 Yue Deng1
Yue Deng1 Jinshui Yang
Jinshui Yang Baozhen Li
Baozhen Li Hongli Yuan
Hongli Yuan- 1State Key Laboratory of Agrobiotechnology, College of Biological Sciences, China Agricultural University, Beijing, China
- 2Departamento de Microbiología, Escuela Nacional de Ciencias Biológicas, Instituto Politécnico Nacional, Mexico City, Mexico
To investigate the diversity of butane-oxidizing bacteria in soils contaminated by long-term light hydrocarbon microseepage and the influence of butane on the soil microbial community, a quantitative study and identification of butane-oxidizing bacteria (BOB) in soils at the Puguang gas field were performed by DNA-based stable isotope probing (DNA-SIP). For the first time, two phylotypes corresponding to the genera Giesbergeria and Ramlibacter were identified as being directly involved in butane oxidation, in addition to the well-known light hydrocarbon degrader Pseudomonas. Furthermore, bmoX genes were strongly labeled by 13C-butane, and their abundances in gas field soils increased by 43.14-, 17.39-, 21.74-, and 30.14-fold when incubated with butane for 6, 9, 12, and 14 days, respectively, indicating that these bmoX-harboring bacteria could use butane as the sole carbon and energy source and they play an important role in butane degradation. We also found that the addition of butane rapidly shaped the bacterial community and reduced the diversity of bmoX genes in the gas field soils. These findings improve our understanding of BOB in the gas field environment and reveal the potential for their applications in petroleum exploration and bioremediation.
Introduction
The MPOG is a surface geochemical exploration method based on the microseepage theory that light hydrocarbons (such as C1–C4 alkanes) penetrate and migrate upward to the surface from subsurface oil and gas accumulations and that these hydrocarbons are utilized by alkane-oxidizing bacteria in the surface soil (Dayal et al., 2014). These bacteria are mostly enriched due to the continuous supply of hydrocarbon gases (Rasheed et al., 2013). Therefore, the anomalous distribution and activity of light hydrocarbons (C1–C4 alkanes) degraders can serve as an indicator for oil and gas deposits (Veena Prasanna et al., 2013; Rasheed et al., 2017). Although the distribution, abundance, and community composition of methanotrophs are well-studied in oil and gas fields (Zhang et al., 2010; Kadnikov et al., 2012; Xu et al., 2013b), the anomalously high density of BOB is considered the most promising indicator for petroleum prospecting, as butane can only come from oil and gas reservoirs (Zhang et al., 2013).
Most of the previous studies have used culture-dependent methods to investigate the BOB in oil- and gas-contaminated environments (Zhang et al., 2013), which led to the isolation of Gram-positive CNMR (Corynebacterium, Nocardia, Mycobacterium, and Rhodococcus) strains and Gram-negative strains of Thauera butanivorans (McLee et al., 1972; Takahashi et al., 1980; Ashraf et al., 1994; Saunders et al., 1999; Cooley et al., 2009). These BOB can also utilize ethane and propane as carbon sources, but they cannot use methane (Zhang et al., 2013). As one of the greenhouse gas and a precursor to many atmospheric pollutants (such as alkyl nitrates and ozone) (Collins et al., 2002; Katzenstein et al., 2003), butane is emitted into the biosphere at a rate of 10 Tg per year via hydrocarbon seeps, mud volcanoes, and hydrothermal vents (Jaekel et al., 2014). Consequently, increasing concentrations of butane in the atmosphere is the cause of many environmental problems, such as global warming, ozone enrichment, and photochemical smog formation (Chan et al., 2006). Therefore, BOB are significant in both petroleum exploration and bioremediation. Nevertheless, the diversity and biogeography of BOB in oil and gas fields are still not sufficiently explored (Cappelletti et al., 2015).
Soluble di-iron monooxygenases (SDIMOs) are essential enzymes for diverse bacteria to initiate the oxidation of hydrocarbons (Coleman et al., 2006). Based on component arrangement, substrate specificity, and sequence similarity, SDIMOs are classified into aromatic/alkene monooxygenases (group 1), phenol monooxygenases (group 2), soluble methane monooxygenases (group 3), alkene monooxygenases (group 4), THF/propane monooxygenases (group 5), and an additional unclassified group (group 6) (Leahy et al., 2003; Coleman et al., 2006). The butane metabolism pathway in Pseudomonas butanovora ATCC43655 has been studied in detail (Arp, 1999). In this pathway, n-butane is first oxidized to butanol, then to butyraldehyde and finally to butyrate that can be directly assimilated into cell materials. This pathway is activated by butane monooxygenases (BMOs), which are non-heme iron monooxygenases (Sluis et al., 2002). The substrates for BMO usually range from C2 to C9 alkanes (Halsey et al., 2006; Doughty et al., 2008). The bmoX gene, encoding the alpha hydroxylase subunit of BMO (BmoX) classified into the group 3 SDIMOs (Coleman et al., 2006), is a conserved gene segment and its insertional mutation in a P. butanovora strain results in the loss of the ability to grow on butane (Sluis et al., 2002). Zhang Y. et al. (2016) reported that the expression level of bmoX genes can reflect the butane-oxidizing activity of BOB. Despite the importance of the bmoX gene in butane oxidation, its sequence diversity and distribution in BOB species have been rarely studied.
Isolation method is fundamental but most of the microorganisms in nature are uncultivable (Amann et al., 1995), therefore, this method underestimates the prokaryotic diversity and may ignore important microbes (Oren, 2004). Moreover, it is difficult to describe complex interactions within microbial communities in complex environments by means of isolation method (Jones et al., 2011). SIP, a culture-independent method, is a powerful tool for identifying active microorganisms that are able to metabolize specific substrates in complex systems (Radajewski et al., 2000). In this strategy, substrates are labeled with heavy isotopes, such as 13C and 15N, and used to feed microorganisms in environmental samples. The stable isotope is then assimilated into cellular components (such as DNA, RNA, and protein). Subsequently, the components labeled with heavy atoms are separated from the unlabeled biomass by density gradient centrifugation. Microbes participating in the metabolism of the labeled substrates can be identified and characterized by analyzing the labeled DNA (Gutierrez et al., 2013). In recent years, SIP has been widely used to identify bacteria that are capable of metabolizing alkane hydrocarbons and aromatic hydrocarbons (Song et al., 2015, 2016; Paul et al., 2017; Posman et al., 2017). However, the successful application of SIP to aerobic BOB has not been reported.
In the present study, to investigate the diversity of BOB in hydrocarbon-contaminated soils and the influence of butane on the soil microbial community, DNA-SIP was applied to Puguang gas field soils. In addition, the abundance of bmoX genes in butane-amended microcosms were determined by real-time quantitative PCR, and the diversity of bmoX genes were analyzed with clone libraries. The results provide more insight into BOB in oil and gas field environments and suggest the potential use of BOB in petroleum exploration and bioremediation.
Materials and Methods
Sample Collection
Soil samples were collected as mentioned previously (Deng et al., 2016). Briefly, gas field soils (G) with long-term microseepage were collected from a site adjacent to gas pumping wells at the Puguang gas field (31°31′48′′N, 107°46′12′′E) in Sichuan Province, China. Non-gas field soils (NG) served as background samples and were taken from sites (31°30′22′′N, 107°40′16′′E) that were approximately 10 km away from the gas field. Petroleum and gas reservoirs had never been discovered below the NG site. A geographical map indicating the sampling sites is provided in the Supplementary Figure S1. Considering the aerobic character of BOB and to avoid the effects of human cultivation, all the soils were taken from a depth of 30–50 cm in five replicates, then mixed thoroughly and transported to the laboratory in pre-sterilized plastic bags. The samples were stored at 4°C and –20°C for further analysis.
Setup of Butane-Degrading Microcosms
Microcosms were constructed with 10 g of fresh soils in 50-ml pre-sterilized serum bottles. After sealing the bottles with butyl rubber stoppers and fixing the stoppers in place with aluminum crimp caps, 6 ml of air was taken out from the bottles with a gas-tight syringe (SGE, Australia), and then equal volume of unlabeled butane (99.99%, Beiwen Gas Manufacturer, Beijing, China) or 13C-butane (99%, CGN, Beijing, China) was injected into the bottles. Four treatments were prepared, including unlabeled butane-amended treatments (12C-BT), 13C-labeled butane-amended treatments (13C-BT), negative controls without butane addition (NB), and sterile controls with unlabeled butane addition (12C-ST). In addition, soils were autoclaved at 121°C for 30 min on three consecutive days to prepare sterile controls (Carter et al., 2007). Twelve serum bottles were prepared for each treatment and cultured in the dark at room temperature. Three of twelve were harvested at the desired time (6, 9, 12, and 14 days) and pooled, then they were stored at –20°C for DNA extraction. The residual butane in the microcosms was measured at 0, 6, 9, 12, and 14 days by gas chromatography–flame ionization detection as described in detail below.
Butane Analysis
Previous studies have demonstrated that there are no significant differences in the removal rates of 13C-labeled and unlabeled substrates (Jiang et al., 2015; Song et al., 2015). Therefore, only the soil samples amended with unlabeled butane were selected to analyze the butane degradation in this study. To collect gas samples from microcosms, 10-ml penicillin bottles containing 2 ml of saturated sodium chloride solution were prepared. At 0, 6, 9, 12, and 14 days, 0.2 ml aliquots of gas (in triplicate) from each treatment were taken out from the top of the serum bottles and injected to the penicillin bottles using a gas-tight syringe. The penicillin bottles were inverted immediately to prevent the escape of butane and transported to the National Research Center for Geoanalysis at the Chinese Academy of Geological Sciences. The butane concentration in each penicillin bottle was determined by gas chromatography (Finnigan Trace GC Ultra; Thermo-Finnigan, Germany) equipped with a flame ionization detector and a 30-m length, 0.32-mm inner diameter column (Agilent HP-AL/S, Santa Clara, CA, United States) as described by Deng et al. (2016). Data were analyzed by unpaired two-tailed Student’s t-tests using the statistical software SPSS (SPSS, Inc., Chicago, IL, United States). The differences between treatments were considered significant at p < 0.05.
DNA Extraction, Centrifugation, and Fractionation
Total genomic DNA was extracted from 0.5 g soil samples collected from 12C-BT, 13C-BT, and NB at each time point in duplicate using the E.Z.N.A. Soil DNA Kit (Omega, Norcross, GA, United States), according to the manufacturer’s protocol. The DNA concentration of each extract was determined using P300 Nanophotometer Spectrophotometers (IMPLEN, München, Germany). Then, DNA was stored at -20°C for further analysis.
Subsequently, the DNA of 12C-BT and 13C-BT were processed by density gradient centrifugation to separate the unlabeled and 13C-labeled DNA as previously described (Zheng et al., 2014). Briefly, approximately 2 μg DNA was mixed well with 4.9 ml of CsCl stock solution, and then gradient buffer (pH 8.0; 100 mM Tris–HCl; 100 mM KCl; and 1.0 mM EDTA) was added to a final volume of 5 ml. The average buoyant density (BD) of the mixture was determined with an AR200 digital hand-held refractometer (Reichert Inc., Buffalo, NY, United States), and adjusted to 1.725 g/ml using CsCl solution or gradient buffer, if necessary. The mixture was added to a 5.1 ml Quick-Seal polyallomer tubes and centrifuged at 190,000 g for 44 h at 20°C with a Vti 65.2 vertical rotor (Beckman Coulter, Inc., Palo Alto, CA, United States). After centrifugation, the DNA solution was fractionated into fifteen aliquots of 340 μl each using an NE-1000 single syringe pump (New Era Pump Systems, Inc., Farmingdale, NY, United States). The BD of each fraction was measured and the CsCl was removed by PEG precipitation (polyethylene glycol 6000). Finally, the DNA pellet was purified with 70% ethanol and dissolved in 30 μl sterile water.
High-Throughput Sequencing and Data Analysis
The V3–V4 regions of bacterial 16S rRNA genes in unfractionated DNA of 12C-BT, 13C-BT, and NB, along with fractionated DNA from fractions 4 to 12 (the BD was between 1.712 and 1.737 g/ml) of each treatment, were amplified using the 340F/805R primer set. Sequencing of the amplicons was performed on the Illumina HiSeq platform by Beijing Microread Genetics Co., Ltd. (Beijing, China).
The acquired sequences were assigned to each sample with unique barcodes. Paired-end reads were merged using FLASH (Edgar, 2013), and filtered by Quantitative Insights Into Microbial Ecology (QIIME) to remove uncorrectable barcodes, primer mismatches, or ambiguous bases. Chimeric sequences were detected and removed as described previously (Edgar et al., 2011). Sequences were analyzed by the QIIME software package and the UPARSE pipeline, and assigned to operational taxonomic units (OTUs) based on 97% cutoff (Stackebrandt and Goebel, 1994; Edgar, 2013). Sequences with the highest abundance in each OTU were selected as representatives for the OTUs and annotated using the Greengenes database (vision 13.8). The relative abundance of each OTU was determined and the top 100 OTUs with high levels of relative abundance were selected for further analysis (Li et al., 2017). Butane degraders in samples were determined by OTUs enriched in the heavy fractions from 13C-BT compared with 12C-BT samples. In this study, three OTUs encoded as OTU 5, OTU 6, and OTU 49 were enriched in the butane-amended gas field samples, and these were aligned to Giesbergeria spp., Pseudomonas spp., and Ramlibacter spp., respectively.
Detrended correspondence analysis (DCA) of bacterial composition based on the Chi-square distance matrix was performed using the vegan package version 2.2-1 in the R computing environment. Because some DNA samples failed to be sequenced, only the samples with qualified sequences are presented in the Section “Results.”
Real-Time Quantitative PCR of bmoX Genes and 16S rRNA Genes
Copy numbers of bmoX genes and 16S rRNA genes in unfractionated DNA of 12C-BT as well as fractionated DNA of 13C-BT and 12C-BT were determined by real-time quantitative PCR (qPCR). The primers for bmoX genes were bmoX-F: 5′-TGG TTC GAG CAC AAC TAY CCN GGN TGG-3′ and bmoX-R: 5′-TGC GGC TGC GCG ATC AGC GTY TTN CCR TC-3′ (Zhang et al., 2013). These primers were designed according to the conserved fragments among 2 bmoX, 6 prmA, 2 prm1A, and 5 mmoX sequences downloaded from the National Center for Biotechnology Institute (NCBI) database. They had been verified to amplify the bmoX genes from the standard butane-oxidizing bacterium P. butanovora ATCC43655 (Sluis et al., 2002). The primers for the 16S rRNA genes were 785F: 5′-GGA TTA GAT ACC CBR GTA GTC-3′ and 1061R: 5′-TCA CGR CAC GAG CTG ACG AC-3′ (Bogaert et al., 2011). The qPCR procedures for the bmoX genes and the 16S rRNA genes were the same except for the primers. Each PCR mixture contained 10 μl of SYBR Premix Ex Taq (TaKaRa Bio, Beijing, China), 0.2 μl of ROX Reference Dye II, 0.2 μl of each primer, 1 μl of DNA template, and 8.4 μl of deionized water in a final volume of 20 μl. The amplification reactions were performed in a 96-well plate on a QuantStudio 6 Flex System (Applied Biosystems) as follows: denaturation at 95°C for 30 s, followed by 40 cycles of 5 s at 95°C, 30 s at 60°C, and 30 s at 72°C, at which the SYBR green signal intensities were determined. After the PCR stage, melting curves were obtained by increasing the temperature from 60 to 95°C. To prepare the standard curve, bmoX genes amplified from the 12C-BT of gas field soils were ligated to the pGEM®-T vectors (Promega, Madison, WI, United States) and transformed into E. coli DH5α (Biomed, Beijing, China). A positive clone of E. coli containing bmoX genes was picked out, then the DNA of pGEM-T vectors was extracted from E. coli using the StarPrep Plasmid Miniprep Kit (GenStar, Beijing, China). The extract served as a standard DNA template. A decimal dilution series of the DNA template over five orders of magnitude was set up. The standard curve of 16S rRNA genes was made in a similar way to that of bmoX genes. All the DNA samples were run in triplicate and the average value was determined as the copy numbers for each sample. Data were analyzed by unpaired two-tailed Student’s t-tests using the statistical software SPSS (SPSS, Inc., Chicago, IL, United States). The differences between treatments were considered significant at p < 0.05.
Cloning, Sequencing, and Phylogenetic Analysis of bmoX Genes
The bmoX genes in 12C-BT of gas field (G) samples harvested at each time point were amplified with the primers bmoX-F/bmoX-R described above. The 20-μl reaction mixture that included 10 μl of 2×Taq PCR StarMix (GenStar, Beijing, China), 0.2 μl of each primer, 2 μl of genomic DNA, and 7.6 μl of deionized water was run on a Veriti 96-well Thermal Cycler (Thermo Fisher Scientific, MA, United States) with the following conditions: initial denaturation at 95°C for 10 min, 35 cycles of repeated denaturation at 95°C for 30 s, annealing for 30 s (annealing temperature decreased by 0.5°C per cycle for 20 cycles, then held at 46°C for 15 cycles), and extension at 72°C for 1 min, followed by a final extension step at 72°C for 10 min. The PCR amplicons were gel purified using a Universal DNA Purification kit (TIANGEN Biotech, Beijing, China) and ligated into pGEM®-T vectors. The products were transformed into E. coli DH5α following the manufacturer’s guidelines. The transformed clones were spread on Luria-Bertani (LB) medium with ampicillin (50 μg/ml) for blue/white screening. The white strains were considered as positive clones and randomly picked to verify DNA insertion by PCR with M13F/M13R primers (Deng et al., 2017). Then, the positive clones with inserts were grown on LB plates for 12 h and sent to the Beijing Genomics Institute (Beijing, China) for sequencing. The sequences (vector sequences removed) were compared with the GenBank nucleotide database library via BLASTX. The deduced protein sequences obtained from the bmoX genes were grouped into operational protein families (OPFs) based on a cutoff value of 87% sequence identity. The α-diversity and coverage values were compared for all clone libraries at the cutoff levels of 83, 87, 90, and 95%, respectively. Finally, the value of 87% was selected. The representative sequences of all the OPFs that comprised not less than two sequences were aligned by ClustalW with their reference sequences for distinctive groups of SDIMOs, and a phylogenetic tree was reconstructed by MEGA 6.0 using a neighbor-joining method with 1000 bootstrap replications.
Nucleotide Sequence Accession Number
The sequences obtained by Illumina HiSeq sequencing are available in the NCBI Sequence Read Archive (SRA) under the accession number SRP135746. The bmoX gene sequences used for tree construction were deposited in GenBank with accession numbers KY399888 to KY399891, KY399894, KY399898, KY399901, KY399902, KY399903, KY399907, KY399915, MG557980, MG557981, and MG557982.
Results
Butane Degradation in Microcosms
Detailed data on the butane biodegradation in the samples from the gas field (G) and non-gas field (NG) are shown in Supplementary Table S1. The residual butane was 86.0 and 75.4% in the sterile controls of the G and NG microcosms, respectively, after 14 days of incubation; these decreases in butane levels were regarded as being due to physical loss during the relatively long incubation time. Butane biodegradation was calculated from the difference between the total butane reduction and the physical loss in the microcosms. Significant butane biodegradation was observed in the unsterilized microcosms of the G samples, where 16.1, 19.9, and 20.7% butane were degraded after 9, 12, and 14 days of incubation, respectively. In contrast, no significant butane biodegradation was found in the unsterilized NG microcosms.
Microbial Community Structure Analysis After Butane Incubation
To study the effects of butane on the bacterial community structures in the soil samples, DNA extracted from the samples amended with unlabeled butane (12C-BT) and negative controls incubated without butane (NB) were subjected to sequencing before centrifugation and fractionation, and then, DCA was conducted. DCA axis 1 explains 51.95% of the variability, and DCA axis 2 explains 13.86%, with a cumulative percentage of 65.81% (Figure 1). The DCA plot shows a clear separation between the G samples and NG samples, indicating that their bacterial community structures are different from each other. The G samples incubated with 12C-butane rapidly deviate from the original sample and cluster in another location of the DCA plot, which suggests an early alteration in the microbial community. There was a slight change of the microbial community of the NG samples after incubation with butane, which is reflected by their grouping around the original NG sample. These results demonstrate that butane can rapidly shape the microbial community of gas field soils that undergo long-term microseepage, but butane has limited effects on non-gas field soils within a short period of time.
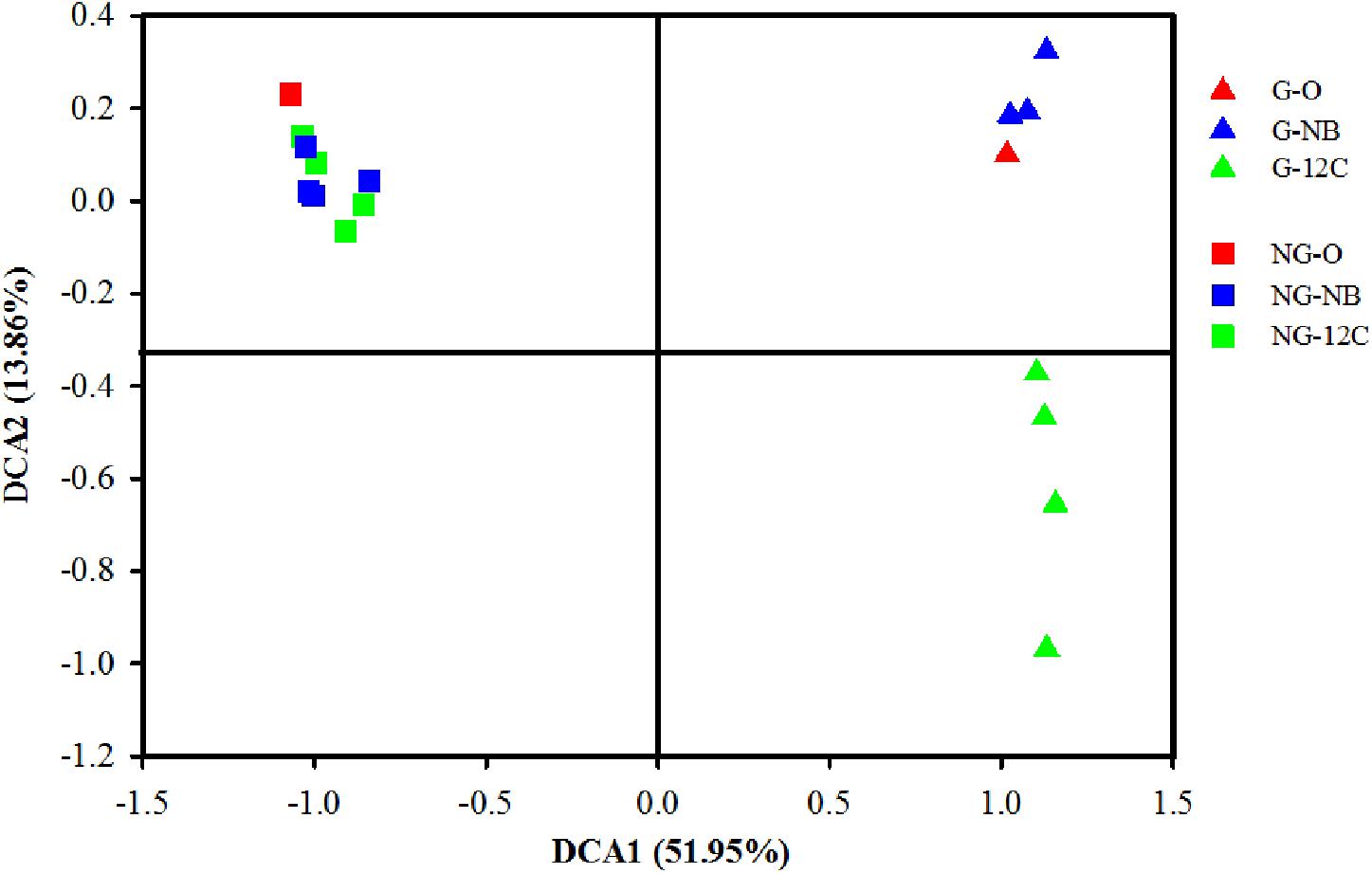
FIGURE 1. Detrended correspondence analysis (DCA) of bacterial community structures of 12C-BT and NB samples. G, samples from gas field; NG, samples from non-gas field; O, original samples without incubation; NB, incubated without butane; 12C, incubated with 12C-butane.
The relative abundance levels of the top 10 phyla in all of the samples are shown in Figure 2A. Proteobacteria was the most predominant phylum in all samples, and its relative abundance in the original G sample was 47.5%, twice as high as that in the original NG sample (22.3%). Notably, the relative abundance of Proteobacteria in the G samples sharply increased in the early days and reached the highest value at day 9 (78.9%), then it showed a slight decrease at day 12 and 14. As to the members of the Proteobacteria, only Betaproteobacteria and Gammaproteobacteria were enriched in the G samples after incubated with butane (Figure 2B). The relative abundance of Betaproteobacteria in the G samples increased by 1.9, 13.8, 10.0, and 6.9% after incubated with butane for 6, 9, 12, and 14 days, respectively. The relative abundance of Gammaproteobacteria in the G samples had a greater change, increasing by 29.0, 32.4, 24.8, and 11.9% after incubated with butane for 6, 9, 12, and 14 days, respectively. These results suggest that Betaproteobacteria and Gammaproteobacteria play important roles in the early stage of butane degradation. However, only slight modifications in the relative abundances of all phyla were observed in the NG samples and negative controls, which was consistent with the results of the DCA (Figure 1).
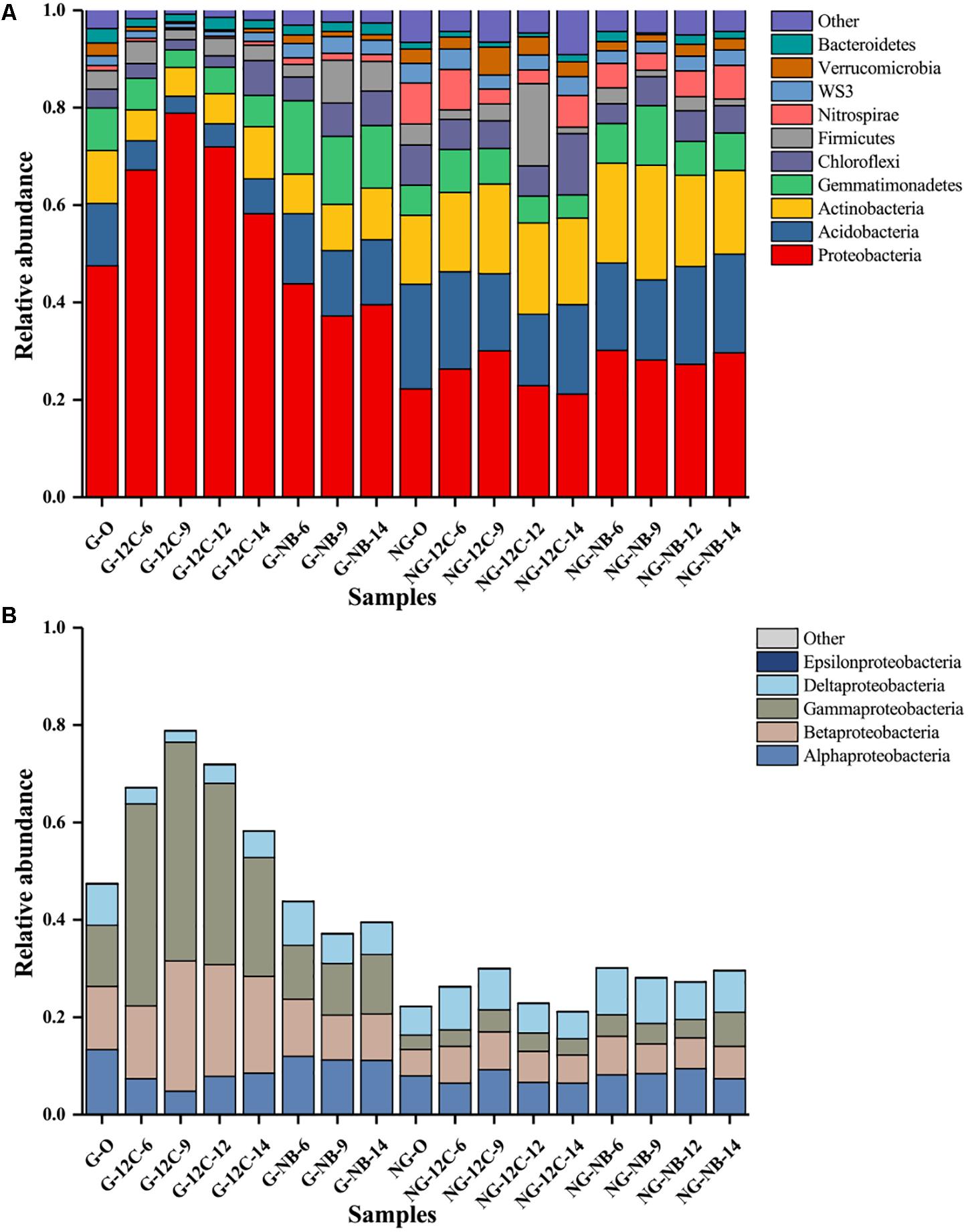
FIGURE 2. Relative abundance of the top 10 phyla (A) and proteobacterial classes (B) in samples based on 16S rRNA genes sequences. G, samples from gas field; NG, samples from non-gas field; O, original samples without incubation; NB, incubated without butane; 12C, incubated with 12C-butane.
BOB in Soil Samples
The 16S rRNA genes in each fraction of the 12C-BT and 13C-BT samples were quantified, and the results are shown in Supplementary Figure S2. An apparent shift to higher BD was observed in the 13C-BT compared to 12C-BT of the G samples after incubation for 12 days, and the increase of 13C-labeled 16S rRNA genes (in the heavy fractions) at day 12 and 14 in G was significant (p < 0.05) in comparison with those at day 6 and 9 in the same samples and those in the NG samples, indicating that 13C-butane was assimilated by BOB in the G samples.
The bacteria responsible for butane assimilation were determined by comparing the relative abundances of specific microorganisms represented by OTUs in the fractions of the 13C-BT and 12C-BT samples. The results showed that OTU 5 was enriched at 12 and 14 days in the G microcosms supplied with 13C-butane, and its relative abundance at heavy buoyant densities (>1.723 g/ml) in the 13C-BT samples greatly exceeded that in the 12C-BT samples at day 12 and 14 (Figure 3). Similarly, the relative abundance levels of OTU 6 and OTU 49 at the heavy BDs were also higher in the 13C-BT samples than in the 12C-BT samples at 12 and 14 days. Compared to the 12C-BT samples, the enrichment of OTU 5, OTU 6, and OTU 49 in the heavy fractions of the 13C-BT samples indicated that bacteria represented by these OTUs played a key role in butane oxidation. However, these OTUs were not enriched at heavy BDs in either the 13C-BT or 12C-BT of the NG samples (Supplementary Figure S3). Using the Greengenes database, OTU 5, OTU 6, and OTU 49 were matched to Giesbergeria spp., Pseudomonas spp., and Ramlibacter spp., respectively.
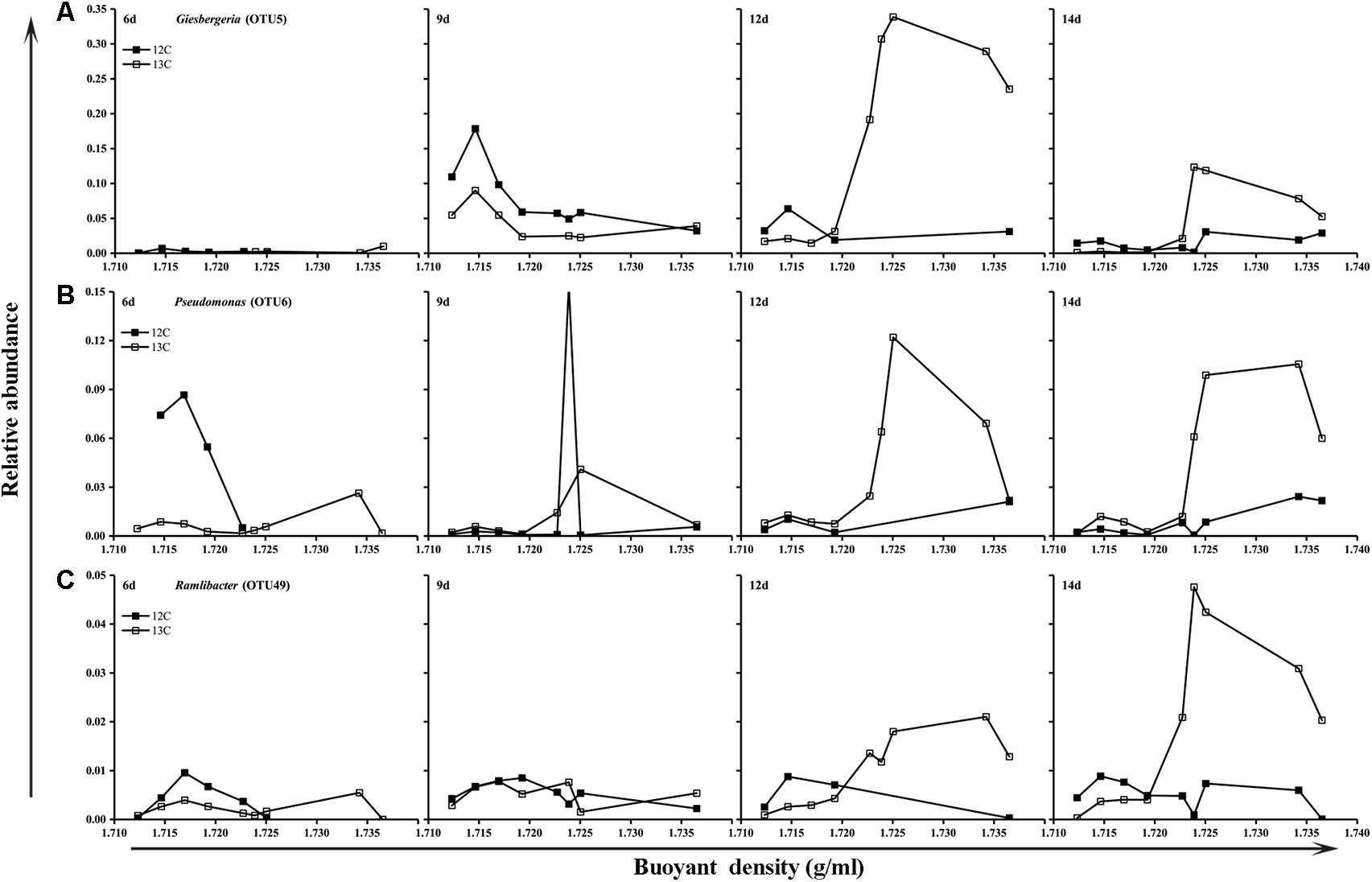
FIGURE 3. Relative abundance of Giesbergeria (A), Pseudomonas (B), and Ramlibacter (C) against the buoyant density gradients in gas field samples (G).
Occurrence of bmoX Genes
The bmoX genes in the unfractionated G and NG samples were quantified by real-time quantitative PCR. To minimize the effects of environmental disturbance in soil samples, we normalized the abundance of the bmoX genes to the total abundance of the 16S rRNA genes, and the absolute copy numbers of the bmoX genes and 16S rRNA genes are shown in Supplementary Figures S4, S5, respectively. The results (Figure 4) show that the abundance of the bmoX genes increased in the gas field soils after incubation with butane, as the ratio increased by 43.14-, 17.39-, 21.74-, and 30.14-fold at days 6, 9, 12, and 14, respectively. In contrast, the bmoX genes in the non-gas fields samples showed a slight increase at day 6 (by 5.80-fold), followed by decreases at day 9 (by 0.76-fold), day 12 (by 0.21-fold), and day 14 (by 0.42-fold). Thereafter, the bmoX genes in each fraction were also quantitatively analyzed. For the G samples (Figure 5), the majority of bmoX genes in the 12C-BT samples were found in the light fractions (BD < 1.7285 g/ml) at 6, 9, 12, and 14 days, and the highest point (BD ≈ 1.7227 g/ml) remained the same during the incubation process. In the 13C-BT samples, the bmoX genes were also concentrated in the light fractions at day 6 and 9, and these results were identical to those of the 12C-BT samples. However, after being incubated for 12 and 14 days, the bmoX genes were significantly transferred to the heavy fractions (BD > 1.7319 g/ml), and the copy numbers of the bmoX genes reached the highest point at the BD values of 1.7354 and 1.7377 g/ml, suggesting that the bmoX genes were labeled and spundown during the isopycnic ultracentrifugation of the total DNA. For the NG samples, there were no significant differences between the 13C-BT and 12C-BT in the whole incubation process (Supplementary Figure S6).
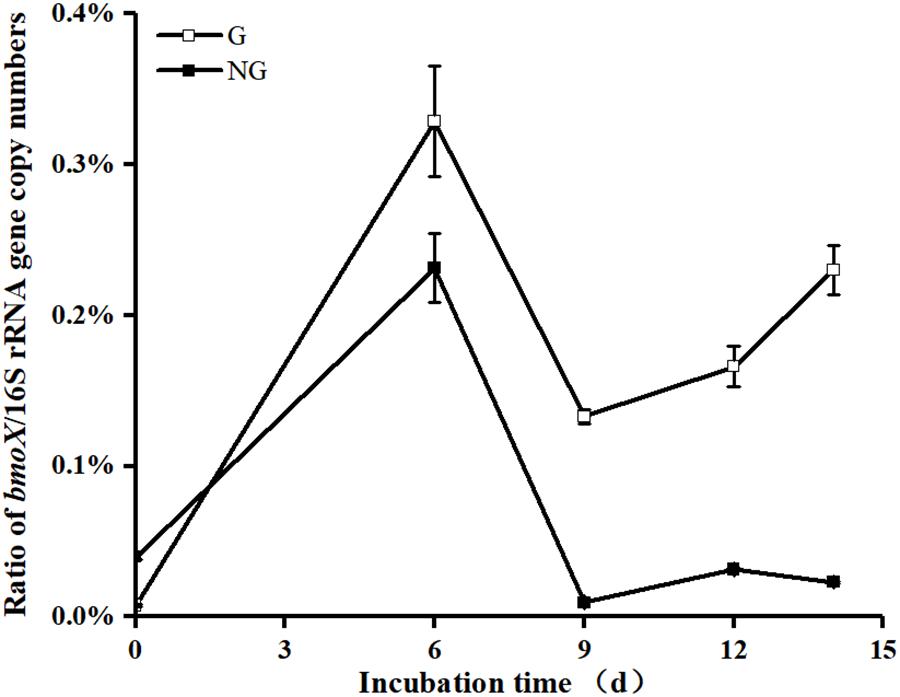
FIGURE 4. Comparison showing the significantly greater (p < 0.05) ratio of gene copy numbers of bmoX/16S rRNA gene in unfractionated DNA from gas field soil samples (G) than that from non-gas field soil samples (NG) after incubated with butane for 9, 12, and 14 days. Data are averages of three replicates. Error bars represent SD, some error bars are smaller than the symbols.
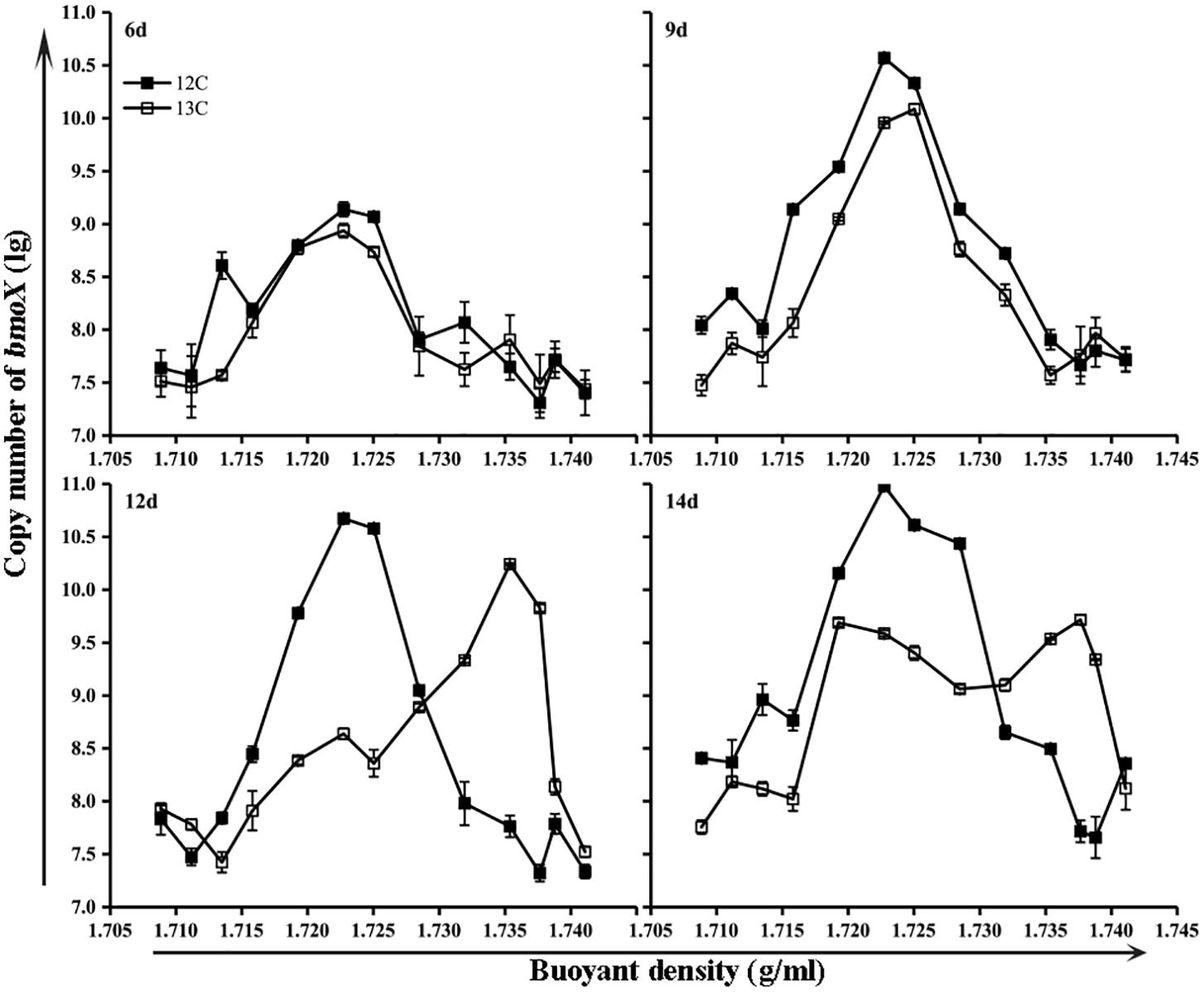
FIGURE 5. Gene copies of bmoX genes (lg) in fractions of gas field soil samples (G) incubated with 12C-butane and 13C-butane showing that the bmoX genes were labeled after 12 days of incubation, since the bmoX gene numbers of the 13C-BT samples were significantly increased (p < 0.05) in the heavy fractions (1.7354 and 1.7377 g/ml) at day 12 and 14 in comparison with those at day 6 and 9. Data are averages of three replicates. Error bars represent SD, some error bars are smaller than the symbols.
Phylogenetic Characterization of bmoX Genes
To study the diversity of the bmoX genes in the G samples at the phylogenetic level, five clone libraries (G-0, G-6, G-9, G-12, and G-14) were constructed to represent samples from each time point and 330 sequences were obtained. Both the asymptotic behavior of the rarefaction curves (Supplementary Figure S7) and the coverage values (Table 1) of all libraries indicated a satisfactory sampling effort. The number of OPFs (87% sequence similarity cutoff), the α-diversity, and OPF richness (Chao 1) were calculated for each sample (Table 1). The highest and lowest numbers of observed OPFs were 22 in sample G-0 and 2 in G-12, respectively. Interestingly, the α-diversity and richness decreased sharply at day 6, and then, both rose again at day 14. The deduced protein sequences of all of the OPFs represented by two or more sequences were selected to construct the phylogenetic tree (Figure 6). The deduced protein sequences of the bmoX genes in this study had 60–98% similarities with the sequences within group 3 SDIMOs, group 5 SDIMOs, and unclassified monooxygenases from Alphaproteobacteria, Betaproteobacteria, and Actinobacteria. Most of the clones (79.4%) were defined as OPF4 that showed high affinity to methane monooxygenase (Mmo-like) from Burkholderiaceae (WP_007590207, 90%) and propane monooxygenase hydroxylase large subunit (PrmA-like) from Paraburkholderia hospital (SEI28097.1, 90%). The G-0 sequences were distributed over 13 OPFs, such as OPF4, OPF25 with 98% identity to PrmA-like sequences from Mycobacterium sp. ENV421 (AFO66440), and OPF20 matched to PrmA-like sequences from uncultured bacteria (AKG54434, 94%). Almost all clones of G-6, G-9, G-12, and G-14 were clustered into the OPF4, indicating that incubation with butane reduced the diversity of bmoX genes in the samples.
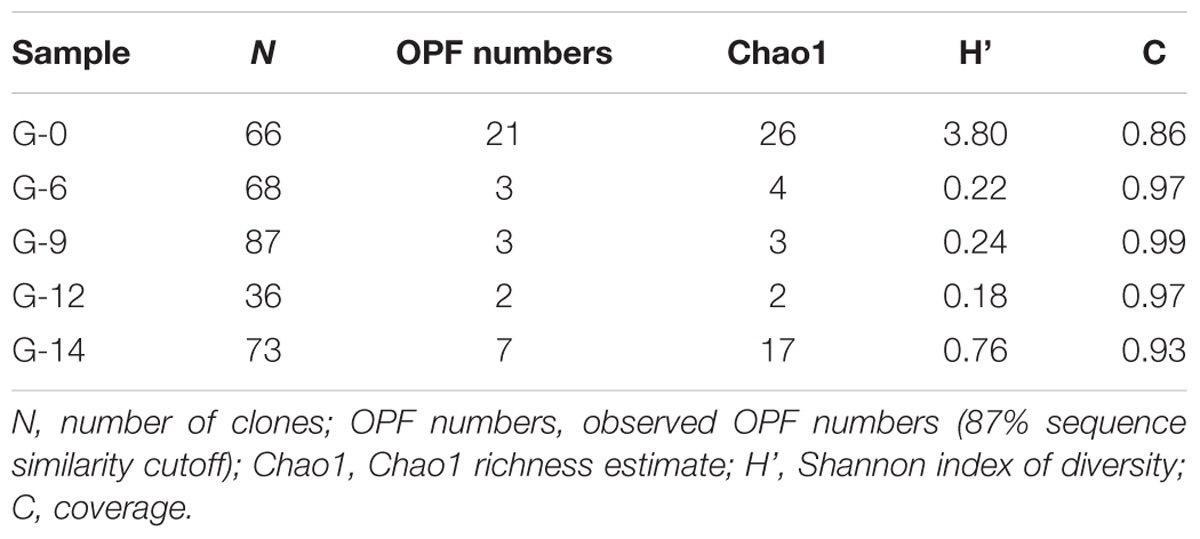
TABLE 1. The diversity indexes and coverage of bmoX gene clone libraries of G samples incubated with 12C-butane for 0, 6, 9, 12, and 14 days (abbreviated as G-0, G-6, G-9, G-12, and G-14, respectively).
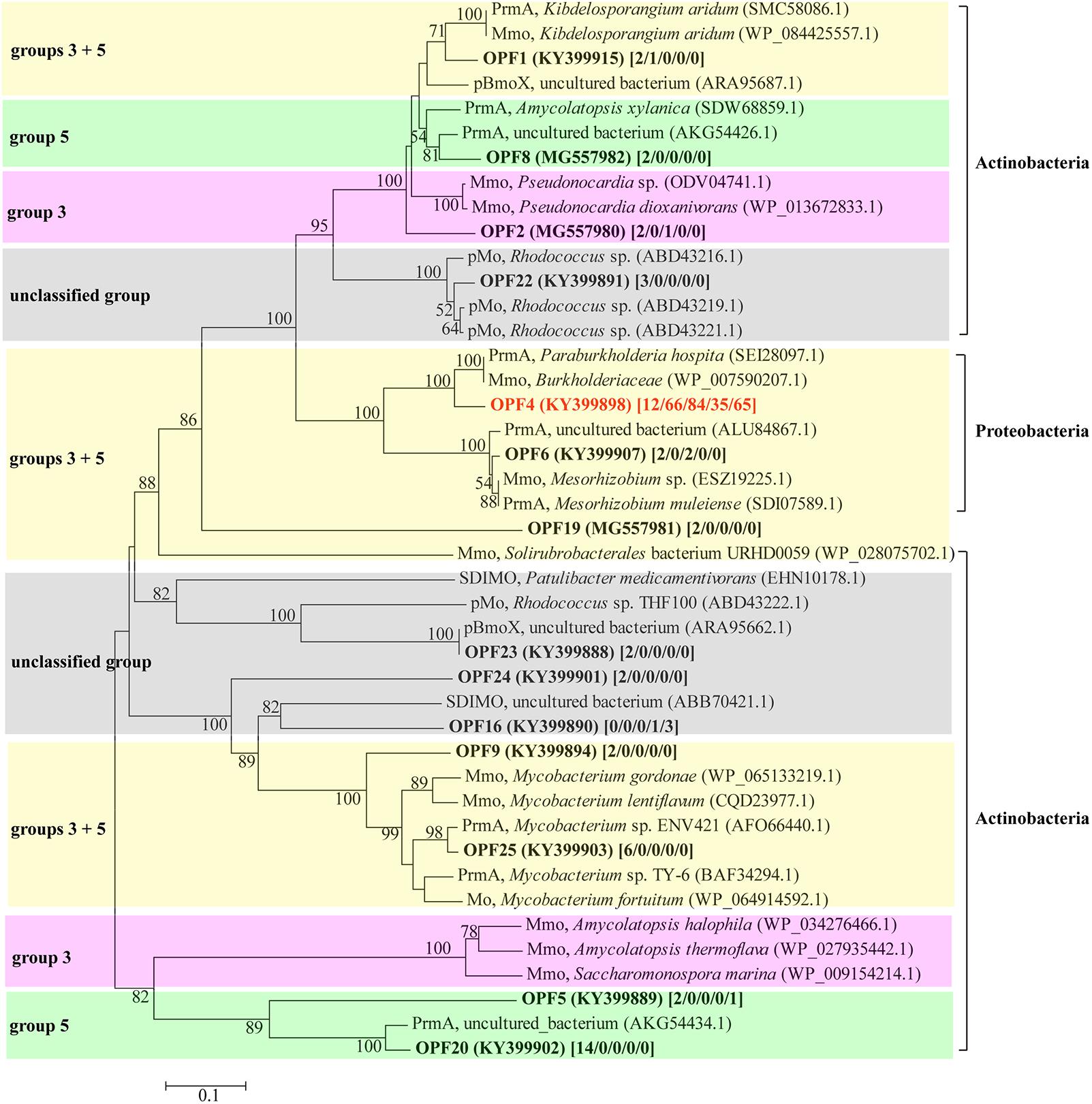
FIGURE 6. Phylogenetic tree of representative deduced bmoX sequences generated from different clone libraries of gas field soil samples (G) and reference sequences from GenBank. The tree was created using the neighbor-joining method with 1000 bootstrap replications; bootstrap values over 50% are shown at the branch nodes; the scale bars represents 10% sequence divergence. The GenBank accession number of all sequences used to build the tree is given in parentheses. The number of clones for each OPF is given in brackets [G-0/G-6/G-9/G-12/G-14]. Abbreviations for SDIMOs were as follows: Mmo, methane monooxygenase; PrmA, propane monooxygenase hydroxylase large subunit; pBmoX, putative butane monooxygenase alpha subunit; Mo, monooxygenase; pMo, putative monooxygenase. Sequences in clusters with pink background belong to group 3 SDIMOs; sequences in clusters with green background belong to group 5 SDIMOs; clusters with yellow background include both sequences belonging to group 3 and group 5 SDIMOs; clusters with gray background are unclassified SDIMOs and monooxygenases.
Discussion
Revealing the bacteria related to butane metabolism and their responses to light hydrocarbon microseepage are important for the environment and for microbial ecology. However, the characterization of BOB and the quantification of their functional genes in oil and gas fields in situ are still challenging. In the present study, we analyzed BOB in soils from Puguang gas field by DNA-SIP; additionally, the expression and diversity of bmoX genes were investigated.
Long-term microseepage of gaseous hydrocarbons may lead to anomalous enrichment of alkane-oxidizing bacteria, which causes the microbial community structure in the surface soil of oil and gas reservoirs to be different from those in non-oil and gas soils (Wu et al., 2014; Deng et al., 2016). In this study, the clear separation of the samples from the gas field and the non-gas field in the DCA plot indicates the great disparity of their microbial community structures (Figure 1), which is similar to the results of previous studies (Xu et al., 2013a,b). The rapid change in the microbial community in the butane-incubated G samples, but not the NG samples (Figure 1), demonstrates that BOB already exist in the G samples as microorganisms adapted well to a long-term and continuous supply of light hydrocarbons. Thus, they can rapidly respond to butane input. An anomalously high abundance of hydrocarbon-oxidizing bacteria (HOB) in the surface soil is usually used as an indicator for the MPOG (Rasheed et al., 2012; Veena Prasanna et al., 2013), and a good connection between the HOB abundance and the existence of oil and gas reservoirs has been observed (Rasheed et al., 2013). Additionally, the abundance levels of n-alkane-degrading bacteria in oil and gas soil samples are significantly higher than those in background soils (Xu et al., 2013a). Although our results showed an obvious microbial dissimilarity between gas field and non-gas field soils, whether MPOG can be applied in the Puguang gas field still requires further evidence from more samples.
The predominance and dramatic increase in the relative abundances of Proteobacteria, especially Betaproteobacteria and Gammaproteobacteria, in the G samples at day 6 and 9 (Figures 2A,B) suggests that Betaproteobacteria and Gammaproteobacteria might be mainly responsible for degradation in the early stage of butane degradation. The slight decrease in their relative abundances in the G samples at day 12 and 14 might be due to the gradual occupation of the niche by other slow-response butane degraders or other bacteria coexisting with BOB in the consortium. Some bacteria from Betaproteobacteria and Gammaproteobacteria are capable of degrading short-chain hydrocarbons (Redmond et al., 2010), as well as other alkanes (Wallisch et al., 2014) and arenes (Deng et al., 2017), and they have also been detected in other microseepage ecosystems, such as the sedimentary basin in Brazil (Miqueletto et al., 2011). The identification of three OTUs belonging to the genera Pseudomonas, Giesbergeria, and Ramlibacter by the coupling of DNA-SIP and high-throughput sequencing (Figure 3) further proved the important roles of Betaproteobacteria and Gammaproteobacteria in butane biodegradation.
Gammaproteobacteria species from the genus Pseudomonas is widespread in the natural environment and is able to metabolize a wide range of organic compounds (Salgado-Brito et al., 2007; Dong et al., 2015; Baruah et al., 2016; Hemidouche et al., 2016), including light hydrocarbons (Zhang Y. et al., 2016). For this reason, Pseudomonas was entrusted with the task of biodegrading hydrocarbon pollutants in contaminated sites, as well as serving as a biological indicator of oil and gas reservoirs. Deng et al. (2016) proposed Pseudomonas as a universal biomarker for subterranean oil deposits because its relative abundance remarkably increased after being incubated with butane. Our results provide firm evidence for the degradation of butane by Pseudomonas-related bacteria in the soil of the Puguang gas field using DNA-SIP.
The identification of Giesbergeria and Ramlibacter as BOB by DNA-SIP in this study is a novel finding. The genus Giesbergeria was first defined by Grabovich et al. (2006), and only five species have been reported to belong to this genus, with the species Giesbergeria voronezhensis isolated from an active sludge1. Genus Ramlibacter was first described by Heulin et al. (2003), and four species in this genus have been reported2. Its species Ramlibacter tataouinensis was made famous for its morphological transition capacity (Gommeaux et al., 2005). Both Giesbergeria and Ramlibacter belong to Burkholderiales, an order in Betaproteobacteria with many members that are able to degrade hydrocarbons. Abbasian et al. (2016) reported that Bacillales, Flavobacteriales, Pseudomonadales, and Burkholderiales are enriched in crude oil-contaminated soil. Species within Burkholderiales and Sphingobacteriales are the pivotal benzo[a]pyrene degraders in Mt. Maoer soils (Song et al., 2015). Clone libraries of the bmoX gene in this study also indicate that some bacteria of the family Burkholderiaceae (order Burkholderiales) might be responsible for butane degradation (Figure 6). However, previous studies have not reported that Giesbergeria and Ramlibacter directly participate in butane oxidation because they have not been associated with butane metabolism previously. Our results prove for the first time that some representatives within these two genera are primarily responsible for butane oxidation in light hydrocarbon microseepage soil. The data presented in Figure 6 revealed the diversity of bomX genes amplified in this study in both the SDIMO groups and the phylogenetic lineages, and demonstrated the inconsistence among the groups of SDIMOs and their sequence phylogeny, because the enzymes in the same group of SDIMOs, like those in groups 3 and 5, were divided into different phylogenetic lineages. The two unclassified lineages might imply the existence of novel SDIMOs and supported the suggestion of group 6 for SDIMOs by Coleman et al. (2006) since a group 6 SDIMO (ABB70421.1) was included in one of the unclassified lineage.
The bmoX genes are extremely important in butane oxidation (Sluis et al., 2002). We also found that bmoX genes were associated with butane degradation in G samples, as the increased abundance of bmoX genes in the G samples (Figure 4) was positively correlated with the significant butane degradation taking place in the G microcosms (Supplementary Table S1). Zhang Y. et al. (2016) observed a similar phenomenon, i.e., bmoX genes were upregulated as the butane degradation rate increased. High concentrations of oil might be toxic to microbes (Samanta et al., 2002), and therefore, many studies have reported that oil contamination decreases the diversity of microorganisms and the relevant functional genes in a variety of environments (Liang et al., 2011; Bell et al., 2013; Yang et al., 2014). In this study, we also found that the diversity of bmoX genes decreased after the microcosms were incubated with butane (Table 1). In contrast, higher bacterial diversity was found by Deng et al. (2016) in next-to-well samples polluted by trace petroleum hydrocarbons, compared to the background area. This difference indicates that the effects of pollutants on bacterial communities might vary depending on the hydrocarbon concentrations or soil features.
Previous studies have suggested that light hydrocarbon-oxidizing-genes, such as pmoA and prmA, could also be utilized as biological indicators for MPOG (Zhang C.Y. et al., 2016). With our results, BOB were enriched by adding butane in the G samples, but not in the NG samples (Figure 3 and Supplementary Figure S3), while bomX richness was increased in both soil samples by adding butane (Figure 4 and Supplementary Figure S4). These data might indicate that some bomX-harboring bacteria exist in NG soil, which could be enriched by adding butane. However, they are not BOB since no significant butane removal was detected in the corresponding sample. Therefore, bomX or its homologs may be also included in other metabolic pathways, other than butane oxidization. Before these issues are clarified in further studies, it is premature to use bmoX as indicator gene for oil and gas exploration. The first step in butane removal is the oxidization of butane into butanol by monooxygenases, therefore, the accumulation of butanol inhibited butane degradation. Moreover, the degradation ability of BOB is related to their tolerance to butanol (Zhang Y. et al., 2016). The decrease of bomX abundance in NG sample after 6 days of incubation (Supplementary Figure S4) might be explained by the possibility that butane stimulated the growth of bomX-harboring bacteria in NG sample, which oxidize the butane into butanol. However, these bacteria may have relatively low butane-degrading ability and low tolerance to butanol. Therefore, the accumulation of butanol inhibited their further increase. To confirm this estimation, further study on detection of butanol in the microcosm is needed. In addition, the persistence of high abundance of bomX in G samples is consistent with the enrichment of BOB and confirmed their participation in butane degradation.
An unexpected result in this study was the different microbial characterization results that were obtained from 16S rRNA genes analysis compared with bmoX genes analysis. Studies on BOB and bmoX genes have been relatively rare compared to other light HOB reported. Previously amplified bmoX genes have been detected in P. butanovora that is able to grow with butane (Sluis et al., 2002) and in a butane-oxidizing bacterium Arthrobacter sp. PG-3-2 isolated from the Puguang gas field (Zhang et al., 2013). Since there are only few bmoX gene sequences in the NCBI database3 (Sluis et al., 2002; Brzostowicz et al., 2005), we were unable to assign exact species identities to the obtained bmoX gene sequences. Moreover, the bmoX gene primers used in this study were designed with reference to the homologous SDIMOs-encoding genes of bmoX genes (Zhang et al., 2013), therefore some sequences generated by the clone libraries in this study might not be the genes for butane degradation. Horizontal gene transfer, which allows bacteria acquire exogenous genes from foreign species, is necessary for bacteria to survive in and adapt to harsh environmental conditions (Lawrence, 1997; Molbak et al., 2003). It has been pointed that horizontal gene transfer plays a vital role in the bioremediation of soils contaminated with petroleum hydrocarbons (Shahi et al., 2017). Li et al. (2017) also found that the phenanthrene degraders revealed by the 16S rRNA genes and the polycyclic aromatic hydrocarbons-ring hydroxylating dioxygenase (PAH-RHD) genes were different, and they speculated that horizontal gene transfer and hybridization might have occurred. Considering the high sequence similarity and homology between the bmoX genes and other SDIMOs-encoding (especially to methane monooxygenase and propane monooxygenase) genes, they may be recombined and shared extensively among different genera via horizontal gene transfer.
Conclusion
This is the first study to utilize DNA-SIP for detecting the bacteria responsible for butane oxidation in gas field soils. Proteobacteria species from the classes Betaproteobacteria and Gammaproteobacteria were primarily responsible for butane oxidation in the Puguang gas field samples. Pseudomonas, Giesbergeria, and Ramlibacter were further proven to be the main BOB in gas field soils; the butane-degrading abilities of Giesbergeria and Ramlibacter are a novel observation. In addition, the diversity of bmoX genes in the G samples reduced after incubated with butane. These results expand our knowledge on BOB in light hydrocarbon-contaminated environments and provide valuable guidance for petroleum exploration and bioremediation.
Author Contributions
YD, CD, and HY conceived and designed the experiments. YD performed all the experiments and analysis and wrote the paper. JY and BL participated in the soil samples collection. EW revised the manuscript. HY supervised the overall work, discussed the results, and revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 31270533).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Professor Zhongjun Jia for his help on the DNA-SIP experiment.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01576/full#supplementary-material
Abbreviations
BOB, butane-oxidizing bacteria; DNA-SIP, DNA-based stable isotope probing; MPOG, microbial prospecting of oil and gas; SDIMO, soluble di-iron monooxygenase.
Footnotes
- ^ http://www.bacterio.net/giesbergeria.html
- ^ http://www.bacterio.net/ramlibacter.html
- ^ https://www.ncbi.nlm.nih.gov/nuccore/?term=bmox
References
Abbasian, F., Lockington, R., Megharaj, M., and Naidu, R. (2016). The biodiversity changes in the microbial population of soils contaminated with crude oil. Curr. Microbiol. 72, 663–670. doi: 10.1007/s00284-016-1001-4
Amann, R. I., Ludwig, W., and Schleifer, K. H. (1995). Phylogenetic identification and in-situ detection of individual microbial-cells without cultivation. Microbiol. Rev. 59, 143–169.
Arp, D. J. (1999). Butane metabolism by butane-grown ‘Pseudomonas butanovora’. Microbiology 145(Pt 5), 1173–1180. doi: 10.1099/13500872-145-5-1173
Ashraf, W., Mihdhir, A., and Murrell, J. C. (1994). Bacterial oxidation of propane. FEMS Microbiol. Lett. 122, 1–6. doi: 10.1111/j.1574-6968.1994.tb07134.x
Baruah, R., Kalita, D. J., Saikia, B. K., Gautam, A., Singh, A. K., and Deka Boruah, H. P. (2016). Native hydrocarbonoclastic bacteria and hydrocarbon mineralization processes. Int. Biodeterior. Biodegradation 112, 18–30. doi: 10.1016/j.ibiod.2016.04.032
Bell, T. H., Yergeau, E., Maynard, C., Juck, D., Whyte, L. G., and Greer, C. W. (2013). Predictable bacterial composition and hydrocarbon degradation in Arctic soils following diesel and nutrient disturbance. ISME J. 7, 1200–1210. doi: 10.1038/ismej.2013.1
Bogaert, D., Keijser, B., Huse, S., Rossen, J., Veenhoven, R., Van Gils, E., et al. (2011). Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS One 6:e17035. doi: 10.1371/journal.pone.0017035
Brzostowicz, P. C., Walters, D. M., Jackson, R. E., Halsey, K. H., Ni, H., and Rouviere, P. E. (2005). Proposed involvement of a soluble methane monooxygenase homologue in the cyclohexane-dependent growth of a new Brachymonas species. Environ. Microbiol. 7, 179–190. doi: 10.1111/j.1462-2920.2004.00681.x
Cappelletti, M., Presentato, A., Milazzo, G., Turner, R. J., Fedi, S., Frascari, D., et al. (2015). Growth of Rhodococcus sp. strain BCP1 on gaseous n-alkanes: new metabolic insights and transcriptional analysis of two soluble di-iron monooxygenase genes. Front. Microbiol. 6:393. doi: 10.3389/fmicb.2015.00393
Carter, D. O., Yellowlees, D., and Tibbett, M. (2007). Autoclaving kills soil microbes yet soil enzymes remain active. Pedobiologia 51, 295–299. doi: 10.1016/j.pedobi.2007.05.002
Chan, L.-Y., Chu, K.-W., Zou, S.-C., Chan, C.-Y., Wang, X.-M., Barletta, B., et al. (2006). Characteristics of nonmethane hydrocarbons (NMHCs) in industrial, industrial-urban, and industrial-suburban atmospheres of the Pearl River Delta (PRD) region of south China. J. Geophys. Res. Atmos. 111:D11304. doi: 10.1029/2005jd006481
Coleman, N. V., Bui, N. B., and Holmes, A. J. (2006). Soluble di-iron monooxygenase gene diversity in soils, sediments and ethene enrichments. Environ. Microbiol. 8, 1228–1239. doi: 10.1111/j.1462-2920.2006.01015.x
Collins, W. J., Derwent, R. G., Johnson, C. E., and Stevenson, D. S. (2002). The oxidation of organic compounds in the troposphere and their global warming potentials. Clim. Change 52, 453–479. doi: 10.1023/A:1014221225434
Cooley, R. B., Dubbels, B. L., Sayavedra-Soto, L. A., Bottomley, P. J., and Arp, D. J. (2009). Kinetic characterization of the soluble butane monooxygenase from Thauera butanivorans, formerly ‘Pseudomonas butanovora’. Microbiology 155, 2086–2096. doi: 10.1099/mic.0.028175-0
Dayal, A. M., Mani, D., Madhavi, T., Kavitha, S., Kalpana, M. S., Patil, D. J., et al. (2014). Organic geochemistry of the Vindhyan sediments: implications for hydrocarbons. J. Asian Earth Sci. 91, 329–338. doi: 10.1016/j.jseaes.2014.03.010
Deng, C., Yu, X., Yang, J., Li, B., Sun, W., and Yuan, H. (2016). Universal indicators for oil and gas prospecting based on bacterial communities shaped by light-hydrocarbon microseepage in China. J. Microbiol. Biotechnol. 26, 1320–1332. doi: 10.4014/jmb.1602.02045
Deng, Y., Yang, F., Deng, C., Yang, J., Jia, J., and Yuan, H. (2017). Biodegradation of BTEX aromatics by a haloduric microbial consortium enriched from a sediment of Bohai Sea, China. Appl. Biochem. Biotechnol. 183, 893–905. doi: 10.1007/s12010-017-2471-y
Dong, C., Bai, X., Sheng, H., Jiao, L., Zhou, H., and Shao, Z. (2015). Distribution of PAHs and the PAH-degrading bacteria in the deep-sea sediments of the high-latitude Arctic Ocean. Biogeosciences 12, 2163–2177. doi: 10.5194/bg-12-2163-2015
Doughty, D. M., Kurth, E. G., Sayavedra-Soto, L. A., Arp, D. J., and Bottomley, P. J. (2008). Evidence for involvement of copper ions and redox state in regulation of butane monooxygenase in Pseudomonas butanovora. J. Bacteriol. 190, 2933–2938. doi: 10.1128/JB.01409-07
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Gommeaux, M., Barakat, M., Lesourd, M., Thiery, J., and Heulin, T. (2005). A morphological transition in the pleomorphic bacterium Ramlibacter tataouinensis TTB310. Res. Microbiol. 156, 1026–1030. doi: 10.1016/j.resmic.2005.05.010
Grabovich, M., Gavrish, E., Kuever, J., Lysenko, A. M., Podkopaeva, D., and Dubinina, G. (2006). Proposal of Giesbergeria voronezhensis gen. nov., sp. nov. and G. kuznetsovii sp. nov. and reclassification of [Aquaspirillum] anulus, [A.] sinuosum and [A.] giesbergeri as Giesbergeria anulus comb. nov., G. sinuosa comb. nov. and G. giesbergeri comb. nov., and [Aquaspirillum] metamorphum and [A.] psychrophilum as Simplicispira metamorpha gen. nov., comb. nov. and S. psychrophila comb. nov. Int. J. Syst. Evol. Microbiol. 56, 569–576. doi: 10.1099/ijs.0.64027-0
Gutierrez, T., Singleton, D. R., Berry, D., Yang, T., Aitken, M. D., and Teske, A. (2013). Hydrocarbon-degrading bacteria enriched by the deepwater horizon oil spill identified by cultivation and DNA-SIP. ISME J. 7, 2091–2104. doi: 10.1038/ismej.2013.98
Halsey, K. H., Sayavedra-Soto, L. A., Bottomley, P. J., and Arp, D. J. (2006). Site-directed amino acid substitutions in the hydroxylase alpha subunit of butane monooxygenase from Pseudomonas butanovora: implications for substrates knocking at the gate. J. Bacteriol. 188, 4962–4969. doi: 10.1128/JB.00280-06
Hemidouche, S., Favier, L., Sadaoui, Z., and Amrane, A. (2016). Degradation of clofibric acid by a phenol resistant Pseudomonas aeruginosa strain. J. Biotechnol. 231:S71. doi: 10.1016/j.jbiotec.2016.05.259
Heulin, T., Barakat, M., Christen, R., Lesourd, M., Sutra, L., De Luca, G., et al. (2003). Ramlibacter tataouinensis gen. nov., sp. nov., and Ramlibacter henchirensis sp. nov., cyst-producing bacteria isolated from subdesert soil in Tunisia. Int. J. Syst. Evol. Microbiol. 53, 589–594. doi: 10.1099/ijs.0.02482-0
Jaekel, U., Vogt, C., Fischer, A., Richnow, H. H., and Musat, F. (2014). Carbon and hydrogen stable isotope fractionation associated with the anaerobic degradation of propane and butane by marine sulfate-reducing bacteria. Environ. Microbiol. 16, 130–140. doi: 10.1111/1462-2920.12251
Jiang, L., Song, M., Luo, C., Zhang, D., and Zhang, G. (2015). Novel phenanthrene-degrading bacteria identified by dna-stable isotope probing. PLoS One 10:e0130846. doi: 10.1371/journal.pone.0130846
Jones, M. D., Crandell, D. W., Singleton, D. R., and Aitken, M. D. (2011). Stable-isotope probing of the polycyclic aromatic hydrocarbon-degrading bacterial guild in a contaminated soil. Environ. Microbiol. 13, 2623–2632. doi: 10.1111/j.1462-2920.2011.02501.x
Kadnikov, V. V., Mardanov, A. V., Beletsky, A. V., Shubenkova, O. V., Pogodaeva, T. V., Zemskaya, T. I., et al. (2012). Microbial community structure in methane hydrate-bearing sediments of freshwater Lake Baikal. FEMS Microbiol. Ecol. 79, 348–358. doi: 10.1111/j.1574-6941.2011.01221.x
Katzenstein, A. S., Doezema, L. A., Simpson, I. J., Blake, D. R., and Rowland, F. S. (2003). Extensive regional atmospheric hydrocarbon pollution in the southwestern United States. Proc. Natl. Acad. Sci. U.S.A. 100, 11975–11979. doi: 10.1073/pnas.1635258100
Lawrence, J. G. (1997). Selfish operons and speciation by gene transfer. Trends Microbiol. 5, 355–359. doi: 10.1016/S0966-842X(97)01110-4
Leahy, J. G., Batchelor, P. J., and Morcomb, S. M. (2003). Evolution of the soluble diiron monooxygenases. FEMS Microbiol. Rev. 27, 449–479. doi: 10.1016/s0168-6445(03)00023-8
Li, J., Zhang, D., Song, M., Jiang, L., Wang, Y., Luo, C., et al. (2017). Novel bacteria capable of degrading phenanthrene in activated sludge revealed by stable-isotope probing coupled with high-throughput sequencing. Biodegradation 28, 423–436. doi: 10.1007/s10532-017-9806-9
Liang, Y., Van Nostrand, J. D., Deng, Y., He, Z., Wu, L., Zhang, X., et al. (2011). Functional gene diversity of soil microbial communities from five oil-contaminated fields in China. ISME J. 5, 403–413. doi: 10.1038/ismej.2010.142
McLee, A. G., Kormendy, A. C., and Wayman, M. (1972). Isolation and characterization of n-butane-utilizing microorganisms. Can. J. Microbiol. 18, 1191–1195. doi: 10.1139/m72-186
Miqueletto, P. B., Andreote, F. D., Dias, A. C., Ferreira, J. C., Dos Santos Neto, E. V., and De Oliveira, V. M. (2011). Cultivation-independent methods applied to the microbial prospection of oil and gas in soil from a sedimentary basin in Brazil. AMB Express 1:35. doi: 10.1186/2191-0855-1-35
Molbak, L., Licht, T. R., Kvist, T., Kroer, N., and Andersen, S. R. (2003). Plasmid transfer from Pseudomonas putida to the indigenous bacteria on alfalfa sprouts: characterization, direct quantification, and in situ location of transconjugant cells. Appl. Environ. Microbiol. 69, 5536–5542. doi: 10.1128/aem.69.9.5536-5542.2003
Oren, A. (2004). Prokaryote diversity and taxonomy: current status and future challenges. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 623–638. doi: 10.1098/rstb.2003.1458
Paul, B. G., Ding, H., Bagby, S. C., Kellermann, M. Y., Redmond, M. C., Andersen, G. L., et al. (2017). Methane-oxidizing bacteria shunt carbon to microbial mats at a marine hydrocarbon seep. Front. Microbiol. 8:186. doi: 10.3389/fmicb.2017.00186
Posman, K. M., Derito, C. M., and Madsen, E. L. (2017). Benzene degradation by a Variovorax species within a coal tar-contaminated groundwater microbial community. Appl. Environ. Microbiol. 83:e02658-16. doi: 10.1128/AEM.02658-16
Radajewski, S., Ineson, P., Parekh, N. R., and Murrell, J. C. (2000). Stable-isotope probing as a tool in microbial ecology. Nature 403, 646–649. doi: 10.1038/35001054
Rasheed, M. A., Lakshmi, M., Kalpana, M. S., Dayal, A. M., and Patil, D. J. (2013). The microbial activity in development of hydrocarbon microseepage: an indicator for oil and gas exploration. Geosci. J. 17, 329–338. doi: 10.1007/s12303-013-0026-y
Rasheed, M. A., Lakshmi, M., Kalpana, M. S., Patil, D. J., and Dayal, A. M. (2017). Recognition of hydrocarbon microseepage using microbial and adsorbed soil gas indicators in the petroliferous region of Krishna-Godavari Basin, India. Curr. Sci. 112:560. doi: 10.18520/cs/v112/i03/560-568
Rasheed, M. A., Lakshmi, M., Kalpana, M. S., Rao, P. L. S., Patil, D. J., Sudarshan, V., et al. (2012). Geo-microbial and geochemical evidences in the near surface soils of Jamnagar sub-basin, Saurashtra, Gujarat, India: implications to hydrocarbon resource potential. Geosci. J. 16, 455–467. doi: 10.1007/s12303-012-0038-z
Redmond, M. C., Valentine, D. L., and Sessions, A. L. (2010). Identification of novel methane-, ethane-, and propane-oxidizing bacteria at marine hydrocarbon seeps by stable isotope probing. Appl. Environ. Microbiol. 76, 6412–6422. doi: 10.1128/AEM.00271-10
Salgado-Brito, R., Neria, M. I., Mesta-Howard, A. M., Díaz Cedillo, F., and Wang, E. T. (2007). Oxidation of solid paraffin (C11–40) by Pseudomonas aeruginosa MGP-1. Ann. Microbiol. 57, 321–328. doi: 10.1007/BF03175067
Samanta, S. K., Singh, O. V., and Jain, R. K. (2002). Polycyclic aromatic hydrocarbons: environmental pollution and bioremediation. Trends Biotechnol. 20, 243–248. doi: 10.1016/s0167-7799(02)01943-1
Saunders, D. F., Burson, K. R., and Thompson, C. K. (1999). Model for hydrocarbon microseepage and related near-surface alterations. Am. Assoc. Pet. Geol. Bull. 83, 170–185.
Shahi, A., Ince, B., Aydin, S., and Ince, O. (2017). Assessment of the horizontal transfer of functional genes as a suitable approach for evaluation of the bioremediation potential of petroleum-contaminated sites: a mini-review. Appl. Microbiol. Biotechnol. 101, 4341–4348. doi: 10.1007/s00253-017-8306-5
Sluis, M. K., Sayavedra-Soto, L. A., and Arp, D. J. (2002). Molecular analysis of the soluble butane monooxygenase from ‘Pseudomonas butanovora’. Microbiology 148, 3617–3629. doi: 10.1099/00221287-148-11-3617
Song, M., Jiang, L., Zhang, D., Luo, C., Wang, Y., Yu, Z., et al. (2016). Bacteria capable of degrading anthracene, phenanthrene, and fluoranthene as revealed by DNA based stable-isotope probing in a forest soil. J. Hazard. Mater. 308, 50–57. doi: 10.1016/j.jhazmat.2016.01.009
Song, M., Luo, C., Jiang, L., Zhang, D., Wang, Y., and Zhang, G. (2015). Identification of benzo[a]pyrene-metabolizing bacteria in forest soils by using DNA-based stable-isotope probing. Appl. Environ. Microbiol. 81, 7368–7376. doi: 10.1128/AEM.01983-15
Stackebrandt, E., and Goebel, B. M. (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int. J. Syst. Bacteriol. 44, 846–849. doi: 10.1099/00207713-44-4-846
Takahashi, J., Ichkawa, Y., Sagae, H., Komura, I., Kanou, H., and Yamada, K. (1980). Isolation and identification of n-butane-assimilating bacterium. Agric. Biol. Chem. 44, 1835–1840. doi: 10.1080/00021369.1980.10864232
Veena Prasanna, M., Rasheed, M. A., Patil, D. J., Dayal, A. M., and Rajeswara Reddy, B. (2013). Geo-microbiological studies in conjunction with different geo-scientific studies for the evaluation of hydrocarbon prospects in Proterozoic Vindhyan Basin, India. J. Pet. Sci. Eng. 108, 239–249. doi: 10.1016/j.petrol.2013.04.010
Wallisch, S., Gril, T., Dong, X., Welzl, G., Bruns, C., Heath, E., et al. (2014). Effects of different compost amendments on the abundance and composition of alkB harboring bacterial communities in a soil under industrial use contaminated with hydrocarbons. Front. Microbiol. 5:96. doi: 10.3389/fmicb.2014.00096
Wu, X. Y., Xu, X. M., Wu, C. F., Fu, S. Y., Deng, M. C., Feng, L., et al. (2014). Responses of microbial communities to light-hydrocarbon microseepage and novel indicators for microbial prospecting of Oil/gas in the Beihanzhuang oilfield, Northern Jiangsu, China. Geomicrobiol. J. 31, 697–707. doi: 10.1080/01490451.2013.843619
Xu, K., Tang, Y., Ren, C., Zhao, K., and Sun, Y. (2013a). Diversity and abundance of n-alkane-degrading bacteria in the near-surface soils of a Chinese onshore oil and gas field. Biogeosciences 10, 2041–2048. doi: 10.5194/bg-10-2041-2013
Xu, K., Tang, Y., Ren, C., Zhao, K., Wang, W., and Sun, Y. (2013b). Activity, distribution, and abundance of methane-oxidizing bacteria in the near surface soils of onshore oil and gas fields. Appl. Microbiol. Biotechnol. 97, 7909–7918. doi: 10.1007/s00253-012-4500-7
Yang, Y., Wang, J., Liao, J., Xie, S., and Huang, Y. (2014). Distribution of naphthalene dioxygenase genes in crude oil-contaminated soils. Microb. Ecol. 68, 785–793. doi: 10.1007/s00248-014-0457-7
Zhang, C. Y., He, Z., Zhang, S., Yin, M. Y., Ning, Z., and Liu, Y. C. (2016). A DNA-based analysis of a microbial technique for the prospecting of oil and gas applied to a known oil field, China. Geomicrobiol. J. 34, 63–70. doi: 10.1080/01490451.2016.1139641
Zhang, F., She, Y., Zheng, Y., Zhou, Z., Kong, S., and Hou, D. (2010). Molecular biologic techniques applied to the microbial prospecting of oil and gas in the Ban 876 gas and oil field in China. Appl. Microbiol. Biotechnol. 86, 1183–1194. doi: 10.1007/s00253-009-2426-5
Zhang, Y., Deng, C. P., Shen, B., Yang, J. S., Wang, E. T., and Yuan, H. L. (2016). Syntrophic interactions within a butane-oxidizing bacterial consortium isolated from Puguang gas field in China. Microb. Ecol. 72, 538–548. doi: 10.1007/s00248-016-0799-4
Zhang, Y., Tang, X.-J., Shen, B., Yu, X.-J., Wang, E.-T., and Yuan, H.-L. (2013). Identification and characterization of the butane-utilizing bacterium, Arthrobacter sp. PG-3-2, harboring a novel bmox gene. Geomicrobiol. J. 30, 85–92. doi: 10.1080/01490451.2011.653086
Keywords: butane-oxidizing bacteria, DNA-SIP, real-time quantitative PCR, bmoX gene, light hydrocarbon microseepage
Citation: Deng Y, Deng C, Yang J, Li B, Wang E and Yuan H (2018) Novel Butane-Oxidizing Bacteria and Diversity of bmoX Genes in Puguang Gas Field. Front. Microbiol. 9:1576. doi: 10.3389/fmicb.2018.01576
Received: 20 March 2018; Accepted: 25 June 2018;
Published: 17 July 2018.
Edited by:
Simona Rossetti, Istituto di Ricerca sulle Acque (IRSA), ItalyReviewed by:
Martina Cappelletti, Università degli Studi di Bologna, ItalyJochen Ait Mueller, Helmholtz-Zentrum für Umweltforschung, Germany
Copyright © 2018 Deng, Deng, Yang, Li, Wang and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongli Yuan, aGx5dWFuQGNhdS5lZHUuY24=; eXVhbmFlbW9pbEBjYXUuZWR1LmNu