 Carol Davies-Sala
Carol Davies-Sala Saumya Jani
Saumya Jani Angeles Zorreguieta
Angeles Zorreguieta Marcelo E. Tolmasky
Marcelo E. Tolmasky- 1Center for Applied Biotechnology Studies, College of Natural Sciences and Mathematics, California State University, Fullerton, Fullerton, CA, United States
- 2Fundación Instituto Leloir, IIBBA-CONICET, Buenos Aires, Argentina
- 3Facultad de Ciencias Exactas y Naturales de la Universidad de Buenos Aires, University of Buenos Aires, Buenos Aires, Argentina
The bacterial ribonuclease P or RNase P holoenzyme is usually composed of a catalytic RNA subunit, M1, and a cofactor protein, C5. This enzyme was first identified for its role in maturation of tRNAs by endonucleolytic cleavage of the pre-tRNA. The RNase P endonucleolytic activity is characterized by having structural but not sequence substrate requirements. This property led to development of EGS technology, which consists of utilizing a short antisense oligonucleotide that when forming a duplex with a target RNA induces its cleavage by RNase P. This technology is being explored for designing therapies that interfere with expression of genes, in the case of bacterial infections EGS technology could be applied to target essential, virulence, or antibiotic resistant genes. Acinetobacter baumannii is a problematic pathogen that is commonly resistant to multiple antibiotics, and EGS technology could be utilized to design alternative therapies. To better understand the A. baumannii RNase P we first identified and characterized the catalytic subunit. We identified a gene coding for an RNA species, M1Ab, with the expected features of the RNase P M1 subunit. A recombinant clone coding for M1Ab complemented the M1 thermosensitive mutant Escherichia coli BL21(DE3) T7A49, which upon transformation was able to grow at the non-permissive temperature. M1Ab showed in vitro catalytic activity in combination with the C5 protein cofactor from E. coli as well as with that from A. baumannii, which was identified, cloned and partially purified. M1Ab was also able to cleave a target mRNA in the presence of an EGS with efficiency comparable to that of the E. coli M1, suggesting that EGS technology could be a viable option for designing therapeutic alternatives to treat multiresistant A. baumannii infections.
Introduction
Ribonuclease P, or RNase P, is a ubiquitous ribozyme that was first identified for its participation in the maturation of the precursor tRNA (pre-tRNA) by endonucleolytic cleavage at the 5′-end of the molecule (Robertson et al., 1972). Later, it was shown that RNase P participates in other biological processes like the synthesis of other RNA species such as transfer messenger RNA, precursors to 4.5S RNA, some multicistronic mRNAs, small non-coding RNA genes, phage-related RNAs, and others (Bothwell et al., 1976; Alifano et al., 1994; Komine et al., 1994; Hartmann et al., 1995; Reiner et al., 2006; Yang and Altman, 2007; Jarrous and Gopalan, 2010; Altman, 2011; Klemm et al., 2016). The RNase P holoenzyme is a ribonucleoprotein composed by the RNA molecule responsible for its catalytic activity, known as M1 in Escherichia coli (M1Ec), and one or more proteins that act as cofactors (Guerrier-Takada et al., 1983; Marvin and Engelke, 2009; Jarrous, 2017). In particular, bacterial RNase P holoenzymes usually contain only one small cofactor protein, called C5 in E. coli (C5Ec) (Reiter et al., 2010; Mondragon, 2013). Numerous studies on the RNase P enzymes from different organisms belonging to all three life domains showed many common structural features among the RNA components and a common core with similar secondary structure (Chen and Pace, 1997; Mondragon, 2013). Three types of RNA components of RNase P were identified in bacteria, the most common are known as types A (for ancestral) and B (for Bacillus); type C includes RNA molecules from green non-sulfur bacteria (Haas and Brown, 1998). For a comparative structural diagram among all three types see the review by Mondragon (Mondragon, 2013). The RNA components of bacterial RNase P include two domains, one of them called C (catalytic) that recognizes the acceptor stem and the 3′-CCA sequence of the substrate RNA and mediates the endonucleolytic cleavage. The other domain is called S (specificity) and is responsible for substrate recognition (Mondragon, 2013). Crystallographic studies on bacterial RNase P are limited, only the three-dimensional structures of the intact Thermotoga maritima RNA component and a fragment including the C domain of that of Bacillus stearothermophilus are available (Kazantsev et al., 2005; Torres-Larios et al., 2005). However, although not at the structural level, the E. coli RNase P, which consists of the 377-nucleotides catalytic RNA subunit M1Ec and the 119-amino acids cofactor protein C5Ab, is one of the best characterized (Guerrier-Takada et al., 1983). RNase P requires a particular structure in the substrate RNA that includes a double-stranded region (acceptor stem) followed by a single-stranded stretch that includes the RCCA sequence at the 3′ end, which facilitates interaction with the enzyme (Figure 1). One of the two complementary segments of the acceptor stem is called “external guide sequence” (EGS) and is instrumental in guiding RNase P to cleave the opposite strand during the maturation process (Figure 1). With the exception of the acceptor stem, most other regions of the substrate RNA can be deleted without completely abolishing RNase P activity and, although essential for cleavage, the EGS is not required to be tethered to the rest of the molecule (Forster and Altman, 1990; Gopalan et al., 2002; Figure 1). Furthermore, cleavage is not dependent on the sequence of the substrate (Figure 1). This finding was the foundation of the EGS technology, a gene silencing strategy in which a short oligomer (EGS) interacts with a target RNA, usually mRNA, and elicits its cleavage, interfering with expression of the gene (Lundblad and Altman, 2010). EGS Technology approaches have been explored as alternatives to design therapeutic tools or antibiotic adjuvants for treatment of multidrug resistant bacterial infections (Guerrier-Takada et al., 1997; Soler Bistue et al., 2007, 2009; Ko et al., 2008; Shen et al., 2009; Sawyer et al., 2013).
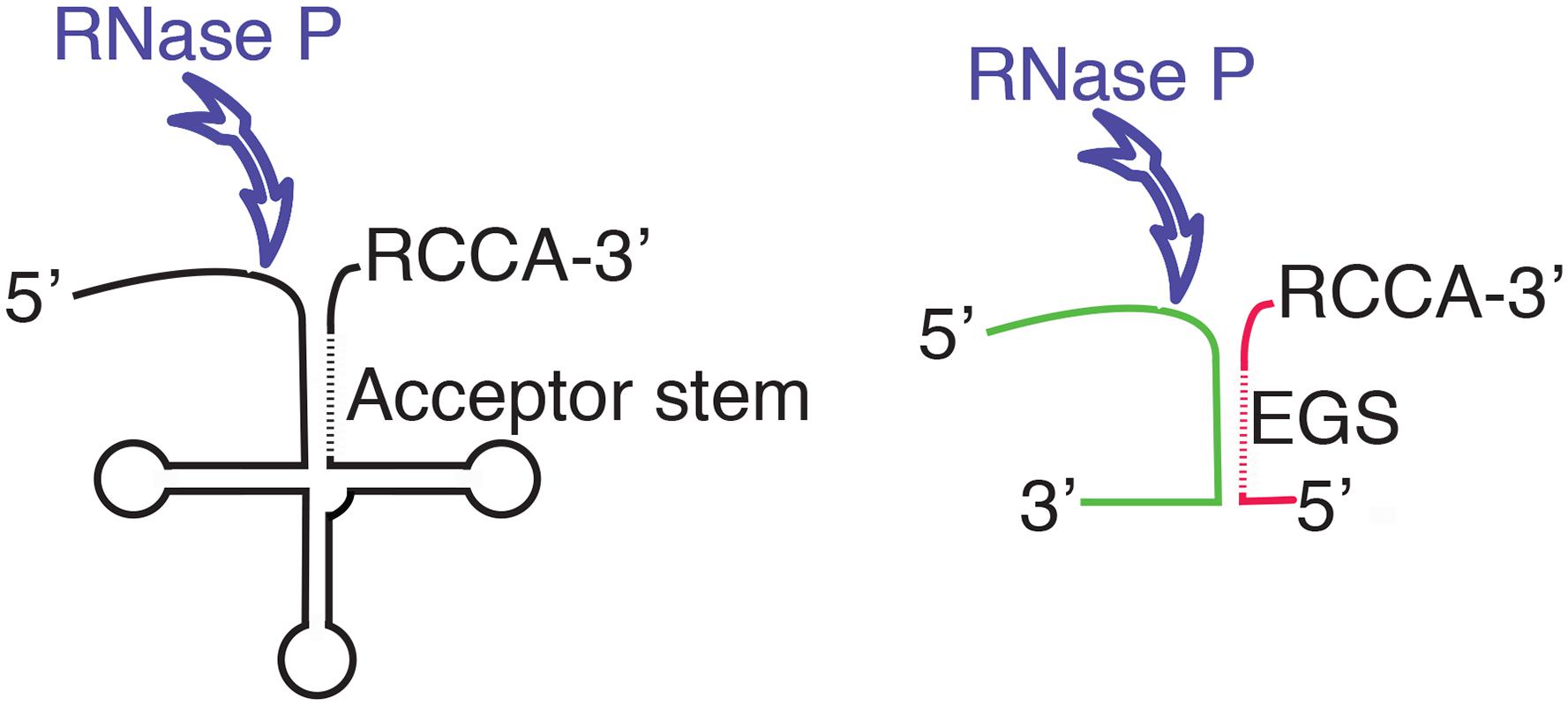
FIGURE 1. Diagram showing the structure of the pre-tRNA and the location of the endonucleolytic cleavage by RNase P (left). The diagram to the right shows a complex between two RNA molecules with a complementary region that form the appropriate structure to be recognized as substrate by RNase P. The different colors illustrate the fact that while there are structural requirements, RNase P does not show specificity of sequence. The antisense regions in the acceptor stem and the EGS are shown as dashed lines. The RCCA sequence facilitates the interaction between the substrate and RNase P.
Acinetobacter baumannii is a nosocomial pathogen that belongs to the ESKAPE group of pathogens and causes a wide range of severe infections, mainly among immunocompromised patients (Wong et al., 2017; Harding et al., 2018). Treatment of these infections is complicated by the multidrug-resistant nature of most strains, a characteristic that led to inclusion of A. baumannii in the priority list of antibiotic-resistant bacteria by the World Health Organization (Doi et al., 2017; Tacconelli et al., 2018; Tacconelli and Magrini, 2018). As a consequence, there is a high need for new treatments of A. baumannii and development of EGSs that inhibit expression of essential functions or antibiotic-resistance genes could be an alternative (Sala et al., 2012). A better understanding of the A. baumannii RNase P will be beneficial for EGS Technology development of therapies to treat multidrug resistant infections caused by this bacterium.
Materials and Methods
Bacterial Strains and Plasmids
Acinetobacter baumannii ATCC 17978 (Smith et al., 2007) was used as source of genomic DNA and the primers were designed using the complete genome sequence (Accession No. CP000521.1). E. coli DH5α (Taylor et al., 1993) and BL21(DE3) (Studier et al., 1990) were used as hosts for cloning experiments. E. coli BL21(DE3) T7A49 is a thermosensitive M1 mutant (Guerrier-Takada et al., 1995). Plasmid pM1Ab was generated by inserting a DNA fragment including the A. baumannii rnpB gene (rnpBAb) under the control of the T7 promoter into the cloning vector pCR2.1 (Life Technologies). This fragment was an amplicon generated using A. baumannii ATCC 17978 chromosomal DNA as template and the primers 5′-GCAAGCTTTAATACGACTCACTATAGGGGAAGTGAGCCGGATGGTC-3′ (the T7 promoter is underlined) and 5′-GCAGGATCCAGGTGAAGTGAGCCTATAAGCC-3′. The sequence of EGSA2 was 5′-CGAUAUGAGAUCGACCA-3′. Plasmid pC5Ab was generated by ligating an amplicon including the A. baumannii rnpA gene (rnpAab) to NdeI and XhoI-digested pET22(+) (Novagen). The amplicon was obtained using the primers 5′-GCCCATATGGTGCATCAACCCCATTTTTT-3′ and 5′-GAACTCGAGATTCTGCGAGGTTGGGACA-3′. The recombinant plasmid pC5Ab codes for the A. baumannii C5 (C5Ab) protein fused to a His-tag at the C-terminus under the control of the T7 promoter.
General Procedures
Plasmid DNA preparations were carried out using the Wizard® Plus SV Minipreps DNA Purification System (Promega). Endonuclease restriction and ligase treatments were performed according to the recommendations of the supplier (New England Biolabs). Polymerase chain reactions were carried out using Taq DNA Polymerase (Invitrogen) and the primers indicated (purchased from IDT Technologies), following the supplier’s recommendations. In vitro synthesis of RNA molecules was done using a MEGAshortscript high-yield transcription T7 kit according to the protocols provided by the supplier (Ambion). Denaturing polyacrylamide gel electrophoresis was performed as described previously (Sarno et al., 2003) on 6% polyacrylamide 19:1 (acrylamide–bis-acrylamide), gels containing 7 M urea using a Tris-Borate-EDTA buffer (TBE) or glycerol-tolerant gel (GTG) buffer. Electrophoresis of an aliquot of the M1Ab purified transcript is shown in Supplementary Figure S1A. DNA and RNA sequence analyses were carried out using Basic local alignment search tool (BLAST) (Altschul et al., 1990), MUSCLE (Edgar, 2004), and Bcheck (Yusuf et al., 2010). E. coli M1 (M1Ec) and C5 (C5Ec) were purified as described before (Guerrier-Takada et al., 1983).
Overexpression of C5Ab was carried out culturing E. coli BL21 (DE3; pC5Ab) in LB broth containing 100 μg of ampicillin/ml at 37°C until the optical density at 600 nm (OD600) was 0.8. At this point isopropyl β-D-galactopyranoside (IPTG) was added to a concentration of 0.5 mM, the culture was incubated for 90 m at 28°C, and the cells were harvested by centrifugation and suspended in phosphate buffer saline (PBS) buffer (20 mM sodium phosphate and 300 mM sodium chloride) pH 7.4. Cells were lysed by French press at 1000 psi (three passages) followed by centrifugation at 4000 g at 4°C for 15 min to remove cell debris. The supernatant was centrifuged at 13500 g at 4°C for 20 min and the supernatant was subjected to one last centrifugation at 100000 g for 1 h at 4°C. The supernatant containing the soluble fraction was used for purification of C5Ab. The protein was purified by immobilized metal affinity chromatography (IMAC) using a nickel-charged nitrilotriacetic acid (NTA) resin column (Thermo Scientific HisPur Ni-NTA Spin Columns) following the recommendations of the supplier. Once eluted using PBS containing 250 mM imidazole and 6 M Guanidine HCl, pH 7.4, the samples were dialyzed against PBS at 4°C. Proteins were analyzed using sodium dodecyl sulfate 15% polyacrylamide gel electrophoresis (SDS-PAGE)(Laemmli, 1970) followed by staining with Coomassie Brilliant Blue. Nucleotide and amino acid sequence analyses and comparisons were performed using the Clustal Omega and the BLAST programs (Altschul et al., 1990; Sievers et al., 2011). The prediction of physical and chemical parameters, search of specific domains, and modeling were carried out using the ProtParam, Pfam, and SWISS-MODEL, respectively (Gasteiger et al., 2005; Finn et al., 2014; Bienert et al., 2017). An SDS-PAGE analysis of the purified C5Ab protein is shown in Supplementary Figure S1B.
In vitro RNase P Assays
Unimolecular substrate: the reaction contained pre-tRNATyr (80 pmol), M1Ec or M1Ab (40 pmol), C5EC or C5Ab (50 pmol; when indicated) in C5 buffer (20 mM HEPES-KOH pH = 8, 400 mM ammonium acetate, 10 mM magnesium acetate, 5% glycerol) in a total volume of 10 μl. Incubation was performed at 37°C for 90 m. Bimolecular substrate: aac(6)′-Ib mRNA (40 pmol) was incubated with EGS (50 pmol) at room temperature for 15 min. Simultaneously, M1Ab (40 pmol) was incubated with C5Ec (50 pmol) in C5 buffer at 37°C for 15 min. Both fractions were combined and incubated for 90 min at 37°C. The reaction was stopped by heating and subjected to phenol/chloroform extraction followed by ethanol precipitation as described before (Jani et al., 2018). The products were resuspended in 1 volume of gel loading buffer and analyzed by 6% denaturing TBE-PAGE or GTG-PAGE (Soler Bistue et al., 2009). RNA bands were visualized by staining with ethidium bromide and UV transillumination.
M1 Heterologous Complementation Assays
Escherichia coli BL21(DE3) T7A49 and E. coli BL21(DE3) T7A49 (pM1Ab) were incubated in LB broth for 24 h at 28 or 42°C, with or without 0.1 mM IPTG. Bacterial growth was determined by measuring OD600. Assays were carried out in duplicate and repeated three times. Statistical significance was analyzed by one-way ANOVA with Dunnett’s multiple comparison test. P < 0.05 was considered statistically significant.
Results and Discussion
A BLAST search using as query the E. coli rnpB nucleotide sequence (Accession No. NCBI Gene ID 947634) and the complete genome of A. baumannii ATCC 17978 as subject (Accession No. GenBank: CP000521.1) identified a region (coordinates 987928–988235) with 80% identity to the E. coli rnpB that was called rnpBAb. A comparative analysis of the nucleotide sequence of this region to the rnpB genes from E. coli and Klebsiella pneumoniae (Accession No. GenBank: M32719.1) (Lawrence et al., 1987) permitted us to determine the rnpBAb promoter region as well as the first and last nucleotide of the RNA molecule encoded, called M1Ab (Figure 2A). Inspection of the M1Ab sequence shows that it belongs to the type A group of RNase P catalytic subunits and includes the conserved regions (Chen and Pace, 1997; Mondragon, 2013). Further analysis using Bcheck (Yusuf et al., 2010) identified the M1Ab sequence as an RNase P RNA. The high level of identity between the sequence of rnpBAb and those from the rnpB genes from E. coli and K. pneumoniae extends from the -10 nucleotide in the promoter region (Lawrence et al., 1987) to the 3′ end of the RNA molecule. Conversely, the -35 and spacer regions show divergence (Figure 2B). This could be a consequence of adaptation to different properties between the RNA polymerases from A. baumannii and the two Enterobacteriaceae. Future comparative studies of expression and activity levels in all three bacteria may lead to a better understanding of the significance of these differences.

FIGURE 2. Alignment of the nucleotide sequences of the coding and promoter regions of RNA subunit (M1) of the ribonuclease P from Escherichia coli, Klebsiella pneumoniae, and Acinetobacter baumannii. (A) Multiple alignment of the coding sequences of M1 performed using MUSCLE. The yellow rectangles show nucleotides belonging to the universally Conserved Regions present in RNA subunits of RNase P (Chen and Pace, 1997), each region is indicated in roman numbers. (B) Alignment of the promoter regions.
The rnpBAb gene was cloned under the control of the T7 promoter and the 355-nt M1Ab RNA was synthesized in vitro as described in section “Materials and Methods” (Supplementary Figure S1A). The synthesized product was tested to determine its RNase P activity using as substrate pre-tRNATyr. Figure 3 shows that both M1Ab and the M1Ec cleaved the substrate with similar efficiency in the presence of the cofactor protein C5Ec and were inactive in the absence of the protein in the conditions used in the assay. Previous work carried out with M1 showed that at certain magnesium concentrations in vitro, cleavage occurs in the absence of C5 (Guerrier-Takada et al., 1983). The results of the experiment shown in Figure 3 not only confirmed that the M1Ab RNA is the A. baumannii RNase P catalytic subunit, but also that it is active in the presence of a heterologous cofactor as it is the C5Ec protein. To confirm the activity of M1Ab in vivo, we carried out an experiment using the M1 thermosensitive mutant E. coli BL21(DE3) T7A49, which does not grow at the non-permissive temperature (42°C). This strain was transformed with the plasmid pM1Ab and the transformant strain was cultured at 28 and 42°C in the presence or absence of IPTG. Figure 4 shows that the E. coli BL21(DE3) T7A49(pM1Ab) acquired the ability to grow at 42°C when expression of M1Ab was induced by addition of IPTG, indicating that RNase P function was restored. This result showed that, as it was the case for the in vitro reaction, M1Ab could interact with C5Ec and produce a functional RNase P in vivo.
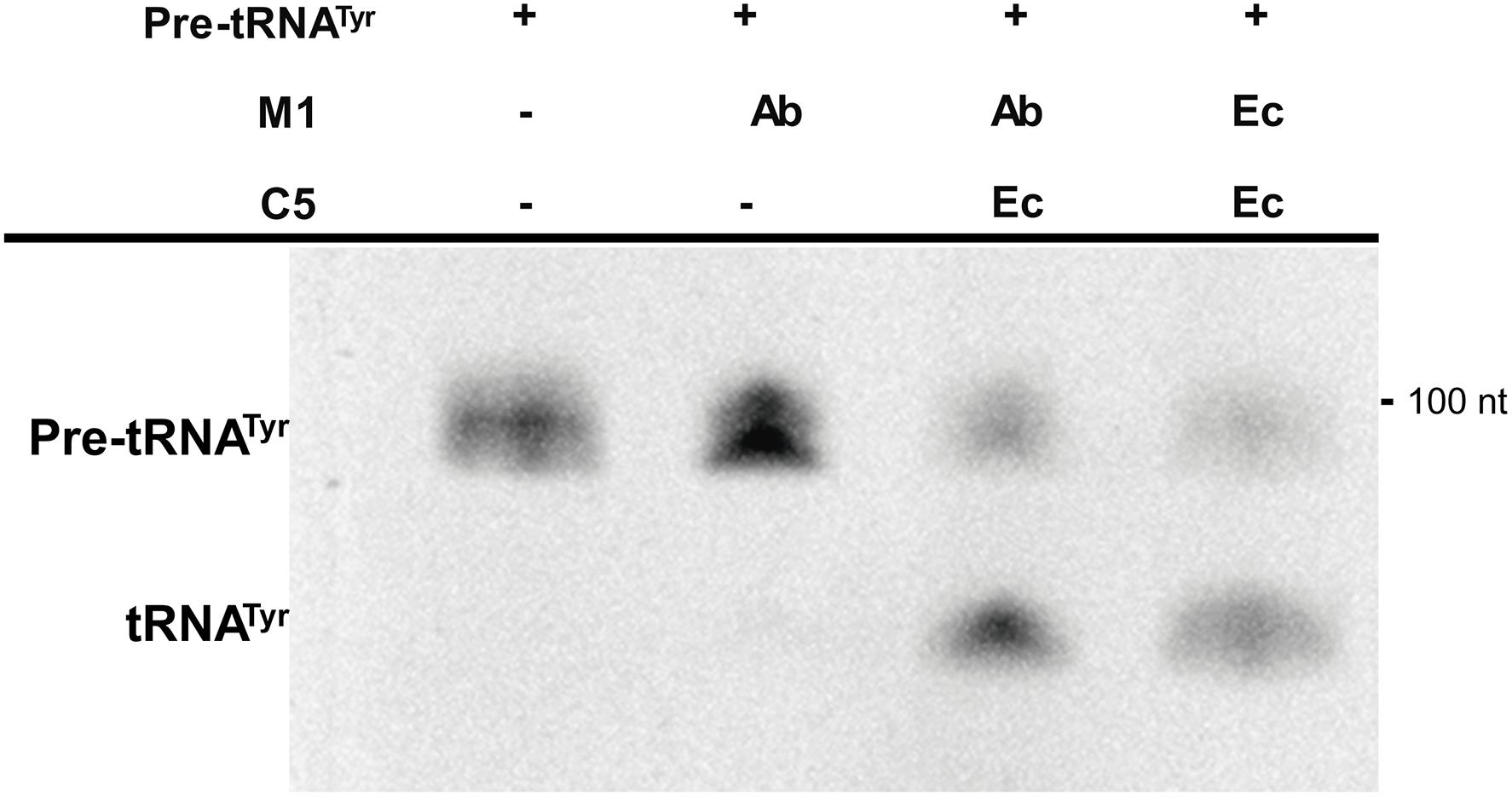
FIGURE 3. In vitro activity of the RNA subunit of the ribonuclease P from Acinetobacter baumannii (M1Ab). The unimolecular substrate pre-RNAtTyr, was used for the reaction performed as described in section “Materials and Methods”. The reaction was analyzed on denaturing PAGE. The location and size of the substrate and the cleavage product are indicated to the left and right, respectively. Ab, A. baumannii and Ec, E. coli. C5
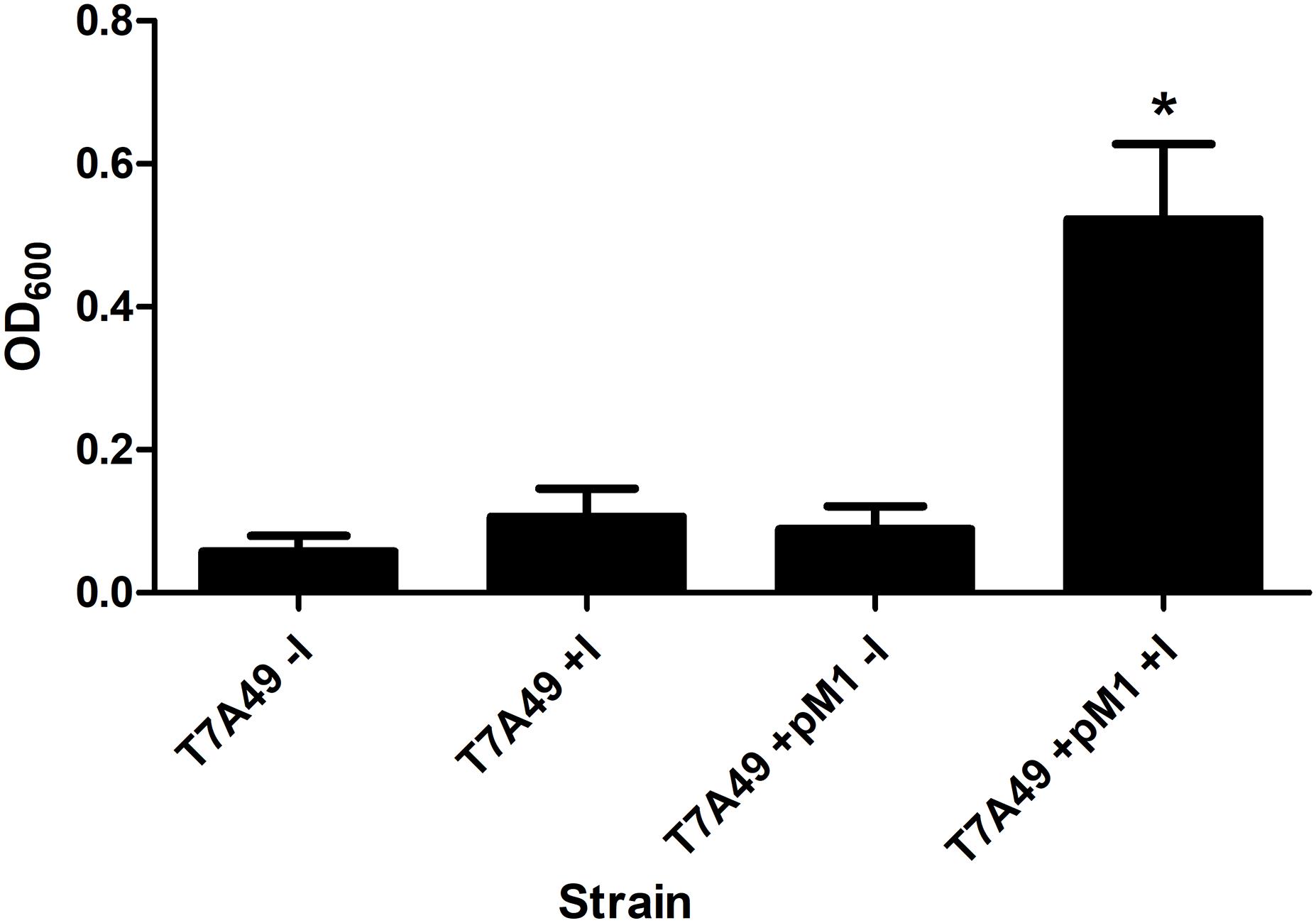
FIGURE 4. Heterologous complementation of strain E. coli BL21(DE3) T7A49. The M1EC thermosensitive mutant was transformed with pM1Ab, a plasmid encoding M1Ab under the control of a T7 promoter. E. coli BL21(DE3) T7A49 (T7A49) and E. coli BL21(DE3) T7A49 (pM1Ab) (T7A49M1) were cultured in the presence (IPTG) or absence of 0.1 mM IPTG Growth was assessed measuring OD600. Statistical analysis was carried out using one-way ANOVA and Dunnett’s multiple comparison test. ∗indicates statistical significance (P < 0.05).
Studies on the RNase P showed that most of the pre-tRNA substrate molecule could be removed without affecting its activity (Gopalan et al., 2002; Lundblad and Altman, 2010). Furthermore, bimolecular complexes were also substrates as long as they form the appropriate structure regardless of the nucleotide sequence (Gopalan et al., 2002; Lundblad and Altman, 2010). These findings originated what is known as EGS technology, which takes advantage of the host RNase P activity to induce degradation of a target mRNA in the presence of an antisense oligonucleotide known as EGS (Gopalan et al., 2002; Lundblad and Altman, 2010; Davies-Sala et al., 2015). This technology could be an option for designing antimicrobials that target essential A. baumannii functions or adjuvants that inhibit expression of resistance genes and would be used in combination with the appropriate antibiotic to restore its therapeutic power. We assessed the ability of M1Ab to elicit cleavage of a target mRNA in the presence of an EGS (bimolecular RNA substrate) in comparison to that of M1EC. For this we used a bimolecular substrate consisting of the aac(6’)-Ib mRNA, which codes for an acetyltransferase that catalyzes inactivation of several aminoglycosides of clinical relevance (Ramirez and Tolmasky, 2010; Ramirez et al., 2013), and an EGS, EGSA2, that elicits cleavage of the mRNA by the E. coli RNase P holoenzyme (Soler Bistue et al., 2009). Figure 5 shows that the reactions carried out with both M1EC and M1Ab produced the same level of degradation of the aac(6’)-Ib mRNA strongly suggesting that EGS technology could be an alternative for novel treatments of A. baumannii infections.
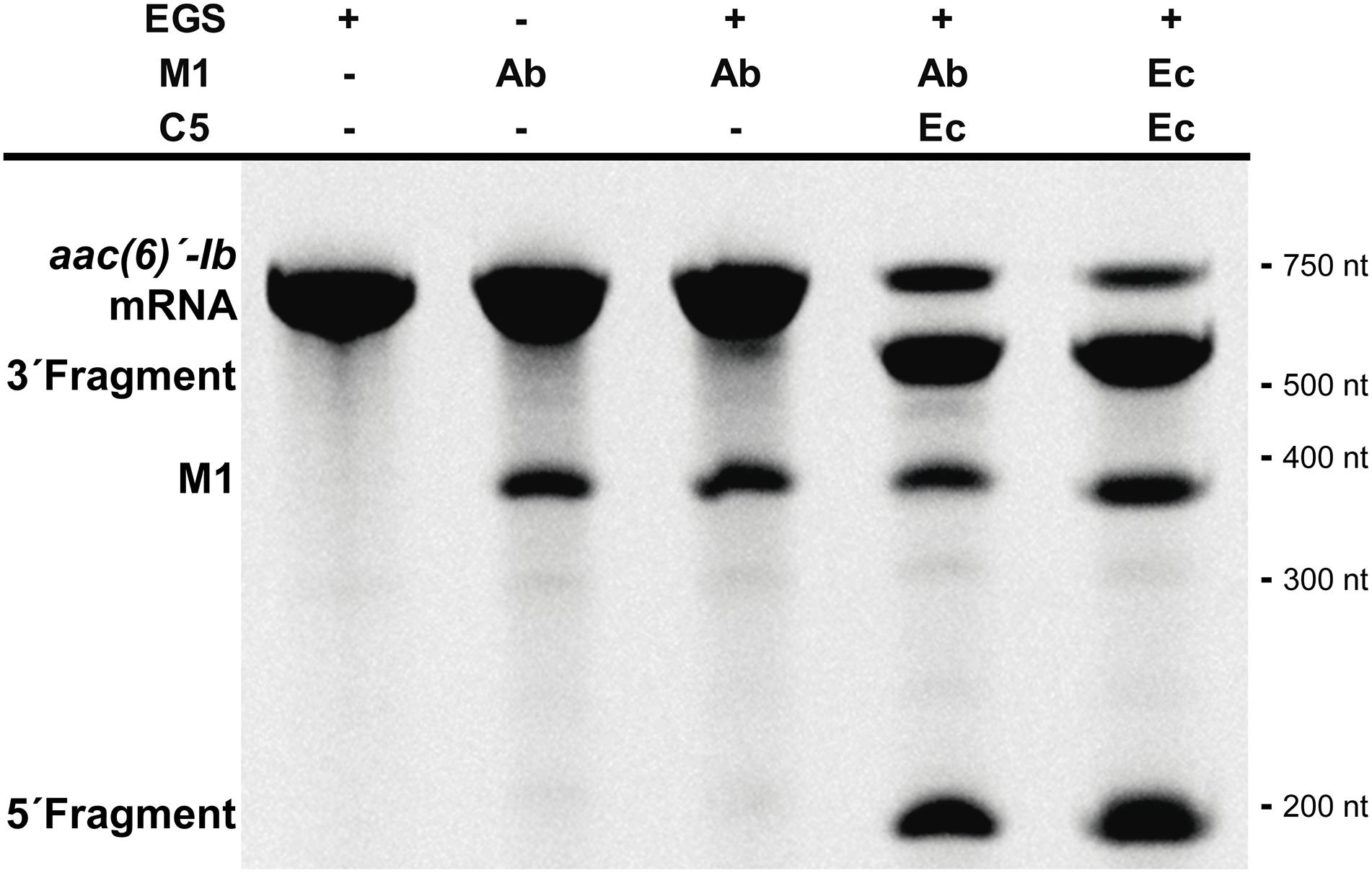
FIGURE 5. Acinetobacter baumannii RNase P RNA subunit and its application for EGS technology. In vitro activity of the M1Ab, RNA subunit of ribonuclease P of A. baumannii in presence of a bimolecular substrate. The substrate is composed of mRNA aac(6)′-Ib and EGSA2. The reaction was analyzed on denaturing PAGE. On the left is shown the location of the substrate, M1Ab and cleavage products. Molecular size standards are shown to the right. Ab, A. baumannii and Ec, E. coli. C5
Further analysis of the A. baumannii ATCC17978 genome sequence permitted us to identify an open reading frame potentially coding for C5Ab, the A. baumannii RNase P cofactor protein. Amino acid sequence comparison between the C5Ec and C5Ab proteins showed low similarity throughout most of the sequence. However, a shared conserved 30-amino acid central core characteristic of C5 proteins was identified (Figure 6A, highlighted in yellow). The C5Ab predicted isoelectric point was 10.8, characteristic of nucleic acids-binding proteins. Pfam analysis predicted this protein to possess a domain (amino acids 2–86) corresponding to cofactors of RNase P family proteins. The C5Ab protein was used to reconstitute the A. baumannii holoenzyme. Figure 6B shows that M1Ab or M1Ec were activated in the presence of C5Ab when tested using pre-tRNAtyr as substrate.
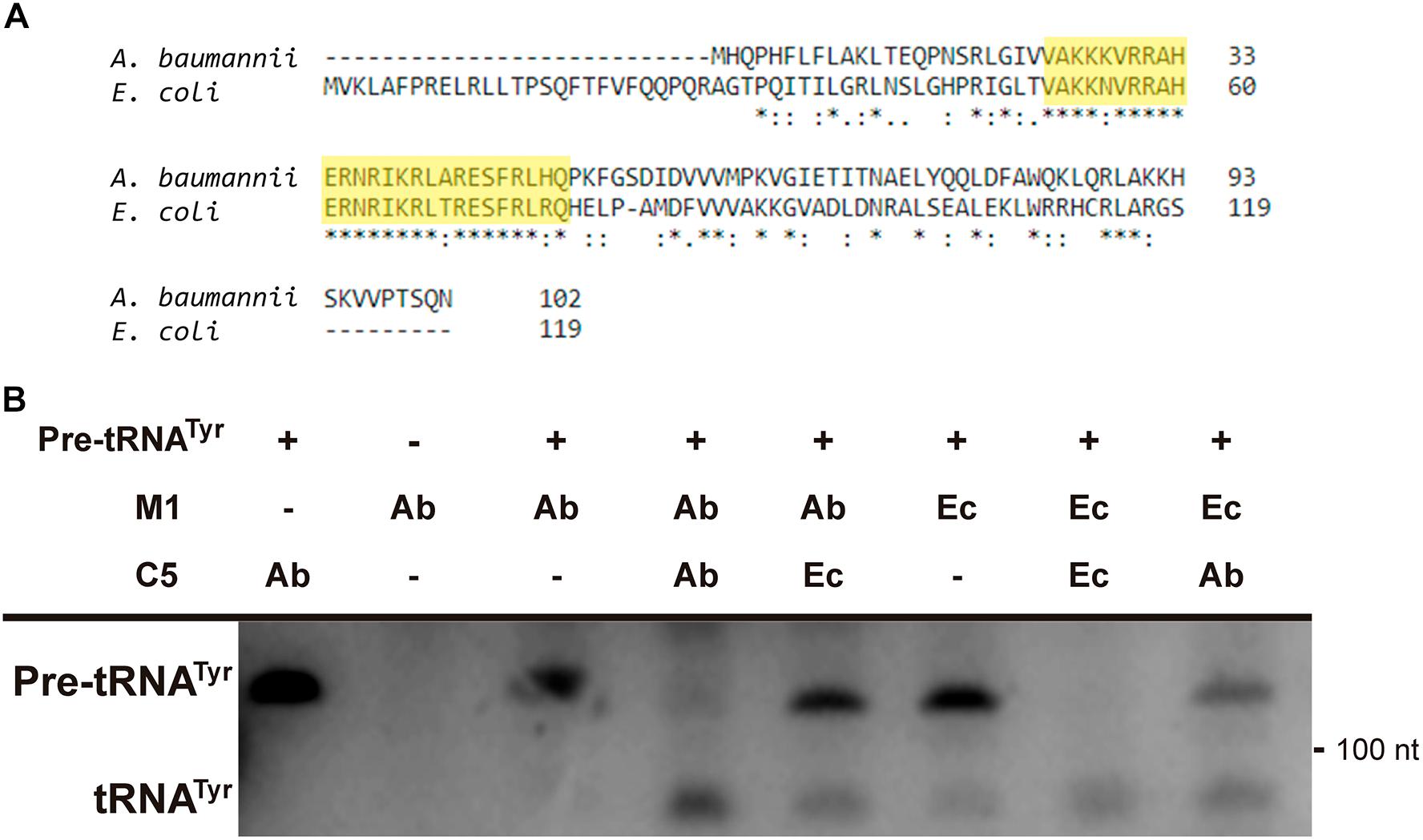
FIGURE 6. C5Ab protein. (A) C5Ec and C5Ab amino acid sequence comparison. (B) In vitro activity of the reconstituted RNase P holoenzymes. The substrate pre-RNAtTyr was incubated with the subunits indicated on top in the conditions described in section “Materials and Methods”. The reaction was analyzed on denaturing PAGE, the fourth and fifth wells were run in the same gel but were not contiguous to the first three. The location and size of the substrate and the cleavage product are indicated to the left and right, respectively. Ab, A. baumannii and Ec, E. coli.
In conclusion, the results described in this study indicate that we identified the A. baumannii ATCC 17978 RNase P gene coding for the catalytic subunit, M1Ab, and showed that its activity is comparable to that of the E. coli M1 subunit. M1Ab was functional in the presence of C5Ec as well as C5Ab, the latter of which was partially purified after the gene was identified and cloned. Furthermore, the M1Ab ability to cleave otherwise non-substrate target mRNAs in the presence of an adequate EGS indicates that EGS technology could be a viable option for designing therapeutic alternatives to treat multiresistant A. baumannii infections. However, numerous challenges remain to be addressed before this technique can be reduced to practice. Non-hydrolyzable, but active analogs must be designed to ensure stability. Also, the compound must efficiently penetrate the cells once it reached the site of infection. Promising but still preliminary results have been obtained testing conjugates between nuclease resistant hybrid locked nucleic acids (LNA)/DNA oligomers and the cell penetrating peptide (RXR)4XB (where R stands for arginine, X for 6-aminohexanoic acid, and B for beta-alanine) (Jackson et al., 2016; Jani et al., 2018). Although previous reports indicate that antisense compounds containing LNA and DNA nucleotides show low toxicity (Wahlestedt et al., 2000), once a specific compound is identified as a candidate for treatment of A. baumannii infections, its cytotoxicity will have to be determined.
Author Contributions
AZ, CD-S, and MT conceived and designed the experiments. CD-S and SJ performed the experiments. AZ, CD-S, MT, and SJ analyzed the data. AZ, CD-S, and MT wrote the paper.
Funding
This work was funded by Public Health Service Grant 2R15AI047115-05A1 (to MT) from the National Institutes of Health and X-240 Universidad de Buenos Aires (to AZ). AZ is a career member of Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET). CD-S was supported by a fellowship from CONICET.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02408/full#supplementary-material
References
Alifano, P., Rivellini, F., Piscitelli, C., Arraiano, C. M., Bruni, C. B., and Carlomagno, M. S. (1994). Ribonuclease E provides substrates for ribonuclease P-dependent processing of a polycistronic mRNA. Genes Dev. 8, 3021–3031. doi: 10.1101/gad.8.24.3021
Altman, S. (2011). Ribonuclease P. Philos. Trans. R. Soc. Lond. B Biol. Sci. 366, 2936–2941. doi: 10.1098/rstb.2011.0142
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bienert, S., Waterhouse, A., de Beer, T. A., Tauriello, G., Studer, G., Bordoli, L., et al. (2017). The SWISS-MODEL repository-new features and functionality. Nucleic Acids Res. 45, D313–D319. doi: 10.1093/nar/gkw1132
Bothwell, A. L., Garber, R. L., and Altman, S. (1976). Nucleotide sequence and in vitro processing of a precursor molecule to Escherichia coli 4.5 S RNA. J. Biol. Chem. 251, 7709–7716.
Chen, J. L., and Pace, N. R. (1997). Identification of the universally conserved core of ribonuclease P RNA. RNA 3, 557–560.
Davies-Sala, C., Soler-Bistue, A., Bonomo, R. A., Zorreguieta, A., and Tolmasky, M. E. (2015). External guide sequence technology: a path to development of novel antimicrobial therapeutics. Ann. N. Y. Acad. Sci. 1354, 98–110. doi: 10.1111/nyas.12755
Doi, Y., Bonomo, R. A., Hooper, D. C., Kaye, K. S., Johnson, J. R., Clancy, C. J., et al. (2017). Gram-negative bacterial infections: research priorities, accomplishments, and future directions of the antibacterial resistance leadership group. Clin. Infect. Dis. 64(Suppl_1), S30–S35. doi: 10.1093/cid/ciw829
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Forster, A. C., and Altman, S. (1990). External guide sequences for an RNA enzyme. Science 249, 783–786. doi: 10.1126/science.1697102
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., et al. (2005). “Protein identification and analysis tools on the ExPASy server,” in The Proteomics Protocols Handbook, ed. J. M. Walker (New York, NY: Humana Press), 571–607. doi: 10.1385/1-59259-890-0:571
Gopalan, V., Vioque, A., and Altman, S. (2002). RNase P: variations and uses. J. Biol. Chem. 277, 6759–6762. doi: 10.1074/jbc.R100067200
Guerrier-Takada, C., Gardiner, K., Marsh, T., Pace, N., and Altman, S. (1983). The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35, 849–857. doi: 10.1016/0092-8674(83)90117-4
Guerrier-Takada, C., Li, Y., and Altman, S. (1995). Artificial regulation of gene expression in Escherichia coli by RNase P. Proc. Natl. Acad. Sci. U.S.A. 92, 11115–11119. doi: 10.1073/pnas.92.24.11115
Guerrier-Takada, C., Salavati, R., and Altman, S. (1997). Phenotypic conversion of drug-resistant bacteria to drug sensitivity. Proc. Natl. Acad. Sci. U.S.A. 94, 8468–8472. doi: 10.1073/pnas.94.16.8468
Haas, E. S., and Brown, J. W. (1998). Evolutionary variation in bacterial RNase P RNAs. Nucleic Acids Res. 26, 4093–4099. doi: 10.1093/nar/26.18.4093
Harding, C. M., Hennon, S. W., and Feldman, M. F. (2018). Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 16, 91–102. doi: 10.1038/nrmicro.2017.148
Hartmann, R. K., Heinrich, J., Schlegl, J., and Schuster, H. (1995). Precursor of C4 antisense RNA of bacteriophages P1 and P7 is a substrate for RNase P of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 5822–5826. doi: 10.1073/pnas.92.13.5822
Jackson, A., Jani, S., Sala, C. D., Soler-Bistue, A. J., Zorreguieta, A., and Tolmasky, M. E. (2016). Assessment of configurations and chemistries of bridged nucleic acids-containing oligomers as external guide sequences: a methodology for inhibition of expression of antibiotic resistance genes. Biol. Methods Protoc. 1:bw001. doi: 10.1093/biomethods/bpw001
Jani, S., Jackson, A., Davies-Sala, C., Chiem, K., Soler-Bistue, A., Zorreguieta, A., et al. (2018). Assessment of external guide sequences’ (EGS) efficiency as inducers of RNase P-mediated cleavage of mRNA target molecules. Methods Mol. Biol. 1737, 89–98. doi: 10.1007/978-1-4939-7634-8_6
Jarrous, N. (2017). Roles of RNase P and its subunits. Trends Genet. 33, 594–603. doi: 10.1016/j.tig.2017.06.006
Jarrous, N., and Gopalan, V. (2010). Archaeal/eukaryal RNase P: subunits, functions and RNA diversification. Nucleic Acids Res. 38, 7885–7894. doi: 10.1093/nar/gkq701
Kazantsev, A. V., Krivenko, A. A., Harrington, D. J., Holbrook, S. R., Adams, P. D., and Pace, N. R. (2005). Crystal structure of a bacterial ribonuclease P RNA. Proc. Natl. Acad. Sci. U.S.A. 102, 13392–13397. doi: 10.1073/pnas.0506662102
Klemm, B. P., Wu, N., Chen, Y., Liu, X., Kaitany, K., Howard, M. J., et al. (2016). The diversity of Ribonuclease P: protein and RNA catalysts with analogous biological functions. Biomolecules 6:27. doi: 10.3390/biom6020027
Ko, J. H., Izadjoo, M., and Altman, S. (2008). Inhibition of expression of virulence genes of Yersinia pestis in Escherichia coli by external guide sequences and RNase P. RNA 14, 1656–1662. doi: 10.1261/rna.1120508
Komine, Y., Kitabatake, M., Yokogawa, T., Nishikawa, K., and Inokuchi, H. (1994). A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 91, 9223–9227. doi: 10.1073/pnas.91.20.9223
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Lawrence, N. P., Richman, A., Amini, R., and Altman, S. (1987). Heterologous enzyme function in Escherichia coli and the selection of genes encoding the catalytic RNA subunit of RNase P. Proc. Natl. Acad. Sci. U.S.A. 84, 6825–6829. doi: 10.1073/pnas.84.19.6825
Lundblad, E. W., and Altman, S. (2010). Inhibition of gene expression by RNase P. N. Biotechnol. 27, 212–221. doi: 10.1016/j.nbt.2010.03.003
Marvin, M. C., and Engelke, D. R. (2009). RNase P: increased versatility through protein complexity? RNA Biol. 6, 40–42.
Mondragon, A. (2013). Structural studies of RNase P. Annu. Rev. Biophys. 42, 537–557. doi: 10.1146/annurev-biophys-083012-130406
Ramirez, M. S., Nikolaidis, N., and Tolmasky, M. E. (2013). Rise and dissemination of aminoglycoside resistance: the aac(6’)-Ib paradigm. Front. Microbiol. 4:121. doi: 10.3389/fmicb.2013.00121
Ramirez, M. S., and Tolmasky, M. E. (2010). Aminoglycoside modifying enzymes. Drug Resist. Updat. 13, 151–171. doi: 10.1016/j.drup.2010.08.003
Reiner, R., Ben-Asouli, Y., Krilovetzky, I., and Jarrous, N. (2006). A role for the catalytic ribonucleoprotein RNase P in RNA polymerase III transcription. Genes Dev. 20, 1621–1635. doi: 10.1101/gad.386706
Reiter, N. J., Osterman, A., Torres-Larios, A., Swinger, K. K., Pan, T., and Mondragon, A. (2010). Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature 468, 784–789. doi: 10.1038/nature09516
Robertson, H. D., Altman, S., and Smith, J. D. (1972). Purification and properties of a specific Escherichia coli ribonuclease which cleaves a tyrosine transfer ribonucleic acid presursor. J. Biol. Chem. 247, 5243–5251.
Sala, C. D., Soler-Bistue, A. J., Korprapun, L., Zorreguieta, A., and Tolmasky, M. E. (2012). Inhibition of cell division induced by external guide sequences (EGS Technology) targeting ftsZ. PLoS One 7:e47690. doi: 10.1371/journal.pone.0047690
Sarno, R., Ha, H., Weinsetel, N., and Tolmasky, M. E. (2003). Inhibition of aminoglycoside 6’-N-acetyltransferase type Ib-mediated amikacin resistance by antisense oligodeoxynucleotides. Antimicrob. Agents Chemother. 47,3296–3304. doi: 10.1128/AAC.47.10.3296-3304.2003
Sawyer, A. J., Wesolowski, D., Gandotra, N., Stojadinovic, A., Izadjoo, M., Altman, S., et al. (2013). A peptide-morpholino oligomer conjugate targeting Staphylococcus aureus gyrA mRNA improves healing in an infected mouse cutaneous wound model. Int. J. Pharm. 453, 651–655. doi: 10.1016/j.ijpharm.2013.05.041
Shen, N., Ko, J. H., Xiao, G., Wesolowski, D., Shan, G., Geller, B., et al. (2009). Inactivation of expression of several genes in a variety of bacterial species by EGS technology. Proc. Natl. Acad. Sci. U.S.A. 106, 8163–8168. doi: 10.1073/pnas.0903491106
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Smith, M. G., Gianoulis, T. A., Pukatzki, S., Mekalanos, J. J., Ornston, L. N., Gerstein, M., et al. (2007). New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21, 601–614. doi: 10.1101/gad.1510307
Soler Bistue, A. J., Ha, H., Sarno, R., Don, M., Zorreguieta, A., and Tolmasky, M. E. (2007). External guide sequences targeting the aac(6’)-Ib mRNA induce inhibition of amikacin resistance. Antimicrob. Agents Chemother. 51,1918–1925. doi: 10.1128/AAC.01500-06
Soler Bistue, A. J., Martin, F. A., Vozza, N., Ha, H., Joaquin, J. C., Zorreguieta, A., et al. (2009). Inhibition of aac(6’)-Ib-mediated amikacin resistance by nuclease-resistant external guide sequences in bacteria. Proc. Natl. Acad. Sci. U.S.A. 106, 13230–13235. doi: 10.1073/pnas.0906529106
Studier, F. W., Rosenberg, A. H., Dunn, J. J., and Dubendorff, J. W. (1990). Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60–89. doi: 10.1016/0076-6879(90)85008-C
Tacconelli, E., Carrara, E., Savoldi, A., Harbarth, S., Mendelson, M., Monnet, D. L., et al. (2018). Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. doi: 10.1016/S1473-3099(17)30753-3
Tacconelli, E., and Magrini, N. (2018). Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics. Geneva: World Health Organization.
Taylor, R. G., Walker, D. C., and McInnes, R. R. (1993). E. coli host strains significantly affect the quality of small scale plasmid DNA preparations used for sequencing. Nucleic Acids Res. 21, 1677–1678. doi: 10.1093/nar/21.7.1677
Torres-Larios, A., Swinger, K. K., Krasilnikov, A. S., Pan, T., and Mondragon, A. (2005). Crystal structure of the RNA component of bacterial ribonuclease P. Nature 437, 584–587. doi: 10.1038/nature04074
Wahlestedt, C., Salmi, P., Good, L., Kela, J., Johnsson, T., Hokfelt, T., et al. (2000). Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 97, 5633–5638. doi: 10.1073/pnas.97.10.5633
Wong, D., Nielsen, T. B., Bonomo, R. A., Pantapalangkoor, P., Luna, B., and Spellberg, B. (2017). Clinical and pathophysiological overview of Acinetobacter infections: a century of challenges. Clin. Microbiol. Rev. 30, 409–447. doi: 10.1128/CMR.00058-16
Yang, L., and Altman, S. (2007). A noncoding RNA in Saccharomyces cerevisiae is an RNase P substrate. RNA 13, 682–690. doi: 10.1261/rna.460607
Keywords: RNase P, Acinetobacter, ESKAPE, ribozyme, EGS technology, antisense
Citation: Davies-Sala C, Jani S, Zorreguieta A and Tolmasky ME (2018) Identification of the Acinetobacter baumannii Ribonuclease P Catalytic Subunit: Cleavage of a Target mRNA in the Presence of an External Guide Sequence. Front. Microbiol. 9:2408. doi: 10.3389/fmicb.2018.02408
Received: 17 April 2018; Accepted: 20 September 2018;
Published: 08 October 2018.
Edited by:
Rustam Aminov, University of Aberdeen, United KingdomReviewed by:
Rosalia Cavaliere, University of Technology Sydney, AustraliaLisa Sedger, University of Technology Sydney, Australia
Copyright © 2018 Davies-Sala, Jani, Zorreguieta and Tolmasky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo E. Tolmasky, bXRvbG1hc2t5QGZ1bGxlcnRvbi5lZHU=
†Present address: Carol Davies-Sala, Instituto de Investigaciones en Ingeniería Genética y Biología; Molecular “Dr. Héctor N. Torres”, CONICET, Buenos Aires, Argentina