 Guangyun Tan
Guangyun Tan Hongxiao Song1
Hongxiao Song1- 1Department of Immunology, Institute of Translational Medicine, The First Hospital of Jilin University, Changchun, China
- 2Department of Microbiology, Immunology and Molecular Genetics, University of California, Los Angeles, Los Angeles, CA, United States
- 3Center of System Medicine, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 4Suzhou Institute of Systems Medicine, Suzhou, China
Chronic hepatitis B virus (HBV) infection imposes a severe burden on global public health. Currently, there are no curative therapies for millions of chronic HBV-infected patients (Lok et al., 2017). Interferon (IFN; including pegylated IFN) is an approved anti-HBV drug that not only exerts direct antiviral activity, but also augments immunity against HBV infection. Through a systematic review of the literature, here we summarize and present recent progress in research regarding the interactions between IFN and HBV as well as dissect the antiviral mechanisms of IFN. We focus on inhibition of HBV replication by IFN-stimulated genes (ISGs) as well as inhibition of IFN signaling by HBV and viral proteins. Finally, we briefly discuss current IFN-based HBV treatment strategies. This review may help to better understand the mechanisms involved in the therapeutic action of IFN as well as the crosstalk between IFN and HBV, and facilitate the development of both direct-acting and immunology-based new HBV drugs.
Introduction
More than 50 years have passed since the identification of human hepatitis B virions, initially named “Dane particles,” in the late 1960s; and hepatitis B virus (HBV) infection remains a serious global health problem (Hirschman et al., 1969; Cossart and Field, 1970; Dane et al., 1970; Zanen-Lim, 1976; Glebe, 2007). Three-quarters of the world’s population live in HBV endemic regions, and one-third of them have been exposed to HBV. Despite the implementation of HBV vaccination for nearly 30 years, 240 million of 2 billion HBV-infected people worldwide have become chronically infected. Approximately 39 million chronic HBV infections occur in the region of Southeast Asia (Childs et al., 2018), and there are an estimated 100 million HBV surface antigen (HBsAg)-positive people in both China and Africa who are at high risk for developing liver cirrhosis and hepatocellular carcinoma (HCC) (El-Serag and Rudolph, 2007; Levrero and Zucman-Rossi, 2016). The World Health Organization estimates that there were 1.34 million HBV infection-related deaths in 2015, which is comparable to the annual deaths caused by tuberculosis and is higher than HIV-related mortality (Breakwell et al., 2017).
Interferons (IFNs) are a group of signaling proteins made and released by host cells in response to various pathogens, including bacteria, viruses, and parasites (De Andrea et al., 2002; Lee and Baldridge, 2017). Based on the receptor type to which they bind, human IFNs are classified into type I, II, and III (Isaacs and Lindenmann, 1957; Wheelock, 1965; Sheppard et al., 2003; Vilcek, 2006; Table 1). IFN-α and pegylated IFN-α have been approved for the treatment of chronic hepatitis B (De Andrea et al., 2002; Liang et al., 2015). In addition, IFN-α has been reported to inhibit HBV replication in in vitro systems (Wieland et al., 2003; Pollicino et al., 2013; Lin and Young, 2014). HBV infection can activate the innate immune response in the liver and trigger the local production of type I or type III IFN (Wieland et al., 2004; Shlomai et al., 2014b; Sato et al., 2015). However, it also has been reported that the host’s response to HBV infection does not induce production of either type I or III IFN (Cheng et al., 2017).
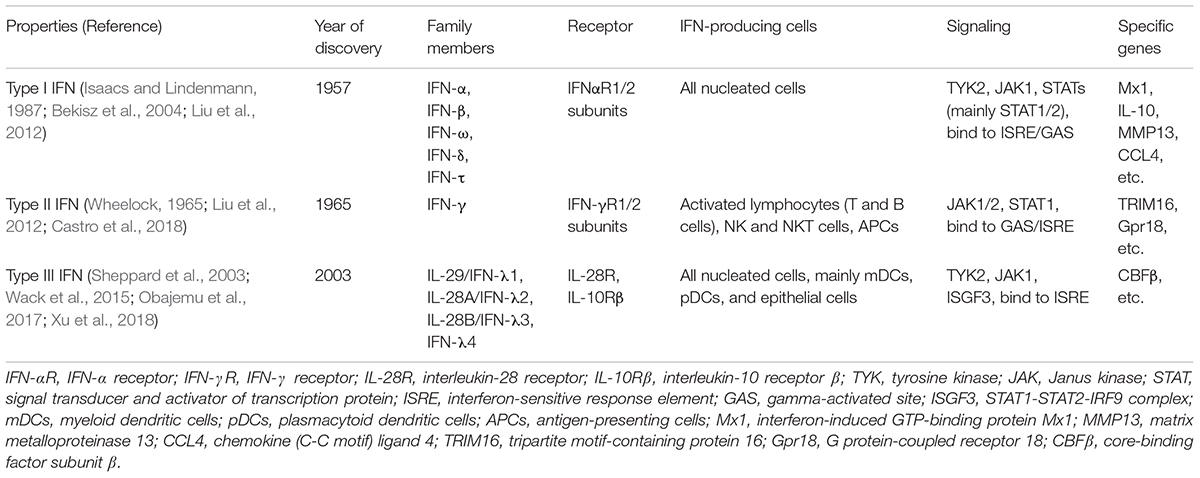
TABLE 1. Current classification of human interferons.
Many IFN-stimulated genes (ISGs) have been shown to inhibit HBV infection and replication through different pathways, and many studies have tried to understand why IFN signaling is weak in an HBV-infected liver. In this review, we discuss the crosstalk between IFN and HBV based on the results generated in our lab and those published by others, which may shed new light on the interaction between IFN and HBV as well as provide insight into new HBV drug discovery and development. This review consists of three sections: (1) the HBV life cycle and the classical IFN induction pathway in response to viral infection; (2) a systematic discussion of the crosstalk between IFN and HBV, with a focus on two-way inhibition: the inhibition of HBV infection and replication by ISGs as well as the inhibition of IFN signaling by HBV and HBV proteins; and (3) a brief discussion of the current IFN-based HBV treatment strategies.
HBV Genome Organization and Life Cycle
Hepatitis B virus belongs to the Hepadnaviridae family and contains a 3.2-kb partially double-stranded DNA (Evans and Seeger, 2007). Once entered into blood circulation, HBV binds to its receptor, currently known as sodium taurocholate co-transporting polypeptide (NTCP), a multiple transmembrane transporter predominantly expressed in hepatic cells (Yan et al., 2012), resulting in viral entry. In the cytoplasm, the virus is caught by the endosomes. In response to endosome maturation, the viral envelope fuses with the endosome membrane and releases the free nucleocapsid into the cytoplasm (Hayes et al., 2016). The nucleocapsid is translocated to the nucleus where relaxed circular DNA is released. And under the help of cellular replicative machinery (Levrero et al., 2009; Koniger et al., 2014), the relaxed circular DNA is converted into a covalently closed circular DNA (cccDNA), which further interacts with histone proteins to form a minichromosome. HBV synthesizes the capsid in the cytoplasm. It appears that cccDNA in infected cells is difficult to eliminate (Tong and Revill, 2016).
The HBV genome contains four overlapping open reading frames (S, C, P, and X) (Figure 1). HBV cccDNA functions as a transcription template, encoding four major viral RNA species: pregenomic RNA (pgRNA, 3.5 kb), preS1 HBs RNA (2.4 kb), preS2/S HBs RNA (2.1 kb), and HBV X protein RNA (HBx, 0.7 kb) (Figure 1; Tuttleman et al., 1986; Newbold et al., 1995). The viral RNA transcription is initiated by activating four promoters (the core, pre-S1, pre-S2/S, and X promoters) with host-specific transcriptional factors such as hepatocyte nuclear factors (NFs) and epigenetic modifications (Pollicino et al., 2006; Kim et al., 2016). The HBV genome also contains two enhancer sites (En I and En II) for upregulation of transcription (Hu and Siddiqui, 1991). Viral mRNAs are then transferred to the cytoplasm where they function as templates either for the synthesis of viral proteins or for the synthesis of minus-strand DNA (reverse transcription) (Shlomai et al., 2014a). The HBV mRNAs encode seven proteins depending on the locations of the initiation codon in the RNA transcripts: preC mRNA for HBV e antigen (HBeAg) (3.5 kb), pgRNA for HBV c antigen (HBcAg) and Pol (3.5 kb), preS1 for large S (2.4 kb), preS2/S mRNAs for middle and small surface proteins (MS and HBsAg, 2.1 kb), and HBx mRNA for HBxAg (0.7 kb) (Figure 1; Liang, 2009). Once viral protein synthesis reaches a certain level in the cytoplasm, pgRNA is selectively packaged into the “HBcAg house” along with Pol protein, which form the immature nucleocapsid, and then pgRNA is reversely transcribed to the minus-strand DNA catalyzed by the reverse transcriptase domain of Pol. A variable length of plus-strand HBV DNA is synthesized using the minus strand as the template to generate the HBV genome. The mature HBV capsid is either enveloped with HBsAg and released from hepatocytes or recycled to the nucleus for amplification of the cccDNA pool (Figure 2; Rehermann and Nascimbeni, 2005).
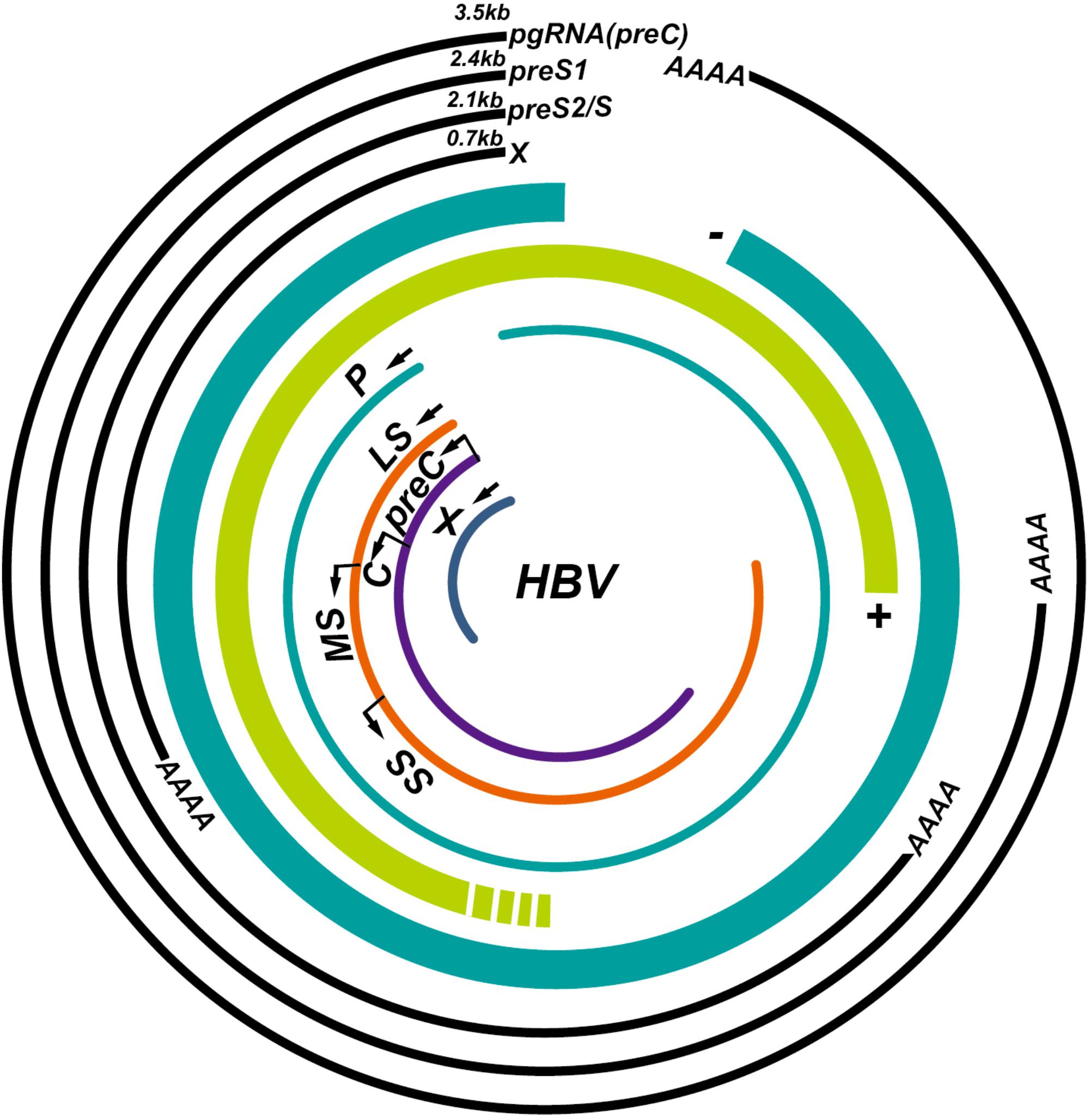
FIGURE 1. Illustration of the hepatitis B virus (HBV) genome and its overlapping open reading frames and transcripts. HBV is a partially double-stranded DNA virus that transcribes four major transcripts: pgRNA (3.5 kb), preS1 (2.4 kb), preS2/S (2.1 kb), and X (0.7 kb) and synthesizes seven proteins: P, polymerase; LS, large S; MS, middle S; SS, small S; preC, pre-Core; C, core; X, HBx.
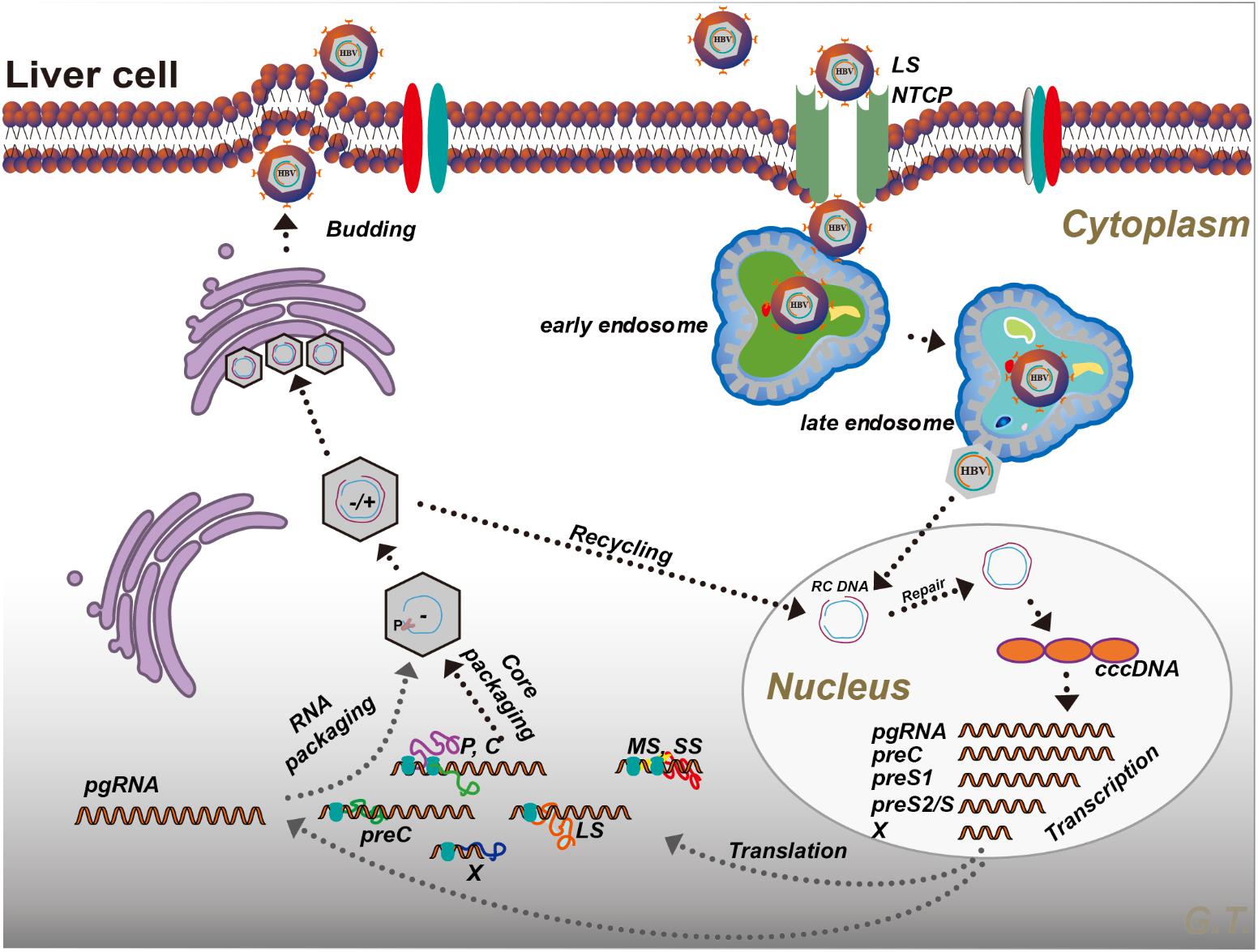
FIGURE 2. Hepatitis B virus life cycle. Please see the text for details. P, polymerase; LS, large S; MS, middle S; SS, small S; preC, pre-Core; C, core; X, HBx; RC DNA, relaxed circular DNA; NTCP, sodium taurocholate co-transporting polypeptide; cccDNA, covalently closed circular DNA.
Classical IFN and ISG Induction Pathways in Response to Viral Infection
In response to viral infection, IFN production can be efficiently induced in many types of cells by the activation of pattern-recognition receptors. Activation of type I IFN family genes constitutes one of the earliest transcriptional responses, perhaps representing the most important innate immune response to viral infection. Most nucleated vertebrate cells are able to both produce and respond to type I IFN. The detection of intracellular microbial DNA and RNA in macrophages and/or infected cells is critical to elicit an appropriate innate immune response. During viral infection, virus-derived nucleic acids (both RNA and DNA) are mainly sensed by certain cytosolic pattern-recognition receptors. For instance, viral RNA is recognized by retinoic acid inducible gene I (RIG-I)-like receptors, including RIG-I (Pichlmair et al., 2006; Myong et al., 2009) and melanoma differentiation associated protein 5 (Takeuchi and Akira, 2008, 2009), leading to its association with the mitochondrial antiviral-signaling protein (MAVS). DNA is sensed by DEAD-box protein (DDX) 41, cyclic GMP-AMP synthase (cGAS), and γ-IFN-inducible protein 16 (IFI16) (Unterholzner et al., 2010; Zhang et al., 2011; Sun et al., 2013; Bhat and Fitzgerald, 2014), all of which lead to the activation of stimulator of IFN genes (STING). Both MAVS recruitment and STING activation after RNA or DNA viral infection lead to TANK-binding kinase 1 (TBK1) phosphorylation and activation of IFN regulatory factors (IRFs), which are primarily responsible for IFN production (Sato et al., 2000). Cytosolic RNA polymerase III is specialized to detect foreign and aberrant DNA, which is converted into RNA. RNA synthesized in the cytosol contains 5′-triphosphate, which can be detected by RIG-I. Therefore, RNA polymerase III can act as a DNA sensor that triggers the innate immune response through the RIG-I pathway (Chiu et al., 2009; Figure 3).
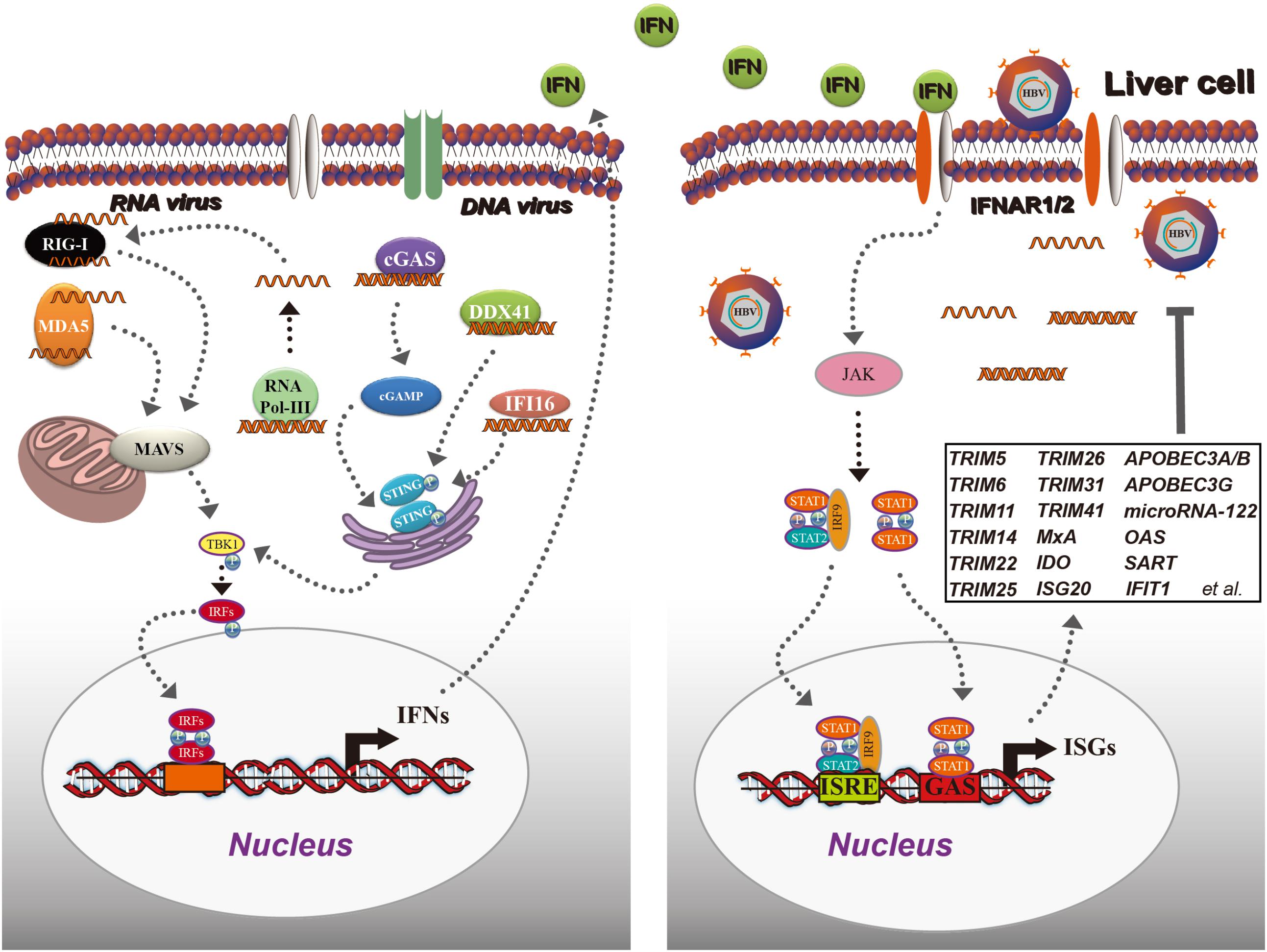
FIGURE 3. Classical interferon (IFN) and IFN-stimulated gene (ISG) induction in response to viral infection. In the cytoplasm, viral DNA is recognized by cGAS, DDX41, or IFI16, leading to the activation of STING, which further recruits TBK1 for IRF activation and translocates to the nucleus to initiate IFN production. Viral RNA is detected by RIG-I and MDA5, and recruits another adaptor protein: MAVS (IPS-1), resulting in TBK1 activation, IRF phosphorylation, and IFN induction. Then, IFNs are secreted from the cells and bind to IFNAR1/2, leading to JAK/STAT activation. The activated ISGF3 trimer or STAT1 dimer bind to the ISRE or GAS sequence and promote the expression of ISGs. ISGs inhibit HBV as shown in the black box. cGAS, cyclic GMP-AMP synthase; DDX41, DEAD-box protein 41; IFI16, γ-IFN-inducible protein 16; STING, stimulator of IFN genes; TBK1, TANK-binding kinase 1; IFNAR1, type I IFN receptor 1; JAK, Janus tyrosine kinase; STAT, signal transducer and activator of transcription.
The antiviral function of type I IFN is cascaded via binding to type I IFN receptor (IFNAR1), activating the Janus tyrosine kinase (JAK)/signal transducer and activator of transcription (STAT) pathway, and subsequently inducing approximately 300 ISGs (Ivashkiv and Donlin, 2014). Different ISGs can inhibit different stages of the viral life cycle (Liu et al., 2013; Chen et al., 2014). The HBV replication steps that can be inhibited by ISGs are illutrstated in Figure 3.
How HBV Interacts with IFN Induction and Signaling
As reported previously, the lack of a functional innate DNA-sensing pathway in hepatocytes allows HBV to establish infection and to replicate (Thomsen et al., 2016). As a consequence of co-evolution, viruses have acquired the ability to counter the antiviral response induced by type I IFN. First, some host factors that are pro-HBV replication or antagonize the innate immunity are induced after HBV infection. For instance, NF-κB essential modulator (NEMO) has an essential role in the production of IFN-β by phosphorylating and activating IFN regulatory factor 3 (IRF3) in a polyubiquitin-dependent manner (Clark et al., 2013); and rubicon, a virus-induced protein, has been reported to bind to NEMO, which inhibits the ubiquitination of NEMO and further inhibits type I IFN production (Wan et al., 2017). In addition, HBV-induced parkin can recruit a linear ubiquitin assembly complex (LUBAC) to mitochondria and modulate K-63 and the linear ubiquitin chain on the MAVS signalosome, which abrogates IFN-β synthesis (Khan et al., 2016). These two reports clearly indicate the mechanism of HBV in regulating some of the critical factors in IFN production. Moreover, HBV is also reported to inhibit IFN signaling by regulating IFNAR1, and HBV-activated collagen triple helix repeat containing 1 (CTHRC1) reduces IFNAR1/2 expression and downregulates the activity of type I IFN (Bai et al., 2015). However, how IFNAR1/2 is reduced has not been clarified. Interestingly, another report has shown that HBV can induce matrix metalloproteinase 9 (MMP9), which binds to IFNAR1, promotes its degradation via ubiquitination, and suppresses IFN/JAK/STAT signaling (Chen et al., 2017).
Second, HBV or viral proteins also have been clearly reported to inhibit IFN production. Major vault protein (MVP) is a novel virus-induced cellular protein that upregulates type I IFN production. The middle domain of MVP (amino acid residues 310–620) is essential for MyD88 binding; interestingly, both HBsAg and HBeAg have been shown to suppress the interaction between MVP and MyD88 (Kawai et al., 2004), which leads to limiting the downstream IFN signaling (Liu S. et al., 2015). In addition, HBV Pol has been shown to interact with STING and to decrease the Lys63-linked polyubiquitination of STING dramatically, resulting in inhibiting the activation of STING-stimulated IRF3 and decreasing the induction of IFN-β (Liu Y. et al., 2015). Moreover, Pol has been reported to evade the innate immune response in the early phase of infection, thus interrupting the interaction between the inhibitor of NF-κB kinase (IKK) 𝜀 and DDX3 DEAD box RNA helicase as well as inhibit TBK1/IKK𝜀 activity (Wang and Ryu, 2010). Interestingly, HBV viral particles have appeared to readily inhibit the innate immune response through virion/cell interactions (Luangsay et al., 2015), which might partially explain the “stealthy” characteristics of HBV. In vitro, upon infection of primary hepatocytes with HBV, both type I and III IFN transcripts were suppressed to 10% of the levels observed in uninfected cultures, suggesting an active role for HBV in suppressing innate immune activation from the side (Ortega-Prieto et al., 2018).
Another viral protein is HBx, which is a 17-kDa protein, consisting of an N-terminal negative regulatory domain and a C-terminal transactivation or co-activation domain (Gong et al., 2013). HBx may function as a transactivator and appears to regulate the expression of cellular genes; in addition, it is implicated in the development of HCC (Zhang et al., 2017). HBx interacts with several cellular proteins and impacts HBV replication through these interactions. The best-characterized HBx binding partner is damage-specific DNA binding protein 1 (DDB1), and the interaction between HBx and DDB1 is essential for HBV replication (Hodgson et al., 2012). HBx induces cytokine signaling 3 and protein phosphatase 2A suppression, impairing IFN signaling (Tsunematsu et al., 2017). HBx has been reported to elevate MSL2 expression and degrade APOBEC3B, leading to cccDNA accumulation and hepatocarcinogenesis (Gao et al., 2017). HBx also has been shown to induce a single CpG methylation in the 5′-UTR of the tripartite motif 22 (TRIM22) gene and inhibit IFN-induced TRIM22 expression (Lim et al., 2017). These reports demonstrate that HBx has a negative impact on interrupting IFN signaling and producing both IFN and ISGs. Several reports have shown that HBx targets the structural maintenance of chromosomes (SMC)5/6 complex to enhance HBV gene expression from episomal cccDNA (Murphy et al., 2016; Livingston et al., 2017). It has been reported that the SMC complex is not induced by IFN. However, SMC5/6 forms the foundation of a multi-subunit DNA repair complex and is a restriction factor that selectively blocks extrachromosomal DNA transcription (Uhlmann, 2016). Moreover, it has been demonstrated that HBx binds to the DDB1 subunit of the DDB1-containing E3 ubiquitin ligase and forms an HBx-DDB1-cullin 4-ROC1 E3 ligase complex for ubiquitin-mediated degradation of SMC5/6 complexes (Decorsiere et al., 2016), which lift the restriction of HBV gene expression from episomal cccDNA. A disruption of the interaction between HBx and host proteins might increase the efficiency of IFN treatment (Figure 4).
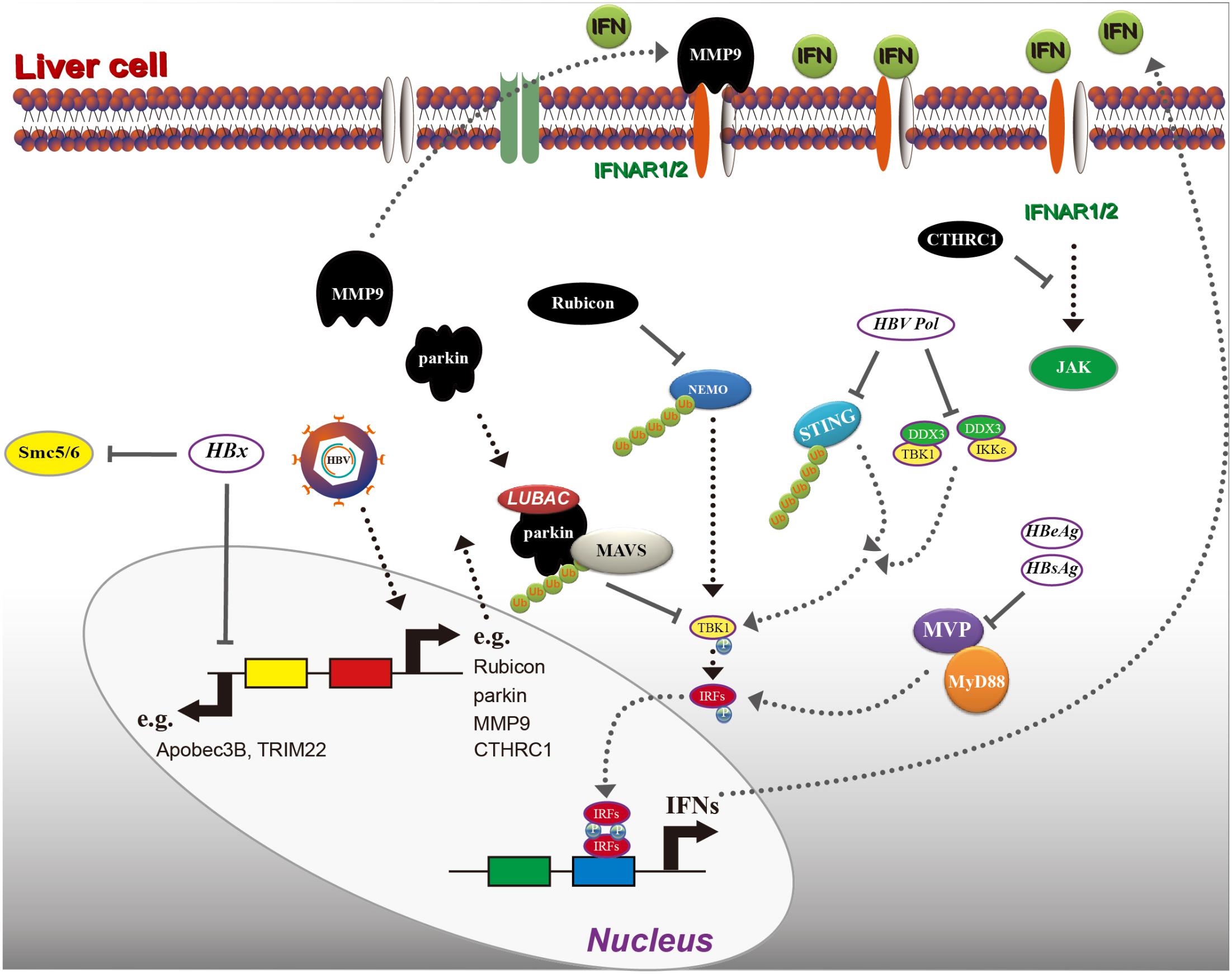
FIGURE 4. Hepatitis B virus interrupts interferon (IFN) signaling. As indicated, HBV induces several host factors, including rubicon, parkin, MMP9, and CTHRC1. These proteins inhibit IFN production in a different mechanism, and HBV proteins including HBx, Pol (polymerase), HBsAg, and HBeAg can also block IFN induction or directly inhibit ISG induction. Please see the text for details. MVP, major vault protein; MMP9, matrix metalloproteinase 9; NEMO, nuclear factor-κB essential modulator; LUBAC, linear ubiquitin assembly complex; RIG-I, retinoic acid inducible gene I; MAVS, mitochondrial antiviral-signaling protein.
Hepatitis B virus DNA has been reported to be recognized by cGAS, resulting in activation of the cGAS-STING pathway, which can suppress HBV replication by inducing ISG56 (IFIT1) (Dansako et al., 2016). However, IFN induction is not presented in this report. In addition, HBV pgRNA can be recognized by RIG-I, leading to induction of type III but not type I IFN (Sato et al., 2015). HBV replication generates both DNA and RNA species; therefore, many more studies are required to elucidate the complicated mechanisms involved in the immune response to HBV infection.
Hepatitis B virus pgRNA represents another target by the host immunity as it is recognized by RIG-I, thus eliciting type III IFN induction (Sato et al., 2015), which can also inhibit HBV replication (Hong et al., 2007; Pagliaccetti et al., 2010; Tian et al., 2014). In our recent study, we have found that type III IFN inhibits HBV replication through inducing core-binding factor beta (CBFβ). It is known that CBFβ suppresses the formation of the HBx-DDB1-SMC5/6 complex, which blocks HBx transcriptional activity (Xu et al., 2018).
Taken together, there is complicated crosstalk between HBV and IFN/ISGs. Therefore, a thorough understanding of the mechanisms responsible for the HBV-mediated inhibition of IFN is needed for designing and developing immunology-based HBV therapy.
How ISGs Inhibit HBV Replication
Many ISGs are known to inhibit HBV replication. One of them is the APOBEC gene, which encodes a family of cytidine deaminases that edit DNA and/or RNA bases by deaminating cytidine and converting it to uridine. A total of 11 members of this family are encoded by the human genome (Vieira and Soares, 2013), among which APOBEC3A/B has been shown to be induced by IFN, resulting in cytidine deamination, apurinic/apyrimidinic site formation, and finally cccDNA degradation that prevents HBV reactivation in HBV-infected cells (Lucifora et al., 2014). We also have reported that APOBEC3G inhibits HBV replication and that IFN-induced APOBEC3G is STAT3 but not STAT1 dependent. However, A3G expression is reduced in HBV-infected patients, which might due to the inhibition of STAT3 by HBsAg (Xu et al., 2016).
In addition, ISG20 inhibits HBV replication through directly binding to the epsilon stem-loop structure of pgRNA (Liu et al., 2017). Indoleamine-pyrrole 2,3-dioxygenase, an IFN-induced enzyme catalyzing tryptophan degradation, has been found to reduce the intracellular HBV DNA level efficiently by tryptophan deprivation without altering the steady-state level of viral RNA (Mao et al., 2011). Myxovirus resistance protein 1 (MxA) interacts with the HBV core protein, which might interfere with capsid assembly (Li et al., 2012). The 2′,5′-linked oligoadenylate is synthesized by a family of enzymes named oligoadenylate synthetases, a type I IFN-inducible gene. RNase L binds to 2′,5′-linked oligoadenylate to activate it, thus inhibiting HBV RNAs (Park et al., 2014). In addition, squamous cell carcinoma antigen recognized by T cells (SART) is involved in regulating ISG expression against HBV infection after IFN treatment (Li et al., 2016). Interestingly, miR-122 also has been reported to be induced by IFN, thus inhibiting HBV replication by targeting suppressor of cytokine signaling 3, a molecule contributing to inactivation of the IFN signaling pathway (Gao et al., 2015; Figure 3).
In recent years, several TRIM-containing family members, which are also categorized as ISGs, have been found to be important in regulating IFN signaling and inhibiting HBV replication (Zhang et al., 2013). They comprise a really interesting new gene (RING) domain, one or two B-box domains, and an associated coiled-coil domain in the amino-terminal region and share RING finger E3 ubiquitin ligase activity (Torok and Etkin, 2001; Ozato et al., 2008; Rajsbaum et al., 2014). As reported by us and others, TRIM expression is induced in response to IFN stimulation and is required to control viral infections (Uchil et al., 2013; Versteeg et al., 2013; Liu et al., 2016; Tan G. et al., 2017). We have found that TRIM25 expression is elevated under the induction of type I IFN in an interleukin-27-dependent manner, thus inhibiting HBV replication through augmenting IFN production (Tan G. et al., 2017). TRIM22 also has been shown to inhibit HBV core promoter activity (Gao et al., 2009). Among the TRIMs, TRIM14 has recently emerged as an attractive gene that regulates IFN signaling. TRIM14 recruits ubiquitin-specific protease 14 to cleave the lysine (Lys) 48-linked ubiquitin chains of cGAS at Lys414 and facilitates the activation of type I IFN signaling, thus inhibiting p62-mediated autophagic degradation of cGAS (Chen et al., 2016). TRIM14 undergoes Lys-63-linked polyubiquitination at Lys-365 and recruits the NEMO to the MAVS signalosome, activating both the IRF3 and NF-κB pathways, which enhance the innate immune response (Zhou et al., 2014). In addition, RIG-I-mediated innate antiviral immunity has been reported to require TRIM14 to form a Werner helicase interacting protein–TRIM14–phosphoprotein phosphatase 6C mitochondrial signalosome (Tan P. et al., 2017). TRIM31 also has been shown to inhibit HBV replication and regulate IFN signaling. Mechanistically, the interaction between TRIM31 and MAVS catalyzes the Lys63 (K63)-linked polyubiquitination of Lys10, Lys311, and Lys461 on MAVS, which forms prion-like aggregates of MAVS in response to viral infection (Liu et al., 2016). In addition, additional TRIMs (e.g., TRIM56, Tsuchida et al., 2010; Wang et al., 2011; Shen et al., 2012 and TRIM38, Hu et al., 2016), potential regulators of HBV replication, have been shown to regulate IFN production (Figure 3). TRIMs might be potential targets for HBV therapy.
Rational for IFN-Based Treatment of Chronic Hepatitis B
Interferon-α has been used for treating chronic hepatitis B for over two decades. Three guidelines are recommended by the American Association for the Study of Liver Diseases (AASLD): (1) risk for virological relapse, hepatic decompensation, liver cancer, and death; (2) financial concerns; and (3) preference of the patient (Ghany, 2017). Because of parenteral administration, severe side effects, and less effective inhibition of HBV DNA replication, IFN-α is not a first-line drug recommended by the guidelines, nor has it been widely used in the clinic (European Association For The Study Of The Liver, 2012). Injectable formulations of pegylated IFN-α (peg-IFN-α-2a and α-2b) are available for HBV therapy (Tang et al., 2018). peg-IFN has a longer half-life compared with standard IFN, so less frequent dosing is required and more sustained viral suppression has been shown in clinical trials compared with standard IFN (Cooksley et al., 2003). The injection of pegylated IFN-α is administered once a week in a dose of 180 μg for 48 weeks (Liaw et al., 2011). Its use is limited by adverse effects including flu-like symptoms, thyroid dysfunction, gastrointestinal symptoms, neutropenia, and so on (Marcellin et al., 2004; Janssen et al., 2005; Lau et al., 2005). According to the three major liver societies, the AASLD, the European Association for the Study of the Liver (EASL), and the Asian Pacific Association for the Study of the Liver (APASL), the IFN-α treatment guidelines are all similar (reviewed in Sato et al., 2015).
Currently, 7 therapies are approved, including standard and peg-IFN, and 5 nucleos(t)ide analogs (NAs) (NUCs: ADV, adefovir; ETV, entecavir; LMV, lamivudine; TBV, telbivudine; TDF, tenofovir), in which Peg-IFN, ETV, and TDF was recommended by the guidelines of major liver associations (Lok and McMahon, 2009; European Association For The Study Of The Liver, 2012; Liaw et al., 2012). Despite the adverse effects, IFN shows a better efficacy in inducing higher percentages of HBeAg loss as well as HBsAg loss compared to nucleoside or NAs (Konerman and Lok, 2016). Several reports showed that the outcome of peg-IFN combining with LMV treatment was similar compared to peg-IFN alone (Marcellin et al., 2004; Janssen et al., 2005; Lau et al., 2005; Buster et al., 2008). It was also reported that TDF plus peg-IFN treatment increased the loss of HBsAg than those receiving TDF or peg-IFN alone (Marcellin et al., 2016). Peg-IFN-α-2b combination with ADV therapy inhibited viral productivity by 99% and subsequent ADV monotherapy by 76%, while with only a small number of patients involved (24 patients), this clinical trial is limited (Lutgehetmann et al., 2008). Switch to TBV after 24 weeks of Peg-IFNα-2a therapy was shown to promote HBeAg seroconversion that merits investigation in HBeAg-positive CHB patients (Luo et al., 2017). Sequential approaches with peg-IFN and NUCs were also tested, however, no significant benefit was noted (Xie et al., 2014; Brouwer et al., 2015). It was reported that patients started to resistant to NUCs after long term therapy, interestingly, after subcutaneous peg-IFN add-on treatment, anti-HBs antibody appeared, which suggested peg-IFN might benefit HBsAg seroconversion (Ouzan et al., 2013; Stanzione et al., 2016). And peg-IFN monotherapy showed greater advantages in HBeAg seroconversion compared to peg-IFN-LMV combination or LMV monotherapy (32 and 27% vs. 19%) (Lau et al., 2005). The combination of high-dose peg-IFN and NAs resulted in sustained loss of serum HBV DNA and other HBV markers in an immunodeficient mouse model (Uchida et al., 2017), suggesting that HBV replication can be effectively inhibited by antivirals in the absence of a cellular immune response. A recombinant human serum albumin (rHSA)-IFN-α2a fusion protein is under development and aims to extend the in vivo half-life of IFN-α. The data from clinical trials reveal that rHSA/IFNα2a is well tolerated and effective at inhibiting HBV DNA (Ding et al., 2017).
Interferon treatment stayed a good alternative for people with a good prognosis (young age, low viral load, and female gender) (Sokal et al., 1998; Roberts, 2000). And due to different HBV genotype, HBeAg positive or negative stage of liver disease Peg-IFN therapy shows different response and outcome (Konerman and Lok, 2016), therefore, precision treatment should be created for every individual patient.
Recently, a combination therapy of IFN-λ with entecavir was assessed for its efficacy in treating chronic hepatitis B patients, and the trial data showed a more profound decrease in the levels of HBV DNA, HBsAg, and HBeAg compared with entecavir monotherapy (European Association For The Study Of The Liver, 2012; Phillips et al., 2017). Since only 13 patients were involved in this clinical trial, the results need to be further confirmed with many more chronic hepatitis B patient samples. Despite the severe side effects, the NAs may induce IFN-λ3 production, which triggers the expression of ISGs and reduces the HBsAg level (Murata et al., 2018). If we can clarify the exact inhibition mechanism involved in this process, a cure for HBV infection will be just around the corner.
GS-9620, a toll-like receptor 7 agonist, which was first evaluated in HBV-infected chimpanzees in 2013 (Lanford et al., 2013), has demonstrated a prolonged inhibition of HBV replication via a type I IFN-dependent mechanism (Niu et al., 2017). In combination with a NA, GS-9620 appears to increase T-cell and natural killer cell responses and to reduce the ability of natural killer cells to suppress T cells in chronic hepatitis B patients, raising the possibility that GS-9620 might potentially be used as an adjunct drug in HBV treatment to augment the immune response to HBV (Boni et al., 2018). However, comparing the data from chimpanzees and woodchuck models (Lanford et al., 2013; Menne et al., 2015), a much lower efficacy of GS-9620 in clinical trials in humans has been reported, since no significant effect was shown on serum HBsAg levels, tempering the previous expectation (Agarwal et al., 2018; Boni et al., 2018). The reason for this finding might be that the animals received higher doses (in terms of concentration and/or frequency) than those used in CHB patients.
Hepatitis B virus cccDNA resides as an episomal template in nuclei of infected hepatocytes. Although the average cccDNA copy number per cell is low (10 copies in a duck liver cell and 0.1–1 copies in a human liver cell) (Zhang et al., 2003; Volz et al., 2013), persistent or residual cccDNA is the reason why recurrent HBV infection occurs in resolved patients. A complete cure of HBV infection is a functional cure plus cccDNA elimination (Durantel, 2017). The impact of IFN treatment on cccDNA is a subject for debate. For instance, in some reports, cccDNA remains at a similar level in IFN-treated primary human hepatocytes (Niu et al., 2017; Shen et al., 2017; Mutz et al., 2018). However, other reports have shown a significant decrease in the cccDNA level after IFN treatment (Chuaypen et al., 2017; Mu et al., 2017). In addition, IFN-α has been shown to repress HBV cccDNA transcriptional activity by an epigenetic modification (Belloni et al., 2012). Moreover, a novel site-specific PEGylated recombinant human IFN-β (TRK-560) demonstrates a stronger antiviral potency by inducing a higher expression of ISGs and a stronger stimulation of immune cell chemotaxis compared with PEG-IFN-α2a. Furthermore, the reduction of cccDNA under TRK-560 treatment in vivo was significantly higher than that by PEG-IFN-α2a treatment (Tsuge et al., 2017). However, the cccDNA synthetic process involves several host enzymes that could be difficult to target as they are also required for other biological functions. Eradication of HBV from an infected liver requires elimination of cccDNA (Nassal, 2015), which appears to be unrealistic at this moment.
Conclusion and Future Outlook
Evolution has equipped the human immune system with effective strategies to defend against viral infections. IFN production and signaling represent the most studied as well as the earliest immune response. Upon HBV infection, both HBV DNA and pgRNA can be recognized by both a DNA sensor (cGAS) and an RNA sensor (RIG-I). However, only type III but not type I IFN is induced in infected hepatocytes, which might reflect a lack of or inactive DNA sensing pathways in hepatocytes (Cheng et al., 2017). IFN-α has demonstrated the ability to reduce the levels of HBV DNA, HBsAg, and cccDNA efficiently. Specific and efficient activation of the IFN pathway in HBV patients might represent one treatment strategy to eradicate HBV from an infected liver. However, the challenge is that the antiviral function of IFN is compromised by the presence of HBV. As discussed above, most crosstalk between IFN and HBV is mediated by the interactions between ISG proteins and HBV proteins. For example, rubicon binds to NEMO, MMP9 binds to IFNAR, Pol interacts with STING, parkin recruits LUBAC, and HBx targets SMC5/6. All these protein interactions are deleterious to IFN signaling and benefit HBV infection. Therefore, small molecules designed to interfere with these interactions may interrupt the inhibition of IFN signaling by HBV and viral products.
Author Contributions
GT planned the research and wrote the paper. HS and FX gave suggestions and revised the manuscript. GC conceived the research project and revised the manuscript.
Funding
This work was supported by National Natural Science Foundation of China, Jilin Province Science and Technology Department of the Youth Fund Project, and Jilin University Bethune Training Program (Grant Nos. 81401290, 20160520161JH, and 470110000456 to GT).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agarwal, K., Ahn, S. H., Elkhashab, M., Lau, A. H., Gaggar, A., Bulusu, A., et al. (2018). Safety and efficacy of vesatolimod (GS-9620) in patients with chronic hepatitis B who are not currently on antiviral treatment. J. Viral. Hepat. doi: 10.1111/jvh.12942 [Epub ahead of print].
Bai, L., Zhang, W., Tan, L., Yang, H., Ge, M., Zhu, C., et al. (2015). Hepatitis B virus hijacks CTHRC1 to evade host immunity and maintain replication. J. Mol. Cell Biol. 7, 543–556. doi: 10.1093/jmcb/mjv048
Bekisz, J., Schmeisser, H., Hernandez, J., Goldman, N. D., and Zoon, K. C. (2004). Human interferons alpha, beta and omega. Growth Factors 22, 243–251. doi: 10.1080/08977190400000833
Belloni, L., Allweiss, L., Guerrieri, F., Pediconi, N., Volz, T., Pollicino, T., et al. (2012). IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Invest. 122, 529–537. doi: 10.1172/JCI58847
Bhat, N., and Fitzgerald, K. A. (2014). Recognition of cytosolic DNA by cGAS and other STING-dependent sensors. Eur. J. Immunol. 44, 634–640. doi: 10.1002/eji.201344127
Boni, C., Vecchi, A., Rossi, M., Laccabue, D., Giuberti, T., Alfieri, A., et al. (2018). TLR7 agonist increases responses of HBV-specific T cells and natural killer cells in patients with chronic hepatitis B treated with nucleos(T)Ide analogues. Gastroenterology 154, 1764.e7–1777.e7. doi: 10.1053/j.gastro.2018.01.030
Breakwell, L., Tevi-Benissan, C., Childs, L., Mihigo, R., and Tohme, R. (2017). The status of hepatitis B control in the African region. Pan Afr. Med. J. 27(Suppl. 3):17. doi: 10.11604/pamj.supp.2017.27.3.11981
Brouwer, W. P., Xie, Q., Sonneveld, M. J., Zhang, N., Zhang, Q., Tabak, F., et al. (2015). Adding pegylated interferon to entecavir for hepatitis B e antigen-positive chronic hepatitis B: a multicenter randomized trial (ARES study). Hepatology 61, 1512–1522. doi: 10.1002/hep.27586
Buster, E. H., Flink, H. J., Cakaloglu, Y., Simon, K., Trojan, J., Tabak, F., et al. (2008). Sustained HBeAg and HBsAg loss after long-term follow-up of HBeAg-positive patients treated with peginterferon alpha-2b. Gastroenterology 135, 459–467. doi: 10.1053/j.gastro.2008.05.031
Castro, F., Cardoso, A. P., Goncalves, R. M., Serre, K., and Oliveira, M. J. (2018). Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front. Immunol. 9:847. doi: 10.3389/fimmu.2018.00847
Chen, J., Xu, W., Chen, Y., Xie, X., Zhang, Y., Ma, C., et al. (2017). Matrix metalloproteinase 9 facilitates hepatitis B virus replication through binding with type I interferon (IFN) receptor 1 to repress IFN/JAK/STAT signaling. J. Virol. 91:e01824-16. doi: 10.1128/JVI.01824-16
Chen, M., Meng, Q., Qin, Y., Liang, P., Tan, P., He, L., et al. (2016). TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 64, 105–119. doi: 10.1016/j.molcel.2016.08.025
Chen, Y., Wang, S., Yi, Z., Tian, H., Aliyari, R., Li, Y., et al. (2014). Interferon-inducible cholesterol-25-hydroxylase inhibits hepatitis C virus replication via distinct mechanisms. Sci. Rep. 4:7242. doi: 10.1038/srep07242
Cheng, X., Xia, Y., Serti, E., Block, P. D., Chung, M., Chayama, K., et al. (2017). Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 66, 1779–1793. doi: 10.1002/hep.29348
Childs, L., Roesel, S., and Tohme, R. A. (2018). Status and progress of hepatitis B control through vaccination in the South-East Asia Region, 1992-2015. Vaccine 36, 6–14. doi: 10.1016/j.vaccine.2017.11.027
Chiu, Y. H., Macmillan, J. B., and Chen, Z. J. (2009). RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591. doi: 10.1016/j.cell.2009.06.015
Chuaypen, N., Sriprapun, M., Praianantathavorn, K., Payungporn, S., Wisedopas, N., Poovorawan, Y., et al. (2017). Kinetics of serum HBsAg and intrahepatic cccDNA during pegylated interferon therapy in patients with HBeAg-positive and HBeAg-negative chronic hepatitis B. J. Med. Virol. 89, 130–138. doi: 10.1002/jmv.24601
Clark, K., Nanda, S., and Cohen, P. (2013). Molecular control of the NEMO family of ubiquitin-binding proteins. Nat. Rev. Mol. Cell Biol. 14, 673–685. doi: 10.1038/nrm3644
Cooksley, W. G., Piratvisuth, T., Lee, S. D., Mahachai, V., Chao, Y. C., Tanwandee, T., et al. (2003). Peginterferon alpha-2a (40 kDa): an advance in the treatment of hepatitis B e antigen-positive chronic hepatitis B. J. Viral. Hepat. 10, 298–305. doi: 10.1046/j.1365-2893.2003.00450.x
Cossart, Y. E., and Field, A. M. (1970). Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1, 695–698. doi: 10.1016/S0140-6736(70)92460-8
Dane, D. S., Cameron, C. H., and Briggs, M. (1970). Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1, 695–698. doi: 10.1016/S0140-6736(70)90926-8
Dansako, H., Ueda, Y., Okumura, N., Satoh, S., Sugiyama, M., Mizokami, M., et al. (2016). The cyclic GMP-AMP synthetase-STING signaling pathway is required for both the innate immune response against HBV and the suppression of HBV assembly. FEBS J. 283, 144–156. doi: 10.1111/febs.13563
De Andrea, M., Ravera, R., Gioia, D., Gariglio, M., and Landolfo, S. (2002). The interferon system: an overview. Eur. J. Paediatr. Neurol. 6, A41–A46. doi: 10.1053/ejpn.2002.0573
Decorsiere, A., Mueller, H., van Breugel, P. C., Abdul, F., Gerossier, L., Beran, R. K., et al. (2016). Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 531, 386–389. doi: 10.1038/nature17170
Ding, Y., Lou, J., Chen, H., Li, X., Wu, M., Li, C., et al. (2017). Tolerability, pharmacokinetics and antiviral activity of rHSA/IFNalpha2a for the treatment of chronic hepatitis B infection. Br. J. Clin. Pharmacol. 83, 1056–1071. doi: 10.1111/bcp.13184
Durantel, D. (2017). New treatments to reach functional cure: Virological approaches. Best Pract. Res. Clin. Gastroenterol. 31, 329–336. doi: 10.1016/j.bpg.2017.05.002
El-Serag, H. B., and Rudolph, K. L. (2007). Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology 132, 2557–2576. doi: 10.1053/j.gastro.2007.04.061
European Association For The Study Of The Liver (2012). EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J. Hepatol. 57, 167–185. doi: 10.1016/j.jhep.2012.02.010
Evans, J. D., and Seeger, C. (2007). Differential effects of mutations in NS4B on West Nile virus replication and inhibition of interferon signaling. J. Virol. 81, 11809–11816. doi: 10.1128/JVI.00791-07
Gao, B., Duan, Z., Xu, W., and Xiong, S. (2009). Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 50, 424–433. doi: 10.1002/hep.23011
Gao, D., Zhai, A., Qian, J., Li, A., Li, Y., Song, W., et al. (2015). Down-regulation of suppressor of cytokine signaling 3 by miR-122 enhances interferon-mediated suppression of hepatitis B virus. Antiviral Res. 118, 20–28. doi: 10.1016/j.antiviral.2015.03.001
Gao, Y., Feng, J., Yang, G., Zhang, S., Liu, Y., Bu, Y., et al. (2017). HBx-elevated MSL2 Modulates HBV cccDNA through inducing degradation of APOBEC3B to enhance hepatocarcinogenesis. Hepatology 66, 1413–1429. doi: 10.1002/hep.29316
Ghany, M. G. (2017). Current treatment guidelines of chronic hepatitis B: the role of nucleos(t)ide analogues and peginterferon. Best Pract. Res. Clin. Gastroenterol. 31, 299–309. doi: 10.1016/j.bpg.2017.04.012
Glebe, D. (2007). Recent advances in hepatitis B virus research: a German point of view. World J. Gastroenterol. 13, 8–13. doi: 10.3748/wjg.v13.i1.8
Gong, D. Y., Chen, E. Q., Huang, F. J., Leng, X. H., Cheng, X., and Tang, H. (2013). Role and functional domain of hepatitis B virus X protein in regulating HBV transcription and replication in vitro and in vivo. Viruses 5, 1261–1271. doi: 10.3390/v5051261
Hayes, C. N., Zhang, Y., Makokha, G. N., Hasan, M. Z., Omokoko, M. D., and Chayama, K. (2016). Early events in hepatitis B virus infection: From the cell surface to the nucleus. J. Gastroenterol. Hepatol. 31, 302–309. doi: 10.1111/jgh.13175
Hirschman, R. J., Shulman, N. R., Barker, L. F., and Smith, K. O. (1969). Virus-like particles in sera of patients with infectious and serum hepatitis. JAMA 208, 1667–1670. doi: 10.1001/jama.1969.03160090027006
Hodgson, A. J., Hyser, J. M., Keasler, V. V., Cang, Y., and Slagle, B. L. (2012). Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology 426, 73–82. doi: 10.1016/j.virol.2012.01.021
Hong, S. H., Cho, O., Kim, K., Shin, H. J., Kotenko, S. V., and Park, S. (2007). Effect of interferon-lambda on replication of hepatitis B virus in human hepatoma cells. Virus Res. 126, 245–249. doi: 10.1016/j.virusres.2007.03.006
Hu, K. Q., and Siddiqui, A. (1991). Regulation of the hepatitis B virus gene expression by the enhancer element I. Virology 181, 721–726. doi: 10.1016/0042-6822(91)90906-R
Hu, M. M., Yang, Q., Xie, X. Q., Liao, C. Y., Lin, H., Liu, T. T., et al. (2016). Sumoylation promotes the stability of the DNA sensor cGAS and the Adaptor STING to regulate the kinetics of response to DNA virus. Immunity 45, 555–569. doi: 10.1016/j.immuni.2016.08.014
Isaacs, A., and Lindenmann, J. (1957). Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 147, 258–267. doi: 10.1098/rspb.1957.0048
Isaacs, A., and Lindenmann, J. (1987). Virus interference. I. The interferon. By A. Isaacs and J. Lindenmann, 1957. J. Interferon Res. 7, 429–438. doi: 10.1089/jir.1987.7.429
Ivashkiv, L. B., and Donlin, L. T. (2014). Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49. doi: 10.1038/nri3581
Janssen, H. L., van Zonneveld, M., Senturk, H., Zeuzem, S., Akarca, U. S., Cakaloglu, Y., et al. (2005). Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet 365, 123–129. doi: 10.1016/S0140-6736(05)17701-0
Kawai, T., Sato, S., Ishii, K. J., Coban, C., Hemmi, H., Yamamoto, M., et al. (2004). Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5, 1061–1068. doi: 10.1038/ni1118
Khan, M., Syed, G. H., Kim, S. J., and Siddiqui, A. (2016). Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. PLoS Pathog. 12:e1005693. doi: 10.1371/journal.ppat.1005693
Kim, D. H., Kang, H. S., and Kim, K. H. (2016). Roles of hepatocyte nuclear factors in hepatitis B virus infection. World J. Gastroenterol. 22, 7017–7029. doi: 10.3748/wjg.v22.i31.7017
Konerman, M. A., and Lok, A. S. (2016). Interferon treatment for hepatitis B. Clin. Liver Dis. 20, 645–665. doi: 10.1016/j.cld.2016.06.002
Koniger, C., Wingert, I., Marsmann, M., Rosler, C., Beck, J., and Nassal, M. (2014). Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. U.S.A. 111, E4244–E4253. doi: 10.1073/pnas.1409986111
Lanford, R. E., Guerra, B., Chavez, D., Giavedoni, L., Hodara, V. L., Brasky, K. M., et al. (2013). GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 144, 1508.e10–1517.e10. doi: 10.1053/j.gastro.2013.02.003
Lau, G. K., Piratvisuth, T., Luo, K. X., Marcellin, P., Thongsawat, S., Cooksley, G., et al. (2005). Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N. Engl. J. Med. 352, 2682–2695. doi: 10.1056/NEJMoa043470
Lee, S., and Baldridge, M. T. (2017). Interferon-lambda: a potent regulator of intestinal viral infections. Front. Immunol. 8:749. doi: 10.3389/fimmu.2017.00749
Levrero, M., Pollicino, T., Petersen, J., Belloni, L., Raimondo, G., and Dandri, M. (2009). Control of cccDNA function in hepatitis B virus infection. J. Hepatol. 51, 581–592. doi: 10.1016/j.jhep.2009.05.022
Levrero, M., and Zucman-Rossi, J. (2016). Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 64(1 Suppl.), S84–S101. doi: 10.1016/j.jhep.2016.02.021
Li, N., Zhang, L., Chen, L., Feng, W., Xu, Y., Chen, F., et al. (2012). MxA inhibits hepatitis B virus replication by interaction with hepatitis B core antigen. Hepatology 56, 803–811. doi: 10.1002/hep.25608
Li, Y., Zhu, C., Wang, F., Zhu, T., Li, J., Liu, S., et al. (2016). Expression of interferon effector gene SART1 correlates with interferon treatment response against hepatitis B infection. Mediators Inflamm. 2016:3894816. doi: 10.1155/2016/3894816
Liang, T. J. (2009). Hepatitis B: the virus and disease. Hepatology 49(5 Suppl.), S13–S21. doi: 10.1002/hep.22881
Liang, T. J., Block, T. M., McMahon, B. J., Ghany, M. G., Urban, S., Guo, J. T., et al. (2015). Present and future therapies of hepatitis B: From discovery to cure. Hepatology 62, 1893–1908. doi: 10.1002/hep.28025
Liaw, Y. F., Jia, J. D., Chan, H. L., Han, K. H., Tanwandee, T., Chuang, W. L., et al. (2011). Shorter durations and lower doses of peginterferon alfa-2a are associated with inferior hepatitis B e antigen seroconversion rates in hepatitis B virus genotypes B or C. Hepatology 54, 1591–1599. doi: 10.1002/hep.24555
Liaw, Y. F., Kao, J. H., Piratvisuth, T., Chan, H. L., Chien, R. N., Liu, C. J., et al. (2012). Asian-Pacific consensus statement on the management of chronic hepatitis B: a 2012 update. Hepatol. Int. 6, 531–561. doi: 10.1007/s12072-012-9365-4
Lim, K. H., Park, E. S., Kim, D. H., Cho, K. C., Kim, K. P., Park, Y. K., et al. (2017). Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5′-UTR of TRIM22. Gut 67, 166–178. doi: 10.1136/gutjnl-2016-312742
Lin, F. C., and Young, H. A. (2014). Interferons: success in anti-viral immunotherapy. Cytokine Growth Factor Rev. 25, 369–376. doi: 10.1016/j.cytogfr.2014.07.015
Liu, B., Zhang, M., Chu, H., Zhang, H., Wu, H., Song, G., et al. (2016). The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol. 18, 214–224. doi: 10.1038/ni.3641
Liu, S., Peng, N., Xie, J., Hao, Q., Zhang, M., Zhang, Y., et al. (2015). Human hepatitis B virus surface and e antigens inhibit major vault protein signaling in interferon induction pathways. J. Hepatol. 62, 1015–1023. doi: 10.1016/j.jhep.2014.11.035
Liu, Y., Li, J., Chen, J., Li, Y., Wang, W., Du, X., et al. (2015). Hepatitis B virus polymerase disrupts K63-linked ubiquitination of sting to block innate cytosolic DNA-sensing pathways. J. Virol. 89, 2287–2300. doi: 10.1128/JVI.02760-14
Liu, S. Y., Aliyari, R., Chikere, K., Li, G., Marsden, M. D., Smith, J. K., et al. (2013). Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105. doi: 10.1016/j.immuni.2012.11.005
Liu, S. Y., Sanchez, D. J., Aliyari, R., Lu, S., and Cheng, G. (2012). Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. U.S.A. 109, 4239–4244. doi: 10.1073/pnas.1114981109
Liu, Y., Nie, H., Mao, R., Mitra, B., Cai, D., Yan, R., et al. (2017). Interferon-inducible ribonuclease ISG20 inhibits hepatitis B virus replication through directly binding to the epsilon stem-loop structure of viral RNA. PLoS Pathog. 13:e1006296. doi: 10.1371/journal.ppat.1006296
Livingston, C. M., Ramakrishnan, D., Strubin, M., Fletcher, S. P., and Beran, R. K. (2017). Identifying and characterizing interplay between hepatitis B virus X protein and Smc5/6. Viruses 9:E69. doi: 10.3390/v9040069
Lok, A. S., and McMahon, B. J. (2009). Chronic hepatitis B: update 2009. Hepatology 50, 661–662. doi: 10.1002/hep.23190
Lok, A. S., Zoulim, F., Dusheiko, G., and Ghany, M. G. (2017). Hepatitis B cure: from discovery to regulatory approval. J. Hepatol. 67, 847–861. doi: 10.1016/j.jhep.2017.05.008
Luangsay, S., Gruffaz, M., Isorce, N., Testoni, B., Michelet, M., Faure-Dupuy, S., et al. (2015). Early inhibition of hepatocyte innate responses by hepatitis B virus. J. Hepatol. 63, 1314–1322. doi: 10.1016/j.jhep.2015.07.014
Lucifora, J., Xia, Y., Reisinger, F., Zhang, K., Stadler, D., Cheng, X., et al. (2014). Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343, 1221–1228. doi: 10.1126/science.1243462
Luo, X. D., Chen, X. F., Zhou, Y., and Chen, X. P. (2017). Comparison of 208-week sequential therapy with telbivudine and entecavir in HBeAg-positive chronic hepatitis B patients with suboptimal responses to 24 weeks of Peg-IFNalpha-2a therapy: an open-labelled, randomized, controlled, “real-life” trial. J. Viral. Hepat. 24(Suppl. 1), 36–42. doi: 10.1111/jvh.12790
Lutgehetmann, M., Volzt, T., Quaas, A., Zankel, M., Fischer, C., Dandri, M., et al. (2008). Sequential combination therapy leads to biochemical and histological improvement despite low ongoing intrahepatic hepatitis B virus replication. Antivir. Ther. 13, 57–66.
Mao, R., Zhang, J., Jiang, D., Cai, D., Levy, J. M., Cuconati, A., et al. (2011). Indoleamine 2,3-dioxygenase mediates the antiviral effect of gamma interferon against hepatitis B virus in human hepatocyte-derived cells. J. Virol. 85, 1048–1057. doi: 10.1128/JVI.01998-10
Marcellin, P., Ahn, S. H., Ma, X., Caruntu, F. A., Tak, W. Y., Elkashab, M., et al. (2016). Combination of tenofovir disoproxil fumarate and peginterferon alpha-2a increases loss of hepatitis B surface antigen in patients with chronic hepatitis B. Gastroenterology 150, 134.e10–144.e10. doi: 10.1053/j.gastro.2015.09.043
Marcellin, P., Lau, G. K., Bonino, F., Farci, P., Hadziyannis, S., Jin, R., et al. (2004). Peginterferon alfa-2a alone, lamivudine alone, and the two in combination in patients with HBeAg-negative chronic hepatitis B. N. Engl. J. Med. 351, 1206–1217. doi: 10.1056/NEJMoa040431
Menne, S., Tumas, D. B., Liu, K. H., Thampi, L., AlDeghaither, D., Baldwin, B. H., et al. (2015). Sustained efficacy and seroconversion with the Toll-like receptor 7 agonist GS-9620 in the Woodchuck model of chronic hepatitis B. J. Hepatol. 62, 1237–1245. doi: 10.1016/j.jhep.2014.12.026
Mu, D., Yuan, F. C., Chen, Y., Jiang, X. Y., Yan, L., Jiang, L. Y., et al. (2017). Baseline value of intrahepatic HBV DNA over cccDNA predicts patient’s response to interferon therapy. Sci. Rep. 7:5937. doi: 10.1038/s41598-017-05242-y
Murata, K., Asano, M., Matsumoto, A., Sugiyama, M., Nishida, N., Tanaka, E., et al. (2018). Induction of IFN-lambda3 as an additional effect of nucleotide, not nucleoside, analogues: a new potential target for HBV infection. Gut 67, 362–371. doi: 10.1136/gutjnl-2016-312653
Murphy, C. M., Xu, Y., Li, F., Nio, K., Reszka-Blanco, N., Li, X., et al. (2016). Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. 16, 2846–2854. doi: 10.1016/j.celrep.2016.08.026
Mutz, P., Metz, P., Lempp, F. A., Bender, S., Qu, B., Schoneweis, K., et al. (2018). HBV bypasses the innate immune response and does not protect HCV from antiviral activity of interferon. Gastroenterology 154, 1791.e22–1804.e22. doi: 10.1053/j.gastro.2018.01.044
Myong, S., Cui, S., Cornish, P. V., Kirchhofer, A., Gack, M. U., Jung, J. U., et al. (2009). Cytosolic viral sensor RIG-I is a 5′-triphosphate-dependent translocase on double-stranded RNA. Science 323, 1070–1074. doi: 10.1126/science.1168352
Nassal, M. (2015). HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64, 1972–1984. doi: 10.1136/gutjnl-2015-309809
Newbold, J. E., Xin, H., Tencza, M., Sherman, G., Dean, J., Bowden, S., et al. (1995). The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 69, 3350–3357.
Niu, C., Li, L., Daffis, S., Lucifora, J., Bonnin, M., Maadadi, S., et al. (2017). Toll-like receptor 7 agonist GS-9620 induces prolonged inhibition of HBV via a type I interferon-dependent mechanism. J. Hepatol. 68, 922–931. doi: 10.1016/j.jhep.2017.12.007
Obajemu, A. A., Rao, N., Dilley, K. A., Vargas, J. M., Sheikh, F., Donnelly, R. P., et al. (2017). IFN-lambda4 attenuates antiviral responses by enhancing negative regulation of IFN signaling. J. Immunol. 199, 3808–3820. doi: 10.4049/jimmunol.1700807
Ortega-Prieto, A. M., Skelton, J. K., Wai, S. N., Large, E., Lussignol, M., Vizcay-Barrena, G., et al. (2018). 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat. Commun. 9:682. doi: 10.1038/s41467-018-02969-8
Ouzan, D., Penaranda, G., Joly, H., Khiri, H., Pironti, A., and Halfon, P. (2013). Add-on peg-interferon leads to loss of HBsAg in patients with HBeAg-negative chronic hepatitis and HBV DNA fully suppressed by long-term nucleotide analogs. J. Clin. Virol. 58, 713–717. doi: 10.1016/j.jcv.2013.09.020
Ozato, K., Shin, D. M., Chang, T. H., and Morse, H. C. III. (2008). TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8, 849–860. doi: 10.1038/nri2413nri2413
Pagliaccetti, N. E., Chu, E. N., Bolen, C. R., Kleinstein, S. H., and Robek, M. D. (2010). Lambda and alpha interferons inhibit hepatitis B virus replication through a common molecular mechanism but with different in vivo activities. Virology 401, 197–206. doi: 10.1016/j.virol.2010.02.022
Park, I. H., Kwon, Y. C., Ryu, W. S., and Ahn, B. Y. (2014). Inhibition of hepatitis B virus replication by ligand-mediated activation of RNase L. Antiviral Res. 104, 118–127. doi: 10.1016/j.antiviral.2014.01.021
Phillips, S., Mistry, S., Riva, A., Cooksley, H., Hadzhiolova-Lebeau, T., Plavova, S., et al. (2017). Peg-interferon lambda treatment induces robust innate and adaptive immunity in chronic hepatitis B patients. Front. Immunol. 8:621. doi: 10.3389/fimmu.2017.00621
Pichlmair, A., Schulz, O., Tan, C. P., Naslund, T. I., Liljestrom, P., Weber, F., et al. (2006). RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001. doi: 10.1126/science.1132998
Pollicino, T., Bellinghieri, L., Restuccia, A., Raffa, G., Musolino, C., Alibrandi, A., et al. (2013). Hepatitis B virus (HBV) induces the expression of interleukin-8 that in turn reduces HBV sensitivity to interferon-alpha. Virology 444, 317–328. doi: 10.1016/j.virol.2013.06.028
Pollicino, T., Belloni, L., Raffa, G., Pediconi, N., Squadrito, G., Raimondo, G., et al. (2006). Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 130, 823–837. doi: 10.1053/j.gastro.2006.01.001
Rajsbaum, R., Garcia-Sastre, A., and Versteeg, G. A. (2014). TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol. 426, 1265–1284. doi: 10.1016/j.jmb.2013.12.005S0022-2836(13)00746-8
Rehermann, B., and Nascimbeni, M. (2005). Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 5, 215–229. doi: 10.1038/nri1573
Roberts, E. A. (2000). Why treat chronic hepatitis B in childhood with interferon alpha? Gut 46, 591–593. doi: 10.1136/gut.46.5.591a
Sato, M., Suemori, H., Hata, N., Asagiri, M., Ogasawara, K., Nakao, K., et al. (2000). Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13, 539–548. doi: 10.1016/S1074-7613(00)00053-4
Sato, S., Li, K., Kameyama, T., Hayashi, T., Ishida, Y., Murakami, S., et al. (2015). The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 42, 123–132. doi: 10.1016/j.immuni.2014.12.016
Shen, F., Li, Y., Yang, W., Sozzi, V., Revill, P. A., Liu, J., et al. (2017). Hepatitis B virus sensitivity to interferon-alpha in hepatocytes is more associated with cellular interferon response than with viral genotype. Hepatology 67, 1237–1252. doi: 10.1002/hep.29609
Shen, Y., Li, N. L., Wang, J., Liu, B., Lester, S., and Li, K. (2012). TRIM56 is an essential component of the TLR3 antiviral signaling pathway. J. Biol. Chem. 287, 36404–36413. doi: 10.1074/jbc.M112.397075
Sheppard, P., Kindsvogel, W., Xu, W., Henderson, K., Schlutsmeyer, S., Whitmore, T. E., et al. (2003). IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4, 63–68. doi: 10.1038/ni873
Shlomai, A., de Jong, Y. P., and Rice, C. M. (2014a). Virus associated malignancies: the role of viral hepatitis in hepatocellular carcinoma. Semin. Cancer Biol. 26, 78–88. doi: 10.1016/j.semcancer.2014.01.004
Shlomai, A., Schwartz, R. E., Ramanan, V., Bhatta, A., de Jong, Y. P., Bhatia, S. N., et al. (2014b). Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. U.S.A. 111, 12193–12198. doi: 10.1073/pnas.1412631111
Sokal, E. M., Conjeevaram, H. S., Roberts, E. A., Alvarez, F., Bern, E. M., Goyens, P., et al. (1998). Interferon alfa therapy for chronic hepatitis B in children: a multinational randomized controlled trial. Gastroenterology 114, 988–995. doi: 10.1016/S0016-5085(98)70318-X
Stanzione, M., Stornaiuolo, G., Rizzo, V., Pontarelli, A., and Gaeta, G. B. (2016). HBsAg seroconversion after pegylated interferon alfa 2a rescue in a lamivudine-resistant patient with HBeAg-negative chronic hepatitis B and favourable IL28-B genotype. Infez. Med. 24, 144–146.
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. doi: 10.1126/science.1232458
Takeuchi, O., and Akira, S. (2008). MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 20, 17–22. doi: 10.1016/j.coi.2008.01.002
Takeuchi, O., and Akira, S. (2009). Innate immunity to virus infection. Immunol. Rev. 227, 75–86. doi: 10.1111/j.1600-065X.2008.00737.x
Tan, G., Xiao, Q., Song, H., Ma, F., Xu, F., Peng, D., et al. (2017). Type I IFN augments IL-27-dependent TRIM25 expression to inhibit HBV replication. Cell. Mol. Immunol. 15, 272–281. doi: 10.1038/cmi.2016.67
Tan, P., He, L., Cui, J., Qian, C., Cao, X., Lin, M., et al. (2017). Assembly of the WHIP-TRIM14-PPP6C mitochondrial complex promotes RIG-I-mediated antiviral signaling. Mol. Cell 68, 293.e5–307.e5. doi: 10.1016/j.molcel.2017.09.035
Tang, L. S. Y., Covert, E., Wilson, E., and Kottilil, S. (2018). Chronic hepatitis B infection: a review. JAMA 319, 1802–1813. doi: 10.1001/jama.2018.3795
Thomsen, M. K., Nandakumar, R., Stadler, D., Malo, A., Valls, R. M., Wang, F., et al. (2016). Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 64, 746–759. doi: 10.1002/hep.28685
Tian, S., Hui, X., Fan, Z., Li, Q., Zhang, J., Yang, X., et al. (2014). Suppression of hepatocellular carcinoma proliferation and hepatitis B surface antigen secretion with interferon-lambda1 or PEG-interferon-lambda1. FASEB J. 28, 3528–3539. doi: 10.1096/fj.14-250704
Tong, S., and Revill, P. (2016). Overview of hepatitis B viral replication and genetic variability. J. Hepatol. 64(1 Suppl.), S4–S16. doi: 10.1016/j.jhep.2016.01.027
Torok, M., and Etkin, L. D. (2001). Two B or not two B? Overview of the rapidly expanding B-box family of proteins. Differentiation 67, 63–71. doi: 10.1046/j.1432-0436.2001.067003063.x
Tsuchida, T., Zou, J., Saitoh, T., Kumar, H., Abe, T., Matsuura, Y., et al. (2010). The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33, 765–776. doi: 10.1016/j.immuni.2010.10.013
Tsuge, M., Uchida, T., Hiraga, N., Kan, H., Makokha, G. N., Abe-Chayama, H., et al. (2017). Development of a novel site-specific pegylated interferon beta for antiviral therapy of chronic hepatitis B virus. Antimicrob. Agents Chemother. 61:e00183-17. doi: 10.1128/AAC.00183-17
Tsunematsu, S., Suda, G., Yamasaki, K., Kimura, M., Izumi, T., Umemura, M., et al. (2017). Hepatitis B virus X protein impairs alpha-interferon signaling via up-regulation of suppressor of cytokine signaling 3 and protein phosphatase 2A. J. Med. Virol. 89, 267–275. doi: 10.1002/jmv.24643
Tuttleman, J. S., Pourcel, C., and Summers, J. (1986). Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 47, 451–460. doi: 10.1016/0092-8674(86)90602-1
Uchida, T., Imamura, M., Hayes, C. N., Hiraga, N., Kan, H., Tsuge, M., et al. (2017). Persistent loss of hepatitis B virus markers in serum without cellular immunity by combination of peginterferon and entecavir therapy in humanized mice. Antimicrob. Agents Chemother 61:AAC.00725-17. doi: 10.1128/AAC.00725-17
Uchil, P. D., Hinz, A., Siegel, S., Coenen-Stass, A., Pertel, T., Luban, J., et al. (2013). TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J. Virol. 87, 257–272. doi: 10.1128/JVI.01804-12JVI.01804-12
Uhlmann, F. (2016). SMC complexes: from DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 17, 399–412. doi: 10.1038/nrm.2016.30
Unterholzner, L., Keating, S. E., Baran, M., Horan, K. A., Jensen, S. B., Sharma, S., et al. (2010). IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004. doi: 10.1038/ni.1932
Versteeg, G. A., Rajsbaum, R., Sanchez-Aparicio, M. T., Maestre, A. M., Valdiviezo, J., Shi, M., et al. (2013). The E3-ligase TRIM family of proteins regulates signaling pathways triggered by innate immune pattern-recognition receptors. Immunity 38, 384–398. doi: 10.1016/j.immuni.2012.11.013
Vieira, V. C., and Soares, M. A. (2013). The role of cytidine deaminases on innate immune responses against human viral infections. Biomed Res. Int. 2013:683095. doi: 10.1155/2013/683095
Vilcek, J. (2006). Fifty years of interferon research: aiming at a moving target. Immunity 25, 343–348. doi: 10.1016/j.immuni.2006.08.008
Volz, T., Allweiss, L., Ben, M. M., Warlich, M., Lohse, A. W., Pollok, J. M., et al. (2013). The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 58, 861–867. doi: 10.1016/j.jhep.2012.12.008
Wack, A., Terczynska-Dyla, E., and Hartmann, R. (2015). Guarding the frontiers: the biology of type III interferons. Nat. Immunol. 16, 802–809. doi: 10.1038/ni.3212
Wan, Y., Cao, W., Han, T., Ren, S., Feng, J., Chen, T., et al. (2017). Inducible Rubicon facilitates viral replication by antagonizing interferon production. Cell. Mol. Immunol. 14, 607–620. doi: 10.1038/cmi.2017.1
Wang, H., and Ryu, W. S. (2010). Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog. 6:e1000986. doi: 10.1371/journal.ppat.1000986
Wang, J., Liu, B., Wang, N., Lee, Y. M., Liu, C., and Li, K. (2011). TRIM56 is a virus- and interferon-inducible E3 ubiquitin ligase that restricts pestivirus infection. J. Virol. 85, 3733–3745. doi: 10.1128/JVI.02546-10
Wheelock, E. F. (1965). Interferon-Like Virus-Inhibitor Induced in Human Leukocytes by Phytohemagglutinin. Science 149, 310–311. doi: 10.1126/science.149.3681.310
Wieland, S., Thimme, R., Purcell, R. H., and Chisari, F. V. (2004). Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. U.S.A. 101, 6669–6674. doi: 10.1073/pnas.0401771101
Wieland, S. F., Vega, R. G., Muller, R., Evans, C. F., Hilbush, B., Guidotti, L. G., et al. (2003). Searching for interferon-induced genes that inhibit hepatitis B virus replication in transgenic mouse hepatocytes. J. Virol. 77, 1227–1236. doi: 10.1128/JVI.77.2.1227-1236.2003
Xie, Q., Zhou, H., Bai, X., Wu, S., Chen, J. J., Sheng, J., et al. (2014). A randomized, open-label clinical study of combined pegylated interferon Alfa-2a (40KD) and entecavir treatment for hepatitis B “e” antigen-positive chronic hepatitis B. Clin. Infect. Dis. 59, 1714–1723. doi: 10.1093/cid/ciu702
Xu, F., Song, H., Li, N., and Tan, G. (2016). HBsAg blocks TYPE I IFN induced up-regulation of A3G through inhibition of STAT3. Biochem. Biophys. Res. Commun. 473, 219–223. doi: 10.1016/j.bbrc.2016.03.082
Xu, F., Song, H., Xiao, Q., Li, N., Zhang, H., Cheng, G., et al. (2018). Type III interferon-induced CBFbeta inhibits HBV replication by hijacking HBx. Cell. Mol. Immunol. doi: 10.1038/s41423-018-0006-2
Yan, H., Zhong, G., Xu, G., He, W., Jing, Z., Gao, Z., et al. (2012). Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 3:e00049. doi: 10.7554/eLife.00049
Zanen-Lim, O. G. (1976). Virus-like particles demonstrated by freeze-squeeze technique in acute-phase serum of patients with HBAg-negative hepatitis. Lancet 1, 18–20. doi: 10.1016/S0140-6736(76)92911-1
Zhang, S., Gao, S., Zhao, M., Liu, Y., Bu, Y., Jiang, Q., et al. (2017). Anti-HBV drugs suppress the growth of HBV-related hepatoma cells via down-regulation of hepatitis B virus X protein. Cancer Lett. 392, 94–104. doi: 10.1016/j.canlet.2017.02.003
Zhang, S., Guo, J. T., Wu, J. Z., and Yang, G. (2013). Identification and characterization of multiple TRIM proteins that inhibit hepatitis B virus transcription. PLoS One 8:e70001. doi: 10.1371/journal.pone.0070001
Zhang, Y. Y., Zhang, B. H., Theele, D., Litwin, S., Toll, E., and Summers, J. (2003). Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. U.S.A. 100, 12372–12377. doi: 10.1073/pnas.2033898100
Zhang, Z., Yuan, B., Bao, M., Lu, N., Kim, T., and Liu, Y. J. (2011). The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965. doi: 10.1038/ni.2091
Keywords: interferon, HBV, ISGs, HBV drug, cccDNA, TRIMs
Citation: Tan G, Song H, Xu F and Cheng G (2018) When Hepatitis B Virus Meets Interferons. Front. Microbiol. 9:1611. doi: 10.3389/fmicb.2018.01611
Received: 26 April 2018; Accepted: 28 June 2018;
Published: 18 July 2018.
Edited by:
Lisa Sedger, University of Technology Sydney, AustraliaCopyright © 2018 Tan, Song, Xu and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangyun Tan, dGd5MDQyNUBqbHUuZWR1LmNu Genhong Cheng, Z2NoZW5nQG1lZG5ldC51Y2xhLmVkdQ==