 Ingrid M. Rubin1
Ingrid M. Rubin1 Thomas A. Hansen1
Thomas A. Hansen1 Anne Mette Klingenberg1
Anne Mette Klingenberg1 Andreas M. Petersen1,2,3
Andreas M. Petersen1,2,3 Peder Worning1
Peder Worning1 Henrik Westh1,3
Henrik Westh1,3 Mette D. Bartels1*
Mette D. Bartels1*- 1Department of Clinical Microbiology, Hvidovre Hospital, Hvidovre, Denmark
- 2Department of Gastroenterology, Hvidovre Hospital, Hvidovre, Denmark
- 3Institute of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark
This study describes a sporadically occurring 4-year outbreak of methicillin-resistant Staphylococcus aureus (MRSA) originating from a surgical ward. Whole-genome sequencing (WGS) identified the outbreak clone as spa type t267, sequence type ST97, and SCCmec IVa. Prompted by the finding of four patients within 6 months in the same ward with this unusual MRSA type, an outbreak was suspected. Subsequent MRSA screening in the ward in February 2017 identified three-additional patients and two health care workers (HCWs) with t267/ST97-IVa. WGS linked these 9 isolates to 16 previous isolates in our WGS database and the outbreak thus included 23 patients and two HCWs. Twenty-one patients had a connection to the surgery ward during the period 2013–2017, but half of them had MRSA diagnosed in the community long after discharge. The community debut of several patients MRSA infections weeks to months after hospital discharge made the identification of a hospital source difficult and it was the SNP relatedness of the isolates that led us to identify the common denominator of hospitalization. An index patient was not identified, but our hypothesis is that HCWs with unrecognized long-term MRSA colonization could have caused sporadic nosocomial transmission due to intermittent breaches in infection prevention and control practice.
Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) has been a global medical challenge since its emergence in 1961, 2 years after methicillin was introduced to treat penicillin-resistant S. aureus (Jevons, 1961). In human medicine, the focus has traditionally been on the hospital-acquired clones (HA-MRSA), but new clones have emerged both in livestock and in community settings (DeLeo et al., 2010; Gonçalves da Silva et al., 2017). Outbreaks in hospitals and nursing homes are today caused by both HA-MRSA and community-associated MRSA (CA-MRSA) clones (DeLeo et al., 2010; Di Ruscio et al., 2017; Henderson and Nimmo, 2017) and it has been suggested no longer to regard HA-MRSA and CA-MRSA as separate entities (Zarfel et al., 2013). Hospital admission of unknown MRSA carriers, lack of MRSA admittance screening and spread of MRSA among nursing home residents could all be part of the explanation of this mélange of HA-MRSA and CA-MRSA in hospitals (Gonzalez et al., 2006). General contact procedures, an important part of infection control, will keep patients safe from transmission of MRSA from health care workers (HCWs) (Harris A.D. et al., 2013, Harris et al., 2017). By typing MRSA isolates and drawing interferences on transmission based on genetic relatedness, transmission pathways can be tracked (Harris S.R. et al., 2013). Today the most precise typing of bacteria is by whole-genome sequencing (WGS). WGS has proven an excellent tool, with good inter-laboratory reproducibility in hospital outbreaks (Bartels et al., 2013; Leopold et al., 2014; SenGupta et al., 2014).
Here, we describe a prolonged outbreak of MRSA initiated in a hospital ward that was confirmed by WGS. Tracing of the clone led us to 23 patients and two HCWs, who in most cases had a common denominator in one of the hospitals surgery wards.
Materials and Methods
Setting
This retrospective study analyzed an outbreak spanning the period between June 2013 and February 2017 that occurred at Hvidovre Hospital, Copenhagen, Denmark. The MRSA isolates studied were routinely found in clinical samples or after the outbreak was discovered in February 2017 through MRSA screening of patients and staff.
Data Set
All MRSA isolates are investigated by WGS as part of the routine at the Department of Clinical Microbiology, Hvidovre Hospital. In order to define the outbreak, we investigated the relatedness to other t267/ST97/SCCmec IVa isolates from the period 2013–2017 in the Capital Region of Denmark and these isolates were included in the SNP analysis.
Whole-Genome Sequencing and Analysis
Each MRSA isolate was initially confirmed with an in-house multiplex real-time polymerase chain reaction (PCR) that detects the presence of nuc, femA, mecA, and mecC. Since January 2013 all MRSA isolates have been WGS on a MiSeq (Illumina, United States). DNA extraction were performed on all MRSA isolates and libraries were made with 2 × 150 bp paired-end Nextera XT DNA sample preparation kit (Illumina, United States) and sequenced on a MiSeq (Illumina, United States). The reads were mapped to a USA300 reference sequence (US300_TCH1516) using stampy (Lunter and Goodson, 2011) with an expected substitution rate of 0.01 (Didelot et al., 2012) for single nucleotide variants detection. Variants were called using SAMtools v0.1.12 (Li, 2011) mpileup command with options -M0 -Q30 -q30 -o40 -e20 -h100 -m2 -D -S. The genome was assembled using Velvet v1.0.11 (Zerbino and Birney, 2008) or Spades (Bankevich et al., 2012). Phylogeny was inferred by neighbor-joining analysis.
Ethical Considerations
Permission to link the sequencing of MRSA from routine clinical samples to patient data without individual patient consent was obtained from the Danish Data Protection Agency (no. AHH-2017-095, I-Suite nr. 06029). Permission to look up the patients admission data without individual consent was obtained from the Hospital Board (no. WZ17038300-2018-14).
Results
Documentation of the Outbreak
In January 2017, an MRSA infected patient was found in one of the hospital’s surgery wards. From the sample of abdominal pus two distinct MRSA types were found based on antimicrobial susceptibility patterns. WGS of the two isolates identified a t267/ST97-IVa and a t002/ST5-IVg. Looking 6 months back in our MRSA WGS database, we identified three other patients admitted to the same ward that had tested positive for MRSA t267/ST97. Searching our WGS database back to 2013 we found this to be an unusual MRSA type in our region with just 34 isolates from 2013 to January 2017 (0.7%). A neighbor-joining tree was then constructed with all 34 isolates showing 20 isolates with a close connection of ≤50 SNPs. These isolates also shared the same SCCmec type, namely IVa. Thus, the suspicion of an MRSA outbreak was confirmed. This led to a several week-long screening of patients, HCWs and other staff members in the ward resulting in the finding of three-additional patients and two HCWs with t267/ST97-IVa. Furthermore, screening at the ward revealed that two HCWs carried other MRSA types (t045/ST5-IVc/e, t002/ST5-IVg) and one patient with known contact to pigs carried the livestock-associated t034/ST398-V.
In our final outbreak cluster (Figure 1) we ended up with 25 persons. Eighteen had been admitted to the surgical ward, two were HCWs, two were family members of MRSA positive patients, and one had shared a room at another ward in another hospital with an MRSA positive patient, who had previously been admitted to the surgery ward. Finally, two patients had no relation either to another patient or to the ward to the best of our knowledge. We have data on hospitalization at the surgery ward for most patients but the surgery ward has many sub-departments and we lack data on room allocations as well as the exact department admitted to. Our data show that 13 patients had an overlap in admission period with at least one other patient (Supplementary Figure S1).
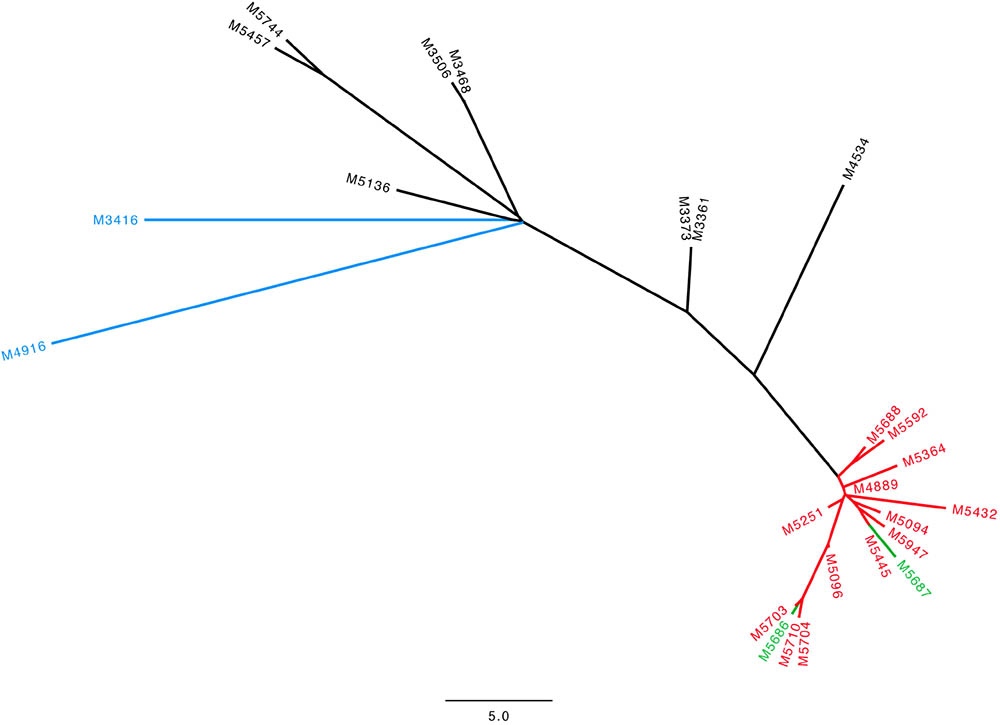
FIGURE 1. Neighbor-joining tree for the 25 isolates in our outbreak cluster with t267/ST97 SCCmec IVa. Scale bar indicates the SNP distance. Green: HCWs, Red: patients admitted in 2016 and 2017, Black: patients admitted 2013–2015, Blue: patients with no identified relation to the surgical ward.
Nine patients had been diagnosed with MRSA t267/ST97-IVa at their General Practitioner and three more had been diagnosed at the Emergency Room, resulting in 12 cases first identified in the community, here defined as community-onset MRSA (CO-MRSA). Eight patients had their MRSA diagnosed at hospital wards HA-MRSA. Five persons had been diagnosed by screening of the hospital ward (including the two HCWs) (Table 1).
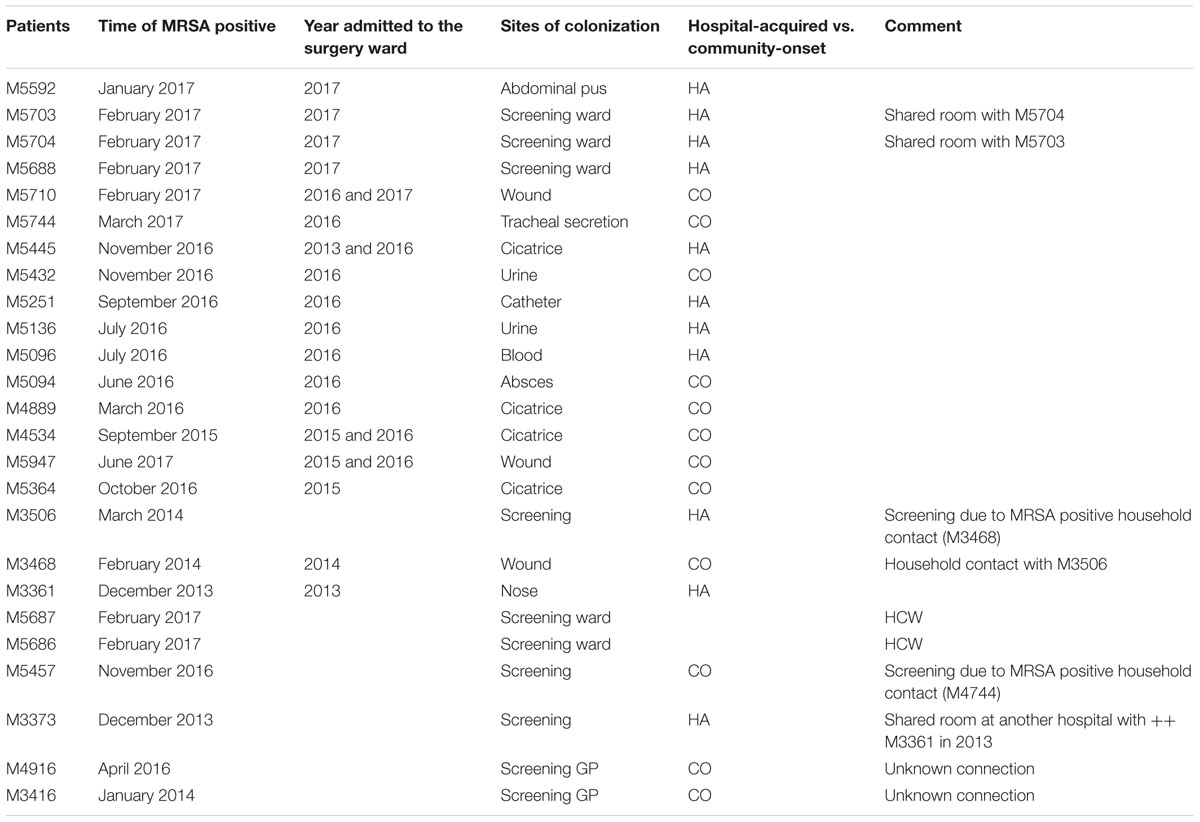
TABLE 1. Demographics.
Due to the finding of other MRSA types among our HCWs, we also studied the phylogenetic tree of all the t045/ST5-IVc/e and t002/ST5-IVg isolates in our WGS database. There were only three t045/ST5 isolates and the two others had another SCCmec type (Supplementary Figure S2). Our database contained 266 t002/ST5 isolates of which 40 harbored SCCmec IVg (Supplementary Figure S3).
SNP Analysis and Characterization of the Outbreak Clone
The 25 t267/ST97/IVa isolates showed a low diversity with a maximum of 50 SNP differences over the 4-year period. Within our outbreak cluster, a further sub-cluster could be distinguished with 15 isolates with a maximum of 11 SNPs. This sub-cluster consisted of 13 patients admitted in 2016 and 2017 as well as the two HCWs.
All outbreak isolates belonged to spa type t267 and ST97 except for two isolates with an unnamed ST that was a single locus variant (SLV) of ST97. All isolates had SCCmec IVa and no isolates had Panton–Valentine leukocidin (PVL) or the arginine catabolic mobile element (ACME). The 14 non-outbreak related isolates of t267/ST97 had SCCmec IVc/e (Gonzalez et al., 2006), SCCmec IVa (Henderson and Nimmo, 2017), and SCCmec V (Gonçalves da Silva et al., 2017). The outbreak isolates were resistant to methicillin and susceptible to erythromycin, clindamycin, gentamicin, fusidic acid, linezolid, mupirocin, trimethoprim/sulfamethoxazole, and rifampicin.
Infection Control Measures
An outbreak group with representatives from the surgical ward, the Department of Cleaning, the Department of Clinical Microbiology, and the Infection Control Organization was established. In order to exclude shortcomings of general infection control precautions, various initiatives were launched. The ward had daily visits by the infection control and prevention nurse, where behavior was observed and adherence to procedures monitored, and on this basis an increased focus was placed on the use of protective equipment such as gloves and plastic aprons, which among other things led to an increased availability of protective equipment in the department. Furthermore, focus was on infection control precautions for both HCWs and patients and in particular on how patients could be motivated for better hand hygiene.
The ward was cleaned by standard hospital cleaning, followed by manual disinfection with bleach. The HCWs with the outbreak clone were successfully decolonized.
Discussion
It is a global trend that patients are hospitalized for increasingly shorter time periods. Therefore, hospital acquisition of an MRSA might not be suspected or identified due to clinical onset long after discharge. Healthcare-associated MRSA outbreaks are rare in Denmark (Andersen and Knudse, 2016) and have been predominantly associated with outbreaks in Neonatal Wards (Ramsing et al., 2013; Bartels et al., 2015; Franck et al., 2017). The extent to which the community serves as a reservoir to the spread of MRSA into hospitals is largely unknown, but globally several reports on CA-MRSA in hospitals have emerged (DeLeo et al., 2010; Thurlow et al., 2012). With repeated introduction of CA-MRSA into hospitals (Cho and Chung, 2017; Coll et al., 2017), a better action plan is needed to tackle and curb community-associated carriage (Bartels et al., 2010). If the level of MRSA carriage increases in the general population then it will also increase in patients and HCWs. In this situation the sporadic spread of MRSA between HCWs and patients with unknown carrier-state, that we find in low MRSA prevalence countries might contribute to more intermittent transmission despite general infection control procedures.
Here, we report a 4-year long outbreak of MRSA type t267/ST97 SCCmec IVa, that was discovered as the MRSA was found in four patients in the same ward within 6 months. This, for us, rare MRSA has sporadically been found around the world and has been described both as a CA-MRSA (Monecke et al., 2011) and as a LA-MRSA in pigs and associated with bovine mastitis (Menegotto et al., 2012; Pantosti, 2012; Feltrin et al., 2015). We have routinely WGS all MRSA isolates since January 2013 and a phylogenetic tree clustered 25 isolates together and patient records revealed that the common denominator was the surgery ward. The outbreak isolates differed up to 50 SNPs, with a subcluster of 13 patients from 2016 or 2017 and the two HCWs whose isolates differed by no more than 11 SNPs. This gives an evolution of the core genome of about 5–6 SNPS per year in our sub-cluster comparable to the 6–9 SNPs per year described in one study (Holden et al., 2013) and more than the 3–4 SNPs per year in other studies (Harris et al., 2010; Senn et al., 2016). The remaining 14 isolates with the same MRSA type had no link to the ward in question and the SNP distance to the outbreak isolates was between 50 and 249 SNPs. Furthermore, most of these isolates had a different SCCmec, which also indicates another clone.
Since the outbreak was ongoing for such a prolonged period at one of our busiest surgical wards, the actual number of people infected or colonized with the MRSA clonal isolate is probably higher than the rather modest number we report. Only individuals in the community who had a clinical infection would have been identified through a sample so MRSA carriers can very well have eluded the system. There could be various explanations to the increase in the number of positive MRSA patients in 2016–2017 in this study. Of course, we found quite a few through the screening in 2017 that might not have been found if the screening had not been performed. Another aspect could be that there were unknown MRSA positive HCWs that had quit the ward before the screening in 2017, but contributed to the increase in cases in 2016 and 2017. Nevertheless, the finding of relatively few patients over a 4-year period indicates there is relatively little spread between HCWs and patients. Previous studies have concluded that nosocomial outbreaks caused by HCWs represent rare events, and therefore screening of personnel should not be performed regularly (Danzmann et al., 2013). Another study proposes three possible scenarios on the role of HCWs: being vectors of transmission, persistent reservoirs, or innocent by-standers, and concludes by suggesting aggressive screening and eradication policies in outbreak investigations (Albrich and Harbarth, 2008). Another aspect of the role of HCWs, and one that is enhanced by our findings, is that due to good infection control measures, HCWs with unrecognized long-term colonization cause only sporadic transmission. This is further supported by the finding of two other MRSA types in the HCWs during the ward screening, with no documented outbreaks caused by them. In support of the conclusion that there had been bacterial transmission from HCWs to patients is the fact that after the HCWs were declared free of MRSA, no more outbreak isolates have been seen at the ward as of May 2018. Of course, transmission could also have occurred between patients, since 13 patients had an overlap in admission time with at least one other patient. However, we lack data on room and sub-department allocation to support this. Due to good infection control measurements transmission only occurred rarely as the outbreak was going on unnoticed.
To automatically detect potential outbreaks the combination of detailed epidemiological data together with WGS is crucial (Roer et al., 2017). In the future an electronic system linking a patients isolate to the last seen most related isolate, will enable earlier detection of outbreaks (Mellmann et al., 2016).
Conclusion
Whole-genome sequencing can enhance the detection of prolonged hospital outbreaks of MRSA. Furthermore, we bring to light the fact that increasingly shorter hospital stays delays outbreak detection if continuous analysis of WGS is not performed. In this study, we identified an MRSA outbreak in February 2017 and with the use of WGS we could trace the outbreak 4 years back in time.
We hypothesize that HCWs with an unknown MRSA carrier-state might cause sporadic transmission and sustain an unknown outbreak over many years. However, we believe that HCWs with a known MRSA carrier-state usually do not cause transmission due to their personally increased awareness of the importance of infection control standard precautions.
Author Contributions
TH and PW performed the bioinformatics analyses. All authors contributed to the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01494/full#supplementary-material
FIGURE S1 | Timeline of hospitalization dates at the surgery ward and date of first positive MRSA sample. Thirteen patients had an overlap with at least one other patient. M5136 had an admission in June 2016, but the exact dates were not available. Sample date denotes the date when MRSA was first discovered in the patient.
FIGURE S2 | Neighbor-joining tree for the three isolates with t045/ST5-IVc/e. Scale bar indicates the SNP distance. M5696 is the HCW. The difference between the isolates was more than 200 SNPs.
FIGURE S3 | NJ-tree of the t002/ST5-IVg isolates. Scale bar indicates the SNP distance. Of the 40 isolates we choose to portray the 25 isolates with less than 100 SNPs difference. The isolates connected with the HCW at the surgical ward are highlighted and the isolate of the HCW in question is M5667. M6160 is a household contact. RH410 is the patient she nursed at the ward, who in turn was also positive for t267, and part of our outbreak cluster. M5746 is an unknown connection, but with no known link to the surgery ward. Between these four isolates there are four SNPs at the most.
References
Albrich, W. C., and Harbarth, S. (2008). Health-care workers: source, vector, or victim of MRSA? Lancet Infect. Dis. 8, 289–301. doi: 10.1016/S1473-3099(08)70097-5
Andersen, P. H., and Knudse, L. K. (2016). MRSA. Available at: https://www.ssi.dk/Aktuelt/Nyhedsbreve/EPI-NYT/2017/Uge%2023
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bartels, M. D., Boye, K., Oliveira, D. C., Worning, P., Goering, R., and Westh, H. (2013). Associations between dru types and SCCmec cassettes. PLoS One 8:e61860. doi: 10.1371/journal.pone.0061860
Bartels, M. D., Kristoffersen, K., Boye, K., and Westh, H. (2010). Rise and subsequent decline of community-associated methicillin resistant Staphylococcus aureus ST30-IVc in Copenhagen, Denmark through an effective search and destroy policy. Clin. Microbiol. Infect. 16, 78–83. doi: 10.1111/j.1469-0691.2009.02829.x
Bartels, M. D., Larner-Svensson, H., Meiniche, H., Kristoffersen, K., Schønning, K., Nielsen, J. B., et al. (2015). Monitoring meticillin resistant Staphylococcus aureus and its spread in Copenhagen, Denmark, 2013, through routine whole genome sequencing. Euro Surveill. 20:21112. doi: 10.2807/1560-7917.ES2015.20.17.21112
Cho, S. Y., and Chung, D. R. (2017). Infection prevention strategy in hospitals in the era of community-associated methicillin-resistant Staphylococcus aureus in the Asia-Pacific region: a review. Clin. Infect. Dis. 64, S82–S90. doi: 10.1093/cid/cix133
Coll, F., Harrison, E. M., Toleman, M. S., Reuter, S., Raven, K. E., Blane, B., et al. (2017). Longitudinal genomic surveillance of MRSA in the UK reveals transmission patterns in hospitals and the community. Sci. Transl. Med. 9:eaak9745.
Danzmann, L., Gastmeier, P., Schwab, F., and Vonberg, R.-P. (2013). Health care workers causing large nosocomial outbreaks: a systematic review. BMC Infect. Dis. 13:98. doi: 10.1186/1471-2334-13-98
DeLeo, F. R., Otto, M., Kreiswirth, B. N. B., and Chambers, H. F. H. (2010). Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375, 1557–1568. doi: 10.1016/S0140-6736(09)61999-1
Di Ruscio, F., Bjørnholt, J. V., Larssen, K. W., Leegaard, T. M., Moen, A. E. F., and de Blasio, B. F. (2017). Epidemiology and spa-type diversity of meticillin-resistant Staphylococcus aureus in the community and healthcare settings in Norway. J. Hosp. Infect. doi: 10.1016/j.jhin.2017.12.019 [Epub ahead of print].
Didelot, X., Bowden, R., Wilson, D. J., Peto, T. E. A., and Crook, D. W. (2012). Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 13, 601–612. doi: 10.1038/nrg3226
Feltrin, F., Alba, P., Kraushaar, B., Ianzano, A., Argudín, M. A., Di Matteo, P., et al. (2015). A livestock-associated, multidrug-resistant, methicillin-resistant Staphylococcus aureus clonal complex 97 lineage spreading in dairy cattle and pigs in Italy. Appl. Environ. Microbiol. 82, 816–821. doi: 10.1128/AEM.02854-15
Franck, K. T., Gumpert, H., Olesen, B., Larsen, A. R., Petersen, A., Bangsborg, J., et al. (2017). Staphylococcal aureus Enterotoxin C and enterotoxin-like L associated with post-partum mastitis. Front. Microbiol. 8:173. doi: 10.3389/fmicb.2017.00173
Gonçalves da Silva, A., Baines, S. L., Carter, G. P., Heffernan, H., French, N. P., Ren, X., et al. (2017). A phylogenomic framework for assessing the global emergence and evolution of clonal complex 398 methicillin-resistant Staphylococcus aureus. Microb. Genomics 3:e000105. doi: 10.1099/mgen.0.000105
Gonzalez, B. E., Rueda, A. M., Shelburne, S. A. III, Musher, D. M., Hamill, R. J., and Hulten, K. G. (2006). community-associated strains of methicillin-resistant Staphylococccus aureus as the cause of healthcare-associated infection. Infect. Control Hosp. Epidemiol. 27, 1051–1056. doi: 10.1086/507923
Harris, A. D., Morgan, D. J., Pineles, L., Perencevich, E. N., and Barnes, S. L. (2017). Deconstructing the relative benefits of a universal glove and gown intervention on MRSA acquisition. J. Hosp. Infect. 96, 49–53. doi: 10.1016/j.jhin.2017.03.011
Harris, A. D., Pineles, L., Belton, B., Johnson, J. K., Shardell, M., Loeb, M., et al. (2013). Universal glove and gown use and acquisition of antibiotic-resistant bacteria in the ICU: a randomized trial. JAMA 310, 1571–1580. doi: 10.1001/jama.2013.277815
Harris, S. R., Cartwright, E. J. P., Török, M. E., Holden, M. T. G., Brown, N. M., Ogilvy-Stuart, A. L., et al. (2013). Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect. Dis. 13, 130–136. doi: 10.1016/S1473-3099(12)70268-2
Harris, S. R., Feil, E. J., Holden, M. T. G., Quail, M. A., Nickerson, E. K., Chantratita, N., et al. (2010). Evolution of MRSA during hospital transmission and intercontinental spread. Science 327, 469–474. doi: 10.1126/science.1182395
Henderson, A., and Nimmo, G. R. (2017). Control of healthcare- and community-associated MRSA: recent progress and persisting challenges. Br. Med. Bull. 125, 1–17. doi: 10.1093/bmb/ldx046
Holden, M. T. G., Hsu, L.-Y., Kurt, K., Weinert, L. A., Mather, A. E., Harris, S. R., et al. (2013). A genomic portrait of the emergence, evolution, and global spread of a methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 23, 653–664. doi: 10.1101/gr.147710.112
Jevons, M. P. (1961). “Celbenin” -resistant staphylococci. Br. Med. J. 1, 124–125. doi: 10.1136/bmj.1.5219.124-a
Leopold, S. R., Goering, R. V., Witten, A., Harmsen, D., and Mellmann, A. (2014). Bacterial whole-genome sequencing revisited: portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J. Clin. Microbiol. 52, 2365–2370. doi: 10.1128/JCM.00262-14
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Lunter, G., and Goodson, M. (2011). A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21, 936–939. doi: 10.1101/gr.111120.110
Mellmann, A., Bletz, S., Böking, T., Kipp, F., Becker, K., Schultes, A., et al. (2016). Real-time genome sequencing of resistant bacteria provides precision infection control in an institutional setting. J. Clin. Microbiol. 54, 2874–2881. doi: 10.1128/JCM.00790-16
Menegotto, F., González-Cabrero, S., Lorenzo, B., Cubero,Á., Cuervo, W., Gutiérrez, M. P., et al. (2012). Molecular epidemiology of methicillin-resistant Staphylococcus aureus in a Spanish hospital over a 4-year period: clonal replacement, decreased antimicrobial resistance, and identification of community-acquired and livestock-associated clones. Diagn. Microbiol. Infect. Dis. 74, 332–337. doi: 10.1016/j.diagmicrobio.2012.08.001
Monecke, S., Coombs, G., Shore, A. C., Coleman, D. C., Akpaka, P., Borg, M., et al. (2011). A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One 6:e17936. doi: 10.1371/journal.pone.0017936
Pantosti, A. (2012). Methicillin-resistant Staphylococcus aureus associated with animals and its relevance to human health. Front. Microbiol. 3:127. doi: 10.3389/fmicb.2012.00127
Ramsing, B. G. U., Arpi, M., Andersen, E. A., Knabe, N., Mogensen, D., Buhl, D., et al. (2013). First outbreak with mrsa in a danish neonatal intensive care unit: risk factors and control procedures. PLoS One 8:e66904. doi: 10.1371/journal.pone.0066904
Roer, L., Hansen, F., Frølund Thomsen, M. C., Knudsen, J. D., Hansen, D. S., Wang, M., et al. (2017). WGS-based surveillance of third-generation cephalosporin-resistant Escherichia coli from bloodstream infections in Denmark. J. Antimicrob. Chemother. 72, 1922–1929. doi: 10.1093/jac/dkx092
SenGupta, D. J., Cummings, L. A., Hoogestraat, D. R., Butler-Wu, S. M., Shendure, J., Cookson, B. T., et al. (2014). Whole-genome sequencing for high-resolution investigation of methicillin-resistant Staphylococcus aureus epidemiology and genome plasticity. J. Clin. Microbiol. 52, 2787–2796. doi: 10.1128/JCM.00759-14
Senn, L., Clerc, O., Zanetti, G., Basset, P., Prod’hom, G., Gordon, N. C., et al. (2016). The stealthy superbug: the role of asymptomatic enteric carriage in maintaining a long-term hospital outbreak of ST228 methicillin-resistant Staphylococcus aureus. mBio 7:e02039-15. doi: 10.1128/mBio.02039-15
Thurlow, L. R., Joshi, G. S., and Richardson, A. R. (2012). Virulence strategies of the dominant USA300 lineage of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). FEMS Immunol. Med. Microbiol. 65, 5–22. doi: 10.1111/j.1574-695X.2012.00937.x
Zarfel, G., Luxner, J., Folli, B., Leitner, E., Feierl, G., Kittinger, C., et al. (2013). Genomic insights into the emergence and spread of international clones of healthcare-, community- and livestock-associated meticillin-resistant Staphylococcus aureus: blurring of the traditional definitions. J. Clin. Microbiol. 7, 1–10.
Keywords: WGS, outbreak, CO-MRSA, ST97, HCWs
Citation: Rubin IM, Hansen TA, Klingenberg AM, Petersen AM, Worning P, Westh H and Bartels MD (2018) A Sporadic Four-Year Hospital Outbreak of a ST97-IVa MRSA With Half of the Patients First Identified in the Community. Front. Microbiol. 9:1494. doi: 10.3389/fmicb.2018.01494
Received: 16 March 2018; Accepted: 18 June 2018;
Published: 10 July 2018.
Edited by:
Miklos Fuzi, Semmelweis University, HungaryReviewed by:
David Christopher Coleman, Dublin Dental University Hospital, IrelandElizabeth Marion Dickson, NHS Greater Glasgow and Clyde, United Kingdom
Copyright © 2018 Rubin, Hansen, Klingenberg, Petersen, Worning, Westh and Bartels. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mette D. Bartels, bWV0dGUuZGFta2phZXIuYmFydGVsc0ByZWdpb25oLmRr