 Stephanie R. Bollmann
Stephanie R. Bollmann Caroline M. Press
Caroline M. Press Brett M. Tyler
Brett M. Tyler Niklaus J. Grünwald
Niklaus J. Grünwald
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 30 November 2018
Sec. Evolutionary and Genomic Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02841
Modulation of gene expression through RNA interference is well conserved in eukaryotes and is involved in many cellular processes. In the oomycete Phytophthora, research on the small RNA machinery and function has started to reveal potential roles in the pathogen, but much is still unknown. We examined Argonaute (AGO) homologs within oomycete genome sequences, especially among Phytophthora species, to gain a clearer understanding of the evolution of this well-conserved protein family. We identified AGO homologs across many representative oomycete and stramenopile species, and annotated representative homologs in P. sojae. Furthermore, we demonstrate variable transcript levels of all identified AGO homologs in comparison to previously identified Dicer-like (DCL) and RNA-dependent RNA polymerase (RDR) homologs. Our phylogenetic analysis further refines the relationship of the AGO homologs in oomycetes and identifies a conserved tandem duplication of AGO homologs in a subset of Phytophthora species.
The RNA interference (RNAi) machinery, including Argonaute (AGO) proteins, is well conserved among most eukaryotic supergroups (Cerutti and Casas-Mollano, 2006). After small RNA duplex formation by Dicer/Dicer-like (DCR/DCL) enzymes with or without the function of RNA-dependent RNA polymerases (RDR), the duplex is loaded onto an AGO, unwound, and the guide strand is retained to complete the mature RNA-induced silencing complex (RISC) (Schwarz et al., 2003). The AGO effector present in the RISC complex determines whether the target mRNA will be down-regulated through cleavage or translational repression. AGO homologs share a common set of domains, including the N-terminal, Linker 1, PAZ (Piwi Argonaute and Zwille), Linker 2, Mid, and PIWI (originally P-element Induced WImpy testis in Drosophila) domains. The N-domain has been shown to be required for unwinding of the small RNA duplex in the process of forming the mature RISC complex (Kwak and Tomari, 2012) and in some AGOs it is required for cleavage of the target mRNA (Hauptmann et al., 2013). The PAZ domain binds and anchors the 3’ end of the small RNA guide (Lingel et al., 2003, 2004; Song et al., 2003; Yan et al., 2003; Ma et al., 2004) and aids in unwinding the small RNA duplex (Gu et al., 2012). The MID domain binds the 5′ nucleotide of the guide sRNA (Frank et al., 2010, 2012), which, in addition to the structure of the small RNA duplex, is important for the binding specificity seen in some AGOs (Tomari et al., 2007; Jannot et al., 2008; Czech et al., 2009; Okamura et al., 2009; Ghildiyal et al., 2010). The MID-PIWI interface binds and anchors the 5′ phosphate of the guide sRNA (Parker et al., 2005). The PIWI domain is the catalytic domain that allows some AGOs to cleave the target mRNA (slicer activity) which is complementary to the bound guide sRNA (Song et al., 2004; Rivas et al., 2005). The catalytic tetrad (DEDD/H) is required, but not sufficient for slicer activity (Liu et al., 2004; Song et al., 2004; Rivas et al., 2005; Nakanishi et al., 2012; Faehnle et al., 2013; Hauptmann et al., 2013; Sheng et al., 2014).
The number and type of AGO homologs varies from organism to organism (Tolia and Joshua-Tor, 2007). There are four major families of AGO homologs: AGO-like, PIWI, WAGO, and Trypanosoma AGO families (Yigit et al., 2006; Garcia Silva et al., 2010). The AGO-like family is widely conserved among eukaryotes, is predominantly responsible for interaction with micro (miRNAs) and short interfering RNAs (siRNAs), and is involved in regulation of transcription and translation, maintenance of germ cells, alternative splicing, and heterochromatin formation (Azzam et al., 2012; Michalik et al., 2012; Wei et al., 2012; Gagnon et al., 2014; Huang and Li, 2014). The PIWI family is also well conserved, interacts with PIWI-interacting RNAs (piRNAs), and is involved with germline stem cell maintenance and transposon silencing in stem cells (Lin and Spradling, 1997; Brennecke et al., 2007). Worm-specific AGO (WAGO) proteins are limited to nematodes, and act as secondary Argonautes in response to an initial AGO targeted cleavage of an mRNA, resulting in a more specific silencing response (Yigit et al., 2006). The AGO family in Trypanosoma, typified by T. brucei, inhibits transposon activity (Djikeng et al., 2001; Shi et al., 2009).
Research on the small RNA biology in the Stramenopile kingdom, including brown algae, diatoms, and oomycetes, is currently limited. The genus Phytophthora contains prominent oomycetes such as the Irish famine pathogen P. infestans or the sudden oak death pathogen P. ramorum, affecting many hosts and leading to multibillion dollar crop damage and devastation of natural environments (Tyler, 2007; Fry, 2008; Grünwald et al., 2008). With the advancement of genome sequencing technologies, several oomycete genomes have become available for comparative genomic analyses (Tyler et al., 2006; Haas et al., 2009; Baxter et al., 2010; Lévesque et al., 2010; Links et al., 2011). Within the Chromalveolate supergroup, putative miRNAs have been described in the apicomplexan Toxoplasma gondii (Braun et al., 2010) and a miRNA family was found to be conserved across three diverse Phytophthora genomes, with evidence for target cleavage in P. sojae (Fahlgren et al., 2013). Diversity in the number of small RNA biogenesis genes has also been seen. T. gondii has a single homolog each of DCR, RDR, and AGO (Braun et al., 2010), whereas Phytophthora species have two DCR homologs with a diverse origin, a single RDR homolog, and a large and varied number of AGO homologs (Fahlgren et al., 2013; Bollmann et al., 2016). High-throughput sequencing in three Phytophthora species revealed two major size classes of small RNAs, 21-nt and 25/26-nt, which matches well with the existence of two DCR homologs (Vetukuri et al., 2011; Fahlgren et al., 2013).
Phylogenetic analyses of the DCR homologs in Phytophthora revealed a divergent evolutionary origin, with DCL1 clustering with plant and animal DCR-like homologs, and DCL2 clustering more basally in the tree with homologs of Drosha (Bollmann et al., 2016). Additionally, although both DCL1 and DCL2 have two RNase III domains, typical of DCR homologs, the other domains differ greatly. DCL1 is longer and includes the DEAD-box helicase and dsRNA binding domain, whereas DCL2 has a PAZ domain and a dsRNA binding motif. In plants and animals, for comparison, it is common for most if not all of these domains to be present in each homolog. Interestingly, Phytophthora species, similar to Dictyostelium, have a DEAD-box helicase and Helicase-C domains on the N-terminus of the RDR homolog. This possible domain transfer from the DCL2 homolog to RDR may suggest interaction of those proteins in the same pathway, lending support for DCL2 in the 25-26-nt sRNA pathway. The study also showed nuclear localization of both DCR homologs (Bollmann et al., 2016).
A recent study showed the first evidence for specific roles of AGO homologs in Phytophthora (Åsman et al., 2016). Phylogenetic analysis revealed that oomycete AGO homologs cluster with the AGO-like family and that the brown algae homolog is more closely related to oomycetes than to the diatom homologs. This study also confirmed that oomycetes have two well-defined AGO clades (Fahlgren et al., 2013), with AGO1 in a separate clade from the rest of the AGO homologs in the species tested, as well as differences in key residues in the PIWI domain (Åsman et al., 2016). Co-immunoprecipitation assays in P. infestans revealed that Ph_infeAGO1 primarily associates with 20-22-nt sRNAs, with a preference for 5′ C, Ph_infeAGO4 primarily associates with 24-26-nt sRNAs, with a strong preference for 5′ U, and Ph_infeAGO5 has no apparent size preference. In addition, sequencing the precipitated sRNAs revealed that Ph_infeAGO1 associates with the one confirmed miRNA (Fahlgren et al., 2013) and with sRNAs derived from Crinkler genes, protein-coding genes, and Gypsy long terminal repeat retrotransposons. In contrast, Ph_infeAGO4 associates with sRNAs derived from Helitron, Crypton, PiggyBac, and Copia transposons, and Ph_infeAGO5 associates with sRNAs derived from introns and intergenic regions (Åsman et al., 2016). In the P. infestans strain used for the study, there was an early indel in Ph_infeAGO3 which disrupted the protein and did not allow further testing. However, they showed that the N-terminus of Ph_infeAGO3 is Gly-Arg-rich, which in T. brucei and T. gondii has been shown to be important for AGO function (Shi et al., 2004; Musiyenko et al., 2012). Unlike the DCR homologs, Ph_infeAGO1 and Ph_infeAGO4 were shown to be localized to the cytoplasm (Åsman et al., 2016).
The goal of this study was to further refine the phylogenetic analysis of oomycete AGO homologs, both by deeper analysis of the broader eukaryotic tree, and a more in-depth analysis of a wider range of Phytophthora species. Unlike other Stramenopiles, Phytophthora species have diverse numbers of AGO homologs, and clade 7 species in particular show a conserved expansion of a tandem duplication common to Phytophthora species. Through real-time quantitative PCR (RT-qPCR), we documented dynamic transcript levels of AGO genes in P. sojae, in contrast to the steadier levels of transcript levels of the DCR and RDR homologs. We additionally document the syntenic relationships of the tandem duplication of AGO homologs and the presence/absence of key conserved residues in the MID and PIWI domains.
AGO and PIWI homolog sequences were initially identified using TBLASTN (NCBI) searches against species-specific genomic databases. Various queries were used, including full-length sequences or PAZ/PIWI domain sequences from oomycetes such as Phytophthora sojae. Species with partial or fully-sequenced genomes were selected from all available Chromalveolate groups, as well as species from other eukaryotic supergroups in order to assess ancient evolutionary relationships. A sample of AGO and PIWI homologs identified in previous phylogenetic analyses (Cerutti and Casas-Mollano, 2006; Yigit et al., 2006; Murphy et al., 2008) were included along with homologs from Chromalveolates that were previously less represented. The JGI database1 was searched by querying specific organisms for the presence of annotated PAZ and PIWI domains. Oomycete homolog sequences were identified in the VBI Microbial Database2 and the P. infestans database at the Broad Institute3 (now at fungidb.org). Other genomes were made available by permission: P. cinnamomi (JGI; Wayne Reeve, pers. comm.) and P. melonis (Wenwu Ye, pers. comm.). Genomic sequences encompassing the identified Chromalveolate homologs were selected and coding sequences were manually identified and confirmed using Genscan (Burge and Karlin, 1997) or FGENESH4. Protein domains were identified using Pfam database searches (Finn et al., 2010). To be retained in the analyses, an AGO homolog minimally had to contain a PAZ and a PIWI domain. An AGO-like homolog from the Archaea was also identified for use as an outgroup.
P. sojae was used as the model Phytophthora species to validate AGO sequences. Total RNA was isolated from P. sojae strain P6497 mycelium (grown in cleared V8 broth) using TRIzol (Invitrogen, Carlsbad, CA, United States) from 250 μg total RNA with an Oligotex column (Qiagen, Germantown, MD, United States). cDNA was produced using the GeneRacer RACE Ready kit (Invitrogen) following the manufacturer’s guidelines, in order to capture the 5′ and 3′ ends of the mRNAs. Coding sequences from genomic database queries were predicted with Genscan (Burge and Karlin, 1997). Primers were designed using the Primer3 (Rozen and Skaletsky, 2000) plug-in in Geneious5 (Kearse et al., 2012). Primer sequences and locations are detailed in Supplementary Table 1 and Supplementary Figure 1. Genomic DNA isolated from mycelium with a FastPrep kit (MP Biomedicals, Solon, OH, United States) was used for initial primer testing to confirm the predicted genomic sequence and test the efficacy of the primers. All PCR products were amplified with Taq polymerase (Genscript, Piscataway, NJ, United States) following the manufacturer’s guidelines. All primer annealing temperatures were between 56–60°C and extension times were calculated using a rate of 1 kb per minute. As recommended by GeneRacer guidelines, amplification of the 5′ and 3′ UTRs utilized touchdown PCR, typically with a 1-min extension. Amplification of internal sequences typically involved a 60°C annealing temperature and 4-min extension. All PCR products were cloned with the TOPO-TA cloning kit (Invitrogen, Carlsbad, CA, United States). A minimum of two to three clones were sequenced for each product using the BigDye Terminator v. 3.1 Cycle Sequencing Kit on an ABI Prism 3730 Genetic Analyzer (Applied Biosystems, Carlsbad, CA, United States). The sequences were assembled and manually analyzed using Geneious software. P. sojae sequences have been deposited in GenBank under the following accession numbers: PsAGO1L (MH198153), PsAGO1S (MH198154), PsAGO2 (MH198155).
Transcript levels of AGO genes were characterized in several life stages and in a time series of infected host tissue. Total RNA was isolated using TRIzol from P. sojae mycelium, zoospores, and germinated cysts (6 replicates each) or from two independent soybean infection experiments, each with three replicate hypocotyls collected at 3, 6, 12, 24, or 48 h-post-infection. Zoospores were produced by repeated washing of 11 day-old V8-200 plates of mycelium followed by overnight incubation at 14°C. Germinated cysts were produced by exposing zoospores to cleared V8 broth for 1 h. Soybean infection experiments followed the protocol of Qutob et al. (2000) in which infected hypocotyls were incubated at 28°C for 14 h in the light and 25°C for 10 h in the dark. 50 mg of TRIzol-isolated RNA from each replicate was processed through the AllPrep DNA/RNA Mini kit (Qiagen) following the manufacturer’s guidelines with the addition of one DNase I treatment. The Superscript III First-Strand Synthesis System for RT-PCR kit (Invitrogen, Carlsbad, CA, United States) was used to produce cDNA, followed with purification by phenol:chloroform extraction. cDNA was quantified with a Nanodrop ND-1000 (Thermo Scientific, Waltham, MA, United States) to allow for equal quantities of cDNA template in subsequent reactions. Primers were designed as described above. Standard real-time quantitative PCR reactions were performed with Fast SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA, United States) on an ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Carlsbad, CA, United States). Data was normalized to reference genes β-Tubulin (Ps109498) and WS41 (Ps137777; protein of the BAR-domain family) and analyzed with Bestkeeper (Pfaffl et al., 2004), REST (Pfaffl et al., 2002), and SAS (Cary, NC, United States) software.
A phylogenetic analysis was conducted to establish the evolutionary history of AGO genes. Alignments of protein sequences (Supplementary Data Sheets 2, 3 and Supplementary Table 2) were created with MAFFT6 (Kuraku et al., 2013; Katoh et al., 2017). The amino acid sequences encompassing the PAZ through PIWI domains were analyzed across a broad range of species, while all predicted domains were analyzed across the Stramenopiles. To reduce error introduced due to partial sequences and to minimize computational load, amino acid positions that were absent in a majority of the analyzed species were removed from the alignment (Baurain et al., 2010). Any protein sequences with significant gaps in the alignment were removed from the analysis. Species were identified in phylogenetic trees with an abbreviated name in the format “Ge_spec” (the first two letters of the generic name followed by the first four letters of the specific name). Gene number designations do not necessarily follow homology. Previous designations, either from the genomic or predicted transcript database or a previous publication (Cerutti and Casas-Mollano, 2006; Yigit et al., 2006; Murphy et al., 2008), were maintained; otherwise the number assignment was arbitrary when there was no available reference. Phytophthora gene number designations were changed to reflect the evolutionary relationships among homologs.
Condensed domain alignments for the broader phylogeny were used for Bayesian phylogenetic analyses using MrBayes-3.1.2 (Huelsenbeck and Ronquist, 2001; Ronquist and Huelsenbeck, 2003) with the following settings: mixed amino acid model of evolution with invgamma rates, unconstrained brlenspr, sample frequency of 50, 2 runs with 4 chains each, temperature of 0.2, and diagnostic frequency of 1,000. The phylogenetic analysis ran for 8 million generations with a final burn-in of 25%. The final consensus tree from the Bayesian analyses was obtained using maximum likelihood analysis using RAxMLPTHREADS-7.0.0 (Stamatakis, 2014); 1,000 bootstrap replications were performed. Chromalveolate alignments were analyzed in MEGA7 (Kumar et al., 2016) using the Neighbor-Joining method (Saitou and Nei, 1987) as their evolutionary distances were much smaller. The Poisson correction method (Zuckerkandl and Pauling, 1965) was used for estimating evolutionary distances, with removal of ambiguous positions for each sequence pair and a bootstrap test with 1,000 replicates (Felsenstein, 1985).
To infer the evolutionary history of Phytophthora AGO homologs, we conducted a phylogenetic analysis of AGO homologs including species selected across the Chromalveolate supergroup and representatives of eukaryote AGO-like and PIWI-like homologs, including an Archaea PIWI-like homolog as an outgroup (Figure 1). The conserved PAZ through PIWI domains were aligned and used for Bayesian and maximum likelihood phylogenetic analysis. As expected, the ciliate AGO homologs formed a distinct group with PIWI-like homologs from C. elegans, humans, and Dictyostelium. Oomycete AGO homologs formed a separate group with AGO-like homologs from C. elegans, humans, and Arabidopsis. There were several groups that did not clearly cluster into either of these larger groups, namely homologs from diatoms, Apicomplexa, fungi and C. elegans. The Apicomplexan Eimeria tenella AGO and archaea PIWI-like homologs were both outgroups of the tree.
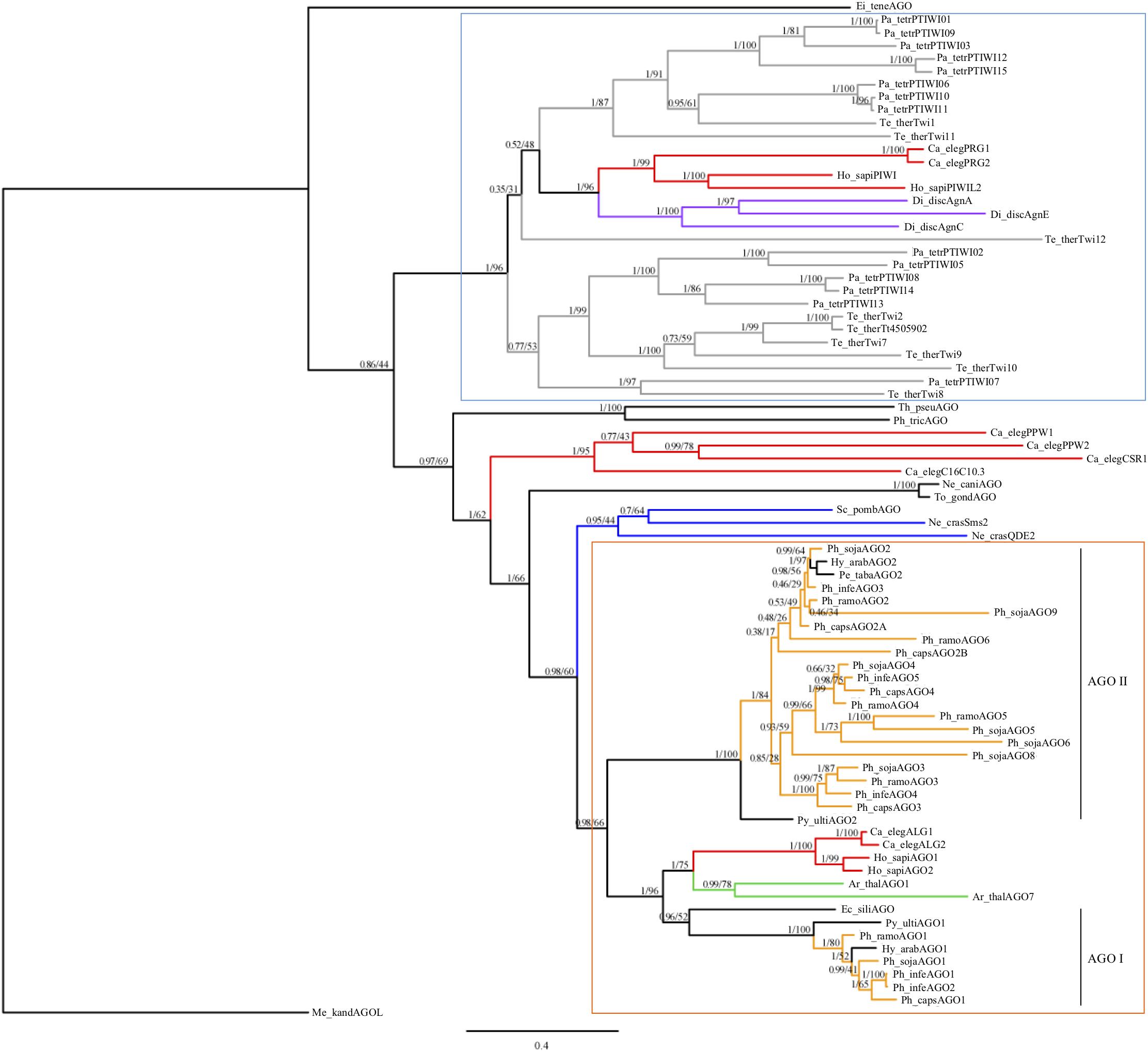
FIGURE 1. Consensus phylogram of AGO homologs based on PAZ through PIWI domains. Bayesian support and maximum likelihood values, respectively, are indicated. Branches are colored to denote major species groups: gray, ciliate; red, animal; purple, amoeba; blue, fungi; light orange, Phytophthora; green, plant. An archaea PIWI-like homolog (Methanopyrus kandleri AV19) was included as an outgroup (Shabalina and Koonin, 2008). Well supported PIWI-like (blue upper box) and AGO-like (orange lower box) clades are indicated, as well as oomycete AGO I and AGO II clades. Species are abbreviated as: Ge_spec (Supplementary Table 2).
Within the oomycetes, the AGO-like sequences divided into two distinct clades (I and II). The AGO I clade contained the AGO1 homologs from Phytophthora, Hyaloperonospora, Pythium, and the single AGO homolog from Ectocarpus siliculosis; this clade grouped with the other eukaryotic AGO-like homologs. In clade I, most of the oomycete AGO1 sequences were present in single copy. The exception was P. infestans, which has a pair of recently duplicated genes, which encoded proteins that are 99.9% identical with a single amino acid difference. The AGO II clade contained exclusively oomycete sequences. This clade contained a single AGO2 homolog in two downy mildew species, including Hyaloperonospora arabidopsidis, Peronospora tabacina, as well as Pythium ultimum. However, there was evidence for multiple duplication and divergence events among the Phytophthora species resulting in sub-groups AGO2 to AGO9. Members of the AGO II clade in general have longer, undefined N-termini that are Gly-Arg-rich, whereas members of the AGO I clade have shorter, undefined N-termini that are Pro-rich.
To further investigate the structural differences between Phytophthora proteins in the AGO I and AGO II clades, we selected the AGO genes from P. sojae for detailed gene annotation. Nine P. sojae homologs of AGO were identified bioinformatically from the genome sequence v1.0 (VBI Microbial Database2), most with good support from RNAseq data at FungiDB7. The original annotation of the reference genome did not predict nine homologs. Ph_sojaAGO5 and Ph_sojaAGO8 were bioinformatically annotated as one gene, and Ph_sojaAGO9 was not predicted at all, as these three homologs are very close together on the chromosome (discussed further below). Detailed bioinformatic analysis of the region allowed separation of the homologs into distinct predicted ORFs.
Of the nine AGO homologs identified, four were predicted to have all six of the conserved AGO domains (N-terminal, Linker 1, PAZ, Linker 2, Mid, and PIWI), and all except Ph_sojaAGO8 displayed both PAZ and PIWI domains. The other five homologs, in which all six conserved domains were not predicted, still showed similar amino acid sequences across all predicted domain regions, albeit the conservation within the PIWI domain was the highest. Supplementary Figure 2 shows the predicted structures of Ph_sojaAGO3 through Ph_sojaAGO9.
We used sequence analysis of full-length cDNA clones to confirm predicted gene structures for three representative homologs: Ph_sojaAGO1 and Ph_sojaAGO2, representing the two major oomycete clades, and Ph_sojaAGO7 which was predicted to be a pseudogene and had a structure which was difficult to resolve. In the case of Ph_sojaAGO1, two different isoforms were found among the cDNA clones. One of these (Ph_sojaAGO1L), represented by 7 independent clones, displayed a more upstream start site as well as the removal of a 204 nt intron (Figure 2 and Supplementary Figure 3). The other isoform (Ph_sojaAGO1S), displayed a start site 351 bp downstream of the Ph_sojaAGO1L start site, with no intron, resulting in a predicted protein that is 64 residues shorter at the N-terminus than Ph_sojaAGO1L. Both Ph_sojaAGO1 and Ph_sojaAGO2 displayed typical UTRs: 5′ UTRs ranged from 10 to 64 bases in length, and 3’ UTRs ranged from 150 to 246 bases in length. The genes follow the general trends of Phytophthora genes, in which only one-third possess an intron (average length of 79 bases), and the average number of introns is 1.5 per gene (Kamoun, 2003; Tyler et al., 2006; Haas et al., 2009). Of the three annotated transcripts, the Ph_sojaAGO1L transcript contains an intron that is 204 bases in length, although the exon/intron boundaries are not the conventional GT..AG bases (Supplementary Figure 3). The GC content of the three transcripts are 62.6% (Ph_sojaAGO1L), 62.1% (Ph_sojaAGO1S), and 62.9% (Ph_sojaAGO2); typical of oomycete transcribed sequences (average of 58%). The sequences surrounding the transcription start site and translation start site also generally follow the trend seen in other Phytophthora genes (Supplementary Figure 3). Figure 2 displays annotated gene structures for Ph_sojaAGO1 and Ph_sojaAGO2, including the two alternative transcription start sites for Ph_sojaAGO1, along with conserved AGO domains in the encoded proteins.
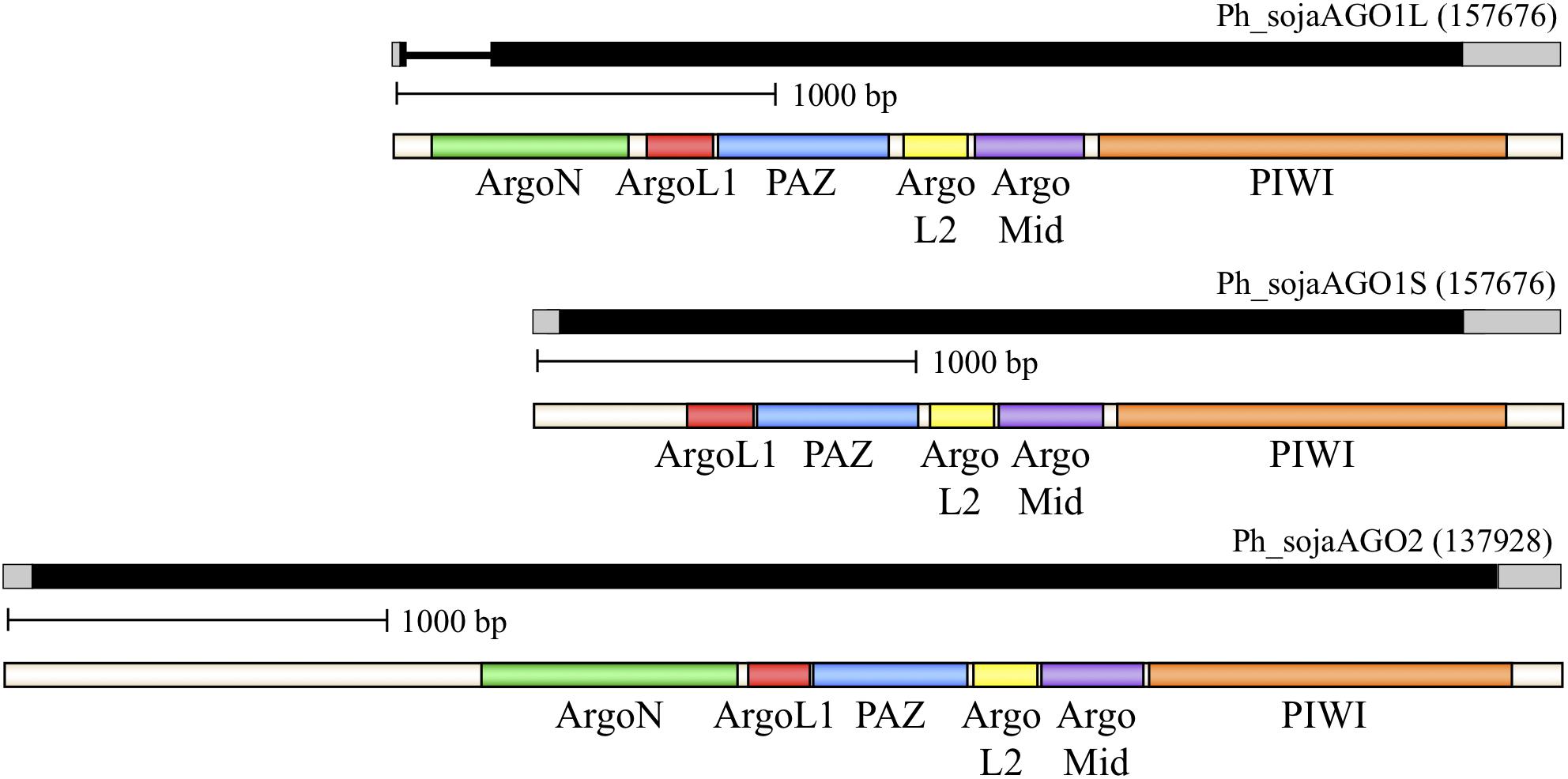
FIGURE 2. Organization of representative P. sojae Argonaute (AGO) homologs. Genes are labeled based on their Gene ID from the EuMicrobeDB database (eumicrobedb.org). In the genomic DNA diagrams, exons and introns are represented as black bars and lines, respectively. 5′ and 3′ UTRs are represented as gray bars. Conserved domains are indicated by colored bars in the mRNA diagrams: ArgoN, N-terminal Argonaute domain; ArgoL1, linker domain between N-terminus and PAZ domain; PAZ, PAZ domain named for the proteins Piwi Argonaute and Zwille; ArgoL2, linker domain between PAZ and PIWI lobes of Argonaute; ArgoMid, part of the PIWI lobe; PIWI, active domain for dsRNA-guided hydrolysis of mRNA.
Ph_sojaAGO7 was confirmed to be a pseudogene. The predicted Ph_sojaAGO7 coding sequence had no introns and had several premature termination codons. The AGO7 pseudogene transcript additionally had an unresolved start position. To allow for phylogenetic analyses, the best predicted transcript with the largest blocks of conserved domain sequences was used, which required many putative introns and had a GC content of 59.4%.
To investigate the respective functional roles of the P. sojae AGO homologs, qRT-PCR analysis was conducted to determine the transcript levels across three different life stages and across an infection time series. Housekeeping genes WS41 and β-tubulin were used as internal controls (Supplementary Figure 4), and DCL1, DCL2, and RDR were also analyzed for comparison. The DCL1, DCL2, and RDR homologs showed fairly stable transcript levels across the stages and time series (Figure 3A and Supplementary Figure 5A), with a slight increase in levels in zoospores, as reported previously (Bollmann et al., 2016). The AGO homologs, in comparison, exhibited a wide range of transcript levels (Figures 3B,C and Supplementary Figures 5B,C). Whereas AGO1 had transcript levels nearly as high as the housekeeping gene WS41, AGO3 exhibited much lower levels, and in fact transcripts could not be detected at the 3 hours post infection (hpi) or 6 hpi treatments. AGO2, AGO4, and AGO7 exhibited similar transcript levels as DCL1, DCL2, and RDR, although only AGO7 showed increased levels in zoospores. AGO5, AGO6, AGO8, and AGO9, similar to AGO3, exhibited much lower transcript levels that were undetectable in some samples. Only AGO6 and AGO7 exhibited increased transcript levels in zoospores comparable to DCL1, DCL2, and RDR.
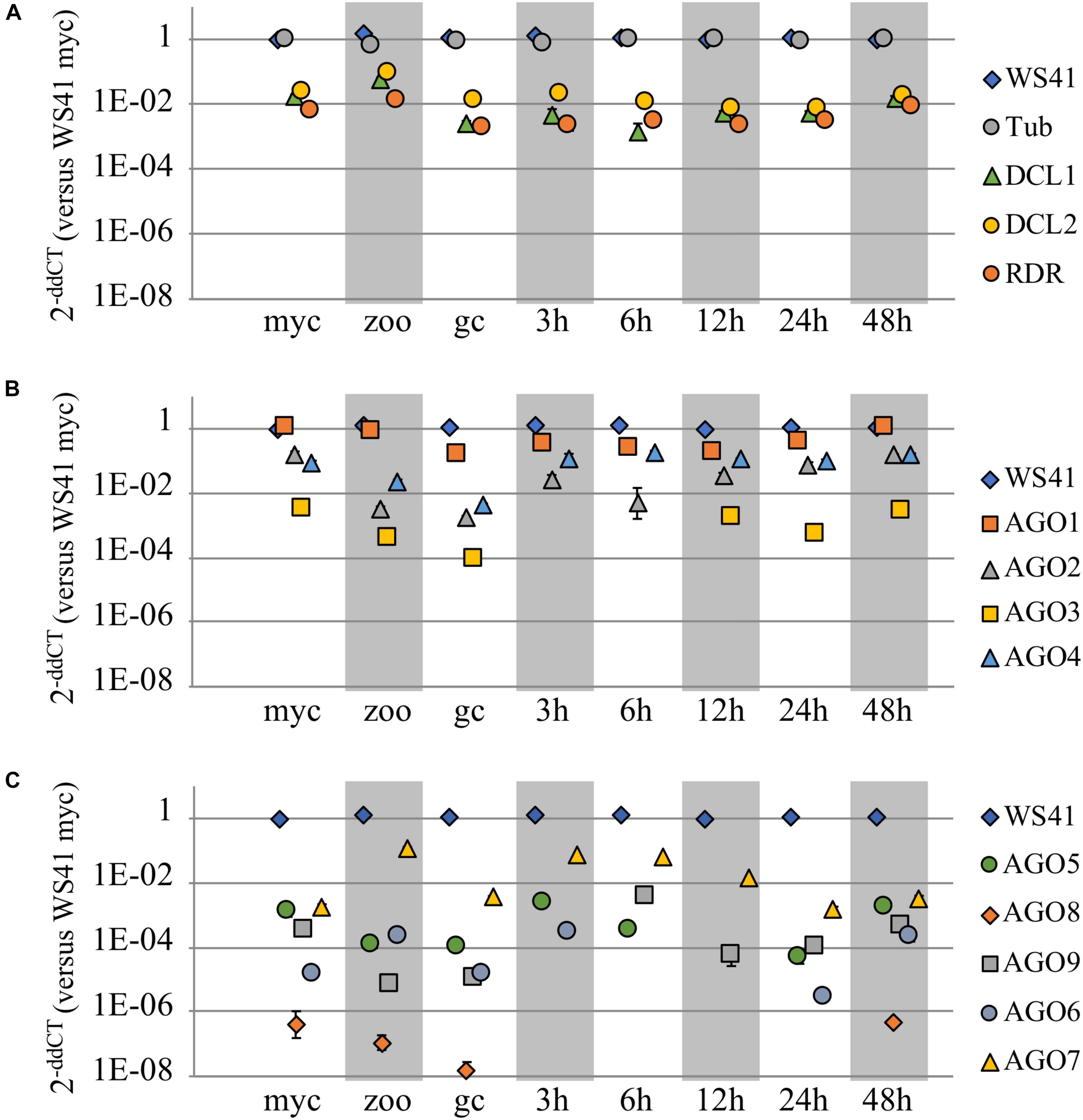
FIGURE 3. Transcript levels of AGO and other small RNA biogenesis genes. Three different life stages (myc, mycelium; zoo, zoospores; gc, germinated cysts) and an infection time series (3, 6, 12, 24, and 48 h post infection) were assayed in replicate by qRT-PCR to determine transcript levels of (A) DCL1, DCL2, RDR, and (B,C) the AGO homologs in P. sojae as compared to housekeeping genes b-tubulin and WS41. Each life stage or time series point is normalized to the housekeeping gene(s). Standard error bars are included, but in most cases the standard error bars are blocked by the symbol due to low value.
In order to analyze the two clades of oomycete AGOs in more detail, the number of oomycete AGO homologs was expanded. AGO proteins were predicted bioinformatically from several newly sequenced Phytophthora genomes, as well as additional representative oomycete genomes from the genera Pythium, Aphanomyces, and Albugo. This set of homologs was less diverse than presented in Figure 1, and therefore a neighbor-joining tree was sufficient for phylogenetic analysis. The additional Phytophthora sequences revealed more details of the relationships between the different subclades. The results of the analysis are shown in Figure 4, where the major Phytophthora AGO groups are collapsed for clarity, while the full tree is shown in Supplementary Figure 6. As seen in Figure 1 and reported in previous studies (Fahlgren et al., 2013; Åsman et al., 2016), two major AGO clades are present in oomycetes: AGO I, comprised primarily of AGO1 and AGO7 homologs, and AGO II, comprised of most other AGO homologs. The two clades were defined as having 100% bootstrap support. AGO I contains representatives from all of the oomycete species examined, including representatives from the Saprolegniales (i.e., Aphanomyces species) and the single AGO homolog found in E. siliculosus. In the AGO II clade, representatives from the Pythiales (Pythium spp), Albuginales (i.e., Albugo) and downy mildews (H. arabidopsidis) were restricted to the AGO2-AGO3 cluster. The AGO4 to AGO9 homologs were restricted to representatives from Phytophthora. Within the different Phytophthora AGO subclades, the trees were largely congruent with the phylogenetic clades previously described for the genus (Blair et al., 2008), (color coded in Supplementary Figure 6).
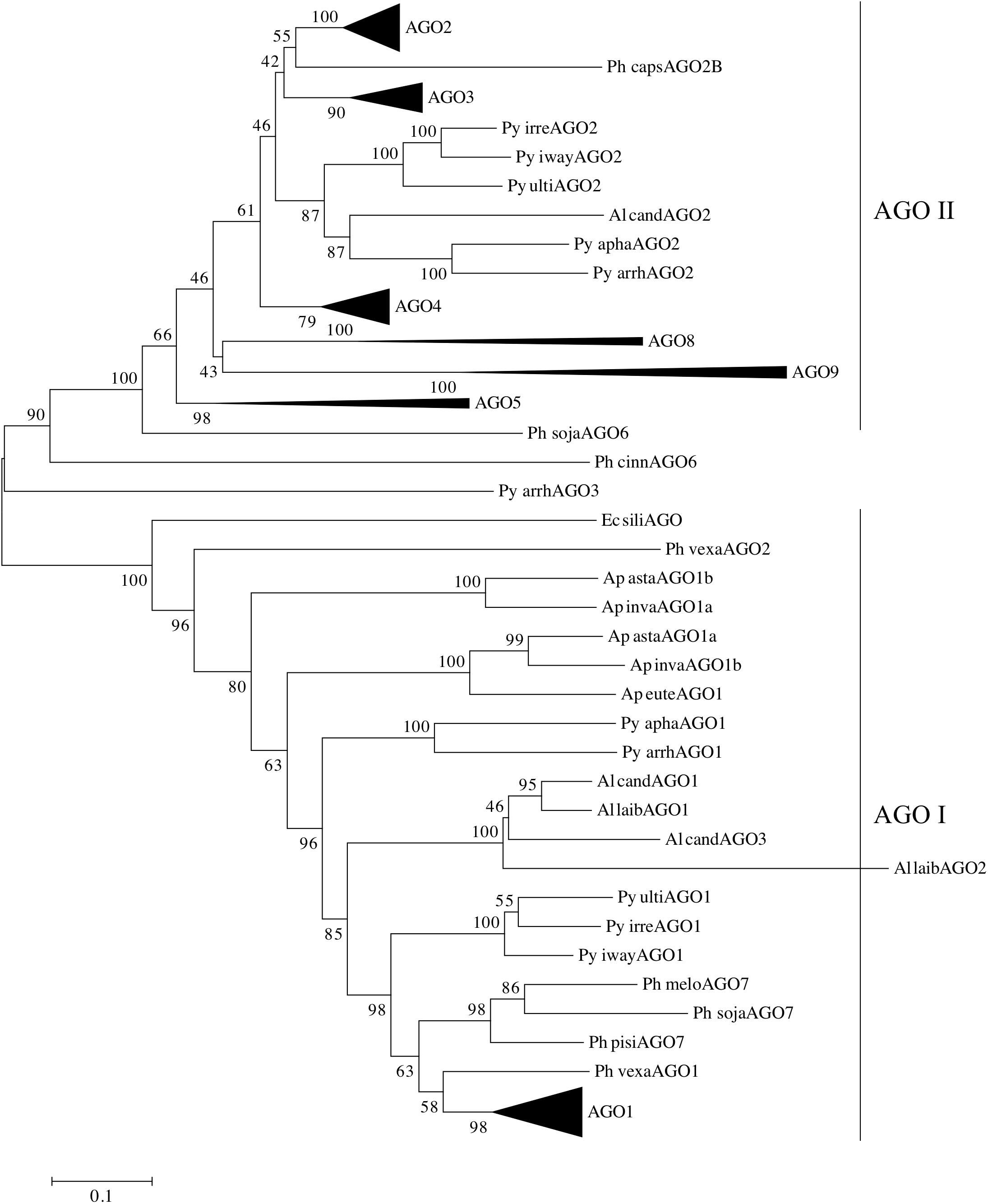
FIGURE 4. Phylogram of AGO homologs based on conserved domains. The neighbor-joining tree is based on evolutionary distances computed with the Poisson correction method using pairwise deletion of ambiguous sites. Bootstrap values (10,000 replicates) are indicated. Triangles denote collapsed ortholog groups as indicated; oomycete AGO I and AGO II clades are also noted. Species are abbreviated as: Ge spec (Supplementary Table 2).
Pythium and Albugo species both had two distinct groups of AGO homologs, one within AGO I, basal to the Phytophthora AGO1 sequences, and one within AGO II basal to the Phytophthora AGO2 and AGO3 groups. In contrast, the multiple AGO sequences within Aphanomyces all grouped together within AGO I, basal to the Albugo and Pythium sequences. Downy mildew (Peronospora and Hyaloperonospora) homologs of AGO1 and AGO2 grouped within the respective Phytophthora subclades within AGO I and AGO II.
The brown algae (Ectocarpus siliculosus) AGO homolog used as an outgroup lay basal to the AGO1 group, along with a few AGO sequences from Aphanomyces, Pythium, and Phytopythium species, which possibly may be encoded by pseudogenes. There was not enough sequence to resolve the position of Phytophthora cinnamomi AGO6 or Pythium arrhenomanes AGO3.
In Phytophthora, the AGO I clade consists primarily of single AGO1 homologs from each species. The exceptions are P. infestans, which has two nearly identical homologs, P. pinifolia with two homologs, and P. cryptogea with three homologs. Basal to the group of Phytophthora AGO1 sequences (Supplementary Figure 7) lie AGO1 homologs from Hyaloperonospora arabidopsidis and Phytopythium vexans, as well as a small group of Phytophthora AGO7 homologs (Figure 4). The remainder of the AGO I clade is made up of AGO1 homologs from Pythium, Albugo and Aphanomyces species.
The AGO II clade consists of multiple subgroups, each with multiple representatives from different Phytophthora species. Subgroups AGO2, AGO3, and AGO4 (Supplementary Figures 8–10) have representative genes from almost every Phytophthora clade present in the analysis. While not every species was represented, this may result from deficiencies in the genome assemblies used for identifying the AGO homologs. In most cases, there was a single representative of each subgroup in each species. Species from Phytophthora clades 3 and 4 appeared to be limited to AGO1 and AGO2 groups, although we cannot rule out the possibility that the sequence assembly had poor coverage over those regions. The AGO5/6/8/9 group, however, was primarily limited to Phytophthora clade 7 species, which exhibited a single homolog in each subgroup. The AGO5, 8, and 9 subgroups (Supplementary Figure 11) had strong support values. In contrast, the two AGO6 sequences, from P. sojae and P. cinnamomi, did not group together strongly and may not have a recent common origin.
Evolutionary relationships among the AGO homologs were further investigated by looking for evidence of synteny within homolog clusters. As shown previously (Fahlgren et al., 2013) in P. sojae, P. infestans, and P. ramorum, AGO2 lies on the same contig as AGO3 and AGO4 (the corresponding P. infestans genes are named PiAGO3, PiAGO4 and PiAGO5, respectively). As shown in Figure 5, our analysis revealed that AGO5, AGO8, and AGO9 are also within this gene cluster in the case of P. sojae. We then analyzed a wider set of Phytophthora genome sequences to look for syntenic AGO gene clusters (Figure 5). In P. infestans and P. ramorum, the AGO4, AGO3, and AGO2 homologs lie in the same order on the same contig, although the spacing between AGO4 and AGO3 homologs is small (3 kb, 4 kb) and the spacing between AGO3 and AGO2 homologs is large (187 kb, 60 kb). All P. infestans homologs lie in the same orientation, whereas in P. ramorum AGO4 lies in the opposite orientation of AGO3 and AGO2. In comparison, P. sojae is more complex and has AGO4, AGO3, AGO9, AGO8, AGO5, and AGO2 in that order. Again, AGO2 is separated from the other homologs by a large distance (130 kb), whereas the total span from the predicted start codon of AGO5 to the predicted stop codon of AGO4 is small (18 kb). The cluster of homologs from AGO5 to AGO4 all lie in the opposite orientation to AGO2 in P. sojae. Phytophthora species in clade 7 dominate the AGO5/8/9 group of homologs. P. melonis and likely P. pisi (truncated contig) show a similar gene order and orientation as P. sojae. P. cinnamomi also has all five tandem homologs in the same orientation, but in a different order than P. sojae: AGO4, AGO5, AGO8, AGO9, and AGO3. P. fragariae and P. rubi (Tabima et al., 2017) have three of the five clustered homologs (AGO4, AGO3, AGO9) in the same orientation, but different orders, and only AGO3 of the tandem set was identified in P. niederhauserii; however, we cannot rule out the possibility that sequences were missed in the genome assembly. Only two other genes grouped within the AGO5/8/9 group of homologs, specifically the AGO5 group, but these genes were not physically as close to the AGO4 and AGO3 genes as in clade 7 species. In P. parasitica, the distance between AGO5 and AGO3 is larger than in any other species, and in P. ramorum the AGO5 gene is on another contig. Surrounding syntenic genes can indicate boundaries of large changes between species, for example the unique inversion of multiple tandemly arranged genes between P. sojae and P. cinnamomi (AGO3,9,8,5; glycosyl hydrolases).
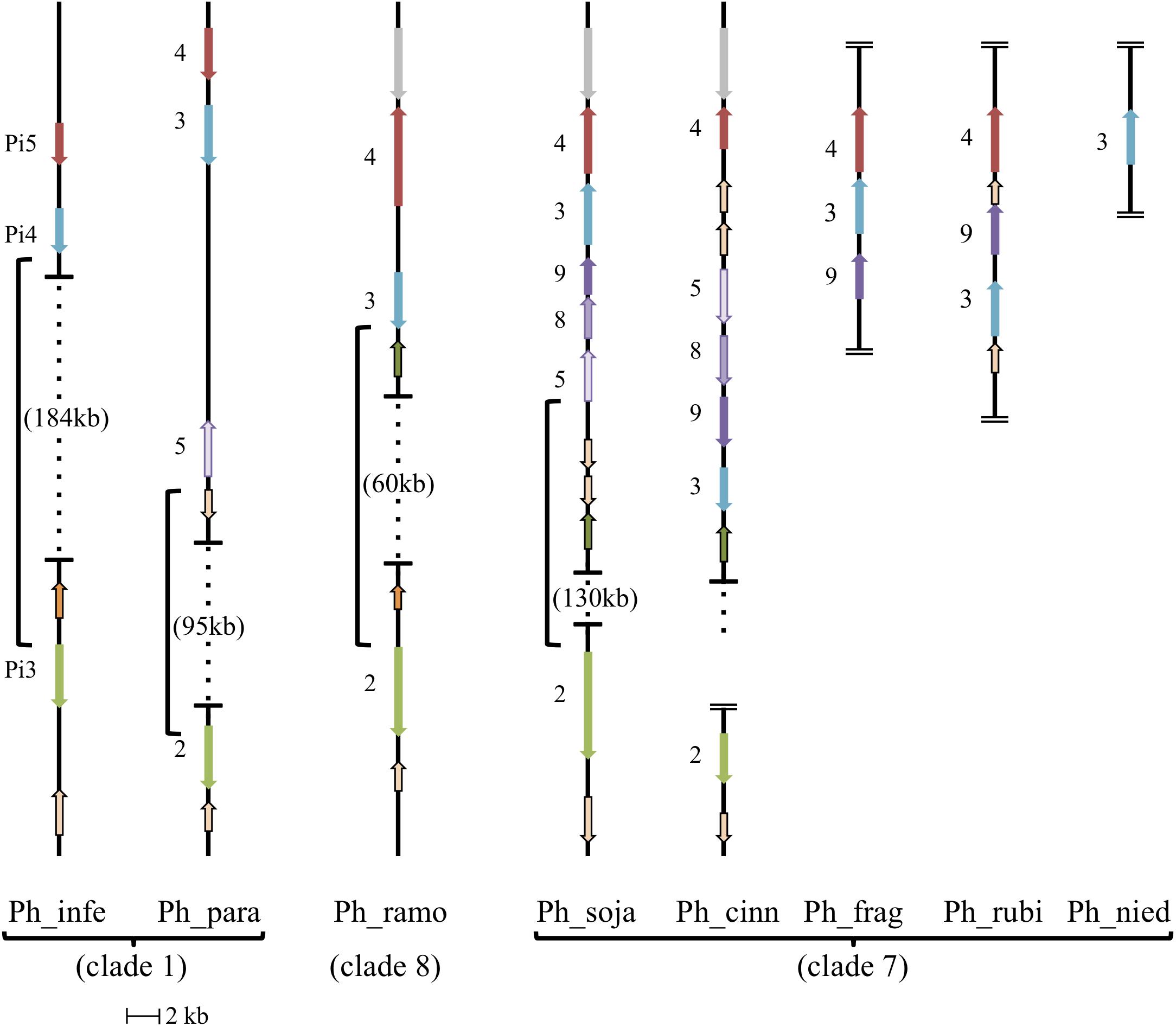
FIGURE 5. Synteny of AGO II clade orthologs. Diagram showing relative positions and orientations of AGO II clade orthologs which lie on the same contig. Horizontal lines with dotted vertical lines in between, have the genomic span between the closest orthologs indicated with braces (open ended dotted line indicates the contig continues, but does not have any further relevant orthologs). Double horizontal lines at the end of a contig indicate the end of the contig, where assembled contigs were too short to include all listed orthologs, in which case placement is hypothesized from related species. All AGO II clade orthologs are indicated with their gene number, and color coordinated to indicate orthology/synteny. Other conserved, syntenic genes surrounding the AGO II clade orthologs are noted: Glycosyl hydrolase family (tan with black border), Alpha/beta hydrolase fold family (green with black border), ER lumen protein retaining receptor family (orange with black border), and unknown protein (syntenic; gray). Phytophthora melonis (Ph_melo) has the same gene order and orientation as Phytophthora sojae (Ph_soja), although a homolog for AGO2 was not identified. Phytophthora pisi (Ph_pisi) likely has the same gene order as Phytophthora sojae, although the contig with AGO4-3-9-8-5 is cut off before the AGO5 homolog, which is missing in gene predictions, and AGO2 is on a different, shorter contig. Phytophthora ramorum (Ph_ramo) also has an AGO5 homolog, however, it is on a different contig.
To assess whether the AGO homologs had the required amino acids for RNA cleavage activity, positions of known conserved residues in the MID and PIWI domains were compared (Figure 6). In the MID domain, the amino acids Y-K-(S/T)Q-(K)K are critical for binding of the 5′ phosphate of the guide sRNA, while other residues in the MID domain can confer specificity of nucleotide binding in some species (Boland et al., 2010; Frank et al., 2010). In the majority of the AGO proteins in our analysis, these residues are perfectly conserved. The exceptions, namely Ph_crypAGO1a, Ph_meloAGO7, Al_laibAGO2, Al_candAGO3, Py_vexaAGO2, Py_arrhAGO3, Ph_idaeAGO2, Ph_agatAGO3, Ph_capsAGO2B, Ph_sojaAGO8, Ph_cinnAGO8, Ph_meloAGO8, Ph_cinnAGO9, and Ph_meloAGO9, may result from incomplete sequences or the predicted proteins potentially being pseudogenes. The AGO5 group all have the residues Y-K-(S/C)Q-(K)R, which may or may not alter function. In the PIWI domain, the amino acids D-E-D-D/H are required, but not sufficient for cleavage of the target mRNA (slicer activity) (Nakanishi et al., 2012; Sheng et al., 2014). In agreement with the report by Åsman et al. (2016), the AGO I clade proteins from our analysis primarily contained the D-E-D-H residue pattern, while the majority of the AGO II clade proteins contained a D-D-D-H residue pattern, which may or may not affect the RNA cleavage ability of the protein. As with the MID domain residues, many of the Phytophthora clade 7 tandem repeat homologs showed variation in the conserved residues, including the AGO5 group (D-D-D-N, D-E-G-N, D-S-D-N), Ph_sojaAGO6 (C-D-D-R), Ph_cinnAGO6 (D-D-S-Q), the AGO8 group (D-_-N-R, D-D-N-R, E-D-N-H, E-D-_-_), and the AGO9 group (S-D-G-_, N-D-G-R, N-D-G-K, S-D-_-R, E-D-S-R). Additional changes include AGO2 from H. arabidopsidis (D-G-D-H), AGO2B from P. capsici (D-D-N-Q), AGO4 from P. rubi (D-E-D-H), AGO3 from P. parasitica (D-D-N-H), and AGO2 from several Pythium species (N-D-D-Q).

FIGURE 6. Alignment of key conserved residues in the Mid and PIWI domains of Chromalveolate AGO homologs. Amino acid sequence regions from MAFFT alignment of Chromalveolate AGO homologs with key residues highlighted. Conserved Mid domain residues: YKQK (S/T before the Q and an extra K before the final K have also been shown to have significance in some cases); conserved PIWI domain residues: DEDD/H. Species names as in Figure 4.
Our analysis has provided a refinement in our understanding of the evolutionary history of oomycete AGO, in particular for the AGO II clade. Our results support the hypothesis that there was one ancestral AGO2 gene followed by formation of paralogs. The AGO I and AGO II clades likely diverged early in oomycete evolution as they are both present in Phytophthora, Peronospora, Pythium, and Albugo species, but not in Aphanomyces or brown algae. The more extensive duplication and divergence of genes within the AGO II clade appears however, to be restricted to the genus Phytophthora. The presence of AGO2, AGO3, and AGO4 on the same genome sequence contig (where the contigs are sufficiently) long further supports this idea. Phytophthora clade 7 in particular shows even more extensive duplication and divergence where expansion of AGO3 and AGO4 has produced tandem arrays of five AGO homologs, with rearrangements of the order and orientation of the genes among species. Gene duplications exist in some of the other Phytophthora species, such as the recent duplication of AGO1 in P. infestans, but currently only in clade 7 is there evidence for a clade-specific, tandemly arrayed gene expansion, although we cannot rule out this phenomenon may be present in other species we did not include.
The different P. sojae AGO genes exhibit considerable variation in gene structure. P. sojae AGO1, AGO2, AGO3, and AGO4 consist of only one exon that encodes all of the predicted conserved AGO domains, whereas AGO5 has two predicted exons and is not predicted to have the Mid domain, and AGO6 has three predicted exons and lacks conserved sequences matching the N-terminal and Mid domains. Ph_sojaAGO9, with five predicted exons, lacks conserved sequence for the Mid domain, and Ph_sojaAGO8 with six predicted exons is only predicted to have the N-terminal, Linker 2, and PIWI domains. We cannot rule out that some gene model predictions are incorrect for those AGO genes which were not cloned, but the RNAseq data that is available supports the predictions, and in most cases the predicted introns are short. Ph_sojaAGO7 likely is a pseudogene, even though it exhibits reasonably high transcript levels compared with the other AGO genes. When cDNA was cloned for Ph_sojaAGO7, there were inconsistent results for the 5′ end of the gene, with each different reading frame represented by multiple clones and variation in initial short introns. Other clones further in the sequence of the gene revealed no other introns, even though each reading frame has many stop codons. Predictions of each reading frame showed representation of all but the N-terminal domain, but the conserved domains were scattered across the different frames. The best computationally predicted gene structure, which included Linker 1, PAZ, and PIWI domains, was used for the phylogenetic analysis. Where conserved domain predictions were absent in P. sojae AGO homologs, it is likely the matching scores of the residues were simply too low to predict the corresponding Pfam model, as the amino acid alignments showed conservation throughout the majority of the sequences. This divergence from the consensus could also be seen in the conservation of key AGO residues required for function. All of the AGO I clade proteins showed conservation of key residues in both the MID and PIWI domains. In contrast, in the AGO II clade, the MID domain residues were better conserved than the PIWI domain residues, especially among proteins encoded by the clade 7 tandem gene clusters. It is unknown if the variation in conserved residues in clade II proteins could be a reflection of differences in function, for example use of translation repression rather than RNA cleavage, or if many of the genes in the expanded clusters of the clade are pseudogenes. We also cannot rule out the possibility that some predicted gene models might be incorrect, leading to misalignment of key residues.
The differences in transcript levels and patterns of the various AGO genes in P. sojae further highlight the divergences in potential function among the genes, though we note that transcript levels do not always accurately reflect protein levels. Whereas the DCL1, DCL2, and RDR genes in P. sojae exhibit fairly stable transcript levels, comparable to housekeeping genes, the AGO genes exhibited very different transcript levels. AGO1 exhibited constitutive transcript levels across tissue types nearly as high as the housekeeping genes tested and well above DCL1, DCL2, and RDR. AGO2 and AGO4 also exhibited quite consistent transcript levels, except for some reduction in zoospores and germinating cysts, and at a level below AGO1 and above DCL1, DCL2, and RDR. AGO3 and AGO5 exhibited levels around 10-fold lower than AGO2 and AGO4, while AGO6 and AGO9 transcripts levels were 100-fold lower and AGO8 levels were 1000-fold lower. In some samples these weakly transcribed AGO genes exhibited no detectable transcripts. The large variation in transcript levels among the different AGO genes also suggests distinct differences in function and/or that some are pseudogenes. The large variation in transcript levels among AGO3 through AGO9 occurred despite the colocalization of the genes in the genome. Surprisingly, transcript levels from the pseudogene AGO7 were almost as high as those from AGO2 and AGO4. None of the AGO genes showed strong tissue-specific transcript levels, except that AGO2, AGO3, AGO4, and AGO9 transcript levels showed some reduction in zoospores and germinating cysts. In summary, at least one AGO clade I gene and one AGO II gene always had transcript levels comparable to housekeeping genes but some AGO paralogs show substantial variation.
As indicated previously (Fahlgren et al., 2013; Jia et al., 2017), the presence of two size classes of small RNA (sRNA) in Phytophthora species correlates with the two major AGO clades. Åsman et al. (2016) demonstrated a physical association between the P. infestans PiAGO1 protein and 20-22 nt sRNAs and a miRNA, and between the AGO3 homolog PiAGO4 and 24-26 nt sRNAs. Interestingly, they found that the AGO2 homolog (named PiAGO3) had an early indel and a truncation in their strain of P. infestans, making it apparently non-functional. Their findings suggest that in Phytophthora, AGO I clade proteins may be specific to the smaller size class of sRNAs, and AGO II clade proteins may be specific to the larger sRNA size class. We and others (Fahlgren et al., 2013; Åsman et al., 2016; Jia et al., 2017) previously found a smaller size class of sRNAs that were derived from Crinkler and RXLR effector genes that are important for Phytophthora infection of the host plant; this is the class associated with PiAGO1 (Åsman et al., 2016). Only one miRNA has been identified in Phytophthora (Fahlgren et al., 2013), but if the AGO I proteins function in all 20-22 nt size sRNA RISC complexes, and not just miRNA pathways, then the high transcript levels of AGO1 seen in P. sojae may be important for other types of small RNA regulation. The transcript level of AGO1 is in fact high in both mycelium and zoospores, but drops six-fold in germinated cysts and remains at levels three to five-fold below the mycelial level until 48 hpi, similar to the pattern seen for DCL1, DCL2, and RDR. Perhaps AGO1 regulation of effector transcript levels is relaxed upon encounter with a host plant. Further analysis of AGO homologs and their partner small RNAs will further reveal how Phytophthora species use small RNAs to regulate their own transcript levels and their interaction with host plants.
All authors contributed to designing and analyzing experiments, specifically SB was the primary contributor with help from CP and NG on design of transcript level assays and NG and BT on phylogenetic analysis. All authors also contributed to the drafting, revising, and final approval of the manuscript and are accountable for its accuracy.
This work was supported in part by grants 2008-35600-18780 and 2011-68004-30154 from the USDA National Institute of Food and Agriculture and CRIS project number 5358-22000-041-00D from the USDA Agricultural Research Service to NG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Christopher M. Sullivan and Brent Kronmiller for excellent technical support and the Center for Genome Research and Biocomputing, Oregon State University for use of the bioinformatics computing cluster. We also thank Wayne Reeve and Igor Grigoriev (P. cinnamomi) and Wenwu Ye (P. melonis) for making unpublished genomes available for this analysis. Mention of trade names or commercial products in this manuscript are solely for the purpose of providing specific information and do not imply recommendation or endorsement by the U.S. Department of Agriculture.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02841/full#supplementary-material
Åsman, A. K. M., Fogelqvist, J., Vetukuri, R. R., and Dixelius, C. (2016). Phytophthora infestans Argonaute 1 binds microRNA and small RNAs from effector genes and transposable elements. New Phytol. 211, 993–1007. doi: 10.1111/nph.13946
Azzam, G., Smibert, P., Lai, E. C., and Liu, J.-L. (2012). Drosophila Argonaute 1 and its miRNA biogenesis partners are required for oocyte formation and germline cell division. Dev. Biol. 365, 384–394. doi: 10.1016/j.ydbio.2012.03.005
Baurain, D., Brinkmann, H., Petersen, J., Rodríguez-Ezpeleta, N., Stechmann, A., Demoulin, V., et al. (2010). Phylogenomic evidence for separate acquisition of plastids in cryptophytes, haptophytes, and stramenopiles. Mol. Biol. Evol. 27, 1698–1709. doi: 10.1093/molbev/msq059
Baxter, L., Tripathy, S., Ishaque, N., Boot, N., Cabral, A., Kemen, E., et al. (2010). Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 330, 1549–1551. doi: 10.1126/science.1195203
Blair, J. E., Coffey, M. D., Park, S.-Y., Geiser, D. M., and Kang, S. (2008). A multi-locus phylogeny for phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol. 45, 266–277. doi: 10.1016/j.fgb.2007.10.010
Boland, A., Tritschler, F., Heimstädt, S., Izaurralde, E., and Weichenrieder, O. (2010). Crystal structure and ligand binding of the MID domain of a eukaryotic Argonaute protein. EMBO Rep. 11, 522–527. doi: 10.1038/embor.2010.81
Bollmann, S. R., Fang, Y., Press, C. M., Tyler, B. M., and Grünwald, N. J. (2016). Diverse evolutionary trajectories for small RNA biogenesis genes in the oomycete genus Phytophthora. Front. Plant Sci. 7:284. doi: 10.3389/fpls.2016.00284
Braun, L., Cannella, D., Ortet, P., Barakat, M., Sautel, C. F., Kieffer, S., et al. (2010). A complex small RNA repertoire is generated by a plant/fungal-like machinery and effected by a metazoan-like Argonaute in the single-cell human parasite Toxoplasma gondii. PLoS Pathog. 6:e1000920. doi: 10.1371/journal.ppat.1000920
Brennecke, J., Aravin, A. A., Stark, A., Dus, M., Kellis, M., Sachidanandam, R., et al. (2007). Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128, 1089–1103. doi: 10.1016/j.cell.2007.01.043
Burge, C., and Karlin, S. (1997). Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94. doi: 10.1006/jmbi.1997.0951
Cerutti, H., and Casas-Mollano, J. A. (2006). On the origin and functions of RNA-mediated silencing: from protists to man. Curr. Genet 50, 81–99. doi: 10.1007/s00294-006-0078-x
Czech, B., Zhou, R., Erlich, Y., Brennecke, J., Binari, R., Villalta, C., et al. (2009). Hierarchical rules for argonaute loading in Drosophila. Mol. Cell. 36, 445–456. doi: 10.1016/j.molcel.2009.09.028
Djikeng, A., Shi, H., Tschudi, C., and Ullu, E. (2001). RNA interference in Trypanosoma brucei: cloning of small interfering RNAs provides evidence for retroposon-derived 24-26-nucleotide RNAs. RNA 7, 1522–1530.
Faehnle, C. R., Elkayam, E., Haase, A. D., Hannon, G. J., and Joshua-Tor, L. (2013). The making of a slicer: activation of human argonaute-1. Cell Reports 3, 1901–1909. doi: 10.1016/j.celrep.2013.05.033
Fahlgren, N., Bollmann, S. R., Kasschau, K. D., Cuperus, J. T., Press, C. M., Sullivan, C. M., et al. (2013). Phytophthora have distinct endogenous small RNA populations that include short interfering and microRNAs. PLoS One 8:e77181. doi: 10.1371/journal.pone.0077181
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., et al. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222. doi: 10.1093/nar/gkp985
Frank, F., Hauver, J., Sonenberg, N., and Nagar, B. (2012). Arabidopsis Argonaute MID domains use their nucleotide specificity loop to sort small RNAs. EMBO J. 31, 3588–3595. doi: 10.1038/emboj.2012.204
Frank, F., Sonenberg, N., and Nagar, B. (2010). Structural basis for 5′-nucleotide base-specific recognition of guide RNA by human AGO2. Nature 465, 818–822. doi: 10.1038/nature09039
Fry, W. (2008). Phytophthora infestans: the plant (and R gene) destroyer. Mol. Plant Pathol. 9, 385–402. doi: 10.1111/j.1364-3703.2007.00465.x
Gagnon, K. T., Li, L., Chu, Y., Janowski, B. A., and Corey, D. R. (2014). RNAi factors are present and active in human cell nuclei. Cell Rep. 6, 211–221. doi: 10.1016/j.celrep.2013.12.013
Garcia Silva, M. R., Tosar, J. P., Frugier, M., Pantano, S., Bonilla, B., Esteban, L., et al. (2010). Cloning, characterization and subcellular localization of a Trypanosoma cruzi argonaute protein defining a new subfamily distinctive of trypanosomatids. Gene 466, 26–35. doi: 10.1016/j.gene.2010.06.012
Ghildiyal, M., Xu, J., Seitz, H., Weng, Z., and Zamore, P. D. (2010). Sorting of Drosophila small silencing RNAs partitions microRNA∗ strands into the RNA interference pathway. RNA 16, 43–56. doi: 10.1261/rna.1972910
Grünwald, N. J., Goss, E. M., and Press, C. M. (2008). Phytophthora ramorum: a pathogen with a remarkably wide host range causing sudden oak death on oaks and ramorum blight on woody ornamentals. Mol. Plant Pathol. 9, 729–740. doi: 10.1111/j.1364-3703.2008.00500.x
Gu, S., Jin, L., Huang, Y., Zhang, F., and Kay, M. A. (2012). Slicing-independent RISC activation requires the argonaute PAZ domain. Curr. Biol. 22, 1536–1542. doi: 10.1016/j.cub.2012.06.040
Haas, B. J., Kamoun, S., Zody, M. C., Jiang, R. H. Y., Handsaker, R. E., Cano, L. M., et al. (2009). Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461, 393–398. doi: 10.1038/nature08358
Hauptmann, J., Dueck, A., Harlander, S., Pfaff, J., Merkl, R., and Meister, G. (2013). Turning catalytically inactive human Argonaute proteins into active slicer enzymes. Nat. Struct. Mol. Biol. 20, 814–817. doi: 10.1038/nsmb.2577
Huang, V., and Li, L.-C. (2014). Demystifying the nuclear function of Argonaute proteins. RNA Biol. 11, 18–24. doi: 10.4161/rna.27604
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Jannot, G., Boisvert, M.-E. L., Banville, I. H., and Simard, M. J. (2008). Two molecular features contribute to the Argonaute specificity for the microRNA and RNAi pathways in C. elegans. RNA 14, 829–835. doi: 10.1261/rna.901908
Jia, J., Lu, W., Zhong, C., Zhou, R., Xu, J., Liu, W., et al. (2017). The 25-26 nt Small RNAs in Phytophthora parasitica are associated with efficient silencing of homologous endogenous genes. Front. Microbiol. 8:773. doi: 10.3389/fmicb.2017.00773
Kamoun, S. (2003). Molecular genetics of pathogenic oomycetes. Eukaryot. Cell 2, 191–199. doi: 10.1128/EC.2.2.191-199.2003
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. doi: 10.1093/bib/bbx108 [Epub ahead of print].
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kuraku, S., Zmasek, C. M., Nishimura, O., and Katoh, K. (2013). aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 41, W22–W28. doi: 10.1093/nar/gkt389
Kwak, P. B., and Tomari, Y. (2012). The n domain of argonaute drives duplex unwinding during RISC assembly. Nat. Struct. Mol. Biol. 19, 145–151. doi: 10.1038/nsmb.2232
Lévesque, C. A., Brouwer, H., Cano, L., Hamilton, J. P., Holt, C., Huitema, E., et al. (2010). Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 11:R73. doi: 10.1186/gb-2010-11-7-r73
Lin, H., and Spradling, A. C. (1997). A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development 124, 2463–2476.
Lingel, A., Simon, B., Izaurralde, E., and Sattler, M. (2003). Structure and nucleic-acid binding of the Drosophila Argonaute 2 PAZ domain. Nature 426, 465–469. doi: 10.1038/nature02123
Lingel, A., Simon, B., Izaurralde, E., and Sattler, M. (2004). Nucleic acid 3’-end recognition by the Argonaute2 PAZ domain. Nat. Struct. Mol. Biol. 11, 576–577. doi: 10.1038/nsmb777
Links, M. G., Holub, E., Jiang, R. H. Y., Sharpe, A. G., Hegedus, D., Beynon, E., et al. (2011). De novo sequence assembly of Albugo candida reveals a small genome relative to other biotrophic oomycetes. BMC Genomics 12:503. doi: 10.1186/1471-2164-12-503
Liu, J., Carmell, M. A., Rivas, F. V., Marsden, C. G., Thomson, J. M., Song, J.-J., et al. (2004). Argonaute 2 is the catalytic engine of mammalian RNAi. Science 305, 1437–1441. doi: 10.1126/science.1102513
Ma, J.-B., Ye, K., and Patel, D. J. (2004). Structural basis for overhang-specific small interfering RNA recognition by the PAZ domain. Nature 429, 318–322. doi: 10.1038/nature02519
Michalik, K. M., Böttcher, R., and Förstemann, K. (2012). A small RNA response at DNA ends in Drosophila. Nucleic Acids Res. 40, 9596–9603. doi: 10.1093/nar/gks711
Murphy, D., Dancis, B., and Brown, J. R. (2008). The evolution of core proteins involved in microRNA biogenesis. BMC Evol. Biol. 8:92. doi: 10.1186/1471-2148-8-92
Musiyenko, A., Majumdar, T., Andrews, J., Adams, B., and Barik, S. (2012). PRMT1 methylates the single Argonaute of Toxoplasma gondii and is important for the recruitment of Tudor nuclease for target RNA cleavage by antisense guide RNA. Cell Microbiol. 14, 882–901. doi: 10.1111/j.1462-5822.2012.01763.x
Nakanishi, K., Weinberg, D. E., Bartel, D. P., and Patel, D. J. (2012). Structure of yeast Argonaute with guide RNA. Nature 486, 368–374. doi: 10.1038/nature11211
Okamura, K., Liu, N., and Lai, E. C. (2009). Distinct mechanisms for MicroRNA strand selection by Drosophila Argonautes. Mol. Cell 36, 431–444. doi: 10.1016/j.molcel.2009.09.027
Parker, J. S., Roe, S. M., and Barford, D. (2005). Structural insights into mRNA recognition from a PIWI domain–siRNA guide complex. Nature 434, 663–666. doi: 10.1038/nature03462
Pfaffl, M. W., Horgan, G. W., and Dempfle, L. (2002). Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30:e36. doi: 10.1093/nar/30.9.e36
Pfaffl, M. W., Tichopad, A., Prgomet, C., and Neuvians, T. P. (2004). Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: best Keeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515. doi: 10.1023/B:BILE.0000019559.84305.47
Qutob, D., Hraber, P. T., Sobral, B. W., and Gijzen, M. (2000). Comparative analysis of expressed sequences in Phytophthora sojae. Plant Physiol. 123, 243–254. doi: 10.1104/pp.123.1.243
Rivas, F. V., Tolia, N. H., Song, J.-J., Aragon, J. P., Liu, J., Hannon, G. J., et al. (2005). Purified Argonaute2 and an siRNA form recombinant human RISC. Nat. Struct. Mol. Biol. 12, 340–349. doi: 10.1038/nsmb918
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Rozen, S., and Skaletsky, H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386.
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. doi: 10.1093/oxfordjournals.molbev.a040454
Schwarz, D. S., Hutvágner, G., Du, T., Xu, Z., Aronin, N., and Zamore, P. D. (2003). Asymmetry in the assembly of the RNAi enzyme complex. Cell 115, 199–208. doi: 10.1016/S0092-8674(03)00759-1
Shabalina, S. A., and Koonin, E. V. (2008). Origins and evolution of eukaryotic RNA interference. Trends Ecol. Evol. 23, 578–587. doi: 10.1016/j.tree.2008.06.005
Sheng, G., Zhao, H., Wang, J., Rao, Y., Tian, W., Swarts, D. C., et al. (2014). Structure-based cleavage mechanism of Thermus thermophilus Argonaute DNA guide strand-mediated DNA target cleavage. PNAS 111, 652–657. doi: 10.1073/pnas.1321032111
Shi, H., Chamond, N., Djikeng, A., Tschudi, C., and Ullu, E. (2009). RNA interference in Trypanosoma brucei: role of the n-terminal RGG domain and the polyribosome association of argonaute. J. Biol. Chem. 284, 36511–36520. doi: 10.1074/jbc.M109.073072
Shi, H., Ullu, E., and Tschudi, C. (2004). Function of the Trypanosome Argonaute 1 protein in RNA interference requires the N-terminal RGG domain and arginine 735 in the Piwi domain. J. Biol. Chem. 279, 49889–49893. doi: 10.1074/jbc.M409280200
Song, J.-J., Liu, J., Tolia, N. H., Schneiderman, J., Smith, S. K., Martienssen, R. A., et al. (2003). The crystal structure of the Argonaute2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nat. Struct. Mol. Biol. 10, 1026–1032. doi: 10.1038/nsb1016
Song, J.-J., Smith, S. K., Hannon, G. J., and Joshua-Tor, L. (2004). Crystal structure of argonaute and its implications for RISC slicer activity. Science 305,1434–1437. doi: 10.1126/science.1102514
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tabima, J. F., Kronmiller, B. A., Press, C. M., Tyler, B. M., Zasada, I. A., and Grünwald, N. J. (2017). Whole genome sequences of the raspberry and strawberry pathogens Phytophthora rubi and P. fragariae. Mol. Plant Microbe Interact. 30, 767–769. doi: 10.1094/MPMI-04-17-0081-A
Tolia, N. H., and Joshua-Tor, L. (2007). Slicer and the argonautes. Nat. Chem. Biol. 3, 36–43. doi: 10.1038/nchembio848
Tomari, Y., Du, T., and Zamore, P. D. (2007). Sorting of Drosophila small silencing RNAs. Cell 130, 299–308. doi: 10.1016/j.cell.2007.05.057
Tyler, B. M. (2007). Phytophthora sojae: root rot pathogen of soybean and model oomycete. Mol. Plant Pathol. 8, 1–8. doi: 10.1111/j.1364-3703.2006.00373.x
Tyler, B. M., Tripathy, S., Zhang, X., Dehal, P., Jiang, R. H. Y., Aerts, A., et al. (2006). Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 313, 1261–1266. doi: 10.1126/science.1128796
Vetukuri, R. R., Avrova, A. O., Grenville-Briggs, L. J., Van West, P., Söderbom, F., Savenkov, E. I., et al. (2011). Evidence for involvement of dicer-like, argonaute and histone deacetylase proteins in gene silencing in Phytophthora infestans. Mol. Plant Pathol. 12, 772–785. doi: 10.1111/j.1364-3703.2011.00710.x
Wei, W., Ba, Z., Gao, M., Wu, Y., Ma, Y., Amiard, S., et al. (2012). A role for small RNAs in DNA double-strand break repair. Cell 149, 101–112. doi: 10.1016/j.cell.2012.03.002
Yan, K. S., Yan, S., Farooq, A., Han, A., Zeng, L., and Zhou, M.-M. (2003). Structure and conserved RNA binding of the PAZ domain. Nature 426, 469–474. doi: 10.1038/nature02129
Yigit, E., Batista, P. J., Bei, Y., Pang, K. M., Chen, C.-C. G., Tolia, N. H., et al. (2006). Analysis of the C. elegans argonaute family reveals that distinct argonautes act sequentially during RNAi. Cell 127, 747–757. doi: 10.1016/j.cell.2006.09.033
Keywords: Phytophthora, argonaute, Chromalveolate, evolution, small RNA, stramenopiles
Citation: Bollmann SR, Press CM, Tyler BM and Grünwald NJ (2018) Expansion and Divergence of Argonaute Genes in the Oomycete Genus Phytophthora. Front. Microbiol. 9:2841. doi: 10.3389/fmicb.2018.02841
Received: 09 August 2018; Accepted: 05 November 2018;
Published: 30 November 2018.
Edited by:
Baolei Jia, Chung-Ang University, South KoreaReviewed by:
Vasudevan Seshadri, National Centre for Cell Science (NCCS), IndiaCopyright © 2018 Bollmann, Press, Tyler and Grünwald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Niklaus J. Grünwald, bmlrLmdydW53YWxkQGFycy51c2RhLmdvdg==
†Present address: Stephanie R. Bollmann, Department of Integrative Biology, Oregon State University, Corvallis, OR, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.