophisticated redox sensory systems and diverse c-di-GMP controlling systems, allowing them to be able to effectively adjust their biofilm lifestyle to changing environments. The virulence factors in Comamonas were found to be highly species-specific. The conserved strategies used by potentially pathogenic Comamonas for surface adherence, motility control, nutrient acquisition and stress tolerance were also revealed.
The Comamonas genus belongs to the Burkholderiales order in the Betaproteobacteria class. They are a group of Gram-negative, non-fermentative and rod-shaped bacteria (Willems and Vos, 2006). While most members of this genus are aerobic chemoheterotrophs, some of them, including C. nitrativorans, C. koreensis, and C. denitrificans, are facultative anaerobes capable of using nitrate or ferric iron (Fe3+) as an alternative electron acceptor (Gumaelius et al., 2001; Wu et al., 2009). The Comamonas genus has been reported as one of the major members of microbial communities in various natural and engineered environments (Supplementary Table S1; Auguet et al., 2015; Guo et al., 2015; Sotres et al., 2016). Although Comamonas spp. are considered as non-pathogenic or rare opportunistic pathogens to human, some Comamonas species have been suggested to be involved in some invasive infections, like appendicitis, bacteraemia and meningitis (Tsui et al., 2011; Opota et al., 2014; Zhou et al., 2018). Several cases of Comamonas-associated infection, in particular, infections caused by C. testosteroni, C. kerstersii, and C. aquatica have been reported in recent years (Farshad et al., 2012; Almuzara et al., 2013; Orsini et al., 2014).
The versatile catabolic capabilities of Comamonas have been studied extensively in the last decade (Boon et al., 2000; Wu et al., 2006; Liu et al., 2007). They have been shown to be capable of catabolizing a wide range of organic substrates, including amino acids, carboxylic acids, steroids and aromatic compounds. In contrast, little is known about the ecological roles and adaptation of Comamonas to different environments as well as the virulence of potentially pathogenic Comamonas strains.
The development of genome sequencing provides a great opportunity to describe Comamonas genomic traits at the genus level. The objective of this study was to elucidate the core- and pan-genomic feature of the Comamonas genus which sheds light on their potential ecological role in different habitats, including natural and engineered environments as well as medical settings. Virulence factors of the potentially pathogenic strains were also identified. Specifically, we analyzed the entire gene set (pangenome) of the Comamonas genus using all the available genome sequences. The genetic distribution was revealed and the conserved gene set (core genome) across all the genomes was identified. To investigate their ecological functionality, the key metabolic features, signaling systems, and potential virulence factors were analyzed at the genus level.
The genome sequences of thirty-four Comamonas strains were retrieved from the IMG database1 (Table 1). Among them, the complete genome sequences for C. kerstersii 8943, C. testosteroni CNB-1 and C. testosteroni TK102 are available. Gene clustering was performed by using GET_HOMOLOGUES. The clustering was based on BLASTP and OrthoMCL algorithm (inflation parameter = 1.5). The similar genes required a minimum of 75% coverage with respect to the shortest sequence in the alignment and E-value of 1e-8 (Udaondo et al., 2017). Due to the unavailability of 16S rRNA gene in some draft genomes, the housekeeping gene rpoA was chosen to construct the phylogenetic tree. The rpoA gene (about 1 kb) from each genome was aligned by Muscle MEGA using the maximum likelihood tree method with 100 bootstrap replicates (Kumar et al., 2016).
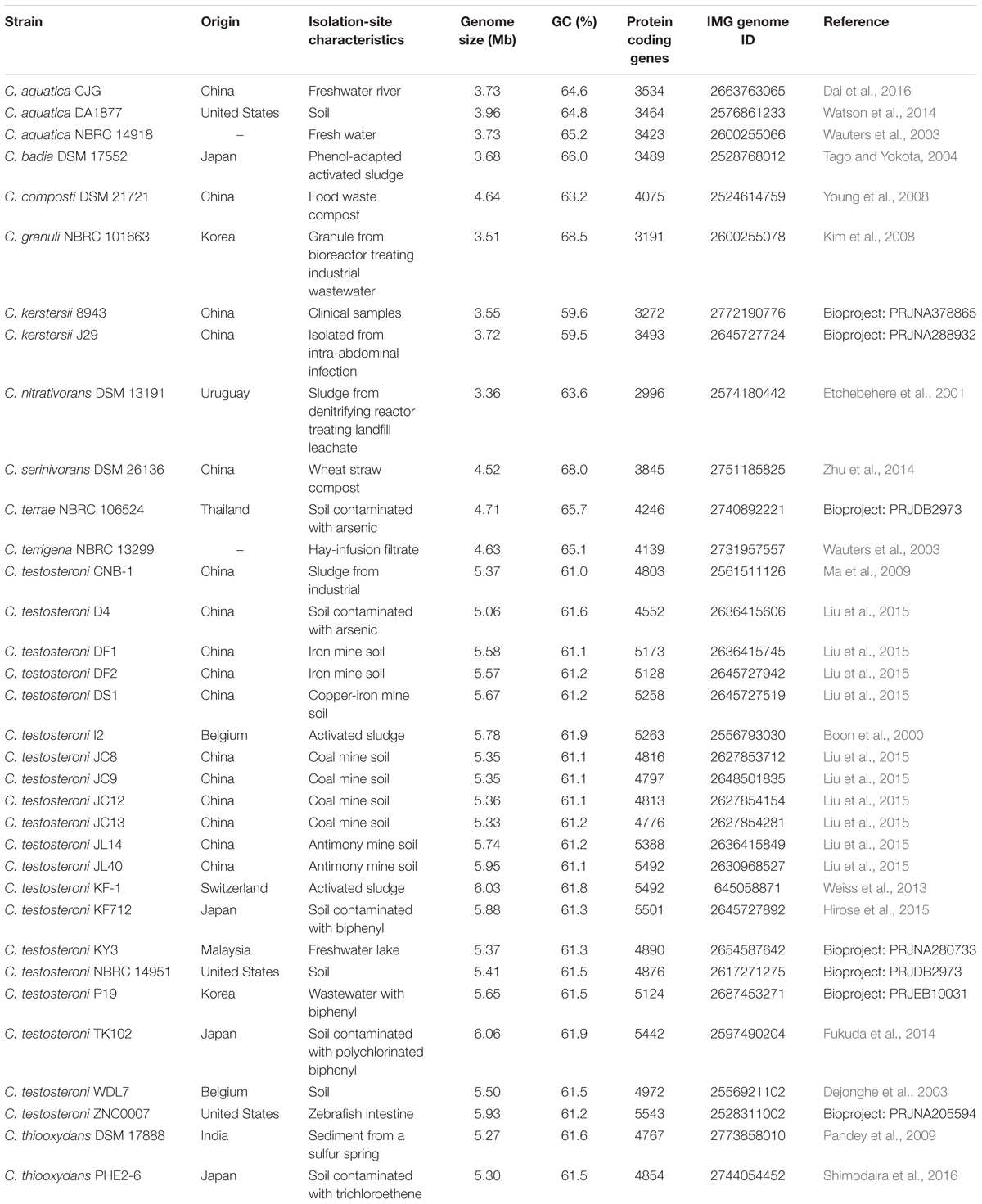
Table 1. Genomes of Comamonas used in the analysis.
To estimate the GC content at the first, second and third nucleotide position of a codon, GC1, GC2 and GC3 were calculated. GC12 represents the average value of the GC content at the first and second positions. ENC value, a measure of codon usage bias for individual gene, was quantified using the SADEG package in R (Khorsand et al., 2017). RSCU value represents the ratio of the observed frequency of the codon relative to the expected frequency under a uniform codon usage for the same amino acid.
Assignments of genes to COG, KEGG and Pfam database were predicted by WebMGA with COG, KEGG and Pfam databases2 (Wu et al., 2011). Virulence related factors were predicted using VFDB3, VRprofile4, and MP35 (Gupta et al., 2014; Chen et al., 2016; Li et al., 2017). The protein reference database of experimentally-verified virulence factors in VFDB was used. Blastp hits with minimum 40% identity with E-value less than 1e-4 were considered as positive results. The threshold for the SVM-HMM classifier in the MP3 database was -0.2. Network visualization was conducted using Cytoscape 3.6.1 (Shannon et al., 2003). Membrane transporters were identified using TransportDB 2.06 (Elbourne et al., 2017). Signal transduction genes in the core genome were analyzed based on annotation of C. testosteroni CNB-1 in MiST27 (Ulrich and Zhulin, 2009). The predicted transporter and signal transduction genes were verified by the alignment in KEGG and Pfam database.
A total of 34 well-annotated genomes from eleven different Comamonas species were chosen to represent the Comamonas genus. C. kerstersii J29 and 8943 were isolated from clinical samples and all the other strains were isolated from natural environments and anthropogenic processes, including mine soil, activated sludge and compost (Table 1). Intriguingly, the fresh water isolate, C. aquatica CJG, also showed potential pathogenic traits such as multiple antibiotic resistance genes as well as the ability to cause sera agglutination (Dai et al., 2016).
The RNA polymerase α subunit (rpoA) gene-based phylogenetic relationship of these 34 strains is shown in Figure 1. The 34 strains separated into three distinct clades. C. badia DSM 17552, C. granuli NBRC 101663 and C. serinivorans DSM 26136 were distinct from the rest. The most sequenced Comamonas, C. testosteroni revealed high similarity with C. thiooxydans. Meanwhile the clinical isolates, C. kerstersii and C. aquatic were closely related to each other. Further comparative analyses revealed that most of the genomes shared an approximate 70% similarity with other species in the genus (Figure 2). In agreement with Figure 1, C. serinivorans DSM 26136 is the most distinct strain, whose genome exhibited a mean similarity of 60.5% with the genomes of the other thirty-three strains. The genomes of the C. kerstersii clinical isolates and the potential pathogenic C. aquatica strains, exhibited a high similarity of approximately 80%, suggesting that they may share similar virulence-related genomic features. Surprisingly, C. thiooxydans strains shared an average similarity of 88.4% with the twenty C. testosteroni strains, which is comparable to the similarity between the C. testosteroni strains (87.6%). Despite a high similarity in the genome and the 16S rRNA gene sequences, C. thiooxydans was defined as a new species because of its thiosulfate oxidation capability (Pandey et al., 2009). Interestingly, all the twenty C. testosteroni strains contain sulfur oxidation gene clusters (SOX), suggesting a potential capability of C. testosteroni in oxidizing thiosulfate. Hence, considering the high similarity in genomic features and the functional SOX gene clusters, we argue that C. thiooxydans may need to be reclassified as C. testosteroni.
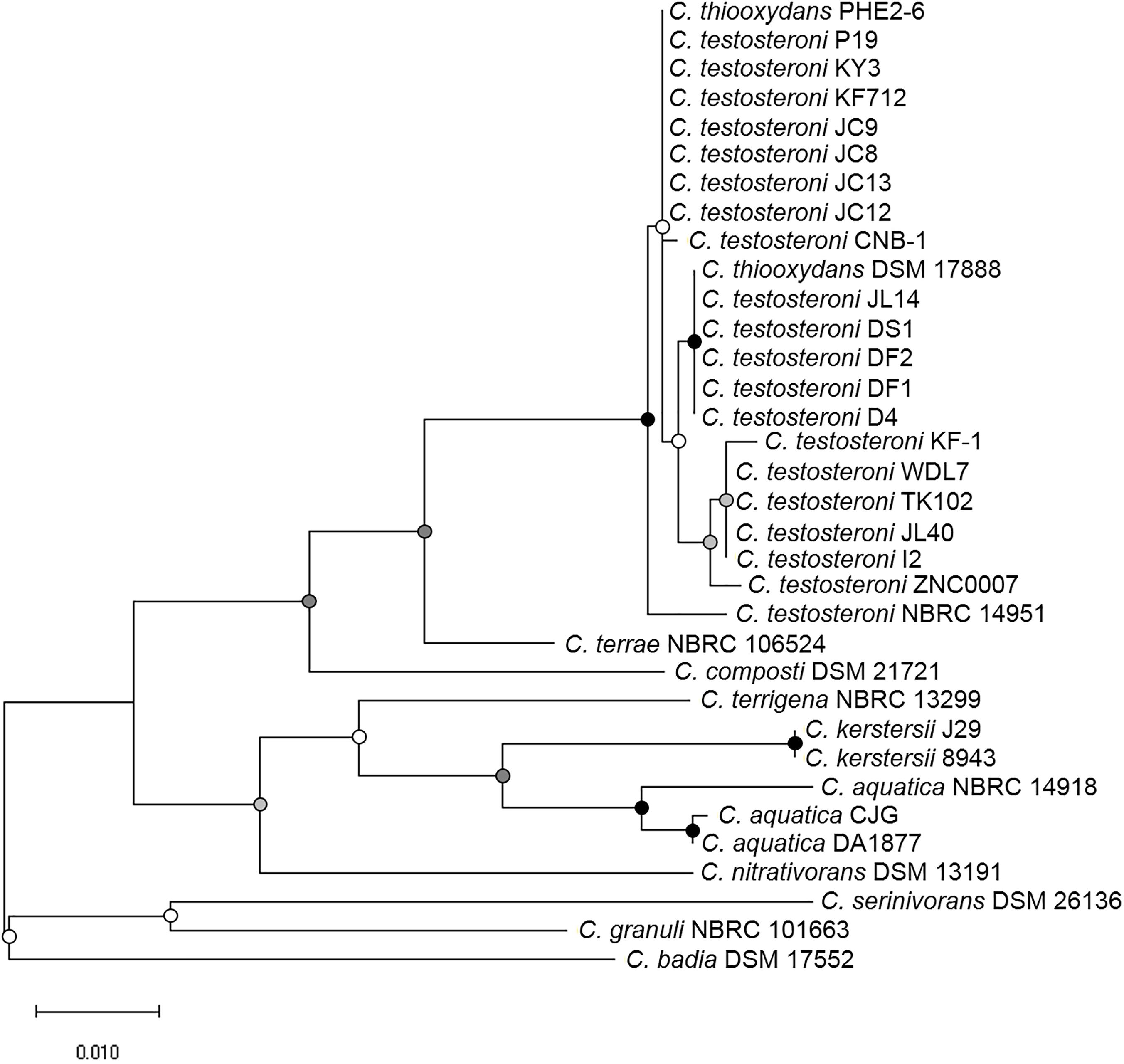
Figure 1. Bootstrapped maximum likelihood phylogenetic tree based on RNA polymerase α subunit (rpoA) gene of Comamonas species used in this study. The 34 strains were clustered into three subgroups: (1) C. badia, C. granuli and C. serinivorans; (2) C. nitrativorans, C. aquatica, C. kerstersii and C. terrigena; (3) C. composti, C. terrae, C. testosteroni, and C. thiooxydans. The scale indicates the number of substitutions per site. Bootstrap values are indicated by dots at node. Black, >80%; gray, 50–80%; white, <50%.
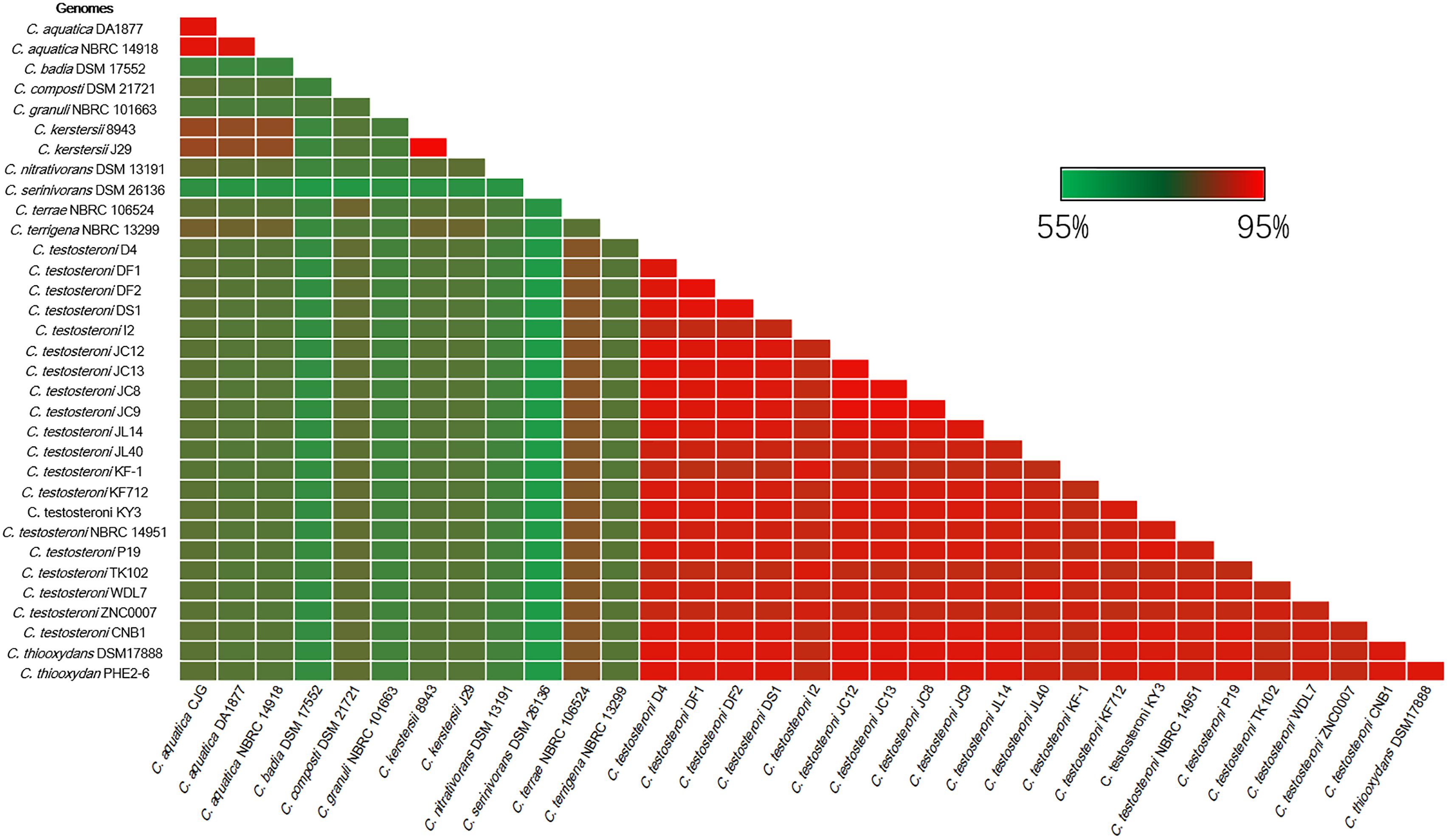
Figure 2. Heatmap indicating percentage of orthologous genes shared by two strains. The identity matrix was calculated based on BLASTP. The average similarity between different species is about 70%. Red gradient bar represents the scale of similarity percentage.
Codon usage is an important determinant in evolution. The base composition of protein-coding genes revealed that only C. kerstersii J29 has a GC content below 60% and C. granuli NBRC 101663 has the highest GC content of 68.44 ± 4.47% (Supplementary Table S2). The reduced GC content for the clinical isolate could be attributed to the conservation of replication expense in nutrient-limiting environments (Rocha and Danchin, 2002). With the lowest effective number of codons (ENC) of 38.86 ± 4.59, genes of C. granuli NBRC 101663 exhibited a strong bias in codon usage pattern. The ENC values for other environmental strains ranged from 40.20 to 49.20, while the average value for C. kerstersii strains was up to 51.46, suggesting more low-codon-biased genes in these strains. Based on the relative synonymous codon usage (RSCU) (Figure 3), Comamonas spp. prefer certain codons. GGC (Gly), CTG (Leu), CGC (Arq), AGC (Ser), and GTG (Val) were more frequently used, which had RSCU values higher than 2 across all the 34 Comamonas strains.
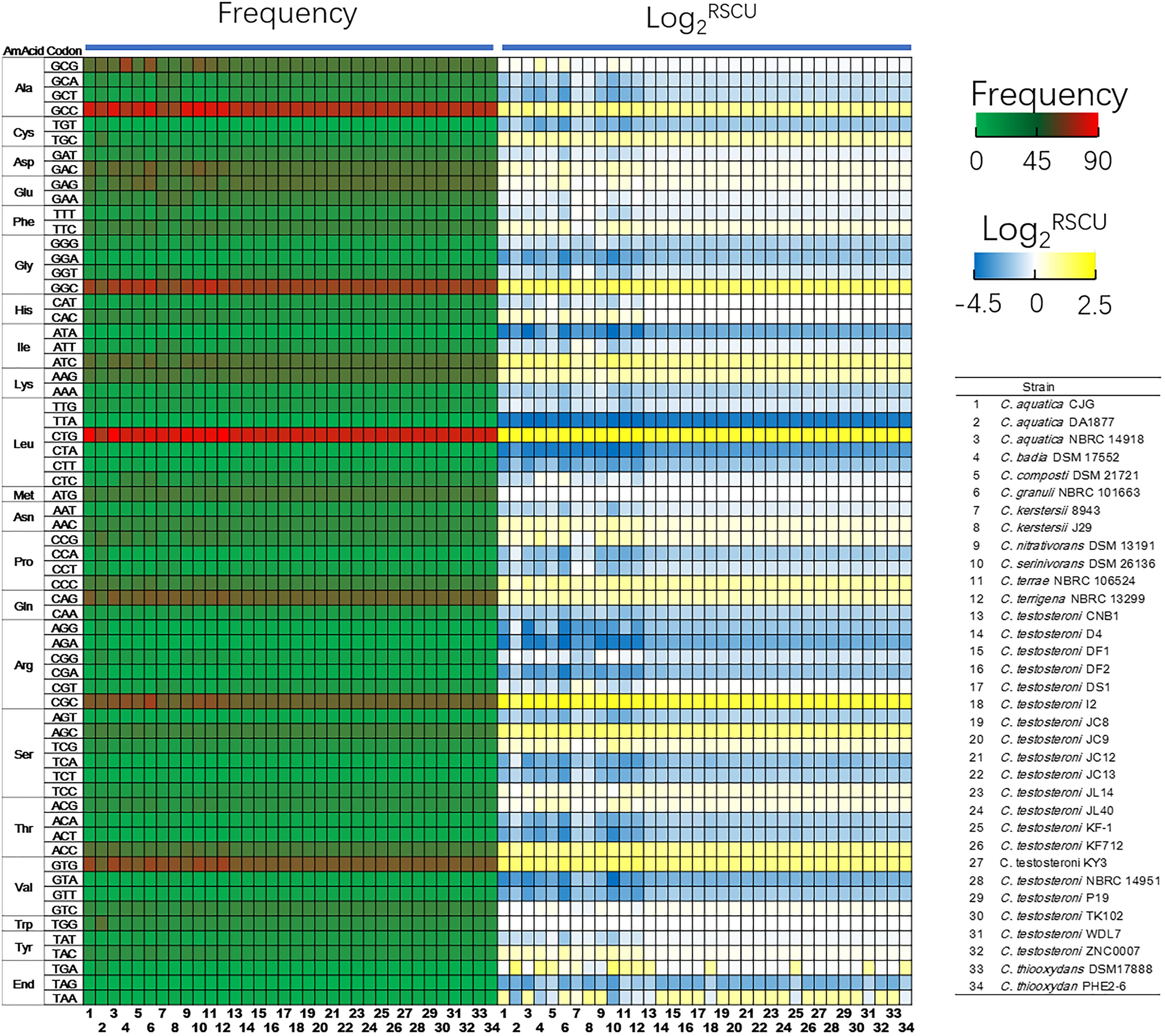
Figure 3. Frequency of codon usage (occurrences per 1000 codons) and relative synonymous codon usage (RSCU) data of Comamonas spp. RSCU is the ratio of the observed frequency of the codon to the expected frequency under equal usage of synonymous codons. The codon is more frequently used when Log2-transformed RSCU value higher than 0.
All the protein-coding genes in the 34 genomes were clustered into 22,599 gene clusters (i.e., the pangenome). Among them, 1,009 gene clusters composed the core genome. Different genes were clustered when the encoded protein sequence coverage was higher than 75% coverage, with a cut-off less than 1E-8 (Udaondo et al., 2017). At different similarity thresholds, the core genome sizes were consisted of 1,190 gene clusters for 25% coverage and 1,146 for 50% coverage (Supplementary Figure S1A). Reduction of similarity thresholds from 75 to 25% resulted in an increase of the core gene clusters percentage in the pangenome from 4.46 to 6.74%. In the pangenome, 7.54% of gene clusters were conserved in more than 32 genomes (>94% of all taxa), 26.24% of gene clusters were shared by 3–31 genomes, and 66.69% were only present in 1 or 2 genomes (<6% of all taxa) (Supplementary Figure S1B). Collectively, these results suggest the presence of a significant amount of conserved and highly specific genes in all the 34 Comamonas strains.
Based on the statistical estimation, the analysis of the increasing number of genomes converged the core genome size to about 1000 gene clusters (Figure 4A). In contrast, an average of approximately 193 strain-specific gene clusters were added into the pangenome with the inclusion of each additional genome (Figure 4B). It indicates that every Comamonas strain encodes a certain number of unique genes and the Comamonas genus possesses an open pangenome, meaning that the analyzed strains contain certain unique genes which are not shared by other strains and the gene pool size would continue to increase with the increasing number of genomes incorporated in the analysis. This pan-genomic feature enables different strains to survive in diverse environmental niches.
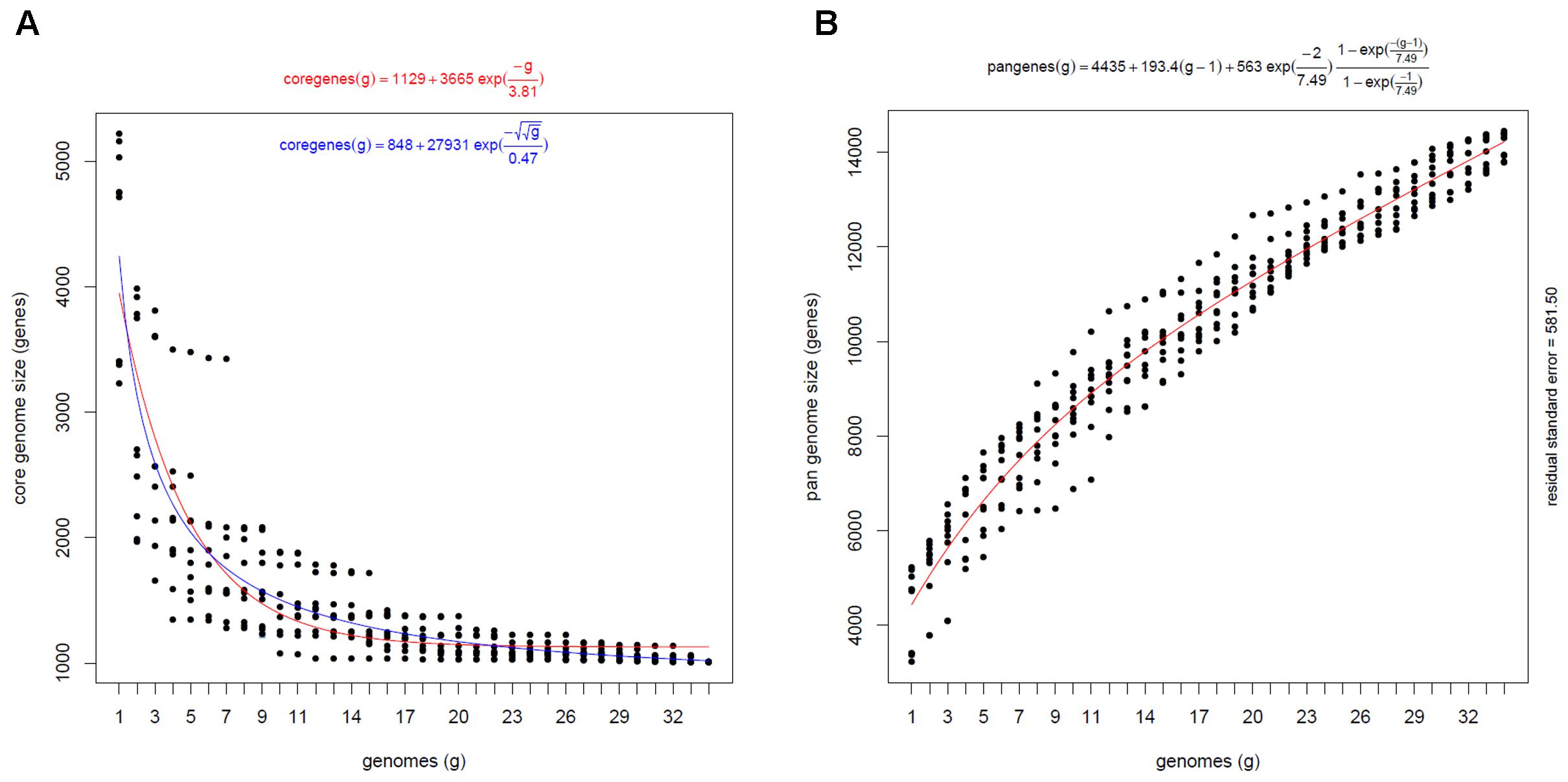
Figure 4. Statistic estimation of the core genome (A) and pangenome (B) size of Comamonas genus. Core genome size was estimated with Tellelin (Red) and Willenbrock (Blue) fit. The number of core gene clusters approximated to 1000. Pangenome size was estimated with Tettelin fit. 193 gene clusters were added to the pool with per genome inclusion. The continuous curve represents the least-squares fit of the corresponding exponential functions.
The analyses of the core genome of the Comamonas genus revealed conserved acetate and pyruvate metabolic pathways across different Comamonas species (Figure 5). In particular, the presence of the poly-β-hydroxybutyrate (PHB) cycle in the central carbon metabolism pathway suggests that Comamonas has the capability of accumulating carbon in the form of PHB granules. Based on BLAST analysis (Supplementary Figure S2), these enzymes were assigned as class I PHA synthases which preferentially utilize short-carbon-chain-length hydroxyalkanoic acid CoA thioesters as substrate (Bernd, 2003). Although many Comamonas strains have been reported to be able to degrade aromatic compounds, no core genes were assigned to the xenobiotic biodegradation function by KEGG annotation. This suggests that the catabolic pathways are not well conserved at the genus level. Our previous studies have demonstrated that a number of catabolic genes are present in the plasmids (Wu et al., 2006, 2015a).
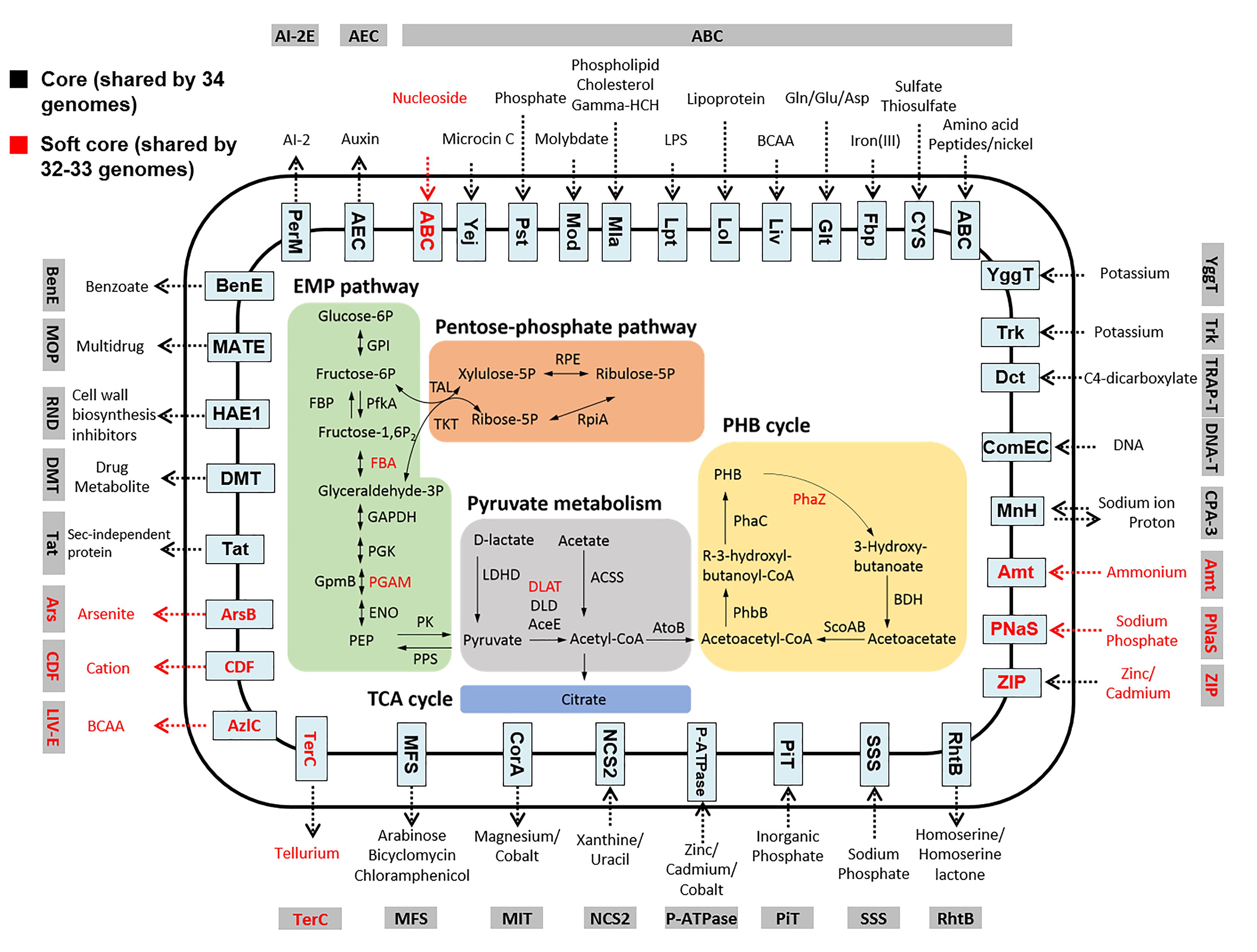
Figure 5. Overview of central carbon metabolism and transporters in Comamonas. Transporters are grouped into different families. Gene products shared by all the 34 genomes (core genome) and 32–33 genomes (soft core genome) are shown in black and red, respectively. Comamonas possess conserved pathways to metabolize phosphorylated glucose and to store the assimilated carbon as PHB. EMP pathway: Embden-Meyerhof-Parnas pathway. Genes are referred in commonly used names. GPI, glucose-6-phosphate isomerase; FBA, fructose-bisphosphate aldolase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PGK, phosphoglycerate kinase; PGAM, 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase; ENO, enolase; PK pyruvate kinase. PEP, Phosphoenolpyruvate.
Although the core genome lacks key genes for glucose phosphorylation and the phosphotransferase system, putative enzymes such as glucose-6-phosphate isomerase (GPI), 6-phosphofructokinase 1 (PfkA) and fructose-bisphosphate aldolase (FBA) encoded in the core genome are capable of metabolizing phosphorylated glucose (e.g., glucose-6P). In addition, 23 of the 34 Comamonas genomes also contain the gene encoding sugar phosphate permease. These transporters may enable Comamonas to uptake phosphorylated sugar from the environment (Chico-Calero et al., 2002). Unlike P. putida and many other environmental bacteria, the core genome of Comamonas does not code putative enzymes for the Entner Doudoroff pathway for glucose metabolism. Similar core genomic features were observed based on the COG functional classification (Supplementary Figure S3). Approximately 140 proteins from each genome were predicted for amino acid metabolism, in comparison to 31 proteins for carbohydrate metabolism. Consistent with the core genome analyses, Comamonas strains have been reported to grow well on various amino acids and organic acids, but rarely catabolize pentoses or hexoses (Willems and Vos, 2006). Consequently, when the same inoculum was introduced to microbial fuel cells, Comamonas was enriched in the acetate-fed but not in the glucose-fed system (Xing et al., 2009).
In previous studies, Comamonas has often been reported to be enriched in nitrate-reducing conditions (Patureau et al., 2000; Guo et al., 2015; Zhong et al., 2015), suggesting a potential role of Comamonas in nitrate reduction. Genomic analyses revealed highly diverse nitrogen metabolism pathways in Comamonas (Figure 6). C. terrae NBRC 106524 is the only strain which does not contain any genes involved in nitrate or nitrite metabolism. The genomes of C. nitrativorans DSM 13191 and C. granuli NBRC 101663 contain all the genes required for complete denitrification. C. aquatica and C. terrigena strains are capable of reducing nitrate to nitrous oxide. Except C. terrae, C. badia, and C. serinivorans, each of the rest thirty-one genomes harbors at least one copy of the nitrate reductase gene. Twenty-nine genomes including those of C. testosteroni, C. thiooxydans, C. composti, C. aquatica, C. terrigena, and C. kerstersii contain an analogous gene cluster for ammonification which comprises one assimilatory nitrate reductase gene (nas) and at least two nitrite reductase genes (nir). Twenty-five among them also contain one dissimilatory nitrate reductase gene. Therefore, Comamonas spp. are capable of assimilatory and dissimilatory reduction of nitrate. The resultant nitrite can be further reduced to ammonia by nitrite reductase (Nir).

Figure 6. Comparison of nitrogen metabolism in Comamonas spp. Most strains are capable of nitrate reduction via ammonification or denitrification. nap, periplasmic nitrate reductase; nir, nitrite reductase; nas, assimilatory nitrate reductase; nar, nitrate reductase; nirA, ferredoxin-nitrite reductase; nor, nitric oxide reductase.
Due to the high abundance in denitrifying microbial communities and denitrification capability of specific isolates, Comamonas spp. were considered to be potential denitrifiers (Lemmer et al., 1997; Etchebehere et al., 2001; Gumaelius et al., 2001). Our results confirmed the genomic potential of the Comamonas genus in nitrate reduction. Previous studies found that the increased ratio of electron donors to electron acceptors can stimulate the ammonification route relative to denitrification (King and Nedwell, 1987). When the competition for nitrate becomes more severe, the decreased nitrate levels would favor ammonifying bacteria over denitrifying bacteria (Dong et al., 2009). Moreover, the ATP synthesis in denitrification has been shown to be lower than in ammonification (Strohm et al., 2007). Therefore, Comamonas strains may gain a competitive advantage from ammonification over other denitrifiers in nitrate-reducing conditions. Generally, Comamonas are considered aerobes, although the strains with the Nar-type nitrate reductases are facultative anaerobes capable of respiring under anoxic conditions (Wu et al., 2015b).
The core genome analysis of the Comamonas genus revealed 39 gene clusters with potential roles in microbial signal transduction, including 25 for the one-component systems, 12 for the two-component systems, and 2 for extracytoplasmic function (ECF). Per-Arnt-Sim (PAS) is the most abundant sensor domain in two-component systems while LysR substrate-binding domain is the most abundant in one-component systems. With PAS domains, Comamonas strains are capable of mediating the responses to changes in redox state and cellular oxygen level (Krell et al., 2010). Other than C. granuli and C. nitrativorans, other Comamonas strains harbor putative genes for three types of oxygen-dependent terminal oxidases, among which the cbb3-type and bd-type are abundant. As the cbb3-type and bd-type oxidases have a high oxygen affinity (Alvarez-Ortega and Harwood, 2007), this combination facilitates growth of Comamonas under conditions with varying oxygen levels. This could be one reason why Comamonas was often found to be highly abundant in bioprocesses with intermittent aeration (e.g., EBPR reactors) (Liu et al., 2005; Zhang et al., 2005; Zengin et al., 2011).
The GGDEF domain proteins are essential for c-di-GMP synthesis, while EAL and HD-GYP domain proteins break down this signaling molecule (Hengge, 2009). Based on Pfam annotation, there is no c-di-GMP controlling gene conserved across the 34 genomes. Except for C. serinivorans, many genes (21–60) which control c-di-GMP are present (Supplementary Figure S4). The percentage of c-di-GMP controlling genes range from 0.5 to 1.7% of total protein-coding genes. C. composti which shared a similar environmental source with C. serinivorans encoded the lowest number of these catalytic proteins. Meanwhile, the ratio of c-di-GMP controlling genes in all the other thirty-two genomes are above 0.75%, which is higher than 0.6% in other environmental bacteria such as E. coli K12 and P. putida KT2440 (Wu et al., 2015a). Among these putative c-di-GMP controlling proteins, about 45% of them have binding capacity to a sensory domain that enables effective c-di-GMP level regulation. Although many genes involved in c-di-GMP signaling were identified among the thirty-three strains, only one conserved gene encoding HD-GYP domain-containing protein was found. It suggests that the c-di-GMP controlling genes are diversified across different species in Comamonas genus.
Quorum sensing (QS) is another central signaling system for biofilm regulation in which bacterial cells communicate through secreted signaling molecules. Although no QS signal synthase genes were found in the core genome, two genes coding for anthranilate synthase (phnA and phnB) were identified. Anthranilate synthase has been reported to be exclusively for synthesizing the precursor of QS signals in Pseudomonas (Farrow and Pesci, 2007). The presence of anthranilate synthase genes in the core genome of Comamonas suggests that Comamonas may produce QS signals through similar pathways.
Since 1987, Comamonas had been found to be associated with certain human infections (Barbaro et al., 1987). To analyze the pathogenic potential of Comamonas, virulence factors in the genomes were identified by BLAST search against the Virulence Factors Database (VFDB)8. On average, each genome of Comamonas contains about 200 virulence factors while no conserved virulence factor was found in the core genome. Principal coordinates analysis (PCoA) of virulence factors in each genome revealed two distinct clusters (Figure 7A). Similar clustering patterns were also observed for the virulence factors predicted in MP3 and VRprofile (Supplementary Figure S5). One consists of the clinically isolated C. kerstersii, potentially pathogenic C. aquatica and environmental C. terrigena, while the other consists of all the C. testosteroni and C. thiooxydans strains. These species, especially C. kerstersii and C. testosteroni, are most frequently isolated from clinical samples (Opota et al., 2014). C. terrigena was also reported to be associated with bacteraemia which reveals a close phylogenetic relationship with C. kerstersii and C. aquatica (Figure 1; Sonnenwirth, 1970). Although the genomic information of clinically isolated C. testosteroni is not available, the close similarity between strains from various origins indicates the species share certain virulence factors. To explore the conserved potential virulence mechanisms for Comamonas, the virulence factors of strains in the two clusters which were identified by VFDB were further analyzed.
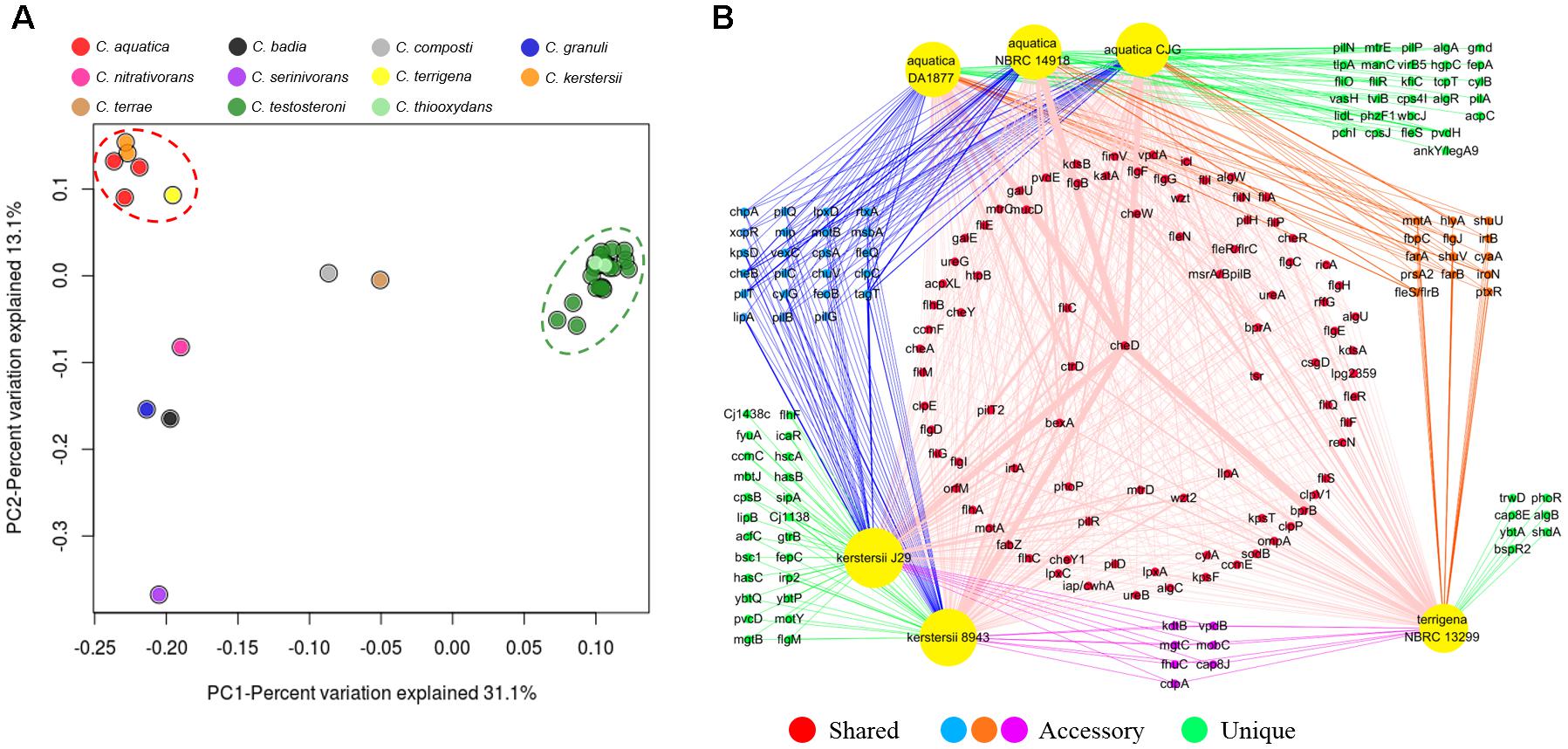
Figure 7. PCoA plot illustrates the differences between virulence gene orthologs in Comamonas spp. (A). The network analysis reveals the shared (present in three species), accessory (present in two species) and unique (present in one species) virulence factors in C. terrigena, C. aquatica, and C. kerstersii (B). The width of each edge is proportional to the number factors in the genome.
There are 87 virulence factors shared by C. kerstersii, C. aquatica, and C. terrigena. 44 factors are conserved by two of them and 60 factors are unique which were only predicted in one species (Figure 7B). The shared offensive virulence factors comprise a number of genes responsible for bacterial motility and adherence. The flagellar and pilus assembly pathways and chemotaxis signaling system are well-conserved in these three species, in which multiple genes encoding methyl-accepting chemotaxis proteins (Tsr) were identified (Supplementary Table S3). The shared biosynthesis genes of capsular polysaccharide, lipopolysaccharide, alginate and adhesins, like IlpA, Hsp60, P60, and P5, suggest the adherence and biofilm formation capability of these species. Although no shared toxin protein was predicted, hlyA or rtxA which encode hemolysin are present in five of these six strains (83.3%). The shared defensive and nonspecific (i.e., neither offensive nor defensive) virulence factors consist of various stress proteins and metabolic enzymes. ClpEP (phagosome), SodB (SOD), RecN (ROS, neutrophils), Urease (acidity), and MsrAB (ROS) are able to protect cellular functions against stress. The ability to source for nutrients is key for survival and multiplication in the host. The putative isocitrate lyase (Icl) enables them to use fatty acids as the carbon and energy source.
For the other cluster composed of C. testosteroni and C. thiooxydans, 108 factors are shared by these 22 genomes (Supplementary Figure S6). Fifty-seven factors are unique and shared by only one or two strains (Supplementary Table S4). Similar to the previous cluster, C. testosteroni and C. thiooxydans also encode a number of genes for motility, polysaccharide and adhesin biosynthesis. The pilus assembly and polysaccharide biosynthesis genes are more abundant in this cluster. Each genome also contains an average of 5 bspR2 genes which encode putative quorum-sensing regulatory proteins. It indicates that C. testosteroni and C. thiooxydans strains may have better biofilm-forming capabilities. Meanwhile, these species may be less cytotoxic than C. kerstersii and C. aquatica, as only 14 out of 22 genomes (63.6%) contain the hemolysin gene. With respect to the defensive and nonspecific virulence factors, more genes shared in this cluster encode putative metal uptake proteins, especially for various siderophores.
In summary, the virulence factors of the Comamonas genus are very diverse. Meanwhile, clinically isolated and potentially pathogenic strains shared specific virulence factors, including polysaccharide biosynthesis for adherence and anti-phagocytosis, motility system and metabolic enzymes for adaptation in vivo. Although no related pathogenic effect on healthy people caused by Comamonas strains has been reported, all the clinically isolated Comamonas strains and a number of environmental Comamonas contain hemolysin genes. Therefore, the core- and pan-genomic analyses revealed that potential virulence may be species-specific as certain virulence factors are conserved in pathogenic-like strains.
Comamonas is a group of ubiquitous bacteria present in various natural and engineered environments. Some of them are also involved in a number of clinical cases. It has been suggested that Comamonas strains may share specific genomic features at the genus level and play certain ecological roles to different habitats. The pan-genomic analysis shows the diverse genomic features that contribute to the wide adaptation of the genus to various environments. The core genome reveals central metabolic pathways that enable Comamonas to utilize various nutrient sources and store excess resources as PHB. The conserved dissimilatory and assimilatory nitrate reductases in Comamonas explain their presence in nitrate reducing environments and suggest an important role in the nitrogen biogeochemical cycle. They also encode sophisticated redox sensory systems and effective c-di-GMP controlling systems, allowing them to adjust their biofilm lifestyle under dynamic conditions. The virulence factors in Comamonas are found to be highly species-specific. The conserved mechanisms for potentially pathogenic Comamonas are related to surface adherence, motility control, nutrient acquisition and stress tolerance.
YW and BC conceived this study and wrote the manuscript. YW and NZ performed the analyses. All authors read and approved the final manuscript.
This research was supported by the Fundamental Research Funds for the Central Universities (Program No. 52902-0900201674), the National Natural Science Foundation of China (41807024), and the National Research Foundation and MOE Singapore under its Research Centre of Excellence Programme, Singapore Centre for Environmental Life Sciences Engineering (SCELSE) (M4330005.C70), Nanyang Technological University, Singapore.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Daphne Ng for her assistance in revising the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.03096/full#supplementary-material
Almuzara, M. N., Cittadini, R., Ocampo, C. V., Bakai, R., Traglia, G., Ramirez, M. S., et al. (2013). Intra-abdominal infections due to Comamonas kerstersii. J. Clin. Microbiol. 51, 1998–2000. doi: 10.1128/JCM.00659-13
Alvarez-Ortega, C., and Harwood, C. S. (2007). Responses of Pseudomonas aeruginosa to low oxygen indicate that growth in the cystic fibrosis lung is by aerobic respiration. Mol. Microbiol. 65, 153–165. doi: 10.1111/j.1365-2958.2007.05772.x
Auguet, O., Pijuan, M., Guasch-Balcells, H., Borrego, C. M., and Gutierrez, O. (2015). Implications of downstream nitrate dosage in anaerobic sewers to control sulfide and methane emissions. Water Res. 68, 522–532. doi: 10.1016/j.watres.2014.09.034
Barbaro, D. J., Mackowiak, P. A., Barth, S. S., and Southern, P. M. (1987). Pseudomonas testosteroni infections: eighteen recent cases and a review of the literature. Rev. Infect. Dis. 9, 124–129. doi: 10.1093/clinids/9.1.124
Bernd, H. (2003). Polyester synthases: natural catalysts for plastics. Biochem. J. 376, 15–33. doi: 10.1042/bj20031254
Boon, N., Goris, J., De Vos, P., Verstraete, W., and Top, E. M. (2000). Bioaugmentation of activated sludge by an indigenous 3-chloroaniline-degrading Comamonas testosteroni strain, I2gfp. Appl. Environ. Microbiol. 66, 2906–2913. doi: 10.1128/AEM.66.7.2906-2913.2000
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Chico-Calero, I., Suárez, M., González-Zorn, B., Scortti, M., Slaghuis, J., Goebel, W., et al. (2002). Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc. Natl. Acad. Sci. 99, 431–436. doi: 10.1073/pnas.012363899
Dai, W., Zhu, Y., Wang, X., Sakenova, N., Yang, Z., Wang, H., et al. (2016). Draft genome sequence of the bacterium Comamonas aquatica CJG. Genome Announc. 4:e001186-16. doi: 10.1128/genomeA.01186-16
Dejonghe, W., Berteloot, E., Goris, J., Boon, N., Crul, K., Maertens, S., et al. (2003). Synergistic degradation of linuron by a bacterial consortium and isolation of a single linuron-degrading Variovorax strain. Appl. Environ. Microbiol. 69, 1532–1541. doi: 10.1128/AEM.69.3.1532-1541.2003
Dong, L. F., Smith, C. J., Papaspyrou, S., Stott, A., Osborn, A. M., and Nedwell, D. B. (2009). Changes in benthic denitrification, nitrate ammonification, and anammox process rates and nitrate and nitrite reductase gene abundances along an estuarine nutrient gradient (the colne estuary, United Kingdom). Appl. Environ. Microbiol. 75, 3171–3179. doi: 10.1128/AEM.02511-08
Elbourne, L. D., Tetu, S. G., Hassan, K. A., and Paulsen, I. T. (2017). TransportDB 2.0: a database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 45, D320–D324. doi: 10.1093/nar/gkw1068
Etchebehere, C., Errazquin, M. I., Dabert, P., Moletta, R., and Muxí, L. (2001). Comamonas nitrativorans sp. nov., a novel denitrifier isolated from a denitrifying reactor treating landfill leachate. Int. J. Syst. Evol. Microbiol. 51, 977–983. doi: 10.1099/00207713-51-3-977
Farrow, J. M., and Pesci, E. C. (2007). Two distinct pathways supply anthranilate as a precursor of the Pseudomonas quinolone signal. J. Bacteriol. 189, 3425–3433. doi: 10.1128/JB.00209-07
Farshad, S., Norouzi, F., Aminshahidi, M., Heidari, B., and Alborzi, A. (2012). Two cases of bacteremia due to an unusual pathogen, Comamonas testosteroni in Iran and a review literature. J. Infect. Dev. Ctries. 6, 521–525.
Fukuda, K., Hosoyama, A., Tsuchikane, K., Ohji, S., Yamazoe, A., Fujita, N., et al. (2014). Complete genome sequence of polychlorinated biphenyl degrader Comamonas testosteroni TK102 (NBRC 109938). Genome Announc. 2:e00865-14. doi: 10.1128/genomeA.00865-14
Gumaelius, L., Magnusson, G., Pettersson, B., and Dalhammar, G. (2001). Comamonas denitrificans sp. nov., an efficient denitrifying bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 51, 999–1006. doi: 10.1099/00207713-51-3-999
Guo, Y., Gong, H., and Guo, X. (2015). Rhizosphere bacterial community of Typha angustifolia L. and water quality in a river wetland supplied with reclaimed water. Appl. Microbiol. Biotechnol. 99, 2883–2893. doi: 10.1007/s00253-014-6182-9
Gupta, A., Kapil, R., Dhakan, D. B., and Sharma, V. K. (2014). MP3: a software tool for the prediction of pathogenic proteins in genomic and metagenomic data. PLoS One 9:e93907. doi: 10.1371/journal.pone.0093907
Hengge, R. (2009). Principles of c-di-GMP signalling in bacteria. Nat. Rev. Microbiol. 7:263. doi: 10.1038/nrmicro2109
Hirose, J., Yamazoe, A., Hosoyama, A., Kimura, N., Suenaga, H., Watanabe, T., et al. (2015). Draft genome sequence of the polychlorinated biphenyl-degrading bacterium Comamonas testosteroni KF712 (NBRC 110673). Genome Announc. 3:e01214-15. doi: 10.1128/genomeA.01214-15
Khorsand, B., Sadeghnezhad, E., Zahiri, J., Sharifi, M., and Zare-mayvan, H. (2017). Stability Analysis in Differentially Expressed Genes. R Package, Version 1.0.0.
Kim, K., Ten, L. N., Liu, Q., Im, W., and Lee, S. (2008). Comamonas granuli sp. nov., isolated from granules used in a wastewater treatment plant. J. Microbiol. 46, 390–395. doi: 10.1007/s12275-008-0019-0
King, D., and Nedwell, D. (1987). The adaptation of nitrate-reducing bacterial communities in estuarine sediments in response to overlying nitrate load. FEMS Microbiol. Ecol. 3, 15–20. doi: 10.1111/j.1574-6968.1987.tb02333.x
Krell, T., Lacal, J., Busch, A., Silva-Jiménez, H., Guazzaroni, M.-E., and Ramos, J. L. (2010). Bacterial sensor kinases: diversity in the recognition of environmental signals. Annu. Rev. Microbiol. 64, 539–559. doi: 10.1146/annurev.micro.112408.134054
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lemmer, H., Zaglauer, A., Neef, A., Meier, H., and Amann, R. (1997). Denitrification in a methanol-fed fixed-bed reactor. Part 2: composition and ecology of the bacterial community in the biofilms. Water Res. 31, 1903–1908. doi: 10.1016/S0043-1354(97)00027-4
Li, J., Tai, C., Deng, Z., Zhong, W., He, Y., and Ou, H.-Y. (2017). VRprofile: gene-cluster-detection-based profiling of virulence and antibiotic resistance traits encoded within genome sequences of pathogenic bacteria. Brief. Bioinform. 19, 566–574. doi: 10.1093/bib/bbw141
Liu, L., Jiang, C. Y., Liu, X. Y., Wu, J. F., Han, J. G., and Liu, S. J. (2007). Plant-microbe association for rhizoremediation of chloronitroaromatic pollutants with Comamonas sp. strain CNB-1. Environ. Microbiol. 9, 465–473.
Liu, L., Zhu, W., Cao, Z., Xu, B., Wang, G., and Luo, M. (2015). High correlation between genotypes and phenotypes of environmental bacteria Comamonas testosteroni strains. BMC Genom. 16:110. doi: 10.1186/s12864-015-1314-x
Liu, Y., Zhang, T., and Fang, H. H. (2005). Microbial community analysis and performance of a phosphate-removing activated sludge. Bioresour. Technol. 96, 1205–1214. doi: 10.1016/j.biortech.2004.11.003
Ma, Y., Zhang, Y., Zhang, J., Chen, D., Zhu, Y., Zheng, H., et al. (2009). The complete genome of Comamonas testosteroni reveals its genetic adaptations to changing environments. Appl. Environ. Microbiol. 75, 6812–6819. doi: 10.1128/AEM.00933-09
Opota, O., Ney, B., Zanetti, G., Jaton, K., Greub, G., and Prod’hom, G. (2014). Bacteremia caused by Comamonas kerstersii in a patient with diverticulosis. J. Clin. Microbiol. 52, 1009–1012. doi: 10.1128/JCM.02942-13
Orsini, J., Tam, E., Hauser, N., and Rajayer, S. (2014). Polymicrobial bacteremia involving Comamonas testosteroni. Case Rep. Med. 2014:578127. doi: 10.1155/2014/578127
Pandey, S. K., Narayan, K. D., Bandyopadhyay, S., Nayak, K. C., and Das, S. K. (2009). Thiosulfate oxidation by Comamonas sp. S23 isolated from a sulfur spring. Curr. Microbiol. 58, 516–521. doi: 10.1007/s00284-009-9357-3
Patureau, D., Zumstein, E., Delgenes, J., and Moletta, R. (2000). Aerobic denitrifiers isolated from diverse natural and managed ecosystems. Microb. Ecol. 39, 145–152. doi: 10.1007/s002480000009
Rocha, E. P., and Danchin, A. (2002). Base composition bias might result from competition for metabolic resources. Trends Genet. 18, 291–294. doi: 10.1016/S0168-9525(02)02690-2
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shimodaira, J., Yonezuka, K., Tabata, M., Nagase, S., Kasai, D., Hosoyama, A., et al. (2016). Draft genome sequence of Comamonas thiooxydans strain PHE2-6 (NBRC 110656), a chlorinated-ethene-degrading bacterium. Genome Announc. 4:e00487-16. doi: 10.1128/genomeA.00487-16
Sonnenwirth, A. C. (1970). Bacteremia with and without meningitis due to yersinia enterocolitica, edwards1ella tarda, comamonas terrigena, and Pseudomonas maltophilia. Ann. N. Y. Acad. Sci. 174, 488–502. doi: 10.1111/j.1749-6632.1970.tb45575.x
Sotres, A., Cerrillo, M., Viñas, M., and Bonmatí, A. (2016). Nitrogen removal in a two-chambered microbial fuel cell: establishment of a nitrifying–denitrifying microbial community on an intermittent aerated cathode. Chem. Eng. J. 284, 905–916. doi: 10.1016/j.cej.2015.08.100
Strohm, T. O., Griffin, B., Zumft, W. G., and Schink, B. (2007). Growth yields in bacterial denitrification and nitrate ammonification. Appl. Environ. Microbiol. 73, 1420–1424. doi: 10.1128/AEM.02508-06
Tago, Y., and Yokota, A. (2004). Comamonas badia sp. nov., a floc-forming bacterium isolated from activated sludge. J. Gen. Appl. Microbiol. 50, 243–248. doi: 10.2323/jgam.50.243
Tsui, T., Tsao, S., Liu, K., Chen, T., Wang, Y., Teng, Y., et al. (2011). Comamonas testosteroni infection in Taiwan: reported two cases and literature review. J. Microbiol. Immunol. Infect. 44, 67–71. doi: 10.1016/j.jmii.2011.01.013
Udaondo, Z., Duque, E., and Ramos, J.-L. (2017). The pangenome of the genus Clostridium. Environ. Microbiol. 19, 2588–2603. doi: 10.1111/1462-2920.13732
Ulrich, L. E., and Zhulin, I. B. (2009). The MiST2 database: a comprehensive genomics resource on microbial signal transduction. Nucleic Acids Res. 38, D401–D407. doi: 10.1093/nar/gkp940
Watson, E., MacNeil, L. T., Ritter, A. D., Yilmaz, L. S., Rosebrock, A. P., Caudy, A. A., et al. (2014). Interspecies systems biology uncovers metabolites affecting C. elegans gene expression and life history traits. Cell 156, 759–770. doi: 10.1016/j.cell.2014.01.047
Wauters, G., De Baere, T., Willems, A., Falsen, E., and Vaneechoutte, M. (2003). Description of Comamonas aquatica comb. nov. and Comamonas kerstersii sp. nov. for two subgroups of Comamonas terrigena and emended description of Comamonas terrigena. Int. J. Syst. Evol. Microbiol. 53, 859–862. doi: 10.1099/ijs.0.02450-0
Weiss, M., Kesberg, A. I., LaButti, K. M., Pitluck, S., Bruce, D., Hauser, L., et al. (2013). Permanent draft genome sequence of Comamonas testosteroni KF-1. Stand. Genomic Sci. 8, 239–254. doi: 10.4056/sigs.3847890
Willems, A., and Vos, P. (2006). “Comamonas,” in The Prokaryotes, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt (New York, NY: Springer), 723–736. doi: 10.1007/0-387-30745-1_31
Wu, C.-Y., Zhuang, L., Zhou, S.-G., Li, F.-B., and Li, X.-M. (2009). Fe (III)-enhanced anaerobic transformation of 2, 4-dichlorophenoxyacetic acid by an iron-reducing bacterium Comamonas koreensis CY01. FEMS Microbiol. Ecol. 71, 106–113. doi: 10.1111/j.1574-6941.2009.00796.x
Wu, J., Jiang, C., Wang, B., Ma, Y., Liu, Z., and Liu, S. (2006). Novel partial reductive pathway for 4-chloronitrobenzene and nitrobenzene degradation in Comamonas sp. strain CNB-1. Appl. Environ. Microbiol. 72, 1759–1765. doi: 10.1128/AEM.72.3.1759-1765.2006
Wu, S., Zhu, Z., Fu, L., Niu, B., and Li, W. (2011). WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12:444. doi: 10.1186/1471-2164-12-444
Wu, Y., Arumugam, K., Tay, M. Q. X., Seshan, H., Mohanty, A., and Cao, B. (2015a). Comparative genome analysis reveals genetic adaptation to versatile environmental conditions and importance of biofilm lifestyle in Comamonas testosteroni. Appl. Microbiol. Biotechnol. 99, 3519–3532. doi: 10.1007/s00253-015-6519-z
Wu, Y., Shukal, S., Mukherjee, M., and Cao, B. (2015b). Involvement in denitrification is beneficial to the biofilm lifestyle of Comamonas testosteroni: a mechanistic study and its environmental implications. Environ. Sci. Technol. 49, 11551–11559. doi: 10.1021/acs.est.5b03381
Xing, D., Cheng, S., Regan, J. M., and Logan, B. E. (2009). Change in microbial communities in acetate-and glucose-fed microbial fuel cells in the presence of light. Biosens. Bioelectron. 25, 105–111. doi: 10.1016/j.bios.2009.06.013
Young, C., Chou, J., Arun, A., Yen, W., Sheu, S., Shen, F., et al. (2008). Comamonas composti sp. nov., isolated from food waste compost. Int. J. Syst. Evol. Microbiol. 58, 251–256. doi: 10.1099/ijs.0.65277-0
Zengin, G. E., Artan, N., Orhon, D., Satoh, H., and Mino, T. (2011). Effect of aspartate and glutamate on the fate of enhanced biological phosphorus removal process and microbial community structure. Bioresour. Technol. 102, 894–903. doi: 10.1016/j.biortech.2010.09.023
Zhang, T., Liu, Y., and Fang, H. H. (2005). Effect of pH change on the performance and microbial community of enhanced biological phosphate removal process. Biotechnol. Bioeng. 92, 173–182. doi: 10.1002/bit.20589
Zhong, F., Wu, J., Dai, Y., Yang, L., Zhang, Z., Cheng, S., et al. (2015). Bacterial community analysis by PCR-DGGE and 454-pyrosequencing of horizontal subsurface flow constructed wetlands with front aeration. Appl. Microbiol. Biotechnol. 99, 1499–1512. doi: 10.1007/s00253-014-6063-2
Zhou, Y.-H., Ma, H.-X., Dong, Z.-Y., and Shen, M.-H. (2018). Comamonas kerstersii bacteremia in a patient with acute perforated appendicitis: a rare case report. Medicine 97:e9296. doi: 10.1097/MD.0000000000009296
Keywords: Comamonas, metabolic system, biofilm, virulence, nitrate reduction
Citation: Wu Y, Zaiden N and Cao B (2018) The Core- and Pan-Genomic Analyses of the Genus Comamonas: From Environmental Adaptation to Potential Virulence. Front. Microbiol. 9:3096. doi: 10.3389/fmicb.2018.03096
Received: 01 August 2018; Accepted: 29 November 2018;
Published: 12 December 2018.
Edited by:
Haiwei Luo, The Chinese University of Hong Kong, ChinaReviewed by:
Adriana Ribeiro Carneiro Folador, Universidade Federal do Pará, BrazilCopyright © 2018 Wu, Zaiden and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Cao, YmluY2FvQG50dS5lZHUuc2c=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.