 Luan L. Chu
Luan L. Chu Dipesh Dhakal
Dipesh Dhakal Hee J. Shin
Hee J. Shin Hye J. Jung
Hye J. Jung Tokutaro Yamaguchi
Tokutaro Yamaguchi Jae K. Sohng
Jae K. Sohng- 1Department of Life Science and Biochemical Engineering, Sun Moon University, Asan, South Korea
- 2Department of BT Convergence Pharmaceutical Engineering, Sun Moon University, Asan, South Korea
Flavonoids are one of the predominant groups of plant polyphenols, and these compounds have significant effects on human health and nutrition. Sulfated flavonoids have more favorable attributes compared to their parent compounds such as increased solubility, stability, and bioavailability. In this research, we developed a microbial system to produce sulfated naringenin using Escherichia coli expressing a sulfotransferase (ST) from Arabidopsis thaliana (At2g03770). This wild-type strain was used as a model system for testing clustered regularly interspaced short palindromic repeats (CRISPR) interference (CRISPRi) metabolic engineering strategies. Using synthetic sgRNA to mediate transcriptional repression of cysH, a gene encoding 3′-phosphoadenosine-5′-phosphosulfate (PAPS) ST, which is involved in sulfur metabolism, resulted in an increase in intracellular PAPS accumulation by over 3.28-fold without impairing cell growth. Moreover, naringenin 7-sulfate production by engineering E. coli with its cysH gene repressed in the open reading frame through CRISPRi was enhanced by 2.83-fold in compared with the wild-type control. To improve the efficiency of biotransformation, the concentration of , glucose, and substrate were optimized. The bioproductivity of naringenin 7-sulfate was 135.49 μM [∼143.1 mg (47.7 mg L-1)] in a 3-L fermenter at 36 h. These results demonstrated that the CRISPRi system was successfully applied for the first time in E. coli to develop an efficient microbial strain for production of a sulfated flavonoid. In addition, antibacterial and anticancer activities of naringenin 7-sulfate were investigated and found to be higher than the parent compound.
Introduction
Flavonoids are major natural phenolic compounds found in plants (Buer et al., 2010). One of the most predominant citrus flavonoids is naringenin and it was found to exhibit various biological effects on human health and nutrition. Naringenin showed anti-estrogenic, antioxidant (Bugianesi et al., 2002), anti-obesity and anti-diabetic activities (Hossain et al., 2016). It has been demonstrated to control non-alcoholic steatohepatitis and associated inflammation (Jadeja and Devkar, 2014). Like flavonoids, most of the naringenin in nature accumulates in a glycosylated form (Gattuso et al., 2007), which improves solubility, storage, and stability of the parent compounds (De Bruyn et al., 2015). In addition to glycosylation, many the sulfate conjugate of flavonoids discovered in the different type of plants (Barron et al., 1998). However, the biological and physiological properties of sulfated flavonoids have not been studied (Totta et al., 2005; Wang et al., 2014).
Sulfation plays important roles in detoxification of xenobiotics (Yi et al., 2006; Chen et al., 2015) and regulating the activity of animal hormones (Geyer et al., 2017). Enzymatic sulfation is catalyzed by a family of sulfotransferases (STs) that transfer the sulfonate group of 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to a hydroxyl group or amino group of acceptor compounds with the parallel formation of 3′-phosphoadenosine-5′-phosphate (PAP) (Paul et al., 2012). Although the sequences of many STs have been deposited to various genome databases of plant (Gidda and Varin, 2006; Marsolais et al., 2007), bacteria (Hossain et al., 2011), and mammalian (Wong et al., 2010; Shimohira et al., 2017), the enzymatic and microbial synthesis of sulfonated natural products still are limited. One of the major problems of in vitro sulfation reactions is the use of the costly and unstable sulfate donor, PAPS, which impedes industrial scale-up of the process. Moreover, while chemical synthesis of sulfated compounds is tedious and time-consuming in terms of involving multiple steps (Zhang et al., 2012), PAPS has a poor availability in the cytosol of microbial cells like Escherichia coli and Streptomyces for production or modification of natural compounds (Sekowska et al., 2000; Nishikawa et al., 2017). Recently, genetic engineering has been used for production or post-modification of the natural compound via improving the availability of the common precursor in the biosynthetic pathway. For example, metabolic engineering of E. coli in the superpathway of methionine and S-adenosyl-L-methionine (SAM) biosynthesis, lead to improved SAM availability, and finally increased anthocyanin O-methylation (Cress et al., 2017). The sugar pathway was overexpressed to improve the cytoplasmic pool of NDP-sugars and subsequently expressed glycosyltransferase was used for the biosynthesis of glycosylated flavonoids in E. coli (Kim et al., 2015; Pandey et al., 2016). However, the same approach has not been applied to the biosynthesis of sulfated flavonoids.
Genetic editing using the clustered regularly interspaced short palindromic repeat (CRISPR) system has been widely used in diverse organisms including bacteria (Huang et al., 2015; Wu et al., 2015), fungal kingdoms (Wang et al., 2017), plant (Khan et al., 2017), animals (Gandhi et al., 2017), and human cell lines (Bertomeu et al., 2017). In the type II CRISPR, an RNA-guide DNA endonuclease (Cas9) is targeted to specific DNA sequences through a chimeric guide RNA (gRNA), which is a fusion between a precursor CRISPR RNAs (crRNA) and trans-activating CRISPR RNAs (tracrRNAs) (Huang et al., 2015). This gRNA is capable of recognizing sequences target sites for marker-free integration or gene disruption that are followed by the protospacer-adjacent motif (PAM) sequence NGG, N being any nucleotide (Cress et al., 2015). Recently, CRISPR interference (CRISPRi) has been developed which contains a mutation in the Cas9 protein (D10A and H840A) resulted in a lack of endonuclease activity (dCas9), but DNA binding capability remained for inhibition of transcription (Bikard et al., 2013). CRISPRi has been applied to down-regulation of genes in certain pathways via metabolic engineering of E. coli for production of value natural compounds (Wu et al., 2015; Liang et al., 2016; Gao et al., 2017). However, it has not been reported to regulate a gene in E. coli sulfur metabolism.
In this study, we expressed a ST from Arabidopsis thaliana (At2g03770) for biosynthesis of sulfated naringenin in E. coli BL21 (DE3). Naringenin has been shown to be substrate specificity of At2g03770 (Hashiguchi et al., 2013). Furthermore, we employed a CRISPRi system as a tool for improving PAPS availability by knockdown PAPS ST (cysH) in the sulfur metabolism (Figure 1), resulted in enhancement the final products. This is the first research that a metabolic engineering approach for conjugating a sulfate moiety generated in the cytoplasm of E. coli. The biosynthesized compound was also tested for its potential bioactivities against various pathogens and cancer cell lines.
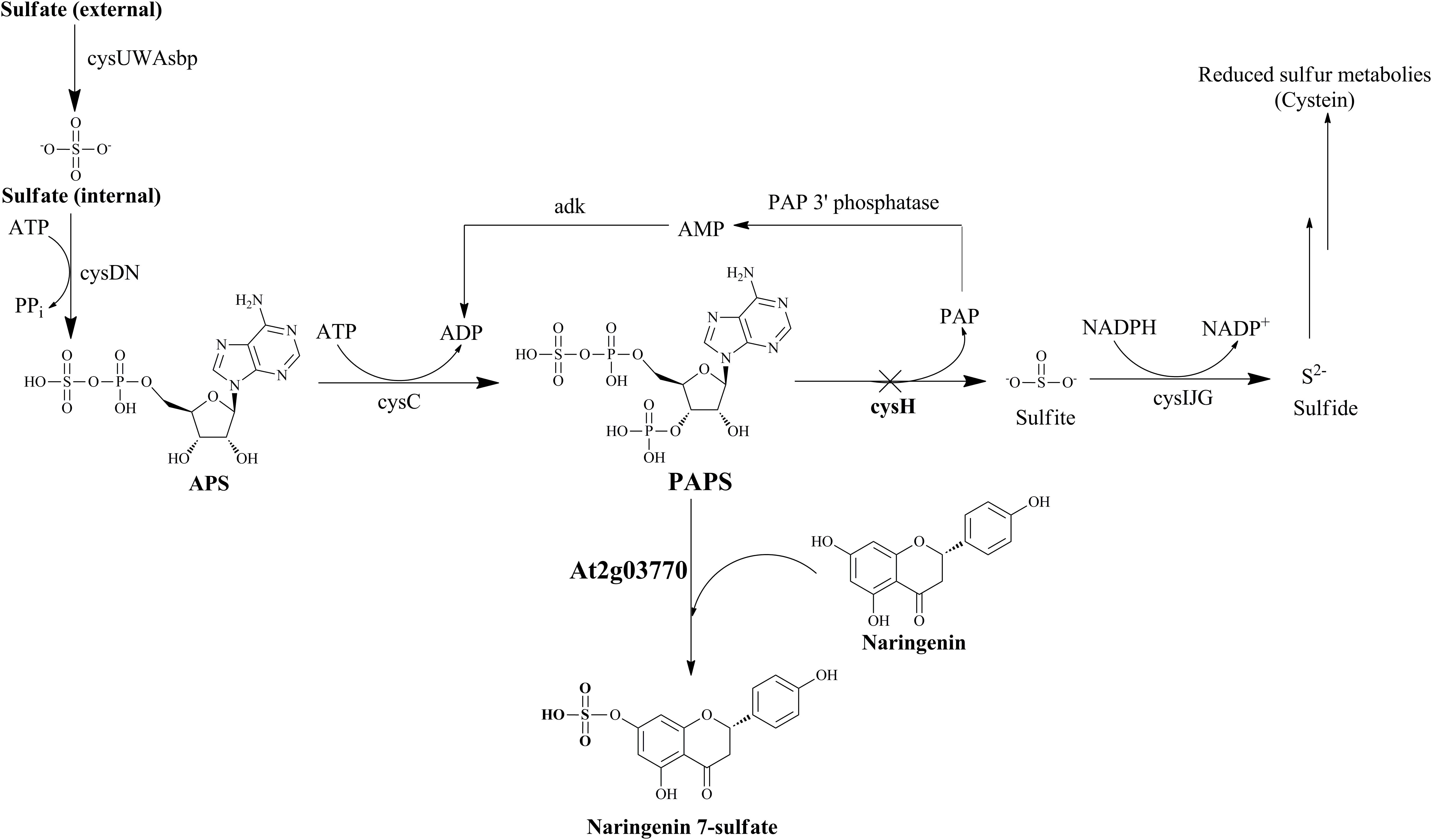
FIGURE 1. Schematic of sulfur metabolic pathway with strategies for biosynthesis of naringenin 7-sulfate in E. coli. Cross show the repressed gene in this pathway by using CRISPRi system. Operon cysUWAsbp, a sulfate permease; operon cysDN, a ATP sulfurylase; cysC, APS kinase; cysH, PAPS sulfotransferase; operon cysIJG, sulfite reductase; adk, adenylate kinase; APS, adenosine 5′-phosphosulfate; PAP, 3′-phosphoadenosine-5′-phosphate.
Materials and Methods
Chemicals and Reagents
Restriction enzymes, shrimp alkaline phosphatase, T4 polynucleotide kinase, and T4 DNA ligase were obtained from New England Biolabs (Hertfordshire, United Kingdom). Standard naringenin, adenosine 3′-phosphate 5′-phosphosulfate lithium salt hydrate (PAPS), adenosine 5′-triphosphate disodium salt hydrate (ATP), adenosine 5′-diphosphate (ADP), deuterium oxide (D2O), and dimethyl sulfoxide-d6 (DMSO-d6) were purchased from Sigma-Aldrich (St. Louis, MO, United States). Isopropyl-β-D-thiogalactopyranoside (IPTG) was obtained from GeneChem Inc. (Daejeon, South Korea). All other chemicals were of the highest grade commercially available.
Plasmid and Strains Constructions
All the E. coli strains and plasmids used in this study are supported in Table 1. PCR primers utilized for gene amplification and cloning were synthesized by GenoTech Corp. (Daejeon, South Korea). At2g03770 were codon-optimized for E. coli, synthesized, cloned into the plasmid vector pUC57 by General Biosystems, Inc. (NC, United States). The BamHI and XhoI sites were added to the 5′ and 3′ ends of the gene. The synthesized gene was restricted by BamHI and XhoI digestion, subcloned into the expression vector pET28a (+) to create the pET28a(+)-At2g03770. Then, the recombinant plasmid was transformed into E. coli BL21 (DE3) to form the strain used for production (wild-type) (Table 1).

TABLE 1. Strains and plasmids used in this study.
Plasmids used for CRISPRi/dCas9-mediated transcriptional repression was obtained from Addgene (Plasmid #65006) (Supplementary Figure S1) and constructed as previously reported (Cress et al., 2015). The specific targeting spacer of cysH from the E. coli K12 genomic DNA was identified near promoter (cysH1) and open reading frame region (cysH2) to prevent RNAP binding and elongation, respectively. The primer pairs crRNA1-Fw/Rv and crRNA2-Fw/Rv were used to construct CRISPRi-1 and CRISPRi-2 listed in Supplementary Table S1. Both primers were synthesized, phosphorylated with T4 polynucleotide kinase, and annealed (Cress et al., 2017). The products insert were then ligated into BsaI-digested, dephosphorylated, gel-purified CRISPRi plasmid backbone. E. coli DH5α was used for cloning experiments. All CRISPRi plasmid arrays possessing synthetic the specific targeting spacer were verified by colony PCR with primer pairs cPCR-Fw/Rv (Supplementary Table S2) and sequencing. Plasmids were constructed with CRISPRi-1 and CRISPRi-2 were transformed into the wild-type strain through calcium chloride and a heat-shock method (Dagert and Ehrlich, 1979), forming variant S1 and S2 strains (Table 1).
Culture Conditions and Recombinant Protein Expression
Wild-type E. coli was cultured in Luria Bertani (LB) liquid medium. The sample was kept in a 250 mL flask on a 37°C with shaking incubator 200 rpm. Kanamycin (50 μg mL-1) was used for plasmid selection and maintenance. A total of 250 μL of the pre-inoculum of wild-type E. coli was transferred to 50 mL fresh LB liquid medium containing an appropriate amount of antibiotic and then incubated at 37°C with 200 rpm shaking. Protein expression was induced by the different concentration of IPTG (0.1, 0.5, and 1.0 mM) when the optical cell density at 600 nm (OD600 nm) reached 0.6–0.8. The cell growth was continued at 20°C for 20 h and harvested by centrifugation at 842 × g for 15 min. The sample was washed twice with 50 mM Tris–HCl (pH 7.5) buffer containing 10% glycerol and prepared for sonication in 1 mL of the same buffer. Following centrifugation at 13,475 × g for 30 min at 4°C, the resulting soluble and insoluble protein fractions were analyzed by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Analysis of Cell Growth and mRNA-Expression Levels
The seed culture of the wild-type and S1, S2 strains were transferred to 50 mL fresh LB medium in a 250 mL flask and OD600 nm of each flask was diluted to 0.1. All the culture containing chloramphenicol (34 μg mL-1) or kanamycin (50 μg mL-1) were growth under sharking at 37°C with 200 rpm shaking. After 6 h, 0.1 mM IPTG was added to induce the CRISPRi system and the growth was continued at 20°C with 200 rpm shaking for 48 h.
Total RNA was isolated by using RNeasy Plus Mini Kit (Qiagen, United States). The QuantiTect Reverse Transcription Kit (Qiagen, United States) was used to synthesize the cDNA from 1 μg total RNA sample. Power SYBR Green Master Mix (Thermo Fisher Scientific, United States) was performed by quantitative real-time PCR (qRT-PCR) on StepOnePlus real-time PCR system (Applied Biosystems, United States). The 16S rRNA gene was used to an endogenous control with primer pairs 16S rRNA-Fw/16S rRNA-Rv (Supplementary Table S2). The comparative 2-Ct experiment was used to calculate relative gene expression (Livak and Schmittgen, 2001) by using wild-type as the reference sample with primer pairs cysH-Fw/cysH-Rv. The results were analyzed using StepOne Software v2.3 (Applied Biosystems, United States).
Intracellular PAPS Collection and Quantification
Three recombinant strains including wild-type, S1 and S2 were grown, induced with 0.1 mM IPTG and expressed in 50 mL LB medium at 20°C with 200 rpm shaking for 12 h. Then, the cells were harvested by centrifugation, suspended, and incubated in 250 mL flasks containing 50 mL of M9 minimal salt medium pH 7.4 at 28°C for 12 h. After that, the samples were taken, chilled immediately on ice and centrifuged at 842 × g at 4°C for 15 min. One milliliter of 6% perchloric acid was used for cell lysis and 0.3 mL of 3 M potassium carbonate was added to neutralize while vortexing the sample (Fowler et al., 2009). After centrifugation removed the cell residue, the supernatant containing was collected, filtered through a 0.22-μm syringe filter and injected into UFLC-PDA to determine the products yield. The determination of microbial biomass was carried out by dry cell weight using a 0.22-μm syringe filter. An aliquot of 1 mL cell culture was filtered, washed with distilled water, and dried in a conventional oven. DCW was represented by the weight difference between empty membranes and those with cell residues (Wu et al., 2015).
Sulfated Naringenin Production and Extraction
The wild-type and S1, S2 strains harboring heterologous a ST and CRISPRi were first expressed in 50 mL fresh LB medium by 0.1 mM IPTG at 20°C for 12 h. Next, the cells were harvested by centrifugation, washed twice with 100 mM phosphate buffer pH 7.4, and incubated at 28°C with shaking in 250 mL flasks containing 50 mL M9 minimal salt medium pH 7.4 (Yan et al., 2005). Appropriate antibiotics and IPTG were added at the same time. Subsequently, 100 μM substrate was supplemented to the same samples and kept on 48 h. We carried out fermentation in 3-L of optimal media under an optimal condition for large-scale production of naringenin derivatives. The processing of fermentation was followed as previously described (Chu et al., 2016). Furthermore, the culture samples were extracted with twice volume of ethyl acetate. The extracts were concentrated by freezing rotary evaporator, suspended in methanol, and then injected into UFLC-PDA to determine the products yield.
HPLC Analysis
The UFLC-PDA system containing a reversed-phase column [Mightysil RP-18 GP 250-4.6 (5 μm) Cica-Reagent; Kanto Chemical Co., Inc., Japan] was used to separate the samples. The mobile phase containing solvent A (HPLC-grade water consisting 0.05% TFA) and solvent B (100% HPLC-grade methanol) was maintained at 30°C. A calibration PAPS and naringenin standards were created with various concentration.
For quantification of PAPS, the intracellular PAPS was analyzed at a UV absorbance of 254 nm (Uesugi et al., 1997; Xu et al., 2012). The mobile phase including solvents A and B was maintained at 1 mL min-1 for 10 min. The program was followed by 10% B for 1 min, 20% B for 1 min, 30% B for 30 s, 35% B for 30 s, 40% B for 1 min, 50% B for 4 min, 90% B for 1 min, and 10% B for 1 min.
Quantification of production was based on the peak areas obtained at 290 nm. The conversion percentage of the substrate was determined after integrating substrate and product peaks. Solvents A and B were run at 1 mL min-1 for a 30 min. The gradient of the mobile phase was carried out and followed by 20% B for 5 min, 50% B for 5 min, 70% B for 5 min, 90% B for 5 min, and 10% B for 10 min.
The purification of naringenin derivatives was performed by preparative HPLC (Shimadzu, Tokyo, Japan) with C18 column [YMC-Pack ODS-AQ (150 mm × 20 mm I.D., 10 μm)] connected to a UV detector at 290 nm using a 40 min binary program with 5% B for 5 min, 40% B for 5 min, 40% B for 5 min, 90% B for 5 min, and 10% B for 10 min at a flow rate of 10 mL min-1.
Mass Spectrometry and Nuclear Magnetic Resonance
High-resolution quadruple time-of-flight electrospray ionization-mass spectrometry (HR–QTOF ESI/MS) analysis was carried out with an ACQUITY column coupled an SYNAPT G2-S (UPLC, Waters Corp., Billerica, MA, United States). A reversed-phase column [Acquity BEH C18 2.1 mm × 100 mm (1.7 μm)] was used to separate the samples. ESI/MS detection of the samples: positive ion mode ESI+, acquisition range: 50–1,400 m/z, capillary voltage: 2.5 kV, cone voltage: 30 V, source temperature: 120°C, desolvation gas temp: 600°C, cone gas flow: 20 L h-1, desolvation gas flow: 800 L h-1. The analyses were performed at a flow rate of 0.35 mL min-1 using the same mobile phase with a gradient of 30% B for initial, 90% B for 4 min, 100% B for 3 min, 100% B for 2.5 min, and 37.5% B for 2.5 min. MassLynx software version 4.1 (Waters Corp.) was used for analysis the chromatograms.
The purified products were dried, lyophilized, and recorded on Bruker Biospin 300 MHz spectrometer in DMSO-d6 for proton 1H-NMR.
Antibacterial Activity
Nine Gram-positive bacteria and six Gram-negative bacteria listed in Table 2 were used for testing the antibacterial of naringenin and its derivative. All strains were cultured in LB medium at 37 ± 1°C. We applied the paper disc diffusion method using ampicillin (current antibiotic standard) as an antibacterial positive control for screening the antibacterial agent (Rios et al., 1998). Each inoculates consisted of 106 colony forming units (CFU mL-1) and was spread on MHA plates for bio-assay. Sterile filter paper discs (6 mm in diameter) consisting of 10 μL of 100 mM of compounds dissolved in MeOH were spotted on the agar surface. The plates were incubated at 37 ± 1°C and checked for 36 h.

TABLE 2. Inhibition zone diameter (mm) of naringenin and naringenin 7-sulfate against nine Gram-positive bacteria and six Gram-negative bacteria.
Anticancer Activities
Three cancer cell lines containing A375SM melanoma, MCF-7 breast cancer, AGS gastric cancer, and 267B1 cellosaurus were observed from the Korean Cell Line Bank (KCLB, Seoul, South Korea). Minimum essential medium (MEM) added 10% fetal bovine serum (FBS) (Gibco, Grand Island, NY, United States) was used for culture the cell lines at 37°C in a humidified 5% CO2 incubator. Cells seeded at 2 × 103 cell well-1 in 96-well plates (SPL Life Sciences, Gyeonggi, South Korea) were treated with each compound in serial dilution (400, 200, 100, and 50 μM) for 72 h. Taxol, a current anticancer agent, was also carried out to provide an additional point of comparison. We examined cell growth using an MTT colorimetric assay (Lee et al., 2017).
Statistical Analysis
Values are mean ± standard deviation (SD). All the results were the average of three independent experiments with SD (n = 3). Differences with p-values <0.05 and <0.005 were indicated a considered significant.
Results
Escherichia coli Expression of Recombinant A. thaliana SULTs
To optimize conditions for the expression of recombinant ST At2g03770 from A. thaliana, we induced with various IPTG concentrations (0.1, 0.5, and 1.0 mM) under 20°C in LB liquid medium. The SDS-PAGE analysis of the soluble and insoluble fraction of At2g03770 indicated the highest expression level as determined at 0.1 mM IPTG. The recombinant protein, At2g03770 (37.72 kDa), was overexpressed in E. coli BL21 (DE3) and obtained in the soluble fraction (Supplementary Figure S2).
Construction of CRISPRi System in E. coli
Gene cysH was selected for studying CRISPRi system on the sulfur metabolism of E. coli. The knockdown of cysH might to the increase of the intracellular PAPS pool enhancing production of sulfated substrates. Therefore, two 66 bp complementary offset oligonucleotides containing 30 bp protospacer (target) sequence were designed (Supplementary Table S1) and inserted to pCRISPathBrick, resulted in CRISPRi-1 and CRISPRi-2 systems formation (Table 1). The length of the cysH gene is 735 bp, while the length of the target sequence is 30 bp. To transcriptional interference, cysH1 was designed 56 bp upstream from start codon (from position -76 to -57), after the PAM sequence AGG (from position -58 to -56). At the same time, cysH2 was designed 10 bp downstream of the start codon (from position 40 to 11), after PAM sequence AGG (from position 41 to 43) (Figure 2A). The unique properties of E. coli BL21 genomic of each two protospacer were confirmed via nucleotide BLAST1. The plasmids were assembled and then biotransformation into E. coli DH5α. Colony PCR (cPCR) was performed with cPCR-Rv and cPCR-Fw primers (Supplementary Table S2). The cPCR products were confirmed through 2% agarose gel with 50 bp DNA ladder marker (ELPIS-Biotech. Inc., South Korea). The length of amplifying obtained from clones of pCRISPathBrick is 85 bp, while a 66 bp increase in PCR obtained from clones of CRISPRi-1 and CRISPRi-2 (Supplementary Figure S3). The plasmids CRISPRi-1 and CRISPRi-2 were transformed into wild-type strain, forming production strains S1 and S2, respectively (Table 1).
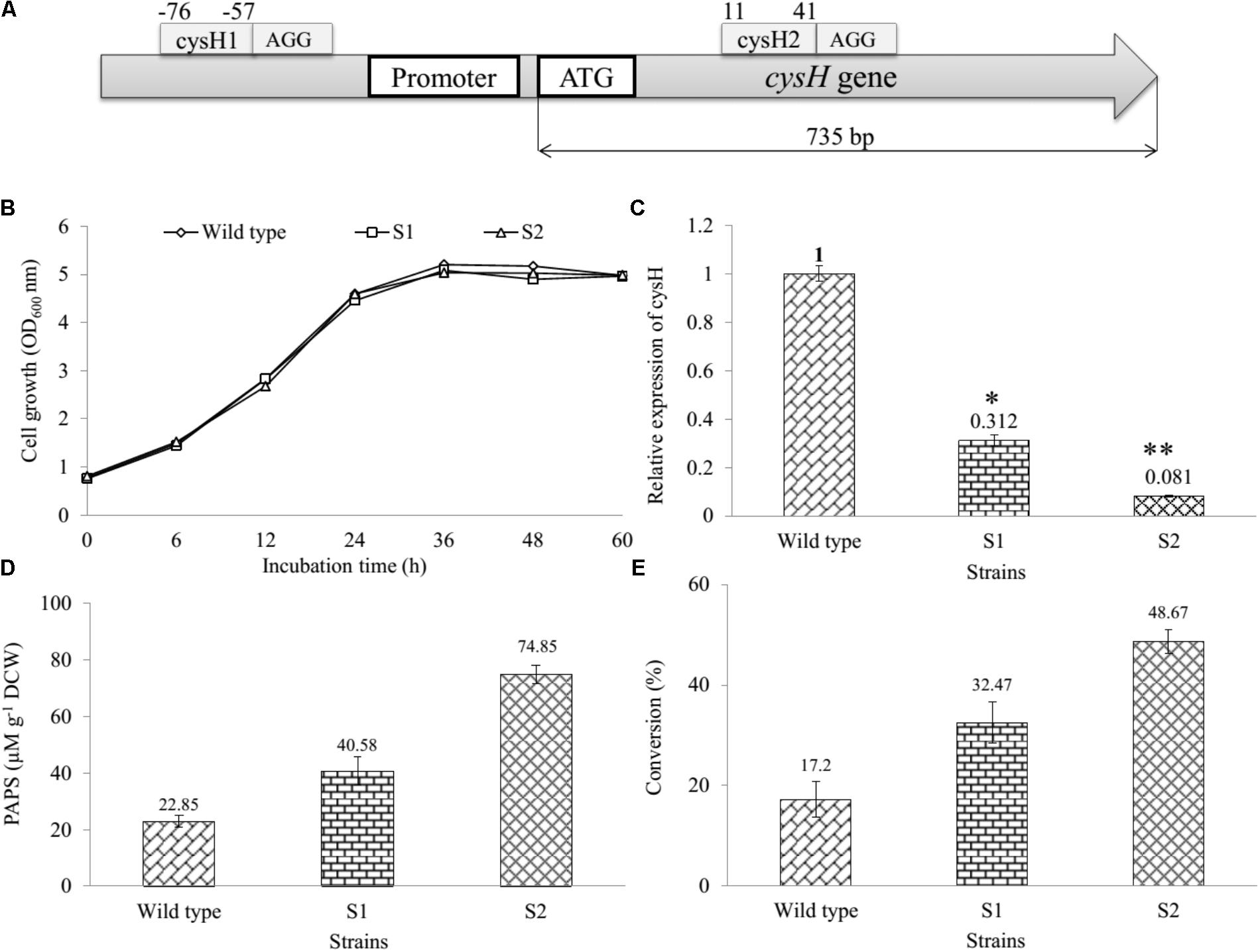
FIGURE 2. Establishment and assessment of CRISPRi in sulfur metabolic pathway of E. coli. (A) The binding at different position on cysH gene using CRISPRi system. Sites selected cysH1 (from position –76 to –57) after the PAM sequence AGG and cysH2 (from position 11 to 44) after PAM sequence AGG. (B) Comparison of cell growth in LB broth medium at OD600, (C) qRT-PCR analysis, (D) concentration of PAPS, and (E) the percentages of bioconversion between recombinant engineered E. coli carried out CRISPRi system targeting cysH gene and wild-type (∗p < 0.05, ∗∗p < 0.005).
Effect of CRISPRi System on Cell Density and Gene Expression
We determined and compared the growth curves of wild-type, S1, and S2 strains by using a spectrophotometer (Thermo Fisher Scientific, United States) via the OD600 nm. The data showed that the S1 and S2 had to resemble with the wild-type on the rate of cell growth (Figure 2B), indicating that cysH gene inhibition by CRISPRi did not affect the cell growth. In addition, the relative qRT-PCR data of knockdown S1 and S2 strains were analyzed through cysH mRNA quantification using 16S rRNA as an endogenous control and wild-type as the reference sample. 16S rRNA, known as a housekeeping gene, is one of the most commonly used endogenous control in E. coli (Zhou et al., 2011). The data showed qRT-PCR cycle threshold values for 16S rRNA gene expressed at almost similar levels in all three samples containing wild-type, S1, and S2 with 23.852 ± 0.371, 22.558 ± 0.031, and 21.732 ± 0.115, respectively. This results demonstrated that the suitability of 16S rRNA gene for the specific case of this experiment (Supplementary Figure S4). Importantly, the value of relative expression of cysH gene transcription in wild-type was 1, while this figure for S1 and S2 were 0.312 and 0.081, respectively (Figure 2C). The result indicated that both the prevent RNA polymerase binding (CRISPRi-1) and elongation (CRISPRi-2) have high efficiency of the reducing transcriptional expression level of cysH.
The Knockdown of cysH Gene Increased PAPS and Naringenin Derivatives
The intracellular PAPS in three strains were extracted using the method described above. All the samples consisting authentic ADP, ATP, and PAPS standard were determined by using UHPLC-PDA coupled HR-QTOF ESI/MS. UHPLC-PDA analysis showed the ADP standard appeared at retention time tR ∼ 0.83 min, while tR of ATP and PAPS standard were 2.71 and 1.27 min. Interestingly, the intracellular PAPS was detected at tR ∼ 1.27 min in the cytosol of wild-type as well as mutant strains (Supplementary Figure S5). These peaks were confirmed by HR-QTOF ESI/MS and the results were shown in Supplementary Table S3 and Supplementary Figures S6, S7. These results suggested that UHPLC-PDA coupled HR-QTOF ESI/MS not only the distinguishing between PAPS from potential interferents ADP and ATP but also the detection of the intracellular PAPS in the cytosol of E. coli. Finally, the intracellular concentration of PAPS was analyzed and shown in Figure 2D. While the figure for S1 and S2 strains went up to 40.58 and 74.85 μM g-1 DCW, the concentration of PAPS for wild-type obtained a 22.85 μM g-1 DCW at the same time. This result demonstrated that cysH is the most required target to inhibit the PAPS consumption.
Moreover, we used the three recombinant strains to produce the sulfated derivative from naringenin. The UFLC-PDA chromatograms of extract from the whole cell of all strains showed a new peak at retention time tR ∼ 18.248 min (P1) in comparison with naringenin standard at tR ∼ 19.979 min under UV absorbance at 290 nm (Supplementary Figure S8). These peaks were further analyzed by HR-QTOF ESI/MS. The found mass of naringenin was ∼273.0780 [M + H]+ m/z+ equivalent to molecular formula C15H13O5 with λmax ∼ 287 nm, for which calculated mass was ∼273.0763 (Supplementary Figure S9A). The found mass of P1 at ∼353.0330 with λmax: 277; 335 nm, corresponding to the exact mass of the mono-sulfate derivative of naringenin with molecular formula C15H12O8S for [M + H]+ m/z+ ∼ 353.0331 (Supplementary Figure S9B). The structural identifies of the product could be verified via NMR in the future experiment. The percentages of bioconversion of naringenin to mono-sulfated naringenin were 17.2% in the wild-type strain, while the figure for both S1 and S2 strains showed an enhancement to 32.47 and 48.67% at the same time (Figure 2E). These results demonstrated that S1 and S2, both mutant strains include the knockdown of the cysH gene were used for the improvement of naringenin derivative. It might be true that S2 could be a good recombinant host system produce derivatives product form naringenin.
Regulating the Concentration of Inorganic Sulfate and Glucose in Medium
For regulation of the concentration MgSO4, we used the M9 minimum media including the various concentrations of MgSO4 (2, 5, 10, 15, and 20 mM) in comparison with the M9 medium without MgSO4. The cell growth and substrate conversion were taken out at 12 h intervals. While the production was noticeably low in the medium without MgSO4, the maximum bioconversion of substrate obtained 98.34% at 48 h with OD600 ∼ 9.94 in M9 medium with 10 mM MgSO4 when 100 μM naringenin was added (Figures 3A,B). Subsequently, the production was carried out by using an M9 medium with 10 mM MgSO4 consisting of the various concentrations of glucose (2, 4, 6, 8, and 10%). The data showed that nearly 100% of 100 μM naringenin was converted to its sulfated derivatives during the addition 2, 4, and 6% glucose at 48 h with OD600 ∼ 9.94, 10.73, and 9.57, respectively (Figures 3C,D). The M9 medium supplementation of 10 mM MgSO4 and 2% glucose were selected for further optimizing the concentration of substrate.
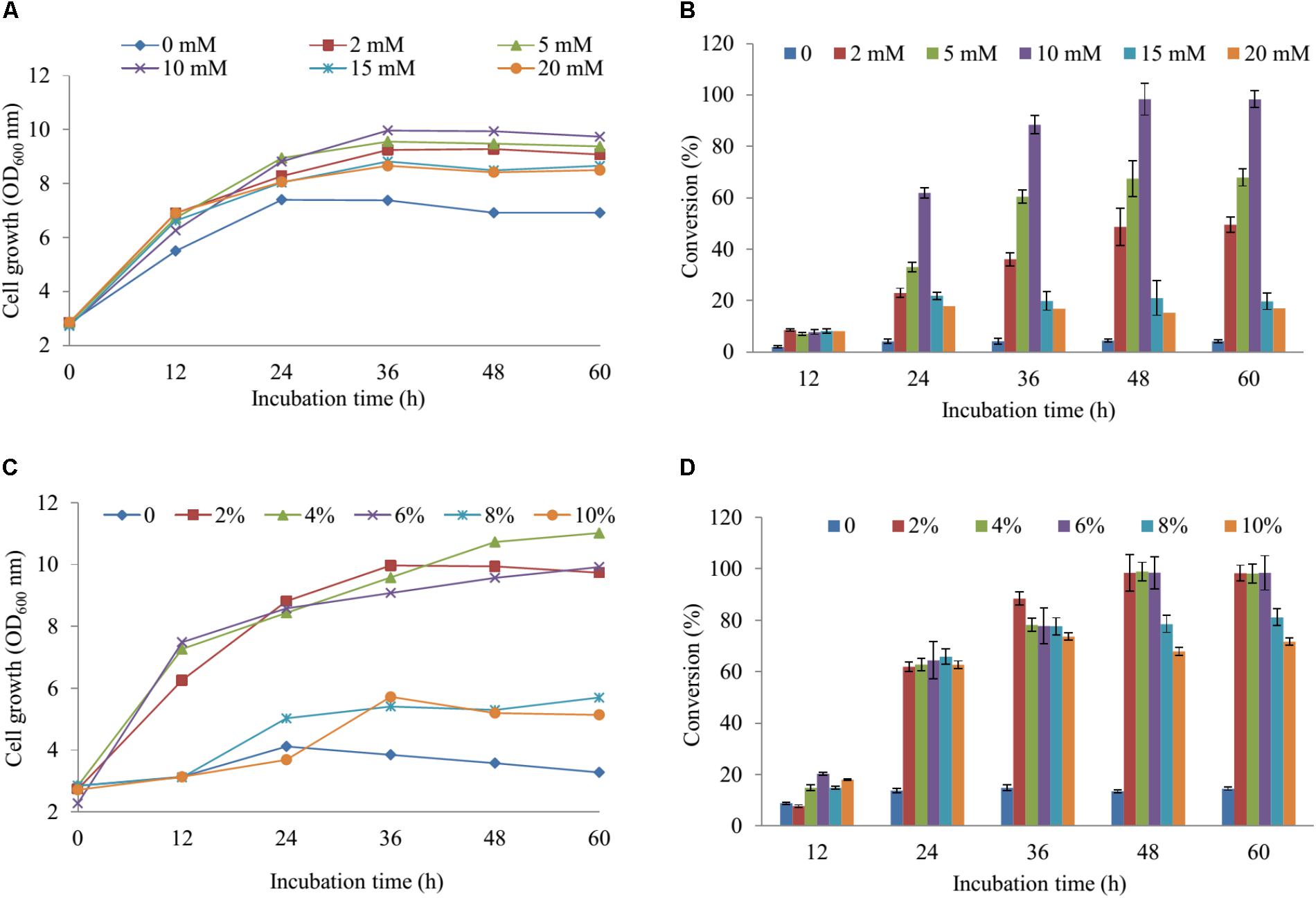
FIGURE 3. The regulation of the concentration MgSO4 and glucose in M9 media. The concentration of effect on cell growth (A) and the production sulfated naringenin (B). The percentages of glucose influent into cell growth (C) and bioconversion of naringenin to its derivative at the various incubation time (D).
Bioconversion With Different Naringenin Concentration and Scale-Up by Fermentation
Five various concentration of naringenin (200, 400, 600, 800, and 1,000 μM) were supplied for biocatalytic reaction system with the S2 strain. The OD600 and conversion rate of naringenin to its derivative were monitored at 12-h intervals. The highest production obtained 189 μM at 48 h with OD600 ∼ 10.24 when 250 μM was fed under M9 medium including 10 mM MgSO4 and 2% glucose (Figures 4A,B).
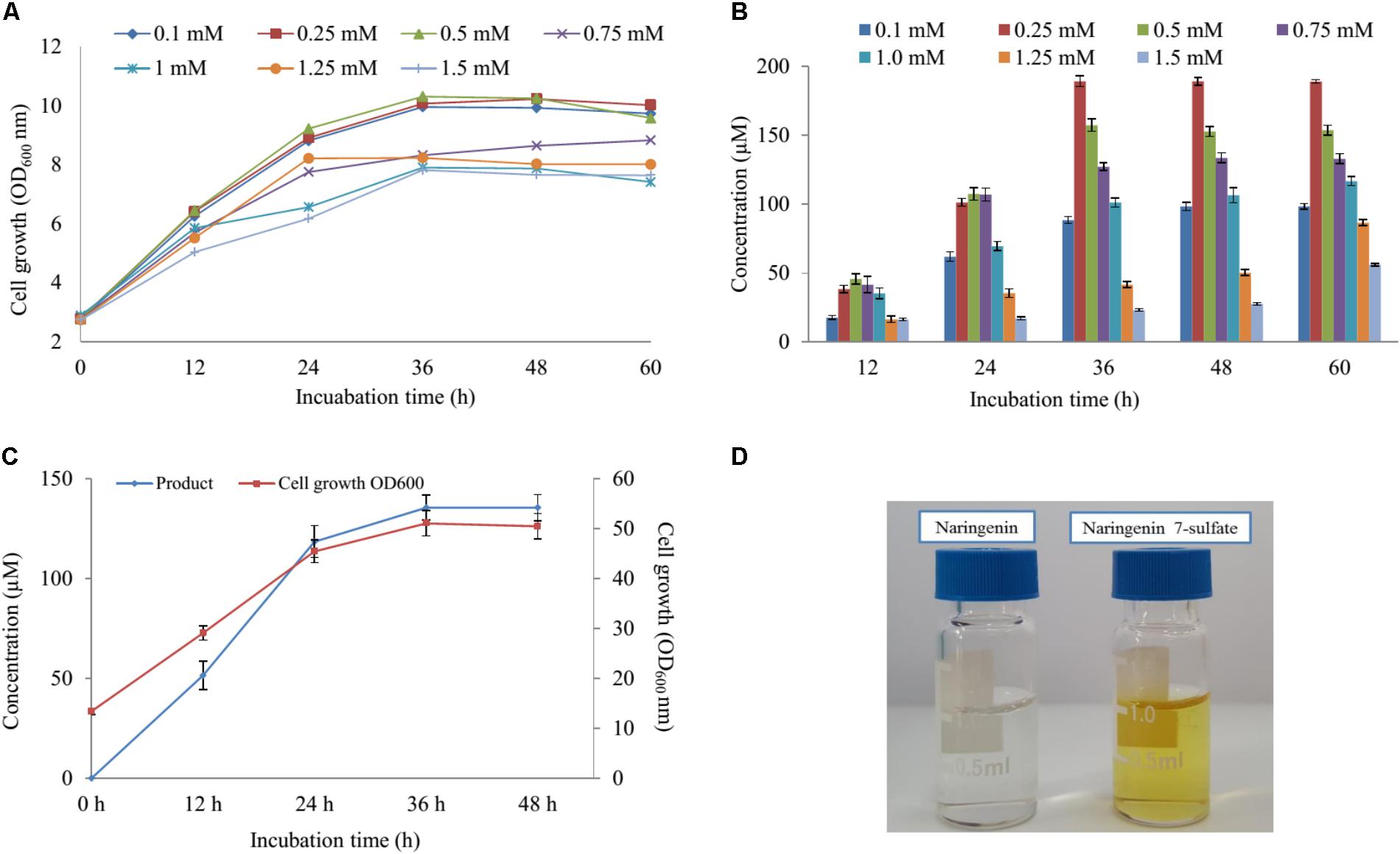
FIGURE 4. The effect of substrate concentration and scale-up production. (A) Cell growth at OD600 nm and (B) the substrate concentration optimization of naringenin in biotransformation. (C) Cell growth at OD600 nm and the scale-up production sulfated naringenin in 3-L fermentation at different time intervals. (D) Comparison of color between substrate and product.
Finally, these optimal conditions were applied for the bioconversion process into the 3-L fermenter (Biotron, South Korea). When OD600 ≥ 6, 0.1 mM IPTG was induced and the temperature decreased to 20°C. After 12 h induction, 250 μM (∼264 mg 3-L) of naringenin was fed to the culture under the pH and temperature were maintained at 7.4 and 30°C, respectively. The samples were taken at 12 h interval and measured by UFLC-PDA. UFLC-PDA analysis revealed that sulfated naringenin was obtained to 135.49 μM (∼143.1 mg 3-L) at 36 h with OD600 ∼ 51.06 (Figure 4C).
Structural Elucidation of Sulfated Naringenin
Previous study exhibited that even though no sulfating activity toward 5-, 3′ and 4′-hydroxyflavone, At2g03770 showed the catalytic activity of 7-hydroxyflavone (Hashiguchi et al., 2013). We re-confirmed the structural compound by 1H-NMR of naringenin standard and purified sulfated product at 300 MHz in DMSO-d6. The 1H-NMR spectrum of sulfated naringenin displayed the absence of OH- group signal at δ = 10.83 ppm for C-7 in comparison with the 1H-NMR spectrum of naringenin (Supplementary Figure S10). Moreover, the 1H-NMR spectrum of this compound exhibited a lower shift at H-6 and H-8 (Supplementary Table S4), indicating that the hydroxyl group at 7 positions of naringenin was substituted by a sulfate group. These data also agreed well with naringenin 7-sulfate obtained in the fungus Cunninghamella elegans NRRL 1392 (Ibrahim, 2000). Based ppm all the results, we could be identified that product was naringenin 7-sulfate. While naringenin was colorless compound, naringenin 7-sulfate was obtained as a yellowish solid (Figure 4D).
Antibacterial and Anticancer of Compounds
Results of disc diffusion assays displayed that naringenin showed only antibacterial activity against Salmonella enterica ATCC 14028 when 10 μL of 100 mM compound was used. In contrast, naringenin 7-sulfate exhibited broad-spectrum antibacterial activity against not only Gram-positive bacteria containing Staphylococcus aureus CCARM 0205, S. aureus CCARM 0204, S. aureus CCARM 3634, Proteus hauseri NBRC 3851, Micrococcus luteus KACC 13377, but also negative–positive bacteria consisting of S. enterica ATCC 14028, Pseudomonas aeruginosa KACC 10232, and Enterobacter cloacae subsp. dissolvens KACC 13002. Noticeably, naringenin 7-sulfate exhibited antibacterial activity against M. luteus KACC 13377 with a zone of inhibition values of 9.5 ± 0.18 mm. Moreover, we detected a zone of inhibition against S. aureus CCARM 0205 (MSSA) by naringenin 7-sulfate and ampicillin seems to share the most similarity with values of 7.0 ± 0.08 and 7.2 ± 0.09 mm. Interestingly, while naringenin and ampicillin did not show antibacterial activity against P. aeruginosa KACC 10232, naringenin 7-sulfate displayed a zone of inhibition values of 7.2 ± 0.12 mm (Table 2 and Supplementary Figure S11). These results indicated that sulfation of naringenin at hydroxyl group of C-7 position could be advantageous for intensifying its antibacterial activity against various bacteria.
Furthermore, the cell viability data showed that naringenin 7-sulfate exhibited good anticancer activities where naringenin did not have anticancer activities against all cell lines. Cell viability of A375SM, MCF-7, AGS, and 267B1 treated 400 μM of naringenin 7-sulfate reduced approximately 48.85, 35.96, 41.78, and 71.20% (p < 0.05), respectively, in comparison with normal test (NT) (Figure 5). These results suggest that naringenin 7-sulfate was relatively less cytotoxic to non-cancer cell than cancer cell lines. In addition, the data showed naringenin 7-sulfate was less cytotoxic than naringenin in the non-cancer cell at 400 μM. As expected, taxol exhibited highly strong anticancer activity at nanomolar ranges of concentration. Taxol also inhibited A375SM and AGS cancer cell lines a little better than 267B1 non-cancer cell line with inhibition of 65.47, 63.40, and 49.90% at 100 nM, respectively (Figure 5). In conclusion, naringenin 7-sulfate could be the more potent anticancer agent with lower cytotoxicity against non-cancer cell than naringenin.
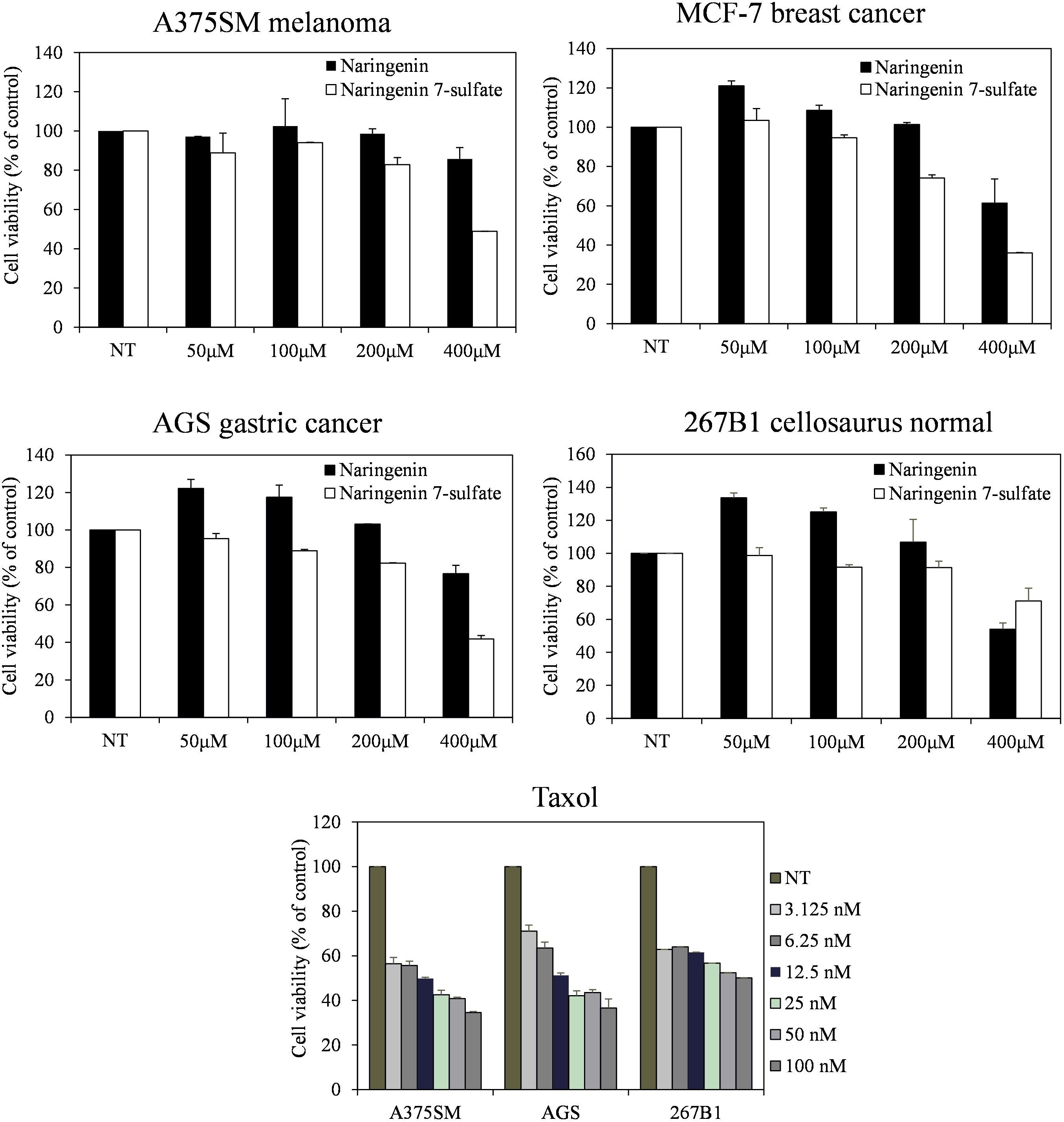
FIGURE 5. Effect of naringenin and naringenin 7-sulfate on the cell viability of three cancer cell lines.
Discussion
Sulfotransferases can be found in various life form from prokaryotes to eukaryotes (Suiko et al., 2017). In a model plant organism, A. thaliana, 18 different STs classified into seven groups are present (Klein and Papenbrock, 2004). Among all the A. thaliana STs, At2g03770 has been exploited as catalysis the sulfation for a variety of flavonoids (Hashiguchi et al., 2013). In this research, we produced 17.2 μM naringenin derivatives via overexpressed At2g03770 in E. coli. Although the yield of substantial amount of At2g03770 product appears in insoluble fraction might be led to the lower harvest of sulfated product (Supplementary Figure S2), the result indicated that heterologous expression STs from A. thaliana could be efficient produce sulfated compounds in whole cell biotransformations. Moreover, metabolic engineering in E. coli also might be applied for the enhanced production of prospective compounds extensively.
In E. coli sulfur metabolism, operon cysUWAsbp codes for a sulfate permease, an ATP binding cassette (ABC)-type transporter (Sekowska et al., 2000), carried the sulfate into the internal cell. Subsequently, ATP sulfurylase coded by operon cysDN, catalyzed an adenylylation of sulfate in the form of APS. After being activated by APS kinase cysC, APS is phosphorylated to PAPS (Hatzios and Bertozzi, 2011). PAPS is the universal sulfate donor in the reaction of sulfation performed by STs on the bacterial metabolite. On another hand, consumption of PAPS in sulfate assimilation pathway start catalyzed with the cysH-encoded PAPS ST. This reaction is continuously reduced for the biosynthesis of essential reduced sulfur metabolites (Figure 1). Therefore, CRISPRi has been introduced into background strain consist of expressed ST At2g03770, targeting strategy to inhibit PAPS consumption and subsequently increase sulfated naringenin. This is the first time the production of sulfated flavonoids has published with the CRISPRi system in E. coli.
CRISPRi recently has applied to the enhanced biosynthesis of flavonoid (Cress et al., 2015) and O-methylated anthocyanin (Cress et al., 2017) through transcription regulation of metabolic pathway in E. coli. Compared to the traditional gene deletion methods, CRISPRi showed the great tool for targeted gene inhibition in microorganisms. Firstly, the construction of CRISPRi plasmid is simple and saving time because dCas9 protein and sgRNA expressed in one vector (Supplementary Figure S1). The efficiency of two different nucleotide sequence targeted on the cysH gene was not the same. The proportion suppression of spacer sequence bound to open reading frame region (cysH2) was 1.34-fold higher than the figure for spacer sequence bound to near promoter (cysH1), at 91.9 and 68.8%, respectively (Figure 2C). The reason behind the variation in gene expression level could be the corresponding to the distance of cysH2 and cysH1 to transcription start site, 10 bp of cysH2 in compared with 56 bp of cysH1 (Figure 2A). Moreover, the regulation of sulfate metabolic pathway by CRISPRi system result in improving PAPS pool without affecting cell growth (Figure 2B). Ultimately, CRISPRi system not only reduces the metabolic burden associated with high-copy overexpression of heterologous ST At2g03770, but also increase production of target molecules. In the whole cell biocatalysis system, production of naringenin derivatives increased by up to 2.83-fold (Figure 2E). This result suggested that S2 strain not only led to the accumulation of intracellular PAPS but also improved the efficiency of sulfation of naringenin.
Recently, the analysis of polar molecules such as ATP, ADP, and PAPS was carried out via liquid chromatography-mass spectrometry (Johnsen et al., 2011; Dowood et al., 2016). In this study, UHPLC-PDA coupled HR-QTOF ESI/MS allowed the detection PAPS, although the quantity of PAPS was lowed in the cytoplasm of E. coli. The method not only is high accuracy but also simple to perform with general solvent and saving time. Moreover, this method was very significant for the separation between PAPS and nucleoside triphosphates as ATP and ADP, because these compounds were the same physical properties (Dowood et al., 2016).
The cultivation of the wild-type and S1, S2 strains was the initial phase of rapid cell growth at 37°C, followed by a growth arrest phase at 20°C, and the biosynthesized compound produced at 28°C. The growth-arrested E. coli induced by using low temperature led to improved the conformational quality and the solubility of STs from A. thaliana and CRISPRi systems (Vera et al., 2007; Cress et al., 2015), which associated with an increased sustained viability and an extended production phase (Rosano and Ceccarelli, 2014). This experiment has a significant impact on the optimal production of the target compound. We decided to the production sulfated naringenin in M9 medium containing MgSO4 and glucose as sulfur and carbon sources, respectively. Almost 100% of conversion rate from a substrate to its derivative was obtained in media consisting of 10 mM MgSO4 and 2–6% glucose, indicating that sulfated compound all accumulated in the extracellular fraction. One possible reason behind this result could be organic anion molecules as sulfated naringenin has been eliminated into extracellular by E. coli multidrug resistance ATP binding cassette transporters (Chang and Roth, 2001). On the other hand, sulfated naringenin including the negative charged led to might not to cross the cell membrane of E. coli. Furthermore, even though the media lacking MgSO4 resulted in the cell growth was low, the production still obtained around 4.42% (Figures 3A,B). The reason could be because of when the growth medium absent inorganic sulfur, E. coli can induce a series of sulfate starvation-inducible genes led to utilize organosulfur compound as a source of sulfur (van der Ploeg et al., 2001). Moreover, the media absence glucose also caused the production was low as well as the cell growth inefficient (Figures 3C, 4D). These results demonstrated that both and glucose are an essential factor produce sulfated naringenin by At2g03770-expressed in E. coli cells. However, the high concentration of (above 15 mM) and glucose (over 6%), as well as more than 0.5 mM of the substrate, could be a reason for inhibition of sulfation product in E. coli whole cell (Figures 3, 4). Finally, the engineered E. coli S2 strain has been applied successfully for the large-scale production of naringenin 7-sulfate, which obtained at 135.49 μM [∼143.1 mg (47.7 mg L-1)] in a 3-L fermenter (Figure 4C). These results indicate that the system is efficient and could be applied for other flavonoids to generate the libraries of molecules with various sulfation approaches.
We also evaluated the antibacterial activity of naringenin and its derivative. Five Gram-positive bacteria S. aureus CCARM 0205, S. aureus CCARM 0204, S. aureus CCARM 3634, P. hauseri NBRC 3851, M. luteus KACC 13377 and three Gram-negative bacteria S. enterica, P. aeruginosa, E. cloacae were sensitive with naringenin 7-sulfate. This result indicated that negatively charged sulfate group might be improved to naringenin for antibacterial activity, however, the mechanism of this compound against bacterial pathogens have been not reported. In addition, naringenin derivative exhibited the most potential anticancer activity against three tested cancer cell lines. The compound showed the similarity in features between its and taxol the less cytotoxic to non-cancer cell line in comparison with cancer cell lines (Figure 5). This is the first report of the activity of naringenin 7-sulfate against A375SM, MCF-7 and AGS cancer cell lines. The previous research has shown that the substituent at the C-7 position of naringenin containing thiophenecarboxylate, phenyl carbonate, isobutyrate, methyl benzoate, and allyloxy inhibited effects on HCT116 human colon cancer cell line via block G1 cell cycle progression by interaction with cyclin-dependent kinase 2 (CDK2) (Yoon et al., 2013). This possible mechanism behind the anticancer activity of naringenin 7-sulfate against three tested cancer cell lines, however, the exact mechanisms of action of this compound must confirm in further studies.
In summary, we targeted to improving production of sulfated flavonoids in engineered E. coli. This is the first report on CRISPRi mediated inhibition in the sulfur metabolism of E. coli. Repression of key reduced sulfur metabolite enzyme cysH by over 91%, causing increase intracellular PAPS accumulation and enhancement of naringenin 7-sulfate over 3.28- and 2.83-fold compared to control, respectively. Further media culture optimization led to obtained at 135.49 μM [∼143.1 mg (47.7 mg L-1)] in a 3-L fermenter. This engineered E. coli opened prospects for the biosynthesis of the sulfated flavonoids. In addition, naringenin 7-sulfate exhibited antibacterial activity against both Gram-positive and Gram-negative bacteria. This compound could be also used as a cancer drug when shown to anticancer activity against A375SM melanoma, MCF-7 breast, and AGS gastric cancer cell lines.
Author Contributions
LC designed, performed the majority of the experiment work, analyzed data, and wrote the manuscript. DD and TY helped in analyzing data. HS and HJ did the majority of anticancer activities. JS and LC were responsible for the original concept and supervised the work. All authors read and approved the final manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (NRF-2017R1A2A2A05000939).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01671/full#supplementary-material
Footnotes
References
Barron, D., Varin, L., Ibrahim, R. K., Harvorne, J. B., and Williams, C. A. (1998). Sulphated flavonoids-an update. Phytochemistry 27, 2375–2395. doi: 10.1016/0031-9422(88)87003-1
Bertomeu, T., Coulombe-Huntington, J., Chatr-Aryamontri, A., Bourdages, K., Coyaud, E., Raught, B., et al. (2017). A high resolution genome-wide CRISPR/Cas9 viability screen reveals structural features and contextual diversity of the human cell-essential proteome. Mol. Cell. Biol. 38, e302–e317. doi: 10.1128/MCB.00302-17
Bikard, D., Jiang, W., Samai, P., Hochschild, A., Zhang, F., and Marraffini, L. A. (2013). Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 41, 7429–7437. doi: 10.1093/nar/gkt520
Buer, C. S., Imin, N., and Djordjevic, M. A. (2010). Flavonoids: new roles for old molecules. J. Integr. Plant Biol. 52, 98–111. doi: 10.1111/j.1744-7909.2010.00905.x
Bugianesi, R., Catasta, G., Spigno, P., D’Uva, A., and Maiani, G. (2002). Naringenin from cooked tomato paste is bioavailable in men. J. Nutr. 132, 3349–3352. doi: 10.1093/jn/132.11.3349
Chang, G., and Roth, C. B. (2001). Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 293, 1793–1800. doi: 10.1126/science.293.5536.1793
Chen, B. H., Wang, C. C., Hou, Y. H., Mao, Y. C., and Yang, Y. S. (2015). Mechanism of sulfotransferase pharmacogenetics in altered xenobiotic metabolism. Expert Opin. Drug Metab. Toxicol. 11, 1053–1071. doi: 10.1517/17425255.2015.1045486
Chu, L. L., Pandey, R. P., Jung, N., Jung, H. J., Kim, E. H., and Sohng, J. K. (2016). Hydroxylation of diverse flavonoids by CYP450 BM3 variants: biosynthesis of eriodictyol from naringenin in whole cells and its biological activities. Microb. Cell Fact. 15:135. doi: 10.1186/s12934-016-0533-4
Cress, B. F., Leitz, Q. D., Kim, D. C., Amore, T. D., Suzuki, J. Y., Linhardt, R. J., et al. (2017). CRISPRi-mediated metabolic engineering of E. coli for O-methylated anthocyanin production. Microb. Cell Fact. 16:10. doi: 10.1186/s12934-016-0623-3
Cress, B. F., Toparlak,Ö. D., Guleria, S., Lebovich, M., Stieglitz, J. T., Englaender, J. A., et al. (2015). CRISPathBrick: modular combinatorial assembly of Type II-A CRISPR arrays for dCas9-mediated multiplex transcriptional repression in E. coli. ACS Synth. Biol. 4, 987–1000. doi: 10.1021/acssynbio.5b00012
Dagert, M., and Ehrlich, S. D. (1979). Prolonged incubation in calcium chloride improves the competence of Escherichia coli cells. Gene 6, 23–28. doi: 10.1016/0378-1119(79)90082-9
De Bruyn, F., Maertens, J., Beauprez, J., Soetaert, W., and De Mey, M. (2015). Biotechnological advances in UDP-sugar based glycosylation of small molecules. Biotechnol. Adv. 33, 288–302. doi: 10.1016/j.biotechadv.2015.02.005
Dowood, R. K., Adusumalli, R., Tykesson, E., Johnsen, E., Lundanes, E., Prydz, K., et al. (2016). Determination of 3′-phosphoadenosine-5′-phosphosulfate in cells and Golgi fractions using hydrophilic interaction liquid chromatography-mass spectrometry. J. Chromatogr. A 1470, 70–75. doi: 10.1016/j.chroma.2016.10.001
Fowler, Z. L., Gikandi, W. W., and Koffas, M. A. (2009). Increased malonyl coenzyme a biosynthesis by tuning the Escherichia coli metabolic network and its application to flavanone production. Appl. Environ. Microbiol. 75, 5831–5839. doi: 10.1128/AEM.00270-09
Gandhi, S., Piacentino, M. L., Vieceli, F. M., and Bronner, M. E. (2017). Optimization of CRISPR/Cas9 genome editing for loss-of-function in the early chick embryo. Dev. Biol. 432, 86–97. doi: 10.1016/j.ydbio.2017.08.036
Gao, C., Wang, S., Hu, G., Guo, L., Chen, X., Xu, P., et al. (2017). Engineering Escherichia coli for malate production by integrating modular pathway characterization with CRISPRi-guided multiplexed metabolic tuning. Biotechnol. Bioeng. 115, 661–672. doi: 10.1002/bit.26486
Gattuso, G., Barreca, D., Gargiulli, C., Leuzzi, U., and Caristi, C. (2007). Flavonoid composition of citrus juices. Molecules 12, 1641–1673. doi: 10.3390/12081641
Geyer, J., Bakhaus, K., Bernhardt, R., Blaschka, C., Dezhkam, Y., Fietz, D., et al. (2017). The role of sulfated steroid hormones in reproductive processes. J. Steroid Biochem. Mol. Biol. 172, 207–221. doi: 10.1016/j.jsbmb.2016.07.002
Gidda, S. K., and Varin, L. (2006). Biochemical and molecular characterization of flavonoid 7-sulfotransferase from Arabidopsis thaliana. Plant Physiol. Biochem. 44, 628–636. doi: 10.1016/j.plaphy.2006.10.004
Hashiguchi, T., Sakakibara, Y., Hara, Y., Shimohira, T., Kurogi, K., Akashi, R., et al. (2013). Identification and characterization of a novel kaempferol sulfotransferase from Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 434, 829–835. doi: 10.1016/j.bbrc.2013.04.022
Hatzios, S. K., and Bertozzi, C. R. (2011). The regulation of sulfur metabolism in Mycobacterium tuberculosis. PLOS Pathog. 7:e1002036. doi: 10.1371/journal.ppat.1002036
Hossain, M. K., Dayem, A. A., Han, J., Yin, Y., Kim, K., Saha, S. K., et al. (2016). Molecular mechanisms of the anti-obesity and anti-diabetic properties of flavonoids. Int. J. Mol. Sci. 17:569. doi: 10.3390/ijms17040569
Hossain, M. M., Moriizumi, Y., Tanaka, S., Kimura, M., and Kakuta, Y. (2011). Molecular cloning, expression, and functional analysis of a predicted sulfotransferase STF9 from Mycobacterium avium. Mol. Cell. Biochem. 350, 155–162. doi: 10.1007/s11010-010-0693-1
Huang, H., Zheng, G., Jiang, W., Hu, H., and Lu, Y. (2015). One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim. Biophys. Sin. 47, 231–243. doi: 10.1093/abbs/gmv007
Ibrahim, A. R. (2000). Sulfation of naringenin by Cunninghamella elegans. Phytochemistry 53, 209–212. doi: 10.1016/S0031-9422(99)00487-2
Jadeja, R. N., and Devkar, R. V. (2014). “Polyphenols in chronic diseases and their mechanisms of action,” in Polyphenols in Human Health and Disease, Vol. 1, ed. R. R. Watson (Cambridge: Academic Press), 615–623.
Johnsen, E., Wilson, S. R., Odsbu, I., Krapp, A., Malerod, H., Skarstad, K., et al. (2011). Hydrophilic interaction chromatography of nucleoside triphosphates with temperature as a separation parameter. J. Chromatogr. A 1218, 5981–5986. doi: 10.1016/j.chroma.2011.01.066
Khan, A. A., El-Sayed, A., Akbar, A., Mangravita-Novo, A., Bibi, S., Afzal, Z., et al. (2017). A highly efficient ligation-independent cloning system for CRISPR/Cas9 based genome editing in plants. Plant Methods 13:86. doi: 10.1186/s13007-017-0236-9
Kim, S. Y., Lee, H. R., Park, K. S., Kim, B. G., and Ahn, J. H. (2015). Metabolic engineering of Escherichia coli for the biosynthesis of flavonoid-O-glucuronides and flavonoid-O-galactoside. Appl. Microbiol. Biotechnol. 99, 2233–2242. doi: 10.1007/s00253-014-6282-6
Klein, M., and Papenbrock, J. (2004). The multi-protein family of Arabidopsis sulphotransferases and their relatives in other plant species. J. Exp. Bot. 55, 1809–1820. doi: 10.1093/jxb/erh183
Lee, S., Kwon, M. C., Jang, J. P., Sohng, J. K., and Jung, H. J. (2017). The ginsenoside metabolite compound K inhibits growth, migration and stemness of glioblastoma cells. Int. J. Oncol. 51, 414–424. doi: 10.3892/ijo.2017.4054
Liang, J. L., Guo, L. Q., Lin, J. F., He, Z. Q., Cai, F. J., and Chen, J. F. (2016). A novel process for obtaining pinosylvin using combinatorial bioengineering in Escherichia coli. World J. Microbiol. Biotechnol. 32:102. doi: 10.1007/s11274-016-2062-z
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Marsolais, F., Boyd, J., Paredes, Y., Schinas, A. M., Garcia, M., Elzein, S., et al. (2007). Molecular and biochemical characterization of two brassinosteroid sulfotransferases from Arabidopsis, AtST4a (At2g14920) and AtST1 (At2g03760). Planta 225, 1233–1244. doi: 10.1007/s00425-006-0413-y
Nishikawa, M., Masuyama, Y., Nunome, M., Yasuda, K., Sakaki, T., and Ikushiro, S. (2017). Whole-cell-dependent biosynthesis of sulfo-conjugate using human sulfotransferase expressing budding yeast. Appl. Microbiol. Biotechnol. 102, 723–732. doi: 10.1007/s00253-017-8621-x
Pandey, R. P., Parajuli, P., Chu, L. L., Kim, S. Y., and Sohng, J. K. (2016). Biosynthesis of a novel fisetin glycoside from engineered Escherichia coli. J. Ind. Eng. Chem. 43, 13–19. doi: 10.1016/j.jiec.2016.07.054
Paul, P., Suwan, J., Liu, J., Dordick, J. S., and Linhardt, R. J. (2012). Recent advances in sulfotransferase enzyme activity assays. Anal. Bioanal. Chem. 403, 1491–1500. doi: 10.1007/s00216-012-5944-4
Rios, J. L., Recio, M. C., and Villar, A. (1998). Screening methods for natural products with antimicrobial activity: a review of the literature. J. Ethnopharmacol. 23, 127–149. doi: 10.1016/0378-8741(88)90001-3
Rosano, G. L., and Ceccarelli, E. A. (2014). Recombinant protein expression in Escherichia coli: advances and challenges. Front. Microbiol. 5:172. doi: 10.3389/fmicb.2014.00172
Sekowska, A., Kung, H. F., and Danchin, A. (2000). Sulfur metabolism in Escherichia coli and related bacteria: facts and fiction. J. Mol. Microbiol. Biotechnol. 2, 145–177.
Shimohira, T., Kurogi, K., Hashiguchi, T., Liu, M. C., Suiko, M., and Sakakibara, Y. (2017). Regioselective production of sulfated polyphenols using human cytosolic sulfotransferase-expressing Escherichia coli cells. J. Biosci. Bioeng. 124, 84–90. doi: 10.1016/j.jbiosc.2017.02.006
Suiko, M., Kurogi, K., Hashiguchi, T., Sakakibara, Y., and Liu, M. C. (2017). Updated perspectives on the cytosolic sulfotransferases (SULTs) and SULT-mediated sulfation. Biosci. Biotechnol. Biochem. 81, 63–72. doi: 10.1080/09168451.2016.1222266
Totta, P., Acconcia, F., Virgili, F., Cassidy, A., Weinberg, P. D., Rimbach, G., et al. (2005). Daidzein-sulfate metabolites affect transcriptional and antiproliferative activities of estrogen receptor-beta in cultured human cancer cells. J. Nutr. 135, 2687–2693. doi: 10.1093/jn/135.11.2687
Uesugi, T., Sano, K., Uesawa, Y., Ikegami, Y., and Mohri, K. (1997). Ion-pair reversed-phase high-performance liquid chromatography of adenine nucleotides and nucleoside using triethylamine as a counterion. J. Chromatogr. B Biomed. Sci. Appl. 703, 63–74. doi: 10.1016/S0378-4347(97)00430-1
van der Ploeg, J. R., Eichhorn, E., and Leisinger, T. (2001). Sulfonate-sulfur metabolism and its regulation in Escherichia coli. Arch. Microbiol. 176, 1–8. doi: 10.1007/s002030100298
Vera, A., Gonzalez-Montalban, N., Aris, A., and Villaverde, A. (2007). The conformational quality of insoluble recombinant proteins is enhanced at low growth temperatures. Biotechnol. Bioeng. 96, 1101–1106. doi: 10.1002/bit.21218
Wang, N., Ren, D., Deng, S., and Yang, X. (2014). Differential effects of baicalein and its sulfated derivatives in inhibiting proliferation of human breast cancer MCF-7 cells. Chem. Biol. Interact. 221, 99–108. doi: 10.1016/j.cbi.2014.08.003
Wang, S., Chen, H., Tang, X., Zhang, H., Chen, W., and Chen, Y. Q. (2017). Molecular tools for gene manipulation in filamentous fungi. Appl. Microbiol. Biotechnol. 101, 8063–8075. doi: 10.1007/s00253-017-8486-z
Wong, C. C., Meinl, W., Glatt, H. R., Barron, D., Stalmach, A., Steiling, H., et al. (2010). In vitro and in vivo conjugation of dietary hydroxycinnamic acids by UDP-glucuronosyltransferases and sulfotransferases in humans. J. Nutr. Biochem. 21, 1060–1068. doi: 10.1016/j.jnutbio.2009.09.001
Wu, J., Du, G., Chen, J., and Zhou, J. (2015). Enhancing flavonoid production by systematically tuning the central metabolic pathways based on a CRISPR interference system in Escherichia coli. Sci. Rep. 5:13477. doi: 10.1038/srep13477
Xu, J., Chen, Y., Li, L., Li, Z., Wang, C., Zhou, T., et al. (2012). An improved HPLC method for the quantitation of 3’-phosphoadenosine 5’-phosphate (PAP) to assay sulfotransferase enzyme activity in HepG2 cells. J. Pharm. Biomed. Anal. 62, 182–186. doi: 10.1016/j.jpba.2011.12.015
Yan, Y., Chemler, J., Huang, L., Martens, S., and Koffas, M. A. (2005). Metabolic engineering of anthocyanin biosynthesis in Escherichia coli. Appl. Environ. Microbiol. 71, 3617–3623. doi: 10.1128/AEM.71.7.3617-3623.2005
Yi, L., Dratter, J., Wang, C., Tunge, J. A., and Desaire, H. (2006). Identification of sulfation sites of metabolites and prediction of the compounds’ biological effects. Anal. Bioanal. Chem. 386, 666–674. doi: 10.1007/s00216-006-0495-1
Yoon, H., Kim, T. W., Shin, S. Y., Park, M. J., Yong, Y., Kim, D. W., et al. (2013). Design, synthesis and inhibitory activities of naringenin derivatives on human colon cancer cells. Bioorg. Med. Chem. Lett. 23, 232–238. doi: 10.1016/j.bmcl.2012.10.130
Zhang, H., Zhang, M., Yu, L., Zhao, Y., He, N., and Yang, X. (2012). Antitumor activities of quercetin and quercetin-5’,8-disulfonate in human colon and breast cancer cell lines. Food Chem. Toxicol. 50, 1589–1599. doi: 10.1016/j.fct.2012.01.025
Keywords: sulfotransferase, CRISPRi, E. coli, PAPS, metabolic engineering
Citation: Chu LL, Dhakal D, Shin HJ, Jung HJ, Yamaguchi T and Sohng JK (2018) Metabolic Engineering of Escherichia coli for Enhanced Production of Naringenin 7-Sulfate and Its Biological Activities. Front. Microbiol. 9:1671. doi: 10.3389/fmicb.2018.01671
Received: 29 March 2018; Accepted: 04 July 2018;
Published: 27 July 2018.
Edited by:
Biswarup Mukhopadhyay, Virginia Tech, United StatesReviewed by:
Blaine Pfeifer, University at Buffalo, United StatesAlvaro R. Lara, Universidad Autónoma Metropolitana, Mexico
Copyright © 2018 Chu, Dhakal, Shin, Jung, Yamaguchi and Sohng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tokutaro Yamaguchi, eWFtYWd1Y2hpQHN1bm1vb24uYWMua3I= Jae K. Sohng, c29obmdAc3VubW9vbi5hYy5rcg==