 Irina A. Garanina
Irina A. Garanina Gleb Y. Fisunov
Gleb Y. Fisunov Vadim M. Govorun1,3
Vadim M. Govorun1,3- 1Federal Research and Clinical Centre of Physical-Chemical Medicine, Moscow, Russia
- 2Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Moscow, Russia
- 3Moscow Institute of Physics and Technology, Dolgoprudny, Russia
Prokaryotes are actively studied objects in the scope of genomic regulation. Microbiologists need special tools for complex analysis of data to study and identification of regulatory mechanism in bacteria and archaea.
We developed a tool BAC-BROWSER, specifically for visualization and analysis of small prokaryotic genomes. BAC-BROWSER provides tools for different types of analysis to study a wide set of regulatory mechanisms of prokaryotes:
- transcriptional regulation by transcription factors (TFs), analysis of TFs, their targets, and binding sites.
- other regulatory motifs, promoters, terminators and ribosome binding sites
- transcriptional regulation by variation of operon structure, alternative starts or ends of transcription.
- non-coding RNAs, antisense RNAs
- RNA secondary structure, riboswitches
- GC content, GC skew, codon usage
BAC-browser incorporated free programs accelerating the verification of obtained results: primer design and oligocalculator, vector visualization, the tool for synthetic gene construction. The program is designed for Windows operating system and freely available for download in http://smdb.rcpcm.org/tools/index.html.
Introduction
Prokaryotes are the great source for discovery of new regulatory mechanisms. Their diversity and relative simplicity of genome organization make them suitable objects for different -omics and comparative analysis. Regulation by transcription factors is considered as well described for bacteria, but even for model bacteria Escherichia coli and Bacillus subtilis new TFs and targets are still being identified (Keseler et al., 2013; Gama-Castro et al., 2016; Fang et al., 2017; Belliveau et al., 2018; Gao et al., 2018). Since the low conservation of regulatory networks, regulation of other bacteria, including important pathogens, remains obscure (Lozada-Chávez et al., 2006; Rodionov, 2007; Fisunov et al., 2016; Eckweiler et al., 2018). Operon structure is one of the interesting and poorly studied features of prokaryotes. An operon is defined as a set of genes transcribed en bloc, so operon consists of several genes transcribed and regulated together (Jacob et al., 1960). New data about bacterial transcription showed that operon structure is not stable and can vary in different conditions (Koide et al., 2009; Mao et al., 2015; Junier and Rivoire, 2016). To cope with this discrepancy, genes transcribed and regulated together were called transcriptional units (TU) (Cho et al., 2009; Junier and Rivoire, 2016) and now term operon has more meaning in terms of functional rather than direct transcriptional link between genes (Okuda et al., 2007). Identification of TUs is a new task that has to be resolved to understand transcriptional regulation. Exact mapping of TUs can facilitate identification of new riboswitches, non-coding RNA, antisense transcripts (Sharma et al., 2010; Boutard et al., 2016). The same TU may have multiple TSSs (transcription start sites) and transcription ends. Alternative TSSs in bacteria are found for 15–60% genes and operons (Stazic and Voß, 2016), they are also involved in transcription regulation (Cho et al., 2009; Li et al., 2015). To characterize both poorly studied regulatory mechanisms and classical ways of regulation is needed to use complex information about transcription and translation and combine it with genomics data. For this purpose were developed special tools and programs (Pavlopoulos et al., 2015).
There are many specific tools for TU mapping, TSS and terminator prediction. They use various strategies for data analysis (Amman et al., 2014; Fortino et al., 2014, 2016; Čuklina et al., 2016; Promworn et al., 2017), and some can visualize the results (McClure et al., 2013; Hilker et al., 2016). However, these tools generally concentrated on one or few aspects of regulation. Wide set of tools utilize known algorithms for high-throughput data analysis and genomes visualization but none of them allow analysis of complex operon structure of prokaryotes (Carver et al., 2012; Okonechnikov et al., 2012; Lechat et al., 2013; Dietrich et al., 2014).
We developed new genome browser BAC-BROWSER designed for molecular biologists and microbiologists that combine user-friendly interface with multiple functions for prokaryotic regulation analysis. Using RNA-seq read coverage BAC-BROWSER can identify TSSs and terminators, map and visualize TUs. BAC-BROWSER provides a wide set of algorithms for analysis of genes, proteins, regulatory motifs and structural elements, like hairpins and riboswitches. Furthermore, the program facilitates subsequent verification of computational results with built-in tools for molecular biology analysis. BAC-BROWSER includes free modules for primer design, vector representation, and analysis. Based on BAC-BROWSER prediction we identified TSSs and gene promoters that helped to reveal a new mechanism of transcriptional regulation and reconstruct transcriptional control network for three bacteria species (Mazin et al., 2014; Fisunov et al., 2016).
Implementation
General Information
BAC-BROWSER software was written in VB.NET 9.0. The program loads genomes in Fasta, Genbank, and GFF format.
Analysis of Coverage and Transcription Unit Mapping
BAC-BROWSER intakes coverage data in SAM/BAM format or in own format. BAC-BROWSER format represents the array of values, where each one represents coverage at a given position starting from the first nucleotide. The SAM format can be converted into BAC-BROWSER format. The coverage dataset can be assembled into a single table file, which simplifies loading of data. The coverage can be represented as full read coverage or first nucleotide coverage. In the second method only first nucleotide contributes to the coverage, while the remaining read is skipped. This representation is used for analysis of 5′-end enriched RNA-seq coverage (Mazin et al., 2014).
The algorithm of transcription unit identification from the coverage have been described in Mazin et al. (2014). Briefly, the algorithm calculates coverage derivative and identifies its local extremes, which correspond to local steps in coverage. The steps may originate from TSSs, transcriptional terminators and RNA ends produced by RNA processing as well as coverage noise produced fortuitously. The algorithm further splits coverage into intervals between consecutive steps and identifies statistical significance of coverage difference between adjacent intervals. In current implementation we use standard deviation, which threshold can be manually set. The original implementation described a quasi-Poisson distribution test. Then if the difference is statistically insignificant the step is removed and intervals are joined. The process repeats iteratively until convergence. The final set of intervals can be assigned as transcription units (operons). The borders between the intervals correspond to TSSs and transcriptional terminators, respectively (Mazin et al., 2014). So, this algorithm identifies TSSs, terminators, and TUs. However, its accuracy depends on the quality of data and parameters the user chooses.
The exact identification of TSSs with single-nucleotide resolution requires first nucleotide coverage data from 5′-end enriched RNA-seq library. The method is different from the algorithm for TU identification and also have been published (Mazin et al., 2014). The program scans the coverage for local maxima, which correspond to TSSs. The algorithm accounts for background signal to identify maxima that are above the noise threshold. The latter is dynamically calculated for the local sequence area (parameters are user-adjusted).
It is also possible to export normalized coverage (FPKM) of annotated features for further analysis. A coverage of a single gene is normalized to the total library coverage or to the coverage of CDSs (Anders and Huber, 2010). The latter excludes overrepresented features like rRNAs and tRNAs that may introduce quantitative bias.
Applications for Sequence Analysis, Oligonucleotide Thermodynamics and Secondary Structure Analysis and Motif Search
Standard algorithms for sequence search are implemented in the application (Altschul et al., 1990). At first stage query and subject sequences are splitted into K-words. K-words are indexed and the matrix of matched hits is built. Matched hits serve as seeds to expand alignment. The gaps between seeds are filled by Wunch-Needleman algorithm (Needleman and Wunsch, 1970). A user can adjust thresholds for sequence similarity, minimal alignment length and length of seeds.
Positional weight matrixes (PWMs) are built from a user-supplied set of aligned sequences. For logo plot construction user can apply log-likelihood score or Shannon positional entropy (Li and Tompa, 2006). PWMs are stored in text format and can be easily edited. External or manually constructed PWMs are suitable for search. The program scans a genome and finds PWM matches above the threshold score manually selected by a user. The score is calculated by the standard method as a sum of frequencies of matched nucleotides. For motif de novo search we implemented modified MEME algorithm (Bailey and Elkan, 1994).
Thermodynamic parameters for oligonucleotides are calculated by the modified Allawi and SantaLucia’s the nearest-neighbor method for DNA (Allawi and SantaLucia, 1997). Thermodynamic parameters for RNA hairpins (used in hairpin finder at genomic view window) are calculated by the nearest-neighbor algorithm for RNA (SantaLucia, 1998). Secondary structures and of oligonucleotide dimers are calculated using iterative annealing with subsequent calculation of duplex stability. For calculation of hairpins, the oligonucleotide is split into two halves and iterative annealing of halves is performed. A search of secondary structures in a genome is performed in a sliding window. For each subsequence the algorithm calculates the hairpin stability in the same way as for oligonucleotide (see above), but with RNA thermodynamic parameters.
The identification of repetitive elements is performed in a sliding window. For each subsequence the algorithm identifies if N nucleotides separated by the spacer of K length form direct or inverted repeats.
Tools for Molecular Biology
PCR primer design in the program includes the following methods: automatic primer design for the given sequence and manual primer design. The first method designs a list of primers that correspond to the manual set of parameters like Tm, amplicon length and secondary structure stability. The algorithm makes the list of subsequences from the sequence to which the primers have to be designed. Then the algorithm filters subsequences until the appropriate ones are found. The second method allows designing oligonucleotide directly on DNA sequence within the viewer window. A user can move and resize oligonucleotide on genomic sequence and Tm is calculated on-flight. This method can be used to design oligonucleotides for gene synthesis or probes for real-time PCR. Tm is calculated by the modified Allawi and SantaLucia’s thermodynamics method (Allawi and SantaLucia, 1997). Calculation of oligonucleotide concentration from OD is performed by approximation of Beer-Lambert law by the following equation: OD / (nA × 15200 + nG × 12010 + nC × 7050 + nT × 8400 + M), where nA, nG, nC, and nT is the number of respective nucleotides and M corresponds for absorbance of DNA modifications (if present).
The protein isoelectric point is calculated based on the Henderson-Hasselbach equation and table values of pK for charged amino acids. The program iteratively adjusts pH until the isoelectric point is reached.
Results and Discussion
Sequence and Coverage Analysis
In BAC-BROWSER there are available linear and circular sequence representations. An easy and usable interface enables to manipulate with annotation, gene sequences, mark and retrieve nucleotide fragments or directly analyze them. BAC-BROWSER allows manual correction, addition, and import of annotation. BAC-BROWSER provides simple extraction of a single or multiple genes or protein sequences. The program can upload files with more than one sequence and show it as one genome. Sequence search is available in the application. BAC-BROWSER performs a search of nucleotide and protein sequences with the customizable percent of identity and can do the simultaneous search of several sequences uploaded in Fasta file. The program readily works with short sequences as vectors, viral and bacterial genomes. BAC-browser will work with the human genome and other large genomes, but it is not designed for this and the analysis will take a long time.
In BAC-BROWSER, there are more than 15 tools implemented for sequence and associated coverage analysis:
- genome sequence searching and editing;
- tools for editing genome annotation: annotation amendment, importing and exporting of features, open reading frame (ORF) prediction;
- tools for motif search and analysis like PWM scanning and building, and also de novo motif search;
- tools for repeat and hairpin formation search;
- restriction site search and enzyme mapping;
- BLAST search;
- GC content (Figure 1) and GC skew calculators, and codon usage.
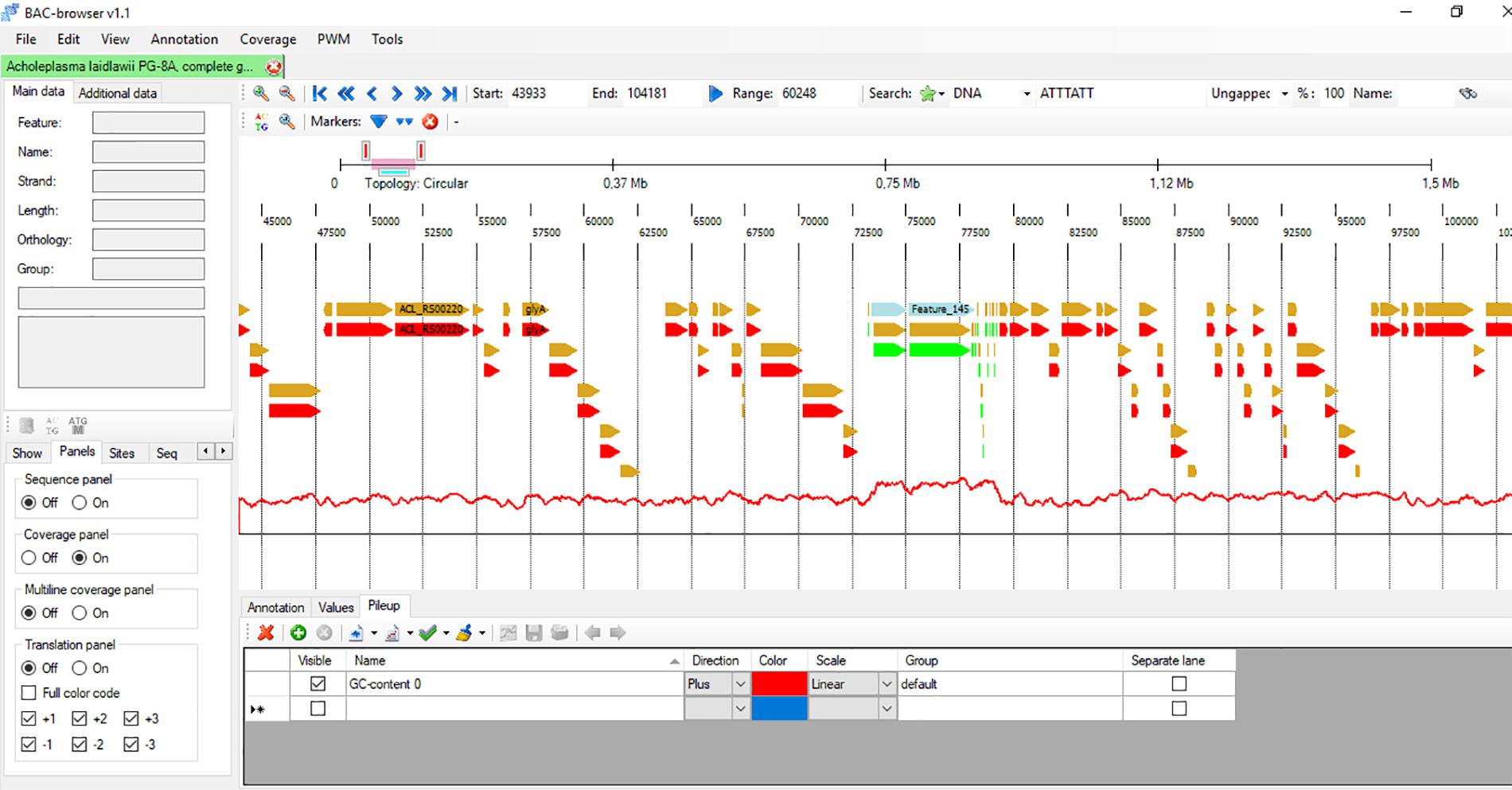
FIGURE 1. The screenshot of BAC-browser. In genome panel the bacterium Acholeplasma laidlawii genome is shown. You can see genes as arrows, the red line under genes shows GC content calculated in 200 nt window. On this example the local GC content increase is observed in the region of 73000–78000 nts there ribosomal RNAs are located.
RNA-seq analysis output in SAM format can be directly loaded by the application. SAM/BAM file user can obtain from programs for RNA-seq analysis such as Bowtie, BWA or TopHat (Li and Durbin, 2009; Langmead and Salzberg, 2012; Kim et al., 2013). Read alignments are automatically converted into nucleotide coverage. BAC-BROWSER can perform coverage normalization and log transformation. A user chooses the type of coverage representation depending on RNA-seq library type. BAC-BROWSER generates standard nucleotide coverage and coverage obtained with first nucleotides of reads. The second type of coverage we recommend to use for 5′ enriched RNA-seq (Sharma et al., 2010; Creecy and Conway, 2015). This type of RNA-seq library preparation used for exact TSSs mapping and quantitate transcript analysis (Mazin et al., 2014; Fisunov et al., 2016). The program also can load other quantitate or qualitative data in simple and universal BAC-BROWSER format. It can be used for methylation, SNP or GC content display. So, BAC-BROWSER provides a simultaneous view on biological data of different types and keeps it in lightweight universal format.
Prediction of Transcriptional Units
In the program we implemented a method for condition-specific TU analysis in bacteria. The program analyses RNA-seq data for fast identification of TSSs, transcription terminators and TUs. This method and algorithm we used in our published work (Mazin et al., 2014), but here we want to compare it with another method and show accuracy on simulated data. For real data testing, we used previously published E. coli dataset (Conway et al., 2014). To get coverage were used processed RNA-seq data. Derived results correspond to results of Conway et al. (2014) and show high sensitivity (0.91) (Table 1).

TABLE 1. Comparison of the results of TU mapping.
For read count simulation was used negative binomial distribution (Frazee et al., 2015). We simulated standard RNA-seq transcriptome reads varying mean coverage of gene from 20 to 400 reads per nucleotide, coverage for each transcript was drawn from the normal distribution. Results of the testing show, that implemented in BAC-BROWSER algorithm for TU identification is sensible for mean transcript coverage and can identify about 90% of TUs (Figure 2).
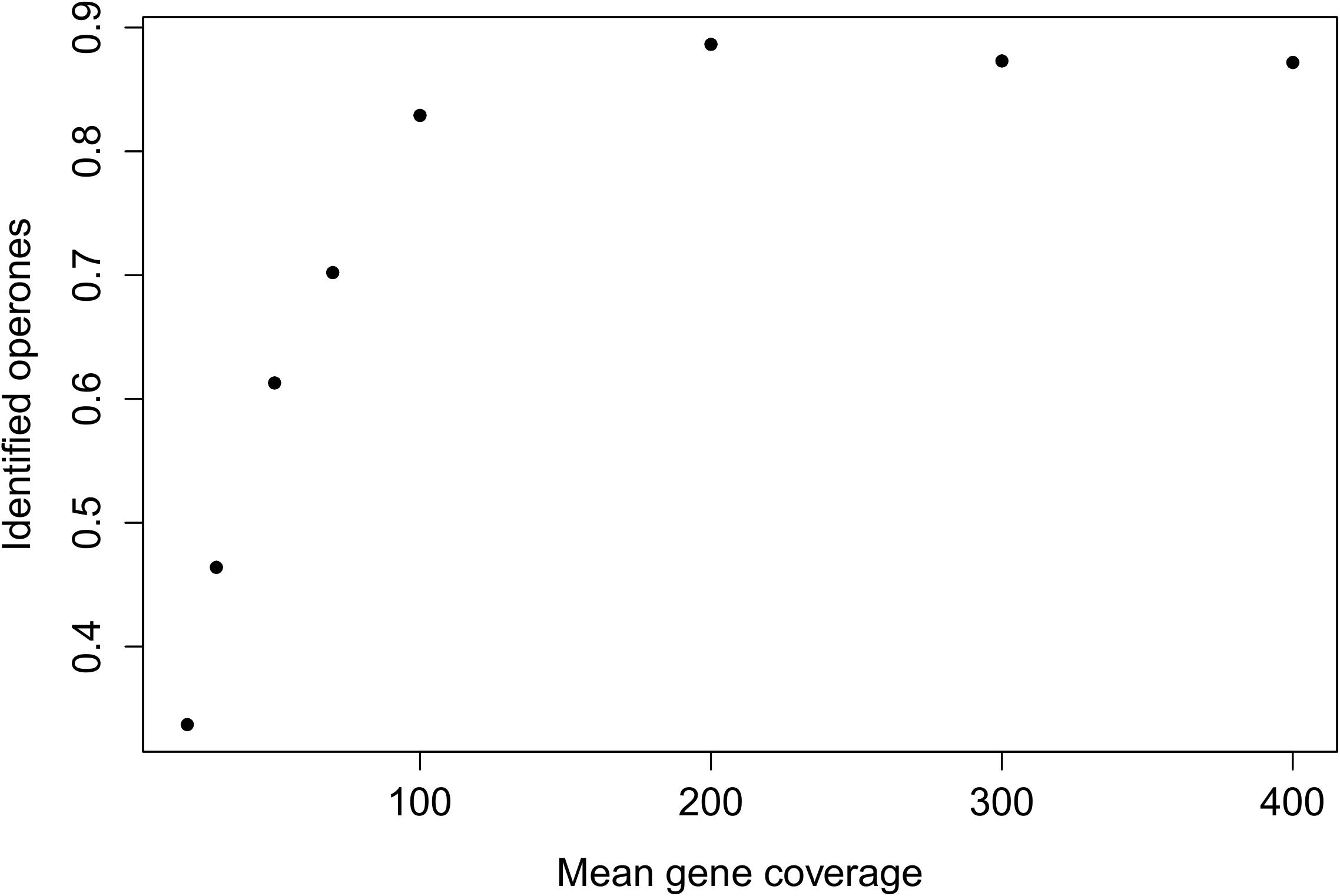
FIGURE 2. Dependence between mean gene coverage in simulated library and fraction of identified operons.
Tools for Molecular Biology
BAC-BROWSER features a set of additional functions for molecular biology: oligonucleotide design, codon usage optimization for amino acid sequences and identification of restriction sites. Oligonucleotide design module includes automatic PCR primer design for a given sequence and manual oligonucleotide design. The program calculates oligonucleotide parameters including dG, Tm and secondary structure. Modifications of 5′ and 3′ ends like fluorophores and quenchers are accounted for. Embedded oligonucleotide calculator can calculate the desired dilutions from OD or molar concentration. Manual oligonucleotide design runs directly in genome view window and allows on-flight calculation of thermodynamic parameters. This tool can be used to design real-time PCR probes, cloning primers and synthetic sequences. The codon optimization module includes calculation of codon usage frequencies for the given genome and re-encoding of the particular amino acid sequence using given codon-usage table. BAC-BROWSER provides the module for in silico 2D electrophoresis analysis. Based on protein properties, the program constructs a theoretical 2D map for all genes in the analyzed genome.
Conclusion
We developed an easy, fast and multifunctional application BAC-BROWSER for prokaryotic genome visualization and analysis. The program combines popular algorithms and methods for better interpretation and analysis of complex data and provides tools for subsequent verification of results with molecular biology methods. The program freely available and will be improved and supplemented in future versions which will be also available for the research community. In particular, we plan to make the web version of the program accessible to users with any operating system. Now BAC-browser has several limitations: it works only in Windows operating system and designed for analysis of small genomes up to 20 Mb in length. The manual describing file formats, usage of the program and parameters of tools is available in http://smdb.rcpcm.org/tools/index.html.
Data Availability
Program in freely available in the SMDB website http://smdb.rcpcm.org/tools/index.html under the GNU GPL license. Operating systems: Windows XP and later versions. Programming language: VB.NET 9.0. Other requirements: none. Any restrictions to use by non-academics: none.
Author Contributions
GF contributed to the software design, software testing, and the drafting of the manuscript. IG contributed to the software testing, bioinformatics analysis, data interpretation, and the drafting of the manuscript. VG contributed to the design of the study and the drafting of the manuscript.
Funding
This work was funded by the Russian Science Foundation grant 14-24-00159 “Systems research of minimal cell on a Mycoplasma gallisepticum model.”
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allawi, H. T., and SantaLucia, J. (1997). Thermodynamics and NMR of internal G.T mismatches in DNA. Biochemistry 36, 10581–10594. doi: 10.1021/bi962590c
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Amman, F., Wolfinger, M. T., Lorenz, R., Hofacker, I. L., Stadler, P. F., and Findeiß, S. (2014). TSSAR: TSS annotation regime for dRNA-seq data. BMC Bioinformatics 15:89. doi: 10.1186/1471-2105-15-89
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Bailey, T. L., and Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36.
Belliveau, N. M., Barnes, S. L., Ireland, W. T., Jones, D. L., Sweredoski, M. J., Moradian, A., et al. (2018). Systematic approach for dissecting the molecular mechanisms of transcriptional regulation in bacteria. Proc. Natl. Acad. Sci. U.S.A. 115, E4796–E4805. doi: 10.1073/pnas.1722055115
Boutard, M., Ettwiller, L., Cerisy, T., Alberti, A., Labadie, K., Salanoubat, M., et al. (2016). Global repositioning of transcription start sites in a plant-fermenting bacterium. Nat. Commun. 7:13783. doi: 10.1038/ncomms13783
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Cho, B.-K., Zengler, K., Qiu, Y., Park, Y. S., Knight, E. M., Barrett, C. L., et al. (2009). The transcription unit architecture of the Escherichia coli genome. Nat. Biotechnol. 27, 1043–1049. doi: 10.1038/nbt.1582
Conway, T., Creecy, J. P., Maddox, S. M., Grissom, J. E., Conkle, T. L., Shadid, T. M., et al. (2014). Unprecedented high-resolution view of bacterial operon architecture revealed by RNA sequencing. mBio 5:e01442-14. doi: 10.1128/mBio.01442-14
Creecy, J. P., and Conway, T. (2015). Quantitative bacterial transcriptomics with RNA-seq. Curr. Opin. Microbiol. 23, 133–140. doi: 10.1016/j.mib.2014.11.011
Čuklina, J., Hahn, J., Imakaev, M., Omasits, U., Förstner, K. U., Ljubimov, N., et al. (2016). Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis - a rich resource to identify new transcripts, proteins and to study gene regulation. BMC Genomics 17:302. doi: 10.1186/s12864-016-2602-9
Dietrich, S., Wiegand, S., and Liesegang, H. (2014). TraV: a genome context sensitive transcriptome browser. PLoS One 9:e93677. doi: 10.1371/journal.pone.0093677
Eckweiler, D., Dudek, C.-A., Hartlich, J., Brötje, D., and Jahn, D. (2018). PRODORIC2: the bacterial gene regulation database in 2018. Nucleic Acids Res. 46, D320–D326. doi: 10.1093/nar/gkx1091
Fang, X., Sastry, A., Mih, N., Kim, D., Tan, J., Yurkovich, J. T., et al. (2017). Global transcriptional regulatory network for Escherichia coli robustly connects gene expression to transcription factor activities. Proc. Natl. Acad. Sci. U.S.A. 114, 10286–10291. doi: 10.1073/pnas.1702581114
Fisunov, G. Y., Garanina, I. A., Evsyutina, D. V., Semashko, T. A., Nikitina, A. S., and Govorun, V. M. (2016). Reconstruction of transcription control networks in mollicutes by high-throughput identification of promoters. Front. Microbiol. 7:1977. doi: 10.3389/fmicb.2016.01977
Fortino, V., Smolander, O.-P., Auvinen, P., Tagliaferri, R., and Greco, D. (2014). Transcriptome dynamics-based operon prediction in prokaryotes. BMC Bioinformatics 15:145. doi: 10.1186/1471-2105-15-145
Fortino, V., Tagliaferri, R., and Greco, D. (2016). CONDOP: an R package for CONdition-dependent operon predictions. Bioinformatics 32, 3199–3200. doi: 10.1093/bioinformatics/btw330
Frazee, A. C., Jaffe, A. E., Langmead, B., and Leek, J. T. (2015). Polyester: simulating RNA-seq datasets with differential transcript expression. Bioinformatics 31, 2778–2784. doi: 10.1093/bioinformatics/btv272
Gama-Castro, S., Salgado, H., Santos-Zavaleta, A., Ledezma-Tejeida, D., Muñiz-Rascado, L., García-Sotelo, J. S., et al. (2016). RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 44, D133–D143. doi: 10.1093/nar/gkv1156
Gao, Y., Yurkovich, J. T., Seo, S. W., Kabimoldayev, I., Drger, A., Chen, K., et al. (2018). Systematic discovery of uncharacterized transcription factors in Escherichia coli K-12 MG1655. Nucleic Acids Res. doi: 10.1093/nar/gky752 [Epub ahead of print].
Hilker, R., Stadermann, K. B., Schwengers, O., Anisiforov, E., Jaenicke, S., Weisshaar, B., et al. (2016). ReadXplorer 2-detailed read mapping analysis and visualization from one single source. Bioinformatics 32, 3702–3708. doi: 10.1093/bioinformatics/btw541
Jacob, F., Perrin, D., Sanchez, C., and Monod, J. (1960). Operon: a group of genes with the expression coordinated by an operator. C. R. Hebd. Seances Acad. Sci. 250, 1727–1729.
Junier, I., and Rivoire, O. (2016). Conserved units of co-expression in bacterial genomes: an evolutionary insight into transcriptional regulation. PLoS One 11:e0155740. doi: 10.1371/journal.pone.0155740
Keseler, I. M., Mackie, A., Peralta-Gil, M., Santos-Zavaleta, A., Gama-Castro, S., Bonavides-Martínez, C., et al. (2013). EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 41, D605–D612. doi: 10.1093/nar/gks1027
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Koide, T., Reiss, D. J., Bare, J. C., Pang, W. L., Facciotti, M. T., Schmid, A. K., et al. (2009). Prevalence of transcription promoters within archaeal operons and coding sequences. Mol. Syst. Biol. 5:285. doi: 10.1038/msb.2009.42
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lechat, P., Souche, E., and Moszer, I. (2013). SynTView - an interactive multi-view genome browser for next-generation comparative microorganism genomics. BMC Bioinformatics 14:277. doi: 10.1186/1471-2105-14-277
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 5, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, J., Qi, L., Guo, Y., Yue, L., Li, Y., Ge, W., et al. (2015). Global mapping transcriptional start sites revealed both transcriptional and post-transcriptional regulation of cold adaptation in the methanogenic archaeon Methanolobus psychrophilus. Sci. Rep. 5:9209. doi: 10.1038/srep09209
Li, N., and Tompa, M. (2006). Analysis of computational approaches for motif discovery. Algorithms Mol. Biol. 1:8. doi: 10.1186/1748-7188-1-8
Lozada-Chávez, I., Janga, S. C., and Collado-Vides, J. (2006). Bacterial regulatory networks are extremely flexible in evolution. Nucleic Acids Res. 34, 3434–3445. doi: 10.1093/nar/gkl423
Mao, X., Ma, Q., Liu, B., Chen, X., Zhang, H., and Xu, Y. (2015). Revisiting operons: an analysis of the landscape of transcriptional units in E. coli. BMC Bioinformatics 16:356. doi: 10.1186/s12859-015-0805-8
Mazin, P. V., Fisunov, G. Y., Gorbachev, A. Y., Kapitskaya, K. Y., Altukhov, I. A., Semashko, T. A., et al. (2014). Transcriptome analysis reveals novel regulatory mechanisms in a genome-reduced bacterium. Nucleic Acids Res. 42, 13254–13268. doi: 10.1093/nar/gku976
McClure, R., Balasubramanian, D., Sun, Y., Bobrovskyy, M., Sumby, P., Genco, C. A., et al. (2013). Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res. 41, e140. doi: 10.1093/nar/gkt444
Needleman, S. B., and Wunsch, C. D. (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48, 443–453. doi: 10.1016/0022-2836(70)90057-4
Okonechnikov, K., Golosova, O., Fursov, M., and Ugene team. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/bioinformatics/bts091
Okuda, S., Kawashima, S., Kobayashi, K., Ogasawara, N., Kanehisa, M., and Goto, S. (2007). Characterization of relationships between transcriptional units and operon structures in Bacillus subtilis and Escherichia coli. BMC Genomics 8:48. doi: 10.1186/1471-2164-8-48
Pavlopoulos, G. A., Malliarakis, D., Papanikolaou, N., Theodosiou, T., Enright, A. J., and Iliopoulos, I. (2015). Visualizing genome and systems biology: technologies, tools, implementation techniques and trends, past, present and future. Gigascience 4:38. doi: 10.1186/s13742-015-0077-2
Promworn, Y., Kaewprommal, P., Shaw, P. J., Intarapanich, A., Tongsima, S., and Piriyapongsa, J. (2017). ToNER: a tool for identifying nucleotide enrichment signals in feature-enriched RNA-seq data. PLoS One 12:e0178483. doi: 10.1371/journal.pone.0178483
Rodionov, D. A. (2007). Comparative genomic reconstruction of transcriptional regulatory networks in bacteria. Chem. Rev. 107, 3467–3497. doi: 10.1021/cr068309
SantaLucia, J. (1998). A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. U.S.A. 95, 1460–1465. doi: 10.1073/pnas.95.4.1460
Sharma, C. M., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiss, S., Sittka, A., et al. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255. doi: 10.1038/nature08756
Keywords: genome browser, viewer, visualization, genome, genes, bacteria, prokaryotes
Citation: Garanina IA, Fisunov GY and Govorun VM (2018) BAC-BROWSER: The Tool for Visualization and Analysis of Prokaryotic Genomes. Front. Microbiol. 9:2827. doi: 10.3389/fmicb.2018.02827
Received: 09 August 2018; Accepted: 05 November 2018;
Published: 21 November 2018.
Edited by:
Andrea Masotti, Bambino Gesù Ospedale Pediatrico (IRCCS), ItalyReviewed by:
Antonella Celluzzi, Bambino Gesù Ospedale Pediatrico (IRCCS), ItalySotirios Vasileiadis, University of Thessaly, Greece
Copyright © 2018 Garanina, Fisunov and Govorun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gleb Y. Fisunov, aGVyci5yb21hbm9mZkBnbWFpbC5jb20=