 Michael Steinert
Michael Steinert Isabell Ramming
Isabell Ramming Simone Bergmann
Simone Bergmann- 1Institut für Mikrobiologie, Technische Universität Braunschweig, Braunschweig, Germany
- 2Department of Infection Biology, Helmholtz Center for Infection Diseases, Braunschweig, Germany
Von Willebrand factor (VWF) is a mechano-sensitive protein with crucial functions in normal hemostasis, which are strongly dependant on the shear-stress mediated defolding and multimerization of VWF in the blood stream. Apart from bleeding disorders, higher plasma levels of VWF are often associated with a higher risk of cardiovascular diseases. Herein, the disease symptoms are attributed to the inflammatory response of the activated endothelium and share high similarities to the reaction of the host vasculature to systemic infections caused by pathogenic bacteria such as Staphylococcus aureus and Streptococcus pneumoniae. The bacteria recruit circulating VWF, and by binding to immobilized VWF on activated endothelial cells in blood flow, they interfere with the physiological functions of VWF, including platelet recruitment and coagulation. Several bacterial VWF binding proteins have been identified and further characterized by biochemical analyses. Moreover, the development of a combination of sophisticated cell culture systems simulating shear stress levels of the blood flow with microscopic visualization also provided valuable insights into the interaction mechanism between bacteria and VWF-strings. In vivo studies using mouse models of bacterial infection and zebrafish larvae provided evidence that the interaction between bacteria and VWF promotes bacterial attachment, coagulation, and thrombus formation, and thereby contributes to the pathophysiology of severe infectious diseases such as infective endocarditis and bacterial sepsis. This mini-review summarizes the current knowledge of the interaction between bacteria and the mechano-responsive VWF, and corresponding pathophysiological disease symptoms.
Introduction
Vascular hemostasis is a live-saving mechanism, which balances coagulation, thrombogenesis, and fibrinolysis in response to vascular injuries and inflammatory processes. Key element of the hemostasis are the Weibel Palade bodies (WPBs), which represent defense vesicles, constitutively produced by the endothelium of the vessel walls. The vesicles are filled with vasoactive substances, immune defense modulators, and proteins involved in coagulation (1, 2). In addition to megakaryocytes, endothelial WPBs are the main source of Von Willebrand factor (VWF). This glycoprotein mediates platelet activation, anchorage of thrombocytes to the subendothelial collagen, and induction of plasma haemostasis via factor VIII (3, 4). Moreover, VWF promotes cell migration in angiogenesis via interaction with different cell surface receptors and induction of signaling pathways (5). The high importance of VWF for balanced hemostasis is conveyed by the appearance of bleeding disorders such as the von Willebrand disease caused by an inherited quantitative or functional VWF deficiency (3).
VWF constantly circulates in the bloodstream at concentrations between 8 and 14.0 μg/mL (3, 6). But, vasoactive hormones such as epinephrine and vasopressin as well as the plasma proteins thrombin, histamine, and numerous other mediators of inflammation and/or thrombosis induce the release of VWF in response to vascular injury or inflammatory stimuli. The released VWF increases the plasma levels of this protein, and some proportion of VWF is temporarily retained on the cell surface and binds to collagen of the exposed subendothelial matrix (7, 8). This subendothelial immobilization is also significantly strengthened by the endothelial glycocalyx in a heparanase-sensitive manner (9). VWF is a mechano-sensitive protein, which responds to shear stress-mediated forces by conformational changes.
Shear stress is defined as the force exerted by the blood flow on blood vessel walls. This stress generates a response in the vascular wall, characterized by release of endothelial mediators, which in turn stimulate structural remodeling through activation of gene expression and protein synthesis (10). The shear stress-derived conformational changes of VWF are crucial for the biological function of VWF in hemostasis. Upon exposition to the shear forces in the bloodstream, the immobilized VWF unfolds to large protein strings, thereby exposing further functionally important binding sites (7, 11, 12). In particular, the defolded A1 binding site mediates adhesion of platelets and recruits them via binding to the platelet glycoprotein GP?bα (11, 13, 14). This VWF-platelet interaction finally results in a factor VIII-induced fibrin-incorporation and in stabilization of generated thrombi.
Elevated VWF-levels are directly associated with cardiovascular diseases (CD) of high-risk groups such as the elderly and diabetes patients (15). Alongside with tissue plasminogen activator (t-PA), and D-dimer of fibrinogen, VWF is characterized as one out of three biomarkers directly associated with atherosclerotic lesions and coronary heart disease (16, 17). This unveils the thrombus-generating activity of elevated VWF-concentrations as one of the dominant causative factors for coronary heart disease (18).
In addition to the role of VWF in CD, VWF serves as a ligand binding site for bacteria, which cause live-threatening local and systemic infection diseases, such as Staphylococcus aureus and Streptococcus pneumoniae (19, 20). S. aureus is a human pathogenic bacterium causing, among others, infective endocarditis and heart valve prosthetic infection (21, 22). In this respect, shear-force-mediated adhesion of staphylococci to VWF is directly associated with coagulation and typical disease symptoms (23, 24). Similarly, S. pneumoniae, a commensal colonizing the upper respiratory epithelium and a major cause of community-acquired pneumoniae in elderly and immunocompromised patients (25, 26), has also been recurringly isolated from heart valve endocardium of patients suffering from subacute endocarditis (27, 28). Furthermore, an increasing amount of clinical case studies report that up to one-third of patients suffer from major adverse cardiac effects (MACE) and vascular impairments within months and even years after recovering from severe pneumococcal infections such as pneumoniae and septicemiae (29–31). The observation of similarities between the association of CD with VWF-release, and symptoms induced by bacterial infections initiated an increasing need to develop infection models and sophisticated visualization techniques in the last decade. With these models, the pathomechanistical function of some crucial bacterial virulence factors in VWF-mediated disease progression could be deciphered.
Bacterial Binding to VWF Under Shear Flow
The release of VWF from endothelial WPBs is induced by host-derived hormones such as epinephrine and histamine and other plasma factors and is also triggered by pathogenic bacteria (32). For example, in 1991, Sporn et al. were the first to observe that the intracellular pathogen Rickettsia rickettsii, the main cause of the Rocky Mountain spotted fever, induces the release of VWF out of WPB of cultured endothelial cells [(33), Table 1]. Moreover, in our previous studies, we demonstrated that luminal VWF secretion from WPB of human lung endothelial cells is significantly increased in response to pneumococcal adherence and the cytotoxic effects of the pneumococcus toxin pneumolysin (45). These results strongly suggest that in vivo, the interaction between circulating bacteria in the bloodstream and the endothelial vasculature might directly lead to elevated VWF plasma levels.

Table 1. Bacterial VWF-binding proteins and function in adhesion and infectious diseases.
In this respect, the scientific question was raised whether the released VWF is directly subverted by the bacteria for their own benefit, i.e., as a binding site at the host endothelium, for platelet aggregation, or interference with the host coagulation. Indeed, Herrmann et al. were the first to demonstrate the binding of S. aureus bacteria to VWF coated surfaces and VWF in suspension (46). A short time later, a heparin-sensitive bacterial binding to soluble VWF was also reported for coagulase-negative Staphylococcus species, often associated with infections of prosthetic devices [(40), Table 1].
Bacterial adhesion to the vascular endothelium is of high importance for the pathology of blood-born infections, since this promotes bacterial settlement, induces inflammatory responses, and facilitates bacterial transmigration and dissemination into deeper tissue sites. It became obvious that blood-flow induced conformational changes of the VWF molecule, which are crucial for the physiological function of VWF in the bloodstream, might also be of high relevance for VWF-mediated bacterial adhesion. For a long time, it remained a technically challenging task to unreveal details of the bacterial interaction with the mechano-sensitive VWF under shear stress condition. But meanwhile, a variety of model systems have been established that enable the simulation of different physiological shear stress situations including sophisticated visualization techniques [for review, please refer to Bergmann and Steinert (47)]. The first experimental studies on the binding of multimerized VWF to platelets were performed with “Cone-and-Plate” viscometers in combination with flow cytometric quantifications (48). Viscometer-generated shear stress application was also combined with ristocetin-incubation of VWF. Ristocetin is an antibiotic produced by Amycolatopsis lurida, and is still used as the Gold standard in diagnostics of von Willebrand-disease (49). Ristocetin binds to VWF in a shear-stress-independent manner, thereby inducing the exposure of the VWF-mediated platelet binding site for thrombocyte recruitment and aggregation (49). Following the objective to quantitatively analyse the specific protein ligand-interaction with VWF under a defined medium flow, several surface-coating technologies have been established that create so-called “functionalized surfaces.” For example, Mascari and Ross have quantified the detachment of staphylococci from collagen in real–time using a parallel plate flow chamber combined with phase-contrast video–microscopy and digital image processing (50). The results provided evidence that staphylococci adhere directly to multimerized VWF strings and attach to collagen of the exposed subendothelium in blood-borne infections (34).
In addition to the biochemical interaction studies, several in vivo mouse infection models employing vwf gene-deficient mice and platelet-depleted mice enable evaluation and monitoring of systemic consequences associated with hemostatic processes. The in vivo analyses revealed that S. aureus bacteria directly attach to cell-bound VWF of the endothelial vasculature (23, 51). Moreover, visualization of bacterial mouse infection via intravital microscopy confirmed that bacteria, which attached to VWF strings, resist shear stress-mediated clearance by the blood flow [(19), Figure 1]. Deeper insight into the pathophysiological consequences of the pneumococcus interaction with VWF was also provided by infection analyses using zebrafish larvae. Danio rerio serves as a suitable in vivo model, sharing high morphologic and functional similarity to the human endothelial tissue and both, intrinsic and extrinsic coagulation pathways (52–54). Microscopic real-time visualization of larval infection confirmed the recruitment of endothelial-derived VWF to circulating pneumococci and VWF-mediated attachment to the endothelial vessel walls (20).
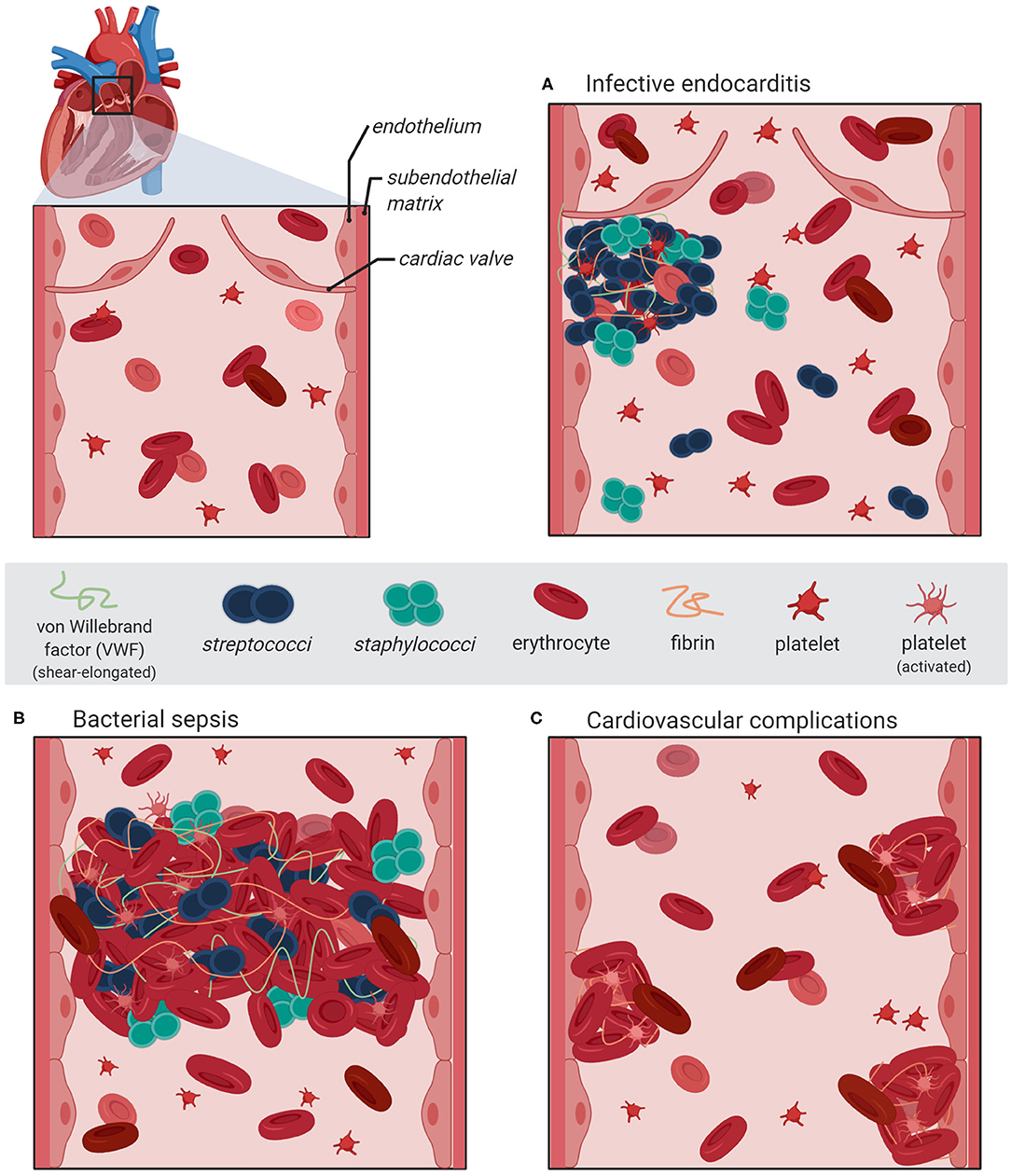
Figure 1. Schematic illustration of bacterial vascular infections and diseases associated with VWF-mediated adhesion of S. aureus and S. pneumoniae to activated endothelium. The section at the upper left represents a magnification of the heart pulmonary arerty with the artrioventricular valve. (A) An infective endocarditis is associated with VWF-mediated bacterial attachment to the activated valve endocard leading to the formation of inflammation-inducing bacterial vegetations containing platelets and fibrin. (B) During sepsis, bacterial adhesion to the inflamed vasculature is promoted via elongated VWF strings and induces the formation thrombi, which might lead to occlusions of the microvasculature. (C) Severe systemic bacterial infections are accompanied with recurring endothel activation leading to a longterm dysbalanced hemostasis, which increases the risk of cardiovascular complications such as major adverse cardiac effects. The figure was generated using bioRender Software.
Bacterial VWF Binding Proteins and Binding Mechanisms
The bacterial interaction with components of the hemostasis in vivo augurs the presence of specific bacterial surface proteins, which mediate binding to VWF. The protein A (SPA) of S. aureus was identified as a bacterial VWF-binding protein. SPA elicits binding activities to both, the soluble and the surface-immobilized VWF [(35), Table 1]. Six years later, the VWF binding sites of protein A were narrowed down to the IgG-binding domain (55). Using single-molecule atomic force microscopy (AFM), Viela et al. further demonstrated that VWF binds very tightly to SPA via a force-sensitive catch bond mechanism, which involves force-induced structural changes in the SPA domains (36, 56). Meanwhile, protein similarities led to the assumption that several bacterial virulence factors may use this binding mechanism to resist clearance by high shear stress during infections (57).
In addition to SPA, a second staphylococcal VWF binding protein (VWbp) with coagulase activity was identified from a phage display-library screen in 2002 [(37, 38), Table 1]. Studies using functionalized surface-technology revealed that in contrast to SPA, VWbp appears to be of significant relevance for VWF recruitment rather than under static conditions (58). Likewise, pneumococci bind VWF under static conditions, and also recruit globular circulating VWF via the surface-exposed enolase [(20), Table 1]. This protein also mediates binding of pneumococci to plasminogen and to extracellular nucleic acids, which both promotes bacterial attachment to epithelial and endothelial cells (59). Moreover, similar to the staphylococcal VWbp, the VWF binding site for the pneumococcal enolase is located within the defolded A1 domain of VWF (19, 20, 60). All bacterial VWF-binding proteins identified so far are listed in Table 1.
In addition to the analyses of perfused VWF protein-coated, functionalized surfaces, the group of Schneider et al. established an air pressure-driven, unidirectional, and continuous pump system manufactured by the company ibidi® (19). In contrast to the formerly described flow systems that are employed to analyse protein-protein-interactions under shear stress conditions, the ibidi® pump technology enables sterile long term cultivation of VWF-producing endothelial cells, which can be incubated with bacteria and microscopically analyzed in real-time. As a result, this air-driven microfluidic pump device enabled the analyses of staphylococcal interaction with VWF on endothelial cell surfaces under shear stress conditions (19) and was also used to establish a pneumococcus cell culture infection model of primary endothelial cells in flow (20, 61). With this system, the attachment of pneumococci to multimerized VWF strings on the endothelial cell surface was successfully visualized and quantitatively evaluated. In accordance with the VWF binding characteristics of S. aureus, VWF binding to pneumococci is heparin-sensitive and depends on the amount of polysaccharide capsule expression (20). It is of note that pneumococcal attachment to VWF strings is also characterized by remarkable bond stability for longer time periods even at high shear flow parameters, which might be promoted by a concerted action of several additional, yet unidentified VWF-binding proteins (20). In addition, results of surface plasmon resonance binding studies and cell culture infections studies in flow revealed that the pneumococcus enolase interacts with both, globular circulating VWF and with VWF strings with comparable avidity. Based on the observation that multi adhesive proteins such as the bacterial enolase are already detected on the surface of various bacterial species, it can be assumed that the bacterial interaction with VWF is part of a general mechanism with pivotal relevance for pathophysiology.
Effect of Staphylococcal and Streptococcal Interaction With VWF on Coagulation and Vascular Diseases
As summarized in Table 1, VWF binding to bacteria has only been studied to detail for staphylococci and streptococci. Taking clinical symptoms into account, different functional aspects of the bacterial interaction with VWF can be directly or indirectly correlated with at least three severe infection diseases: infective endocarditis, bacterial sepsis, and cardiovascular complications.
Infective endocarditis is regarded as a paradigm of bacterial diseases associated with vascular inflammation and VWF-interaction (24). Most of the acute infective endocarditis are caused by S. aureus and are associated with up to 100% mortality rate if left untreated (21, 22). Compared to that, infective endocarditis caused by S. pneumoniae is rare but no less severe (27, 28). Infection of the heart valves is initiated by the attachment of circulating bacteria to the endocardium and the formation of bacterial vegetations, which are embedded in fibrin and platelets (Figure 1A). During disease progress, the vegetations induce further inflammatory processes, which result in ulceration, rupture, and necrosis of the valve cusps (62, 63). Experimental shear stress determination using native porcine aortic valve models revealed that even in a healthy human vasculature, the systolic shear stress at the heart valve leaflet can reach up to 21.3 dyn/cm2 at the aortic site and up to 92 dyn/cm2 at the ventricular site (64, 65). Similar to the activation of specific proinflammatory and procoagulant protein expression patterns of endothelial cells, the hemodynamic forces also promote the activation of endocardial Notch-dependent signaling pathways in the endocardial cells of the atrio-ventricular valve (66). The observed magnitude of shear stress is sensed by the mechano-responsive VWF and induces stretching and multimerization of VWF proteins. Thereby, VWF displays crucial binding sites for bacterial surface adhesins and mediates bacterial attachment to the heart valve. In line with this, visualization of staphylococcal mouse infection via 3D confocal microscopy confirmed the adhesion of fluorescent S. aureus to murine aortic valves (23). The mouse infection studies further demonstrated that following valve damage, VWF and fibrin are both deposited on the damaged valve endocardium and serve as attachment sites for S. aureus [(23, 51), Figure 1A]. Moreover, endothelial cell culture infections and intravital microscopy of bacterial mouse infection confirmed that staphylococci and pneumococci resist shear stress-mediated clearance by the blood flow by binding to VWF strings at the endothelial vessel walls (19, 20, 51). Following disease progress, the VWF-mediated bacterial attachment also promotes the recruitment of large amounts of platelets, capturing S. aureus to the valve surface [(23, 24, 67, 68), Figures 1A,B]. The observation that among the staphylococci, only S. aureus and S. lugdunensis are able to bind VWF might, in part, explain why these bacteria are more effective in causing endocarditis than other staphylococci (41).
Bacterial VWF binding is also involved in the formation of large platelet aggregates within the blood circulation. In this respect, the formation of bacterial-induced platelet aggregates and the depletion of clotting factors from blood represents a crucial pathomechanism, which is directly attributed to disease symptoms typical for bacterial sepsis. For example, staphylococcal sepsis is associated with an increase in coagulation activity and an enhanced thrombosis (Figure 1B; Table 1). It is assumed that the Staphylococcus-induced dysregulated activation of systemic thrombosis leads to thrombotic microangiopathy, which is associated with an accelerated fibrinolysis and bleeding tendency, referred to as disseminated intravascular coagulation [DIC, (69)]. Moreover, this bacterial mechanism is also assumed to directly induce the formation of abscesses [(39, 70–73), Figure 1B]. A similar formation of blood clots, reaching up to 10 μm in diameter, was observed in pneumococcus infection of Danio rerio larvae (20). Based on these data, we suppose that the VWF-mediated bacterial aggregate formation in the blood circulation of the zebrafish cause a partial or complete occlusion of the larval microvasculature. Thus, in severe cases of staphylococcal and pneumococcal septicaemiae, the vascular occlusion of small blood vessels throughout the body represents a life-threatening disease symptom, which might lead to multi-organ failure, resulting in high mortality rates of up to 50% [(74–77), Figure 1B]. Bacterial aggregate formation in sepsis and infective endocarditis, in particular, are also prime examples of the strong connection between the hemostatic system and innate immunity, which is referred to as immune thrombosis (78). It is coincidently proposed that the infection-induced coagulase activity mediates bacterial capture within a fibrin meshwork, which enables this pathogen to disseminate via thromboembolic lesions and to resist opsonophagocytic clearance by host immune cells (73). On the other hand, platelets are the crucial mediators of the innate defense against staphylococci by releasing microbicidal proteins from alpha granules that kill the bacteria (79). On the first view, it appears to be contradictory that bacteria induce a clotting mechanism, which is originally developed as an anti-bacterial immune defense mechanism of the host. However, the biochemical and physiological attributes of the fibrin meshwork formed by staphylocoagulases are thought to be distinct and less solid than those generated by thrombin (80). Therefore, instead of containing the infection, immune thrombosis might rather create the optimal environment for bacteria to survive and to evade the immune defense of the host (24).
It is supposed that the bacterial infection mechanism leading to vascular dysfunction and enhanced activation of inflammation might also be implicated in developing cardiovascular complications (Figure 1C). An increasing number of clinical studies solidify the observation that pneumococci induce vascular inflammation of the endothelial vessel wall, including the aorta (81), and that severe pneumococcal infections such as pneumoniae and septicemiae lead to a higher risk for major adverse cardiac effects (MACE) such as myocardial infarction, ischemic stroke, and arterial thrombosis (29–31).
Since elevated VWF plasma levels are known to be associated with an increased risk for MACE (15), the endothelial VWF release induced by pneumococcal attachment and by pneumolysin activity might be partially responsible for the pathologic effects on the cardio vasculature (45). As further explanation, functional variants of VWF have been identified, which elicit differences in the protein conformation and shear sensitivity. These variants are associated with increased platelet aggregate size and the occurrence of these VWF variants correlates with a higher risk of thromboembolisms including myocardial infarction and stroke (82). In line with these observations, it can be assumed that bacterial interaction with VWF might effect the hemostatic function in various ways, i.e., by sterical hindrance of the platelet binding site, by alteration of the VWF conformation, and by inhibition of dimerization and multimerization activities, thereby increasing the risk for cardiovascular complications.
Conclusions
VWF is a live-saving key component of coagulation and immune thrombosis in response to vascular injury and inflammation. Bacterial interaction with VWF is of high medical and scientific importance since this interaction is directly associated with specific clinical manifestations and long-term complications of infectious diseases. It has been demonstrated that binding of S. aureus and S. pneumoniae to VWF strings is controlled by hydrodynamic flow conditions. So far, at least three bacterial pathomechanisms involving host-derived VWF can be named: (i) binding to multimerized VWF strings mediates bacterial attachment to endothelial surfaces in blood flow–a major prerequisite of bacterial colonization, inflammation, and dissemination. (ii) VWF recruitment facilitates bacterial capture within clotted blood, thereby preventing bacterial clearance via immunothrombosis; (iii) recruitment of intravascular VWF induces bacterial aggregate formation, which leads to occlusion of microcapillaries and impaired blood supply. Although several sophisticated technologies such as microfluidic systems and binding force determinations already provided most valuable insights into the cell biological and biochemical details, the multifactorial complexity of the bacterial interaction with VWF still remains a challenging subject of ongoing scientific research.
Author Contributions
SB and MS contributed to text conception and wrote the text. IR has generated the figure and critically revised the text. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
We acknowledge support by the German Research Foundation and the Open Access Publication Funds of the Technische Universität Braunschweig.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Weibel ER, Palade GE. New cytoplasmic components in arterial endothelia. J Cell Biol. (1964) 23:101–12. doi: 10.1083/jcb.23.1.101
2. Rondaij MG, Sellink E, Gijzen KA, ten Klooster JP, Hordijk PL, van Mourik JA, et al. Small GTP-binding protein Ral is involved in cAMP-mediated release of von willebrand factor from endothelial cells. Arterioscler Thromb Vasc Biol. (2004) 24:1315–20. doi: 10.1161/01.ATV.0000131267.13425.45
3. Ruggeri ZM. Structure of von willebrand factor and its function in platelet adhesion and thrombus formation. Best Pract Res Clin Haematol. (2001) 14:257–79. doi: 10.1053/beha.2001.0133
4. Rondaij MG, Bierings R, Kragt A, van Mourik JA, Voorberg J. Dynamics and plasticity of weibel-palade bodies in endothelial cells. Arterioscler Thromb Vasc Biol. (2006) 26:1002–7. doi: 10.1161/01.ATV.0000209501.56852.6c
5. Randi AM, Laffan MA. Von Willebrand factor and angiogenesis: basic and applied issues. J Thromb Haemost. (2017) 15:13–20. doi: 10.1111/jth.13551
6. Spiel AO, Gilbert JC, Jilma B. Von willebrand factor in cardiovascular disease: focus on acute coronary syndromes. Circulation. (2008) 117:1449–59. doi: 10.1161/CIRCULATIONAHA.107.722827
7. Tischer A, Machha VR, Frontroth JP, Brehm MA, Obser T, Schneppenheim R, et al. Enhanced local disorder in a clinically elusive von willebrand factor provokes high-affinity platelet clumping. J Mol Biol. (2017) 429:2161–77. doi: 10.1016/j.jmb.2017.05.013
8. Vischer UM, Ingerslev J, Wollheim CB, Mestries JC, Tsakiris DA, Haefeli WE, et al. Acute von willebrand factor secretion from the endothelium in vivo: assessment through plasma propeptide (vWf:AgII) Levels. Thromb Haemost. (1997) 77:387–93. doi: 10.1055/s-0038-1655973
9. Kalagara T, Moutsis T, Yang Y, Pappelbaum KI, Farken A, Cladder-Micus L, et al. The endothelial glycocalyx anchors von willebrand factor fibers to the vascular endothelium. Blood Adv. (2018) 2:2347–57. doi: 10.1182/bloodadvances.2017013995
10. Hudlicka O, Brown MM. Adaptation of skeletal muscle microvasculatura to increased or decreased blood flow role of shear stress, nitric oxide and vascular endothelial growth factor. J Vasc Res. (2009) 46:504–12. doi: 10.1159/000226127
11. Springer TA. Von willebrand factor, jedi knight of the bloodstream. Blood. (2014) 124:1412–25. doi: 10.1182/blood-2014-05-378638
12. Löf A, König G, Schneppenheim S, Benoit M, Budde U, Müller JP, et al. Advancing multimer analysis of von willebrand factor by single-molecule AFM imaging. PLoS ONE. (2019) 14:e0210963. doi: 10.1371/journal.pone.0210963
13. Schneider SW, Nuschele S, Wixforth A, Gorzelanny C, Alexander-Katz A, Netz RR, et al. Shear-induced unfolding triggers adhesion of von willebrand factor fibers. Proc Natl Acad Sci USA. (2007) 104:7899–903. doi: 10.1073/pnas.0608422104
14. Flood VH, Gill JC, Christopherson PA, Bellissimo DB, Friedman KD, Haberichter SL, et al. Critical von willebrand factor A1 domain residues influence type VI collagen binding. J Thromb Haemost. (2012) 10:1417–24. doi: 10.1111/j.1538-7836.2012.04746.x
15. Jager A, van Hinsbergh VW, Kostense PJ, Emeis JJ, Yudkin JS, Nijpels G, et al. Von willebrand factor, C-reactive protein, and 5-year mortality in diabetic and nondiabetic subjects: the hoorn study. Arterioscler Thromb Vasc Biol. (1999) 19:3071–8. doi: 10.1161/01.ATV.19.12.3071
16. Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. (2002) 196:887–96. doi: 10.1084/jem.20012044
17. Vischer UM. Von willebrand factor, endothelial dysfunction, and cardiovascular disease. J Thromb Haemost. (2006) 4:1186–93. doi: 10.1111/j.1538-7836.2006.01949.x
18. Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. (2003) 1:1335–42. doi: 10.1046/j.1538-7836.2003.00260.x
19. Pappelbaum KI, Gorzelanny C, Grassle S, Suckau J, Laschke MW, Bischoff M, et al. Ultralarge von willebrand factor fibers mediate luminal Staphylococcus aureus adhesion to an intact endothelial cell layer under shear stress. Circulation. (2013) 128:50–9. doi: 10.1161/CIRCULATIONAHA.113.002008
20. Jagau H, Behrens IK, Lahme K, Lorz G, Koster RW, Schneppenheim R, et al. Von Willebrand factor mediates pneumococcal aggregation and adhesion in blood flow. Front Microbiol. (2019) 10:511. doi: 10.3389/fmicb.2019.00511
21. Moreillon P, Que YA. Infective endocarditis. Lancet. (2004) 363:139–49. doi: 10.1016/S0140-6736(03)15266-X
22. Fowler RA, Gupta S. Subacute and acute infective endocarditis. Lancet. (2005) 366:1964. doi: 10.1016/S0140-6736(05)67788-4
23. Liesenborghs L, Meyers S, Lox M, Criel M, Claes J, Peetermans M, et al. Staphylococcus aureus endocarditis: distinct mechanisms of bacterial adhesion to damaged and inflamed heart valves. Eur Heart J. (2019) 40:3248–59. doi: 10.1093/eurheartj/ehz175
24. Liesenborghs L, Meyers S, Vanassche T, Verhamme P. Coagulation: At the heart of infective endocarditis. J Thromb Haemost. (2020) 18:995–1008. doi: 10.1111/jth.14736
25. Cartwright K. Pneumococcal disease in western Europe: burden of disease, antibiotic resistance and management. Eur J Pediatr. (2002) 161:188–95. doi: 10.1007/s00431-001-0907-3
26. Weiser JN, Ferreira DM, Paton JC. Streptococcus pneumoniae: transmission, colonization and invasion. Nat Rev Microbiol. (2018) 16:355–67. doi: 10.1038/s41579-018-0001-8
27. Aronin SI, Mukherjee SK, West JC, Cooney EL. Review of pneumococcal endocarditis in adults in the penicillin era. Clin Infect Dis. (1998) 26:165–71. doi: 10.1086/516279
28. de Egea V, Munoz P, Valerio M, de Alarcon A, Lepe JA, Miro JM, et al. Characteristics and outcome of Streptococcus pneumoniae endocarditis in the XXI century: a systematic review of 111 cases (2000-2013). Medicine. (2015) 94:e1562. doi: 10.1097/MD.0000000000001562
29. Musher DM, Rueda AM, Kaka AS, Mapara SM. The association between pneumococcal pneumonia and acute cardiac events. Clin Infect Dis. (2007) 45:158–65. doi: 10.1086/518849
30. Corrales-Medina VF, Alvarez KN, Weissfeld LA, Angus DC, Chirinos JA, Chang CC, et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA. (2015) 313:264–74. doi: 10.1001/jama.2014.18229
31. Rae N, Finch S, Chalmers JD. Cardiovascular disease as a complication of community-acquired pneumonia. Curr Opin Pulm Med. (2016) 22:212–8. doi: 10.1097/MCP.0000000000000261
32. van Mourik JA, Romani de Wit T, Voorberg J. Biogenesis and exocytosis of weibel-palade bodies. Histochem Cell Biol. (2002) 117:113–22. doi: 10.1007/s00418-001-0368-9
33. Sporn LA, Shi RJ, Lawrence SO, Silverman DJ, Marder VJ. Rickettsia rickettsii infection of cultured endothelial cells induces release of large von willebrand factor multimers from weibel-palade bodies. Blood. (1991) 78:2595–602. doi: 10.1182/blood.V78.10.2595.bloodjournal78102595
34. Mascari LM, Ross JM. Quantification of staphylococcal-collagen binding interactions in whole blood by use of a confocal microscopy shear-adhesion assay. J Infect Dis. (2003) 188:98–107. doi: 10.1086/375826
35. Hartleib J, Kohler N, Dickinson RB, Chhatwal GS, Sixma JJ, Hartford OM, et al. Protein A is the von willebrand factor binding protein on Staphylococcus aureus. Blood. (2000) 96:2149–56.
36. Viela F, Prystopiuk V, Leprince A, Mahillon J, Speziale P, Pietrocola G, et al. Binding of Staphylococcus aureus protein A to von willebrand factor is regulated by mechanical force. MBio. (2019) 10:e00555–19. doi: 10.1128/mBio.00555-19
37. Bjerketorp J, Nilsson M, Ljungh A, Flock JI, Jacobsson K, Frykberg L. A novel von willebrand factor binding protein expressed by Staphylococcus aureus. Microbiology. (2002) 148:2037–44. doi: 10.1099/00221287-148-7-2037
38. Bjerketorp J, Jacobsson K, Frykberg L. The von willebrand factor-binding protein (vWbp) of Staphylococcus aureus is a coagulase. FEMS Microbiol Lett. (2004) 234:309–14. doi: 10.1111/j.1574-6968.2004.tb09549.x
39. Cheng AG, McAdow M, Kim HK, Bae T, Missiakas DM, Schneewind O. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog. (2010) 6:e1001036. doi: 10.1371/journal.ppat.1001036
40. Li DQ, Lundberg F, Ljungh A. Binding of von willebrand factor by coagulase-negative staphylococci. J Med Microbiol. (2000) 49:217–25. doi: 10.1099/0022-1317-49-3-217
41. Liesenborghs L, Peetermans M, Claes J, Veloso TR, Vandenbriele C, Criel M, et al. Shear-resistant binding to von willebrand factor Allows Staphylococcus lugdunensis to adhere to the cardiac valves and initiate endocarditis. J Infect Dis. (2016) 213:1148–56. doi: 10.1093/infdis/jiv773
42. Nilsson M, Bjerketorp J, Wiebensjo A, Ljungh A, Frykberg L, Guss B. A von willebrand factor-binding protein from Staphylococcus lugdunensis. FEMS Microbiol Lett. (2004) 234:155–61. doi: 10.1111/j.1574-6968.2004.tb09527.x
43. Carter AM, Moayyedi P, Catto A, Heppell RM, Axon AT, Grant PJ. The influence of Helicobacter pylori status on circulating levels of the coagulation factors fibrinogen, von willebrand factor, factor VII, and factor VIII. Helicobacter. (1996) 1:65–9. doi: 10.1111/j.1523-5378.1996.tb00011.x
44. Byrne MF, Kerrigan SW, Corcoran PA, Atherton JC, Murray FE, Fitzgerald DJ, et al. Helicobacter pylori binds von willebrand factor and interacts with GPIb to induce platelet aggregation. Gastroenterology. (2003) 124:1846–54. doi: 10.1016/S0016-5085(03)00397-4
45. Luttge M, Fulde M, Talay SR, Nerlich A, Rohde M, Preissner KT, et al. Streptococcus pneumoniae induces exocytosis of weibel-palade bodies in pulmonary endothelial cells. Cell Microbiol. (2012) 14:210–25. doi: 10.1111/j.1462-5822.2011.01712.x
46. Herrmann M, Hartleib J, Kehrel B, Montgomery RR, Sixma JJ, Peters G. Interaction of von willebrand factor with Staphylococcus aureus. J Infect Dis. (1997) 176:984–91. doi: 10.1086/516502
47. Bergmann S, Steinert M. From single cells to engineered and explanted tissues: new perspectives in bacterial infection biology. Int Rev Cell Mol Biol. (2015) 319:1–44. doi: 10.1016/bs.ircmb.2015.06.003
48. Goto S, Salomon DR, Ikeda Y, Ruggeri ZM. Characterization of the unique mechanism mediating the shear-dependent binding of soluble von willebrand factor to platelets. J Biol Chem. (1995) 270:23352–61. doi: 10.1074/jbc.270.40.23352
49. Just S. Laboratory testing for von willebrand disease: the past, present, and future state of play for von willebrand factor assays that measure platelet binding activity, with or without ristocetin. Semin Thromb Hemost. (2017) 43:75–91. doi: 10.1055/s-0036-1592164
50. Mascari L, Ymele-Leki P, Eggleton CD, Speziale P, Ross JM. Fluid shear contributions to bacteria cell detachment initiated by a monoclonal antibody. Biotechnol Bioeng. (2003) 83:65–74. doi: 10.1002/bit.10650
51. Claes J, Vanassche T, Peetermans M, Liesenborghs L, Vandenbriele C, Vanhoorelbeke K, et al. Adhesion of Staphylococcus aureus to the vessel wall under flow is mediated by von willebrand factor-binding protein. Blood. (2014) 124:1669–76. doi: 10.1182/blood-2014-02-558890
52. Kamei M, Saunders WB, Bayless KJ, Dye L, Davis GE, Weinstein BM. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature. (2006) 442:453–6. doi: 10.1038/nature04923
53. Hanumanthaiah R, Day K, Jagadeeswaran P. Comprehensive analysis of blood coagulation pathways in teleostei: evolution of coagulation factor genes and identification of zebrafish factor VIIi. Blood Cells Mol Dis. (2002) 29:57–68. doi: 10.1006/bcmd.2002.0534
54. Weyand AC, Shavit JA. Zebrafish as a model system for the study of hemostasis and thrombosis. Curr Opin Hematol. (2014) 21:418–22. doi: 10.1097/MOH.0000000000000075
55. O'Seaghdha M, van Schooten CJ, Kerrigan SW, Emsley J, Silverman GJ, Cox D, et al. Staphylococcus aureus protein A binding to von willebrand factor A1 domain is mediated by conserved IgG binding regions. FEBS J. (2006) 273:4831–41. doi: 10.1111/j.1742-4658.2006.05482.x
56. Viela F, Speziale P, Pietrocola G, Dufrene YF. Bacterial pathogens under high-tension: Staphylococcus aureus adhesion to von willebrand factor is activated by force. Microb Cell. (2019) 6:321–3. doi: 10.15698/mic2019.07.684
57. Herman-Bausier P, Labate C, Towell AM, Derclaye S, Geoghegan JA, Dufrene YF. Staphylococcus aureus clumping factor A is a force-sensitive molecular switch that activates bacterial adhesion. Proc Natl Acad Sci USA. (2018) 115:5564–9. doi: 10.1073/pnas.1718104115
58. Thomer L, Schneewind O, Missiakas D. Multiple ligands of von willebrand factor-binding protein (vWbp) promote Staphylococcus aureus clot formation in human plasma. J Biol Chem. (2013) 288:28283–92. doi: 10.1074/jbc.M113.493122
59. Zakrzewicz D, Bergmann S, Didiasova M, Giaimo BD, Borggrefe T, Mieth M, et al. Host-derived extracellular RNA promotes adhesion of Streptococcus pneumoniae to endothelial and epithelial cells. Sci Rep. (2016) 6:37758. doi: 10.1038/srep37758
60. Claes J, Liesenborghs L, Peetermans M, Veloso TR, Missiakas D, Schneewind O, et al. Clumping factor A, von willebrand factor-binding protein and von willebrand factor anchor Staphylococcus aureus to the vessel wall. J Thromb Haemost. (2017) 15:1009–19. doi: 10.1111/jth.13653
61. Jagau H, Behrens IK, Steinert M, Bergmann S. Pneumococcus infection of primary human endothelial cells in constant flow. J Vis Exp. (2019) 152:e60323. doi: 10.3791/60323
62. Guerrero ML, Aldamiz G, Bayon J, Cohen VA, Fraile J. Long-term survival of salvage cardiac transplantation for infective endocarditis. Ann Thorac Surg. (2011) 92:e93–94. doi: 10.1016/j.athoracsur.2011.05.048
63. Thiene G, Basso C. Pathology and pathogenesis of infective endocarditis in native heart valves. Cardiovasc Pathol. (2006) 15:256–63. doi: 10.1016/j.carpath.2006.05.009
64. Yap CH, Saikrishnan N, Tamilselvan G, Yoganathan AP. Experimental measurement of dynamic fluid shear stress on the aortic surface of the aortic valve leaflet. Biomech Model Mechanobiol. (2012) 11:171–82. doi: 10.1007/s10237-011-0301-7
65. Yap CH, Saikrishnan N, Yoganathan AP. Experimental measurement of dynamic fluid shear stress on the ventricular surface of the aortic valve leaflet. Biomech Model Mechanobiol. (2012) 11:231–44. doi: 10.1007/s10237-011-0306-2
66. Gálvez-Santisteban M, Chen D, Zhang R, Serrano R, Nguyen C, Zhao L, et al. Hemodynamic-mediated endocardial signaling controls in vivo myocardial reprogramming. Elife. (2019) 8:e44816. doi: 10.7554/eLife.44816
67. Que YA, Haefliger JA, Piroth L, Francois P, Widmer E, Entenza JM, et al. Fibrinogen and fibronectin binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J Exp Med. (2005) 201:1627–35. doi: 10.1084/jem.20050125
68. Kerdudou S, Laschke MW, Sinha B, Preissner KT, Menger MD, Herrmann M. Fibronectin binding proteins contribute to the adherence of Staphylococcus aureus to intact endothelium in vivo. Thromb Haemost. (2006) 96:183–9. doi: 10.1160/TH06-02-0116
69. Levi M, Ten Cate H. Disseminated intravascular coagulation. N Engl J Med. (1999) 341:586–92. doi: 10.1056/NEJM199908193410807
70. Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. (2009) 23:3393–404. doi: 10.1096/fj.09-135467
71. Friedrich R, Panizzi P, Fuentes-Prior P, Richter K, Verhamme I, Anderson PJ, et al. Staphylocoagulase is a prototype for the mechanism of cofactor-induced zymogen activation. Nature. (2003) 425:535–9. doi: 10.1038/nature01962
72. Kroh HK, Panizzi P, Bock PE. Von willebrand factor-binding protein is a hysteretic conformational activator of prothrombin. Proc Natl Acad Sci USA. (2009) 106:7786–91. doi: 10.1073/pnas.0811750106
73. McAdow M, Kim HK, Dedent AC, Hendrickx AP, Schneewind O, Missiakas DM. Preventing Staphylococcus aureus sepsis through the inhibition of its agglutination in blood. PLoS Pathog. (2011) 7:e1002307. doi: 10.1371/journal.ppat.1002307
74. van der Poll T, Opal SM. Host-pathogen interactions in sepsis. Lancet Infect Dis. (2008) 8:32–43. doi: 10.1016/S1473-3099(07)70265-7
75. van der Poll T, Opal SM. Should all septic patients be given systemic anticoagulation? No. Intensive Care Med. (2017) 43:455–7. doi: 10.1007/s00134-016-4607-x
76. Charalambous BM, Leung MH. Pneumococcal sepsis and nasopharyngeal carriage. Curr Opin Pulm Med. (2012) 18:222–7. doi: 10.1097/MCP.0b013e328352103b
77. van der Linden M, Reinert RR. Serotype distribution in pneumococcal acute otitis media with ruptured tympanic membrane or sepsis in Germany. Eur J Clin Microbiol Infect Dis. (2010) 29:749–54. doi: 10.1007/s10096-010-0945-8
78. Verhamme P, Hoylaerts MF. Hemostasis and inflammation: two of a kind? Thromb J. (2009) 7:15. doi: 10.1186/1477-9560-7-15
79. Yeaman MR, Norman DC, Bayer AS. Platelet microbicidal protein enhances antibiotic-induced killing of and postantibiotic effect in Staphylococcus aureus. Antimicrob Agents Chemother. (1992) 36:1665–70. doi: 10.1128/AAC.36.8.1665
80. Kopec M, Wegrzynowicz Z, Budzynski AZ, Jeljaszewicz J, Latallo ZS, Lipinski B, et al. Formation and properties of fibrin clots resulting from staphylocoagulase (SC) action. Thromb Diath Haemorrh. (1967) 18:475–86. doi: 10.1055/s-0038-1655057
81. Maclennan AC, Doyle DL, Sacks SL. Infectious aortitis due to penicillin-resistant Streptococcus pneumoniae. Ann Vasc Surg. (1997) 11:533–5. doi: 10.1007/s100169900086
Keywords: von Willebrand, Staphylococcus areus, Streptococcus pneumoniae, microfluidic, sepsis
Citation: Steinert M, Ramming I and Bergmann S (2020) Impact of Von Willebrand Factor on Bacterial Pathogenesis. Front. Med. 7:543. doi: 10.3389/fmed.2020.00543
Received: 28 May 2020; Accepted: 30 July 2020;
Published: 03 September 2020.
Edited by:
Silvia Fischer, University of Giessen, GermanyReviewed by:
Volker Huck, University Medical Center Hamburg-Eppendorf, GermanyJonas Emsley, University of Nottingham, United Kingdom
Copyright © 2020 Steinert, Ramming and Bergmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simone Bergmann, c2ltb25lLmJlcmdtYW5uJiN4MDAwNDA7dHUtYnMuZGU=