
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 05 November 2018
Sec. Systems Microbiology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02558
This article is part of the Research TopicDietary Carbohydrate Digestibility and Metabolic Effects in Human HealthView all 11 articles
 Emmanuelle H. Crost1
Emmanuelle H. Crost1 Gwenaelle Le Gall1
Gwenaelle Le Gall1 Jenny A. Laverde-Gomez2Indrani Mukhopadhya2Harry J. Flint2
Jenny A. Laverde-Gomez2Indrani Mukhopadhya2Harry J. Flint2 Nathalie Juge1*
Nathalie Juge1*Dietary and host glycans shape the composition of the human gut microbiota with keystone carbohydrate-degrading species playing a critical role in maintaining the structure and function of gut microbial communities. Here, we focused on two major human gut symbionts, the mucin-degrader Ruminococcus gnavus ATCC 29149, and R. bromii L2-63, a keystone species for the degradation of resistant starch (RS) in human colon. Using anaerobic individual and co-cultures of R. bromii and R. gnavus grown on mucin or starch as sole carbon source, we showed that starch degradation by R. bromii supported the growth of R. gnavus whereas R. bromii did not benefit from mucin degradation by R. gnavus. Further we analyzed the growth (quantitative PCR), metabolite production (1H NMR analysis), and bacterial transcriptional response (RNA-Seq) of R. bromii cultured with RS or soluble starch (SS) in the presence or absence of R. gnavus. In co-culture fermentations on starch, 1H NMR analysis showed that R. gnavus benefits from transient glucose and malto-oligosaccharides released by R. bromii upon starch degradation, producing acetate, formate, and lactate as main fermentation end-products. Differential expression analysis (DESeq 2) on starch (SS and RS) showed that the presence of R. bromii induced changes in R. gnavus transcriptional response of genes encoding several maltose transporters and enzymes involved in its metabolism such as maltose phosphorylase, in line with the ability of R. gnavus to utilize R. bromii starch degradation products. In the RS co-culture, R. bromii showed a significant increase in the induction of tryptophan (Trp) biosynthesis genes and a decrease of vitamin B12 (VitB12)-dependent methionine biosynthesis as compared to the mono-culture, suggesting that Trp and VitB12 availability become limited in the presence of R. gnavus. Together this study showed a direct competition between R. bromii and R. gnavus on RS, suggesting that in vivo, the R. gnavus population inhabiting the mucus niche may be modulated by the supply of non-digestible carbohydrates reaching the colon such as RS.
The human gut is heavily populated by a diverse microbial community (gut microbiota) which plays a crucial role in maintaining human health through, e.g., polysaccharide digestion, metabolite and vitamin production, maturation of the immune system and protection against pathogens (for a review, see Thursby and Juge, 2017). The adult gut microbiota is dominated by members of Firmicutes and Bacteroidetes phyla although organisms from the Actinobacteria, Verrucomicrobia and Proteobacteria phyla also contribute to the structure and function of this microbial community. The microbiota composition varies longitudinally along the gastrointestinal tract but also transversally from the lumen to the mucosa (Donaldson et al., 2016). The colon is the most densely colonized part of the gut, reaching density of 1011–1012 cells per gram. The lumen of the gut is generally considered to host a microbial community which is distinct from that of the mucus layer although partial mixing and dispersal by host factors tend to homogenize the community (Mark Welch et al., 2017; Tropini et al., 2017). Several factors influence the biogeography of symbiotic bacteria within the gut, including the gradient and availability of glycans within discrete physical niches (Koropatkin et al., 2012).
In the colon, bacteria have access to non-digestible polysaccharides from the diet but also to complex oligosaccharides from the host mucins (Koropatkin et al., 2012; Tailford et al., 2015a). It is commonly accepted that diet is the main contributing factor influencing the structure of the gut microbial community in the colon (for a review, see Flint et al., 2017). Dietary alteration in the gut microbiota profile can be temporal (e.g., David et al., 2014) or long-term (e.g., Sonnenburg et al., 2016). One of the largest single source of energy for microbial growth in the human colon is dietary starch that escapes digestion in the upper gut and reaches the colon undigested. The fermentation of these substrates provides nutrients for the gut bacteria and short-chain fatty acids (SCFAs). SCFAs are beneficial for colon health; they are a source of energy for the colonocytes and contribute to the maintenance of gut barrier function, the protection against colorectal cancer development and the control of intestinal inflammation (Flint et al., 2017).
Ruminococcaceae are an important family of Firmicutes bacteria within the colonic microbial communities which have evolved specialized systems to utilize complex carbohydrates. This is in contrast to Bacteroides which have been shown to display diverse and versatile glycan metabolizing capabilities (for a recent review, see Ndeh and Gilbert, 2018). Members of the genus Ruminococcus have been reclassified into three genera and families based on 16S rRNA sequencing, Blautia (Lachnospiraceae), Ruminococcus (Ruminococcaceae) and Clostridium (Clostridiaceae) (Liu et al., 2008). R. bromii is one of the most abundant bacteria constituting the human colonic microbiota and a primary degrader of RS, an important non-digestible dietary polysaccharide (Ze et al., 2012, 2013). Ruminoccocus gnavus was first assigned as a novel species in 1976 (Moore et al., 1976) and recently reclassified into genus Blautia which belongs to Clostridium cluster XIVa, a member of the Lachnospiraceae family but still maintaining its original name (Lawson and Finegold, 2015). R. bromii and R. gnavus are prevalent species of the human gut; They are among the 57 species detected in more than 90% of human fecal samples by metagenomic sequencing (Qin et al., 2010). The median abundances of R. bromii L2-63 and R. gnavus are around 3 and 0.1%, respectively (Qin et al., 2010). In our previous work we showed that R. gnavus ability to grow on host mucin glycoproteins was strain dependent (Crost et al., 2013, 2016), underscoring the importance of analyzing glycan utilization by members of the human gut microbiota at the strain level. The mucin-degrading strain R. gnavus ATCC 29149 utilizes mucin glycan epitopes from the intestinal mucus layer as energy source (Crost et al., 2013, 2016).
It has been proposed that the primary role played by R. bromii is to release energy from RS to other members of the microbial community (Ze et al., 2012). Trophic interactions between members of the microbiota encompass both cooperation and competition. For example, mucin cross-feeding has been reported between gut microbiota species such as infant bifidobacteria and Eubacterium hallii (Bunesova et al., 2018) or Akkermansia muciniphila and non-mucus-degrading bacteria Anaerostipes caccae, Eubacterium hallii, or Faecalibacterium prausnitzii (Belzer et al., 2017; Chia et al., 2018). Examples of cross-feeding have also been reported within the Bifidobacterium genus (Milani et al., 2015; Turroni et al., 2017), and in the presence of primary degraders of RS (with R. bromii) or xylan (with B. ovatus) (Turroni et al., 2010, 2012; Rogowski et al., 2015; Centanni et al., 2017).
Here, we investigated the molecular mechanisms underpinning the trophic interactions between the human gut symbionts R. bromii and R. gnavus on host mucin and dietary starch using a combination of bioinformatics, quantitative PCR (qPCR), NMR-based metabolite profiling and RNA-Seq based transcriptomics of mono- and co-cultures.
D-glucose (Glc), type III pig gastric mucin (PGM), maltose, maltotriose and soluble potato starch (SS) were purchased from Sigma-Aldrich (St Louis, MO, United States). Purified pig gastric mucin (pPGM) was prepared as previously described (Gunning et al., 2013). Maltotetraose was obtained from Carbosynth (Berks, United Kingdom). A retrograded type-III RS derived from high amylose maize was kindly provided by Ingredion (Manchester, United Kingdom).
Ruminococcus gnavus ATCC 29149 was routinely grown in an anaerobic cabinet (Don Whitley, Shipley, United Kingdom) in Brain Heart Infusion broth supplemented with yeast extract and hemin (BHI-YH) as previously described (Crost et al., 2013). R. bromii L2-63 was also grown in an anaerobic cabinet, in anaerobic basal Yeast extract-Casitone-Fatty Acids (YCFA) medium (Duncan et al., 2002) supplemented with 0.5% SS. Growth of both bacteria on single carbon sources utilized YCFA medium supplemented with 0.5% (wt/vol) of Glc or starch (SS or RS), malto-oligosaccharides at a concentration of 27.7 mM Glc units, or 1% (wt/vol) pPGM. The growth assays were performed in 96-well plates with 200 μL of medium/well for screening or in 14 mL-tubes with 10 mL medium/tube for sampling. Growth was determined spectrophotometrically by monitoring changes in optical density (OD) at 595 nm or 600 nm compared to the same medium without bacterium (ΔOD600 nm) hourly for the first 10 h and then at distinct times up to 75 h. Sampling for DNA extraction, RNA extraction or 1H NMR was carried out over growth.
For the isolation of R. gnavus ATCC 29149 and/or R. bromii L2-63 chromosomal DNA, cells from a 2 mL-aliquot of culture were harvested by centrifugation (10,000 g, 5 min, 4°C), at different times of growth. The cell pellet was kept frozen at −20°C until DNA extraction. The DNA extraction was carried out using Gene JET Genomic DNA Purification kit (ThermoFisher Scientific) following the supplier’s procedure for Gram-positive bacteria, except for the elution step which was performed with 100 μL of EB buffer instead of 200 μL. DNA quality and quantity were assessed using the NanoDropTM 2000 spectrophotometer (ThermoFisher Scientific) and the Qubit dsDNA HS assay on Qubit® 2.0 fluorometer (ThermoFisher Scientific). Dilutions at 10 ng/μL were prepared in water then the DNA was diluted further in 5 μg/ml Salmon Sperm DNA to obtain a 1 ng/μL dilution used as template for qPCR (see below).
The 16S rRNA genes of R. gnavus ATCC 2949 and R. bromii L2-63 were amplified with universal primers 27F (5″- AGAGTTTGATCMTGGCTCAG- 3″) and RP2 (5″-ACGGCTACCTTGTTACGACTT-3″). The PCR products were purified, quantified and diluted in water to 16.4 ng/μL which equals to 1010 copies/μL. A series of 10-fold or 20-fold dilutions was then performed from 1010 copies/μL to 102 copies/2 μL using 5 μg/mL Salmon Sperm DNA. Calibration curves were prepared in triplicates for each pair of primers using 107 copies/2 μL to 102 copies/2 μL dilutions of 16S PCR products. The standard curves showed a linear relationship of log input 16S copy number vs. the threshold cycle (CT), with acceptable values for the slopes and the regression coefficients (R2). The dissociation curves were also performed to verify the specificity of the amplicons.
Quantitative PCR was carried out in an Applied Biosystems 7500 Real-Time PCR system (Life Technologies Ltd). Three pairs of primers targeting 16S rRNA gene were used in this study (Supplementary Table S1). Each qPCR reaction (10 μL) was then carried out in triplicates with 2 μL of DNA matrix at 1ng/μL and 0.2 mM of each primer, using the QuantiFast SYBR Green PCR kit (Qiagen) according to supplier’s advice (except for the combined annealing/extension step which was extended to 35 s instead of 30 s).
Total RNA was extracted from 5 mL of mid- to late exponential phase cultures of R. gnavus ATCC 29149 and/or R. bromii L2-63 in YCFA supplemented with a single carbon source (Glc, SS or RS). Four biological replicates were performed for each carbon source. The RNA was stabilized prior to extraction by adding 1/5 vol of phenol (pH 4.3): ethanol (1:9) mixture to 1 vol of culture then incubating 30 min on ice and finally pelleting the cells for 5 min at 10,000 g at 4°C. Cell pellets were stored at −80°C before extraction. The extraction was performed by a method using phenol and chloroform and adapted from Sambrook et al. (1989). Genomic DNA contamination was removed by DNAse treatment using the TURBO DNA-free kit (Life Technologies Ltd., Paisley, United Kingdom) according to the supplier’s recommendations. The purity, quantity and integrity of the DNase-treated RNA were assessed with NanoDrop 2000 Spectrophotometer and with Agilent RNA 600 Nano kit on Agilent 2100 Bioanalyzer (Agilent Technologies, Stockport, United Kingdom). Ribodepletion was then carried out using Ribo-Zero rRNA Removal kit for bacteria according to supplier’s advice (Illumina, Cambridge, United Kingdom); efficiency assessment of the ribodepletion was performed by quantifying RNA before and after rRNA removal using the Qubit RNA HS assay on Qubit 2.0 fluorometer.
The rRNA removal was confirmed with a Nano chip run on a Bioanalyzer 2100 (Agilent). Three out of the four replicates were selected for sequencing for each condition. The resulting ribosomal depleted RNA was then fragmented for 8 min at 94°C using the Elute, Fragment, Prime buffer from Illumina TruSeq RNA kit. These conditions produced final libraries of around 370 bp. The samples were then processed following the standard TruSeq RNA protocol. The 15 Illumina libraries were normalized and equimolar pooled to 11 nM using elution buffer (Qiagen) and run over two lanes of the Illumina HiSeq2500 with a 100 bp paired end read metric.
The library pool was then diluted to 2 nM with NaOH and 5 μL transferred into 995 μL HT1 (Illumina) to give a final concentration of 10 pM. A portion (120 μL) of the diluted library pool was then transferred into a 200 μL-strip tube, spiked with 1% PhiX Control v3 and placed on ice before loading onto the Illumina cBot. The flow cell was clustered using a HiSeq PE Cluster Kit v3 (Illumina PE-401-3001) utilizing the Illumina PE_HiSeq_Cluster_Kit_V3_cBot_recipe_V8.0 method on the Illumina cBot. Following the clustering procedure, the flow-cell was loaded onto the Illumina HiSeq2500 instrument following the manufacturer’s instructions with a 101 cycle paired reads and a 7-cycle index read. The sequencing chemistry used was HiSeq SBS Kit v3 (Illumina FC-401-3001) with HiSeq Control Software 2.2.68 and RTA 1.18.66.3. Reads in bcl format were demultiplexed based on the 6 bp Illumina index by CASAVA 1.8, allowing for a one base-pair mismatch per library, and converted to FASTQ format by bcl2fastq. The RNA-Seq reads were aligned against the combined reference of Ruminococcus_bromii_l2_63.ASM20987v1.31.dna.genome.fa and Ruminococcus_gnavus_atcc_29149.ASM16947v1.31.dna.genome.fa using tophat v2.1.0 with the –max-multihits 1 option. Read counts were obtained using htseq-count v0.6.11. The differential expression analysis was carried out using the DESeq2 (v1.14.0) package (Love et al., 2014). The transcript counts were used as input for DESeq2 and filtered to remove any transcripts with a total count of 0 or 1 over all the samples. Raw counts were normalized to the effective library size separately for R. bromii and R. gnavus before carrying out the differential expression analysis using the DESeq function. An padj cut-off of 0.05 was used to determine differential expressed transcripts.
1H NMR analysis was used to identify the presence, absence, and concentration of several metabolites in the bacterial growth medium of mono- and co-cultures. The spent media were thawed at room temperature and prepared for 1H NMR spectroscopy by mixing 400 μL of spent medium with 200 μL of phosphate buffer (0.26 g NaH2PO4 and 1.41 g K2HPO4) made up in 100% D2O (100 mL), containing 0.1% NaN3 (100 mg), and 1 mM sodium 3-(Trimethylsilyl)-propionate-d4, (TSP; 17 mg) as a chemical shift reference. The samples were mixed, and 500 μL was transferred into a 5-mm NMR tube for spectral acquisition. The 1H NMR spectra were recorded at 600 MHz on a Bruker Avance spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) running Topspin 2.0 software and fitted with a cryoprobe and a 60-slot autosampler. Each 1H NMR spectrum was acquired with 512 scans, a spectral width of 12300 Hz and an acquisition time of 2.7 s and delay time of 3 s. The “noesygppr1d” presaturation sequence was used to suppress the residual water signal with a low-power selective irradiation at the water frequency during the recycle delay. Spectra were transformed with a 0.3-Hz line broadening, manually phased, baseline corrected, and referenced by setting the TSP methyl signal to 0 ppm. Metabolites were identified using information found in the Human Metabolome Database2 and by use of the 2D-NMR methods, COZY, HSQC, and HMBC. The metabolites were quantified using the software Chenomx NMR suite 7.6TM.
Genome mining was performed manually by BLAST using the “align two or more sequences” tool (Boratyn et al., 2013). For each target protein, the query sequence used was the reference protein sequence from NCBI; when no reference protein sequence was available, a sequence from a member of the Clostridiales order was used (Supplementary Table S2). The subject sequences were the sequences corresponding to all putative proteins from R. bromii L2-63 or R. gnavus ATCC 29149 genomes. The search for NanA, E and K in R. bromii L2-63 was performed according to Almagro-Moreno and Boyd (Almagro-Moreno and Boyd, 2009).
The trophic interactions between R. bromii L2-63 and R. gnavus ATCC 29149 on host and dietary carbon sources were determined under anaerobic conditions using YCFA as a suitable minimum medium for both strains (Duncan et al., 2002). The growth of R. bromii L2-63 and R. gnavus ATCC 29149 in mono- or co-cultures was first monitored spectrophotometrically using mucin as sole carbon source. While R. gnavus could utilize this substrate, as previously reported (Crost et al., 2013), R. bromii was unable to grow on mucin as sole carbon source and no growth benefit was observed on this substrate in the presence of R. gnavus (Figure 1A). Mucin degradation by bacteria relies on the expression of glycoside hydrolases (GHs)2 (Lombard et al., 2014) such as sialidases (GH33), α-fucosidases (GH29, GH95), exo- and endo-β-N-acetylglucosaminidases (GH84 and GH85), β-galactosidases (GH2, GH20, GH42), α-N-acetylglucosaminidases (GH89), endo-β1,4-galactosidases (GH98) or α-N-acetylgalactosaminidases (GH101 and GH129) (Tailford et al., 2015a). We previously showed that the ability of R. gnavus to grow on mucin was dependent on the expression of a GH33 intramolecular trans-sialidase (Crost et al., 2013, 2016; Tailford et al., 2015b) and that fucose was released from mucin by the action of GH29 and GH95 fucosidases (Crost et al., 2013). In contrast, the R. bromii L2-63 genome encodes a small number of GHs (Mukhopadhya et al., 2018) compared to R. gnavus ATCC 29149 (Crost et al., 2013) (21 in R. bromii L2-63 vs. 60 in R. gnavus ATCC 29149) and no genes encoding mucin-degrading enzymes were found, in line with the inability of this strain to grow on mucin. In addition, its lack of growth in co-culture with R. gnavus, suggests that R. bromii cannot utilize the monosaccharides released by R. gnavus, in agreement with genomic data suggesting that R. bromii does not harbor genes involved in fucose or sialic acid metabolism (Supplementary Figures S1, S2).
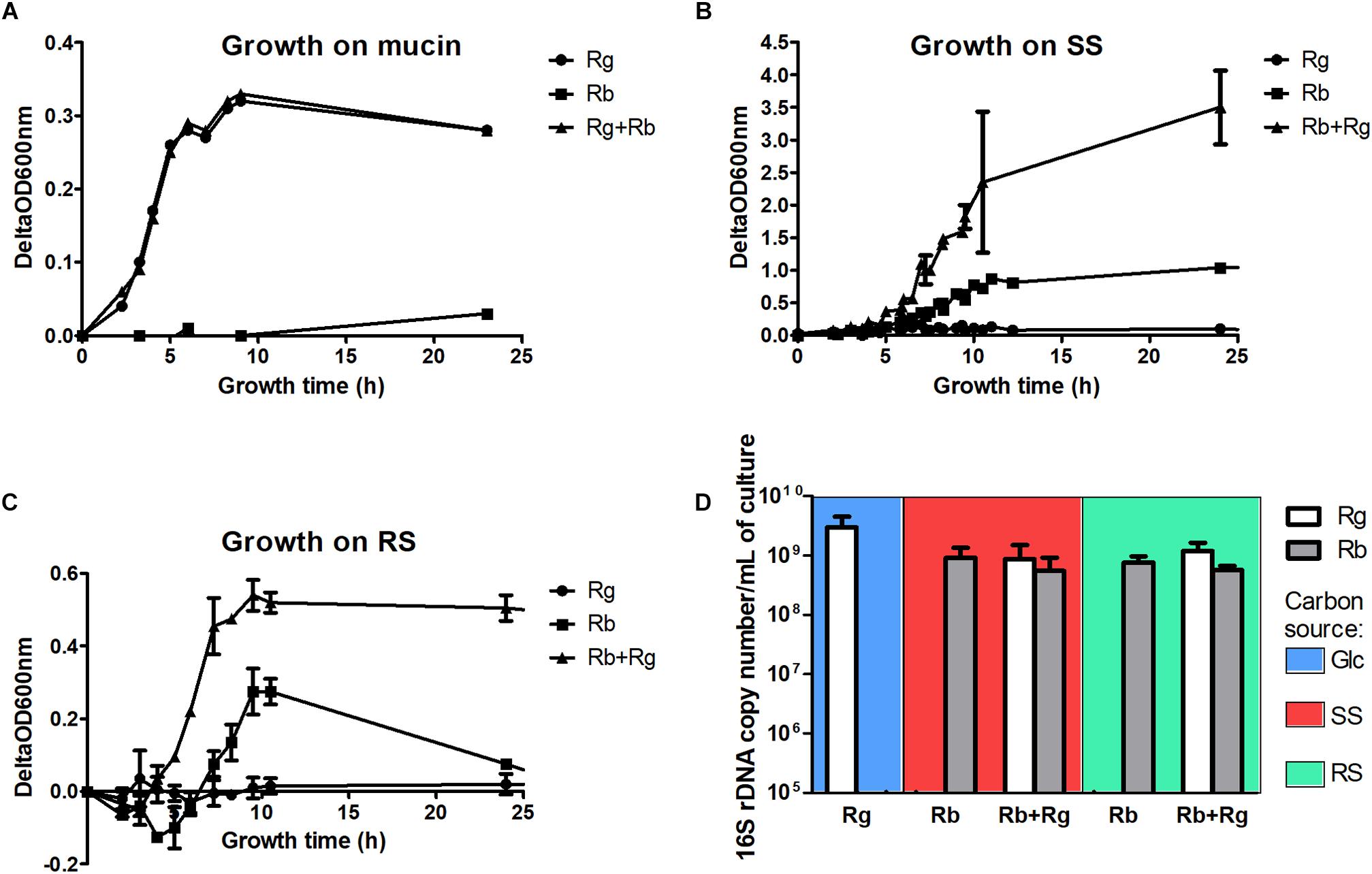
FIGURE 1. Growth curves of the mono- and co-cultures with mucin (A), soluble starch (SS) (B) or resistant starch (RS) (C) as sole carbon source and cell concentrations in the different growth conditions (D). The concentrations were determined by qPCR and expressed as 16S rDNA copy number/mL of culture. The values are averages of 3 replicates for R. gnavus ATCC 29149 grown with Glc or 4 replicates for the other conditions. The error bars correspond to the standard deviations. Cells samples were collected at a time of growth of 7 h for R. gnavus ATCC 29149 grown on Glc, 10 h for R. bromii L2-63 grown on SS and 8 h for the other conditions.
Ruminococcus bromii L2-63 is highly specialized in starch degradation, dedicating 15 of its 21 GH-encoding genes to putative GH13 amylases (Ze et al., 2015). Some of these GH13 amylases revealed an organization in “amylosome”, contributing to R. bromii exceptional ability to degrade dietary RS (Ze et al., 2015; Mukhopadhya et al., 2018). Here we showed that R. bromii was able to utilize both starch substrates (SS and RS) as sole carbon source, in agreement with previous reports (Ze et al., 2012, 2015) whereas no growth was detected with R. gnavus on these substrates despite the presence of 9 GH13-encoding genes in R. gnavus ATCC 29149 genome. Furthermore, while R. bromii growth on SS reached ΔOD600 nm of ∼0.8–1, after 12 h of growth, the presence of R. gnavus increased the density of cells to ΔOD600 nm ∼ 4 after 10 h, suggesting cross-feeding activity (Figure 1B). Due to the presence of insoluble RS particles, the OD600 nm measurements of R. bromii grown on RS result in a two-stage curve reflecting both bacterial growth and bacterial degradation of the RS particles. When R. gnavus and R. bromii were co-cultured with RS, a different profile was observed (Figure 1C), suggesting that cross-feeding also occurs on RS, as confirmed below.
To further assess the behavior of R. bromii L2-63 and R. gnavus ATCC 29149 on starch (SS or RS), the bacteria were quantified by determining 16S rDNA copies per mL of culture by qPCR (Figure 1D). The average 16S rDNA copies of R. bromii when grown in mono- or co-cultures with RS after 8 h was 7.3 × 108 and 5.6 × 108 per mL of culture, respectively. When SS was used as the carbon source, R. bromii 16S rDNA copies per mL of culture was 1 × 109 after 10 h in mono-culture and 5.8 × 108 after 8 h in co-culture with R. gnavus. These analyses indicate that the presence of R. gnavus did not affect R. bromii growth on starch (RS or SS). R. gnavus reached high concentration level in both SS and RS co-cultures with 16S rDNA copies/mL of 6.7 × 108 and 1.2 × 109, respectively, while it was not able to grow in mono-culture on these substrates, confirming that R. gnavus benefits from R. bromii starch degradation by cross-feeding, as also suggested by spectrophotometric measurements. These concentrations were comparable to the growth of R. gnavus on 0.5% Glc as sole carbon reaching 3 × 109 16S rDNA copies/mL after 7 h of growth (Figure 1D).
The production and utilization of starch degradation products was monitored over time by 1H NMR (Figure 2). Maltotetraose, maltose and glucose-1-phosphate were detected in the spent medium of R. bromii mono- or co-cultures (with SS or RS) and their concentration decreased over time. However, while maltotetraose and glucose-1-phosphate were only detected during the exponential phase, maltose was still present at the late stage of growth in mono-cultures. Glc was also released by R. bromii degradation of SS or RS but tended to accumulate in mono-cultures while its concentration decreased over time in the presence of R. gnavus. Interestingly, the concentration of these starch degradation products was higher in R. bromii mono-culture on SS as compared to RS, which may be due to a slower rate of RS degradation allowing a more efficient uptake of the products.
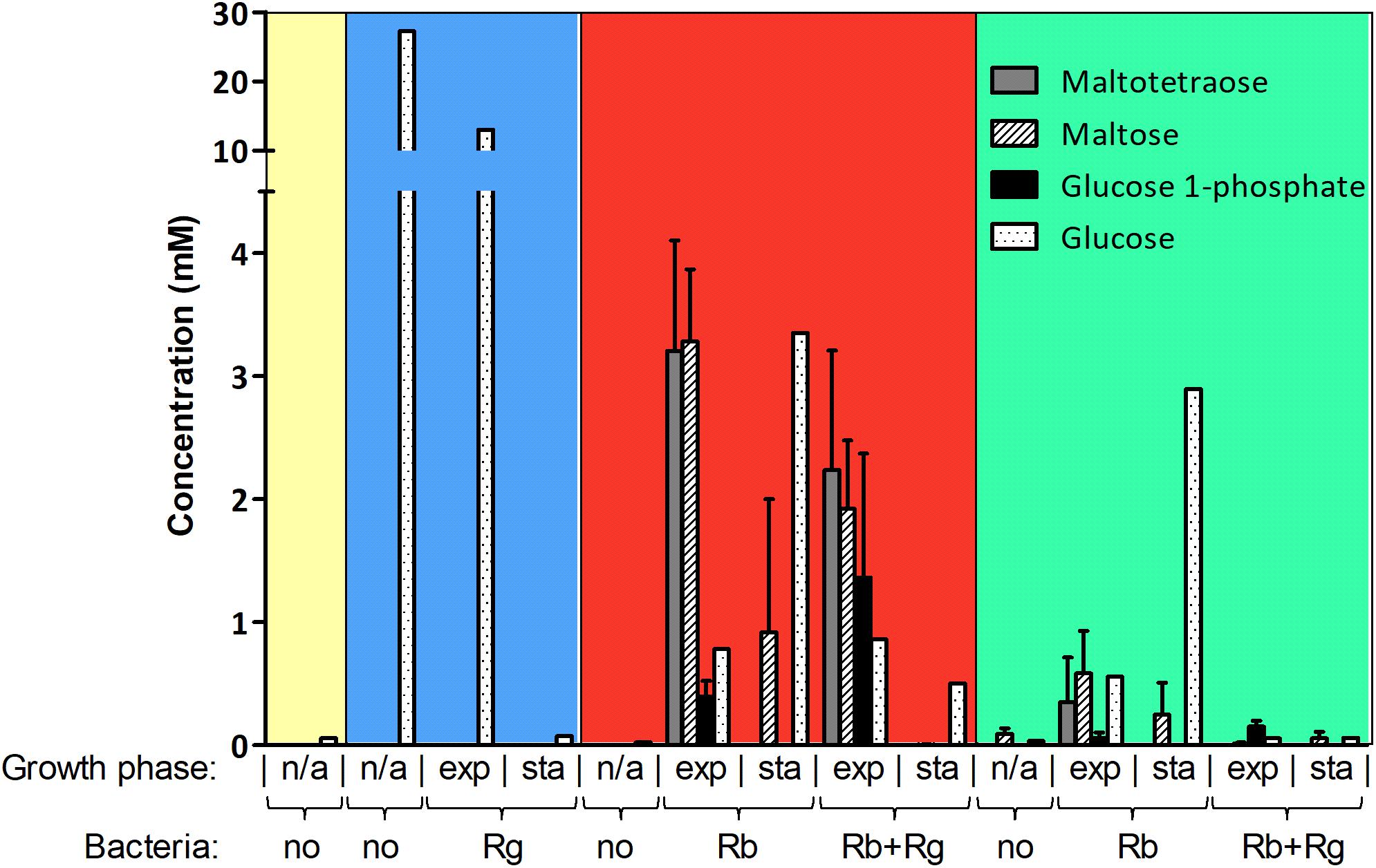
FIGURE 2. Concentration of starch degradation products in the spent media. The concentrations were determined by 1H NMR and the values are averages of 2 to 5 replicates. The error bars correspond to standard deviations. Results presented in the blue, red and green boxes come from growth assays with Glc, SS and RS as sole carbon source, respectively. Results from the YCFA medium alone, without carbon source, are presented inside the yellow box. Abbreviations: exp, exponential; sta, stationary; n/a, non-applicable.
In order to determine which starch degradation products were utilized by R. bromii and R. gnavus, mono-cultures were performed with malto-oligosaccharides and Glc as control. Both strains could utilize maltose, maltotriose and maltotetraose while Glc was only a substrate for R. gnavus (data not shown), suggesting that the release of Glc and malto-oligosaccharides upon R. bromii starch degradation contributed to R. gnavus cross-feeding on SS or RS. These results suggest that both syntrophy and competition could take place when R. gnavus and R. bromii are co-cultured with starch.
Next, we determined the metabolites produced by the bacteria in mono- and co-culture by 1H NMR analysis of the spent media. Acetate was the main SCFA produced by both R. bromii L2-63 and R. gnavus ATCC 29149 in mono- or co-cultures and its production was increased during bacterial growth (Figure 3A). No butyrate or propionate was detected in the growth conditions tested. Formate and ethanol were produced in increasing amounts by both R. bromii L2-63 and R. gnavus ATCC 29149 in mono- or co-cultures during bacterial growth (Figure 3A). Propanol was detected at low concentration at the late stage of growth when R. gnavus was grown with Glc as well as when R. bromii was grown with starch in mono- or co-cultures (Figure 3B). Interestingly, propanediol was only produced when R. gnavus was present (in mono-culture with Glc or in co-cultures with starch), suggesting that propanol is produced via different pathways in R. bromii L2-63 and R. gnavus ATCC 29149 (Figure 3B).
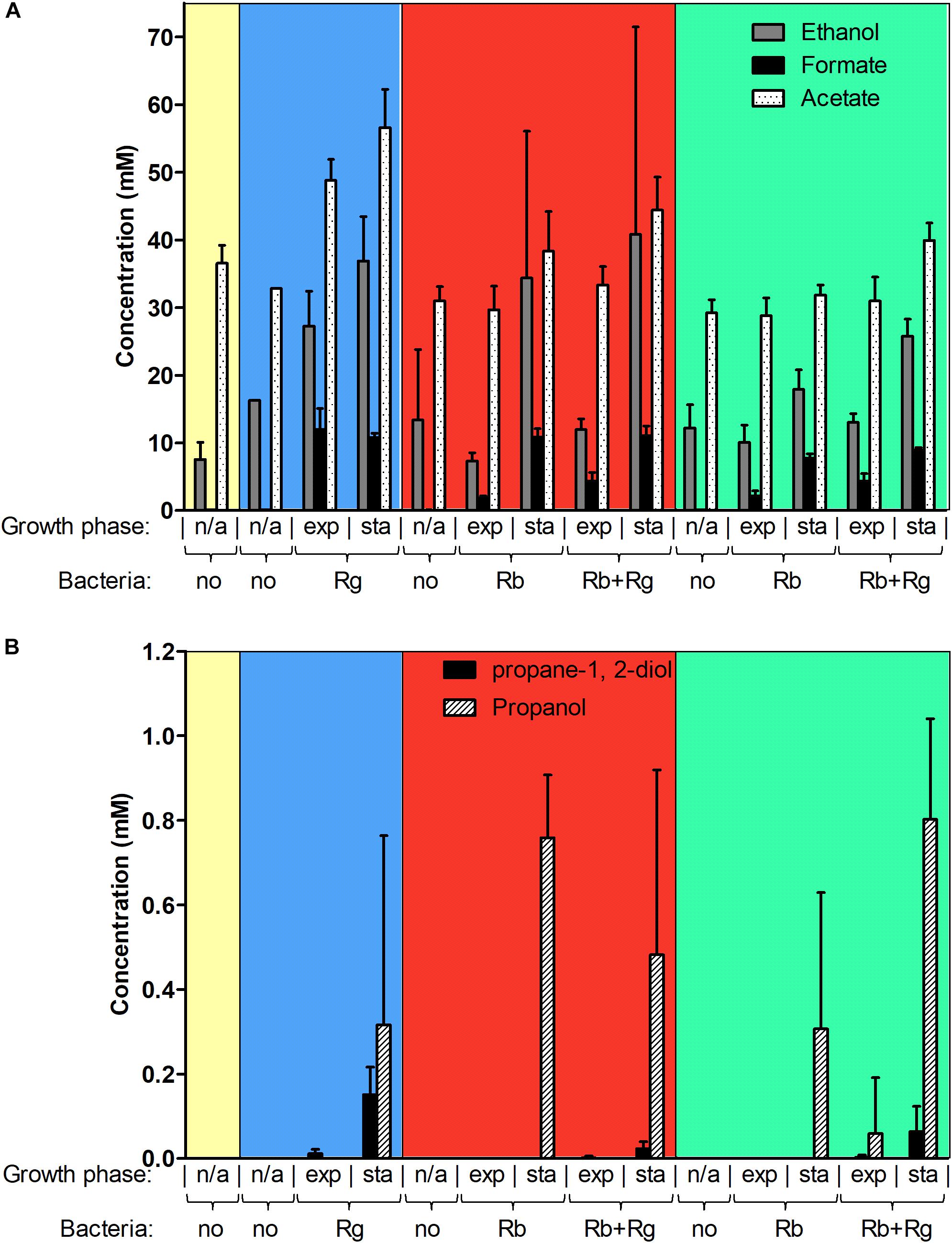
FIGURE 3. Concentration of different metabolites in the spent media. Concentrations of ethanol, formate and acetate are shown in panel (A) while concentrations of propane-1, 2-diol and propanol are shown in panel (B). These concentrations were determined by 1H NMR and the values are averages of 2 to 5 replicates. The error bars correspond to standard deviations. Results presented in the blue, red and green boxes correspond to growth assays with Glc, SS and RS as sole carbon source, respectively. Results from the YCFA medium alone, without carbon source, are presented inside the yellow box. Abbreviations: exp, exponential; sta, stationary; n/a, non-applicable.
Three main propanol biosynthesis pathways have been identified in bacteria, the propane-1, 2-diol pathway, the acrylate pathway and the Wood-Werkman cycle (Reichardt et al., 2014). These pathways all share the last step, i.e., the conversion of propanal into propanol catalyzed by a propanol dehydrogenase (PduQ). Search for putative enzymes involved in propanol production in R. gnavus ATCC 29149 and R. bromii L2-63 genomes, identified genes encoding putative PduQ proteins, RUMGNA_01033 and L2-63_01124, respectively. No gene encoding a putative acryloyl-CoA reductase could be found in the genome of these strains, ruling out the acrylate pathway for propanol production in these bacteria. Although both bacteria encode a putative lactaldehyde reductase and a propanol dehydrogenase, PduCDE homologs could only be found in the R. gnavus genome indicating that propanol can be produced via the propane-1, 2-diol pathway in this bacterium. This pathway is involved in metabolism of the deoxy-sugars fucose and rhamnose (Reichardt et al., 2014). In addition to PduQ, both bacteria encode homologs of the methylmalonyl-CoA carboxytransferase and propanal dehydrogenase needed for propanol production via the Wood-Werkman cycle (Supplementary Figure S3).
To gain further insights into the metabolic pathways underpinning trophic interactions between the two strains, transcriptional analyses of R. bromii L2-63 and R. gnavus ATCC 29149 grown in mono-cultures on starch (RS or SS) or Glc, respectively, or co-cultures on starch (RS or SS) were performed by RNA-Seq. An average of 20 million reads were generated for each sample which is sufficient sequencing depth. In the co-cultures, the reads assigned to R. bromii represented on average around 42% of total reads, for both SS and RS. This result correlates well with the bacterial count determined by qPCR where R. bromii 16S copies represented 46% and 32% of total 16S copies in SS and RS, respectively. Differential gene expression analysis (DESeq 2) was carried out to determine the influence of the carbon source or of the other bacterium on gene transcription.
Interestingly, the transcription of all R. bromii genes was found to be very similar in both mono-cultures irrespective of the type of starch (Figures 4A, 5A), suggesting that the catabolism of RS or SS shares the same metabolic pathway. For example, the dockerin-carrying amylases Amy4, Amy9, Amy10, and Amy12 GH13 enzymes (Ze et al., 2015) were all expressed in the conditions tested in this study. However, the type of starch had an impact on R. bromii gene transcription when in co-culture with R. gnavus, with 11 genes up-regulated with RS compared to SS, suggesting a combined effect of RS and R. gnavus (Figure 5B). These genes belong to 3 different clusters: 3 are part of a cluster of genes potentially involved in sugar metabolism, one is the pduQ gene which is involved in the conversion of propanal into propanol (see above) and 7 genes (ntpABCDEGK) are involved in the formation of a ntp sodium pump operon encoding Vacuolar-type Na + -translocating ATPase (Figure 6 and Supplementary Table S3). Interestingly, sodium and potassium ion gradients also serve as important energy reservoirs of bacterial cells and could be upregulated due to the competitive stress in the co-culture (Murata et al., 1996).
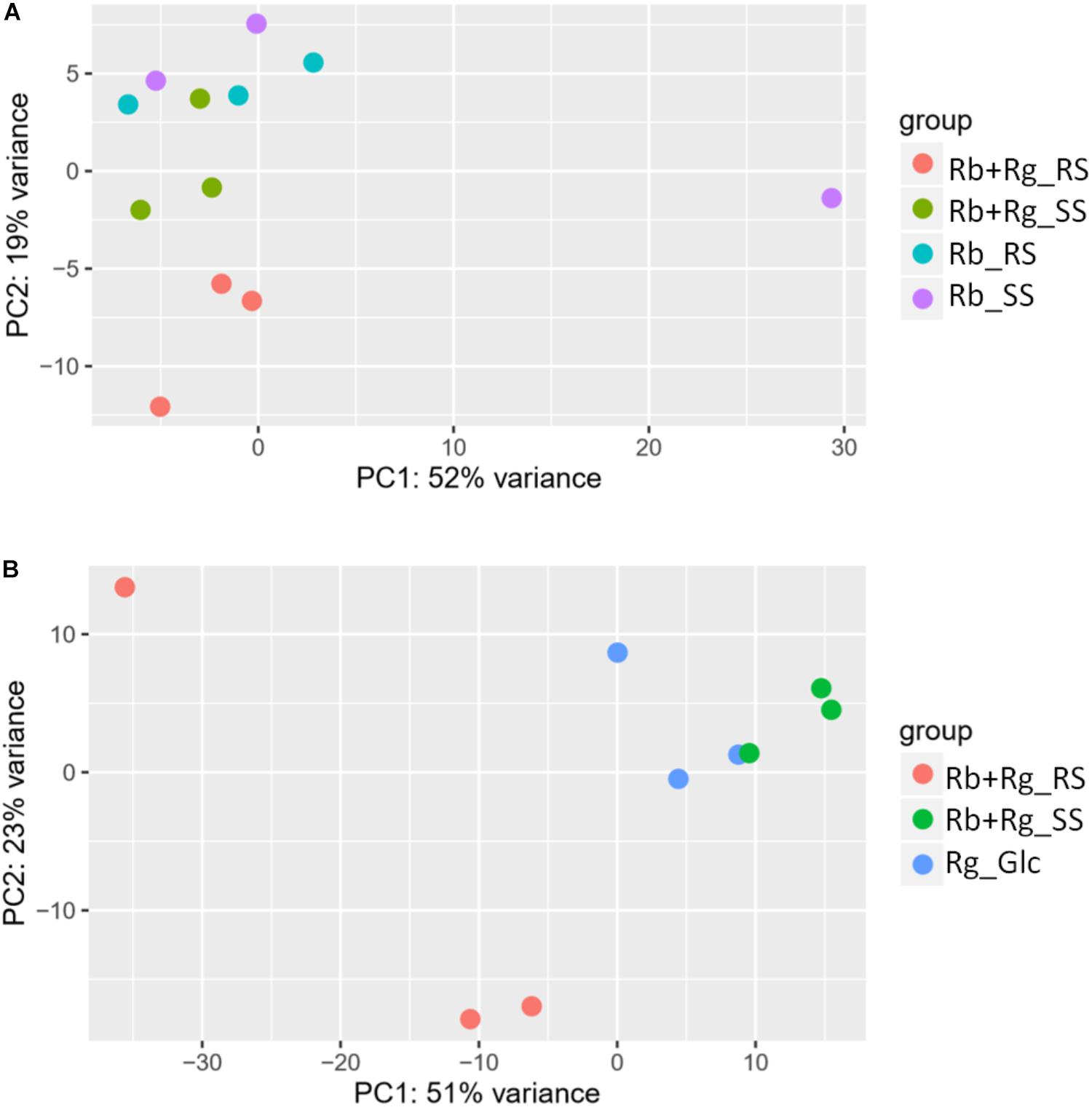
FIGURE 4. Principal component analysis (PCA) plots for transcriptomics data of R. bromii L2-63 genes (A) and R. gnavus ATCC 29149 genes (B).
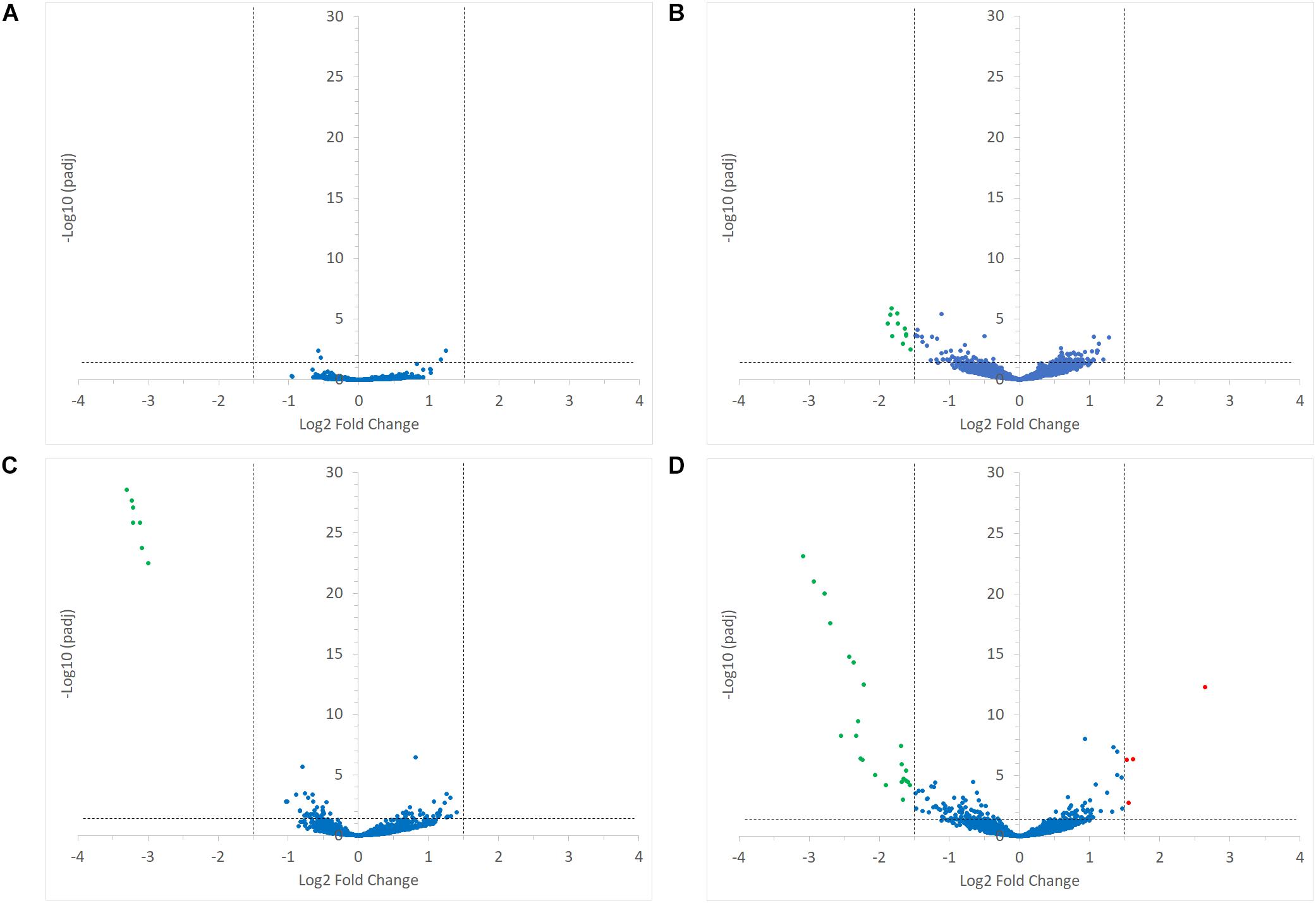
FIGURE 5. Volcano plots representing the differential expression analysis of R. bromii L2-63 genes. Genes are considered to be differentially expressed when Log2 Fold Change < –1.5 or > 1.5 and padj < 0.05; non-differentially expressed genes are shown as blue dots. The impact of starch on R. bromii L2-63 gene transcription in mono-cultures and co-cultures is shown in panel (A,B), respectively. (A) No gene was differentially expressed between both mono-cultures. (B) When comparing the co-cultures, 11 genes were up-regulated with RS as compared to SS (shown as green dots). The impact of R. gnavus ATCC 29149 on R. bromii L2-63 gene transcription with SS and RS as sole carbon source is shown in panel (C,D), respectively. (C) When SS was used as carbon source, 7 genes were up-regulated in the co-culture as compared to the mono-culture (shown as green dots). (D) When RS was used as carbon source, 23 genes were up-regulated in the co-culture (shown as green dots) while 4 genes were upregulated in the mono-culture (shown as red dots).
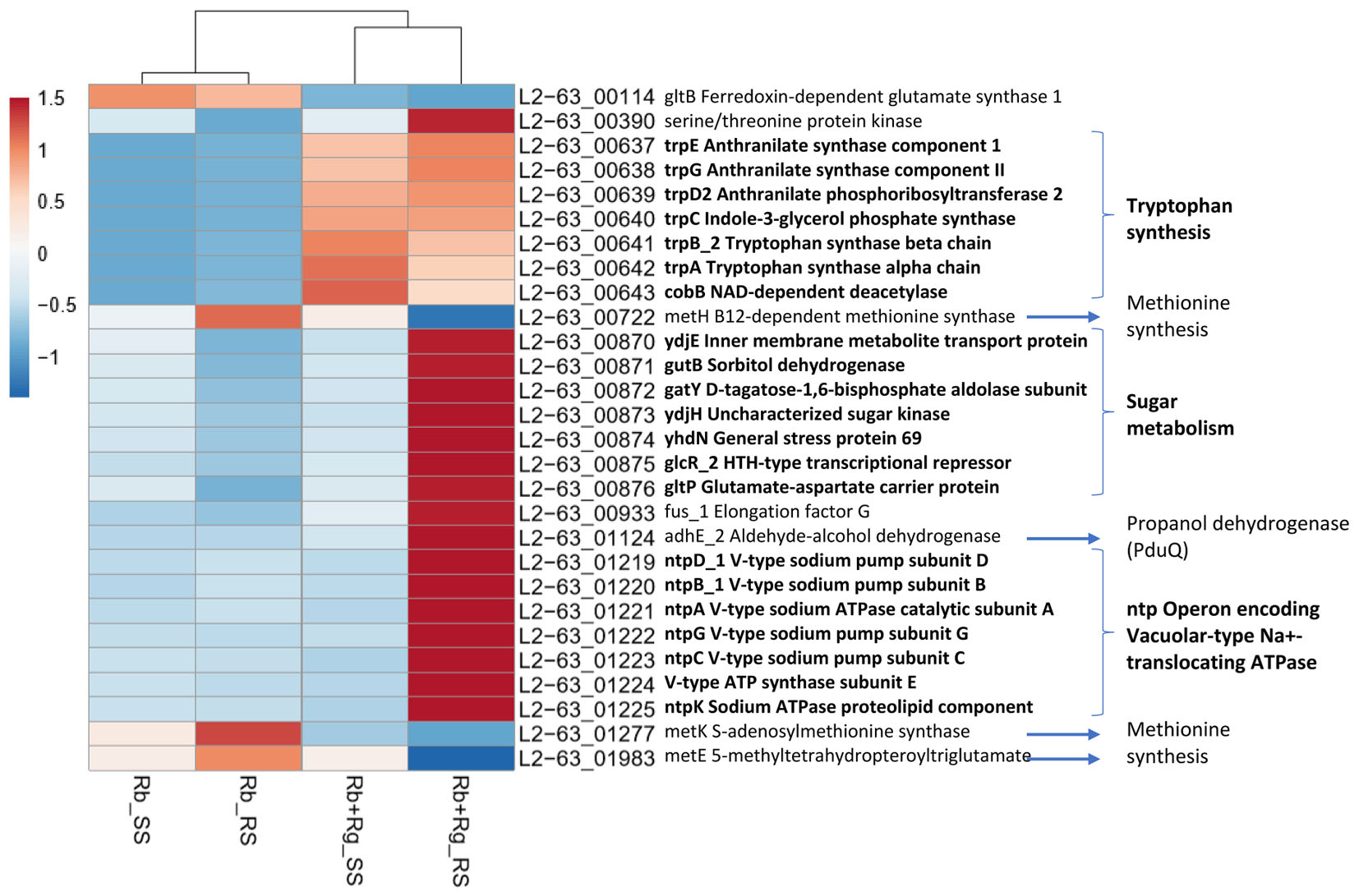
FIGURE 6. Heatmap of the transcription level (in arbitrary unit) of differentially expressed (Log2 Fold Change < –1.5 or > 1.5 and padj < 0.05) R. bromii L2-63 genes in different growth conditions. This heatmap was produced with ClustVis web tool (Metsalu and Vilo, 2015) using the transcript counts as input values.
In co-cultures, R. gnavus had a greater effect than the type of starch on R. bromii gene expression. (Figures 5C,D, 6 and Supplementary Table S3). Seven R. bromii genes were upregulated in the presence of R. gnavus irrespective of the carbon source (Figure 6 and Supplementary Table S3); these genes, which include trpA, B, C, D, E and G, are all involved in the tryptophan biosynthetic pathway and are expressed when tryptophan level is low. In particular trpA, B, C, D, E and G genes were increased by around 13-fold and ninefold in the co-cultures with SS and RS, respectively, as compared to the corresponding mono-cultures. Tryptophan is metabolized by enzymes in the gut mucosa and also by enzymes produced by the gut microbiome. In R. gnavus ATCC 29149, RUMGNA_01526 is capable of decarboxylating tryptophan to tryptamine, an activity that is rare among bacteria, and also shared by the common gut Firmicutes member, Clostridium sporogenes ATCC 15579 (Williams et al., 2014). It is estimated that ∼10% of the human population harbor one of these enzymes. Interestingly, R. gnavus genes involved in tryptophan biosynthesis were not differentially expressed between the three conditions tested, which may be due to R. gnavus higher capacity to acquire and metabolize tryptophan from the medium. NMR data confirmed that the tryptophan level in the spent medium was lower in the co-cultures as compared to the mono-cultures (data not shown). Together these data suggest that tryptophan may become a limiting factor for R. bromii growth on this substrate in the presence of R. gnavus. In addition, 16 genes were found to be specifically upregulated in the RS co-culture as compared to the RS mono-culture, including 4 genes belonging to a cluster of genes involved in sugar metabolism (Figure 6 and Supplementary Table S3). It is worth noting that 10 of the 16 genes were also up-regulated in the RS co-culture as compared to SS co-culture. These results further indicate that the observed transcriptional changes in R. bromii L2-63 were due to a combined effect of R. gnavus and RS.
Interestingly, R. bromii showed a down-regulation of the vitamin B12-dependent methionine synthesis genes (metE, metH, metK) in the RS co-culture as compared to the RS mono-culture (Figure 5D). The downregulation in co-culture could be due to the lack of sufficient VitB12 (cobalamin) amount in the YCFA growth medium to sustain both R. bromii and R. gnavus growth as R. bromii does not have the ability to produce this vitamin (Ze et al., 2015).
The transcription profile of R. bromii, indicate the requirement for R. bromii to adjust its metabolic activity toward tryptophan and vitamin B12, especially when RS was used as sole carbon source, so that its growth remains unaffected in the presence of R. gnavus as shown above.
Ruminococcus gnavus ATCC 29149 showed a much higher number of differentially expressed genes between mono- and co-cultures as compared to R. bromii (Figure 7), especially with RS (Figure 4B). A total of 22 genes were down-regulated in both co-cultures compared to the mono-culture whereas 20 were upregulated including genes encoding several maltose transporters and enzymes involved in degradation of starch-related products such as RUMGNA_01664 to 01673 and RUMGNA_02728 to 02733 (Figure 8 and Supplementary Table S4). These results are in agreement with the NMR findings showing that R. gnavus can utilize malto-oligosaccharides as sole carbon source and from the qPCR analysis showing that R. gnavus can efficiently cross-feed on starch-degradation products when grown with R. bromii. The fact that, upon R. bromii starch degradation, R. gnavus benefits from Glc (which is not a substrate for R. bromii) may explain why R. bromii transcription is not affected on SS (a rapidly degradable starch) in co-culture as this will serve as a preferential nutrient source for R. gnavus. However, the ability of R. gnavus to utilize malto-oligosaccharides, which is a major nutrient source for R. bromii suggests a direct competition between the two strains, which is reflected by R. bromii transcriptomics data on RS. This is corroborated by the results of starch degradation products in the spent media (Figure 2) which showed Glc presence in SS co-culture medium compared to the absence of Glc in the slow degrading RS co-culture medium.
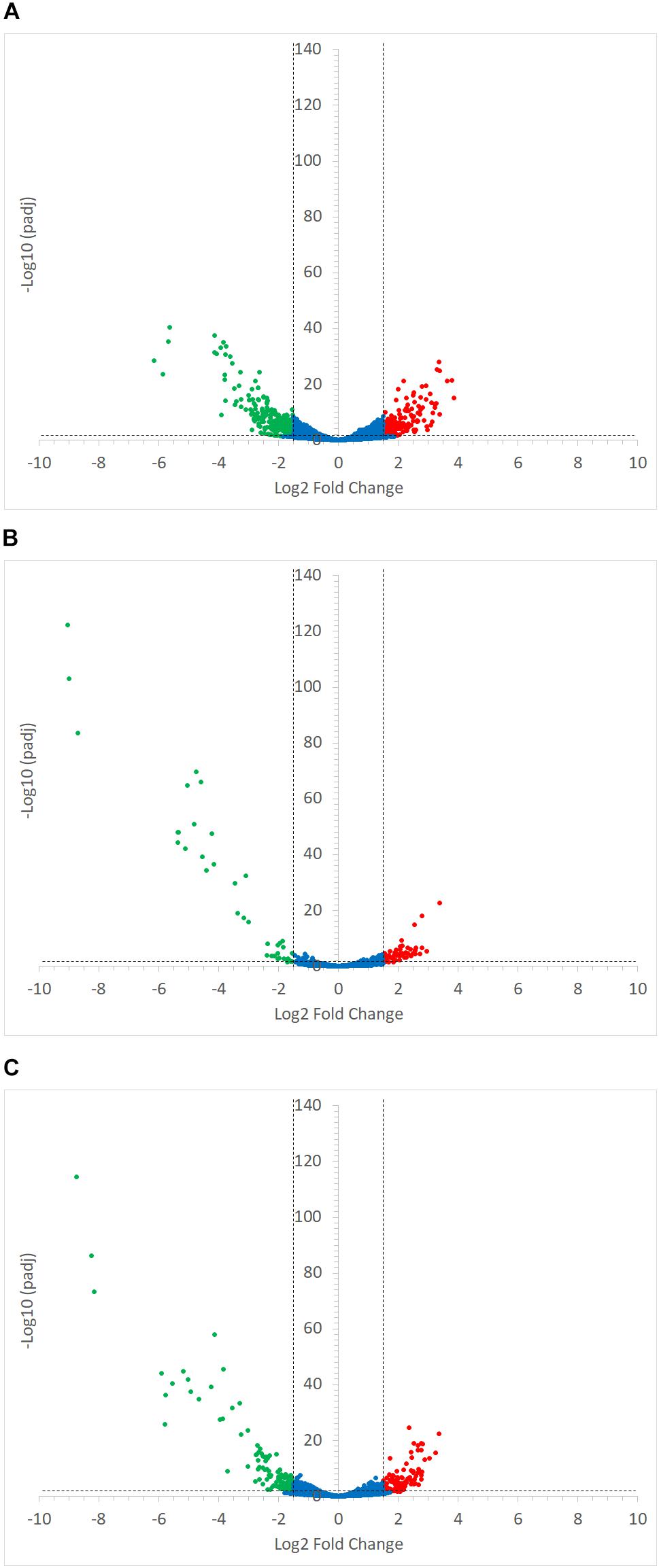
FIGURE 7. Volcano plots representing the differential expression analysis of R. gnavus ATCC 29149 genes. Genes were considered to be differentially expressed when Log2 Fold Change < –1.5 or > 1.5 and padj < 0.05; non-differentially expressed genes are shown as blue dots. Panel (A) shows the impact of starch type on R. gnavus ATCC 29149 gene transcription when co-cultured with R. bromii L2-63; 213 genes were upregulated in the co-culture with RS (shown as green dots) while 212 genes were up-regulated in the co-culture with SS (shown as red dots). The combined effect of the presence of R. bromii L2-63 and the carbon source (starch vs. glucose) is shown in panels (B) and (C) when SS or RS was used in the co-culture, respectively; (B) When SS was used as carbon source, 40 genes were up-regulated in the co-culture (shown as green dots) and 59 genes were up-regulated in the mono-culture (shown as red dots). (C) When RS was used as carbon source, 119 genes were up-regulated in the co-culture (shown as green dots) while 101 genes were up-regulated in the mono-culture (shown as red dots).
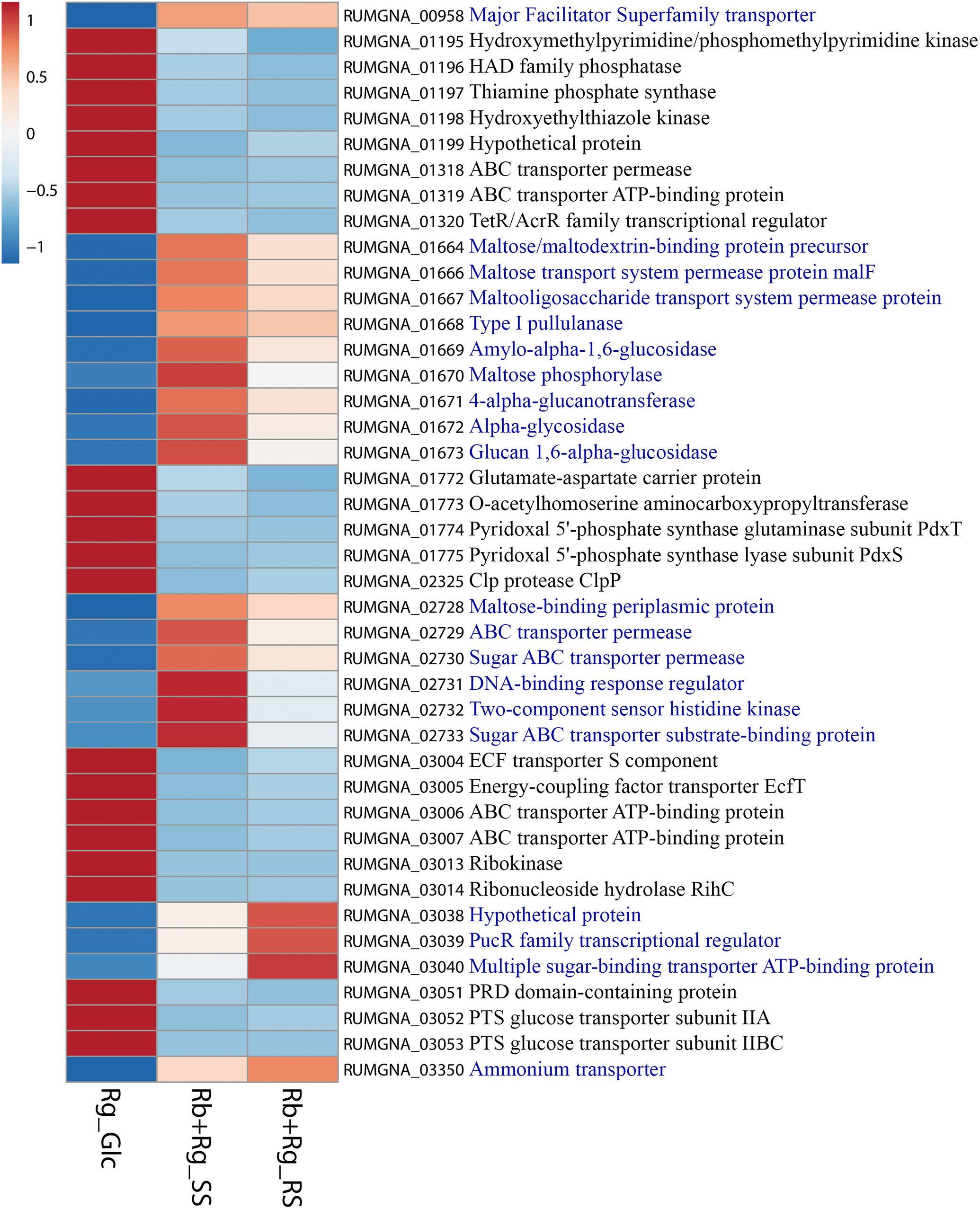
FIGURE 8. Heatmap of the transcription level (in arbitrary unit) of selected differentially expressed (Log2 Fold Change < –1.5 or > 1.5 and padj < 0.05) R. gnavus ATCC 29149 genes in different growth conditions. This heatmap was produced with ClustVis web tool (Metsalu and Vilo, 2015) using the transcript counts as input values. The 20 R. gnavus ATCC 29149 genes with an upregulated transcription in both co-cultures with R. bromii L2-63 on starch compared to the mono-culture on Glc are in blue. The 22 R. gnavus ATCC 29149 genes with an upregulated transcription in the mono-culture on Glc compared to both co-cultures with R. bromii L2-63 on starch are in black.
In summary, we showed that, in vitro, R. gnavus can efficiently cross-feed on starch degradation products released by R. bromii. Cross-feeding plays a crucial role in microbial community shaping in the gut (Hoek and Merks, 2017). This concept involves the ability of bacteria to benefit from substrate degradation products but also from fermentation products and/or cofactors. For example, Anaerostipes caccae L1-92 can utilize both mucin sugars and acetate produced by mucin degradation by Akkermansia muciniphila ATCC BAA-835 to sustain its growth and produce butyrate (Chia et al., 2018). Here, we showed that R. bromii L2-63 could not benefit from degradation products or metabolites released by R. gnavus ATCC 29149 grown on mucin, in line with the unique genomic characteristics of R. bromii strains sequenced to date (Mukhopadhya et al., 2018) and the mucin foraging profile of R. gnavus strains (Crost et al., 2013, 2016). However, we showed that RS cross-feeding initiated by R. bromii promoted growth of R. gnavus leading to the concomitant production of acetate as the main SCFA produced by these strains. Cross-feeding of gut bacteria on starch degradation products has previously been reported in vitro between starch degrader R. bromii or Bifidobacterium longum subsp. suis and bacterial species, potentially sharing the same nutrient niche in the gut such as Anaerostipes hadrus (Ze et al., 2013) or B. thermacidophilum subsp. porcinum (Milani et al., 2015), respectively. Resource sharing is an important ecological feature of microbial communities living in the gut (Tannock et al., 2012; Pereira and Berry, 2017; Centanni et al., 2018). The findings from our study suggest that, although R. gnavus strains are adapted to the mucosal environment owing to their mucin-foraging capacity (Crost et al., 2013, 2016; Tailford et al., 2015b; Owen et al., 2017), their population dynamics within the colon may also be affected by the supply of dietary carbohydrates that reaches the large intestine undigested such as RS. Due to the high prevalence of R. bromii in the human colon, the hydrolysis of RS will cause the release of nutrients such as glucose or metabolites that may reach bacterial species within the mucus layer, potentially promoting the growth of other species to occur, thereby further underscoring the role of R. bromii as a keystone species. These findings open the door to future efforts to explore cross-feeding activities between different nutrient niches in vivo and the use of RS or other complex polysaccharides as a strategy to address dysbiosis of mucus-associated bacteria associated with human diseases.
The RNA-Seq data generated and analyzed for this study have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-7138.
EC carried out most of the practical work (growth assays, DNA extraction, qPCR, RNA extraction, and genome mining) and data analysis. GLG performed the NMR analyses. NJ supervised the research at QIB. IM and HF helped with the analysis of R. bromii transcriptomics. EC and NJ wrote the manuscript with contributions from JL-G, IM, and HF.
The authors gratefully acknowledge the support of the Biotechnology and Biological Sciences Research Council (BBSRC), this research was partly funded by the BBSRC Institute Strategic Programs for The Gut Health and Food Safety (BB/J004529/1), Gut Microbes and Health (BB/R012490/1) and Food Innovation and Health (BB/R012512/1) and by the BB/L008602/1 and BB/LL009951/1 BBSRC responsive mode grants.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer GG and handling Editor declared their shared affiliation.
Next-generation sequencing and library construction was delivered via the BBSRC National Capability in Genomics and Single Cell (BB/CCG1720/1) at the Earlham Institute by members of the Genomics Pipelines Group. We would like to thank Andrea Telatin from the Quadram Institute Bioscience for his help with the RNA-Seq data submission in ArrayExpress.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02558/full#supplementary-material
Padj, adjusted p-value; RS, resistant starch; SS, soluble starch.
Almagro-Moreno, S., and Boyd, E. F. (2009). Insights into the evolution of sialic acid catabolism among bacteria. BMC Evol. Biol. 9:118. doi: 10.1186/1471-2148-9-118
Belzer, C., Chia, L. W., Aalvink, S., Chamlagain, B., Piironen, V., Knol, J., et al. (2017). Microbial metabolic networks at the mucus layer lead to diet-independent butyrate and vitamin B12 production by intestinal symbionts. mBio 8:e00770-17. doi: 10.1128/mBio.00770-17
Boratyn, G. M., Camacho, C., Cooper, P. S., Coulouris, G., Fong, A., Ma, N., et al. (2013). BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41, W29–W33. doi: 10.1093/nar/gkt282
Bunesova, V., Lacroix, C., and Schwab, C. (2018). Mucin cross-feeding of infant Bifidobacteria and Eubacterium hallii. Microb. Ecol. 75, 228–238. doi: 10.1007/s00248-017-1037-4
Centanni, M., Hutchison, J. C., Carnachan, S. M., Daines, A. M., Kelly, W. J., Tannock, G. W., et al. (2017). Differential growth of bowel commensal Bacteroides species on plant xylans of differing structural complexity. Carbohydr. Polym. 157, 1374–1382. doi: 10.1016/j.carbpol.2016.11.017
Centanni, M., Lawley, B., Butts, C. A., Roy, N., Lee, J., Kelly, W. J., et al. (2018). Bifidobacterium pseudolongum has characteristics of a keystone species in bifidobacterial blooms in the ceca of rats fed Hi-Maize starch. Appl. Environ. Microbiol. doi: 10.1128/AEM.00547-18 [Epub ahead of print].
Chia, L. W., Hornung, B. V. H., Aalvink, S., Schaap, P. J., de Vos, W. M., Knol, J., et al. (2018). Deciphering the trophic interaction between Akkermansia muciniphila and the butyrogenic gut commensal Anaerostipes caccae using a metatranscriptomic approach. Antonie Van Leeuwenhoek 111, 859–873. doi: 10.1007/s10482-018-1040-x
Crost, E. H., Tailford, L. E., Le Gall, G., Fons, M., Henrissat, B., and Juge, N. (2013). Utilisation of mucin glycans by the human gut symbiont Ruminococcus gnavus is strain-dependent. PLoS One 8:e76341. doi: 10.1371/journal.pone.0076341
Crost, E. H., Tailford, L. E., Monestier, M., Swarbreck, D., Henrissat, B., Crossman, L. C., et al. (2016). The mucin-degradation strategy of Ruminococcus gnavus: the importance of intramolecular trans-sialidases. Gut Microbes 7, 302–312. doi: 10.1080/19490976.2016.1186334
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. doi: 10.1038/nature12820
Donaldson, G. P., Lee, S. M., and Mazmanian, S. K. (2016). Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 14, 20–32. doi: 10.1038/nrmicro3552
Duncan, S. H., Hold, G. L., Barcenilla, A., Stewart, C. S., and Flint, H. J. (2002). Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int. J. Syst. Evol. Microbiol. 52(Pt 5), 1615–1620. doi: 10.1099/00207713-52-5-1615
Flint, H. J., Duncan, S. H., and Louis, P. (2017). The impact of nutrition on intestinal bacterial communities. Curr. Opin. Microbiol. 38, 59–65. doi: 10.1016/j.mib.2017.04.005
Gunning, A. P., Kirby, A. R., Fuell, C., Pin, C., Tailford, L. E., and Juge, N. (2013). Mining the “glycocode”–exploring the spatial distribution of glycans in gastrointestinal mucin using force spectroscopy. FASEB J. 27, 2342–2354. doi: 10.1096/fj.12-221416
Hoek, M., and Merks, R. M. H. (2017). Emergence of microbial diversity due to cross-feeding interactions in a spatial model of gut microbial metabolism. BMC Syst. Biol. 11:56. doi: 10.1186/s12918-017-0430-4
Koropatkin, N. M., Cameron, E. A., and Martens, E. C. (2012). How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 10, 323–335. doi: 10.1038/nrmicro2746
Lawson, P. A., and Finegold, S. M. (2015). Reclassification of Ruminococcus obeum as Blautia obeum comb. nov. Int. J. Syst. Evol. Microbiol. 65(Pt 3), 789–793. doi: 10.1099/ijs.0.000015
Liu, C., Finegold, S. M., Song, Y., and Lawson, P. A. (2008). Reclassification of Clostridium coccoides, Ruminococcus hansenii, Ruminococcus hydrogenotrophicus, Ruminococcus luti, Ruminococcus productus and Ruminococcus schinkii as Blautia coccoides gen. nov., comb. nov., Blautia hansenii comb. nov., Blautia hydrogenotrophica comb. nov., Blautia luti comb. nov., Blautia producta comb. nov., Blautia schinkii comb. nov. and description of Blautia wexlerae sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 58(Pt 8), 1896–1902. doi: 10.1099/ijs.0.65208-0
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. doi: 10.1093/nar/gkt1178
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Mark Welch, J. L., Hasegawa, Y., McNulty, N. P., Gordon, J. I., and Borisy, G. G. (2017). Spatial organization of a model 15-member human gut microbiota established in gnotobiotic mice. Proc. Natl. Acad. Sci. U.S.A. 114, E9105–E9114. doi: 10.1073/pnas.1711596114
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570. doi: 10.1093/nar/gkv468
Milani, C., Lugli, G. A., Duranti, S., Turroni, F., Mancabelli, L., Ferrario, C., et al. (2015). Bifidobacteria exhibit social behavior through carbohydrate resource sharing in the gut. Sci. Rep. 5:15782. doi: 10.1038/srep15782
Moore, W. E. C., Johnson, J. L., and Holdeman, L. V. (1976). Emendation of Bacteroidaceae and Butyrivibrio and descriptions of Desulfomonas gen. nov. and ten new species in the genera Desulfomonas, Butyrivibrio, Eubacterium, Clostridium, and Ruminococcus. Int. J. Syst. Evol. Microbiol. 26, 238–252. doi: 10.1099/00207713-26-2-238
Mukhopadhya, I., Morais, S., Laverde-Gomez, J., Sheridan, P. O., Walker, A. W., Kelly, W., et al. (2018). Sporulation capability and amylosome conservation among diverse human colonic and rumen isolates of the keystone starch-degrader Ruminococcus bromii. Environ. Microbiol. 20, 324–336. doi: 10.1111/1462-2920.14000
Murata, T., Yamato, I., Igarashi, K., and Kakinuma, Y. (1996). Intracellular Na+ regulates transcription of the ntp operon encoding a vacuolar-type Na+-translocating ATPase in Enterococcus hirae. J. Biol. Chem. 271, 23661–23666.
Ndeh, D., and Gilbert, H. J. (2018). Biochemistry of complex glycan depolymerisation by the human gut microbiota. FEMS Microbiol. Rev. 42, 146–164. doi: 10.1093/femsre/fuy002
Owen, C. D., Tailford, L. E., Monaco, S., Suligoj, T., Vaux, L., Lallement, R., et al. (2017). Unravelling the specificity and mechanism of sialic acid recognition by the gut symbiont Ruminococcus gnavus. Nat. Commun. 8:2196. doi: 10.1038/s41467-017-02109-8
Pereira, F. C., and Berry, D. (2017). Microbial nutrient niches in the gut. Environ. Microbiol. 19, 1366–1378. doi: 10.1111/1462-2920.13659
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Reichardt, N., Duncan, S. H., Young, P., Belenguer, A., McWilliam Leitch, C., Scott, K. P., et al. (2014). Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 8, 1323–1335. doi: 10.1038/ismej.2014.14
Rogowski, A., Briggs, J. A., Mortimer, J. C., Tryfona, T., Terrapon, N., Lowe, E. C., et al. (2015). Glycan complexity dictates microbial resource allocation in the large intestine. Nat. Commun. 6:7481. doi: 10.1038/ncomms8481
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
Sonnenburg, E. D., Smits, S. A., Tikhonov, M., Higginbottom, S. K., Wingreen, N. S., and Sonnenburg, J. L. (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215. doi: 10.1038/nature16504
Tailford, L. E., Crost, E. H., Kavanaugh, D., and Juge, N. (2015a). Mucin glycan foraging in the human gut microbiome. Front. Genet. 6:81. doi: 10.3389/fgene.2015.00081
Tailford, L. E., Owen, C. D., Walshaw, J., Crost, E. H., Hardy-Goddard, J., Le Gall, G., et al. (2015b). Discovery of intramolecular trans-sialidases in human gut microbiota suggests novel mechanisms of mucosal adaptation. Nat. Commun. 6:7624. doi: 10.1038/ncomms8624
Tannock, G. W., Wilson, C. M., Loach, D., Cook, G. M., Eason, J., O’Toole, P. W., et al. (2012). Resource partitioning in relation to cohabitation of Lactobacillus species in the mouse forestomach. ISME J. 6, 927–938. doi: 10.1038/ismej.2011.161
Thursby, E., and Juge, N. (2017). Introduction to the human gut microbiota. Biochem. J. 474, 1823–1836. doi: 10.1042/BCJ20160510
Tropini, C., Earle, K. A., Huang, K. C., and Sonnenburg, J. L. (2017). The gut microbiome: connecting spatial organization to function. Cell Host Microbe 21, 433–442. doi: 10.1016/j.chom.2017.03.010
Turroni, F., Bottacini, F., Foroni, E., Mulder, I., Kim, J. H., Zomer, A., et al. (2010). Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc. Natl. Acad. Sci. U.S.A. 107, 19514–19519. doi: 10.1073/pnas.1011100107
Turroni, F., Milani, C., Duranti, S., Mahony, J., van Sinderen, D., and Ventura, M. (2017). Glycan utilization and cross-feeding activities by Bifidobacteria. Trends Microbiol. 26, 339–350. doi: 10.1016/j.tim.2017.10.001
Turroni, F., Strati, F., Foroni, E., Serafini, F., Duranti, S., van Sinderen, D., et al. (2012). Analysis of predicted carbohydrate transport systems encoded by Bifidobacterium bifidum PRL2010. Appl. Environ. Microbiol. 78, 5002–5012. doi: 10.1128/AEM.00629-12
Williams, B. B., Van Benschoten, A. H., Cimermancic, P., Donia, M. S., Zimmermann, M., Taketani, M., et al. (2014). Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 16, 495–503. doi: 10.1016/j.chom.2014.09.001
Ze, X., Ben David, Y., Laverde-Gomez, J. A., Dassa, B., Sheridan, P. O., Duncan, S. H., et al. (2015). Unique organization of extracellular amylases into amylosomes in the resistant starch-utilizing human colonic firmicutes bacterium Ruminococcus bromii. mBio 6:e01058-15. doi: 10.1128/mBio.01058-15
Ze, X., Duncan, S. H., Louis, P., and Flint, H. J. (2012). Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 6, 1535–1543. doi: 10.1038/ismej.2012.4
Keywords: cross-feeding, gut bacteria, Ruminococcus, mucin, resistant starch
Citation: Crost EH, Le Gall G, Laverde-Gomez JA, Mukhopadhya I, Flint HJ and Juge N (2018) Mechanistic Insights Into the Cross-Feeding of Ruminococcus gnavus and Ruminococcus bromii on Host and Dietary Carbohydrates. Front. Microbiol. 9:2558. doi: 10.3389/fmicb.2018.02558
Received: 20 July 2018; Accepted: 08 October 2018;
Published: 05 November 2018.
Edited by:
Robert Rastall, University of Reading, United KingdomReviewed by:
Glenn Gibson, University of Reading, United KingdomCopyright © 2018 Crost, Le Gall, Laverde-Gomez, Mukhopadhya, Flint and Juge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nathalie Juge, bmF0aGFsaWUuanVnZUBxdWFkcmFtLmFjLnVr; bmF0aGFsaWUuanVnZUBpZnIuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.