 Mauricio Latorre1,2,3,4*
Mauricio Latorre1,2,3,4* Daniela Quenti1
Daniela Quenti1 Dante Travisany2,3
Dante Travisany2,3 Kavindra V. Singh5,6Barbara E. Murray5,6
Kavindra V. Singh5,6Barbara E. Murray5,6 Alejandro Maass2,3,7
Alejandro Maass2,3,7 Verónica Cambiazo1,3
Verónica Cambiazo1,3- 1Laboratorio de Bioinformática y Expresión Génica, Instituto de Nutrición y Tecnología de los Alimentos, Universidad de Chile, Santiago, Chile
- 2Mathomics, Center for Mathematical Modeling, Universidad de Chile, Santiago, Chile
- 3Center for Genome Regulation (Fondap 15090007), Universidad de Chile, Santiago, Chile
- 4Instituto de Ciencias de la Ingeniería, Universidad de O’Higgins, Rancagua, Chile
- 5Division of Infectious Diseases, Department of Internal Medicine, University of Texas Health Science Center at Houston, Houston, TX, United States
- 6Center for Antimicrobial Resistance and Microbial Genomics, McGovern Medical School, University of Texas Health Science Center at Houston (UTHealth), Houston, TX, United States
- 7Department of Mathematical Engineering, Universidad de Chile, Santiago, Chile
The ferric uptake regulator (Fur) plays a major role in controlling the expression of iron homeostasis genes in bacterial organisms. In this work, we fully characterized the capacity of Fur to reconfigure the global transcriptional network and influence iron homeostasis in Enterococcus faecalis. The characterization of the Fur regulon from E. faecalis indicated that this protein (Fur) regulated the expression of genes involved in iron uptake systems, conferring to the system a high level of efficiency and specificity to respond under different iron exposure conditions. An RNAseq assay coupled with a systems biology approach allowed us to identify the first global transcriptional network activated by different iron treatments (excess and limited), with and without the presence of Fur. The results showed that changes in iron availability activated a complex network of transcriptional factors in E. faecalis, among them global regulators such as LysR, ArgR, GalRS, and local regulators, LexA and CopY, which were also stimulated by copper and zinc treatments. The deletion of Fur impacted the expression of genes encoding for ABC transporters, energy production and [Fe-S] proteins, which optimized detoxification and iron uptake under iron excess and limitation, respectively. Finally, considering the close relationship between iron homeostasis and pathogenesis, our data showed that the absence of Fur increased the internal concentration of iron in the bacterium and also affected its ability to produce biofilm. These results open new alternatives in the field of infection mechanisms of E. faecalis.
Introduction
Iron is an essential metal required for several bacterial species (Chandrangsu et al., 2017). Taking advantage of its redox activity, different enzymes use this micronutrient as a cofactor in order to conduct numerous functions such as electron transfer, electrophilic catalysis, nitrogenase, and hydrogenase reactions (De Domenico et al., 2008). At elevated concentrations, iron can be toxic to the cell. Through Fenton and/or Haber–Weiss reactions, iron reacts with hydrogen peroxide and superoxide generating reactive oxygen species (ROS), which are able to damage cell membranes, proteins, and DNA (Eaton and Qian, 2002). In terms of pathogenesis, one of the principal response mechanisms of humans to confront bacterial infection is the seizure of free iron, which prevents the successful acquisition of this metal by pathogenic microbes. In an attempt to limit iron availability to pathogens during infection, the host increases the production of iron storage molecules in the gut, in order to reduce the bioavailability of this metal (Leon-Sicairos et al., 2015).
Changes in iron availability require complex control over mechanisms involved in iron homeostasis (De Domenico et al., 2008). In bacteria, the ferric uptake regulator (Fur) is the primary transcription factor controlling components related to iron homeostasis (Hantke, 2001; Chandrangsu et al., 2017). Fur was first described in Escherichia coli as an iron-responsive repressor of iron uptake systems. The mechanisms of action of this regulator are highly conserved in multiple bacterial pathogens (Carpenter et al., 2009; Lee et al., 2014; Ludwig et al., 2015; Pelliciari et al., 2015), including Helicobacter pylori, Vibrio sp., Pseudomonas sp., Shigella flexneri, and Bacillus subtilis. This transcriptional factor represses its target genes when the intracellular concentration of iron exceeds threshold levels (Fuangthong and Helmann, 2003). In the absence of the metal, Fur-mediated repression is lifted and the iron uptake genes are transcribed (Skaar, 2010) increasing the chances to capture the metal and recover homeostatic levels.
Besides its participation in iron metabolism, Fur contributes to pathogenesis, acting as a positive regulator of genes that encode virulence factors and proteins with roles in oxidative and pH-mediated stress responses, supporting the importance of Fur in the transcriptional regulation of several metabolic processes (Foster, 1991; Troxell and Hassan, 2013; Porcheron and Dozois, 2015). In this context, global gene expression analyses have revealed a novel role of Fur. This protein can function as a transcriptional activator or repressor through direct (promoter interaction) and indirect mechanisms (small regulatory RNAs) (Masse et al., 2007; Yu and Genco, 2012; Oglesby-Sherrouse and Murphy, 2013). In E. coli, besides transcriptional control over iron homeostasis systems, Fur regulates a total of 42 operons related to DNA synthesis, energy metabolism, and biofilm development, revealing the full transcriptional regulatory potential of this regulator (Seo et al., 2014). These data strongly support the fact that the absence of this regulator not only affects iron homeostasis, but also induces global cellular adjustments which impact several cell processes.
Enterococcus faecalis is a facultative anaerobe, Gram-positive bacterium, a common member of the gastro-intestinal tract microbiota. Since the early 90s, it has emerged as a major cause of local or systematic nosocomial infections, including urinary tract and abdominal infections, bacteremia, wound infections, and endocarditis (Arias and Murray, 2012; Guzman Prieto et al., 2016). In previous work, the first global transcriptional response of this bacterium to a non-toxic iron excess condition was described (Lopez et al., 2012). The data allowed authors to conclude that, in E. faecalis, iron acts as a complex stimulus capable of impacting diverse genes encoding for proteins involved in several metabolic processes, suggesting the presence of transcriptional regulators (among them Fur) which play a crucial role in coordinating gene expression under an iron excess condition.
Considering that E. faecalis is one of the principal hospital-acquired pathogens and the importance of the control of iron homeostasis during pathogenesis, we identified and characterized the transcriptional regulator Fur of E. faecalis in this article. To do this, Fur was removed from the bacterium, a strategy that allowed us to determine Fur gene targets. Fur regulon is limited to genes encoding for iron uptake proteins, unlike in other species, where Fur also controls the expression of genes involved in other metabolic processes. Next, using a systems biology approach, we determined the relevance of Fur over the global transcriptional response of E. faecalis under iron deficiency and excess conditions, showing that this bacterium has the capacity to reconfigure its gene network, activating others transcriptional regulators in order to maintain iron homeostasis.
Results and Discussion
Fur Regulon in Enterococcus faecalis
Members of the Fur family of transcriptional regulators are conserved across bacterial species. In particular, the E. faecalis OG1RF genome contains three putative regulators in this family, Fur (iron homeostasis), Per (oxidative stress response), and Zur (zinc homeostasis) (Lopez et al., 2012). The Fur protein (149 aa) is encoded by a monocistronic gene (EF1525) highly conserved (more than 50% identity) across species of the Lactobacillales order (Supplementary Figure S1). Crystal protein models of Fur denote that this regulator acts as a dimer (Pecqueur et al., 2006; Sheikh and Taylor, 2009). Each chain possesses two main domains (A and B) involved in DNA-binding and dimerization; each monomer contains three putative metal binding sites, all of them able to coordinate zinc or iron atoms. In order to in silico validate the predicted gene annotation, we modeled Fur of E. faecalis using the closed conformation of Fur from Vibrio cholerae (over 30% identity and 50% similarity between both species’ protein sequences). The resulting model in E. faecalis describes an asymmetric dimer conformation with the presence of the DNA-binding and dimerization domains (Figure 1). In addition, each monomer contained three metal domains located in the same orientation as the crystal model of V. cholerae, conserving the specific residues involved in iron coordination. The coherent tertiary homology between the model and the crystal strongly suggests that the EF1525 gene from E. faecalis codes for the Fur transcriptional factor.
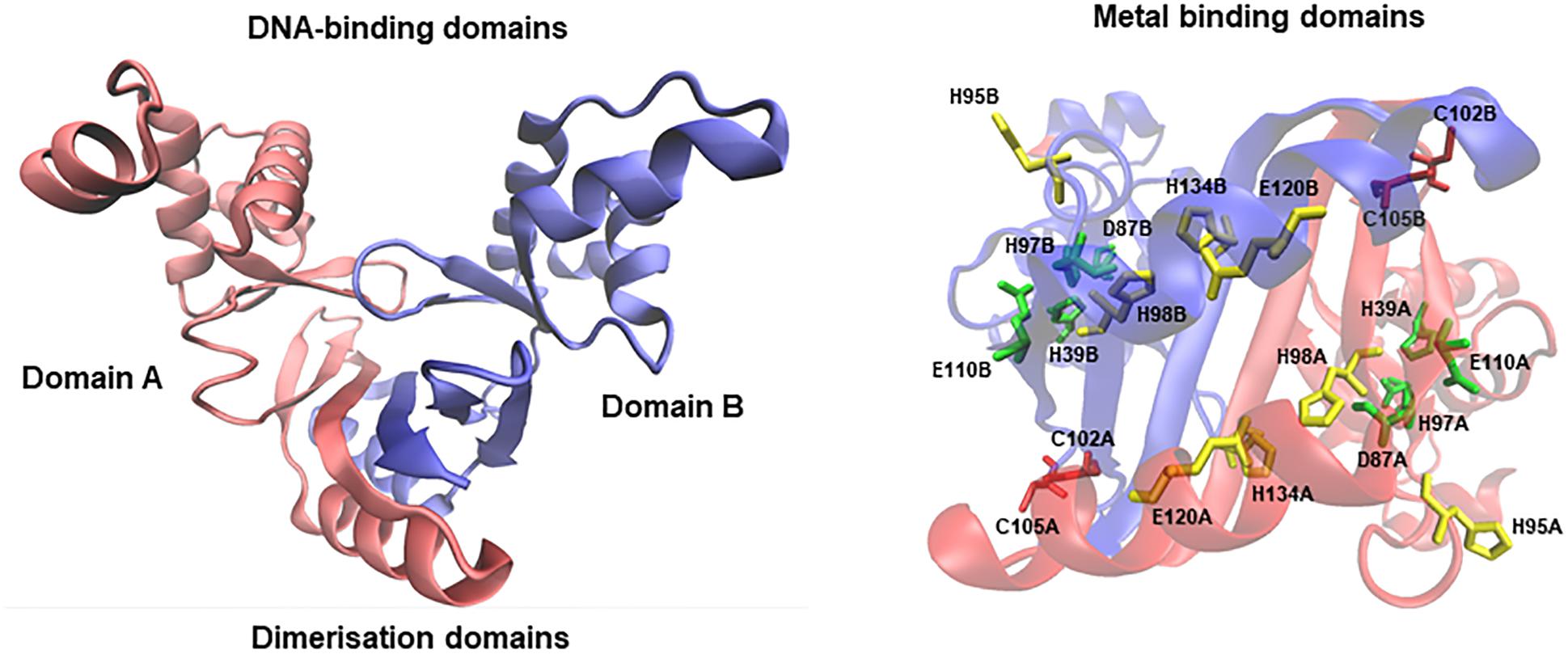
FIGURE 1. Homology model of Fur from E. faecalis. Red and blue chains structures denote domain A and B, respectively. Each domain includes from residues 10 to 142. The stereo view represents the three putative zinc/iron binding domains (conserve residues in red, green, and yellow).
A previous in silico strategy identified three putative transport systems involved in iron homeostasis controlled by Fur in E. faecalis, the operons fhuDCBG (ferrichrome transporter), feoAB (ferrous iron transport), and yclQPN (iron compound ABC transporter) (Latorre et al., 2014). To confirm if these three operons are direct targets of Fur, we generated a null deletion mutant of this protein (Δfur) in E. faecalis, to later determine the transcript abundance of the three iron uptake operons between the wild type and the mutant strain. The qPCR assay showed that all iron uptake genes had at least a fivefold higher expression level in Δfur than in the wild type strain of E. faecalis (Table 1). These results suggest that, in a growing media without adding (excess) or removing (deficiency) iron (i.e., control condition), Fur represses genes involved in iron uptake in E. faecalis, supporting the previous in silico prediction and its transcriptional regulatory action in this bacterium as described in other organisms (Troxell and Hassan, 2013).

TABLE 1. Changes in the relative abundance of Fur regulon genes.
Iron Homeostasis Effects of Fur Absence
Iron homeostasis can be defined as a healthy control of the internal concentrations of this metal (De Domenico et al., 2008). In order to determine the influence of Fur over iron homeostasis in E. faecalis, we evaluated if the absence of the Fur impacted the viability of the bacterium growing under different scenarios of iron exposure.
No differences in the growth rate were observed between the wild type and Δfur strains (Figure 2A), suggesting that the absence of Fur does not impact the viability of the bacterium, even when exposed to an excess or a deficit of the metal (Supplementary Figure S2). This result is consistent with results in other bacterial species, where the removal of Fur has minor effects on growth in vitro (Pasqua et al., 2017). Same phenotype occurs with other metal regulators in E. faecalis, such as CopY (copper homeostasis), where the deletion of this transcription factor does not affect the viability of the bacterium under different copper treatments (Reyes-Jara et al., 2010).
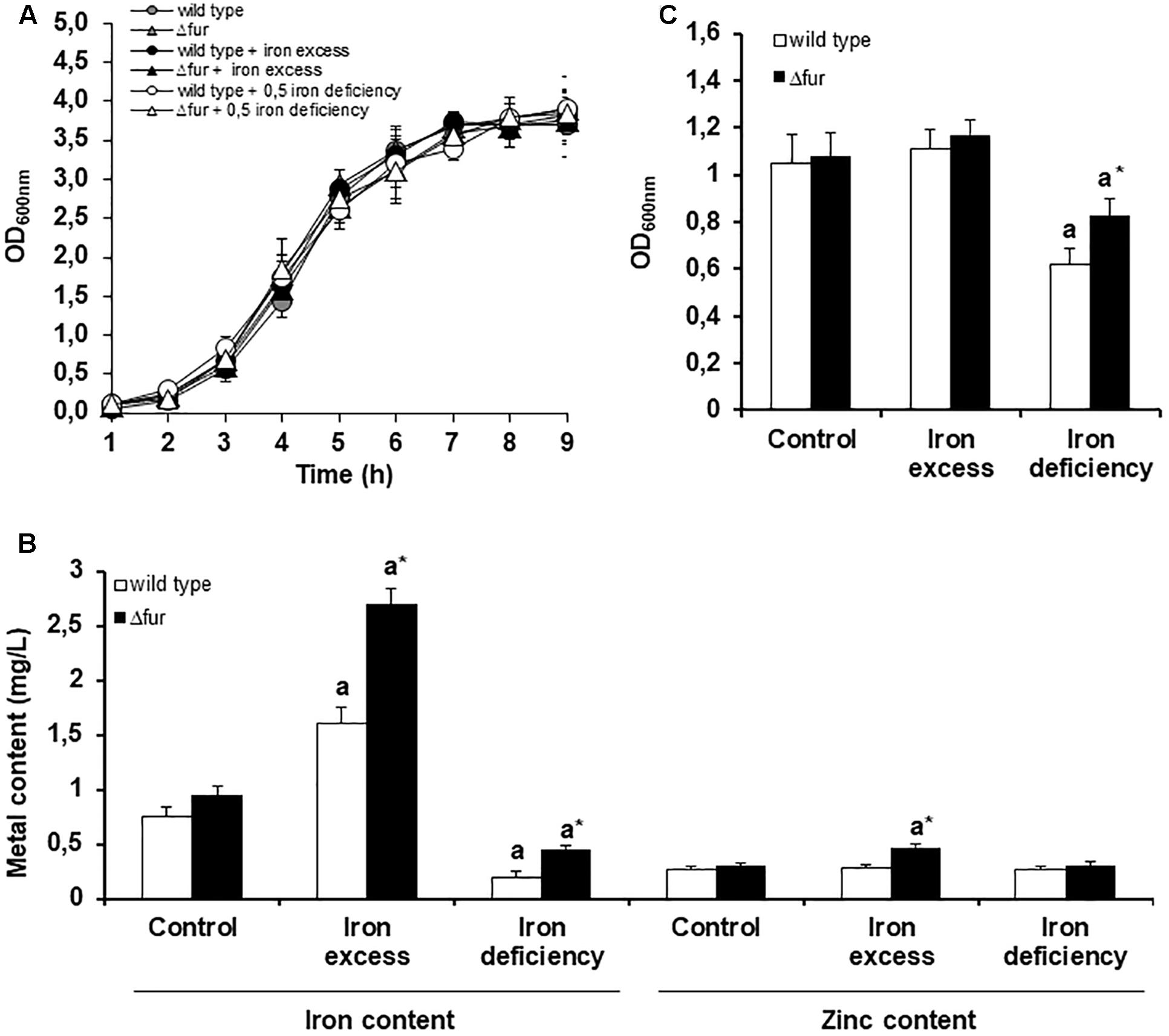
FIGURE 2. Homeostatic response of E. faecalis wild type and Δfur (Fur mutant) under different iron conditions. (A) Growth curve of E. faecalis wild type and Δfur under three iron exposure conditions. (B) Biofilm assay, optic density was measured after 24 h. (C) Bacterial metal content measured after 3 h of iron exposure. Letter a denotes significant difference between the control media and iron excess or deficit for the same strain. Asterisk (∗) marks significant difference between the wild type and Δfur for the same growing condition of iron. Error bars represent standard deviation (SD) values. Three biological replicates (Mann–Whitney test, p < 0.05).
As shown in Figure 2B, exposure of wild type and Δfur mutant strain to excess iron resulted in a significant increase in the internal concentration of iron and zinc. On the contrary, iron deficit reduced the internal concentrations of iron and zinc. Considering that the mutant strain was constantly expressing the iron uptake systems, an increase in the internal concentration of iron was expected. In particular, the increase in zinc in the mutant strain exposed to iron excess can be explained as a requirement of this essential micronutrient as a cofactor in several oxidative stress enzymes and transcriptional factors. A similar phenotype occurs when the bacterium is exposed to other metals with redox activity (Latorre et al., 2014).
The capacity of E. faecalis to produce biofilm is directly correlated with the ability to colonize organs (Nallapareddy et al., 2006; Anderson et al., 2015). In order to study the participation of Fur in the pathogenesis of E. faecalis, a biofilm assay was performed growing the wild type and (fur strains at different conditions of extracellular iron. As shown in Figure 2C, under iron deficit, the (fur strain compared to wild type showed an increase in the production of biofilm. Previous information indicates that Fur mutants in Pseudomonas aeruginosa under low concentrations of iron are able to organize a more mature biofilm, a response likely triggered by an indirect signal effect produced by changes in the internal concentration of the metal (Banin et al., 2005). As showed in Figure 2B, (fur strain contained a higher internal concentration of iron compared to wild type following growth of both under the deficit condition. This difference may be triggering the higher production of biofilms as noted in P. aeruginosa.
Transcriptional Response Activated in Absence of Fur
As mentioned, besides the transcriptional control over genes participating in iron homeostasis, Fur target genes also encode proteins involved in basal metabolism, energy production, and pathogenesis, classifying this regulator as a global transcription factor in several bacteria (Quatrini et al., 2007; Troxell and Hassan, 2013; Seo et al., 2014). To identify new targets of Fur in E. faecalis, we performed RNAseq comparing the global transcriptional abundance between the wild type and Δfur strain, both growing in a control media without an iron excess or deficit condition. Besides the components involved in iron uptake, 65 transcripts significantly changed in abundance (40 upregulated and 25 downregulated) in the absence of Fur (putative genes targets of Fur, Supplementary Table S1). As expected, transport, and basal metabolism were over-represented in relation to the total functions present in the genome, highlighting genes coding for proteins involved in DNA replication, metal transport, and stress response (Figure 3A).
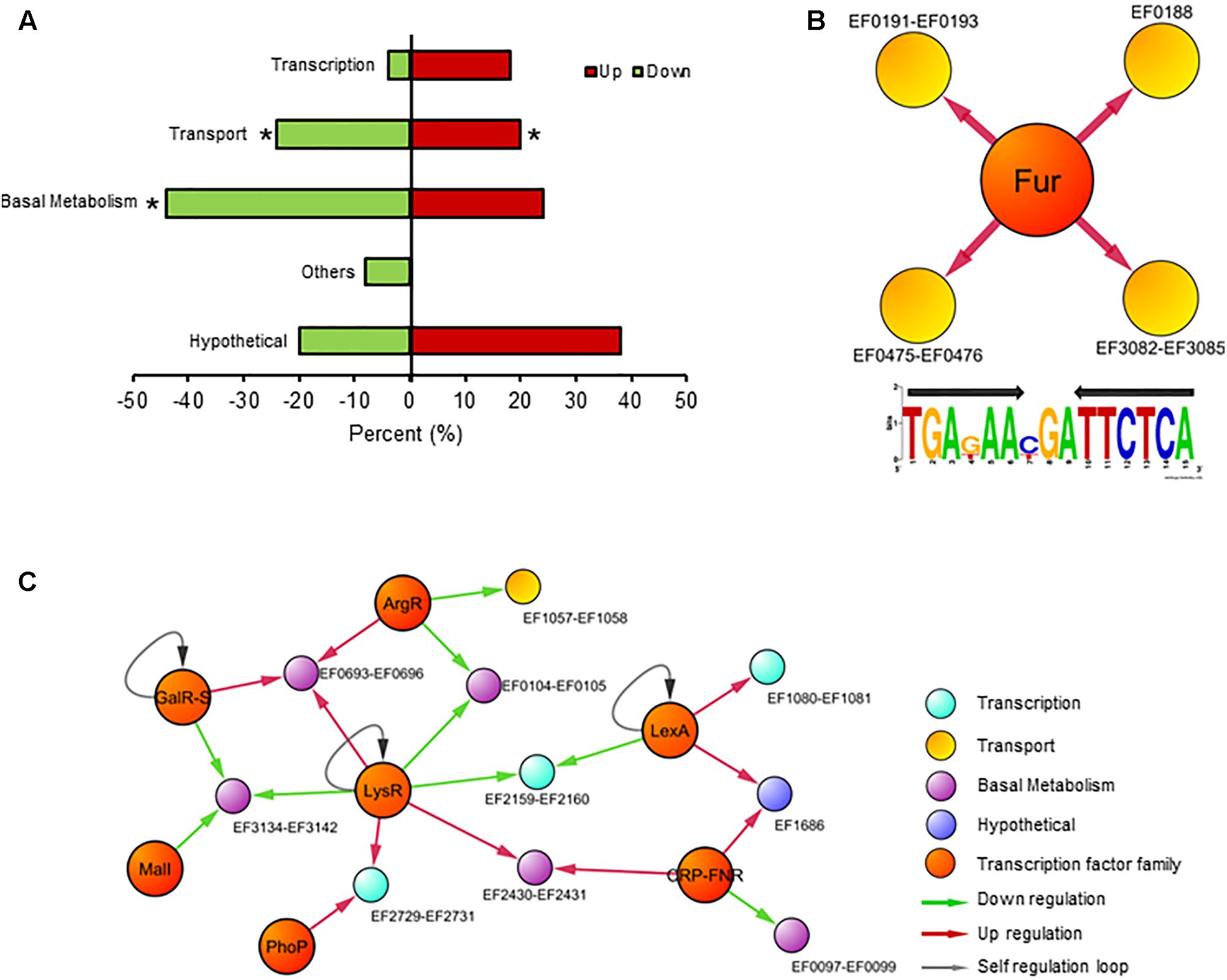
FIGURE 3. Transcriptional response in the absence of Fur. (A) Global transcriptional analysis of E. faecalis wild type and Δfur growing in the iron control condition. RNAseq differentially expressed genes classify by COG super-class categories. Color bars represent the upregulated or downregulated genes in Δfur compared to the wild type. Asterisk denotes significant enrichment in the bacterial genome (p (0.05) compared to total proportion. (B) Fur regulon of E. faecalis. Consensus logo of Fur regulon created with Fur DNA sequence found in promoters of EF0188, EF0191–EF0193, EF475–EF0476, and EF3082–EF3085 operons (the black arrow indicates palindromic sequence). (C) The graph contains 23 nodes (14 operons, 8 transcription factor families) connected by 26 edges (putative binding sites). Color circles represents COG super-class categories.
The next step was to examine whether the classic DNA-binding site sequence of Fur, a palindrome of 15 nucleotides (TGAnAAnnnTTnTCA), was present in the set of 65 genes differentially expressed in the Δfur strain. As mentioned, this sequence is present in all the promoter regions from iron uptake operons in E. faecalis. In a recent article, six new DNA Fur binding motifs were identified in E. coli (Seo et al., 2014). These new DNA Fur binding motifs from E. coli (including the canonical Fur binding site) were searched on the promoter regions of the 65 putative genes targets of Fur. None of these new motifs were found on the first 300 bp upstream from the set of putative Fur target genes, suggesting that this set of 65 genes likely respond to secondary effects induced by the removal of Fur (e.g., an increase in internal iron concentration) and not to direct regulation by this transcription factor. This result suggests that Fur in E. faecalis possessed a high degree of specificity to regulate genes involved in iron homeostasis (feo, fhu, and ycl operons) (Figure 3B). This characteristic is in concordance with previous results obtained in CopY and Zur of E. faecalis, where these two metal regulators are exclusively confined to control the expression of genes involved in copper and zinc homeostasis, respectively (Latorre et al., 2015).
This result suggests the presence of other transcriptional factors controlling these 65 genes, probably activated in order to counteract a putative secondary effect induced by the absence of Fur. In order to identify putative regulators controlling the transcription for these components, the list of differentially expressed genes was integrated into the global regulatory network (Figure 3C). The resulting model contains 7 transcription factor families and 10 transcriptional units (10 operon, 29 genes), covering almost 50% of total transcriptional changes.
Members of the LysR (basal metabolism) and LexA (DNA repairs) families are classic regulators activated during stress conditions (Schell, 1993; Butala et al., 2009), probably sensing in the global regulatory model the increase in the internal iron concentration present in Δfur. In addition, transcription factors related to general metabolism also are represented in the network. For example, the CRP–fumarate nitrate reductase (FNR) family modulates gene expression encoding for L-serine dehydratase (EF0097–EF0098). This enzyme participates in several process such as cell wall biosynthesis, amino acid synthesis and even host colonization, which principal characteristic is utilized by pyridoxal 5′-phosphate (PLP) or [Fe-S]cluster as cofactors (Thoden et al., 2014). In addition, some members of the CRP–FNR family of regulators also contain this iron cluster (Kiley and Beinert, 2003). The production and assembly of [Fe-S] clusters are indirectly controlled by Fur action (Roche et al., 2013), supporting the configuration of the transcriptional network model showed in Figure 3C to E. faecalis.
The absence of Fur impacted the transcriptional regulation of the bacterium in two ways. A direct regulation of the genes involved in iron uptake (three-independent systems), that comprise the Fur regulon in E. faecalis. The upregulation of these genes explained the increase in internal iron concentration produced in Δfur compared to the wild type strain. This change did not disturb the viability of the bacterium, however, it did induce transcriptional changes of genes related to basal metabolism and DNA repair, indicating a secondary effect of the absence of Fur in E. faecalis. In particular, the operons EF1218–EF1223 and EF3204 encoding for spermidine/putrescine transports and cobalamin synthesis proteins, respectively, were upregulated in the Δfur strain (Supplementary Table S1). Both systems participate in oxidative stress protection in bacterial species (Tkachenko et al., 2012; Ferrer et al., 2016), these genes are induced in E. faecalis probably as a response to the increase in internal iron concentration observed in the mutant compared to the wild type strain. In addition, the gene nifU (EF2391) was also is induced in the Δfur strain. The protein NifU provides the molecular scaffold for the assembly of [Fe-S] clusters (Dos Santos et al., 2004). The induction of nifU in the mutant strain reaffirms a previous observation from the network model, that Fur in E. faecalis, besides its participation in the regulation of iron uptake genes, also has an indirect effect over the transcriptional activation of genes encoding for [Fe-S] proteins.
Global Transcriptional Regulatory Network Activated in the Absence of Fur Under Different Iron Conditions
Finally, the capacity of E. faecalis to reconfigure its global transcriptional response in order to cope with the absence of Fur under different scenarios of iron availability was tested. First, global expression data were obtained from E. faecalis wild type and Δfur growing under two different iron treatments, deficiency (0.5 mM BDP for 3 h) and excess (0.5 mM FeCl3 for 3 h) (Figure 4A). In general, the global transcriptional response between the wild type and the mutant was similar after being exposed to the same iron treatments, highlighting the upregulation or downregulation of genes coding for proteins involved in transcription (i.e., gene regulators and nucleotide synthesis), basal metabolism and transport under the iron deficiency condition. A core of 16 genes changed in transcriptional abundance (with respect to the wild type strain growing in control media) under all iron treatments in both strains. In terms of the impact of the absence of Fur on the global expression of E. faecalis growing under iron excess condition, a total of 67 genes responded in this condition. On the other hand, during deficiency of the metal, 78 genes changed their transcript abundance (48 upregulated and 30 downregulated) in the Δfur mutant strain only. In the deficit scenario, the absence of Fur primarily activated nucleotide pathways (Figure 4B), which is consistent with the high number of transcriptional factors activated in this condition.
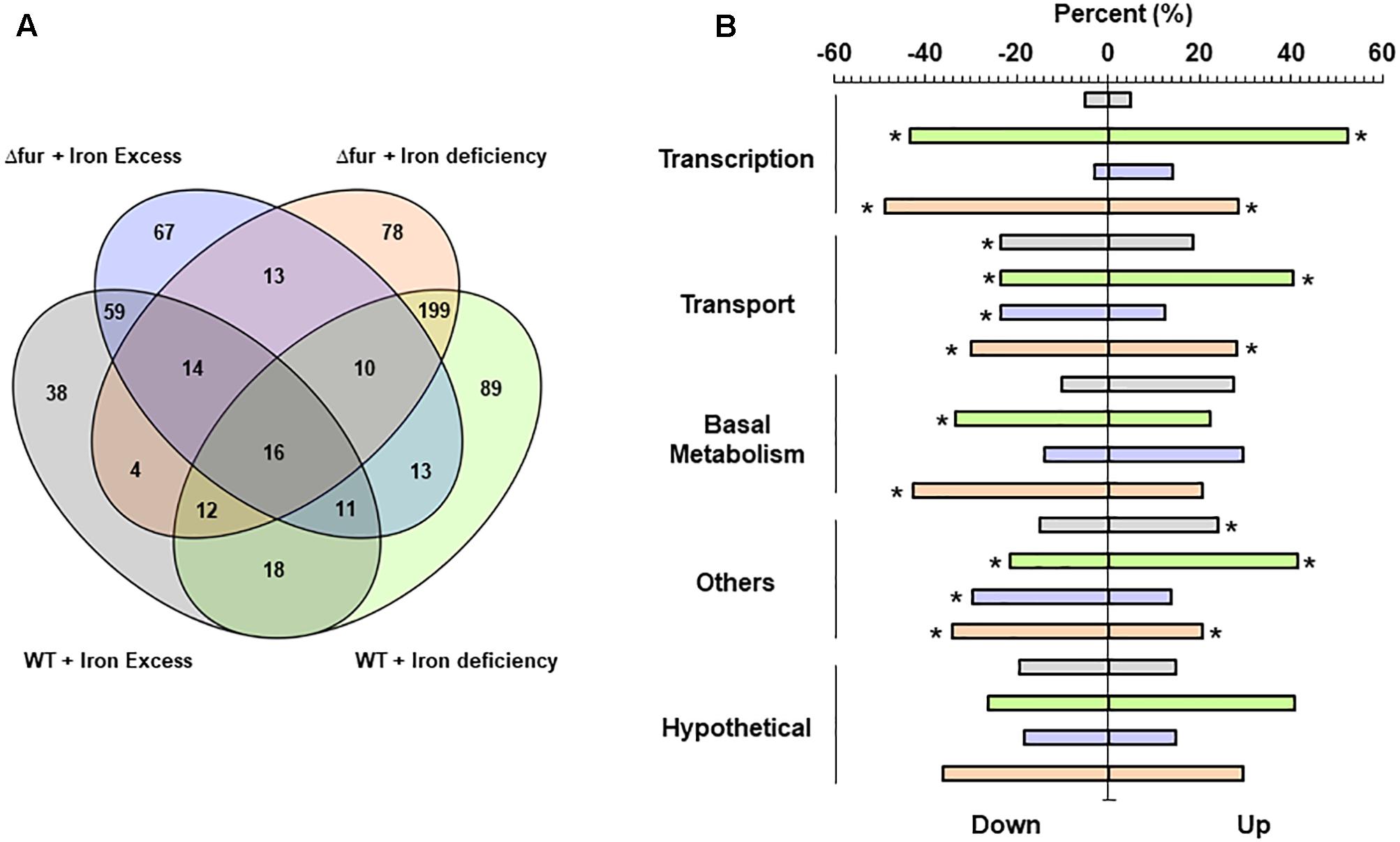
FIGURE 4. Global transcriptional analysis of E. faecalis wild type and Fur mutant growing at different iron treatments. (A) Venn diagrams. Numbers indicate the total genes activated on each condition. (B) RNAseq differentially expressed genes classify by COG super-class categories. Color bars represent the two E. faecalis strains growing under iron excess or deficiency. Asterisk denotes significant enrichment (p (0.05) compared to the total proportion in the bacterial genome.
The exposure to 0.5 mM of iron during 3 h induced changes in a total of 172 and 203 genes in the wild type and Δfur strain, respectively. The global gene expression of E. faecalis OG1RF wild type treated with a concentration of 0.5 mM of iron during 6 h of exposure has been previously described (Lopez et al., 2012). This chronic non-toxic iron treatment activates a total of 475 genes, increasing internal iron content more than four times compared to the control growing condition. As showed in Figure 2B, the wild type strain treated with 0.5 mM of FeCl3 for 3 h duplicated the internal concentration of iron. The internal amount of iron in Δfur tripled under the same condition. These significant differences in the internal concentration were correlated with an increase in the number of genes activated, where E. faecalis likely responds to a gradual rise in the secondary effects generated by excess iron, such as oxidative stress.
In previous work, the global response of E. faecalis to defibrinated horse blood was described using an experimental condition where the bioavailability of iron was reduced, homologating a metal deficiency scenario. Interestingly, from the total activated genes during the exposure to 0.5 mM BDP in the wild type and Δfur, 40% (158 genes) and 50% (176 genes), respectively, changed their transcript abundance during the blood exposure. These results suggest that, under an iron deficiency scenario, E. faecalis activates an important number of common genes, which may be involved in similar global metabolic adjustments in order to maintain the viability and iron homeostasis of the bacterium.
In relation to other bacteria, global expression analysis in Caulobacter crescentus and the human pathogen Vibrio vulnificus growing in iron-limited conditions, indicate that most of the observed changes belong to genes participating in transport, highlighting the TonB and TomB system (Alice et al., 2008; da Silva Neto et al., 2013). In particular, these protein complexes are located in the outer membrane, which is absent in Gram + bacteria such as E. faecalis. Nevertheless, E. faecalis activates a large set of ABC transporters under the iron deficiency condition, a complementary mechanism to Fur regulon that is also able to incorporate iron into the cell (Moeck and Coulton, 1998; Koster, 2001). E. coli and Geobacter sulfurreducens have a similar global transcriptional response to different iron concentrations showed by E. faecalis, activating an important number of genes involved in transport and basal metabolism, components mainly participating in energy production (McHugh et al., 2003; Embree et al., 2014). Another global gene expression study performed in the pathogen Pasteurella multocida, indicates that iron deprivation prompts the expression of a component involved in antibiotic resistance (penicillin-binding proteins) (Paustian et al., 2001). During the iron deficit scenario, E. faecalis wild type and Δfur upregulated the genes EF1055, EF1943, and EF2068, all encoded for drug resistance proteins (Supplementary Table S1). In addition, virulence genes in E. faecalis, such as collagen adhesion protein (ace-EF1099), glucose starvation protein (gls24-EF0079), and the cell wall surface anchor family (Ebp system-EF1092–1093) are up or downregulated under the different iron conditions both in the wild type and Δfur strains.
Figure 5 presents a global transcriptional regulatory network activated by different conditions of iron in the wild type and Δfur. The model contains a total of 181 edges connecting 153 nodes, 21 regulator families, and 139 operon (330 total genes included), covering 60% of the total differentially expressed genes obtained from the RNAseq assays. The topological analyses describe a consistent and reliable network, with an in-degree coefficient value of 3.19 showing a classical power law distribution present in other biological networks (Albert, 2005) and also present in previous models activated by copper and/or zinc in E. faecalis (Latorre et al., 2014, 2015).
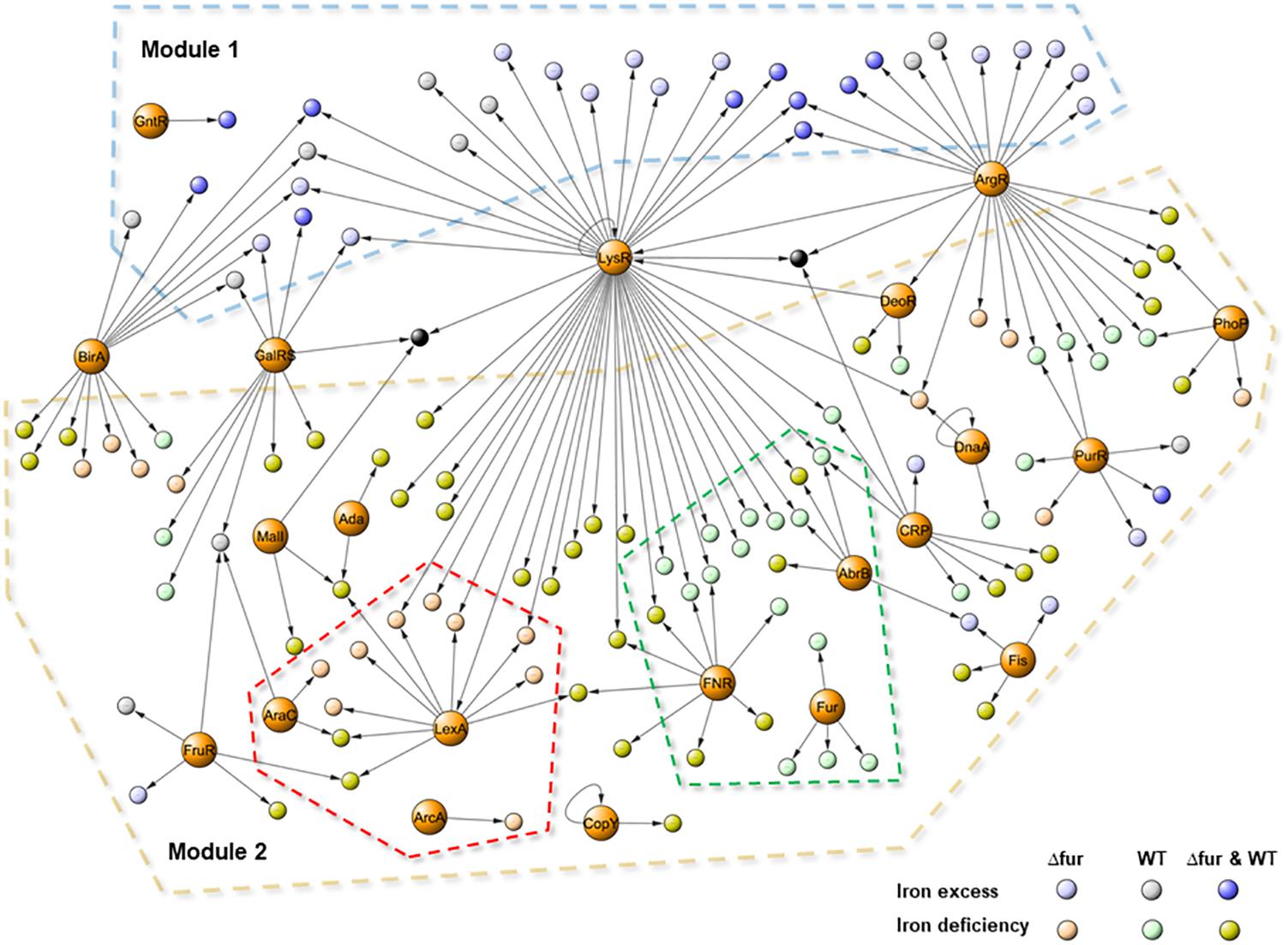
FIGURE 5. Transcriptional regulatory network activated by iron excess and deficiency in E. faecalis wild type and Δfur (Fur mutant). The graph contains 153 nodes (139 activated operons and 21 family of regulators) connected by 181 edges (putative binding sites). Color circles represent differentially expressed responses of each operon in the RNAseq experiment. Dash lines indicate: Module 1 (iron excess) in light blue, Module 2 (iron deficiency) in light brown, wild type sub-network in light green and Δfur in light pink.
Based on the number of gene targets, the network contained three types of transcriptional factors. Global regulators (LysR, ArgR, GalRS, and BirA), controlling 10 or more operons coding for several metabolic process, primarily related to basal metabolism. Local regulators, controlling the expression of a small number of genes coding for proteins participate in the same processes (Ada, DeoR, DnaA, ArcA, GntR, CopY, and Fur), and the rest of the regulator families can be classified as master regulators, controlling between 5 and 10 gene targets functionally related to each other. In terms of transcriptional activation, the model had two main modules. Module 1 contained genes activated during the iron excess condition and Module 2 contained genes activated during the deficit of this metal. Global regulators control genes from both modules; in particular, the ArgR (arginine metabolism) and LysR (general metabolism) families were also present in copper and zinc network models (Latorre et al., 2014, 2015). These regulator families have been shown to respond to different stimuli, explaining the activation under different metal conditions, which is also extremely important to control the global structure of the network.
The model contained two operons activated under all the studied conditions (black nodes in the center of the model). Both operons, EF3134–EF3142 (PTS system) and EF0764 (hypothetical protein), had high regulation complexity, and were controlled by at least three global regulators which may explain induction under iron excess and deficit. In addition, PTS systems have also been correlated with the virulence capacity of E. faecalis (Sauvageot et al., 2017). Activation in different iron conditions opens an interesting field to study the relationship between pathogenesis, metal homeostasis, and gene regulation in E. faecalis.
It has been demonstrated that the transcriptional network activated during copper excess is characterized by the activation of two modules, one specifically related to copper detoxification (cop operon) and a second one involved in general metabolism (Latorre et al., 2014). During iron excess, the activation of Module 1 was mainly controlled by global regulators, supporting the requirements of energy production and synthesis of basal components. As opposed to copper, the specific mechanism involved in iron detoxification in bacteria is currently unclear. Non-specific bivalent cation transporters such as cation diffusion facilitators (CDF) may play a role, however, no transcriptional regulatory factors controlling this element were identified in the network presented in Figure 5.
With respect to the differences between the numbers of genes activated by iron excess or deficit in both strains, Module 2 present this information covering the mayor proportion of the network. In addition to the global regulators, this module contained a set of families of transcriptional factors which were in turn controlling genes induced by iron excess, such as CRP, PurR, FruR, and Fis. These families are classified as global regulators participating in basal metabolism, energy production and DNA topology, and were also activated during the zinc and copper treatments, supporting participation in metal metabolism. Inside Module 2, it was possible to identify two sub-networks. The first one was composed of genes activated in the wild type during iron deficiency (dashed green line in the model). Besides the activation of the four operons involved in iron uptake controlled by Fur, this module contained the regulator FNR, a major repressor of anaerobic metabolism that contains an oxygen-responsive [Fe-S] cluster (Carpenter and Payne, 2014). The inactivation of this protein lies in [Fe-S] stability; the intracellular iron deprivation generated by BDP likely impacted the amount of this metal to produce this cluster, explaining the activation of the gene target of FNR present in the sub-network.
The second sub-network of genes activated only in Δfur by iron deficiency described the specific reconfiguration of the network in order to cope with the absence of Fur. Inside this sub-network, the presence of LexA, which controls the greatest number of genes principally involving in DNA repair and oxidative stress damage, was highlighted. Disruption of LexA in Cyanobacterium synechocystis increases the expression of genes involved in iron uptake (Kizawa et al., 2016). The same phenotype occurred in E. faecalis Δfur, supporting the participation of LexA as a compensative transcriptional mechanism during the iron deficiency condition.
Conclusion
The transcription factor Fur is one of the most studied regulators in bacterial species. Recently, it has been established that Fur is not restricted to iron uptake systems, but is also involved in the control of genes coding for DNA synthesis, energy metabolism, and biofilm formation. Thus, the importance of this regulator beyond the iron homeostasis is clear. In this work, we presented a full transcriptional and metal homeostatic characterization of Fur in E. faecalis. The in silico approach validated through experimental expression analyses indicated that the unique genes controlled by Fur are part of the iron uptake mechanism.
Unlike other bacterial species, E. faecalis allocates specific metal regulators (such as Fur, CopY, and Zur) to the transcriptional control of proteins related to metal homeostasis, conferring to the system a high level of efficiency and specificity under different metal availability scenarios. Interestingly, the results showed that the absence of Fur affects internal iron concentration and activated iron uptake systems. The absence of Fur also impacted the internal concentration of other micronutrients (like zinc) and induced the transcriptional activation of the cop operon (involved in copper homeostasis). These findings strongly suggest cross-talk between iron, copper and zinc at different homeostatic levels.
The systems biology approach allowed the identification of the first transcriptional regulatory network activated by iron in bacterial species described to date. The iron excess condition primarily activated genes and regulators related to basal metabolism. This response was increased when Fur was deleted, activating more elements principally involved in energy production and [Fe-S] proteins, a response that is consistent with the necessity to control the increase of intracellular iron in the mutant strain. On the other hand, iron limitation induced a large set of ABC transporter, optimizing iron acquisition through alternative uptake mechanism such as iron-siderophore transporter.
Besides the direct influence over iron uptake mechanisms, the removal of Fur from the system activated other regulators (which also responds to changes in copper availability), denoting a complex transcriptional reconfiguration in this bacterium to cope with the absence of Fur under different iron availability scenarios, and supporting the idea of a transcriptional interplay between different metals. A core of 16 genes changed in transcriptional abundance under all iron treatments conditions in both strains. This set of elements can be classified as general iron response genes, which are able to respond to changes in the intracellular concentration of the metal, most of which code for transporters (ABC and PTS) and hypothetical proteins.
In P. aeruginosa and Staphylococcus aureus, iron deficiency leads to the activation of several virulence factors (Kim et al., 2003; Oogai et al., 2011). On contrary, virulence factor gene induction is directly correlated with an increase in iron concentration in Pseudomonas syringe (Kim et al., 2009). In E. faecalis, the complex response of the virulence genes under the different iron conditions is consistent with current knowledge, supporting that there is no a direct relationship between the transcriptional response of bacterial virulence factors and iron availability.
The high number of genes changing their transcript abundance, in particular during the iron deficiency condition, coupled with the activation of new gene regulators, supports the idea that the effects of the loss of control in iron uptake by the removal of Fur lead the bacterium to reconfigure its global transcriptional regulation. In this context, the final analysis involved the integration of the RNAseq data into the global transcriptional regulatory network of E. faecalis, a strategy which allowed us to identify common and specific gene regulatory mechanisms that were activated in all studied cases.
Finally, bearing in mind that E. faecalis is one of the principal hospital-associated pathogens, the characterization of Fur in this bacterium opens a new alternative to study the participation of this regulator in pathogenesis. Likewise, considering the direct relationship between iron homeostasis and bacterial infection, the transcriptional network models presented in this work provide a list of putative candidates (genes and regulators) involved in iron adaptation, providing new alternatives to study other mechanisms related to pathogenesis in E. faecalis.
Materials and Methods
Enterococcus faecalis Fur Deletion
Enterococcus faecalis non-polar deletion mutant of fur was constructed using the PheS∗ system (Kristich et al., 2007). Two fragments of ca. 1000 bp located upstream and downstream of target gene were amplified by PCR using: up region: fur_up_s CGCGCGGCCGCTTGTGGATGTTGGCTTACCA and fur_up_a ACCATGAAAATACAGCTCCTTGTTTAGAATGATTACA; down region: fur_down_s AGGAGCTGTATTTTCATGGTATTTGTCAAAGTTGT and fur_down_a CGCGTCGACACTAGCCCAACAATGTTGGC primers. The amplicon was first cloned in pGEM-T Easy (Promega) and then inserted in pCJK47. The resulting constructs were transferred to E. faecalis OG1RF (wild type strain) by electroporation (single cross over insertions). The loss of the plasmid was selected on medium MM9YEG agar supplemented with 10 mM p-Cl-Phe and X-gal 200 lg/ml. Mutant strains (E. faecalis OG1RF Δfur) were verified by sequencing (deletion area) and pulsed with field gel electrophoresis.
Culture Conditions, Growth Curves, and Biofilm Assay
Enterococcus faecalis OG1RF (wild type) and Δfur were grown in N medium (Peptone 1%, yeast extract 0.5%, Na2HPO4 1%, and glucose 1%) (Reyes-Jara et al., 2010). For all experiments, strains were independently cultured overnight in N medium broth at 37°C. The next day, each strain was diluted in parallel cultures to a final OD600 nm of 0.05 and then grown at 37°C and 160 rpm. For growth curves, iron cultures were supplemented with concentrations of 0 (iron normal), 0.25, 0.5, 1, 2, 5, 10, and 15 mM of FeCl3 (iron excess), and 0.25, 0.5, 1, 2, 5, 10, and 15 mM of 2,2′-bipyridyl iron chelator (BPD, iron deficiency) in order to determine non-lethal conditions. Bacterial growth was monitored each hour over 8 h and at 24 h by OD600 nm. Biofilm assay were carried out as previously described (Mohamed et al., 2004). Briefly, strains were grown overnight, diluted 1:100 in 200 μl N medium and inoculated into polystyrene microtiter plates. After 24 h of static incubation at 37°C, attached cells from plates were processed, fixed with Bouin’s fixative for 30 min, stained with 1% crystal violet for 30 min, and rinsed with distilled water. OD600 nm was then determined. Each assay was performed in triplicate.
Intracellular Metal Content
After the iron treatments (control, deficiency or excess), the intracellular metal content for each culture was determinate as described below (Latorre et al., 2011). Both strains growing in the different iron conditions and at the same growth stage (mid-exponential, 3 h of growth cultures according to the growth curves), were centrifuged and then suspended in 200 μl of HNO3 (Merck) and digested during 24 h at 65°C. After the acid lysis, total metal composition was determinate by total reflection X-ray fluorescence (TXRF). The data were expressed as microgram per liter, normalized per total cell culture (absorbance) and represent the average value of three measurements for independent biological replicates. The statistical analysis was done using an ANOVA test (p < 0.05).
Transcriptomic Experiments
For RNAseq assays, total RNA was isolated from three replicate cultures from each strain grown to mid-exponential phase (OD600 nm ∼ 1.5) in N medium (control) and supplemented with 0.5 mM of FeCl3 or 0.5 mM of 2,2′-bipyridyl using the RNeasy kit (Qiagen) as previously described (Reyes-Jara et al., 2010). For RNAseq analysis, each Illumina FASTQ library was first processed by FASTX to remove low complexity and low quality reads. EDGE-pro v1.3.1 software (Magoc et al., 2013) was used to align the reads to E. faecalis OG1RF complete genome1 using Bowtie2 (Langmead and Salzberg, 2012). The gene expression estimation was directly obtained from the alignment output. Protein translation table with coordinates of protein coding genes (.ptt) files were generated using in-house perl scripts and were used as inputs into EDGE-pro. In order to generate the RPKM file, EDGE-pro was run with default parameters except for the read length (-l101). Samples were concatenated through edgeToDeseq.perl script provided by EDGE-pro. DESeq was used to identify the gene expression level. Differentially expressed genes between the wild type and Δfur strains growing in the control condition, and wild type or Δfur 0.5 mM of FeCl3 and 0.5 mM of 2,2′-bipyridyl were considered significant based on a p-value ≤ 0.01 and FDR of 2. Complete data is accessible in the Supplementary Information.
All qPCR assays and data analyses were conducted as described in previous work (Reyes-Jara et al., 2010). All experiments were carried out in triplicate using three-independent RNA samples. The results were expressed as the fold change between wild type strain and Δfur. qPCR expression values were normalized using the gdh reference gene (EF1004). Statistical analyses were assessed using REST2009 (Pfaffl et al., 2002).
Bioinformatics
Fur protein homologous in Lactobacillales order organisms were obtained using BlastP from the National Center for Biotechnology Information (NCBI) website (template E. faecalis Fur EF1525). Global Fur alignments were performed using Clustal W (Hirosawa et al., 1995). The Fur molecular 3D model was generated by SWISS-MODEL program (Guex and Peitsch, 1997), using the crystallographic model information from V. cholerae Fur (PDB Id 2W57) as a template. The final model was displayed with VMD v1.9.4 software (Humphrey et al., 1996).
Transcriptional regulatory networks activated by the different status of iron in E. faecalis wild type and Δfur were performed as previously described in the construction of copper and zinc activated networks. Briefly, each global expression result from RNAseq experiments were integrated into a global transcriptional regulatory network model from E. faecalis and experimentally validated (EfaecalisGTN.gbk file) (Latorre et al., 2014). To relate information between the expression data and the global network we used graphic displays performed using Cytoscape software (Shannon et al., 2003) (Supplementary Table S2). All network topology analyses were carried out using R software (iGraph package).
Author Contributions
ML designed the research and analyzed the data. DQ and DT generated the microbiological and RNAseq data, respectively. AM and KS supported the bioinformatics and mutagenesis analyses, respectively. ML, VC, and BM wrote the paper. ML has responsibility for the manuscript. All authors read and approved the final content.
Funding
This work was supported by FONDECYT N°11150679 and N°1160802, Center for Genome Regulation FONDAP 15090007, Basal Grant of the Center for Mathematical Modeling UMI2807 UCHILE-CNRS N° PFB03 project, Concurso Apoyo a la Formación de Redes Internacionales para Investigadores(as) en Etapa Inicial N° REDI170193.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Ms. Karen Jacques-Palaz for experimental support and Ms. Estela Blanco for editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01580/full#supplementary-material
FIGURE S1 | Protein sequence alignments between E. faecalis Fur (EF1525) and Fur archetypes from different bacterial species. Color circles denote conserve motif involved in iron/zinc binding.
FIGURE S2 | Growth curve of E. faecalis wild type and Δfur under different conditions of iron exposure (excess and deficiency) in three biological replicates (Mann–Whitney test, p < 0.05). Error bars represent standard deviation (SD) values.
TABLE S1 | Differentially expressed genes in E. faecalis wild type and Δfur. The table shows the differentially expressed genes obtained from the RNAseq experiments.
TABLE S2 | Global transcriptional regulatory network activated by iron deficiency or excess in E. faecalis wild type and Δfur.
DATA SHEET S1 | RNAseq complete data.
Footnotes
References
Albert, R. (2005). Scale-free networks in cell biology. J. Cell Sci. 118, 4947–4957. doi: 10.1242/jcs.02714
Alice, A. F., Naka, H., and Crosa, J. H. (2008). Global gene expression as a function of the iron status of the bacterial cell: influence of differentially expressed genes in the virulence of the human pathogen Vibrio vulnificus. Infect. Immun. 76, 4019–4037. doi: 10.1128/IAI.00208-08
Anderson, A. C., Jonas, D., Huber, I., Karygianni, L., Wolber, J., Hellwig, E., et al. (2015). Enterococcus faecalis from food, clinical specimens, and oral sites: prevalence of virulence factors in association with biofilm formation. Front. Microbiol. 6:1534. doi: 10.3389/fmicb.2015.01534
Arias, C. A., and Murray, B. E. (2012). The rise of the Enterococcus: beyond vancomycin resistance. Nat. Rev. Microbiol. 10, 266–278. doi: 10.1038/nrmicro2761
Banin, E., Vasil, M. L., and Greenberg, E. P. (2005). Iron and Pseudomonas aeruginosa biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 102, 11076–11081. doi: 10.1073/pnas.0504266102
Butala, M., Zgur-Bertok, D., and Busby, S. J. (2009). The bacterial LexA transcriptional repressor. Cell. Mol. Life Sci. 66, 82–93. doi: 10.1007/s00018-008-8378-6
Carpenter, B. M., Whitmire, J. M., and Merrell, D. S. (2009). This is not your mother’s repressor: the complex role of fur in pathogenesis. Infect. Immun. 77, 2590–2601. doi: 10.1128/IAI.00116-09
Carpenter, C., and Payne, S. M. (2014). Regulation of iron transport systems in Enterobacteriaceae in response to oxygen and iron availability. J. Inorg. Biochem. 133, 110–117. doi: 10.1016/j.jinorgbio.2014.01.007
Chandrangsu, P., Rensing, C., and Helmann, J. D. (2017). Metal homeostasis and resistance in bacteria. Nat. Rev. Microbiol. 15, 338–350. doi: 10.1038/nrmicro.2017.15
da Silva Neto, J. F., Lourenco, R. F., and Marques, M. V. (2013). Global transcriptional response of Caulobacter crescentus to iron availability. BMC Genomics 14:549. doi: 10.1186/1471-2164-14-549
De Domenico, I., Mcvey Ward, D., and Kaplan, J. (2008). Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 9, 72–81. doi: 10.1038/nrm2295
Dos Santos, P. C., Smith, A. D., Frazzon, J., Cash, V. L., Johnson, M. K., and Dean, D. R. (2004). Iron-sulfur cluster assembly: NifU-directed activation of the nitrogenase Fe protein. J. Biol. Chem. 279, 19705–19711. doi: 10.1074/jbc.M400278200
Eaton, J. W., and Qian, M. (2002). Molecular bases of cellular iron toxicity. Free Radic. Biol. Med. 32, 833–840. doi: 10.1016/S0891-5849(02)00772-4
Embree, M., Qiu, Y., Shieu, W., Nagarajan, H., O’neil, R., Lovley, D., et al. (2014). The iron stimulon and fur regulon of Geobacter sulfurreducens and their role in energy metabolism. Appl. Environ. Microbiol. 80, 2918–2927. doi: 10.1128/AEM.03916-13
Ferrer, A., Rivera, J., Zapata, C., Norambuena, J., Sandoval, A., Chavez, R., et al. (2016). Cobalamin protection against oxidative stress in the acidophilic iron-oxidizing bacterium Leptospirillum Group II CF-1. Front. Microbiol. 7:748. doi: 10.3389/fmicb.2016.00748
Foster, J. W. (1991). Salmonella acid shock proteins are required for the adaptive acid tolerance response. J. Bacteriol. 173, 6896–6902. doi: 10.1128/jb.173.21.6896-6902.1991
Fuangthong, M., and Helmann, J. D. (2003). Recognition of DNA by three ferric uptake regulator (Fur) homologs in Bacillus subtilis. J. Bacteriol. 185, 6348–6357. doi: 10.1128/JB.185.21.6348-6357.2003
Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723. doi: 10.1002/elps.1150181505
Guzman Prieto, A. M., Van Schaik, W., Rogers, M. R., Coque, T. M., Baquero, F., Corander, J., et al. (2016). Global emergence and dissemination of enterococci as nosocomial pathogens: attack of the clones? Front. Microbiol. 7:788. doi: 10.3389/fmicb.2016.00788
Hantke, K. (2001). Iron and metal regulation in bacteria. Curr. Opin. Microbiol. 4, 172–177. doi: 10.1016/S1369-5274(00)00184-3
Hirosawa, M., Totoki, Y., Hoshida, M., and Ishikawa, M. (1995). Comprehensive study on iterative algorithms of multiple sequence alignment. Comput. Appl. Biosci. 11, 13–18. doi: 10.1093/bioinformatics/11.1.13
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–38. doi: 10.1016/0263-7855(96)00018-5
Kiley, P. J., and Beinert, H. (2003). The role of Fe-S proteins in sensing and regulation in bacteria. Curr. Opin. Microbiol. 6, 181–185. doi: 10.1016/S1369-5274(03)00039-0
Kim, B. J., Park, J. H., Park, T. H., Bronstein, P. A., Schneider, D. J., Cartinhour, S. W., et al. (2009). Effect of iron concentration on the growth rate of Pseudomonas syringae and the expression of virulence factors in hrp-inducing minimal medium. Appl. Environ. Microbiol. 75, 2720–2726. doi: 10.1128/AEM.02738-08
Kim, E. J., Sabra, W., and Zeng, A. P. (2003). Iron deficiency leads to inhibition of oxygen transfer and enhanced formation of virulence factors in cultures of Pseudomonas aeruginosa PAO1. Microbiology 149, 2627–2634. doi: 10.1099/mic.0.26276-0
Kizawa, A., Kawahara, A., Takimura, Y., Nishiyama, Y., and Hihara, Y. (2016). RNA-seq profiling reveals novel target genes of LexA in the cyanobacterium Synechocystis sp. PCC 6803. Front. Microbiol. 7:193. doi: 10.3389/fmicb.2016.00193
Koster, W. (2001). ABC transporter-mediated uptake of iron, siderophores, heme and vitamin B12. Res. Microbiol. 152, 291–301. doi: 10.1016/S0923-2508(01)01200-1
Kristich, C. J., Chandler, J. R., and Dunny, G. M. (2007). Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57, 131–144. doi: 10.1016/j.plasmid.2006.08.003
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Latorre, M., Galloway-Pena, J., Roh, J. H., Budinich, M., Reyes-Jara, A., Murray, B. E., et al. (2014). Enterococcus faecalis reconfigures its transcriptional regulatory network activation at different copper levels. Metallomics 6, 572–581. doi: 10.1039/c3mt00288h
Latorre, M., Low, M., Garate, E., Reyes-Jara, A., Murray, B. E., Cambiazo, V., et al. (2015). Interplay between copper and zinc homeostasis through the transcriptional regulator Zur in Enterococcus faecalis. Metallomics 7, 1137–1145. doi: 10.1039/C5MT00043B
Latorre, M., Olivares, F., Reyes-Jara, A., Lopez, G., and Gonzalez, M. (2011). CutC is induced late during copper exposure and can modify intracellular copper content in Enterococcus faecalis. Biochem. Biophys. Res. Commun. 406, 633–637. doi: 10.1016/j.bbrc.2011.02.109
Lee, S. W., Parker, D. L., Geszvain, K., and Tebo, B. M. (2014). Effects of exogenous pyoverdines on Fe availability and their impacts on Mn(II) oxidation by Pseudomonas putida GB-1. Front. Microbiol. 5:301. doi: 10.3389/fmicb.2014.00301
Leon-Sicairos, N., Angulo-Zamudio, U. A., De La Garza, M., Velazquez-Roman, J., Flores-Villasenor, H. M., and Canizalez-Roman, A. (2015). Strategies of Vibrio parahaemolyticus to acquire nutritional iron during host colonization. Front. Microbiol. 6:702. doi: 10.3389/fmicb.2015.00702
Lopez, G., Latorre, M., Reyes-Jara, A., Cambiazo, V., and Gonzalez, M. (2012). Transcriptomic response of Enterococcus faecalis to iron excess. Biometals 25, 737–747. doi: 10.1007/s10534-012-9539-5
Ludwig, M., Chua, T. T., Chew, C. Y., and Bryant, D. A. (2015). Fur-type transcriptional repressors and metal homeostasis in the cyanobacterium Synechococcus sp. PCC 7002. Front. Microbiol. 6:1217. doi: 10.3389/fmicb.2015.01217
Magoc, T., Wood, D., and Salzberg, S. L. (2013). EDGE-pro: estimated degree of gene expression in prokaryotic genomes. Evol. Bioinform. Online 9, 127–136. doi: 10.4137/EBO.S11250
Masse, E., Salvail, H., Desnoyers, G., and Arguin, M. (2007). Small RNAs controlling iron metabolism. Curr. Opin. Microbiol. 10, 140–145. doi: 10.1016/j.mib.2007.03.013
McHugh, J. P., Rodriguez-Quinones, F., Abdul-Tehrani, H., Svistunenko, D. A., Poole, R. K., Cooper, C. E., et al. (2003). Global iron-dependent gene regulation in Escherichia coli. A new mechanism for iron homeostasis. J. Biol. Chem. 278, 29478–29486. doi: 10.1074/jbc.M303381200
Moeck, G. S., and Coulton, J. W. (1998). TonB-dependent iron acquisition: mechanisms of siderophore-mediated active transport. Mol. Microbiol. 28, 675–681. doi: 10.1046/j.1365-2958.1998.00817.x
Mohamed, J. A., Huang, W., Nallapareddy, S. R., Teng, F., and Murray, B. E. (2004). Influence of origin of isolates, especially endocarditis isolates, and various genes on biofilm formation by Enterococcus faecalis. Infect. Immun. 72, 3658–3663. doi: 10.1128/IAI.72.6.3658-3663.2004
Nallapareddy, S. R., Singh, K. V., Sillanpaa, J., Garsin, D. A., Hook, M., Erlandsen, S. L., et al. (2006). Endocarditis and biofilm-associated pili of Enterococcus faecalis. J. Clin. Invest. 116, 2799–2807. doi: 10.1172/JCI29021
Oglesby-Sherrouse, A. G., and Murphy, E. R. (2013). Iron-responsive bacterial small RNAs: variations on a theme. Metallomics 5, 276–286. doi: 10.1039/c3mt20224k
Oogai, Y., Matsuo, M., Hashimoto, M., Kato, F., Sugai, M., and Komatsuzawa, H. (2011). Expression of virulence factors by Staphylococcus aureus grown in serum. Appl. Environ. Microbiol. 77, 8097–8105. doi: 10.1128/AEM.05316-11
Pasqua, M., Visaggio, D., Lo Sciuto, A., Genah, S., Banin, E., Visca, P., et al. (2017). Ferric uptake regulator Fur is conditionally essential in Pseudomonas aeruginosa. J. Bacteriol. 199:e00472-17. doi: 10.1128/JB.00472-17
Paustian, M. L., May, B. J., and Kapur, V. (2001). Pasteurella multocida gene expression in response to iron limitation. Infect. Immun. 69, 4109–4115. doi: 10.1128/IAI.69.6.4109-4115.2001
Pecqueur, L., D’autreaux, B., Dupuy, J., Nicolet, Y., Jacquamet, L., Brutscher, B., et al. (2006). Structural changes of Escherichia coli ferric uptake regulator during metal-dependent dimerization and activation explored by NMR and X-ray crystallography. J. Biol. Chem. 281, 21286–21295. doi: 10.1074/jbc.M601278200
Pelliciari, S., Vannini, A., Roncarati, D., and Danielli, A. (2015). The allosteric behavior of Fur mediates oxidative stress signal transduction in Helicobacter pylori. Front. Microbiol. 6:840. doi: 10.3389/fmicb.2015.00840
Pfaffl, M. W., Horgan, G. W., and Dempfle, L. (2002). Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30:e36. doi: 10.1093/nar/30.9.e36
Porcheron, G., and Dozois, C. M. (2015). Interplay between iron homeostasis and virulence: fur and RyhB as major regulators of bacterial pathogenicity. Vet. Microbiol. 179, 2–14. doi: 10.1016/j.vetmic.2015.03.024
Quatrini, R., Lefimil, C., Veloso, F. A., Pedroso, I., Holmes, D. S., and Jedlicki, E. (2007). Bioinformatic prediction and experimental verification of Fur-regulated genes in the extreme acidophile Acidithiobacillus ferrooxidans. Nucleic Acids Res. 35, 2153–2166. doi: 10.1093/nar/gkm068
Reyes-Jara, A., Latorre, M., Lopez, G., Bourgogne, A., Murray, B. E., Cambiazo, V., et al. (2010). Genome-wide transcriptome analysis of the adaptive response of Enterococcus faecalis to copper exposure. Biometals 23, 1105–1112. doi: 10.1007/s10534-010-9356-7
Roche, B., Aussel, L., Ezraty, B., Mandin, P., Py, B., and Barras, F. (2013). Iron/sulfur proteins biogenesis in prokaryotes: formation, regulation and diversity. Biochim. Biophys. Acta 1827, 455–469. doi: 10.1016/j.bbabio.2012.12.010
Sauvageot, N., Mokhtari, A., Joyet, P., Budin-Verneuil, A., Blancato, V. S., Repizo, G. D., et al. (2017). Enterococcus faecalis uses a phosphotransferase system permease and a host colonization-related ABC transporter for maltodextrin uptake. J. Bacteriol. 199:e00878-16. doi: 10.1128/JB.00878-16
Schell, M. A. (1993). Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47, 597–626. doi: 10.1146/annurev.mi.47.100193.003121
Seo, S. W., Kim, D., Latif, H., O’brien, E. J., Szubin, R., and Palsson, B. O. (2014). Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat. Commun. 5:4910. doi: 10.1038/ncomms5910
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Sheikh, M. A., and Taylor, G. L. (2009). Crystal structure of the Vibrio cholerae ferric uptake regulator (Fur) reveals insights into metal co-ordination. Mol. Microbiol. 72, 1208–1220. doi: 10.1111/j.1365-2958.2009.06718.x
Skaar, E. P. (2010). The battle for iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6:e1000949. doi: 10.1371/journal.ppat.1000949
Thoden, J. B., Holden, H. M., and Grant, G. A. (2014). Structure of L-serine dehydratase from Legionella pneumophila: novel use of the C-terminal cysteine as an intrinsic competitive inhibitor. Biochemistry 53, 7615–7624. doi: 10.1021/bi501253w
Tkachenko, A. G., Akhova, A. V., Shumkov, M. S., and Nesterova, L. Y. (2012). Polyamines reduce oxidative stress in Escherichia coli cells exposed to bactericidal antibiotics. Res. Microbiol. 163, 83–91. doi: 10.1016/j.resmic.2011.10.009
Troxell, B., and Hassan, H. M. (2013). Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front. Cell. Infect. Microbiol. 3:59. doi: 10.3389/fcimb.2013.00059
Keywords: ferric uptake regulator, Enterococcus faecalis, iron homeostasis, transcriptional networks, systems biology
Citation: Latorre M, Quenti D, Travisany D, Singh KV, Murray BE, Maass A and Cambiazo V (2018) The Role of Fur in the Transcriptional and Iron Homeostatic Response of Enterococcus faecalis. Front. Microbiol. 9:1580. doi: 10.3389/fmicb.2018.01580
Received: 03 April 2018; Accepted: 25 June 2018;
Published: 17 July 2018.
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Bradley D. Jones, University of Iowa, United StatesErin R. Murphy, Ohio University, United States
Copyright © 2018 Latorre, Quenti, Travisany, Singh, Murray, Maass and Cambiazo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauricio Latorre, bWF1cmljaW8ubGF0b3JyZUB1b2guY2w=; bWxhdG9ycmVAaW50YS51Y2hpbGUuY2w=