 Wei Chen
Wei Chen Bo Wang
Bo Wang Jordon D. Gruber
Jordon D. Gruber Yong-Mei Zhang
Yong-Mei Zhang Christopher Davies
Christopher Davies
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 20 September 2018
Sec. Microbial Physiology and Metabolism
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.02244
The human opportunistic pathogen Pseudomonas aeruginosa expresses three acyl carrier proteins (ACPs): AcpP, Acp1, and Acp3. The function of AcpP in membrane fatty acid synthesis (FAS) was confirmed recently, but the physiological roles of Acp1 and Acp3 remain unclear. To address this, we investigated the physiological role of Acp3 in P. aeruginosa. We found that expression of Acp3 dramatically increases in the log phase of cell growth and that its transcription is under the control of the QS regulators LasR and RhlR. Deletion of acp3 from P. aeruginosa strain PAO1 results in thicker biofilm formation, increased resistance of the strain to hydrogen peroxide, and higher persistence in a mouse infection model. Tandem affinity purification (TAP) experiments revealed several novel protein-binding partners of Acp3, including KatA, the major catalase in P. aeruginosa. Acp3 was found to repress the catalase activity of KatA and, consistent with inhibition by Acp3, less reactive oxygen species are present in the acp3 deletion strain. Overall, our study reveals that Acp3 has a distinct function from that of the canonical AcpP and may be involved in the oxidative stress response.
Acyl carrier proteins (ACPs) function in the synthesis of fatty acids, polyketides, and non-ribosomal peptides. In addition to the housekeeping ACP used for Type II fatty acid synthesis (FAS), some bacteria contain specialized variants of ACPs (Geiger et al., 1991; Brozek et al., 1996; Epple et al., 1998; Geiger and Lopez-Lara, 2002; Haag et al., 2009; Ramos-Vega et al., 2009; Chan and Vogel, 2010). Some of these divert intermediates of Type II FAS (FAS) to synthesize molecules like lipid A or hemolysin, whereas others are involved in distinct processes such as polyketide synthesis (Byers and Gong, 2007; Chan and Vogel, 2010). Rhizobia, for example, expresses four ACPs (AcpP, NodF, AcpXL, and RkpF), where AcpP is the canonical ACP for FAS and the other three are used for synthesis of cell-surface molecules that facilitate the establishment of symbiosis with their legume hosts (Geiger and Lopez-Lara, 2002). In addition, it was reported recently that in Salmonella enterica, an ACP homologue called IacP acylates a type 3 secretion system (T3SS) translocon protein, SipB, leading to insertion of SipB into host-cell membranes (Viala et al., 2017).
By contrast, Escherichia coli encodes only a single and essential ACP (De Lay and Cronan, 2007). In addition to functioning in Type II FAS, however, it also acts in several other systems. For instance, the protein interacts with SpoT to switch the activity of SpoT from (p)ppGpp degradation to (p)ppGpp synthesis during conditions of fatty acid starvation (Battesti and Bouveret, 2006; Angelini et al., 2012) and also forms a complex with the STAS domain of a SLC26 family anion transporter YchM, linking bicarbonate transport with fatty acid metabolism (Babu et al., 2010). In addition, E. coli ACP plays an essential role in the biosynthesis of membrane-derived oligosaccharides (MDOs), which are important for osmotic regulation (Therisod et al., 1986). Interestingly, the 4-phosphopantetheine moiety of ACP is not required in the MDO transglucosylation function, a clear difference from ACP’s role in fatty acid biosynthesis (Therisod and Kennedy, 1987).
Pseudomonas aeruginosa is an opportunistic pathogen that causes life-threatening nosocomial infections and is of particular concern for immunocompromised and cystic fibrosis (CF) patients (Govan and Deretic, 1996). The P. aeruginosa genome contains three putative acp genes, annotated as PA2966 (acpP), PA1869 (acp1), and PA3334 (acp3). Each contains a conserved serine that is presumed to be the site of 4-phosphopantetheinylation (Raychaudhuri et al., 2005). Recently, AcpP was confirmed as the essential ACP used for membrane FAS, and deletion of acp1 and acp3 had minimal effect on cell growth and in vitro virulence production (Ma et al., 2017). Previous studies have showed that acp1 and acp3 expression is regulated by quorum sensing (QS) (Hentzer et al., 2003; Schuster et al., 2003; Wagner et al., 2003; Chugani et al., 2012) and acp3 is highly induced during infection (Turner et al., 2014). In particular, the 18-fold increase in expression of acp3 in an acute infection model and 30-fold increase in a chronic infection model strongly suggests a key role for Acp3 in pathogenicity (Turner et al., 2014). Beyond these indicators, however, the physiological functions of Acp1 and Acp3 in P. aeruginosa remain unclear.
In this work, we examined the specific physiological role of Acp3 in P. aeruginosa via a combination of transcriptional profiling, western blotting, measuring in vitro and in vivo virulence, and identification of protein-binding partners. We found that acp3 is regulated by the QS system and that expression of Acp3 peaks during the early stationary phase of cell growth. Deletion of acp3 increases biofilm formation, increased resistance of the strain to H2O2 and higher persistence in the mouse lung. We also found that Acp3 binds and represses the activity of the catalase, KatA. Overall, the data suggest a specialized function for Acp3 involving oxidative response mediated by KatA.
Bacterial strains and plasmids used in this study are listed in Table 1. Strain stocks were maintained at -80°C in 10% glycerol. Bacteria were streaked on fresh plates before each experiment. P. aeruginosa and E. coli strains were cultured at 37°C in Miller’s Luria broth (LB) medium (Sigma-Aldrich) unless specified otherwise. Pseudomonas Isolation Agar (PIA) was used for selection of P. aeruginosa. Plasmids used for gene knockout were constructed using the described method (Choi and Schweizer, 2005). Primers used in this study are listed in Supplementary Table S1.
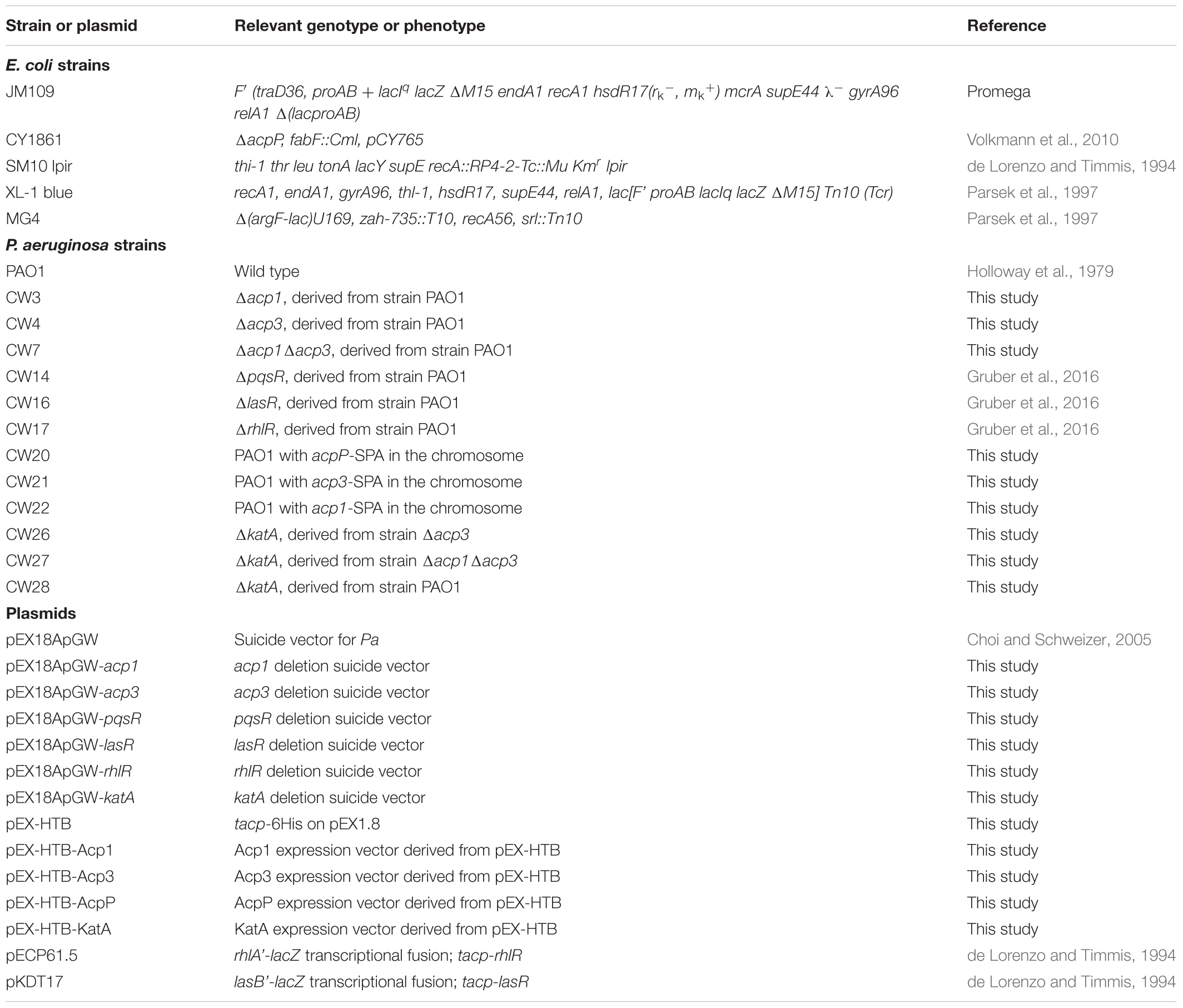
TABLE 1. Strains and plasmids used in this study.
To generate expression constructs of acp1, acp3, acpP, E. coli acp, and katA, the respective genes were amplified and cloned into pEX-HTB vector with restriction and ligation. pEX-HTB is derived from pEX1.8, and encodes a 6x-His tag within its multiple cloning site. All plasmids were confirmed by sequencing. For plasmid maintenance in E. coli cultures, the growth medium was supplemented with 50 μg/ml carbenicillin, 30 μg/ml kanamycin, or 30 μg/ml gentamycin. For P. aeruginosa cultures, 200 μg/ml carbenicillin or 30 μg/ml gentamycin were used, unless otherwise stated.
Unmarked deletion mutants of P. aeruginosa were generated using a published method (Choi and Schweizer, 2005). Briefly, the target gene was replaced by a gentamycin (Gmr) resistance cassette through homologous recombination, followed by removal of the cassette by expression of Flp recombinase from pFLP2 and counter selection with 10% sucrose. All mutants were confirmed by PCR using appropriate flanking primers (Supplementary Table S1) and DNA sequencing. The deleted DNA fragments corresponded to amino acids 5–79 for Acp1 (94% of the protein sequence) and 7–45 for Acp3 (48% of the protein sequence). For katA, the deleted DNA corresponded to amino acids 21–448 (88% of the protein sequence).
Transcript levels of acp genes were measured by real-time PCR using oligonucleotide primers designed with DNAMAN 7.0 software (Lynnon Cooperation). All primer pairs generated one specific product. The expression of gene rpoD was used as the housekeeping control. Cells were harvested at OD600 0.5, 2.0, and 2.6, corresponding to the log, early stationary, and stationary phases, respectively. Total RNA was isolated using RNAqueous kit (Ambion), and genomic DNA was removed by precipitation with LiCl, followed by DNase treatment. cDNA was generated using the High Capability RNA-to-cDNA kit (Ambion), with 1 μg total RNA per reaction. Real-time quantitative PCR was performed in a 96-well plate, using the SensiFAST SYBR® & Fluorescein kit (Bioline). The reaction mixture contained 500 nM gene-specific primers. Amplification and detection were performed on a Bio-Rad MyiQ single-color real time detection system. Experiments were performed in triplicate for each cDNA preparation. No-reverse transcriptase and no-template controls were included for each assay. Data were analyzed by MyiQ software, and the critical threshold cycle (CT) was set automatically. The amounts of acp gene mRNA were normalized against rpoD gene using the ΔΔCT method (Rao et al., 2013).
The expression patterns of ACP proteins in P. aeruginosa were measured by western blotting. Overnight cultures were used to inoculate 100 ml fresh LB broth with an initial OD600 of 0.05, and incubated at 37°C with shaking. Samples were taken every 2 h, and cells lysed in B-PER reagent (Fisher). The total protein concentration was determined by Bradford assay. Fifty micrograms of total protein of the whole cell lysates was then separated by SDS-PAGE on 4–20% precast polyacrylamide gels and transferred to a PVDF membrane. SPA-tagged ACP proteins were detected by monoclonal anti-flag M2 (1:5000) (Sigma) and goat anti-mouse IgG-HRP (1:10,000) (Santa Cruz). Images were quantified by the software ImageJ (Schneider et al., 2012).
For static biofilm formation, cultures of early log phase were diluted into fresh LB to OD600 0.05 and dispensed to a 96-well plate with 100 μl per well. The plate was covered with a transferable solid-phase (TSP) pegged-lid (Nunc) and incubated at 37°C for 18 h without shaking. Biofilms that formed on the pegged lid were washed twice by submerging the lid into a basin of sterilized water and then stained with 0.1% crystal violet (CV) in a new 96-well plate for 15 min with gentle shaking. Excess CV was washed off with water and then bound CV was eluted from the biofilms into 150 μl of 95% ethanol and quantitated spectrophotometrically at 600 nm using a Biotek Synergy HT plate reader.
To construct strains expressing sequential peptide affinity (SPA)-tagged ACPs, a 250-bp DNA fragment encoding a calmodulin-binding peptide and 3× FLAG tag separated by a TEV cleavage site (Babu et al., 2009) was synthesized (Genewiz Inc.) and fused with a Gmr cassette by overlapping PCR, generating a SPA-Gmr fragment. Gene-specific upstream and downstream fragments were fused with this SPA-Gmr fragment by overlapping PCR and cloned into pEX18ApGw to generate pEX18ApGw-acp1/3/P-SPA. Strains with C-terminal SPA-tagged acp were screened and confirmed by PCR and sequencing (Choi and Schweizer, 2005). SPA-tagged ACP proteins were detected by monoclonal anti-Flag M2 (1:5000) (Sigma) and goat anti-mouse IgG-HRP (1:10,000) (Santa Cruz).
Sequential peptide affinity purification was performed as described previously (Babu et al., 2009). Briefly, SPA-tagged strains and wild-type strain PAO1 were grown in 1 l of Terrific Broth in a 2-l flask at 37°C for 12 h with vigorous shaking. Cell pellets from stationary-phase cultures were harvested and lysed by sonication on ice. The cell lysates were treated with universal nuclease (Pierce) to remove nucleic acids and supplemented with protease inhibitor cocktail (Roche) to protect proteins from hydrolysis. The supernatant was mixed with magnetic beads conjugated with anti-Flag M2 antibody (Sigma) and incubated overnight on a rotary mixer at 4°C. Triton X-100 (0.1%) was added to prevent non-specific binding. The magnetic beads were collected by placing the tube in a magnetic rack, washed until the OD280 of flow-through was lower than 0.05, and re-suspended in 200 μl TEV cleavage buffer. TEV protease (40 μl of 1.8 mg/ml) was added and incubated overnight at 4°C with mixing. After the TEV cleavage step was complete, the magnetic beads were collected and the supernatant was transferred to a new tube, to which 400 μl of calmodulin-binding buffer and 1.6 μl of 1 M CaCl2 were added. The solution was then added to calmodulin Sepharose beads (GE Healthcare) and incubated for 3 h at 4°C with gentle mixing. The beads were washed four times with 1 ml of calmodulin-wash buffer (with EGTA). The bound proteins were eluted in 300-μl elution buffer. The target proteins were analyzed by MALDI-TOF mass spectrometry. The entire experiment was repeated once and only proteins identified in both experiments are reported.
A His-KatA expression plasmid, pEXHTB-katA, and pEXHTB empty vector control were each transformed into strain CW21 (PAO1 with Acp3-FLAG). Cell pellets were harvested from 1 l of Terrific Broth after 12-h growth and cell lysates were prepared as described above. His-tagged proteins were purified by affinity chromatography using Ni2+-NTA agarose (Fisher). Flag-tagged-Acp3 in the elution was detected by western blotting with anti-Flag M2 antibody. Anti-ScKatA antibody was used to detect P. aeruginosa KatA (Shin et al., 2008).
The sensitivity of different strains against hydrogen peroxide was determined by the microbroth dilution method in LB. One hundred microliters of a twofold serially diluted H2O2 solution was aliquoted to a 96-well plate. Cultures of early log phase were diluted in fresh LB to OD600 of 0.01 and mixed with H2O2 solution in equal volume. After incubation for 10 h at 37°C, cell density was measured at OD600. To further address the relationship between ACP and H2O2 resistance, pEX-HTB-derived ACP-expressing plasmids were introduced back into the Δacp1/Δacp3 double mutant. Cultures were incubated in LB supplemented with 2 mM H2O2 at 37°C with vigorous shaking, and OD600 was measured after 5 h.
Catalase activity was measured as described previously (Ou and Wolff, 1996). A standard curve was established by mixing serially diluted H2O2 with FOX 1 reagent (250 μM ammonium ferrous sulfate, 100 μM xylenol orange, 0.1 M sorbitol, and 25 mM H2SO4). Cell pellets from 24-h cultures were washed once by PBS and lysed in B-PER reagent (Fisher). After centrifugation, the total protein in the supernatant was determined by Bradford assay. Ten microliters of supernatant was mixed with 990 μl of 200 μM H2O2 in 50 mM potassium phosphate (pH 7.0) and incubated for 1–30 min at room temperature. Fifty microliters of the mixture was then reacted with 950 μl of FOX 1 reagent at room temperature for 30 min. The reaction mixture was centrifuged and the absorbance of the resultant supernatant was measured at 560 nm. The concentration of remaining H2O2 was calculated based on the standard curve. The catalase unit activity was defined as the decomposition of 1 μmol of H2O2 in 1 min by 1 mg of total protein. Three independent experiments were performed. For comparison between replicates, relative catalase activity was calculated.
Cell pellets from 10 ml of overnight cultures of wild-type strain and acp mutants were harvested, washed, and suspended in 1 ml of PBS. Cell suspensions were treated with 20 μM of 2′,7′-dichlorofluorescein diacetate (DCFH2, dissolved in DMSO) at 37°C for 20 min in the dark. Cells treated with an equal volume of DMSO were used as a negative control. The treated cells were washed once and suspended in 1 ml PBS. Two hundred microliters of suspension was transferred to a black opaque 96-well plate and fluorescence was measured using a BioTek microplate reader at 535 nm emission with excitation at 485 nm. The fluorescence data were normalized to the cell density of each sample. Three independent experiments were performed, and relative fluorescence was calculated by normalization to the wild-type strain for each experiment.
Cells from overnight cultures of the P. aeruginosa wild-type strain PAO1 or isogenic acp mutant stains were harvested by centrifugation, washed twice and suspended in sterile PBS to 1 × 108 CFU per 40 μl. Female C57Bl/6J mice (6–8 weeks old, Jackson Laboratories) were inoculated with 40-μl culture via oropharyngeal aspiration under anesthesia with isofluorane. Each treatment group contained 10 mice. At 48 h post infection, lungs were harvested, weighed, and homogenized in 1 ml of sterile PBS for bacterial enumeration. P. aeruginosa were grown and counted on PIA. The data were analyzed using a Kruskal–Wallis test with post hoc Dunn’s multiple-comparison tests to compare the bacterial loads among different groups. All procedures were in accordance with a protocol approved by the MUSC Institutional Animal Care and Use Committee.
As a preliminary step to investigate the function of Acp3, we used qRT-PCR to quantify the levels of all three acp transcripts in log phase (OD600 0.5), early stationary phase (OD600 2.0), and stationary phase (OD600 2.6). The basal levels of the three transcripts were very different in log phase, with acp3 being the least abundant and acpP the most abundant (Figure 1A). Transcript levels of acp1 and acp3 increased 9.6-fold and 106-fold, respectively, from log phase to early stationary phase. The level of acp3 transcript then decreased in stationary phase, whereas that of acp1 transcript level remained high. By contrast, the level of acpP expression remained high throughout, consistent with the expression profile of other known housekeeping acpP genes (Zhang et al., 2002).
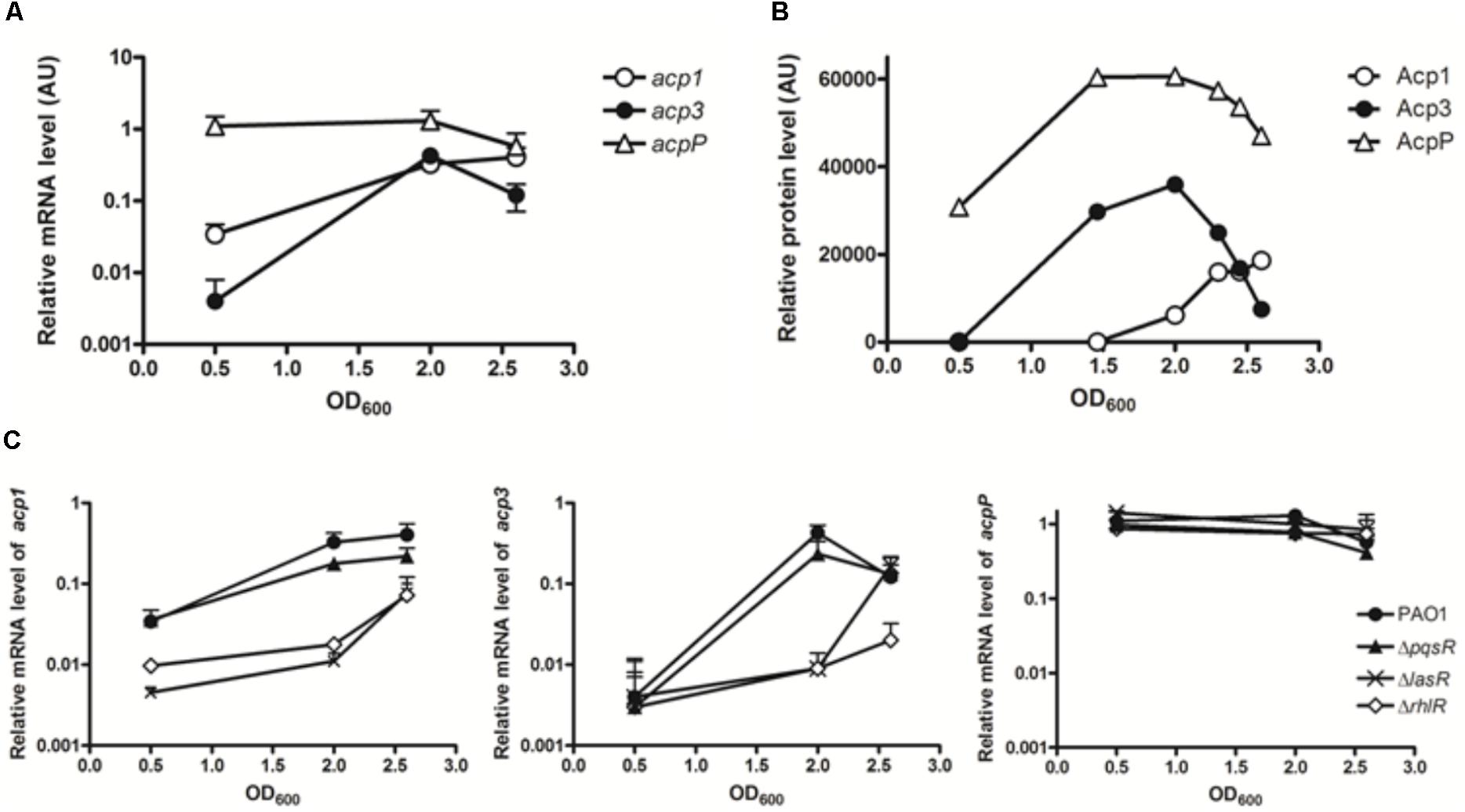
FIGURE 1. Expression profiles of the three acp genes in PAO1. (A) Transcripts levels were measured by qRT-PCR in log phase (OD600 0.5), early stationary phase (OD600 2.0), and stationary phase (OD600 2.6). (B) Expression patterns of three ACP proteins in PAO1 were determined by western blotting with anti-Flag antibody. Samples were taken every 2 h post of inoculation. The immunoblot image was quantified with software ImageJ. (C) Regulatory connection between Acp1 and Acp3 and quorum sensing. Levels of acp1 and acp3 transcripts are lowered in the QS-deficient mutants, ΔlasR and ΔrhlR, but not ΔpqsR. acp1, left panel; acp3, middle panel; acpP, right panel.
To determine the endogenous protein levels of the three ACPs, we constructed strains of PAO1 in which the proteins were expressed as fusions with a SPA tag at the C terminus. The SPA tag contains a calmodulin-binding peptide and three FLAG domains. Semi-quantitative western blotting with an anti-Flag M2 antibody was used to measure protein levels at different growth stages. Commensurate with its function in membrane FAS, AcpP was constitutively expressed and also exhibited the highest expression level of the three ACPs (Figure 1B). The protein level of Acp3 mirrored its transcriptional pattern, indicating that acp3 expression is mainly regulated transcriptionally. The protein level of Acp1 was the lowest of the three ACPs in log phase, but increased as growth progressed to the stationary phase, which suggests it plays a role during late-stage growth.
Previous microarray data have indicated that expression of acp1 and acp3, but not acpP, are under the control of QS signals in P. aeruginosa (Hentzer et al., 2003; Schuster et al., 2003; Wagner et al., 2003). By contrast, a separate study using RNA-seq did not show activation of acp3 expression by QS (Chugani et al., 2012). To determine whether the transcription of acp1 and acp3 is regulated by QS, we measured their respective transcript levels in the QS deletion mutants ΔlasR, ΔrhlR, and ΔpqsR (Gruber et al., 2016). Significantly reduced levels were observed for both acp1 and acp3 in the ΔlasR and ΔrhlR mutants, with at least a 10-fold decrease at an OD of 2.0 for both, but were unchanged in the ΔpqsR mutant (Figure 1C). Thus, it appears that expression of acp1 and acp3 are regulated by both LasR and RhlR systems, but not PqsR. By contrast, the level of the acpP transcript was unaffected in all QS mutants.
Next, we investigated whether deletion of acp3 had any effect on biofilm formation or in vivo pathogenicity. Compared to wild type, strains with single deletions of acp1 or acp3 enhanced static biofilm formation, but the effect was more pronounced in the double mutant (Figure 2A). This suggests that Acp1 and Acp3 each contribute to the virulence of P. aeruginosa. Additionally, all acp deletion strains exhibited higher bacterial loads in the lungs of C57Bl/6J mice, suggesting higher persistence in vivo, although the difference between the acp1 mutant and PAO1 was not significant (Figure 2B). The strongest effect was observed with the acp1/acp3 double mutant, for which the average colony-forming units (CFU) increased approximately three log units. Overall, these data suggest that deletion of acp3 increases the pathogenicity of P. aeruginosa.
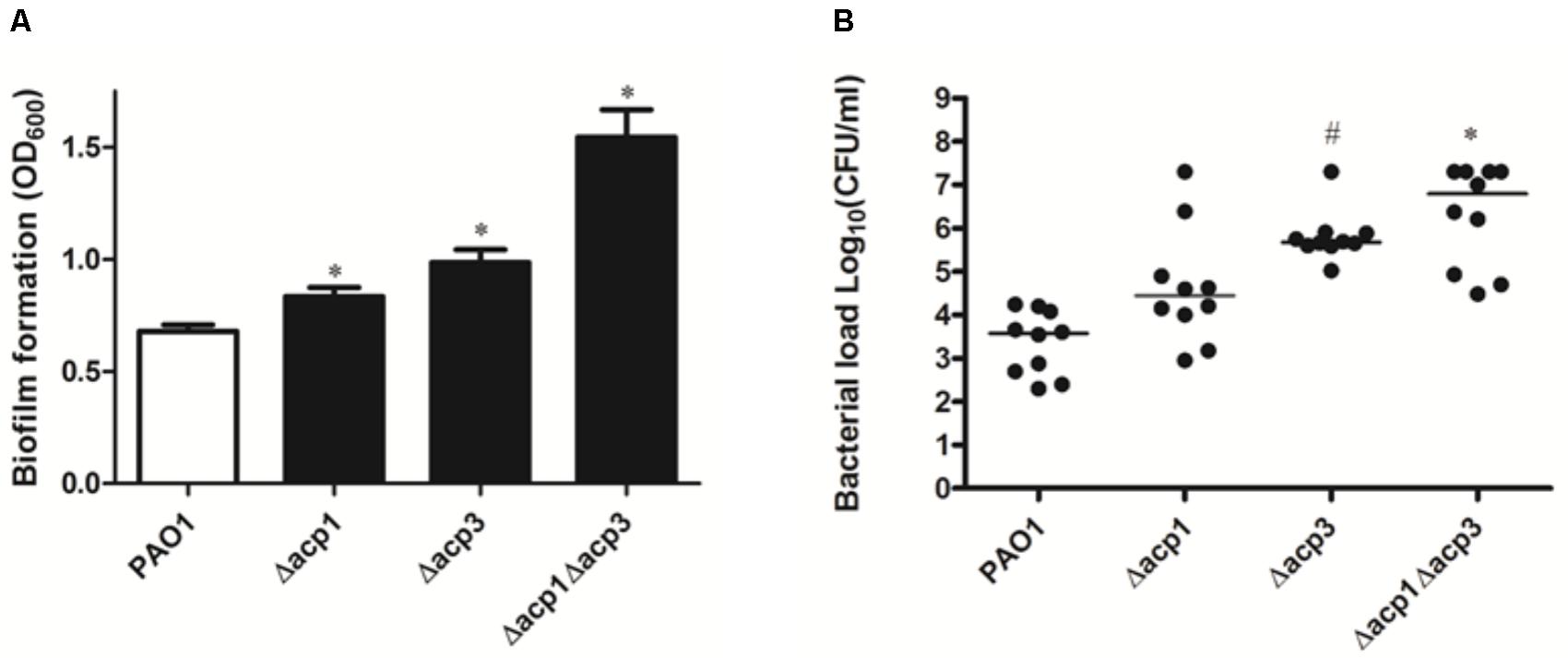
FIGURE 2. Deletion of acp3 increases biofilm formation and persistence of P. aeruginosa PAO1 in a mouse lung infection model. (A) Biofilm formation increased in acp mutants. Biofilm formation by PAO1 and acp mutants was determined using a pegged lid 96-well plate. Data are presented as means ± SEs from three independent experiments. Student’s t-test was performed between PAO1 and acp mutant. ∗ indicates p < 0.01. (B) Determination of pathogenicity of acp mutants in vivo. Overnight cultures of wild-type and acp mutants were used to infect mice lungs. Forty-eight hours post inoculation, mice were sacrificed, lungs were removed and homogenized, and bacteria were counted on a PIA plate. Two independent experiments were performed. Data are presented as log (CFU/ml) analyzed using a Kruskal–Wallis test with post hoc Dunn’s multiple-comparison tests among different strains. Short bars within the data indicate the median CFU/ml. Significant p values are indicated by #p < 0.05 and ∗p < 0.01.
To probe the physiological role of Acp3 further, we used tandem affinity purification (TAP) to identify its protein-binding partners, and alongside, the same for Acp1 and AcpP. Proteins identified only in strains expressing SPA-tagged ACPs but not PAO1 were considered potential ACP-binding partners. For the most part, the three ACPs have different sets of binding partners, commensurate with distinct physiological roles for these proteins. For AcpP, 10 protein candidates were identified (Table 2), including FabG, FabZ, FabA, GlmU, IscS, and LpxD, which are known to bind E. coli ACP (Butland et al., 2005). Their identification in P. aeruginosa provides further evidence that AcpP is functionally equivalent to E. coli ACP (Ma et al., 2017). Interactions with the Type II FAS enzymes FabG, FabZ, and FabA are consistent with a role for AcpP in fatty acid biosynthesis. New binding partners of AcpP were also found compared to those known for E. coli ACP, including CobL, LipB, SpeE2, and AlgB. One interaction that was expected but not observed was that between AcpP and SpoT, because these are known to form a complex in P. aeruginosa (Battesti and Bouveret, 2009).
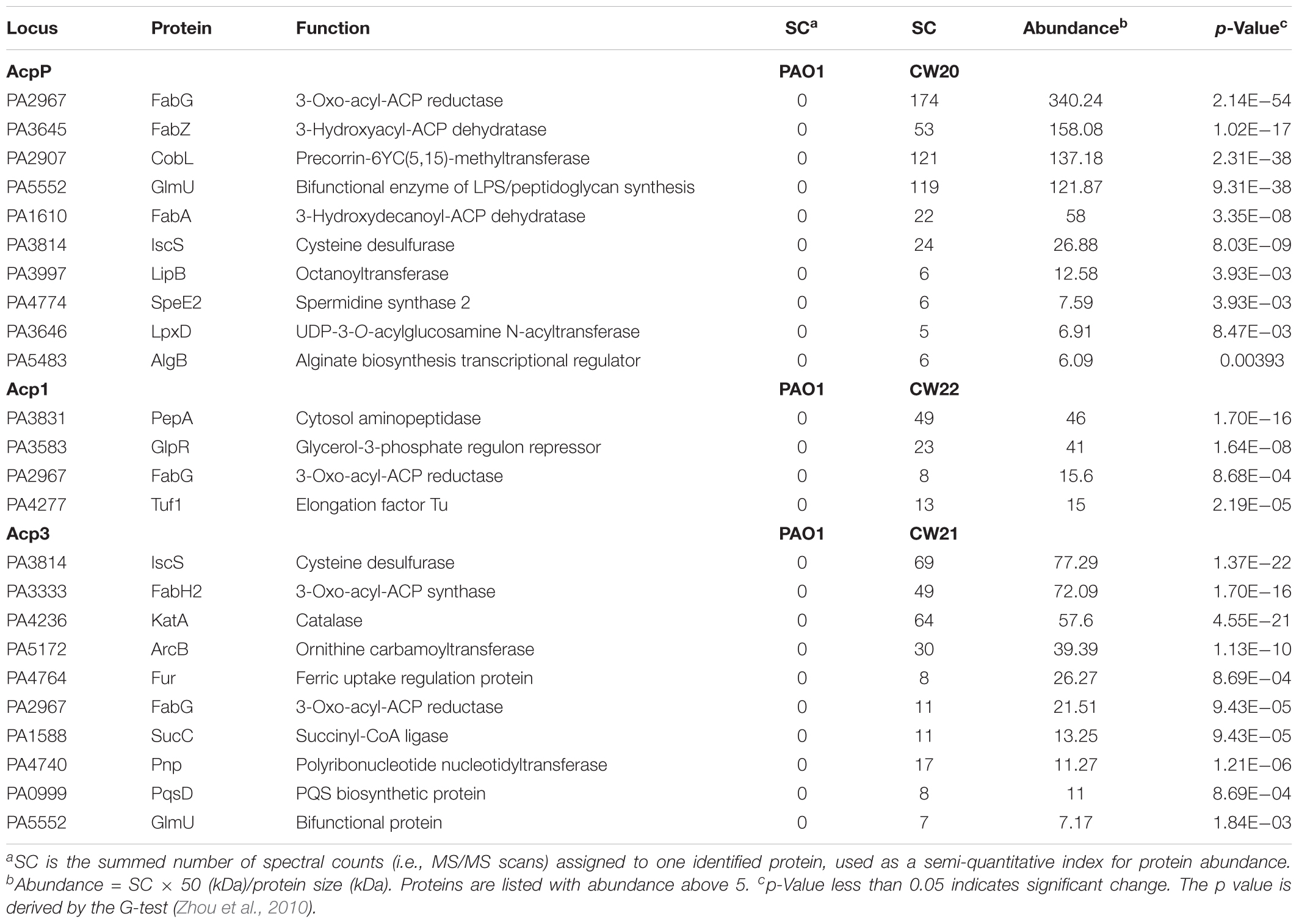
TABLE 2. Binding partners of ACPs in P. aeruginosa identified by tandem affinity purification.
Only four binding partners of Acp1 were identified: PepA, GlpR, Tuf1, and FabG (Table 2). PepA is a leucine aminopeptidase that is homologous to E. coli PepA (Woolwine and Wozniak, 1999), GlpR is annotated as a repressor of the glycerol-3-phosphate regulon and Tuf1 as the elongation factor Tu (Winsor et al., 2016). One reason for there being fewer binding partners for Acp1 compared to AcpP or Acp3 might be the relatively lower expression of the protein at selected sampling time of 12 h (see Figure 1B).
Ten protein-binding partners of Acp3 were identified and most are enzymes of various functions (Table 2). The observed binding with IscS, GlmU, and FabG is in common with AcpP. Of the other proteins identified, novel binding partners include the 3-oxo-acyl-ACP synthase, FabH2 (PA3333), and also PqsD. fabH2 is located within one putative operon with acp3 (PA3334) and is of unknown function, in contrast to FabH (PA5814), which initiates fatty acid biosynthesis in P. aeruginosa (Zhang and Rock, 2012). PqsD is essential for the production of the quinolones DHQ and alkyl quinolones (Zhang et al., 2008). Additional partners are ArcB, an ornithine carbamoyltransferase, SucC, which is involved in the biosynthesis of succinate (Kapatral et al., 2000), and Fur, which is a ferric uptake regulator (Ochsner et al., 1995). Finally, KatA, which is the major catalase in P. aeruginosa, was identified as a binding partner of Acp3 and this was investigated further (see below).
One of the most intriguing binding hits for Acp3 was KatA, which is the major catalase in P. aeruginosa and is required for resistance to peroxide and osmotic stress (Lee et al., 2005). To confirm that Acp3 and KatA interact with each other, we performed a reverse pull-down assay in which the pEXHTB plasmid expressing His-tagged KatA was transformed into PAO1 expressing FLAG-tagged Acp3 (CW21/pEXHTB-katA). The immunoblot shows that the FLAG-tagged Acp3 co-eluted with His-tagged KatA from the Ni2+-affinity column (Figure 3, right panel). The same strain transformed with vector expressing just the His tag was used as a comparison control (CW21/pEXHTB), and no Acp3 was detected in the eluate (Figure 3, left panel). This experiment confirms that Acp3 binds KatA specifically.
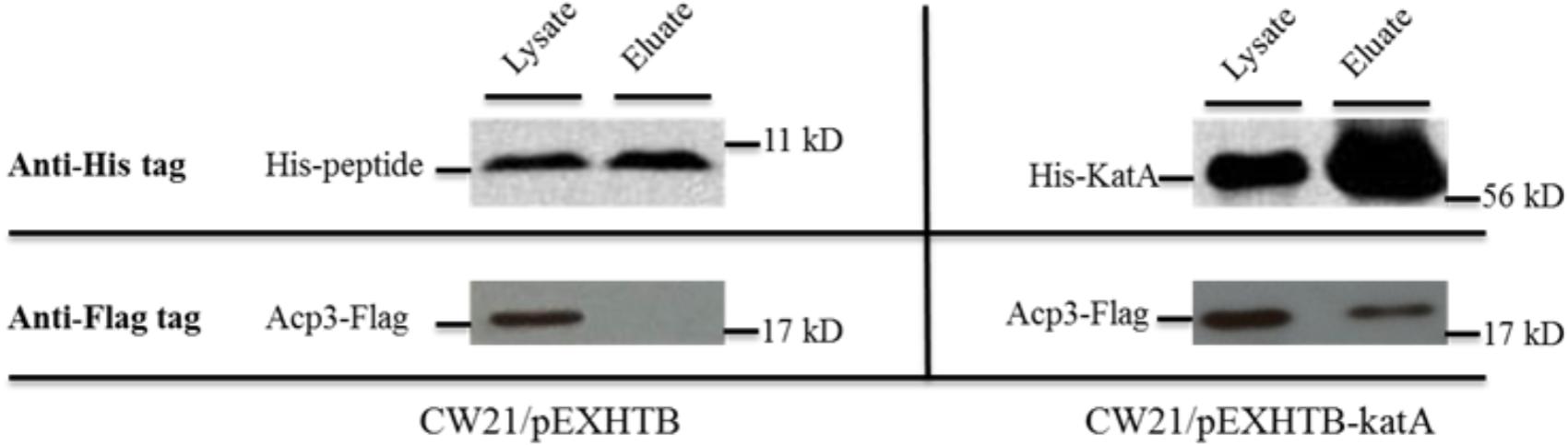
FIGURE 3. Binding of Acp3 and KatA by reverse pull-down assay. His-KatA and Acp3-FLAG were expressed in PAO1. Acp3 co-purified with His-tagged KatA after nickel-affinity chromatography, as detected by immunoblotting with anti-FLAG M2 antibody. Empty vector (pEXHTB) is a negative control. The full image of the immunoblots are shown in Supplementary Figure S1.
One reason for the higher persistence of the acp3 mutant in the mouse infection model could be increased tolerance to reaction oxygen species (ROS) related to the binding of Acp3 with KatA and potential influence on its activity. To examine this, we determined the H2O2 resistance of the acp mutants. Both the Δacp3 and Δacp1/Δacp3 mutants exhibited a twofold higher resistance to killing by H2O2, compared with PAO1 (Figure 4A). The Δacp1 mutant exhibited similar susceptibility as wild type. As a positive control, overexpression of KatA increased resistance in PAO1 by fourfold. To confirm the effect of acp deletion on H2O2 resistance, we complemented the Δacp1/Δacp3 mutant strain with plasmids expressing Acp1 or Acp3. When Acp3 was expressed, susceptibility of the strain to H2O2 was increased, whereas expression of Acp1 had little effect compared to the vector control (Figure 4B). These data show that the presence of Acp3 increases susceptibility of P. aeruginosa to H2O2.
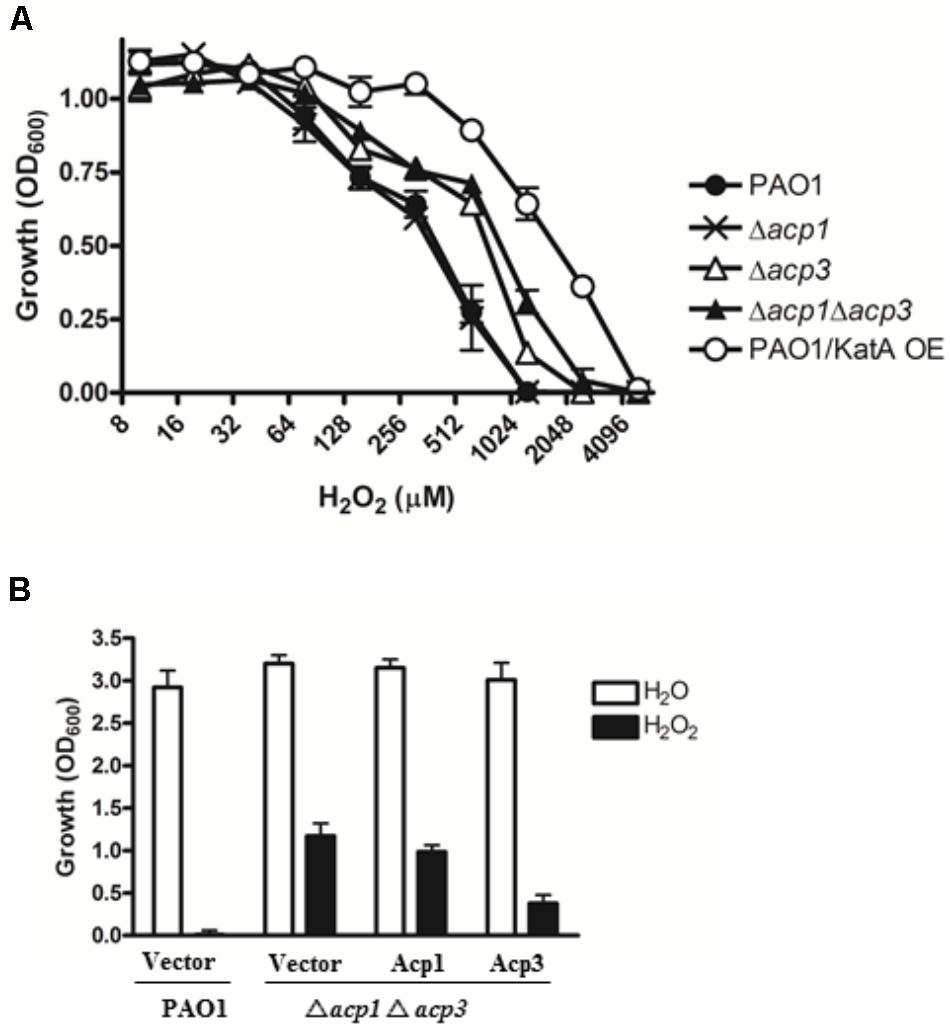
FIGURE 4. The effect of acp gene deletion or overexpression on P. aeruginosa resistance to H2O2. (A) Δacp3 mutant and Δacp1Δacp3 double mutant were more resistant to H2O2 killing than PAO1 and the Δacp1 mutant. PAO1 overexpressing KatA (KatA OE) was used as a positive control. (B) Effect of ACP overexpression on resistance of P. aeruginosa to H2O2 (2 mM). Complementation of the Δacp1Δacp3 double deletion mutant with a plasmid expressing Acp3 increased susceptibility to H2O2. Data are presented as mean ± SEs from three independent determinations.
Since catalases play a major role in protecting bacteria against ROS via decomposition of H2O2 (Mishra and Imlay, 2012), we measured the catalase activity of the acp1 and acp3 deletion strains by the ferrous oxidation xylenol orange (FOX) assay (Ou and Wolff, 1996). Compared with the wild-type PAO1 strain, deletion of acp3 enhanced the catalase activity of P. aeruginosa by 50%, with a lesser increase observed in the acp1 deletion strain (Figure 5). Complementation of Acp3 in the Δacp1/Δacp3 double mutants reduced catalase activity back to wild-type levels. These data are consistent with Acp3 acting as a repressor of KatA activity.
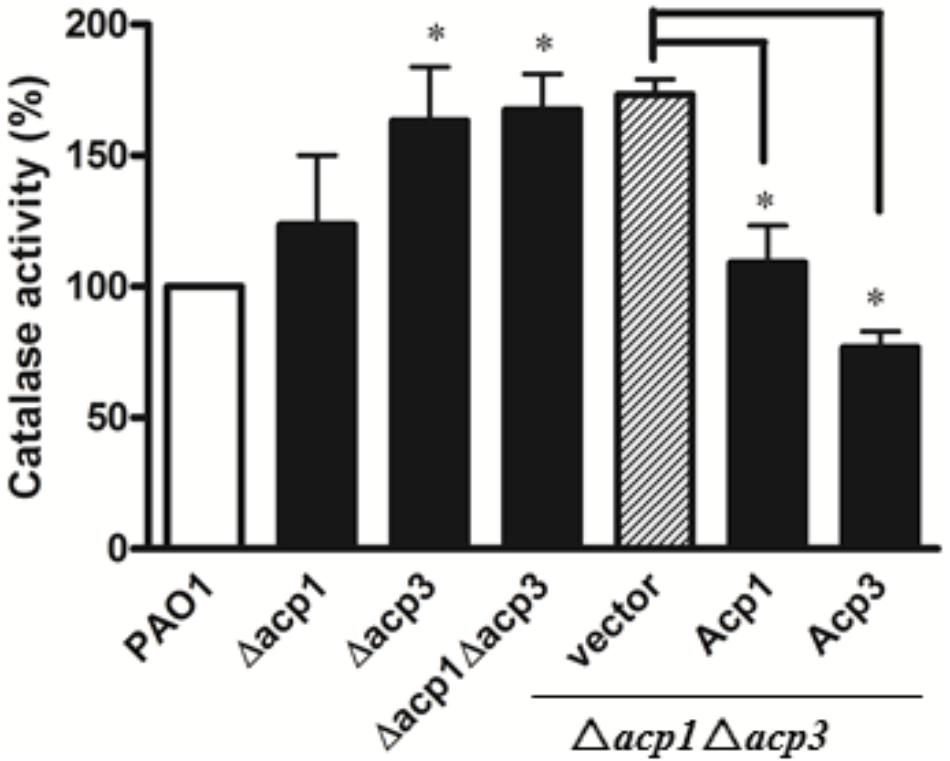
FIGURE 5. The absence of Acp3 increases the catalase activity of P. aeruginosa. Δacp3 and Δacp1Δacp3 mutants exhibited higher catalase activity than wild-type PAO1. Catalase activity reduced after plasmids expressing Acp1 and Acp3 were introduced back into the Δacp1Δacp3 mutant. Data are presented as mean ± SEs from three independent experiments. Student’s t-test was performed between PAO1 and acp mutants. ∗ indicates p < 0.01.
To explore the functional relationship between Acp3 and KatA further, we deleted the katA gene in wild-type and acp mutants. Deletion of katA in PAO1 resulted in a nearly 300-fold reduction in catalase activity (data not shown). Importantly, there was no difference in catalase activity or resistance to H2O2 between the ΔkatA and Δacp3/ΔkatA strains (Figure 6). This shows that the H2O2 resistance of the acp3 mutant is mediated through KatA.
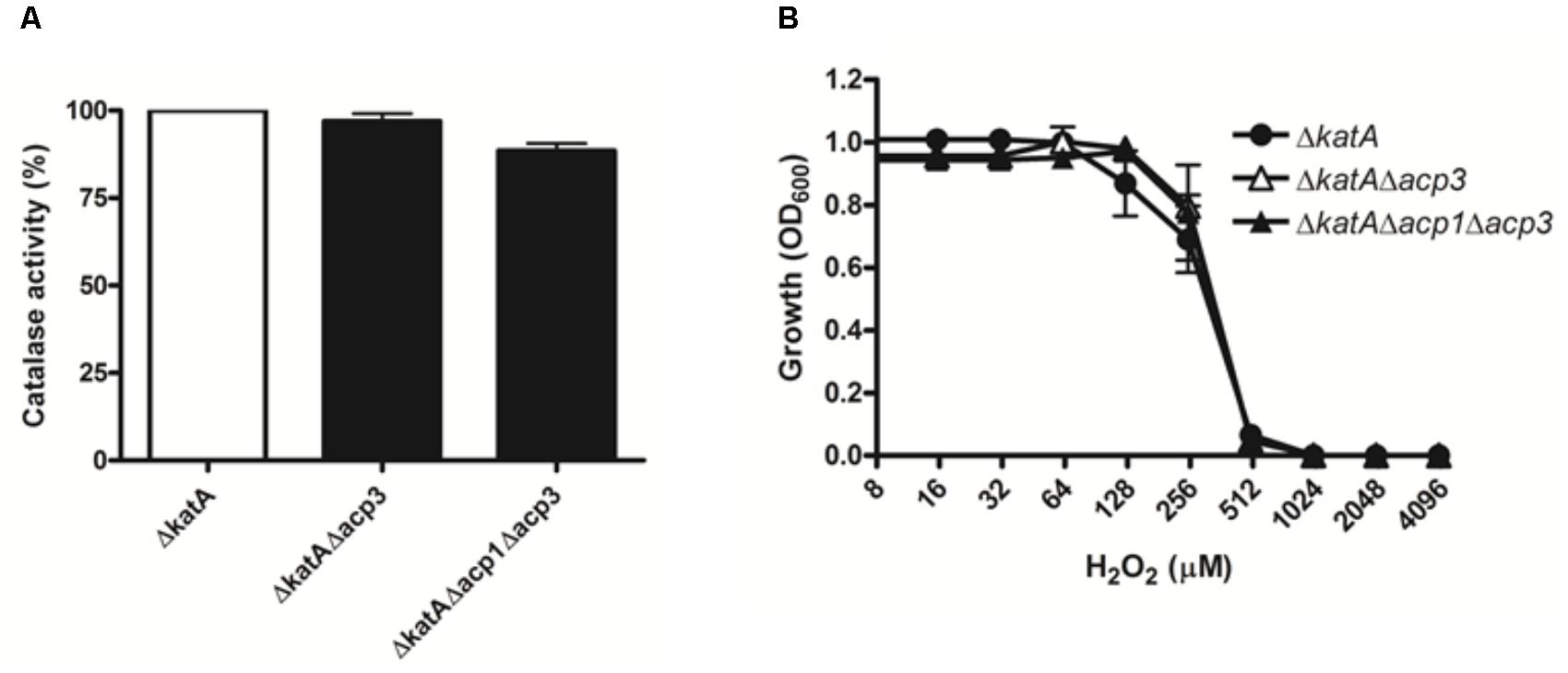
FIGURE 6. The H2O2 resistance of acp3 mutant is mediated by KatA. In contrast to the effect of acp3 deletion on catalase activity and susceptibility to H2O2 in the wild-type background, ΔkatA, ΔkatAΔacp3, and ΔkatAΔacp1Δacp3 mutant strains exhibited similar (A) Catalase activity and (B) resistance against H2O2, suggesting that the increased catalase activity and resistance to H2O2 in the Δacp3 strain is dependent on KatA. Data are presented as mean ± SEs from three independent experiments.
To address whether the absence of Acp3 affects the expression level of katA, we measured the level of katA transcript in the acp3 deletion strain, as well as for several genes that respond to oxidative stress, specifically katB, ahpC, sodB, and trxB2 (Salunkhe et al., 2005). Compared to PAO1, deletion of acp3 did not alter transcription levels of katA or any of the other selected genes (Figure 7A). Similarly, protein levels of KatA were the same in acp1 or acp3 deletion strains compared to PAO1 (Figure 7B). These data suggest that the activity of KatA is inhibited by a direct interaction with Acp3 and not by transcriptional or translational regulation.
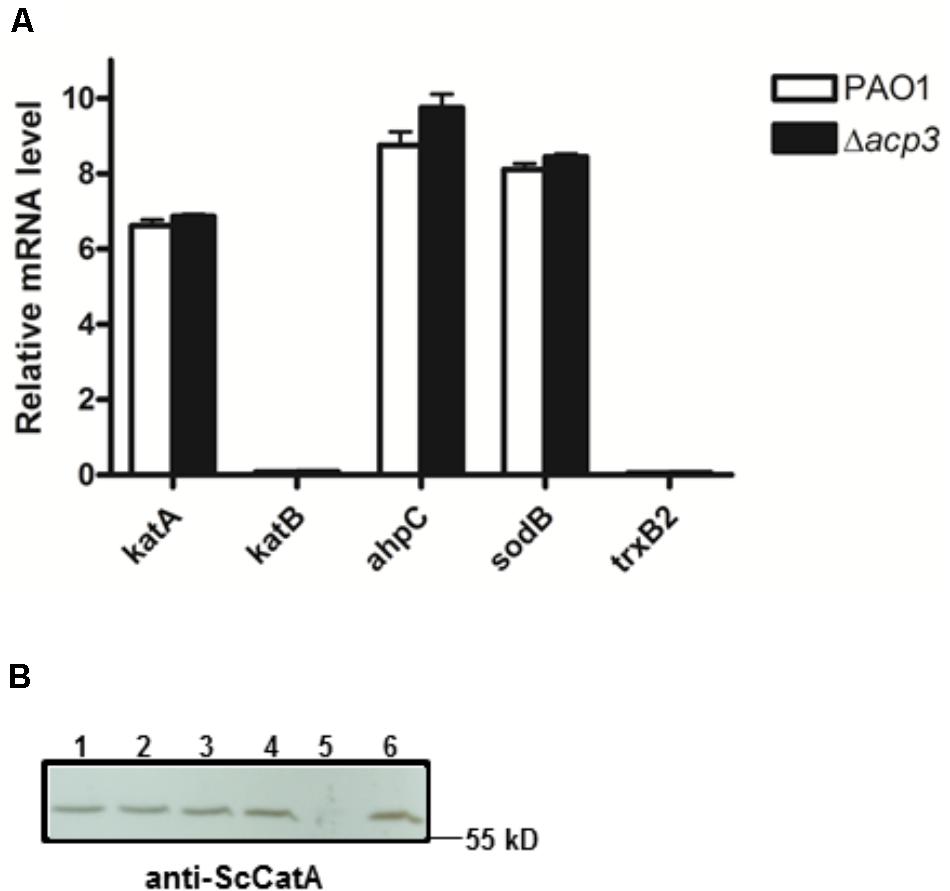
FIGURE 7. Deletion of acp3 does not affect the transcript and protein levels of KatA in P. aeruginosa. (A) Transcriptional levels of selected genes involved in oxidative stress response were determined by qRT-PCR. Data are presented as mean ± SEs from three independent experiments. (B) Western blot analysis of KatA protein levels in PAO1 (lane 1), Δacp1 (lane 2), Δacp3 (lane 3), and Δacp1Δacp3 (lane 4). The ΔkatA strain (lane 5) and purified His-KatA (line 6) were used as negative and positive controls, respectively. Cells from overnight cultures were used to prepare total RNA and the total protein samples for the analyses.
To determine whether inhibition of KatA activity by Acp3 correlates with levels of endogenous ROS, we used DCFH2, a non-specific fluorescence probe, to measure ROS levels in P. aeruginosa cells (Brömme et al., 2008). Significantly less ROS was detected in the Δacp3 mutant compared with wild type, commensurate with a higher level of KatA activity (Figure 8). In addition, the katA deletion mutant contained a much higher level of ROS and the KatA overexpression strain exhibited lower ROS. These data suggest that release of KatA inhibition by the acp3 deletion decreases ROS levels in P. aeruginosa.
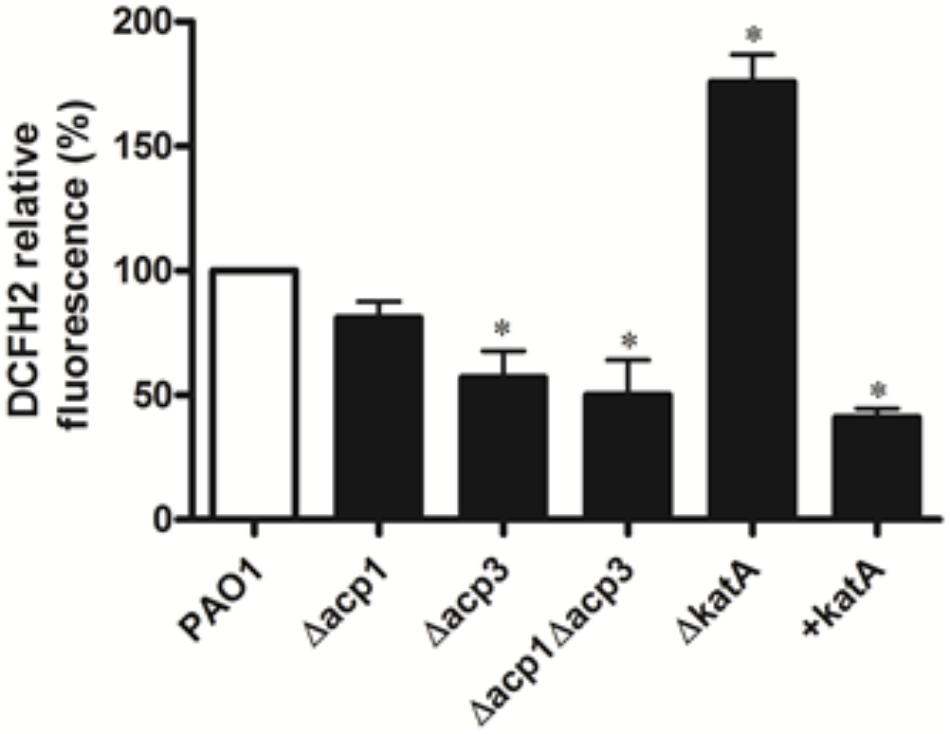
FIGURE 8. Deletion of acp3 gene decreases endogenous ROS. ROS levels were measured by DCFH2 fluorescence. katA-deletion (ΔkatA) and KatA-overexpressing (+katA) strains were used as controls. Data are presented as mean ± SEs from three independent experiments. Student’s t-test was performed between PAO1 and deletion or over-expressing strains. ∗ indicates p < 0.01.
In addition to canonical ACPs that function during FAS, bacteria typically express other ACPs and in many cases, the functions of these homologues are unknown. Following a recent study in which AcpP was confirmed as the essential ACP involved in membrane FAS (Ma et al., 2017), we investigated Acp1 and Acp3 of P. aeruginosa in order to shed light on their physiological roles in this important pathogen. The major finding is that Acp3 impacts virulence and in vivo pathogenicity of P. aeruginosa and also appears to regulate the activity of the catalase KatA in a way that suggests involvement in the cellular response to oxidative stress.
In keeping with the idea that Acp1 or Acp3 have a role in pathogenesis, we first investigated whether their expression is regulated by QS. This is an important mechanism employed by bacterial populations to respond to environmental cues, such as nutrient restriction or the presence of antibiotics. In P. aeruginosa, QS governs biofilm formation, swarming motility, and production of virulence factors such as pyocyanin, elastases, and rhamnolipids (Venturi, 2006). A connection between ACPs and QS is logical because the synthesis of QS signals such as PQS and acyl homoserine lactones (HSLs) requires acyl ACP (Parsek et al., 1997; Raychaudhuri et al., 2005; Dulcey et al., 2013). Furthermore, the higher levels of both transcripts and proteins for ACPs observed during the transition of cell cultures from log to stationary phase could involve QS. Accordingly, we found that levels of acp1 and acp3 transcripts are significantly lower in P. aeruginosa strains lacking LasR or RhlR, which are the receptors for 3-oxo-C12-HSL and C4-HSL, respectively (Gambello and Iglewski, 1991; Ochsner and Reiser, 1995) and thus suggests that expression of Acp1 and Acp3 is governed by the QS machinery.
Our next step was to assess whether Acp1 or Acp3 plays a role in pathogenicity. We found that deletion of acp1 or acp3 has no effect on in vitro virulence of P. aeruginosa, except for thicker biofilm. By contrast, when tested in a mouse infection model, a 1000-fold increase in bacterial load was detected for both acp1 and acp3 deletion mutants, indicating that both Acp1 and Acp3 play a role in P. aeruginosa pathogenicity in vivo. A previous study showed increased expression levels of both acp1 and acp3 genes in acute and chronic wound infection models in mice (Turner et al., 2014), suggesting that Acp1 and Acp3 are important for P. aeruginosa fitness in a disease environment in vivo. The different effects of acp1 and acp3 deletion on virulence in vitro versus in vivo may be due to the differences in the availability of nutrients and oxygen in the growth environment.
To probe their physiological roles further, we used TAP (Babu et al., 2009) to identify protein-binding partners of Acp1 and Acp3. The identification of several enzymes involved in fatty acid biosynthesis as binding partners of AcpP provided validation for the experiment. Acp1 and Acp3 also bound some enzymes of Type II FAS, but more so to proteins from other areas of metabolism. For instance, the top hit for Acp1 was PepA (also known as PhpA), which possesses aminopeptidase activity and whose disruption in an algB mutant increases alginate expression in P. aeruginosa (Woolwine and Wozniak, 1999). PepA also plays a role within the Xer recombinase system of E. coli (where it is called XerB) (Stirling et al., 1989) and it lowers expression of carbamoyl phosphate synthase by binding to the promoter region of the carAB operon in E. coli (Charlier et al., 1995). The association of PepA with Acp1 adds a further dimension to a protein that appears to function in several very different processes. Another binding hit for Acp1 is GlpR, which is a repressor of genes involved in glycerol metabolism (Schweizer and Po, 1996). Since genetic deletion of GlpR increases biofilm formation in P. aeruginosa (Scoffield and Silo-Suh, 2016), it is tempting to speculate that its association with Acp1 may affect virulence.
Among the proteins that bound Acp3 were IscS, FabH2, KatA, and ArcB. The breadth and diversity of these interactions hints at the versatility of Acp3 in its functions. The top hit was IscS, a cysteine desulfurase that provides sulfur for the biosynthesis of FeS clusters, thiamine, biotin, lipoic acid, modified nucleosides in tRNAs, and the molybdenum cofactor (Moco), among others (Hidese et al., 2011; Leimkuhler et al., 2017). Its interaction with Acp3 is not the first evidence of a functional association between ACPs and cysteine desulfurases. Using the same TAP approach, E. coli ACP was also found to bind IscS, with the interaction mediated by a disulfide bond between the 4-phosphopantetheine group and Cys328 of IscS (Gully et al., 2003). It remains to be established whether this group is also required for the interaction in P. aeruginosa.
Interactions between IscS and ACP also occur in humans, albeit indirectly. A multisubunit complex containing NFS1 (the name given to IscS in humans, which shares ∼60% amino acid sequence identity with E. coli IscS), the LYR protein ISD11 and acyl ACP assembles FeS clusters in mitochondria (Van Vranken et al., 2016). Within this complex, ACP interacts with the ISD11 subunit (Herrera et al., 2018), which in turn interacts with NSF1 (Cory et al., 2017). Such Acp–IscS interactions in other species supports the idea that Acp3 may have a role in assembly of FeS clusters in P. aeruginosa.
Another putative binding partner for Acp3 is ArcB, an ornithine carbamoyl transferase (OCT). This enzyme is part of the arginine deiminase system (ADS) found in bacteria that allows anaerobic growth on arginine. In P. aeruginosa, OCT is a dodecameric complex of identical protomers (Marcq et al., 1991) and is allosterically inhibited by spermidine and activated by AMP regulation (Tricot et al., 1993). There appears no involvement of ACP in the reaction mechanism (Tricot et al., 1998) and this putative interaction therefore requires further investigation.
The apparent promiscuity in binding of P. aeruginosa ACPs and especially those for Acp3 is mirrored by E. coli ACP, which binds to a number of protein factors outside of its canonical function in FAS, including IscS, MukB, YbgC, and SpoT (Gully et al., 2003; Gully and Bouveret, 2006). Of these, the interaction with SpoT has been examined in most detail (Battesti and Bouveret, 2006; Angelini et al., 2012). SpoT is a factor that changes its activity from (p)ppGpp hydrolysis to (p)ppGpp synthesis in response to starvation signals, leading to initiation of the stringent response. It has been postulated that the switch in its enzymatic activity toward synthesis may be governed by the binding of ACP to the C-terminal domain of SpoT (Battesti and Bouveret, 2006). Mutations that abrogate binding to SpoT mostly map to hydrophobic residues in helix II of ACP (Angelini et al., 2012) and this region of the protein also mediates interactions with its other protein partners, including FabI, AcpS, and YchM(Parris et al., 2000; Rafi et al., 2006; Babu et al., 2010). Each of the new potential binding partners revealed here for Acp1 and Acp3 offers opportunity to investigate the molecular details of these interactions in a similar way as ACP-SpoT, including establishing whether helix II is a common interface, determining whether an acylated 4-phosphopantetheine prosthetic group is involved in the binding, and understanding their physiological consequences in P. aeruginosa.
With this goal in mind, we decided to investigate further the interaction of Acp3 with KatA, the major catalase of P. aeruginosa (Hassett et al., 1992). katA is one of the oxidative stress response genes expressed during aerobic metabolism or in response to exogenous ROS, and its expression is controlled by the global transcriptional regulator OxyR (Heo et al., 2010). At first sight, the reason for an interaction between Acp3 and KatA is not obvious, but can be rationalized in the context of cellular responses of P. aeruginosa to oxidative stress. Under such conditions, KatA is upregulated in order to dismutate H2O2 and other ROS, and presumably is negatively regulated when ROS levels normalize. It appears that by binding to KatA, Acp3 acts as a negative regulator of its catalase activity. This is supported by the increased resistance of the acp3 deletion strain to H2O2 (because KatA activity is constitutively higher without Acp3). Higher tolerance to ROS may also explain the increased persistence of the acp3 deletion mutant in vivo. The inhibition of KatA by binding of Acp3 is reminiscent of the ACP-SpoT interaction and a further example of how ACPs can regulate enzyme activities.
It is interesting to note that the putative interaction between Acp3 and IscC mentioned above may also be related to oxidative stress. This is because a strain of C. difficile lacking IscS2 (the homologue of IscS) fails to grow when oxygen levels go above 2% (Giordano et al., 2018) suggesting it protects against oxidative stress. However, it needs to be established whether binding of Acp3 to IscS represses its cysteine desulfurase activity similar to KatA, or whether it enhances activity.
Why Acp3 regulates the activity of KatA is unknown. There are probably several potential reasons that could be invoked, but one intriguing possibility relates to the so-called “Insurance Hypothesis.” This is the diversification of phenotype in bacterial communities in response to harsh conditions (Boles et al., 2004). The increased expression of Acp3 prior to stationary phase may serve to reduce catalase activity and thereby increase ROS levels. The subsequent DNA damage and mutation would increase genotypic and phenotypic diversity within the biofilm (Boles et al., 2004), thus allowing the community to survive. While more work is needed to uncover its details, the observed binding of Acp3 to KatA reveals an additional mechanism of control within an already complex regulatory network for KatA and a potential role for this ACP in response to oxidative stress.
WC generated all strains for this work, performed the phenotype analyses and enzymatic assays in vitro, prepared the figures, and contributed to the writing of the manuscript. BW and JG performed the in vivo pathogenicity assays. Y-MZ conceived the study, directed the experiments, and helped draft the manuscript. CD contributed ideas to the project and helped draft the manuscript. All authors contributed to the analysis and interpretation of data. All authors approved the manuscript submitted.
This work was supported in part by the Center for Biomedical Research Excellence (COBRE) in Lipidomics and Pathobiology (NIH P20 RR017677) at the Medical University of South Carolina and by National Natural Science Foundation of China (NSFC 81701973) in the Second Affiliated Hospital of the Southeast University.
Y-MZ is currently employed by Jeneil Biotech Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Dr. You-Hee Cho (Sogang University) for the anti-ScCatA antibody and Dr. Charles Rock (St. Jude Children’s Research Hospital) for assistance with the identification of binding partners of ACPs using LC-MS.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02244/full#supplementary-material
Angelini, S., My, L., and Bouveret, E. (2012). Disrupting the Acyl carrier protein/SpoT interaction in vivo: identification of ACP residues involved in the interaction and consequence on growth. PLoS One 7:e36111. doi: 10.1371/journal.pone.0036111
Babu, M., Butland, G., Pogoutse, O., Li, J., Greenblatt, J. F., and Emili, A. (2009). Sequential peptide affinity purification system for the systematic isolation and identification of protein complexes from Escherichia coli. Methods Mol. Biol. 564, 373–400. doi: 10.1007/978-1-60761-157-8-22
Babu, M., Greenblatt, J. F., Emili, A., Strynadka, N. C., Reithmeier, R. A., and Moraes, T. F. (2010). Structure of a SLC26 anion transporter STAS domain in complex with acyl carrier protein: implications for E. coli YchM in fatty acid metabolism. Structure 18, 1450–1462. doi: 10.1016/j.str.2010.08.015
Battesti, A., and Bouveret, E. (2006). Acyl carrier protein/SpoT interaction, the switch linking SpoT-dependent stress response to fatty acid metabolism. Mol. Microbiol. 62, 1048–1063. doi: 10.1111/j.1365-2958.2006.05442.x
Battesti, A., and Bouveret, E. (2009). Bacteria possessing two RelA/SpoT-like proteins have evolved a specific stringent response involving the acyl carrier protein-SpoT interaction. J. Bacteriol. 191, 616–624. doi: 10.1128/JB.01195-08
Boles, B. R., Thoendel, M., and Singh, P. K. (2004). Self-generated diversity produces “insurance effects” in biofilm communities. Proc. Natl. Acad. Sci. U.S.A. 101, 16630–16635. doi: 10.1073/pnas.0407460101
Brömme, H.-J., Zühlke, L., Silber, R.-E., and Simm, A. (2008). DCFH2 interactions with hydroxyl radicals and other oxidants – Influence of organic solvents. Exp. Gerontol. 43, 638–644. doi: 10.1016/j.exger.2008.01.010
Brozek, K. A., Carlson, R., and Wand Raetz, C. R. (1996). A special acyl carrier protein for transferring long hydroxylated fatty acids to lipid A in Rhizobium. J. Biol. Chem. 271, 32126–32136. doi: 10.1074/jbc.271.50.32126
Butland, G., Peregrin-Alvarez, J. M., Li, J., Yang, W., Yang, X., Canadien, V., et al. (2005). Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433, 531–537. doi: 10.1038/nature03239
Byers, D., and Gong, M. H. (2007). Acyl carrier protein: structure-function relationships in a conserved multifunctional protein family. Biochem. Cell Biol. 85, 649–662. doi: 10.1139/o07-109
Chan, D. I., and Vogel, H. J. (2010). Current understanding of fatty acid biosynthesis and the acyl carrier protein. Biochem. J. 430, 1–19. doi: 10.1042/BJ20100462
Charlier, D., Hassanzadeh, G., Kholti, A., Gigot, D., Pierard, A., and Glansdorff, N. (1995). carP, involved in pyrimidine regulation of the Escherichia coli carbamoylphosphate synthetase operon encodes a sequence-specific DNA-binding protein identical to XerB and PepA, also required for resolution of ColEI multimers. J. Mol. Biol. 250, 392–406. doi: 10.1006/jmbi.1995.0385
Choi, K. H., and Schweizer, H. P. (2005). An improved method for rapid generation of unmarked Pseudomonas aeruginosa deletion mutants. BMC Microbiol. 5:30. doi: 10.1186/1471-2180-5-30
Chugani, S., Kim, B. S., Phattarasukol, S., Brittnacher, M. J., Choi, S. H., Harwood, C. S., et al. (2012). Strain-dependent diversity in the Pseudomonas aeruginosa quorum-sensing regulon. Proc. Natl. Acad. Sci. U.S.A. 109, E2823–E2831. doi: 10.1073/pnas.1214128109
Cory, S. A., Van Vranken, J. G., Brignole, E. J., Patra, S., Winge, D. R., Drennan, C. L., et al. (2017). Structure of human Fe-S assembly subcomplex reveals unexpected cysteine desulfurase architecture and acyl-ACP-ISD11 interactions. Proc. Natl. Acad. Sci. U.S.A. 114, E5325–E5334. doi: 10.1073/pnas.1702849114
De Lay, N. R., and Cronan, J. E. (2007). In vivo functional analyses of the type II acyl carrier proteins of fatty acid biosynthesis. J. Biol. Chem. 282, 20319–20328. doi: 10.1074/jbc.M703789200
de Lorenzo, V., and Timmis, K. N. (1994). Analysis and construction of stable phenotypes in gram-negative bacteria with Tn5- and Tn10-derived minitransposons. Methods Enzymol. 235, 386–405. doi: 10.1016/0076-6879(94)35157-0
Dulcey, C. E., Dekimpe, V., Fauvelle, D. A., Milot, S., Groleau, M. C., Doucet, N., et al. (2013). The end of an old hypothesis: the Pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem. Biol. 20, 1481–1491. doi: 10.1016/j.chembiol.2013.09.021
Epple, G., van der Drift, K. M., Thomas-Oates, J. E., and Geiger, O. (1998). Characterization of a novel acyl carrier protein, RkpF, encoded by an operon involved in capsular polysaccharide biosynthesis in Sinorhizobium meliloti. J. Bacteriol. 180, 4950–4954.
Gambello, M. J., and Iglewski, B. H. (1991). Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J. Bacteriol. 173, 3000–3009. doi: 10.1128/jb.173.9.3000-3009.1991
Geiger, O., and Lopez-Lara, I. M. (2002). Rhizobial acyl carrier proteins and their roles in the formation of bacterial cell-surface components that are required for the development of nitrogen-fixing root nodules on legume hosts. FEMS Microbiol. Lett. 208, 153–162. doi: 10.1111/j.1574-6968.2002.tb11075.x
Geiger, O., Spaink, H. P., and Kennedy, E. P. (1991). Isolation of the Rhizobium leguminosarum NodF nodulation protein: NodF carries a 4’-phosphopantetheine prosthetic group. J. Bacteriol. 173, 2872–2878. doi: 10.1128/jb.173.9.2872-2878.1991
Giordano, N., Hastie, J. L., Smith, A. D., Foss, E. D., Gutierrez-Munoz, D. F., and Carlson, P. E. Jr. (2018). Cysteine desulfurase IscS2 plays a role in oxygen resistance in clostridium difficile. Infect. Immun. 86, e326–e318. doi: 10.1128/IAI.00326-18
Govan, J. R., and Deretic, V. (1996). Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60, 539–574.
Gruber, J. D., Chen, W., Parnham, S., Beauchesne, K., Moeller, P., Flume, P. A., et al. (2016). The role of 2,4-dihydroxyquinoline (DHQ) in Pseudomonas aeruginosa pathogenicity. PeerJ 4:e1495. doi: 10.7717/peerj.1495
Gully, D., and Bouveret, E. (2006). A protein network for phospholipid synthesis uncovered by a variant of the tandem affinity purification method in Escherichia coli. Proteomics 6, 282–293. doi: 10.1002/pmic.200500115
Gully, D., Moinier, D., Loiseau, L., and Bouveret, E. (2003). New partners of acyl carrier protein detected in Escherichia coli by tandem affinity purification. FEBS Lett. 548, 90–96. doi: 10.1016/S0014-5793(03)00746-4
Haag, A. F., Wehmeier, S., Beck, S., Marlow, V. L., Fletcher, V., James, E. K., et al. (2009). The Sinorhizobium meliloti LpxXL and AcpXL proteins play important roles in bacteroid development within alfalfa. J. Bacteriol. 191, 4681–4686. doi: 10.1128/JB.00318-09
Hassett, D. J., Charniga, L., Bean, K., Ohman, D. E., and Cohen, M. S. (1992). Response of Pseudomonas aeruginosa to pyocyanin: mechanisms of resistance, antioxidant defenses, and demonstration of a manganese-cofactored superoxide dismutase. Infect. Immun. 60, 328–336.
Hentzer, M., Wu, H., Andersen, J. B., Riedel, K., Rasmussen, T. B., Bagge, N., et al. (2003). Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 22, 3803–3815. doi: 10.1093/emboj/cdg366
Heo, Y. J., Chung, I. Y., Cho, W. J., Lee, B. Y., Kim, J. H., Choi, K. H., et al. (2010). The major catalase gene (katA) of Pseudomonas aeruginosa PA14 is under both positive and negative control of the global transactivator OxyR in response to hydrogen peroxide. J. Bacteriol. 192, 381–390. doi: 10.1128/JB.00980-09
Herrera, M. G., Pignataro, M. F., Noguera, M. E., Cruz, K. M., and Santos, J. (2018). Rescuing the rescuer: on the protein complex between the human mitochondrial Acyl carrier protein and ISD11. ACS Chem. Biol. 13, 1455–1462. doi: 10.1021/acschembio.8b00184
Hidese, R., Mihara, H., and Esaki, N. (2011). Bacterial cysteine desulfurases: versatile key players in biosynthetic pathways of sulfur-containing biofactors. Appl. Microbiol. Biotechnol. 91, 47–61. doi: 10.1007/s00253-011-3336-x
Holloway, B. W., Krishnapillai, V., and Morgan, A. F. (1979). Chromosomal genetics of Pseudomonas. Microbiol. Rev. 43, 73–102.
Kapatral, V., Bina, X., and Chakrabarty, A. M. (2000). Succinyl coenzyme A synthetase of Pseudomonas aeruginosa with a broad specificity for nucleoside triphosphate (NTP) synthesis modulates specificity for NTP synthesis by the 12-kilodalton form of nucleoside diphosphate kinase. J. Bacteriol. 182, 1333–1339. doi: 10.1128/JB.182.5.1333-1339.2000
Lee, J. S., Heo, Y. J., Lee, J. K., and Cho, Y. H. (2005). KatA, the major catalase, is critical for osmoprotection and virulence in Pseudomonas aeruginosa PA14. Infect. Immun. 73, 4399–4403. doi: 10.1128/IAI.73.7.4399-4403.2005
Leimkuhler, S., Buhning, M., and Beilschmidt, L. (2017). Shared sulfur mobilization routes for tRNA thiolation and molybdenum cofactor biosynthesis in prokaryotes and eukaryotes. Biomolecules 7:5. doi: 10.3390/biom7010005
Ma, J. C., Wu, Y. Q., Cao, D., Zhang, W. B., and Wang, H. H. (2017). Only Acyl carrier protein 1 (AcpP1) functions in Pseudomonas aeruginosa fatty acid synthesis. Front. Microbiol. 8:2186. doi: 10.3389/fmicb.2017.02186
Marcq, S., Diaz-Ruano, A., Charlier, P., Dideberg, O., Tricot, C., Pierard, A., et al. (1991). Molecular size and symmetry of Pseudomonas aeruginosa catabolic ornithine carbamoyltransferase. An X-ray crystallography analysis. J. Mol. Biol. 220, 9–12. doi: 10.1016/0022-2836(91)90375-G
Mishra, S., and Imlay, J. (2012). Why do bacteria use so many enzymes to scavenge hydrogen peroxide? Arch. Biochem. Biophys. 525, 145–160. doi: 10.1016/j.abb.2012.04.014
Ochsner, U., and Reiser, A. J. (1995). Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 92, 6424–6428. doi: 10.1073/pnas.92.14.6424
Ochsner, U. A., Vasil, A. I., and Vasil, M. L. (1995). Role of the ferric uptake regulator of Pseudomonas aeruginosa in the regulation of siderophores and exotoxin A expression: purification and activity on iron-regulated promoters. J. Bacteriol. 177, 7194–7201. doi: 10.1128/jb.177.24.7194-7201.1995
Ou, P., and Wolff, S. P. (1996). A discontinuous method for catalase determination at ‘near physiological’ concentrations of H2O2 and its application to the study of H2O2 fluxes within cells. J. Biochem. Biophys. Methods 31, 59–67. doi: 10.1016/0165-022X(95)00039-T
Parris, K. D., Lin, L., Tam, A., Mathew, R., Hixon, J., Stahl, M., et al. (2000). Crystal structures of substrate binding to Bacillus subtilis holo-(acyl carrier protein) synthase reveal a novel trimeric arrangement of molecules resulting in three active sites. Structure 8, 883–895. doi: 10.1016/S0969-2126(00)00178-7
Parsek, M. R., Schaefer, A. L., and Greenberg, E. P. (1997). Analysis of random and site-directed mutations in rhII, a Pseudomonas aeruginosa gene encoding an acylhomoserine lactone synthase. Mol. Microbiol. 26, 301–310. doi: 10.1046/j.1365-2958.1997.5741935.x
Rafi, S., Novichenok, P., Kolappan, S., Stratton, C. F., Rawat, R., Kisker, C., et al. (2006). Structure of acyl carrier protein bound to FabI, the FASII enoyl reductase from Escherichia coli. J. Biol. Chem. 281, 39285–39293. doi: 10.1074/jbc.M608758200
Ramos-Vega, A. L., Davila-Martinez, Y., Sohlenkamp, C., Contreras-Martinez, S., Encarnacion, S., Geiger, O., et al. (2009). SMb20651 is another acyl carrier protein from Sinorhizobium meliloti. Microbiology 155, 257–267. doi: 10.1099/mic.0.022079-0
Rao, X., Huang, X., Zhou, Z., and Lin, X. (2013). An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 3, 71–85.
Raychaudhuri, A., Jerga, A., and Tipton, P. A. (2005). Chemical mechanism and substrate specificity of rhli, an acylhomoserine lactone synthase from Pseudomonas aeruginosa. Biochemistry 44, 2974–2981. doi: 10.1021/bi048005m
Salunkhe, P., Topfer, T., Buer, J., and Tummler, B. (2005). Genome-wide transcriptional profiling of the steady-state response of Pseudomonas aeruginosa to hydrogen peroxide. J. Bacteriol. 187, 2565–2572. doi: 10.1128/JB.187.8.2565-2572.2005
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Schuster, M., Lostroh, C. P., Ogi, T., and Greenberg, E. P. (2003). Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J. Bacteriol. 185, 2066–2079. doi: 10.1128/jb.185.7.2066-2079.2003
Schweizer, H., and Po, P. C. (1996). Regulation of glycerol metabolism in Pseudomonas aeruginosa: characterization of the glpR repressor gene. J. Bacteriol. 178, 5215–5221. doi: 10.1128/jb.178.17.5215-5221.1996
Scoffield, J., and Silo-Suh, L. (2016). Glycerol metabolism promotes biofilm formation by Pseudomonas aeruginosa. Can. J. Microbiol. 62, 704–710. doi: 10.1139/cjm-2016-0119
Shin, D. H., Choi, Y. S., and Cho, Y. H. (2008). Unusual properties of catalase A (KatA) of Pseudomonas aeruginosa PA14 are associated with its biofilm peroxide resistance. J. Bacteriol. 190, 2663–2670. doi: 10.1128/JB.01580-07
Stirling, C. J., Colloms, S. D., Collins, J. F., Szatmari, G., and Sherratt, D. J. (1989). xerB, an Escherichia coli gene required for plasmid ColE1 site-specific recombination, is identical to pepA, encoding aminopeptidase A, a protein with substantial similarity to bovine lens leucine aminopeptidase. EMBO J. 8, 1623–1627.
Therisod, H., and Kennedy, E. P. (1987). The function of acyl carrier protein in the synthesis of membrane-derived oligosaccharides does not require its phosphopantetheine prosthetic group. Proc. Natl. Acad. Sci. U.S.A. 84, 8235–8238. doi: 10.1073/pnas.84.23.8235
Therisod, H., Weissborn, A. C., and Kennedy, E. P. (1986). An essential function for acyl carrier protein in the biosynthesis of membrane-derived oligosaccharides of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 83, 7236–7240. doi: 10.1073/pnas.83.19.7236
Tricot, C., Nguyen, V. T., and Stalon, V. (1993). Steady-state kinetics and analysis of pH dependence on wild-type and a modified allosteric Pseudomonas aeruginosa ornithine carbamoyltransferase containing the replacement of glutamate 105 by alanine. Eur. J. Biochem. 215, 833–839. doi: 10.1111/j.1432-1033.1993.tb18099.x
Tricot, C., Villeret, V., Sainz, G., Dideberg, O., and Stalon, V. (1998). Allosteric regulation in Pseudomonas aeruginosa catabolic ornithine carbamoyltransferase revisited: association of concerted homotropic cooperative interactions and local heterotropic effects. J. Mol. Biol. 283, 695–704. doi: 10.1006/jmbi.1998.2133
Turner, K. H., Everett, J., Trivedi, U., Rumbaugh, K. P., and Whiteley, M. (2014). Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet. 10:e1004518. doi: 10.1371/journal.pgen.1004518
Van Vranken, J. G., Jeong, M. Y., Wei, P., Chen, Y. C., Gygi, S. P., Winge, D. R., et al. (2016). The mitochondrial acyl carrier protein (ACP) coordinates mitochondrial fatty acid synthesis with iron sulfur cluster biogenesis. eLife 5:e17828. doi: 10.7554/eLife.17828
Venturi, V. (2006). Regulation of quorum sensing in Pseudomonas. FEMS Microbiol. Rev. 30, 274–291. doi: 10.1111/j.1574-6976.2005.00012.x
Viala, J. P., Prima, V., Puppo, R., Agrebi, R., Canestrari, M. J., Lignon, S., et al. (2017). Acylation of the Type 3 secretion system translocon using a dedicated Acyl carrier protein. PLoS Genet. 13:e1006556. doi: 10.1371/journal.pgen.1006556
Volkmann, G., Murphy, P. W., Rowland, E. E., Cronan, J. E. Jr., Liu, X. Q., Blouin, C., et al. (2010). Intein-mediated cyclization of bacterial acyl carrier protein stabilizes its folded conformation but does not abolish function. J. Biol. Chem. 285, 8605–8614. doi: 10.1074/jbc.M109.060863
Wagner, V. E., Bushnell, D., Passador, L., Brooks, A. I., and Iglewski, B. H. (2003). Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J. Bacteriol. 185, 2080–2095. doi: 10.1128/jb.185.7.2080-2095.2003
Winsor, G. L., Griffiths, E. J., Lo, R., Dhillon, B. K., Shay, J. A., and Brinkman, F. S. (2016). Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 44, D646–D653. doi: 10.1093/nar/gkv1227
Woolwine, S. C., and Wozniak, D. J. (1999). Identification of an Escherichia coli pepA homolog and its involvement in suppression of the algB phenotype in mucoid Pseudomonas aeruginosa. J. Bacteriol. 181, 107–116.
Zhang, Y.-M., Frank, M. W., Zhu, K., Mayasundari, A., and Rock, C. O. (2008). PqsD is responsible for the synthesis of 2,4-dihydroxyquinoline, an extracellular metabolite produced by Pseudomonas aeruginosa. J. Biol. Chem. 283, 28788–28794. doi: 10.1074/jbc.M804555200
Zhang, Y. M., Marrakchi, H., and Rock, C. O. (2002). The FabR (YijC) transcription factor regulates unsaturated fatty acid biosynthesis in Escherichia coli. J. Biol. Chem. 277, 15558–15565. doi: 10.1074/jbc.M201399200
Zhang, Y.-M., and Rock, C. O. (2012). Will the initiator of fatty acid synthesis in Pseudomonas aeruginosa please stand up? J. Bacteriol. 194, 5159–5161. doi: 10.1128/jb.01198-12
Keywords: acyl carrier protein, P. aeruginosa, virulence, catalase, oxidative stress
Citation: Chen W, Wang B, Gruber JD, Zhang Y-M and Davies C (2018) Acyl Carrier Protein 3 Is Involved in Oxidative Stress Response in Pseudomonas aeruginosa. Front. Microbiol. 9:2244. doi: 10.3389/fmicb.2018.02244
Received: 18 April 2018; Accepted: 03 September 2018;
Published: 20 September 2018.
Edited by:
Christian Sohlenkamp, Universidad Nacional Autónoma de México, MexicoReviewed by:
Tim McDermott, Montana State University, United StatesCopyright © 2018 Chen, Wang, Gruber, Zhang and Davies. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yong-Mei Zhang, eW16aGFuZzY2QGhvdG1haWwuY29t Christopher Davies, ZGF2aWVzQG11c2MuZWR1
†Present address: Yong-Mei Zhang, Jeneil Biotech Inc., Saukville, WI, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.