 Marion Holzapfel
Marion Holzapfel Guillaume Girault1
Guillaume Girault1 Anne Keriel
Anne Keriel Claire Ponsart
Claire Ponsart David O’Callaghan
David O’Callaghan Virginie Mick
Virginie Mick- 1EU/OIE/FAO and National Reference Laboratory for Brucellosis, Animal Health Laboratory, Anses/Paris-Est University, Maisons-Alfort, France
- 2VBMI, INSERM, U1047, Université de Montpellier, Nîmes, France
- 3CNR Laboratoire Expert Brucella, Service de Microbiologie, CHU Caremeau, Nîmes, France
Brucella spp. are responsible for brucellosis, a widespread zoonosis causing reproductive disorders in animals. Species-classification within this monophyletic genus is based on bacteriological and biochemical phenotyping. Traditionally, Brucella species are reported to have a preferential, but not exclusive mammalian host. However, this concept can be challenged since many Brucella species infect a wide range of animal species. Adaptation to a specific host can be a driver of pathogen variation. It is generally thought that Brucella species have highly stable and conserved genomes, however the degree of genomic variation during natural infection has not been documented. Here, we investigated potential genetic diversity and virulence of Brucella melitensis biovar 3 field isolates obtained from a single outbreak but from different host species (human, bovine, small ruminants). A unique MLVA-16 pattern suggested all isolates were clonal. Comparative genomic analyses showed an almost non-existent genetic diversity among isolates (only one SNP; no architectural rearrangements) and did not highlight any signature specific to host adaptation. Similarly, the strains showed identical capacities to enter and replicate in an in vitro model of macrophage infection. In our study, the absence of genomic variability and similar virulence underline that B. melitensis biovar 3 is a broad-host-range pathogen without the need to adapt to different hosts.
Introduction
Brucellosis, caused by bacteria of the Brucella genus, is a widespread zoonosis causing reproductive disorders in animals and a debilitating infection in humans. Brucella classification is based on bacteriological and biochemical phenotypes (Ficht, 2010). Traditionally, different species were reported to have a preferential mammalian host, however in reality, spill over into a wide range of animal species has been reported. B. melitensis, for example, primarily responsible for disease in domestic small ruminants (sheep and goats), also causes natural infections in bovids, camelids and wild ruminants (e.g., ibex) (Office International des Epizooties [OIE], 2016) (Garin-Bastuji et al., 2014), and is the major species responsible for human infections.
Comparative genomics have allowed us to identify the key steps in the evolution of Brucella from a soil organism to a stealth pathogen. This has involved the acquisition of a battery of virulence factors followed by a toning down of virulence (Wattam et al., 2014). What is not understood is how the different species have adapted to their ‘preferential’ host, or the time line of this adaptation. Despite the strong sequence homogeneity within the genus (>94% nucleotide identity) (Wattam et al., 2009), genome-wide studies suggest genetic variation, especially pseudogenization, i.e., due to IS711 insertion and point mutations within marine Brucella genomes (Suárez-Esquivel et al., 2017), might play a key role in Brucella host preference as the bacteria evolved with their respective preferential hosts (Chain et al., 2005). In a recent report, Ke and collaborators suggested that B. melitensis rapidly accumulates mutations during in vivo passage, however this observation is not supported by the high levels of sequence conservation seen between the genome sequences available in Genbank (Ke et al., 2012).
In this study we investigate seven clonal isolates of B. melitensis biovar 3 (bv3) isolated from different mammalian hosts during a well characterized outbreak in southern France. We used in vitro virulence assays and comparative genomics to determine whether genetic variation is required for the ability to infect different hosts.
Materials and Methods
Bacterial Strains
Seven isolates (Supplementary Table S1) collected over a 1-year period (2000–2001) from a restricted geographical area (Drôme, South-East France) from different host species: human (n = 1); bovine (n = 2); small ruminants (sheep and goat) (n = 4) were biotyped using standard procedures (Office International des Epizooties [OIE], 2016). Records regarding this epidemiological event available at the National Reference Laboratory (NRL) for brucellosis were examined. At least one abortion occurred amongst infected animals, probably in small ruminant. Epidemiological and serological investigations conducted to the slaughtering of the herd.
B. melitensis reference strains 16M (bv1) and Ether (bv3) (Supplementary Table S1) were included in this study.
Molecular Analyses and Comparative Genomics
Genomic DNA was extracted using High Pure DNA Template Preparation kit (Roche Diagnostics, France), according to the manufacturer’s recommendations. Real Time-PCR and MLVA-16 assays were performed as previously described (Al Dahouk et al., 2007; Bounaadja et al., 2009).
Genome sequencing was performed on an Illumina HiSeq2500 platform (2 × 250 bp paired-end reads). Raw reads were trimmed using Trimmomatic (Bolger et al., 2014) and quality controls were conducted with FastQC (Andrews, 2010). Mapping assemblies against reference concatenated genomes (i.e., both chromosomes I and II are concatenated together in a single file) of Ether and 16M were performed using Bionumerics v7.6.2 [Burrows-Wheeler Aligner BWA (Li and Durbin, 2009)], as well as de novo assembly using SPAdes v3.9. Annotations were performed with Prokka (Seemann, 2014) and alignments with progressiveMAUVE (Darling et al., 2010) and BLAST Ring Image Generator (BRIG) 0.95 (Alikhan et al., 2011). Genome assemblies were evaluated with QUAST 4.6.3 (Gurevich et al., 2013).
Single Nucleotide Polymorphism (SNP) and clustering (maximum-parsimony) analyses were performed using Bionumerics v7.6.2 (wgSNP-module). Applied filters removed indels, repeated regions and rRNAs with minimum ten bp-distance between SNPs. Non-synonymous, synonymous and intergenic SNPs were identified against 16M. Local alignment of interesting regions were done with ClustalW (Thompson et al., 1994) via MEGA 6 (Tamura et al., 2013).
To functionally analyze genomes, Roary v3.6.1 (Page et al., 2015) was used to generate matrices of presence/absence of core (i.e., genes present in all genomes included in the study) and accessory (i.e., not core genes) genes among seven investigated genomes, or among seven genomes vs. 16M. Scoary (Brynildsrud et al., 2016) was used to associate the presence/absence of genes/SNPs to given clustering according to host-range.
Sequences were deposited in European-Nucleotide-Archive (Study PRJEB26921) (Supplementary Table S1).
Macrophage Infection Assays
Murine J774.1 macrophages (ATCC® TIB-67TM) were cultivated and infected with Brucella using a standard gentamycin protection protocol as described previously (Soler-Llorens et al., 2016). All experiments were performed in triplicate and independently repeated three times.
The ability of infection of different field isolates was represented as the Brucella penetration index (BPI) and the Brucella multiplication index (BMI). BPI was defined as the percentage of the inoculum inside macrophages at 2 h post-inoculation (PI); BMI as the ratio of CFU in cell lysates at 48 h PI to CFU at 2 h PI. Results were analyzed using a non-parametric Kruskal-Wallis test (GraphPad Prism program) with significant p-values < 0.05.
Results
Seven investigated isolates were confirmed as B. melitensis bv3 using classical microbiological and molecular methods. All seven isolates had identical phenotypes. All isolates had an identical MLVA-16 pattern: Bruce06 3U; Bruce08 5U; Bruce11 3U; Bruce12 13U; Bruce42 1U; Bruce43 1U; Bruce45 3U; Bruce55 3U; Bruce18 7U; Bruce19 42U; Bruce21 8U; Bruce04 8U; Bruce07 5U; Bruce09 12U; Bruce16 8U; Bruce30 3U. This suggests that the isolates were clonal.
Comparative Genomics
The global genomic architecture of the seven strains was consistent with those of published B. melitensis genomes (i.e., presence of two chromosomes). The field isolates had a genome size of approximately 3.3 Mbp, a G-C content similar to that of the Ether genome (57.24% vs. 57.22%) with 3,210–3,218 predicted protein-encoding genes (QUAST). After filtering, a total of 2,748 SNPs were identified among all studied strains vs. 16M, but only one SNP was observed amongst the seven field strains, in an ovine isolate (Figure 1). This C-to-T substitution at position 267 of the hisN gene coding for histidinol-phosphatase (BME_RS10415) is a silent mutation. Manual analysis also showed a bovine isolate harbored another SNP in hisN (A-to-G at nucleotide 599) that was discarded by automated analysis; this lead to an Asp200Gly modification of HisN.
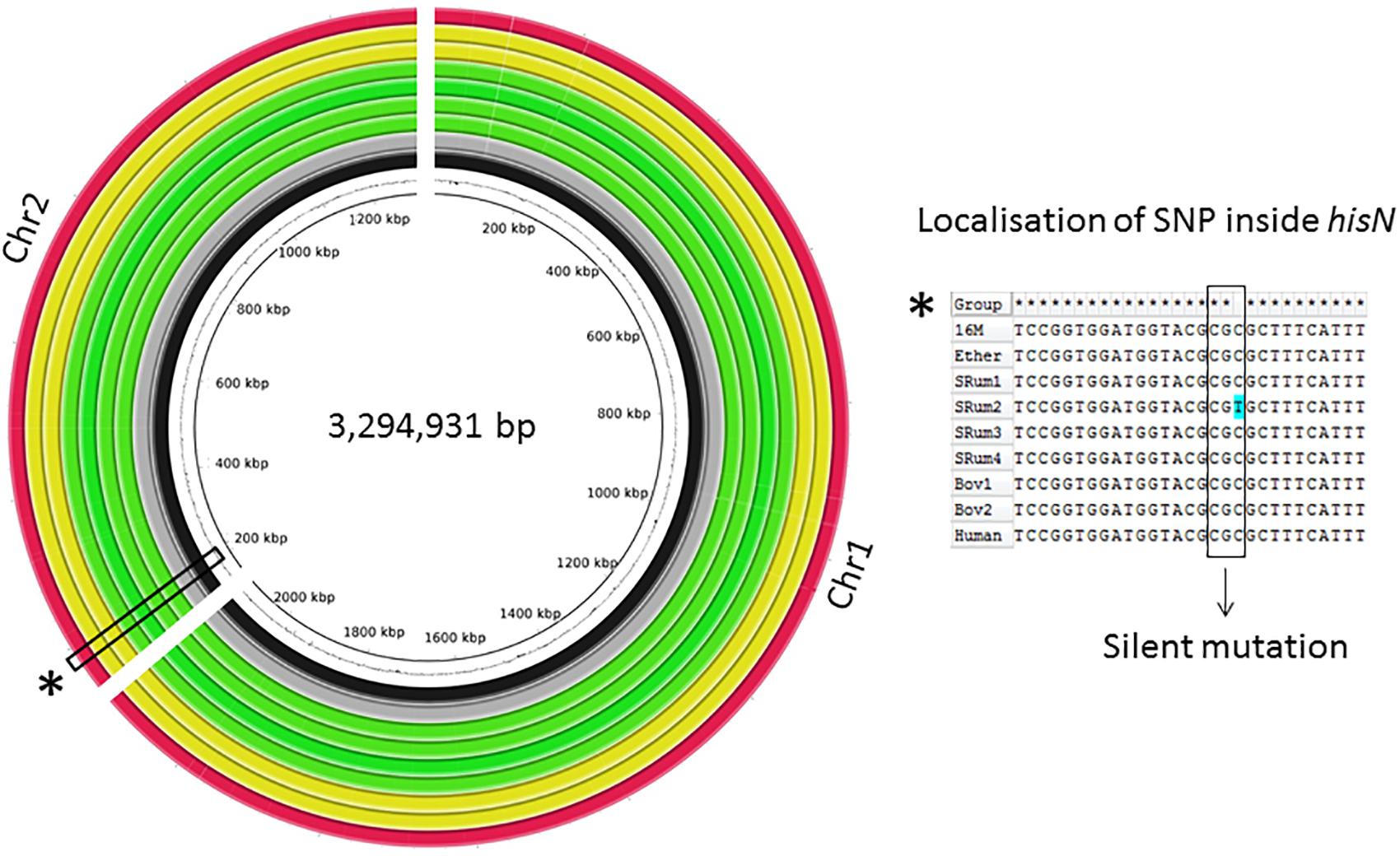
FIGURE 1. Circular alignment representation of investigated Brucella melitensis bv3 genomes. Genomes of seven clonal B. melitensis bv3 isolates were de novo assembled. BRIG alignment of investigated genomes and Ether against 16M are shown. Rings from inside to outside: GC-% of 16M, 16M (black), Ether (gray), B. melitensis bv3 isolated from small ruminants (n = 4) (green), from bovine (n = 2) (yellow) and from human (n = 1) (red). Alignment of the hisN region flanking the unique SNP between seven isolates generated with ClustalW.
Genome-wide comparison from de novo assemblies showed a perfect collinearity of the seven genomes as well as with those of 16M and Ether, highlighting absence of genomic rearrangements (Figure 1). Further comparison regarding the pan-genome, i.e., among seven genomes, highlighted a total amount of 3,196 predicted genes, including 3,141 core genes and 55 accessory genes. The strains showed a full complement of previously described virulence factors (He, 2012).
Macrophage Infection Assays
The virulence of the isolates was compared within an in vitro murine macrophage infection model. The seven bv3 isolates showed similar ability to infect and multiply in macrophages as the reference strain. In our conditions, B. melitensis bv3 small ruminant isolates had slightly but significantly lower BPI values than 16M (p = 0.02), unlike human and bovine isolates (p > 0.99 and p = 0.19, respectively) (Figure 2B). BPI values showed no significant difference among isolates whatever host-species (p = 0.34 for human vs. small ruminants to >0.99 for other comparisons) (Figure 2B). At 48 h PI, BMI observed between 16M vs. isolates (p > 0.99 for all comparisons) and among isolates did not show significant difference (p = 0.26 bovine vs. human; p = 0.37 bovine vs. small ruminants; p > 0.99 human vs. small ruminants) correlated with host-species (Figure 2C). Indeed, multiplication inside macrophages was similar to the intracellular growth of reference 16M (Figure 2A), and statistically identical among tested isolates.
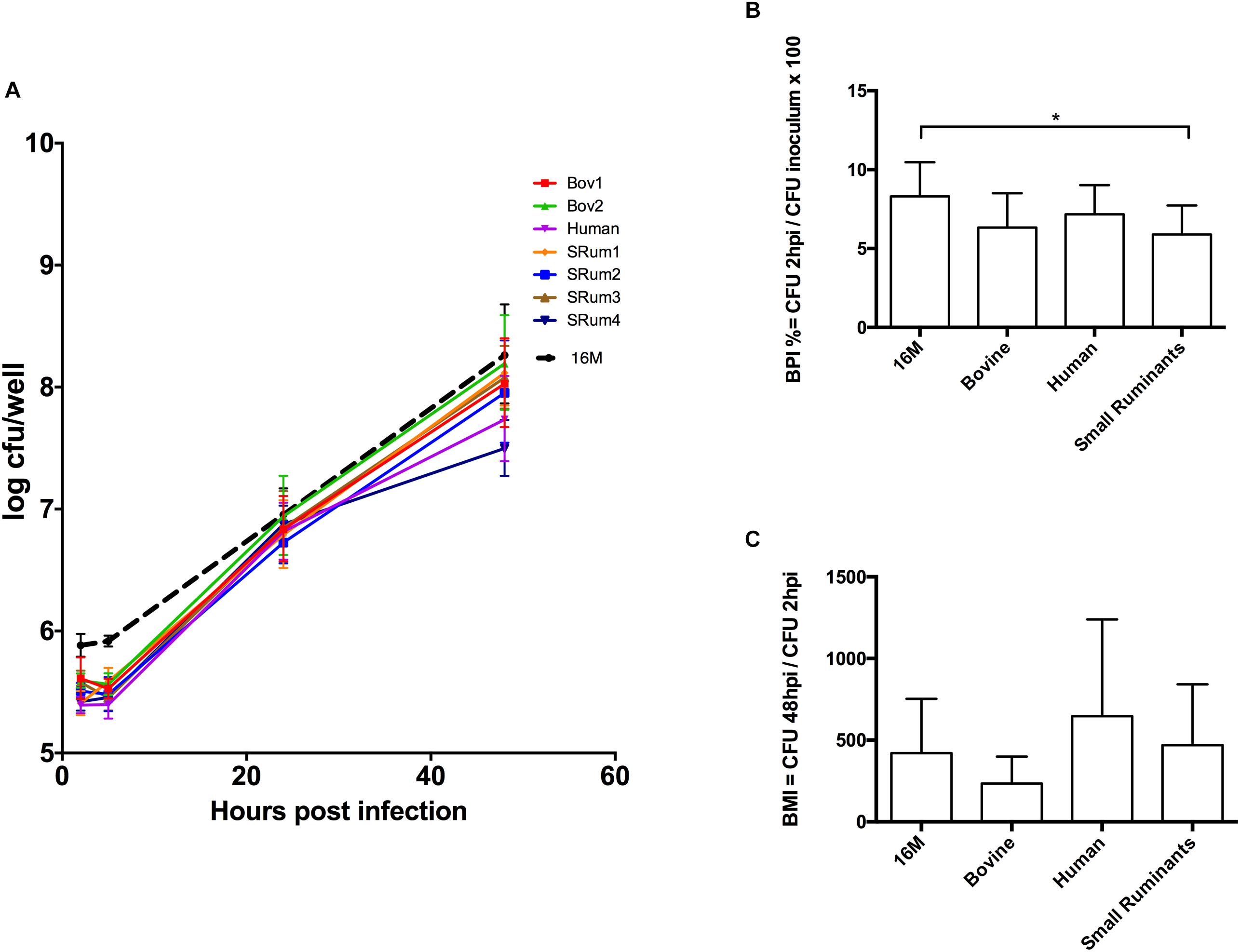
FIGURE 2. Macrophage infection assay with clonal B. melitensis bv3 isolates from different host-range. (A) Intracellular growth of Brucella isolates inside murine macrophages J774A.1. Each point is performed in triplicates from three independent experiments and represents the log10 of the mean ± SD CFU/well. (B) Adhesion/Invasion capacity of Brucella isolates. (C) Brucella multiplicity index. Isolates are grouped into three host-categories – bovine (n = 2), human (n = 1) and small ruminants (n = 4)– and compared to the reference B. melitensis biovar 1str. 16M. Error bars denote standard errors. Statistical significance of each group was determined by Kruskal-Wallis test. ∗p < 0.05. Bov1/2, B. melitensis bv3 isolated from bovine; Human, B. melitensis bv3 isolated from human; SRum1/2/3/4, B. melitensis bv3 isolated from small ruminants.
Discussion
Brucella species have been classically assigned to preferential, but not exclusive animal hosts. Hence, B. melitensis is associated with infection of small ruminants and B. abortus with cattle. There have, however been numerous reports of B. melitensis causing abortion in cattle (Verger et al., 1989; Alvarez et al., 2011; El-Diasty et al., 2018). In this study, we characterized seven B. melitensis bv3 field isolates, considered as epidemiologically related because isolated from a same outbreak (same geographical area, same time-period), but from different mammalian hosts. As it was not possible to distinguish between the strains phenotypically and the strains all had the same single identical pattern in MLVA-16, we considered the seven strains as clonal, according to the definition of a “clone” from Tenover et al. (1995) isolates that are indistinguishable from each other by a variety of genetic tests (e.g., PFGE, multilocus enzyme electrophoresis, or ribotyping) or that are so similar that they are presumed to be derived from a common parent. Consequently, this study mimics an in vivo experimental infection aiming to determine whether genetic modifications are induced during the short-time adaptation of B. melitensis to a transition of environment, i.e., different host species, host being considered as driver of pathogen variation. Adaptation is defined as the process of change by which an organism or species becomes better suited to its environment (Ryall et al., 2012).
Comparison with the genomes of two B. melitensis reference strains showed perfect synteny. Our in-depth comparative genomics analyses revealed an almost non-existent genetic diversity among the seven Brucella field isolates, with only one SNP confirming that there is no signature of adaptation to different host-species. Most importantly, our data is in complete contrast with the study from Ke et al. (2012) who reported over 5,000 SNPs after 13 weeks mouse passage of B. melitensis 16M and suggested that Brucella accumulates a large number of adaptive mutations during chronic infection.
The seven field isolates showed identical virulence in murine macrophages. B. melitensis clearly has the intrinsic capacity to infect a wide range of mammalian hosts without the need to adapt. It is more likely that the traditional host preference was due to the epidemiology and lack of contact between infected small ruminants and other livestock. Domestic dogs and cats, and feral swine are occasionally infected with Brucella through contact with infected livestock (Stoffregen et al., 2007; Lucero et al., 2008; Hinic et al., 2010; Wareth et al., 2017). B. melitensis bv3 has also been isolated from Nile catfish, probably infected by contaminated animal waste in the water contamination, showing the broad spectrum of hosts (El-Tras et al., 2010). Some Brucella species have a more restricted host range. B. suis bv2, which is endemic in wild boar and hares in much of Europe, shows low levels of pathogenicity for ruminants and for humans (Mailles et al., 2017). Genome analysis showed a large inversion and a number of INDELs and SNPs, although none were predicted to affect known virulence factors (Ferreira et al., 2017). The clearest example of host restriction is B. ovis, which is almost only pathogenic for sheep, with some studies reporting clinical sign in Red Deer (Ridler et al., 2012). Here a clear process of genome degradation and accumulation of pseudogenes is evident (Tsolis et al., 2009). Genomic decay seems to be associated with a host-restricted pattern, whereas intact genes are present in broad-host-range pathogens (Kirzinger and Stavrinides, 2012). The absence of pseudogenes and/or of genomic rearrangement among genomes studied here underlines that B. melitensis bv3 remains a broad-host-range pathogen.
In conclusion, this study shows that B. melitensis does not require any adaptive mutations when infecting different mammalian hosts. We cannot exclude the possibility that adaptation to a different host requires differential expression of specific genes that cannot be detected with the simple experimental models that we use.
Author Contributions
VM and DO conceived the study. CP obtained funding. MH and GG performed genomics analyses. MH and AK performed macrophage infection assays. MH, GG, CP, and VM performed data interpretation. MH, DO, and VM wrote the paper. All authors read and approved the manuscript content.
Funding
MH was supported by a Ph.D. fellowship the (CIFRE grant) French Direction Générale de l’Armement (DGA and IDVet). This work was supported by European Union Reference Laboratory for Brucellosis, INSERM, Université de Montpellier and Santé Publique France.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to express their gratitude to Maëllys Kevin and Benoit Durand for critical and supportive comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02505/full#supplementary-material
TABLE S1 | Field Brucella isolates investigated in this study and reference strains from public databases.
References
Al Dahouk, S., Fleche, P. L., Nockler, K., Jacques, I., Grayon, M., Scholz, H. C., et al. (2007). Evaluation of Brucella MLVA typing for human brucellosis.J. Microbiol. Methods 69, 137–145. doi: 10.1016/j.mimet.2006.12.015
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Alvarez, J., Saez, J. L., Garcia, N., Serrat, C., Perez-Sancho, M., Gonzalez, S., et al. (2011). Management of an outbreak of brucellosis due to B. melitensis in dairy cattle in Spain. Res. Vet. Sci. 90, 208–211. doi: 10.1016/j.rvsc.2010.05.028
Andrews, S. (2010). FastQC: A Quality Control tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bounaadja, L., Albert, D., Chenais, B., Henault, S., Zygmunt, M. S., Poliak, S., et al. (2009). Real-time PCR for identification of Brucella spp.: a comparative study of IS711, bcsp31 and per target genes. Vet. Microbiol. 137, 156–164. doi: 10.1016/j.vetmic.2008.12.023
Brynildsrud, O., Bohlin, J., Scheffer, L., and Eldholm, V. (2016). Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 17:238. doi: 10.1186/s13059-016-1108-8
Chain, P. S., Comerci, D. J., Tolmasky, M. E., Larimer, F. W., Malfatti, S. A., Vergez, L. M., et al. (2005). Whole-genome analyses of speciation events in pathogenic Brucellae. Infect. Immun. 73, 8353–8361. doi: 10.1128/IAI.73.12.8353-8361.2005
Darling, A. E., Mau, B., and Perna, N. T. (2010). Progressivemauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
El-Diasty, M., Wareth, G., Melzer, F., Mustafa, S., Sprague, L. D., and Neubauer, H. (2018). Isolation of Brucella abortus and Brucella melitensis from seronegative cows is a serious impediment in brucellosis control. Vet. Sci. 5:E28. doi: 10.3390/vetsci5010028
El-Tras, W. F., Tayel, A. A., Eltholth, M. M., and Guitian, J. (2010). Brucella infection in fresh water fish: evidence for natural infection of Nile catfish, Clarias gariepinus, with Brucella melitensis. Vet. Microbiol. 141, 321–325. doi: 10.1016/j.vetmic.2009.09.017
Ferreira, A. C., Correa de Sa, M. I., Dias, R., and Tenreiro, R. (2017). MLVA-16 typing of Brucella suis biovar 2 strains circulating in Europe. Vet. Microbiol. 210, 77–82. doi: 10.1016/j.vetmic.2017.09.001
Ficht, T. A. (2010). Brucella taxonomy and evolution. Future Microbiol. 5, 859–866. doi: 10.2217/fmb.10.52
Garin-Bastuji, B., Hars, J., Drapeau, A., Cherfa, M. A., Game, Y., Le Horgne, J. M., et al. (2014). Reemergence of Brucella melitensis in wildlife, France. Emerg. Infect. Dis. 20, 1570–1571. doi: 10.3201/eid2009.131517
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
He, Y. (2012). Analyses of Brucella pathogenesis, host immunity, and vaccine targets using systems biology and bioinformatics. Front. Cell Infect. Microbiol. 2:2. doi: 10.3389/fcimb.2012.00002
Hinic, V., Brodard, I., Petridou, E., Filioussis, G., Contos, V., Frey, J., et al. (2010). Brucellosis in a dog caused by Brucella melitensis rev 1. Vet. Microbiol. 141, 391–392. doi: 10.1016/j.vetmic.2009.09.019
Ke, Y., Yang, X., Wang, Y., Bai, Y., Xu, J., Song, H., et al. (2012). Genome sequences of Brucella melitensis 16m and its two derivatives 16m1w and 16m13w, which evolved in vivo. J. Bacteriol. 194:5489. doi: 10.1128/JB.01293-12
Kirzinger, M. W., and Stavrinides, J. (2012). Host specificity determinants as a genetic continuum. Trends Microbiol. 20, 88–93. doi: 10.1016/j.tim.2011.11.006
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Lucero, N. E., Ayala, S. M., Escobar, G. I., and Jacob, N. R. (2008). Brucella isolated in humans and animals in latin America from 1968 to 2006. Epidemiol. Infect. 136, 496–503. doi: 10.1017/S0950268807008795
Mailles, A., Ogielska, M., Kemiche, F., Garin-Bastuji, B., Brieu, N., Burnusus, Z., et al. (2017). Brucella suis biovar 2 infection in humans in France: emerging infection or better recognition? Epidemiol. Infect. 145, 2711–2716.
Office International des Epizooties [OIE] (2016). Brucellosis (Brucella abortus, B. melitensis and B. suis)(Infection with abortus, B., B. melitensis and B. suis). Paris: OIE Terrestrial Manual.
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scal prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Ridler, A. L., West, D. M., and Collett, M. G. (2012). Pathology of Brucella ovis infection in red deer stags (Cervus elaphus). New Zealand Vet. J. 60, 146–149. doi: 10.1080/00480169.2011.638269
Ryall, B., Eydallin, G., and Ferenci, T. (2012). Culture history and population heterogeneity as determinants of bacterial adaptation: the adaptomics of a single environmental transition. Microbiol. Mol. Biol. Rev. 76, 597–625. doi: 10.1128/MMBR.05028-11
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Soler-Llorens, P. F., Quance, C. R., Lawhon, S. D., Stuber, T. P., Edwards, J. F., Ficht, T. A., et al. (2016). A Brucella spp. Isolate from a pac-man frog (Ceratophrys ornata) reveals characteristics departing from classical Brucellae. Front. Cell Infect. Microbiol. 6:116. doi: 10.3389/fcimb.2016.00116
Stoffregen, C., Olsen, S. C., Wheeler, C. J., Bricker, B. J., Palmer, M. V., Jensen, A. E., et al. (2007). Diagnostic characterization of a feral swine herd enzootically infected with Brucella. J. Vet. Diagn. Invest. 19, 227–237.
Suárez-Esquivel, M., Baker, K. S., Ruiz-Villalobos, N., Hernandez-Mora, G., Barquero-Calvo, E., Gonzalez-Barrientos, R., et al. (2017). Brucella genetic variability in wildlife marine mammals populations relates to host preference and ocean distribution. Genome Biol. Evol. 9, 1901–1912. doi: 10.1093/gbe/evx137
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tenover, F. C., Arbeit, R. D., Goering, R. V., Mickelsen, P. A., Murray, B. E., Persing, D. H., et al. (1995). Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J. Clin. Microbiol. 33, 2233–2239.
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680.
Tsolis, R. M., Seshadri, R., Santos, R. L., Sangari, F. J., Lobo, J. M., de Jong, M. F., et al. (2009). Genome degradation in Brucella ovis corresponds with narrowing of its host range and tissue tropism. PLoS One 4:e5519. doi: 10.1371/journal.pone.0005519
Verger, J. M., Garin-Bastuji, B., Grayon, M., and Mahé, A. M. (1989). La brucellose bovine à Brucella melitensis en France. Ann. Rech. Vet. 20, 93–102.
Wareth, G., Melzer, F., El-Diasty, M., Schmoock, G., Elbauomy, E., Abdel-Hamid, N., et al. (2017). Isolation of Brucella abortus from a dog and a cat confirms their biological role in re-emergence and dissemination of bovine brucellosis on dairy farms. Transbound. Emerg. Dis. 64, e27–e30. doi: 10.1111/tbed.12535
Wattam, A. R., Williams, K. P., Snyder, E. E., Almeida, NF Jr, Shukla, M., Dickerman, A. W., et al. (2009). Analysis of ten Brucella genomes reveals evidence for horizontal gene transfer despite a preferred intracellular lifestyle. J. Bacteriol. 191, 3569–3579. doi: 10.1128/JB.01767-08
Keywords: brucellosis, Brucella melitensis, host preference, whole genome sequencing, comparative genomics, macrophage infection, adaptation
Citation: Holzapfel M, Girault G, Keriel A, Ponsart C, O’Callaghan D and Mick V (2018) Comparative Genomics and in vitro Infection of Field Clonal Isolates of Brucella melitensis Biovar 3 Did Not Identify Signature of Host Adaptation. Front. Microbiol. 9:2505. doi: 10.3389/fmicb.2018.02505
Received: 18 July 2018; Accepted: 02 October 2018;
Published: 22 October 2018.
Edited by:
Michel Stanislas Zygmunt, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Jeffrey T. Foster, Northern Arizona University, United StatesMenachem Banai, Kimron Veterinary Institute, Israel
Copyright © 2018 Holzapfel, Girault, Keriel, Ponsart, O’Callaghan and Mick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Virginie Mick, dmlyZ2luaWUubWlja0BhbnNlcy5mcg==