 Xin Pang
Xin Pang Ya-jie Tang
Ya-jie Tang Xiao-hua Ren
Xiao-hua Ren Qian-ming Chen
Qian-ming Chen Ya-ling Tang
Ya-ling Tang Xin-hua Liang
Xin-hua Liang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 26 June 2018
Sec. Microbial Immunology
Volume 9 - 2018 | https://doi.org/10.3389/fmicb.2018.01353
Microbiota has been widely considered to play a critical role in human carcinogenesis. Recent evidence demonstrated that microbiota, epithelial barrier and inflammation has made up a tightly interdependent triangle during the process of carcinogenesis. Hence, we discussed the triangle relationship of microbiota dysbiosis, epithelial barrier dysfunction and dysregulated immune responses to elucidate the mechanisms by which microbiota induces carcinogenesis, especially highlighting the reciprocal crosstalk between transforming growth factor-β signaling and every side of the tumorigenic triangle. This sophisticated interaction will provide insight into the basic mechanisms of carcinogenesis and may bring new hope to cancer prevention and therapeutic intervention.
The microorganism is now seen as a “new organ” in human. The “new organ,” which contains an enormous number of cells and genetic material, inhabits in body surfaces and cavities connecting with exterior in physiological conditions (Marchesi et al., 2016; Zitvogel et al., 2016). One of the characteristics of microbiota is its individuality as it is shaped by genetics, life and dietary styles, medication use and microbial exposure of hosts (Zitvogel et al., 2016). The complex community of microorganisms acts as an integrated ecosystem to provide benefits with healthy hosts. On the contrary, deviating from microbiota equilibrium has been regarded as an important factor in diseases intertwined with gene and environment (Bultman, 2014).
The suspicion that microbiota imbalance is involved in the course of carcinogenesis has been in the Spot-LIGHT in the past few decades. Cancers driven by infectious agents were estimated to comprise of 15–20% of the global cancer burden in 2012, among which Helicobacter pylori (H. pylori), human papilloma viruses (HPV), hepatitis B (HBV) and C viruses (HCV), and Epstein-Barr virus matter a lot (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 2012; Bhatt et al., 2017). Microbiota dysbiosis refers to microbial maladaptation or imbalance of quantity and quality, especially imbalance in microbial richness and function, which makes commensal bacterium become pathogenic bacterium in host with genetical defect or dysregulated inflammation (Bhatt et al., 2017). It is reported that dysbiosis is associated with a large amount of malignancies and is implicated in the formation of cancers such as colorectal cancer (CRC) (Schwabe and Jobin, 2013; Perez-Chanona and Trinchieri, 2016). Accumulating evidence showed that the mechanisms of microbiota dysbiosis-driving oncogenesis may include: (I) integrating oncogenes of oncovirus into host genomes or compromising genomic instability, (II) triggering cancer by promoting inflammation and dampening immune-surveillance of transformed cells, (III) subverting the integrity of epithelial barrier, (IV) contributing to the resistance to cell death and disturbing signaling pathways related to carcinogenesis. Intriguingly, this indicated that microbiota, epithelial barrier and inflammation make up a tightly interdependent triangle during the process of carcinogenesis driven by microbiota (Fox and Wang, 2007; Couturier-Maillard et al., 2013; Hu et al., 2013; Francescone et al., 2014; Ojesina et al., 2014). However, the molecular mechanisms of the interaction of microbial dysbiosis, barrier failure and dysregulated inflammation regulating carcinogenesis have remained unclear (Schwabe and Jobin, 2013; Garrett, 2015).
Transforming growth factor-β (TGF-β) signaling pathway acts as a tumor-suppressor in normal epithelium or early stage of oncogenesis. However, like a coin with two sides, tumor-promoter role of TGF-β comes along that facilitates cell proliferation and immune-evasion (Zhang et al., 2006; Pardali and Moustakas, 2007; White et al., 2010). Mechanistically, TGF-β maintains homeostasis and blocks tumor formation through enhancing cell cycle arrest and apoptosis via canonical TGF-β-SMADs pathways. As TGFβR on tumor cells mutated, TGF-β accumulated in extracellular matrix, shaping a tumor friendly microenvironment that favoring proliferation and invasion of tumor cells. And TGF-β expression was correlated with angiogenesis, myofibroblast formation and inflammatory cells recruitment via non-canonical PI3K/Akt, the nuclear factor κ light-chain-enhancer of activated B cells (NF-κB), or ERK signaling (Iglesias et al., 2000; Lu et al., 2006; Bian et al., 2009; Freudlsperger et al., 2013). Notably, TGF-β realigns immune cells and abrogates the function of tumor-suppressing immune cells, such as dendritic cells (DCs) and T effector cells, to help tumor cells escape from immune-surveillance (Yang, 2010). Intriguingly, plenty of evidence showed that the mechanism of TGF-β signaling in favor of tumorigenesis also correlates with dysregulated inflammation microenvironment driven by microbiota.
In this review, we will outline current understanding of the mechanisms by which microbiota induces cancer and the impact of TGF-β signaling pathway in carcinogenesis driven by microbiota. As we learn more about the intricate relationship between microbiota and cancer, it may help indicate therapeutic targets and biomarkers of cancer (Zeller et al., 2014).
The well-maintained mucosal barrier in gut or oral cavity, consisted of the mucous layer, the stratum corneum, immune cells, secretory immunoglobulin A (sIgA) and antibacterial peptides, is a multi-level defender to separate human tissues from microbiota (Schwabe and Jobin, 2013). Anatomic disruption, microbial composition and mucus production changes can bring about epithelial barrier dysfunction and microenvironment disruption (Schwabe and Jobin, 2013). They grant pathogens and commensal microorganisms access to the epithelium, and then their metabolites, toxins, as well as proteins may regulate tumorigenesis. Intestinal microbiota dysbiosis and its interaction with a functionally impaired mucosal barrier lead to ulcerative colitis. In up to 30% of affected patients after 35 years of ulcerative colitis has the occurrence of CRC (Rogler, 2014). Mucins, the highly glycosylated proteins, were the major component of the mucus that protected underlying epithelium. Absence of mucins increased proliferation and migration, as well as decreased apoptosis of intestinal epithelial cells (IECs). Muc2-knockout mice frequently developed into intestine adenomas and gradually progressed to CRC (Velcich et al., 2002). Bacterial translocation referred to the migration of bacteria and bacterial products to mesenteric lymph nodes or extra-intestinal sites from the gut. Increased translocation of intestinal bacteria contributed to hepatic inflammation and fibrosis, as well as promoted hepatocellular carcinoma (HCC) (Wiest and Garcia-Tsao, 2005; Dapito et al., 2012). While gut sterilization by antibiotic management could reduce HCC in the late stage of hepatocarcinogenesis of mice (Dapito et al., 2012). These indicated that destruction of host/flora equilibrium of epithelial barrier was a critical step in microbial associated diseases, including cancer (Sellers and Morton, 2014; Taur and Pamer, 2016).
Microbiota not only provides nutrients and helps to abstract calories for the hosts, but also produces detrimental metabolites favoring noxious inflammation and tumor formation (Bhatt et al., 2017). Evidence is emerging that sulfides, nitrosamines, hydrogen peroxide and deoxycholic acid (DCA) are the main detrimental metabolites of microbes related to carcinogenesis. Some Firmicutes and Bacteroides sp. ferment excessive protein of the host into sulfides and nitrosamines, which may induce DNA alkylation and mutations in host cells. Glycosulfatase in Bacteroides could catalyze sulfomucins to release sulfides, contributing to mucins degradation and carcinogenesis (Carbonero et al., 2012; Bhatt et al., 2017). And Enterococcus faecalis can produce extracellular superoxide, which also produced by host cells in inflammatory conditions, spontaneously deriving hydrogen peroxide in colon (Huycke et al., 1996, 2002). Hydrogen peroxide can diffuse into epithelial cells passively and form hydroxyl radical which leads to DNA double-strand break, base modification and DNA-protein cross-linking by iron-catalyzed reactions (Huycke and Gaskins, 2004; Wang et al., 2008; Sears and Garrett, 2014). Clostridium scindens converts primary bile acids in colon into secondary deoxycholic acid such as DCA. DCA perturbates cell membranes and releases arachidonic acid, which converted to prostaglandins and reactive oxygen species (ROS), exacerbating DNA damage and initiating carcinogenesis (Ridlon et al., 2016). Hydrogen sulfide and acetaldehyde are not inert bystanders too, resulting in inflammation and genomic instability, and promoting CRC development (Huycke and Gaskins, 2004; Windey et al., 2012; Schwabe and Jobin, 2013). Thus, intestinal bacteria have the potential to stimulate excessive proteins to form detrimental metabolites, genotoxins and tumor-promoters, through DNA double-strand breaks and activation of the DNA damage (Table 1).
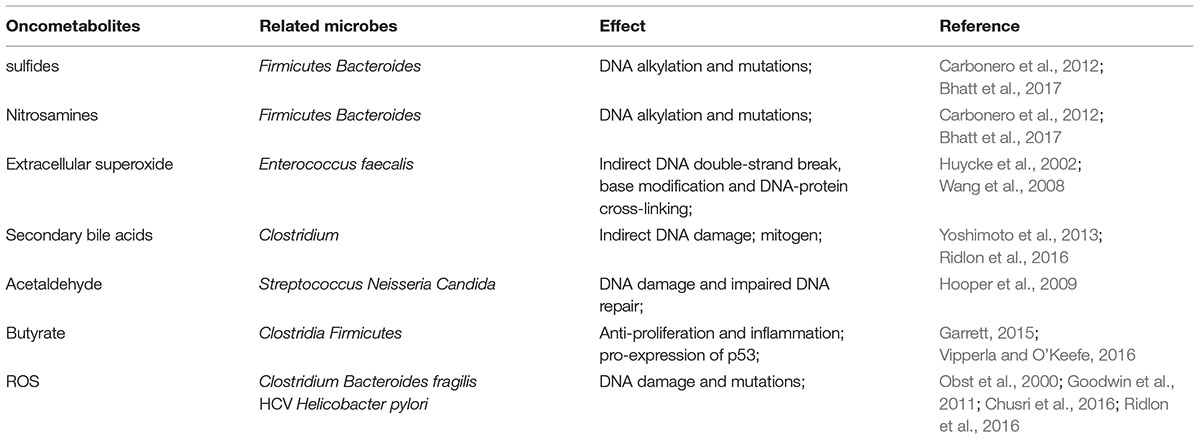
TABLE 1. Oncometabolites from bacteria leading to noxious inflammation and tumor formation.
Evidence supports the role of bacterial derived toxins in driving cell transformation mainly by their genotoxicity and interference with host-derived signaling pathways (Bhatt et al., 2017). Colibactin is the product of “pks,” a genomic island of Escherichia coli strains of phylogenetic group B2. Infection of eukaryotic cells with pks+ E. coli induces DNA double-strand break and activation of DNA damage signaling cascade, resulting in breakage–fusion–bridge cycles and the increase of anchorage-independent growth, which contribute to CRC development (Nougayrede et al., 2006; Cuevas-Ramos et al., 2010). Arthur et al. (2012) demonstrated that azoxymethane (AOM)-treated Interleukin-10 (IL-10) knockout mice developed invasive carcinoma in mono-colonized with the pks+ E. coli. On the contrary, AOM-treated IL-10 knockout mice mono-colonized with E. coli depleted pks showed declined tumor multiplicity and invasion (Arthur et al., 2012). Cytolethal distending toxin (CDT) derived from Gram-negative bacteria is also genotoxic factor that has intrinsic DNase activity. Nuclease CdtB travels to host cells with the help of CdtA and CdtC subunits, where it creates DNA lesions and results in intestinal hyperplasia of mice (Nesic et al., 2004; Shen et al., 2009). Bacteroides fragilis toxin (BFT) is a virulent factor that damages host DNA by eliciting ROS production, and is closely associated with signal transducer and activator of transcription-3 (STAT3)- and T helper 17 (Th17)-dependent inflammation and CRC (Wu et al., 2009; Garrett, 2015). Furthermore, Goodwin et al. (2011) identified that BFT upregulated the expression of spermine oxidase in colonic epithelial cells which catalyzed polyamine catabolism and ROS production, resulting in CRC formation (Goodwin et al., 2011). Thereby, a diverse array of virulence factors of bacterium and their pathways may manipulate basic host cell functions, such as proliferation and invasion, to contribute to carcinogenesis.
Recently, an association between virulence factors and host pathways in the course of carcinogenesis has been recognized, especially protein toxins. FadA encoded by Fusobacterium nucleatum adheres to lectins and E-cadherin on the surface of epithelial cells (Rubinstein et al., 2013). Similarly, H. pylori bears cagA that encodes CagA, an antigenic effector protein interacting with E-cadherin (Abreu and Peek, 2014). The competitive binding of FadA/CagA and E-cadherin impairs the complex between E-cadherin and β-catenin, leading to the activation of β-catenin signaling, which regulates downstream genes, such as c-MYC, cdx1 and Ccnd1 to promote cells proliferation of CRC (Murata-Kamiya et al., 2007; Rubinstein et al., 2013). Besides, AvrA secreted by gall bladder cancer associated Salmonella, has also been suggested to activate β-catenin signaling by suppressing β-catenin ubiquitination and increasing its phosphorylation. β-catenin activated by AvrA increased the expression of cyclin D1 and matrix metalloproteinase (MMP)-7, promoting colonic carcinogenesis (Samaras et al., 2010; Lu et al., 2014). Hence, protein toxins may play an important role in enhancing transformation of epithelial cells by affecting genomic stability and proliferative signaling.
Transforming growth factor-β signaling has been shown to play an important role in microbiota-epithelial interaction. Firstly, special detrimental metabolites increased TGF-β expression. Clostridium exacerbated the production of TGF-β by IECs, possibly through its metabolites, such as butyrate, acetate and propionate. In concert with the increased level of TGF-β, Clostridium promoted the expression of MMP2, 9 and 13 on the surface of IECs, which rendered latent TGF-β activation in the colon (Atarashi et al., 2011; Bauche and Marie, 2017). Chusri et al. (2016) showed that HCV enhanced ROS induction which then phosphorylated JNK and NF-κB in turn. Consequently, TGF-β expression was up-regulated by activated NF-κB (Chusri et al., 2016). Lin et al. (2010) found that HCV induced ROS production also activated p38 MAPK and p42/44 ERK pathways to phosphorylate NF-κB and upregulate TGF-β.
Secondly, special virus proteins interfere with TGF-β signaling components. HCV core proteins activate TGF-β through inducing thrombospondin-1 in extracellular matrix, which binds to the Leu-Ser-Lys-Leu amino acids sequence and alters conformation of latency associated protein (Benzoubir et al., 2013). HBV-encoded pX oncoprotein and HBV X protein facilitated TGF-β signaling via potentiating nuclear translocation of SMAD4 transcription complex and stabilizing p-SMAD2/3, respectively (Lee et al., 2001; Liu y. et al., 2016). It also activated c-Jun N-terminal kinase/pSMAD3L pathway and inhibited canonical TGFβRI/pSMAD3C to add epithelial proliferation in HCC cell lines (Wu et al., 2016). Additionally, HBV upregulates SMAD7, an inhibitor of TGF-β signaling, leading to resistance of host cells to TGF-β induced apoptosis in HCC (Liu et al., 2015). HPV oncoprotein E7 inhibited binding of SMAD complex to SMAD binding sequence, which attenuated TGF-β signal transduction in E7 expressing cell lines and contributed to tumorigenesis (Lee et al., 2002). Similarly, HPV E5 blocked TGF-β signaling via decreasing SMAD2 phosphorylation and SMAD4 nuclear translocation (French et al., 2013).
Finally, TGF-β influences flora-barrier interaction in association with DCs and regulatory T cells (Tregs). Infection with Citrobacter rodentium induces apoptosis of IECs. DCs took part in the phagocytosis of these apoptotic cells and were activated to synthesize TGF-β, IL-6, and IL-23 (Brereton and Blander, 2010). And microbiota induced TGF-β facilitates the expression of fibroblast growth factor 2 (FGF2) in Treg cells. FGF2 cooperates with IL-17 to repair damaged epithelium and control microbes outgrowth, to suppress colitis and colon carcinoma associated with colitis (Song et al., 2015) (Figure 1).
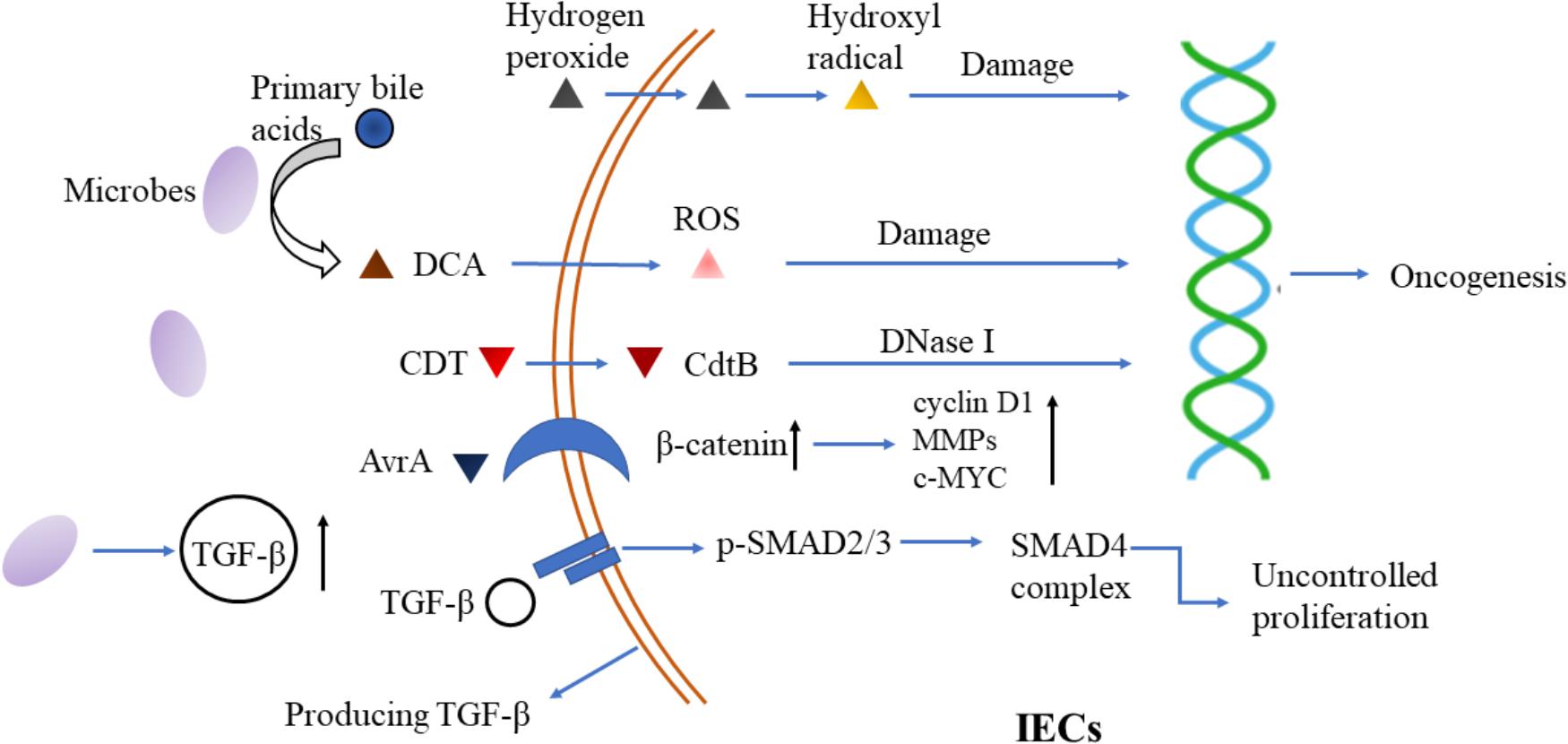
FIGURE 1. Microbiota-epithelial interaction and its interference by TGF-β signaling. Microbial metabolites (hydroxyl radical and ROS for example) and bacterial derived toxins (such as CDT) induce DNA damage and trigger oncogenesis in epithelial cells. Additionally, bacterial virulence factors, such as AvrA, activate β-catenin signaling to promote proliferation of epithelial cells. Besides, microbes stimulate TGF-β production by IECs through their metabolites and specific proteins, which then triggers uncontrolled proliferation in turn. DCA, deoxycholic acid; ROS, reactive oxygen species; CDT, cytolethal distending toxin; MMP, matrix metalloproteinase; TGF-β, transforming growth factor-β; IECs, intestinal epithelial cells.
Inflammatory disorders, including pro-inflammatory and anti-inflammatory responses elicited by microbes, may take place when epithelial barrier deteriorate and bacterial translocate. It has generally been accepted that inflammation is a link between microbiota and cancer progression as described by Virchow (1881) in the 19th century. As a prime example, inflammation induced by H. pylori with gastritis and gastric ulcers is strongly associated with gastric adenocarcinoma (Marshall and Warren, 1984). Inflammation drives a tumor permissive niche, in which the increased production of cytokines, chemokines and growth factors, as well as nitrogen species and reactive oxygen involve in stimulating cell proliferation and/or inhibiting cell apoptosis to facilitate oncogenesis (Akin and Tozun, 2014; Irrazabal et al., 2014; Zitvogel et al., 2015). Moreover, inflammation may alter microbial composition, increase microbial translocation and promote outgrowth of specific bacteria possibly by production of specific metabolites which allow them to thrive (Arthur et al., 2012; Elinav et al., 2013; Schwabe and Jobin, 2013). Patwa et al. (2011) showed that chronic intestinal inflammation induced stress-response genes expression in gut commensal bacterium, which would confer bacterium environmental adaptability. Bacterial antigens are detected and signaled by the Toll-like receptors (TLR) through NF-κB pathway, which is a key mediator triggering cancer-associated inflammation (Karin and Greten, 2005; DiDonato et al., 2012; Bhatt et al., 2017). In turn, microbes also elicit immunosuppression responses and contribute to tumor-immune evasion. F. nucleatum may directly engage TIGIT, a receptor expressed on some T cells and natural killer (NK) cells, to block the antitumor responses of NK cells dependent killing (Gur et al., 2015). Microbial derived butyrate, an inhibitor of histone deacetylase which leads to the activation of forkhead box P3 (FoxP3) regulator and signals through G protein-coupled receptors, could induce naive T cells and dendritic cells to differentiate into Tregs to inhibit anti-tumor immune responses (Gur et al., 2015). These indicated that the association between the microbiota and the host immune system may result in a cause of inflammation and development or progression of cancer.
Increasing evidence showed that TGF-β was an important cytokine in the development of cancer involved in microbiota and immune reaction. Besides of IECs as mentioned above, lamina propria dendritic cells (LPDCs) are also suggested to secrete more TGF-β under stimulation by microbiota-mediated signaling and microbiota-derived products (Bauche and Marie, 2017). In response to Clostridium butyricum, LPDCs induced TGF-β1 production through a cooperation of TLR2-AP-1 and TGF-β-SMAD pathway (Kashiwagi et al., 2015). Also, Atarashi et al. (2008) have demonstrated that commensal bacteria derived ATP increased integrin-alphaV and -β8 expression on inflammatory DCs, which activated latent TGF-β (Boucard-Jourdin et al., 2016). TGF-β level in the gut is directly and indirectly modulated by microbes, which impacts the complex interplay with host and microbiota through its modulation of Tregs, Th17 cells and B cells.
Tregs, abundant within the lamina propria in colon, suppress anti-tumor effector CD4+ and CD8+ T cells responses to maintain immune tolerance to tumor antigens and microbial antigens (Bauche and Marie, 2017). TGF-β and other cytokines, to some degree, decided differentiation of CD4+ T cells into Tregs or Th17. TGF-β can induce Foxp3 expression in CD25- cells from periphery and convert these cells into CD4+CD25+ induced Treg (iTreg) cells (Khattar et al., 2009). High concentrations of TGF-β represses IL-23 receptors (IL-23R) expression so that it contributes to Tregs differentiation. TGF-β induces retinoic acid receptor-related orphan receptor γt (RORγt) co-expression in CD4+ T cells with Foxp3, which could repress RORγt and result in Tregs differentiation. Whereas, the repression could be removed by IL-6, IL-21, and IL-23 (Zhou et al., 2008).
Microbiota and microbiota derived metabolites are also involved in the process of Foxp3 expression. Commensal B. fragilis promotes immunologic tolerance through producing polysaccharide A (PSA), a symbiosis factor. PSA directly stimulates TLR2 on CD4+ T cells to strongly enhance the function and expression of Foxp3, IL-10, and TGF-β (Round et al., 2011). Butyrate may directly result in Foxp3 transcription to induce Tregs among CD4+ T cells through its inhibition of histone deacetylation in conserved non-coding sequence 1 and promoter regions of the Foxp3 locus (Furusawa et al., 2013). On the other hand, butyrate also indirectly enforces Tregs differentiation by inducing IECs secreting TGF-β (Bauche and Marie, 2017).
The differentiation of Th17 cells, important pro-inflammatory cells in many solid tumors, is stimulated by microbes and is influenced by activated TGF-β under inflammatory conditions within intestines (Veldhoen et al., 2006; Ivanov et al., 2008; Wilke et al., 2011). Segmented filamentous bacteria can directly adhere to IECs and induce the production of serum amyloid A proteins 1 and 2, members of acute-phase response proteins family in response to infection or inflammatory, which in turn promote DCs-mediated Th17 cells differentiation in lamina propria and IL-17a secretion (Ivanov et al., 2009; De Simone et al., 2013; Sano et al., 2015). It has been suggested that commensal microbes stimulated CD172α(+) LPDCs inducing Th17 cells partly mediating by TLR5. Once stimulated with flagellin, CD172α(+) LPDCs co-expressed TLR5 can secrete IL-6, IL-23, and TGF-β, which mediate CD4+ T cells to develop into Th17 cells, whereas TLR5-deficient LPDCs do not (Liu H. et al., 2016). While it induces Tregs differentiation at a high concentration, TGF-β at low concentrations is in favors of IL-23R expression and promotes Th17 cells differentiation in cooperation with IL-6 and IL-21 (Zhou et al., 2008). With their distinctively secretion of IL-17 that enhances angiogenesis and induces tumor cells proliferation in immune-deficient hosts or patients with already existed chronic inflammation, Th17 cells are negatively correlated with the prognosis of patients with CRC (Numasaki et al., 2005; Tosolini et al., 2011; Pickup et al., 2013; Song et al., 2014; Gagliani et al., 2015).
Transforming growth factor-β signaling has been reported to regulate responsiveness of B cells to antigens, IgA production and IgA class-switch recombination (CSR) on SMADs-dependent pathways. IgA, a predominant antibody for mucosal surfaces, confers the ability for IgA+ B cells to help maintain the integrity of epithelial barrier and modulate bacterial composition within lumen (Klein et al., 2006). Alcaligenes which inhabits Peyer’s patches and lymphoid follicles, is shown to contribute to CSR through increasing TGF-β as well as IgA-enhancing cytokine IL-6 production, when cocultured with DCs isolated from Peyer’s patches of WT mice (Obata et al., 2010). Alcaligenes has also been reported to bear genes coding for nitric oxide reductase (NOR) which reduces NO. NO is implicated in TGFβRII expression on B cells and regulates TNFα/iNOS-producing DCs mediated IgA CSR (Kukimoto et al., 2000; Tezuka et al., 2007; Bauche and Marie, 2017). In addition, DCs stimulated with microbes induce integrin α4β7 and chemokine receptor 9 (CCR9) to imprint gut-homing specificity on B cells and promote IgA production (Tezuka et al., 2007; Ruane et al., 2016).
Collectively, microbiota modulates the production of TGF-β that impacts the development, proportion and function of immune cell subsets, resulting in inflammation and regulating the interaction between microbiota and hosts in turn (Figure 2).

FIGURE 2. Microbiota-immune interaction and its interference by TGF-β signaling. Bacterial antigens which are detected and signaled by TLRs, could elicit tumor-permissive immune responses by activating Tregs and Th17 cells. In response to microbial metabolites, IECs and DCs secret more TGF-β that impacts the development, proportion and function of Tregs, Th17 cells and B cells, resulting in immune-evasion of tumor cells and regulating the interaction between microbiota and hosts in turn. IECs, intestinal epithelial cells; DCs, dendritic cells; TGF-β, transforming growth factor-β; RORγt, retinoic acid receptor-related orphan receptor γt; IL, Interleukin; Tregs, regulatory T cells; Th17, T helper 17; TLR, Toll-like receptors; PSA, polysaccharide A; IgA, immunoglobulin A.
As stated, gut flora dysbiosis breached epithelial barrier to provoke immune responses indirectly. As a prime example, fiber-fermenting bacteria is diminished by fiber-free diets but Akkermansia muciniphila and Bacteroides caccae are increased which degrade mucus in the lumen. Mucus degrading increased the susceptibility of Citrobacter rodentium and resulted in “leaky gut” to activate intraepithelial lymphocytes and form chronic inflammation environment (Bhatt et al., 2017). On the other side, locally chronic inflammation is a risk factor for initiating carcinogenesis since epithelial cells transformation accumulates. Leukocytes and other phagocytic cells could generate reactive oxygen and nitrogen species during immune responses, which lead to DNA damage and permanent genomic alterations in epithelial cells (Coussens and Werb, 2002). It is noteworthy that chemokines could also regulate neoplastic cells proliferation and metastasis, as well as angiogenesis of in extracellular matrix (Coussens and Werb, 2002). Carcinogenesis induced by chronic inflammation is also required for signaling pathways such as NF-κB and TGF-β secreted by immune cells as mentioned above (Figure 3).
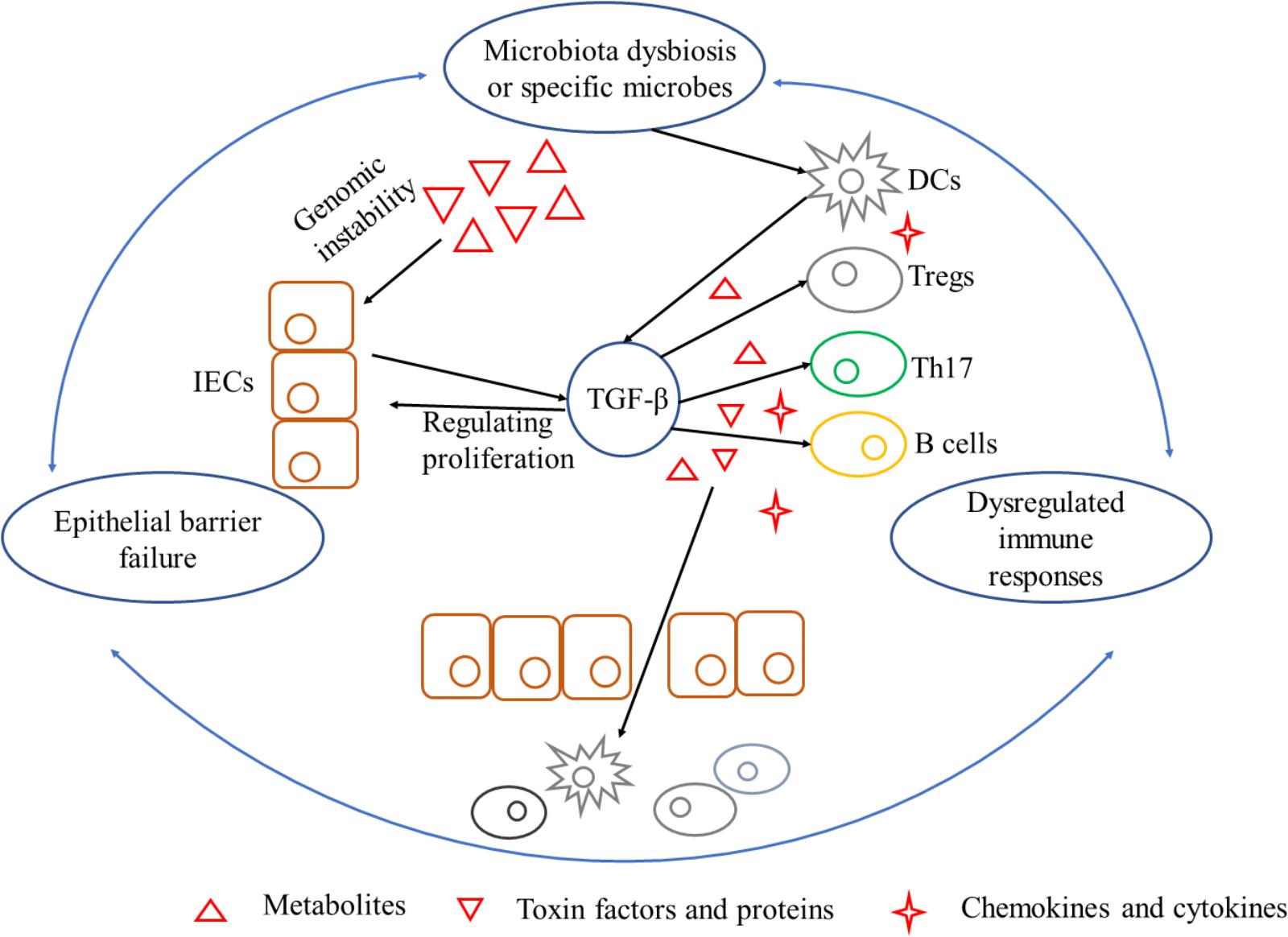
FIGURE 3. A triangle during the process of carcinogenesis driven by microbiota dysbiosis and its interference with TGF-β. Mechanisms by which microbiota dysbiosis induced carcinogenesis include breaking epithelial barrier, inducing DNA damage in epithelial cells, eliciting inflammation and dampening immune-surveillance of transformed cells. Barrier deterioration is a stimulating factor that triggers locally chronic inflammation to aggravate epithelial barrier failure in turn and exacerbate carcinogenesis. TGF-β, produced by microbes stimulated IECs and inflammatory cells, is a pluripotent cytokine that plays an important role in exacerbating unbalance of the triangle. TGF-β can be tumor-permissive by promoting proliferation and inhibiting apoptosis of epithelial cells. On the other hand, TGF-β is in favor of immune-evasion of tumor antigens elicited by special microbes. IECs, intestinal epithelial cells; TGF-β, transforming growth factor-β; DCs, dendritic cells; Tregs, regulatory T cells; Th17, T helper 17.
Colorectal cancer involves in an intricate regulation of tumor cells, microorganism, and non-neoplastic cells (Gagniere et al., 2016). They form an interdependent triangle of microbial dysbiosis, epithelial barrier breach and chronic inflammation in microbiota driven carcinogenesis. And dysregulation of any one side may break the whole equilibrium (Schwabe and Jobin, 2013). Notably, infection of pathogens or commensal bacteria dysbiosis may break epithelial barrier and shape pro-tumorigenic inflammation, leading to cell proliferation. Barrier deterioration, on the one hand, gives microbes and their by-products access to epithelial cells and immune cells (Brennan and Garrett, 2016). On the other hand, perpetual injury triggers locally chronic inflammation and oncogenesis, which then aggravate epithelial barrier failure and microbiota dysbiosis in turn. Bacterial translocation induced inflammation mediates carcinogenesis and inhibits immune-surveillance, as well as contributes to the overgrowing of genotoxic microbes (Arthur et al., 2012). As such, the dysregulation of any one side may break the whole equilibrium, and then forward-amplifying loops formed.
In addition, microbiota functions in anticancer drug metabolism and alters host responses to several drugs. For example, the platinum chemotherapeutic oxaliplatin triggers cancer cell death through forming platinum DNA adducts and intrastrand cross-links. Its efficacy is depended on microbial derived ROS, with augmented intratumoral oxidative stress promoting oxaliplatin associated DNA damage (Iida et al., 2013). Given the crucial effects that microbiota exert on tumor formation and treatment, the differences of microbiota composition, dietary habits, genetic makeup and living environment should be considered when deciding an optimized treatment modality for individuals. It is also called precision medicine treatment involved in precise dosing, symptom management and improved therapeutic responses (Bhatt et al., 2017).
Then, the role of TGF-β signaling pathway is discussed in microbiota driven oncogenesis. Mounting evidence showed that TGF-β was induced by microorganisms, including Clostridium, Bacteroides, and Enterobacteriaceae (Atarashi et al., 2013; Ihara et al., 2017). TGF-β is a pluripotent cytokine that regulates epithelial barrier and immune responses. TGF-β can be tumor-permissive by promoting proliferation and inhibiting apoptosis, but also it is in favor of immune-evasion of tumor antigens elicited by special microbes. In this regard, microbiota increases TGF-β production, as well as reciprocally facilitating the function in oncogenesis. This review indicates that dysregulated microbiota induces carcinogenesis through destabilizing the interdependent interaction of microbiota, epithelial barriers and inflammation, and TGF-β signaling pathway probably plays an important role in exacerbating deviating from homeostasis. As such, it holds great promise for us to look for new therapeutic targets in human cancers relative to microbiota.
XP was mainly responsible for the manuscript writing. X-HR, Y-JT, and Q-MC assisted in writing. Y-LT and X-HL provided suggestions on the ideas and performed the final corrections.
This work was supported by National Natural Science Foundation of China grants (Nos. 81672672, 81772891, 81572650, and 81621062) and by National Program on Key Research Project of China (2016YFC0902700).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abreu, M. T., and Peek, R. M. Jr. (2014). Gastrointestinal malignancy and the microbiome. Gastroenterology 146, 1534.e3–1546.e3. doi: 10.1053/j.gastro.2014.01.001
Akin, H., and Tozun, N. (2014). Diet, microbiota, and colorectal cancer. J Clin. Gastroenterol. 48(Suppl. 1), S67–S69. doi: 10.1097/mcg.0000000000000252
Arthur, J. C., Perez-Chanona, E., Muhlbauer, M., Tomkovich, S., Uronis, J. M., Fan, T. J., et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123. doi: 10.1126/science.1224820
Atarashi, K., Nishimura, J., Shima, T., Umesaki, Y., Yamamoto, M., Onoue, M., et al. (2008). ATP drives lamina propria T(H)17 cell differentiation. Nature 455, 808–812. doi: 10.1038/nature07240
Atarashi, K., Tanoue, T., Oshima, K., Suda, W., Nagano, Y., Nishikawa, H., et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. doi: 10.1038/nature12331
Atarashi, K., Tanoue, T., Shima, T., Imaoka, A., Kuwahara, T., Momose, Y., et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. doi: 10.1126/science.1198469
Bauche, D., and Marie, J. C. (2017). Transforming growth factor beta: a master regulator of the gut microbiota and immune cell interactions. Clin. Transl. Immunol. 6:e136. doi: 10.1038/cti.2017.9
Benzoubir, N., Lejamtel, C., Battaglia, S., Testoni, B., Benassi, B., Gondeau, C., et al. (2013). HCV core-mediated activation of latent TGF-beta via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J. Hepatol. 59, 1160–1168. doi: 10.1016/j.jhep.2013.07.036
Bhatt, A. P., Redinbo, M. R., and Bultman, S. J. (2017). The role of the microbiome in cancer development and therapy. CA Cancer J. Clin. 67, 326–344. doi: 10.3322/caac.21398
Bian, Y., Terse, A., Du, J., Hall, B., Molinolo, A., Zhang, P., et al. (2009). Progressive tumor formation in mice with conditional deletion of TGF-beta signaling in head and neck epithelia is associated with activation of the PI3K/Akt pathway. Cancer Res. 69, 5918–5926. doi: 10.1158/0008-5472.CAN-08-4623
Boucard-Jourdin, M., Kugler, D., Endale Ahanda, M. L., This, S., De Calisto, J., Zhang, A., et al. (2016). beta8 integrin expression and activation of TGF-beta by intestinal dendritic cells are determined by both tissue microenvironment and cell lineage. J. Immunol. 197, 1968–1978. doi: 10.4049/jimmunol.1600244
Brennan, C. A., and Garrett, W. S. (2016). Gut microbiota, inflammation, and colorectal cancer. Annu. Rev. Microbiol. 70, 395–411. doi: 10.1146/annurev-micro-102215-095513
Brereton, C. F., and Blander, J. M. (2010). Responding to infection and apoptosis–a task for TH17 cells. Ann. N. Y. Acad. Sci. 1209, 56–67. doi: 10.1111/j.1749-6632.2010.05747.x
Bultman, S. J. (2014). Emerging roles of the microbiome in cancer. Carcinogenesis 35, 249–255. doi: 10.1093/carcin/bgt392
Carbonero, F., Benefiel, A. C., Alizadeh-Ghamsari, A. H., and Gaskins, H. R. (2012). Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 3:448. doi: 10.3389/fphys.2012.00448
Chusri, P., Kumthip, K., Hong, J., Zhu, C., Duan, X., Jilg, N., et al. (2016). HCV induces transforming growth factor beta1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep. 6:22487. doi: 10.1038/srep22487
Coussens, L. M., and Werb, Z. (2002). Inflammation and cancer. Nature 420, 860–867. doi: 10.1038/nature01322
Couturier-Maillard, A., Secher, T., Rehman, A., Normand, S., De Arcangelis, A., Haesler, R., et al. (2013). NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Invest. 123, 700–711. doi: 10.1172/jci62236
Cuevas-Ramos, G., Petit, C. R., Marcq, I., Boury, M., Oswald, E., and Nougayrede, J. P. (2010). Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 107, 11537–11542. doi: 10.1073/pnas.1001261107
Dapito, D. H., Mencin, A., Gwak, G. Y., Pradere, J. P., Jang, M. K., Mederacke, I., et al. (2012). Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21, 504–516. doi: 10.1016/j.ccr.2012.02.007
De Simone, V., Pallone, F., Monteleone, G., and Stolfi, C. (2013). Role of TH17 cytokines in the control of colorectal cancer. Oncoimmunology 2:e26617. doi: 10.4161/onci.26617
DiDonato, J. A., Mercurio, F., and Karin, M. (2012). NF-kappaB and the link between inflammation and cancer. Immunol. Rev. 246, 379–400. doi: 10.1111/j.1600-065X.2012.01099.x
Elinav, E., Nowarski, R., Thaiss, C. A., Hu, B., Jin, C., and Flavell, R. A. (2013). Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 13, 759–771. doi: 10.1038/nrc3611
Fox, J. G., and Wang, T. C. (2007). Inflammation, atrophy, and gastric cancer. J. Clin. Invest. 117, 60–69. doi: 10.1172/jci30111
Francescone, R., Hou, V., and Grivennikov, S. I. (2014). Microbiome, inflammation, and cancer. Cancer J. 20, 181–189. doi: 10.1097/ppo.0000000000000048
French, D., Belleudi, F., Mauro, M. V., Mazzetta, F., Raffa, S., Fabiano, V., et al. (2013). Expression of HPV16 E5 down-modulates the TGFbeta signaling pathway. Mol. Cancer 12:38. doi: 10.1186/1476-4598-12-38
Freudlsperger, C., Bian, Y., Contag Wise, S., Burnett, J., Coupar, J., Yang, X., et al. (2013). TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 32, 1549–1559. doi: 10.1038/onc.2012.171
Furusawa, Y., Obata, Y., Fukuda, S., Endo, T. A., Nakato, G., Takahashi, D., et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. doi: 10.1038/nature12721
Gagliani, N., Amezcua Vesely, M. C., Iseppon, A., Brockmann, L., Xu, H., Palm, N. W., et al. (2015). Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523, 221–225. doi: 10.1038/nature14452
Gagniere, J., Raisch, J., Veziant, J., Barnich, N., Bonnet, R., Buc, E., et al. (2016). Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 22, 501–518. doi: 10.3748/wjg.v22.i2.501
Goodwin, A. C., Destefano Shields, C. E., Wu, S., Huso, D. L., Wu, X., Murray-Stewart, T. R., et al. (2011). Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 15354–15359. doi: 10.1073/pnas.1010203108
Gur, C., Ibrahim, Y., Isaacson, B., Yamin, R., Abed, J., Gamliel, M., et al. (2015). Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42, 344–355. doi: 10.1016/j.immuni.2015.01.010
Hooper, S. J., Wilson, M. J., and Crean, S. J. (2009). Exploring the link between microorganisms and oral cancer: a systematic review of the literature. Head Neck 31, 1228–1239. doi: 10.1002/hed.21140
Hu, B., Elinav, E., Huber, S., Strowig, T., Hao, L., Hafemann, A., et al. (2013). Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc. Natl. Acad. Sci. U.S.A. 110, 9862–9867. doi: 10.1073/pnas.1307575110
Huycke, M. M., Abrams, V., and Moore, D. R. (2002). Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 23, 529–536. doi: 10.1093/carcin/23.3.529
Huycke, M. M., and Gaskins, H. R. (2004). Commensal bacteria, redox stress, and colorectal cancer: mechanisms and models. Exp. Biol. Med. 229, 586–597. doi: 10.1177/153537020422900702
Huycke, M. M., Joyce, W., and Wack, M. F. (1996). Augmented production of extracellular superoxide by blood isolates of Enterococcus faecalis. J. Infect. Dis. 173, 743–746. doi: 10.1093/infdis/173.3.743
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans (2012). Biological agents. Volume 100 B. A review of human carcinogens. IARC Monogr. Eval. Carcinog. Risks Hum. 100(Pt B), 1–441.
Iglesias, M., Frontelo, P., Gamallo, C., and Quintanilla, M. (2000). Blockade of Smad4 in transformed keratinocytes containing a Ras oncogene leads to hyperactivation of the Ras-dependent Erk signalling pathway associated with progression to undifferentiated carcinomas. Oncogene 19, 4134–4145. doi: 10.1038/sj.onc.1203764
Ihara, S., Hirata, Y., and Koike, K. (2017). TGF-beta in inflammatory bowel disease: a key regulator of immune cells, epithelium, and the intestinal microbiota. J. Gastroenterol. 52, 777–787. doi: 10.1007/s00535-017-1350-1
Iida, N., Dzutsev, A., Stewart, C. A., Smith, L., Bouladoux, N., Weingarten, R. A., et al. (2013). Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 342, 967–970. doi: 10.1126/science.1240527
Irrazabal, T., Belcheva, A., Girardin, S. E., Martin, A., and Philpott, D. J. (2014). The multifaceted role of the intestinal microbiota in colon cancer. Mol. Cell 54, 309–320. doi: 10.1016/j.molcel.2014.03.039
Ivanov, I. I., Atarashi, K., Manel, N., Brodie, E. L., Shima, T., Karaoz, U., et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. doi: 10.1016/j.cell.2009.09.033
Ivanov, I. I., Frutos Rde, L., Manel, N., Yoshinaga, K., Rifkin, D. B., Sartor, R. B., et al. (2008). Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349. doi: 10.1016/j.chom.2008.09.009
Karin, M., and Greten, F. R. (2005). NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5, 749–759. doi: 10.1038/nri1703
Kashiwagi, I., Morita, R., Schichita, T., Komai, K., Saeki, K., Matsumoto, M., et al. (2015). Smad2 and smad3 inversely regulate TGF-beta autoinduction in Clostridium butyricum-activated dendritic cells. Immunity 43, 65–79. doi: 10.1016/j.immuni.2015.06.010
Khattar, M., Chen, W., and Stepkowski, S. M. (2009). Expanding and converting regulatory T cells: a horizon for immunotherapy. Arch. Immunol. Ther. Exp. 57, 199–204. doi: 10.1007/s00005-009-0021-1
Klein, J., Ju, W., Heyer, J., Wittek, B., Haneke, T., Knaus, P., et al. (2006). B cell-specific deficiency for Smad2 in vivo leads to defects in TGF-beta-directed IgA switching and changes in B cell fate. J. Immunol. 176, 2389–2396. doi: 10.4049/jimmunol.176.4.2389
Kukimoto, M., Nishiyama, M., Tanokura, M., and Horinouchi, S. (2000). Gene organization for nitric oxide reduction in Alcaligenes faecalis S-6. Biosci. Biotechnol. Biochem. 64, 852–857. doi: 10.1271/bbb.64.852
Lee, D. K., Kim, B. C., Kim, I. Y., Cho, E. A., Satterwhite, D. J., and Kim, S. J. (2002). The human papilloma virus E7 oncoprotein inhibits transforming growth factor-beta signaling by blocking binding of the Smad complex to its target sequence. J. Biol. Chem. 277, 38557–38564. doi: 10.1074/jbc.M206786200
Lee, D. K., Park, S. H., Yi, Y., Choi, S. G., Lee, C., Parks, W. T., et al. (2001). The hepatitis B virus encoded oncoprotein pX amplifies TGF-beta family signaling through direct interaction with Smad4: potential mechanism of hepatitis B virus-induced liver fibrosis. Genes Dev. 15, 455–466. doi: 10.1101/gad.856201
Lin, W., Tsai, W. L., Shao, R. X., Wu, G., Peng, L. F., Barlow, L. L., et al. (2010). Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 138, 2509–2518, 2518.e1. doi: 10.1053/j.gastro.2010.03.008
Liu, H., Chen, F., Wu, W., Cao, A. T., Xue, X., Yao, S., et al. (2016). TLR5 mediates CD172alpha+ intestinal lamina propria dendritic cell induction of Th17 cells. Sci. Rep. 6:22040. doi: 10.1038/srep22040
Liu, N., Jiao, T., Huang, Y., Liu, W., Li, Z., and Ye, X. (2015). Hepatitis B virus regulates apoptosis and tumorigenesis through the microRNA-15a-Smad7-transforming growth factor beta pathway. J. Virol. 89, 2739–2749. doi: 10.1128/jvi.02784-14
Liu, Y., Xu, Y., Ma, H., Wang, B., Xu, L., Zhang, H., et al. (2016). Hepatitis B virus X protein amplifies TGF-beta promotion on HCC motility through down-regulating PPM1a. Oncotarget 7, 33125–33135. doi: 10.18632/oncotarget.8884
Lu, R., Wu, S., Zhang, Y. G., Xia, Y., Liu, X., Zheng, Y., et al. (2014). Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic beta-catenin signaling pathway. Oncogenesis 3:e105. doi: 10.1038/oncsis.2014.20
Lu, S. L., Herrington, H., Reh, D., Weber, S., Bornstein, S., Wang, D., et al. (2006). Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 20, 1331–1342. doi: 10.1101/gad.1413306
Marchesi, J. R., Adams, D. H., Fava, F., Hermes, G. D., Hirschfield, G. M., Hold, G., et al. (2016). The gut microbiota and host health: a new clinical frontier. Gut 65, 330–339. doi: 10.1136/gutjnl-2015-309990
Marshall, B. J., and Warren, J. R. (1984). Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315. doi: 10.1016/S0140-6736(84)91816-6
Murata-Kamiya, N., Kurashima, Y., Teishikata, Y., Yamahashi, Y., Saito, Y., Higashi, H., et al. (2007). Helicobacter pylori CagA interacts with E-cadherin and deregulates the beta-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 26, 4617–4626. doi: 10.1038/sj.onc.1210251
Nesic, D., Hsu, Y., and Stebbins, C. E. (2004). Assembly and function of a bacterial genotoxin. Nature 429, 429–433. doi: 10.1038/nature02532
Nougayrede, J. P., Homburg, S., Taieb, F., Boury, M., Brzuszkiewicz, E., Gottschalk, G., et al. (2006). Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 313, 848–851. doi: 10.1126/science.1127059
Numasaki, M., Watanabe, M., Suzuki, T., Takahashi, H., Nakamura, A., McAllister, F., et al. (2005). IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J. Immunol. 175, 6177–6189. doi: 10.4049/jimmunol.175.9.6177
Obst, B., Wagner, S., Sewing, K. F., and Beil, W. (2000). Helicobacter pylori causes DNA damage in gastric epithelial cells. Carcinogenesis 21, 1111–1115. doi: 10.1093/carcin/21.5.111
Obata, T., Goto, Y., Kunisawa, J., Sato, S., Sakamoto, M., Setoyama, H., et al. (2010). Indigenous opportunistic bacteria inhabit mammalian gut-associated lymphoid tissues and share a mucosal antibody-mediated symbiosis. Proc. Natl. Acad. Sci. U.S.A. 107, 7419–7424. doi: 10.1073/pnas.1001061107
Ojesina, A. I., Lichtenstein, L., Freeman, S. S., Pedamallu, C. S., Imaz-Rosshandler, I., Pugh, T. J., et al. (2014). Landscape of genomic alterations in cervical carcinomas. Nature 506, 371–375. doi: 10.1038/nature12881
Pardali, K., and Moustakas, A. (2007). Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim. Biophys. Acta 1775, 21–62. doi: 10.1016/j.bbcan.2006.06.004
Patwa, L. G., Fan, T. J., Tchaptchet, S., Liu, Y., Lussier, Y. A., Sartor, R. B., et al. (2011). Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology 141, 1842e1–e10–1851.e1–10. doi: 10.1053/j.gastro.2011.06.064
Perez-Chanona, E., and Trinchieri, G. (2016). The role of microbiota in cancer therapy. Curr. Opin. Immunol. 39, 75–81. doi: 10.1016/j.coi.2016.01.003
Pickup, M., Novitskiy, S., and Moses, H. L. (2013). The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 13, 788–799. doi: 10.1038/nrc3603
Ridlon, J. M., Wolf, P. G., and Gaskins, H. R. (2016). Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 7, 201–215. doi: 10.1080/19490976.2016.1150414
Rogler, G. (2014). Chronic ulcerative colitis and colorectal cancer. Cancer Lett. 345, 235–241. doi: 10.1016/j.canlet.2013.07.032
Round, J. L., Lee, S. M., Li, J., Tran, G., Jabri, B., Chatila, T. A., et al. (2011). The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332, 974–977. doi: 10.1126/science.1206095
Ruane, D., Chorny, A., Lee, H., Faith, J., Pandey, G., Shan, M., et al. (2016). Microbiota regulate the ability of lung dendritic cells to induce IgA class-switch recombination and generate protective gastrointestinal immune responses. J. Exp. Med. 213, 53–73. doi: 10.1084/jem.20150567
Rubinstein, M. R., Wang, X., Liu, W., Hao, Y., Cai, G., and Han, Y. W. (2013). Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195–206. doi: 10.1016/j.chom.2013.07.012
Samaras, V., Rafailidis, P. I., Mourtzoukou, E. G., Peppas, G., and Falagas, M. E. (2010). Chronic bacterial and parasitic infections and cancer: a review. J. Infect. Dev. Ctries. 4, 267–281.
Sano, T., Huang, W., Hall, J. A., Yang, Y., Chen, A., Gavzy, S. J., et al. (2015). An IL-23R/IL-22 circuit regulates epithelial serum amyloid a to promote local effector Th17 responses. Cell 163, 381–393. doi: 10.1016/j.cell.2015.08.061
Schwabe, R. F., and Jobin, C. (2013). The microbiome and cancer. Nat. Rev. Cancer 13, 800–812. doi: 10.1038/nrc3610
Sears, C. L., and Garrett, W. S. (2014). Microbes, microbiota, and colon cancer. Cell Host Microbe 15, 317–328. doi: 10.1016/j.chom.2014.02.007
Sellers, R. S., and Morton, D. (2014). The colon: from banal to brilliant. Toxicol. Pathol. 42, 67–81. doi: 10.1177/0192623313505930
Shen, Z., Feng, Y., Rogers, A. B., Rickman, B., Whary, M. T., Xu, S., et al. (2009). Cytolethal distending toxin promotes Helicobacter cinaedi-associated typhlocolitis in interleukin-10-deficient mice. Infect. Immun. 77, 2508–2516. doi: 10.1128/iai.00166-09
Song, X., Dai, D., He, X., Zhu, S., Yao, Y., Gao, H., et al. (2015). Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity 43, 488–501. doi: 10.1016/j.immuni.2015.06.024
Song, X., Gao, H., Lin, Y., Yao, Y., Zhu, S., Wang, J., et al. (2014). Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 40, 140–152. doi: 10.1016/j.immuni.2013.11.018
Taur, Y., and Pamer, E. G. (2016). Microbiome mediation of infections in the cancer setting. Genome Med. 8:40. doi: 10.1186/s13073-016-0306-z
Tezuka, H., Abe, Y., Iwata, M., Takeuchi, H., Ishikawa, H., Matsushita, M., et al. (2007). Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 448, 929–933. doi: 10.1038/nature06033
Tosolini, M., Kirilovsky, A., Mlecnik, B., Fredriksen, T., Mauger, S., Bindea, G., et al. (2011). Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 71, 1263–1271. doi: 10.1158/0008-5472.can-10-2907
Velcich, A., Yang, W., Heyer, J., Fragale, A., Nicholas, C., Viani, S., et al. (2002). Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 295, 1726–1729. doi: 10.1126/science.1069094
Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M., and Stockinger, B. (2006). TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189. doi: 10.1016/j.immuni.2006.01.001
Vipperla, K., and O’Keefe, S. J. (2016). Diet, microbiota, and dysbiosis: a ‘recipe’ for colorectal cancer. Food Funct. 7, 1731–1740. doi: 10.1039/c5fo01276g
Virchow, R. (1881). An address on the value of pathological experiments. Br. Med. J. 2, 198–203. doi: 10.1136/bmj.2.1075.198
Wang, X., Allen, T. D., May, R. J., Lightfoot, S., Houchen, C. W., and Huycke, M. M. (2008). Enterococcus faecalis induces aneuploidy and tetraploidy in colonic epithelial cells through a bystander effect. Cancer Res. 68, 9909–9917. doi: 10.1158/0008-5472.CAN-08-1551
White, R. A., Malkoski, S. P., and Wang, X. J. (2010). TGFbeta signaling in head and neck squamous cell carcinoma. Oncogene 29, 5437–5446. doi: 10.1038/onc.2010.306
Wiest, R., and Garcia-Tsao, G. (2005). Bacterial translocation (BT) in cirrhosis. Hepatology 41, 422–433. doi: 10.1002/hep.20632
Wilke, C. M., Bishop, K., Fox, D., and Zou, W. (2011). Deciphering the role of Th17 cells in human disease. Trends Immunol. 32, 603–611. doi: 10.1016/j.it.2011.08.003
Windey, K., De Preter, V., and Verbeke, K. (2012). Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 56, 184–196. doi: 10.1002/mnfr.201100542
Wu, S., Rhee, K. J., Albesiano, E., Rabizadeh, S., Wu, X., Yen, H. R., et al. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022. doi: 10.1038/nm.2015
Wu, Y. H., Ai, X., Liu, F. Y., Liang, H. F., Zhang, B. X., and Chen, X. P. (2016). c-Jun N-terminal kinase inhibitor favors transforming growth factor-beta to antagonize hepatitis B virus X protein-induced cell growth promotion in hepatocellular carcinoma. Mol. Med. Rep. 13, 1345–1352. doi: 10.3892/mmr.2015.4644
Yang, L. (2010). TGFbeta and cancer metastasis: an inflammation link. Cancer Metastasis Rev. 29, 263–271. doi: 10.1007/s10555-010-9226-3
Yoshimoto, S., Loo, T. M., Atarashi, K., Kanda, H., Sato, S., Oyadomari, S., et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. doi: 10.1038/nature12347
Zeller, G., Tap, J., Voigt, A. Y., Sunagawa, S., Kultima, J. R., Costea, P. I., et al. (2014). Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 10:766. doi: 10.15252/msb.20145645
Zhang, S., Ekman, M., Thakur, N., Bu, S., Davoodpour, P., Grimsby, S., et al. (2006). TGFbeta1-induced activation of ATM and p53 mediates apoptosis in a Smad7-dependent manner. Cell Cycle 5, 2787–2795. doi: 10.4161/cc.5.23.3523
Zhou, L., Lopes, J. E., Chong, M. M., Ivanov, I. I., Min, R., Victora, G. D., et al. (2008). TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453, 236–240. doi: 10.1038/nature06878
Zitvogel, L., Ayyoub, M., Routy, B., and Kroemer, G. (2016). Microbiome and anticancer immunosurveillance. Cell 165, 276–287. doi: 10.1016/j.cell.2016.03.001
Keywords: microbiota, colorectal cancer, TGF-β signaling, epithelial barrier, immunity
Citation: Pang X, Tang Y-j, Ren X-h, Chen Q-m, Tang Y-l and Liang X-h (2018) Microbiota, Epithelium, Inflammation, and TGF-β Signaling: An Intricate Interaction in Oncogenesis. Front. Microbiol. 9:1353. doi: 10.3389/fmicb.2018.01353
Received: 25 February 2018; Accepted: 05 June 2018;
Published: 26 June 2018.
Edited by:
Juarez Antonio Simões Quaresma, Instituto Evandro Chagas, BrazilReviewed by:
Paolo Puccetti, University of Perugia, ItalyCopyright © 2018 Pang, Tang, Ren, Chen, Tang and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ya-ling Tang, dGFuZ3lhbGluZ0BzY3UuZWR1LmNu Xin-hua Liang, bHhoODg4NjZAc2N1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.