 Paula Gratal
Paula Gratal Ana Lamuedra†Juan Pablo Medina†Ismael Bermejo-Álvarez†
Ana Lamuedra†Juan Pablo Medina†Ismael Bermejo-Álvarez† Raquel Largo
Raquel Largo Gabriel Herrero-Beaumont
Gabriel Herrero-Beaumont Aránzazu Mediero*
Aránzazu Mediero*- Bone and Joint Research Unit, IIS-Fundación Jiménez Díaz UAM, Madrid, Spain
Inflammation triggered by metabolic imbalance, also called metainflammation, is low-grade inflammation caused by the components involved in metabolic syndrome (MetS), including central obesity and impaired glucose tolerance. This phenomenon is mainly due to excess nutrients and energy, and it contributes to the pathogenesis of osteoarthritis (OA). OA is characterized by the progressive degeneration of articular cartilage, which suffers erosion and progressively becomes thinner. Purinergic signaling is involved in several physiological and pathological processes, such as cell proliferation in development and tissue regeneration, neurotransmission and inflammation. Adenosine and ATP receptors, and other members of the signaling pathway, such as AMP-activated protein kinase (AMPK), are involved in obesity, type 2 diabetes (T2D) and OA progression. In this review, we focus on purinergic regulation in osteoarthritic cartilage and how different components of MetS, such as obesity and T2D, modulate the purinergic system in OA. In that regard, we describe the critical role in this disease of receptors, such as adenosine A2A receptor (A2AR) and ATP P2X7 receptor. Finally, we also assess how nucleotides regulate the inflammasome in OA.
Introduction
The biological actions of purine nucleotide and nucleoside signaling have been recognized since 1929. ATP was proposed as responsible for non-adrenergic, non-cholinergic intestinal and bladder transmission. However, it was not until 1972 that G. Burnstock, the founding father of purinergic scientific research, introduced the term “purinergic signaling” (1) and in 1976 specific receptors for extracellular nucleotides were defined (2).
ATP is known for being the universal energy currency. An equilibrium between the intracellular and extracellular amount of ATP is maintained in basal conditions, but in certain physiological and pathological situations, such as apoptosis, infections, mechanical stress, and inflammation (3), cells release ATP from intracellular deposits to the extracellular space. This process is mediated by pannexin (e.g., Pannexin-1) (4) and connexin hemichannels (e.g., Connexin-43) (5–7), but also other ion channels, such as calcium homeostasis modulator 1 (CALHM1) (8), volume-regulated anion channel (VRAC) and maxi-anion channel (MAC) (9), vesicular exocytosis (10) and autophagy-dependent lysosomal exocytosis (11, 12), and through uncontrolled release in apoptotic processes (3). In the extracellular compartment, ATP has an entirely different function and activates purinergic signaling via ion channel and transmembrane purinergic receptors in the cell membrane (3) (Figure 1).
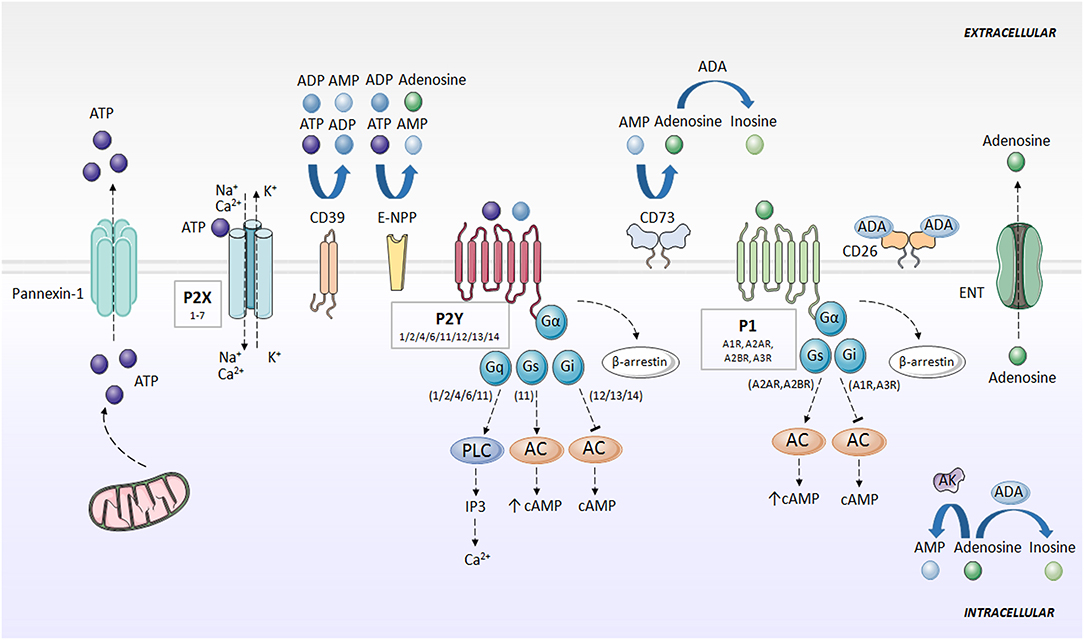
Figure 1. Purinergic signaling and specific receptors for extracellular nucleotides. ATP is released to the extracellular space mainly through pannexins (e.g., Pannexin-1). Then, ATP is rapidly degraded to ADP, AMP, and adenosine by the action of CD39, E-NPP and CD73. Adenosine can be converted to AMP through AK action, or degraded to inosine by ADA. Adenosine is also directly released outside the cell through ENT. P2X receptors activated by ATP, forming an ion channel for Na+, K+, and Ca2+. P2Y receptors are activated by ATP and ADP, and associate with G proteins, promoting intracellular second messengers' cascades, including Ca2+ and cAMP. Adenosine activates P1 receptors, coupled to G-proteins. A1R and A3R interaction with Gi, and A2AR and A2BR with Gs. P1 and P2Y receptors desensitized through internalization by β-arrestins. AC, adenylate cyclase; ADA, adenosine deaminase; ADP, adenosine diphosphate; AK, adenosine kinase; AMP, adenosine monophosphate; ATP, adenosine 5′-triphosphate; cAMP, cyclic adenosine monophosphate; CD26, dipeptidyl peptidase IV; CD39, ecto-nucleoside triphosphate diphosphohydrolase; CD73, ecto-5′-nucleotidase; E-NPP, ecto-nucleotide pyrophosphatase/phosphodiesterase; ENT, nucleoside transporter; G, G protein; IP3, inositol trisphosphate; P1, adenosine receptors; P2X, ionotropic nucleotide receptors; P2Y, metabotropic nucleotide receptors; PLC, phospholipase C.
ATP is rapidly degraded to adenosine, which also exerts an important role in purinergic signaling. Outside the cell, several enzymes hydrolyze ATP and limit nucleotide availability for purinergic signaling. Ecto-nucleoside triphosphate diphosphohydrolase (CD39) converts ATP into ADP, and ADP into AMP. Ecto-nucleotide pyrophosphatase/phosphodiesterase (E-NPP) also converts ATP directly into AMP and hydrolyzes ADP directly to adenosine. Separately, ecto-5'-nucleotidase (CD73) transforms AMP into adenosine (13–16). Additionally, one of the main regulators of the purinergic system in the extracellular space is adenosine deaminase (ADA), which anchores to the plasma membrane through the dipeptidyl peptidase IV (CD26) (17) and metabolizes adenosine to inosine (7, 18). Adenosine kinase (AK) converts adenosine back to AMP (19). Adenosine can also be directly released outside the cell through nucleoside transporters (ENT) (18) (Figure 1).
Pharmacology recognizes two families of purinergic receptors: P1 receptors, selective for adenosine (20), and P2 receptors, selective for nucleotides and dinucleotides of purines and pyrimidines (21).
P1 or adenosine receptors are G-protein coupled receptors (GPCR) currently divided into four subtypes, named A1R, A2AR, A2BR, and A3R, whose activation is dependent on the presence of extracellular adenosine (20). A1R and A3R differ from A2AR and A2BR in the particular G protein they interact with, with A1R and A3R being inhibitors (coupled to Gi), and A2AR and A2BR promoters (coupled to Gs) of cAMP synthesis via adenylate cyclase (AC). The modulation of cAMP levels controls multiple signaling pathways, including mitogen-activated protein kinase (MAPK) and serine-threonine specific kinases (22). Adenosine receptors are desensitized via through their internalization by β-arrestins (18, 23) (Figure 1).
Within the P2 or nucleotide receptors, P2X are ionotropic and P2Y are metabotropic receptors. P2X receptors are activated by ATP, and seven subtypes (1–7) have been identified. P2X receptors are homo- or heterotrimers, which combine differently (e.g., P2X2/3, P2X1/2, P2X1/5, P2X2/6, P2X4/6, and P2X1/4) and form an ion channel for Na+, K+ and Ca2+ (18, 23). P2Y receptors (P2Y1/2/4/6/11/12/13/14) are selective for ATP, ADP, UTP, and UDP, and couple with G proteins (21). Like adenosine receptors, P2Y receptors are internalized by β-arrestin, resulting in desensitization of the purinergic signal (18, 23, 24) (Figure 1).
Purinergic signaling is evolutionarily conserved (25), and is involved in several physiological and pathological processes, such as neurotransmission, cell proliferation, platelet aggregation, vasodilatation, and inflammation (23, 24).
Purinergic signaling contributes to the pathophysiology of several bone and cartilage diseases, such as OA, rheumatoid arthritis (RA), and osteoporosis (18).
Osteoarthritis (OA) is the most frequent and most disabling rheumatic disease in developed countries (26, 27). Around 250 million people suffer from knee OA worldwide, with a 33% prevalence in the population aged over 65 years, and with women being more affected than men (28, 29). OA is characterized by the progressive degeneration of diarthrodial joints (30), involving all the joint tissues, especially articular cartilage (AC), which suffers erosion and progressively becomes thinner. OA has traditionally been considered a “wear and tear” disease, caused by mechanical cartilage breakdown. However, it is now well-accepted that inflammation plays a critical role in the disease's progression both in cartilage and the synovium (31). Among the multifactorial etiology of OA, metabolic syndrome (MetS) and aging are critical risk factors for the onset of the disease, due in part to a state of low-grade chronic inflammation (32). The interaction between tissue damage and destruction by mechanical and biological mechanisms, together with the activation of innate immunity by multiple local and systemic inflammatory factors, are responsible for the chronicity of pathological processes in OA.
In this review, we present the important contribution of MetS and purine metabolism to the chronic pro-inflammatory status during OA progression. Moreover, we provide an integrated view of the mechanisms triggered by low-grade chronic inflammation as a pivotal axis in the pathogenesis of OA.
Metainflammation in OA
MetS is an increasingly prevalent condition, generally diagnosed when more than two of the following risk factors are present: central obesity, lowered HDL cholesterol, high triglycerides, hypertension, and/or impaired glucose tolerance (33–35).
Metainflammation, the inflammation triggered by a metabolic imbalance (36), is a low-grade inflammation generated by components implicated in MetS. This phenomenon is mainly caused by excess nutrients and therefore, by an energy surplus, and it contributes to the pathogenesis of OA (37).
Recent studies have focused on identifying the relationship between MetS components and OA in order to understand its influence on the inflammatory process involved in the pathogenesis of the disease. An epidemiologic association between MetS and OA has been observed in clinical studies, especially with knee OA (38). Animal studies have also demonstrated that OA pathogenesis can be led by metabolic dysregulation (38). Despite increasing evidence of an association between OA and MetS, the mechanisms linking these diseases are not fully understood (39).
Obesity and Metainflammation in OA
Central obesity may lead to OA pathogenesis by promoting systemic and local inflammation and by increasing load and consequent mechanical wear of the joints (40). There is an association between obesity and OA both in weight bearing and non-weight bearing joints, which highlights the key role of chronic inflammation in the disease's development (41).
Increased adipose tissue on the body exerts metainflammation through the production of adipokines and cytokines (42, 43). Some secreted adipokines are leptin, resistin and adiponectin (44). They are able to modulate the immune system and induce synthesis of pro-inflammatory and catabolic mediators, which leads to chondrocyte dysfunction and aggravates OA progression (45). These adipokines are distributed systemically and can be found in high amounts in osteoarthritic synovial fluid (45). They are synthesized by synoviocytes, articular chondrocytes and adipocytes of intra-articular fat tissue (37) (Figure 2).
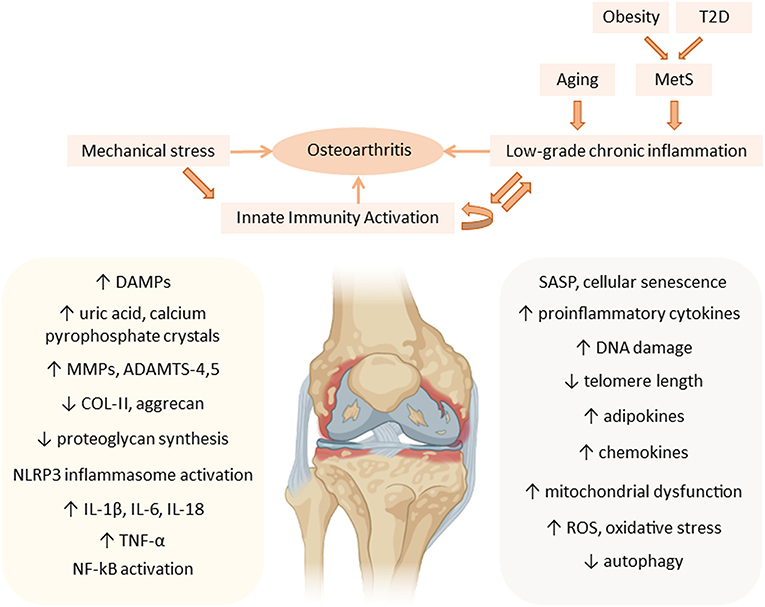
Figure 2. Metainflammation in the pathogenesis of osteoarthritis (OA). Mechanical stress contributes, together with the low-grade chronic inflammation associated with aging and the components of the metabolic syndrome (MetS), to the chronic activation of innate immunity in the joint, mainly affecting articular cartilage, during OA progression. Aging-associated factors and senescence together with metabolic factors, such as an increase in adipokines, maintain a systemic low-grade chronic inflammatory status that contributes to the pathogenesis of OA, increasing the expression of cartilage catabolic enzymes (MMPs, ADAMTS), and pro-inflammatory cytokines (IL-1β, IL-6, IL-18, and TNF-α), thus perpetuating inflammation in the joint. ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; COL-II, type II collagen; DAMPs, danger-associated molecular patterns; DNA, deoxyribonucleic acid; IL, interleukin; MMP, matrix metalloproteinases; MetS, metabolic syndrome; NF-kB, nuclear factor kappa B; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; ROS, reactive oxygen species; SASP, senescence-associated secretory phenotype; T2D, type 2 diabetes; TNF, tumor necrosis factor.
Leptin plays an essential role in energy metabolism, leading to energy consumption. It affects growth factor synthesis and anabolism, and it is present in human chondrocytes, synovium, osteophytes and infrapatellar fat pads (44). In OA, this protein shows a marked expression in cartilage and osteophytes in comparison with healthy tissue (46). Furthermore, leptin contributes to the pathogenesis of OA, since it has a pro-inflammatory and catabolic role in cartilage metabolism, its expression being directly associated with the degree of cartilage destruction, and through the stimulation of growth factor synthesis (46, 47). Leptin induces the synthesis of matrix metalloproteinases (MMPs) in primary cultures of human chondrocytes. Accordingly, in patients with OA its concentration correlates with MMP-1 and MMP-3 levels in the synovium (48, 49). Furthermore, leptin promotes the activation of monocytes, and a subsequent increase in macrophages in the synovium (32).
Resistin is mainly expressed in white adipose tissue, but also by macrophages, osteoblasts, osteoclasts, and chondrocytes (40). Resistin is capable of inducing cytokine and chemokine expression in chondrocytes (50), being a potential regulator of the pro-inflammatory cytokines that activate transcription nuclear factor kappa B (NF-κB) (51). Moreover, it is upregulated during monocyte-macrophage differentiation (52) and tumor necrosis factor (TNF)-α, interleukins (IL)-1β, IL-6, and lipopolysaccharide (LPS) stimulation increases its levels (53). Resistin is also involved in angiogenesis via the induction of endothelial cell growth and migration (54), and can endorse osteoclastogenesis via modulation of bone turnover mediators (51). Thus, bone remodeling and resistin-stimulated pro-inflammatory cytokines in cartilage may promote OA pathogenesis. Nevertheless, further studies are necessary to determine the role of resistin in the onset and progression in vivo of OA (40).
Adiponectin, another adipokine produced by adipose tissue, shows higher concentrations within the joint, which suggests an important role in the onset and progression of OA. The synovial fluid of patients with OA shows a 100-fold increase in adiponectin expression as compared with their plasma (55). In addition, adiponectin receptor 1 (AdipoR1) is expressed in cartilage, bone, and synovial tissues (44). Adiponectin may protect against progression of OA since it induces the downregulation of MMP-13 and upregulation of an associated MMP-13 inhibitor (44). Another study suggests that adiponectin may exert an anti-inflammatory effect on cartilage, upregulating the tissue inhibitor of metalloproteinases 2 (TIMP-2) (40, 55). However, serum adiponectin levels are not associated with OA (56).
Experimental models, such as rats, which developed OA after been fed with a high-fat diet (HFD), show that there is a close relationship between obesity and the local immune response in synovial tissue (57). Synovial resident macrophages play a key role in HFD-induced inflammation, which produces an increase in M1 macrophages compared with M2 polarized cells. This highlights the importance of M1 macrophage subsets in the development of obesity-associated OA (57). Furthermore, HFD aggravates synovial inflammation during OA, by increasing macrophage infiltration and metabolic-mediated remodeling of adipose tissue, together with a significant presence of pro-inflammatory factors (58). Additionally, free fatty acids induce activation of Toll-like receptor 2 (TLR2) or TLR4, trigger macrophage recruitment, and activate the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome (59).
In addition to the secretion of metabolic molecules, obesity contributes to a major mechanical load on the joint. Mechanical loading studies in human and animal models show that abnormal loads can lead to changes in the composition, structure, and mechanical properties of articular cartilage (44), together with inflammatory responses throughout the joint. Mechanoreceptors on the surface of chondrocytes detect increased loading on the knee, triggering intracellular signaling cascades of cytokines, growth factors and MMPs (44).
Osteoarthritic chondrocytes respond differently to mechanical stimulation when compared with cells from healthy joint cartilage (60). The production of catabolic cytokines IL-1β and IL-6, the lack of an increase in mRNA aggrecan levels, and persistent mechanical stimulation are the main differences (61, 62). The mechanotransduction pathway involves recognition of the mechanical stimuli by integrins and activation of integrin-mediated signaling pathways leading to the production of cytokines, in both healthy and osteoarthritic chondrocytes (63).
When we focus on the effect of mechanical loading in OA through the stimulation of pro-inflammatory mediators, in vitro mechanical loading experiments show that injurious compression leads to proteoglycan depletion, destruction of the collagen network and cartilage degradation. In response, pro-inflammatory products are released and are postulated to cause synovitis (64). Interestingly, joint movement induces the expression of the anti-inflammatory cytokine IL-10. Therefore, not only the induction of inflammation, but also the lack of resolution of inflammation could play a role in OA (65).
Type 2 Diabetes and Metainflammation in OA
Type 2 diabetes (T2D) is a metabolic disorder characterized by high blood glucose levels that cause an inadequate β-cell response to the progressive insulin resistance (66). The incidence and prevalence of the most common T2D has nearly doubled in the last two decades, and its presence is reported in a high proportion of knee OA cases (67). Both diseases have many risk factors in common, which may explain the increased prevalence of musculoskeletal diseases in diabetic patients (67).
A considerable association between T2D and OA has been observed in several meta-analyses (68, 69), which is why T2D is the most commonly studied component of MetS as a risk factor for OA (70). Accordingly, epidemiological and experimental evidence suggests that T2D could be an independent risk factor for the onset and progression of OA, that is, the diabetes-induced osteoarthritic phenotype (71, 72). However, other studies show that diabetes mellitus is associated with accelerated degeneration of the cartilage matrix (67) and that T2D is a strong predictor for the development of severe OA, independent of age and body mass index (BMI). This suggests that longstanding diabetes per se is detrimental for knee and hip joints (73). According to this, increased levels of the cartilage oligomeric matrix protein (COMP) can be found in the synovial fluid of T2D subjects. Therefore, treating T2D may minimize glycemic control parameters and inflammation, along with synovial fluid COMP levels and OA progression (74).
Furthermore, inflammation plays a key role in the pathogenesis of T2D (59). T2D is associated with low-grade inflammation of adipose tissue in obesity and auto-inflammation in pancreatic islets (75). Indeed, in T2D, pancreas and adipose tissue, as well as other tissues, are infiltrated by macrophages and other immune cells, switching their profiles from an anti-inflammatory to a pro-inflammatory phenotype (76). Pro-inflammatory cytokines inhibit insulin signaling in peripheral tissues and induce β-cell dysfunction, contributing critically to the pathogenesis of T2D (76).
Inflammation in T2D can be triggered by excessive levels of nutrients, like glucose, which leads to the local production and release of cytokines and chemokines: IL-1β, TNF-α, CC-chemokine ligand 2 (CCL2), CCL3 and CXC-chemokine ligand 8 (CXCL8) in the pancreatic islets and insulin-sensitive tissues. In T2D patients, IL-1 receptor antagonist (IL-1RA) production by β-cells is decreased and IL-1β induces inflammation in pancreatic islets through NF-κB activation, which plays a critical role in the development of OA (59). Consequently, immune cells (macrophages and mast cells) are recruited and contribute to tissue inflammation (59, 77) (Figure 2).
Other authors postulate that high levels of glucose stimulate intercellular adhesion molecule 1 (ICAM-1) overexpression in the endothelium. ICAM-1 is a hallmark of diabetes-related inflammation and a crucial driver for cartilage degradation in T2D models. ICAM-1 enables and facilitates the adhesion and entrance of macrophages from serum or the synovium and facilitates the release of cytokines like IL-1β by the chondrocytes or synoviocytes This event triggers the production of MMP-13 and the recruitment of more macrophages, creating a vicious circle (78).
Senescence and Metainflammation in OA
Aging is a critical risk factor for the onset and progression of many chronic diseases, from metabolic disorders to neurodegenerative diseases, such as Alzheimer's and Parkinson's disease (32, 79). However, the aging process itself does not seem to be the direct cause of these disease states. Aging results from an imbalance between a variety of environmental and cellular stressors, and an insufficient capacity of cellular mechanisms to resolve stress processes. This results in the accumulation of unrepaired damage, which makes individuals more susceptible to developing chronic diseases (80). Also, there is an energy imbalance as a result of an age-associated decrease in the resting metabolic rate, in part due to a loss of lean mass, which does not synthesize enough ATP in older individuals. Finally, there is a dysregulation of signaling cascades that guarantee tissue homeostasis, including hormones, inflammatory mediators and antioxidants, thus triggering an inflammatory state characteristic of the aging process (80) (Figure 1).
This imbalanced situation ultimately leads to decreased adaptation to stress, loss of proteostasis, cellular senescence, inflammatory responses, metabolic disarrangement, DNA damage, detrimental epigenetic modifications, and impaired cell - cell communication. These defective responses trigger a state of low-grade chronic inflammation associated with aging that has been termed “inflammaging,” which plays a major role in the pathogenesis of age-related diseases (32, 79–83).
Aging and age-related diseases share diverse mechanisms that promote low-grade chronic inflammation (32). These mechanisms include inflammatory processes, mainly the chronic activation of innate immunity, but also changes in the adaptive immune system, such as defective T cell functioning, leading to immunosenescence (79). Another prominent mechanism actively contributing to inflammaging is cellular senescence. Senescent cells are unable to proliferate but remain metabolically active and acquire a senescent-associated secretory phenotype (SASP). This SASP is characterized by altered secretion of pro-catabolic enzymes, such as MMP-1, MMP-3, MMP-9, and MMP-13; pro-inflammatory cytokines, such as IL-6, IL-1α and β; chemokines (IL-8, CCL2, CCL5, and CCL19); and the following growth factors: transforming growth factor-β (TGF-β), fibroblast growth factors (FGFs), vascular endothelial growth factor (VEGF) (45, 83). Furthermore, senescent cells exhibit DNA damage, shortening of telomeres, defective autophagy, mitochondrial dysfunction, and increased oxidative stress (32, 41, 83) (Figure 1).
In osteoarthritic cartilage, chondrocytes suffer cellular senescence, known as “chondrosenescence,” as a result of inflammatory signals that increase with aging and mechanical stress (32, 84). An accumulation of senescent cells is also found in the osteoarthritic synovium (41, 83). These factors, together with chondrocyte inability to repair the damaged tissue, are responsible for cartilage degeneration in OA (82). Aging in cartilage is also associated with lower cell density and loss of proteoglycans, joint stiffness, increased chondrocyte size and protein glycosylation (32, 85, 86). Advanced glycation end products (AGEs) increase with age in the cartilage extracellular matrix (ECM) and modify the mechanical properties of cartilage. Their receptor can also interact with TLRs on the surface membrane, thus enhancing innate immune activation (83). Chondrosenescence also occurs through the loss of chondrocyte autophagy. Chondrocytes closely depend on autophagy for the maintenance of cartilage homeostasis, since it is the protective mechanism responsible for the removal of cellular debris and macromolecules through lysosome-mediated degradation (32). In the OA pathogenic context, autophagy reduces inflammation in the joint tissues (87–89). However, the loss of chondrocyte autophagic capacity associated with aging results in further destruction of the cartilage ECM and increased oxidative stress (32, 90). The senescent phenotype in chondrocytes and synoviocytes contributes to perpetuating a local pro-inflammatory state in the joint, due to the release of cytokines and increased cartilage degradation. At the same time, the SASP favors a state of low-grade systemic inflammation through the release of chemokines that also contributes to the pathogenesis of OA (32) (Figure 2).
Immunity and the Purinergic System in OA
As mentioned above, OA displays low-grade chronic inflammation, primarily mediated by innate immunity (45). Articular cartilage damage and degeneration occurs mainly because of an imbalance between the mechanical load it receives and its ability to absorb and distribute it. Altered mechanical joint loading is often associated with being overweight, anatomical misalignment or post-traumatic joint instability, and these are all critical risk factors for joint destruction (91, 92). The progressive destruction of the cartilage matrix leads to the appearance of tissue fragments in the articular cavity, known as “damage-associated molecular patterns” (DAMPs). These signals are detected by “pattern recognition receptors” (PRRs), present in chondrocytes and synoviocytes. Toll-like receptors (TLRs) are the PRRs that mainly mediate this signaling, particularly TLR2 and TLR4 in OA cartilage (41, 93). TLRs trigger a signaling cascade that results in innate immunity activation, by leading the activation of interferon-regulatory factors, NF-κB, and activator protein 1 (AP-1), and inducing pro-inflammatory mediators, such as cyclooxygenase (COX)-2; cytokines, such as IL-6 and TNF-α; components of the inflammasome, such as caspase-1, NLRP3, pro-IL-1 and pro-IL-18; MMPs and aggrecanases, such as ADAMTS-4 and−5 (41, 45). The activity of MMPs and ADAMTS, enzymes that degrade type II collagen and aggrecan in the cartilage matrix, promote the appearance of more DAMPs, thereby perpetuating tissue damage and innate immunity activation.
Varying extracellular concentrations of different purinergic metabolites, such as ATP, ADP and adenosine, are reliable indicators of the tissue status during inflammation, entailing a robust regulatory system of the immune response (94–97).
Adenosine is a potent modulator of inflammation and immune responses (98). Adenosine regulates function, proliferation and activation of immune cells, and promotes the resolution of inflammation dampening the immune response in physiological and pathological situations, acting as a self-limiting signal (96). Platelets, endothelial cells, neutrophils and macrophages are considered an important source that contributes to increasing extracellular adenosine during inflammation (99) (Table 1). It is well-known that adenosine biases TLR4. Adenosine, by A2AR activation, can inhibit LPS-induced TLR4-mediated responses, by inhibiting Th1-polarizing responses (TNF-α, IL-12p70) and activating anti-inflammatory cytokine production and Th2-polarizing responses (IL-6 and IL-10) (111, 112) and inflammation-resolving properties (113). Adenosine produces increased levels of cAMP, and this second messenger inhibits LPS-induced IL-12 production in murine peritoneal macrophages (114), decreases LPS-induced TNF but enhances IL-10 in human monocytes (115). Nevertheless, the effects of cAMP on TLR-mediated cytokine production can be dependent on cell type. Recent evidence suggests that cAMP levels in chondrocytes are an indicator of the metabolic function, with low levels indicating cartilage degradation, and increased levels suggesting an increased cartilage synthesis (116).
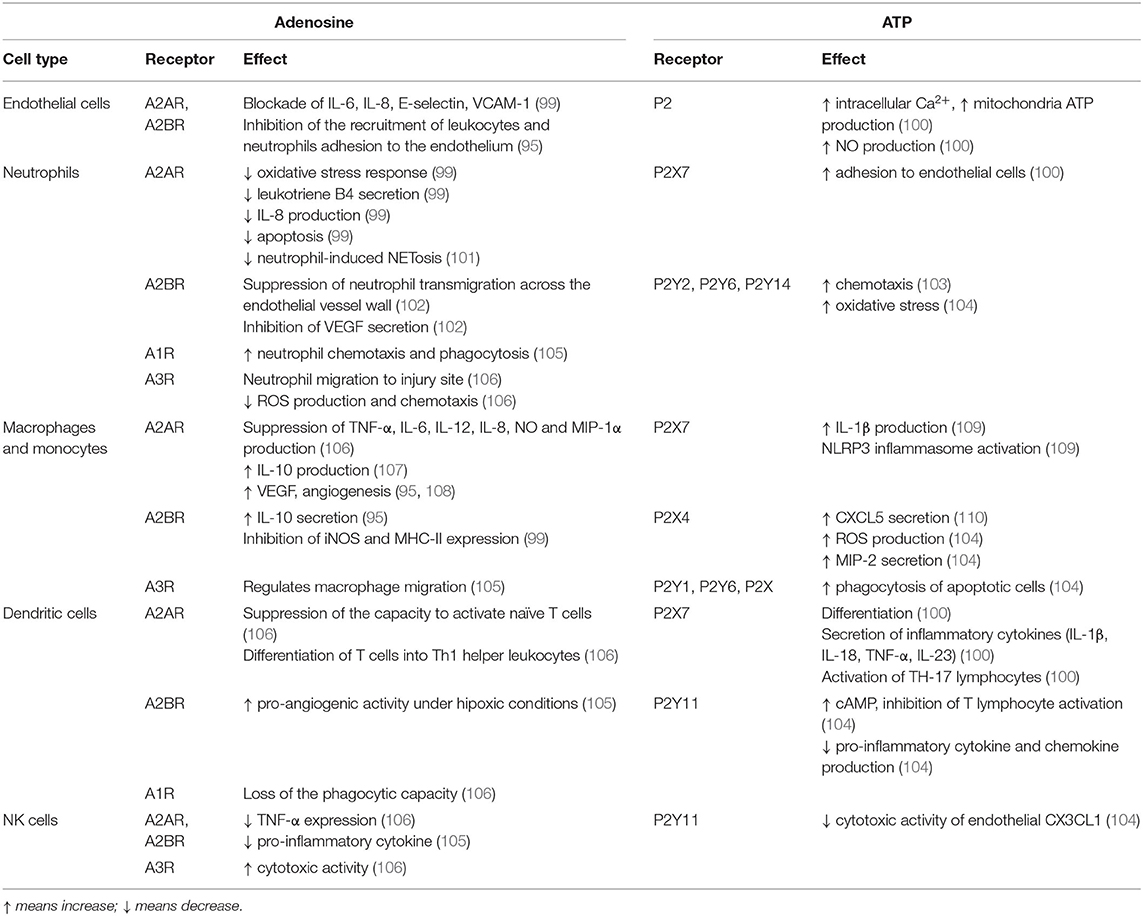
Table 1. Effects of adenosine and ATP through their specific purinergic receptors in inflammation.
As mentioned, TLRs are activated during OA progression, particularly TLR4 (93). Different drugs have been shown to decrease the inflammatory and catabolic response in OA chondrocytes stimulated with different DAMPs by inhibiting the TLR4/MyD88/NF-κB signaling pathway (93). In murine articular chondrocytes, hyaluronan (HA) fragments have been found to induce inflammation via CD44 and TLR4 and NF-kB activation; adenosine can attenuate this inflammation process via A2AR activation (117). Although the exact mechanism has not yet been fully elucidated, it is known that adenosine produces high levels of cAMP that activate protein kinase A (PKA) and inhibit NF-κB (118). No data has been found in the literature, but this mechanism must be altered in OA progression.
In contrast to the anti-inflammatory role of adenosine-mediated purinergic signaling, nucleotide receptors promote inflammatory mechanisms (3). Elevated concentrations of ATP are usually a warning sign of cell death detected by immune cells at sites of active inflammation. Extracellular ATP plays an important role in the innate immune response, upregulating the inflammatory pathways (119). Extracellular ATP (in the millimolar range) predominantly induces proinflammatory effects through activation of the low affinity receptor P2X7 (120), but low (micromolar) extracellular ATP concentrations exert immunosuppressive action through the activation of the high affinity P2Y11 receptor (121, 122).
ATP is considered like a DAMP in OA as it activates TLR4 and leads to NLRP3 inflammasome activation and caspase-1-mediated IL-1β secretion (123) and activates NF-κβ signaling (124), leading to cartilage degradation and synovial inflammation.
Purinergic System in OA and Modulation by Metainflammation
The direct effect on the joint of purinergic system activation in different cell subtypes has been described extensively (18, 125). Therefore, we shall only concentrate on the modulation of the purinergic system in chondrocytes, since they play a key role in OA.
Adenosine and Its Receptors in OA
The presence of adenosine receptors in human articular chondrocytes was first discovered in 1999 (126), and they were further characterized in bovine chondrocytes in the presence or absence of low-frequency low-energy pulsed electromagnetic fields (PEMFs) (127). Since then, much importance has been conferred to the role of adenosine in the regulation of inflammatory processes in cartilage and the maintenance of joint homeostasis, modulating the release of pro-inflammatory mediators and cytokines (128). All four adenosine receptors are expressed in chondrocytes, but A2AR and A3R are particularly relevant (18, 22, 128, 129).
A2AR has a broad range of physiological activities, and is known to play an essential role in the maintenance of articular cartilage homeostasis (130). Biophysical interventions, such as PEMFs, for stimulation of bone and cartilage via A2AR are currently being studied (131, 132). There are different modalities of in vivo adenosine delivery: polydeoxyribonucleotides, liposomes (129), and functionalized nanoparticles (133). All of them are for intra-articular administration and activate adenosine receptors. When cartilage explants are treated with A2AR antagonists (ZM241385 as a specific antagonist; CGS15943 and theophylline as broad receptor antagonists), cartilage matrix degradation occurs, which is evidenced by increased glycosaminoglycans (GAG), MMP-3, MMP-13, Prostaglandin E2 (PGE2) and NO release (134). It has been observed that in mouse articular chondrocytes stimulated with IL-1β, an A2AR agonist (CGS21680) can counteract the upregulation of the inflammatory markers NF-κB, TNF-α, IL-6, MMP-13, and NO (135). It has also been reported that in the presence of IL-1β, osteoarthritic chondrocytes release less adenosine and ATP, suggesting that inflammation reduces purinergic signaling via A2AR. This may be contradictory to another study which reported that in equine articular chondrocytes, LPS stimulates accumulation of extracellular adenosine working as an inflammatory blocker on chondrocytes (136). The divergence between studies could be due to the nature of the stimuli or species-dependent differences. Mice lacking A2AR develop OA, which can be seen in increase in MMP-13 and Col10a1 expression, fibrillation and thinning of cartilage, disordered chondrocytes, less GAG and loss of sulfated proteoglycans and collagen in cartilage (130). These events are not present in the preventive group or in the treatment group, after intra-articular injection of liposomal adenosine in a rat model of post-traumatic OA (130). Adenosine released by osteoarthritic chondrocytes can also signal via A2AR to limit the production of intracellular NO, which is associated with ECM degradation and chondrocyte apoptosis (129, 137) (Table 2).

Table 2. Description of purinergic receptor roles in osteoarthritis (OA), obesity and type 2 diabetes (T2D).
A3R exerts anti-inflammatory effects on different experimental OA models (185). A3R knockout (KO) mice develop progressive loss of articular cartilage. Agonists for A3R downregulate key genes implicated in OA pathology, such as RUNX2 (151). In a recent study CF101, a highly selective A3R agonist, was orally administered twice daily to monosodium iodoacetate OA-induced rats. It was found that CF101 downregulated the signaling pathway of NF-κB, which led to decreased levels of TNF-α. This effect prevented cartilage damage and chondrocyte apoptosis, osteophyte formation, and bone destruction. It also reduced pannus formation and lymphocyte infiltration. On the other hand, all these effects were counteracted by the A3R antagonist MRS1220 (155) (Table 2).
During inflammatory processes, ADA is secreted to the extracellular space altering adenosine levels. Synovial fluid ADA measurement, in association with C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) levels, makes it possible to distinguish OA from other rheumatic diseases, like RA (186). In cartilage, when extracellular adenosine levels decrease, cartilage damage markers appear, e.g., increase in GAG release, and higher production of MMPs, PGE2 or NO (187). In some rheumatic diseases like RA, ADA1 levels are increased, compromising extracellular adenosine levels and possibly contributing to the severity of the disease (188).
Pain intensity correlates with OA progression. At low concentrations, adenosine binds to A1R inducing analgesia by decreasing nociceptor nerve conduction, and to A2AR, which triggers an anti-inflammatory response by secreting anti-inflammatory mediators. However, excessive adenosine concentration can be detrimental (138). In the case of excessive joint motion, which leads to excessive activation of CD73 via hypoxia inducible factor (HIF)-1α, extracellular adenosine accumulates. The consequent activation of the low-affinity A2BR seems to play a harmful role in cartilage homeostasis, possibly due to its capacity of stimulating inflammatory pathways involving MAPK (128, 151). In OA, as a low-grade chronic inflammatory disease, increased extracellular adenosine levels may switch selectivity of receptor binding toward A2BR, generating hyperalgesia (138) (Table 2).
As mentioned above, inflammation reduces signaling via A2AR in chondrocytes. However, in response to metabolic stress and inflammation, adenosine accumulates extracellularly, like in obesity (189). The activation of the adenosine A1R increases lipogenesis (139, 140), adipogenesis (141) and leptin production (142, 143). Accordingly, overexpression of A1R in adipose tissue in mice protects from obesity-induced insulin resistance (144), whereas A1R KO mice show anti-lipolytic effects (139). A1R KO mice exhibit increased fat mass and body weight, and impaired glucose tolerance and insulin sensitivity (145, 146). Contrary to A1R, the activation of A2BR, which is highly expressed in human primary pre-adipocytes (148), can inhibit both adipogenesis and lipogenesis in vitro (141, 190). Moreover, in rodent models, the overexpression of A2BR seems to correlate with parameters of obesity, being upregulated in visceral adipose tissue of mice fed HFD (152). Finally, studies in obese patients show that A2BR expression in subcutaneous fat is positively associated with BMI and other parameters of obesity (149). On the other hand, activation of A2AR in human and murine adipose tissue not only activates lipolysis, but also induces increased energy expenditure and protects mice from diet-induced obesity (148). Finally, the receptor A3R is also expressed in adipocytes, and KO animals for this receptor present less abdominal and total body fat (148) (Table 2).
Furthermore, obesity is closely related to a state of insulin resistance, which is considered to be a key step in the development of diabetes and MetS (190). The stimulation of A1R induces insulin sensitivity and reduces insulin secretion (147). On the other hand, the activation of A2BR contributes to increased insulin resistance by affecting the production of IL-6 and other cytokines (153). Animal studies confirm that A2BR activation increases serum IL-6 levels (154), which may be involved in the development of insulin resistance and improve insulin sensitivity (191). A2BR affects inflammatory processes in adipose tissue through the activation of macrophages, and indirectly inducing the development of insulin resistance (153) (Table 2).
In view of the above, these changes in the purinergic system mediated by MetS could be involved in the development of OA, which would partially explain the relationship between both pathologies. It has been stated that in adipose tissue there is a low stimulation of A1R and A2AR in obesity conditions (148). Interestingly, this situation is similar in chondrocytes, which could induce a loss in cartilage homeostasis, since they play an essential role in the maintenance of tissue, and consequently promote the development of OA (130). Moreover, the activation of A2BR in obesity contributes to inflammatory processes and secretion of pro-inflammatory molecules (148), which possibly contribute to the cartilage alteration seen in OA. According to this, new studies are being developed and show that intra-articular stimulation of A2AR can reverse not only OA induced by anterior cruciate ligament injury, but also obesity-related OA in experimental models of the disease (192).
As described above, T2D is the most commonly studied component of MetS as a risk factor for OA. Metformin, a first-line drug for T2D treatment, is known to affect the energy state of the cell. It has been recently proposed that metformin may be beneficial in obese patients with knee OA (193) and can also inhibit respiratory chain complex 1, activating AMPK and inhibiting AMP deaminase, which results in an increase in extracellular adenosine (194). The increased adenosine levels might activate A2AR and could explain the beneficial effect of metformin in obese patients with knee osteoarthritis, but this needs to be explored further.
P2 Receptor Modulation in OA
Currently, it is known that extracellular nucleotides are fundamental in the regulation of biological processes in many tissues, including the musculoskeletal system, and could work as potential therapeutic targets (195).
We have known that nucleotide receptors are expressed by chondrocytes and are associated with PGE2 release since 1991 (196). However, their role in chondrocytes may be controversial, possibly depending on the amount of extracellular ATP available and the physiological/pathological conditions of the joint (18). Regarding the physiological conditions, prechondrogenic condensation occurs, a phenomenon necessary for chondrogenic differentiation and skeletal patterning. This process of prechondrogenic condensation is mediated by extracellular ATP signaling via the P2X4 receptor, which leads to Ca2+-driven ATP oscillations, ensuring the correct constitution of the joint (197). P2X1 and P2X2 receptors were identified in primary bovine chondrocytes, showing that they can facilitate the release of NO and PGE2, which are involved in inflammatory processes and cartilage resorption, in the presence of high concentrations of ATP (156). Upregulation of proteoglycan levels and reduction in NO release after dynamic compression occurs in bovine articular chondrocytes via P2 and has an anabolic effect on the cartilage (198). P2X2 and P2X3 receptors are also expressed in chondrocytes. ATP levels in OA have been linked to pain intensity (199). Activation of P2X3 receptors in chondrocytes induces NO and PGE2 release, suggesting their function in modulating the inflammatory process and playing an important role in the development of articular hyperalgesia in osteoarthritic joints (162) (Table 2).
The P2X7 receptor is highly expressed in cells of the immune system, and participates in regulating inflammation and pain, although the exact mechanisms are not yet understood (200). Interestingly, the P2X7 receptor needs a high concentration of ATP for full activation, suggesting a specific role under pathological conditions (201). Accumulation of ATP released by osteoarthritic chondrocytes could act as a warning signal via P2X7, aggravating the inflammatory process and pain and causing cell death (168). Low concentrations of extracellular ATP produce a positive response of the P2X7 receptor activation toward cell proliferation. However, when the concentration of ATP is high, as it is supposed to be in cartilage areas affected by OA, there is an overactivation of the P2X7 receptor, leading to cell death (47). The strategy of blocking the P2X7 receptor could benefit the survival of chondrocytes in OA. It has been found that signaling via P2X7 in rabbit articular chondrocytes leads to chondrocyte apoptosis through the activation of the phospholipase A2 (PLA2)/cyclooxygenase-2 (COX-2) pathway (45). Numerous studies have focused on pain mitigation, one of the main consequences of OA, and their results show that targeting purinergic receptors may be considered as a therapeutic alternative to stop or slow down articular cartilage degeneration. Blocking the P2X7 receptor would have an impact on cartilage itself since it has been observed that its inhibition by the selective antagonist AZD9056 exerts pain-relieving and anti-inflammatory effects. This antagonist counteracts the induction of MMP-13 or NF-κB, both upregulated in osteoarthritic chondrocytes, in an OA rat model. In this study, AZD9056 also reversed the upregulation of IL-1β, IL-6, TNF-α, substance P and PGE2 (169) (Table 2).
Extracellular inorganic pyrophosphate, whose levels regulate physiological and pathological mineralization, are also influenced by P2 receptors. Suramin, a putative antagonist of P2 receptors, can block the stimulation generated by ADP on the production of inorganic pyrophosphate in chondrocytes, with the consequent blockade of excessive deposition of calcium pyrophosphate dihydrate (CPPD) in the articular cartilage, which would hinder the correct mineralization of the bone (202). Similarly, and consolidating the idea that high concentrations of extracellular ATP promote cartilage damage, another study on primary articular chondrocytes has shown that P2X7 and P2X4 may be implicated in excessive ATP efflux when the ANK gene is overexpressed, as in OA (163), causing accumulation of CPPD (164). Modulation of these receptors would facilitate the reduction of joint damage and cartilage regeneration (Table 2).
However, activating or blocking P2 receptors in OA does not only act on the inflammatory onset of the disease. As mentioned above, abnormal mechanical loading of the joint exerts changes in cell structure and mechanical properties that lead to OA. Physiological joint loading and articular cartilage compression seem to play a key role in ATP release and P2 signaling. Mechanical forces coming from the ECM are transmitted to rat articular chondrocytes via integrins, activating different molecular mechanisms, such as stimulation of the P2Y pathway, mainly by P2Y1, P2Y2, and P2Y4 receptors (203). A candidate to transfer these forces is the primary cilium, which modulates ATP-induced Ca2+ signaling via P2X (P2X4, P2X7) and P2Y (P2Y1, P2Y2) receptors (204). As an overview, the forces applied to chondrocytes lead to an increase in ATP release, which downregulates the expression of MMPs, such as MMP-13 (205). On the contrary, when sustained pressure is maintained on chondrocytes, ATP release is suppressed, causing cartilage degeneration (206). This mechanical stimulation is essential for maintaining cartilage integrity, and different responses to this input have been observed between healthy and osteoarthritic chondrocytes. Healthy chondrocytes increase the amount of extracellular ATP after mechanical stimulation via the P2Y2 receptor exerting anabolic responses, while osteoarthritic chondrocytes do not increase these ATP levels (63, 176) (Table 2).
From another perspective, the activation of some receptors, such as P2Y1, P2Y2, P2Y4, and P2Y11, induces the adipogenic differentiation of stem cells (170, 174), whereas, other receptors, like P2Y13, P2Y14, and P2X7 show anti-adipogenic effects (170). In addition, the P2Y1 receptor can induce leptin production in murine adipose tissue (175). On the other hand, activation of the P2Y4 receptor inhibits adiponectin expression, and P2Y4 KO mice show increased adiponectin secretion (178). P2X7 KO mice have increased body weight and adipocyte hyperplasia in fat pads (171) (Table 2).
Rat models have revealed that the P2Y1 receptor is present on intra-islet capillaries, and P2X4 receptors appear in β-cells. Moreover, P2X1, P2X3, P2Y1, and P2Y2 receptors are expressed in small pancreatic blood vessels, and β-cells present P2X7 receptors, which are downregulated in T2D. In human islets, the receptor seems to be involved in the secretion of insulin and IL-1RA (165). The P2X7 KO mice show lower β-cell mass, impaired glucose tolerance, and defective insulin and interleukin secretion (172). Furthermore, the P2Y13 receptor is involved in the induction of β-cell apoptosis in the presence of high glucose and free fatty acid levels (184). Extracellular ATP (1 μM) increases insulin secretion in mouse β-cell lines, but at higher ATP concentrations, cell viability decreases, with involvement of the P2Y1 and P2X4 receptors (165) (Table 2).
There is some controversy in the modulation of these receptors both in OA and MetS. Extracellular concentrations of ATP modulate the activation of these receptors, which act differently according to the tissue where they are expressed. The P2X7 receptor is related to the inflammatory processes during OA development, with high levels of extracellular ATP (168). However, in adipose tissue, this receptor shows an anti-adipogenic effect (171). In T2D, the P2X7 receptor has been reported to be decreased (172). Furthermore, activation of the P2Y1 receptor increases in obesity in the adipose tissue (175) and in T2D in β-cells (165). In chondrocytes, the P2Y1 receptor plays a key role in the response to mechanical forces, and when the pressure is maintained, increased ATP release may cause cartilage degeneration (203). A deeper knowledge of these receptors in these three conditions is necessary to better describe the close relationship among them (Table 2).
Mitochondrial Metabolism, Purinergic Signaling, and OA
During chondrosenescence, the glycolysis pathway is overstimulated, trying to generate ATP promptly, which is necessary for repairing damaged cartilage (207). Chondrocytes are mainly glycolysis-dependent cells, but they keep the ability to use mitochondrial respiration in certain cases to enhance cell survival and ECM biosynthesis in periods of nutrient stress to sustain ATP synthesis (208, 209). However, under OA conditions, mitochondrial biogenesis and activity are disrupted, leading to a greater amount of ROS production. This can be linked to AMPK deficiency in OA, a molecule responsible for regulating cellular metabolism and energy balance (210). AMPK is also a chondroprotective molecule that is capable of inhibiting procatabolic responses to inflammation and biomechanical injury via its downstream targets, peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α and forkhead box O (FOXO)3A (211). AMPK/sirtuin (SIRT)-3 signaling protects mitochondria from oxidative stress by deacetylating superoxide dismutase (SOD2) and decreasing ROS. This pathway also prevents mitochondrial DNA damage by activating mitophagy (212). Some approaches have focused on enhancing the AMPK/SIRT1 pathway, treating an OA rat model with quercetin, and finding overall improvement in mitochondrial function, higher ATP levels in mitochondria, increased mitochondrial copy number and attenuation of ROS levels (213). Other studies have observed that pharmacological stimulation of AMPK increases PGC-1α via SIRT1, reversing impairments in mitochondrial biogenesis, oxidative phosphorylation (OXPHOS) and intracellular ATP in human knee OA chondrocytes (214). It is established that extracellular adenosine can contribute to AMPK activation and regulation (215). Adiposomal injections and CGS21680 A2AR agonist have demonstrated FOXO1/3 activation and retention in the nucleus, which is implicated in increased autophagy and cartilage homeostasis. This recent study provides a mechanism in which A2AR can activate SIRT1 and AMPK through PKA (216) (Figure 3).
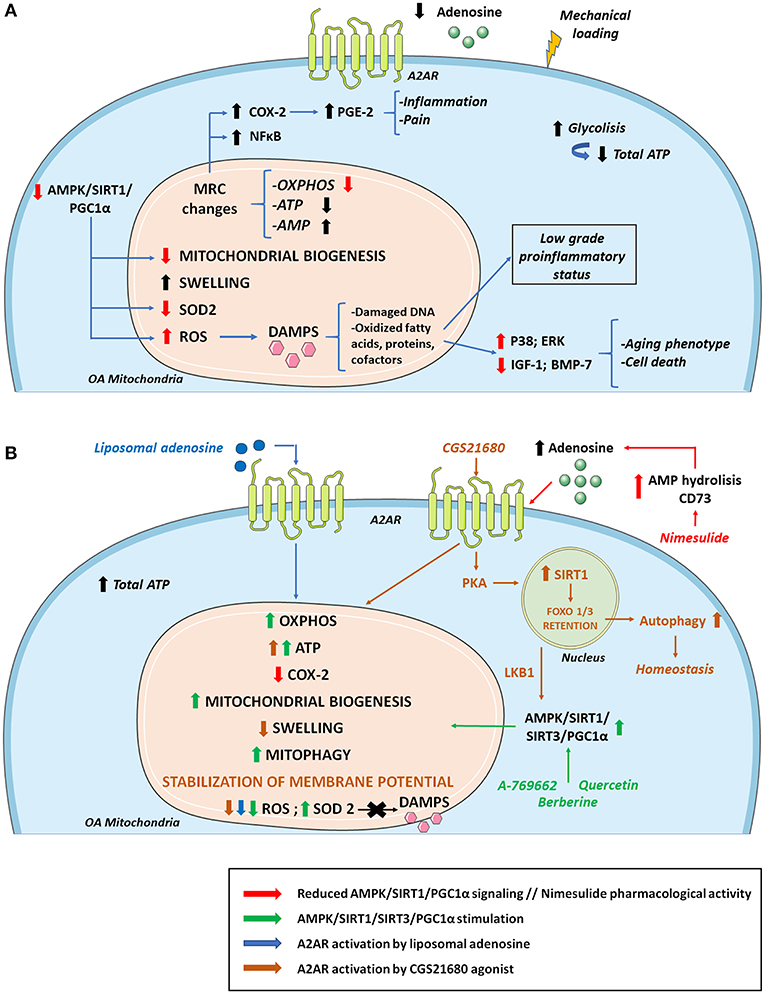
Figure 3. Osteoarthritis (OA) mitochondrial metabolism and its modulation by A2AR and AMPK stimulation. (A) During the development of OA, cellular metabolic alterations occur. There is an increase in glycolysis activity to compensate the energy requirement necessary to repair damaged cartilage. However, total ATP levels decrease due to mitochondrial defects together with sustained mechanical loading. Changes in the MRC and its complexes impair ATP and AMP production. As a result, there are lower extracellular levels of ATP and adenosine which diminish A2AR signaling. These alterations in the MRC cause an increase in proinflammatory molecules and transcription factors such as COX-2 and NF-κB, respectively, aggravating inflammation and pain. Reduced AMPK/SIRT1/PGC-1α signaling compromises mitochondrial biogenesis and OXPHOS. There is also a decrease in antioxidant enzymes such as SOD2 that cause increases in ROS production, contributing to the release of DAMPS that aggravate the inflammatory status, stimulating catabolic pathways (p38; ERK) and reducing anabolic pathways (IGF-1; BMP-7), which supposes cell death and an aging phenotype. (B) Mitochondrial OA impairment can be partly reversed by AMPK and A2AR stimulation. Quercetin, berberine and pharmacological AMPK activator A-769662 can activate AMPK/SIRT1/PGC-1α and AMPK/SIRT3 pathways increasing OXPHOS, ATP, mitochondrial biogenesis, mitophagy, and SOD2 activity. AMPK stimulation also promotes a decrease in ROS production with reduced DAMPs secretion diminishing inflammation and aging phenotype. Regarding ROS level reduction, similar effects are found when A2AR is activated by agonists like liposomal adenosine. Moreover, A2AR agonist CGS21680 stimulates SIRT1 via PKA allowing FOXO1/3 retention and increased autophagy. CGS2168 indirectly activates AMPK through LKB1. Nimesulide increases extracellular adenosine levels favoring a reduction in the expression of COX-2 and the associated inflammation and pain. AMP, adenosine monophosphate; AMPK, AMP activated protein kinase; BMP-7, bone morphogenetic protein 7; DAMPS, damage associated molecular patterns; IGF-1, insulin growth factor 1; LKB1, liver kinase B1; MRC, mitochondrial respiratory chain; OXPHOS, oxidative phosphorylation; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; PKA, protein kinase A; SIRT1, sirtuin 1; SIRT3, sirtuin 3; SOD2, superoxide dismutase.
Age-associated mitochondrial dysfunction and mitochondrial dysfunction in OA contributes to perpetuating the low-grade systemic pro-inflammatory status by further promoting cellular senescence and SASP, and with it, the pathological processes associated with inflammaging (32, 210, 217). In this context, mitochondria increase the production of superoxide anion and hydrogen peroxide, and the amount of AMP increases with respect to ATP, and mitochondrial danger-associated molecular patterns (DAMPs) are released. The increase in ROS, DNA damage, oxidized fatty acids, proteins, and cofactors, ultimately alters cellular function (32). This chondrocyte ROS increase is partly due to mitochondrial dysfunction and reduced SOD2 activity, which is an antioxidant enzyme and a by-product of the mitochondrial electron transport chain. The altered redox environment promote disruptions in signaling, increasing pro-catabolic MAP kinases p38 and ERK and inhibition of anabolic insulin growth factor 1 (IGF-1) or bone morphogenic protein 7 (BMP-7) signaling, leading to an aging phenotype and chondrocyte death (218). In OA, this increase in oxidative stress also disrupts proteoglycan and collagen network, favoring the appearance of residues that further activate innate immunity. The accumulation of ROS accelerates cellular senescence processes and apoptosis by decreasing ATP levels (32). Sustained mechanical loading is essential in OA for reducing ATP levels and respiratory activity, and for increasing ROS production on bovine osteochondral explants (206) (Figure 3).
Inflammation and mitochondrial disruption are closely related and affect OA progression. Inhibition of the mitochondrial respiratory chain induces inflammatory responses. In primary human chondrocytes the inhibition of complexes III and V induces COX-2 expression and PGE2 production, the generation of ROS and the activation of NF-κB (219). Therefore, COX-2 levels are upregulated in inflamed joint tissues and are located in the superficial layers of human cartilage with OA, where damage firstly appears (219). COX-2 overexpression is related to higher PGE2 production in osteoarthritic cartilage, which is partly responsible for inflammation and pain (220). Nimesulide, a COX-2 inhibitor, mediates its anti-inflammatory effects in vivo and in vitro by increasing CD73 activity and AMP hydrolysis so that higher levels of adenosine are available for A2AR activation (221). This mechanism of action positions adenosine signaling as a major player in inflammatory-derived pain mitigation by non-steroidal anti-inflammatory drugs (NSAIDs) and points out an correlation between mitochondrial alterations, inflammation and pain.
Osteoarthritic chondrocytes exhibit a depletion in the mitochondrial production of ATP with a consequent reduction of extracellular adenosine and A2AR stimulation, disrupting chondrocyte homeostasis (222). This ATP level decrease in OA contributes to an increase in glycolysis with pyruvate conversion to lactate and defects in OXPHOS, leading to mitochondrial dysfunction (209). A2AR-null mice develop spontaneous OA with increased mitochondrial dysfunction, increased ROS burden and reduced ATP production via OXPHOS (22, 130). This model also shows mitochondrial depolarization, swelling, fragmentation and mitophagy (222). ROS burden is mitigated in a mouse model of obesity-induced OA by intra-articular injections of liposomal adenosine or CGS21680. This A2AR agonist also increases mitochondrial basal rates of respiration and ATP production in vitro, with increased ATP release in IL-1β-treated human chondrocytic T/C-28a2 cells. A2AR activation also stabilizes mitochondrial membrane potential and reduces mitochondrial swelling after IL-1β exposure (222) (Figure 3).
Purinergic System, Inflammasome and OA
The NLRP3 inflammasome is considered an important component involved in OA pathogenesis (223). Inflammasomes are multi-protein complexes mainly present in the cytosol of myeloid cells, such as neutrophils, monocytes and DCs, but they are also found in non-hematopoietic lineage cells such as endothelial fibroblasts or chondrocytes (224).
Assembly and activation of the inflammasome components by different stimuli (e.g., DAMPs) triggers a regulated pro-inflammatory reaction and induces cellular pyroptosis as a protection mechanism orchestrated by innate immunity to restore tissue homeostasis (225). In the joint, inflammasome dysregulation can lead to synovial inflammation and cartilage destruction, characteristic of OA pathogenesis (226).
It has been established that at least two signals are necessary to induce NLRP3 activation: a first priming signal, where TLRs recognize DAMPs or PAMPs promoting synthesis of inflammasome components mediated through NF-kB; and a second activation step, which leads to the assembly of the scaffold inflammasome proteins and recruitment of pro-caspase-1. This second signal is stimulated by cellular stress or pathogen-derived molecules (227) (Figure 4).
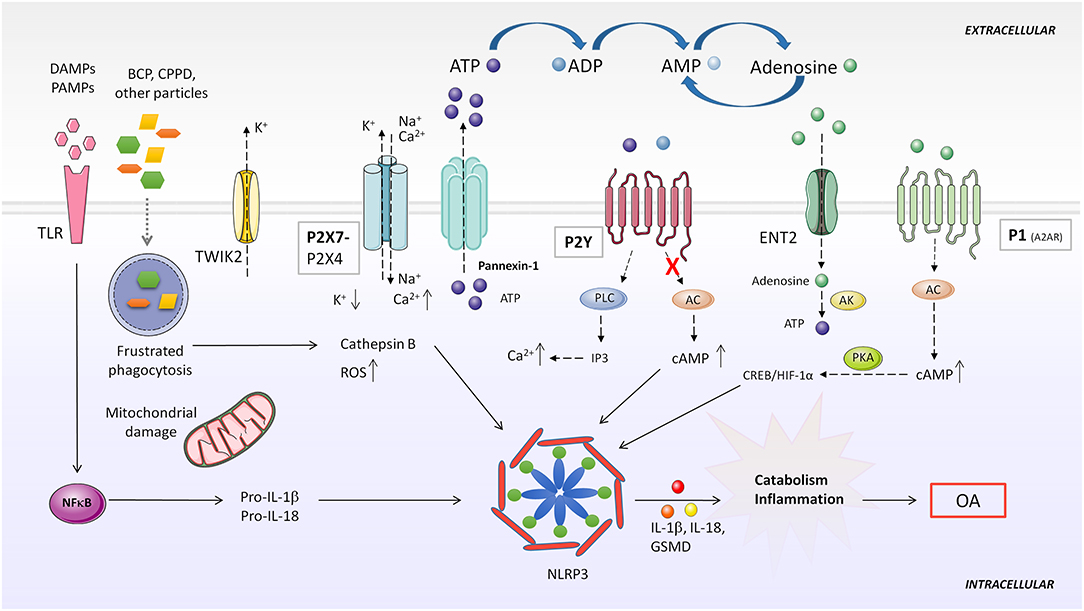
Figure 4. Purinergic system implication in NLRP3 inflammasome activation. DAMPS or PAMPs present in the microenvironment are recognized by TLR receptors, which in turn, upregulate pro-IL-1β and pro-IL-18 expression through NF-κB activity. BCP and CPPD crystals can be both recognized by TLRs and phagocytized by the cell. Impaired particle or crystal phagocytosis by phagosomes leads to cathepsin B release and mitochondrial dysfunction, which induce elevated ROS production and consequent NLRP3 activation. Additionally, frustrated phagocytosis can lead to massive ATP release through Pannexin-1, activatingP2X7 and causing ionic cytoplasmic imbalance. Excessive K+ efflux, especially through TWIK2, and Na+ and Ca2+ influx through P2X7 activates NLRP3 inflammasome. Activation of different P2Y by ATP or ADP increases intracellular Ca2+ concentration. Particles can also impede induction of purinergic AC-cAMP pathway allowing NLRP3 activity. A2AR activation by extracellular adenosine induces HIF-1α upregulation, which activates NLRP3. AC, adenylate cyclase; AK, adenosine kinase; BCP, basic calcium phosphate; CPPD, calcium pyrophosphate dehydrate; DAMP, danger-associated molecular patterns; ENT2, equilibrative nucleoside transporter 2; GSMD, gasdermin D; IP3, inositol 1,4,5-trisphosphate; NLRP3, NOD-like receptor pyrin domain containing 3; PAMP, pathogen-associated molecular patterns; PKA, protein kinase A; PLC, phospholipase C; ROS, reactive oxygen species; TLR, toll like receptor; TWIK2, two-pore domain weak inwardly rectifying K+ channel 2.
Although the exact role of NLRP3 is still controversial, several studies have pointed out that overactivation of the NLRP3 inflammasome is involved in low-grade chronic inflammation during OA development (226, 228). Inflammasome secretion of pro-inflammatory IL-1β leads to synovial inflammation and cartilage degradation (229). IL-18, another cytokine processed by the NLRP3 inflammasome, is elevated in synovial fluid and serum of patients with OA, and has been proven to decrease type II collagen and aggrecan expression, disrupt autophagy and induce apoptosis in chondrocytes (230, 231). A recent work shows that inhibition of the mTOR pathway after IL-18 treatment has protective effects in chondrocytes both in vitro and in vivo (232). Data suggest that IL-1β and IL-18 are mainly produced in the damaged synovium rather than cartilage (233). Analyses of knee joint explants from patients with primary OA have demonstrated that the synovium expresses high levels of NLRP3 inflammasome components (234), and NLRP3 and NLRP1 inflammasomes are overexpressed in synovium derived from knee OA patients (235). In addition, oxidative stress-mediated NLRP3 inflammasome activation was enhanced after inhibiting HO-1 expression (236). Pyroptosis mediated by NLRP3 inflammasome have also been found in chondrocytes in vitro and in vivo (234).
All these data support the idea that the NLRP3 inflammasome is involved in the onset and progression of OA and can even be considered a potential new biomarker for diagnosis and tracking in patients with OA (228).
Usually, CPPD and basic calcium phosphate (BCP) crystal deposits are found in the synovial fluid and cartilage of patients with OA. Both crystals are believed to be involved in joint inflammation and may correlate with disease harshness (237, 238). BCP crystals can promote NLRP3 inflammasome-dependent or -independent inflammation (239). Although some reports challenge this assumption, like one performed in a mouse meniscectomy OA model, overwhelming data support the notion that BCP and CPPD crystals induce synovial inflammation (240). In vivo and in vitro models have shown that these crystals are recognized by innate immune system cells, activating the NLRP3 inflammasome (237, 241). TLRs can recognize CPPD and BCP crystals, and their phagocytosis may induce ATP release, K+ efflux and ROS production (242), suggesting that the purinergic system plays an important role in crystal-mediated inflammasome activation processes in OA.
ATP, its derived metabolites and their recognition by P2X7, P2Y or adenosine receptors are a fine-tuned regulatory mechanism of inflammasome activity, sustaining a chronic inflammatory condition in different diseases. However, their exact role during OA development remains uncertain. Below we address different mechanisms of inflammasome activation mediated by purinergic signaling. This may open a new insight for understanding OA pathogenesis.
Adenosine, Its Receptors and the Inflammasome
Extracellular adenosine activates inflammasome-mediated IL-1β production through A2AR, A2BR, and A3R (243). Adenosine influx through nucleoside transporter ENT2 was converted to ATP by AK, increasing intracellular ATP concentration, which led to ATP secretion from macrophages and consequent upregulation of NLRP3 and IL-1β secretion. On the other hand, ADA reduced IL-1β secretion mediated by nanoparticle-induced NLRP3 inflammasome activation in macrophages (243). Another paper demonstrated that after LPS or ATP stimulation of mouse peritoneal macrophages, adenosine prolonged inflammasome activation, upregulating NLRP3 and IL-1β and enhancing caspase-1 activity. Adenosine stimulated A2AR, activating the downstream cAMP/PKA and CREB/HIF-1α pathway (Figure 4). In this study, adenosine was able to overcome a tolerant unresponsive state acquired by macrophages after previous LPS stimulation, re-inducing high levels of IL-1β secretion (244).
ATP, Its Receptors and the Inflammasome
In normal conditions, excessive extracellular ATP is degraded by ecto-ATPases to adenosine. However, when homeostasis is disrupted, high levels of extracellular ATP released to the milieu from stimulated or necrotic cells acts as a warning signal, being detected by the P2X7 receptor. This results in a massive efflux of K+ across the porous cytoplasmic membrane (245). The P2X7 receptor mediates NLRP3 inflammasome activation in different inflammatory diseases and conditions, and is highly expressed in leukocytes and in other cell types (246–248). Pannexin-1 forms hemichannels across the plasma membrane permeable to ATP, releasing it to the extracellular milieu (249). Pannexin-1 directly interacts with the P2X7 receptor in different cell lines, and this has been associated with NLRP3 inflammasome activation (249–251) (Figure 4).
Riteau et al. demonstrated that several sterile particles induced endogenous ATP release from human macrophages in a P2X7 receptor-dependent way. They proposed that ATP release through Pannexin-/Connexin hemichannel activation could be a consequence of cathepsin B leakage from lysosome disruption. Extracellular ATP could then act in an autocrine/paracrine way, activating the P2X7 receptor and amplifying ATP release and inflammasome activation response (252) (Figure 4).
An alternative mechanism was described by Di et al. through which the P2X7 receptor could mediate K+ efflux in macrophages. The two-pore domain weak inwardly rectifying K+ channel 2 (TWIK2) is involved in NLRP3 inflammasome activation in an LPS-induced lung inflammation mouse model. They proposed that ATP activated P2X7 receptor-favored influx of Na+ and Ca2+ cations, modulating membrane potential and promoting efflux of K+ through the TWIK2 channel (253).
ROS are considered another well-known stimulus through which the NLRP3 inflammasome can be activated. It has been reported that Ca2+ influx mediated by the activated P2X7 receptor can phosphorylate NADPH oxidase complexes in different cell types, including macrophages, stimulating the production of ROS (246) (Figure 4).
Although the P2X7 receptor has been described as the main purinergic receptor involved in NLRP3 inflammasome activation, a recent study in rat urothelial cells showed that treating cells with both P2X7 and P2X4 inhibitors eliminated intracellular caspase-1 activity, suggesting the involvement of the P2X4 receptor in inflammasome activity (254).
Riteau et al. also observed that at low ATP doses, the addition of the metabolites ADP and UTP, which act specifically on P2Y receptors, enhanced particle-mediated IL-1β production (252). Oxidized ATP, an inhibitor of P2X and P2Y receptors, was able to decrease IL-1β production in a dose-dependent manner, but P2X7 receptor selective inhibition did not impair IL-1β production. Interestingly, P2Y1 and P2Y2 receptors, together with A2AR and A2BR expression are upregulated and IL-1β synthesis increased by ATP metabolites and their stable forms. As these purinergic receptors couple to G proteins, in this study the authors suggested that the signaling pathway involved in this process was mediated by phospholipase C (PLC)-IP3 activation or inhibition of AC-cAMP. Therefore, an increase in cytoplasmic Ca2+ or a decrease in cAMP levels were sensed by the NLRP3 inflammasome, increasing its activity (243).
Conclusions and Future Directions
In this review, we have described the relevance of the purinergic system in osteoarthritic cartilage and how MetS components associated with OA influence this system (18, 160). It is well-established that A2AR activity mediated by adenosine is involved in articular cartilage homeostasis maintenance, and is a key modulator during OA (134). Obesity-triggered stimulation of this receptor could contribute to cartilage loss and OA development in obese patients (141).
On the other hand, a more in-depth study of the contribution of nucleotide receptors in OA and MetS is needed to understand better the underlying mechanisms that activate purinergic signaling in the pathogenesis of OA and elucidate confounding data. For example, the P2X7 receptor mediates ATP related inflammation during OA (169, 251), but this receptor exerts an anti-adipogenic effect (148). Therefore, it remains unclear whether the link between obesity and OA might be lie in ATP receptors.
Apart from the activation or inhibition of adenosine/ATP receptors, other components of the purinergic system are involved in OA progression and may be potential modulators of metabolic inflammation. AMPK is an energy sensor involved in inflammation, metabolism and T2D, which is able to adapt cellular metabolism in response to the cellular nutritional and environmental stage (255). AMPK activity is constitutively present in healthy articular chondrocytes and decreases with age and during OA progression (213, 256). AMPK activity has been described to prevent articular cartilage degeneration during mouse aging (257), and is also required to maintain mitochondrial function and prevent OA (213). As we have described earlier in this review, metformin can activate AMPK, resulting in an increase in extracellular adenosine (194). It is known that metformin is beneficial in obese patients with knee OA, but the contribution of AMPK to this effect requires further study.
Another component that needs further analysis is the contribution of ADA to this disease. ADA is an indicator of cellular immunity, and its levels are increased in the following diseases: RA, psoriasis, sarcoidosis, cancer and tuberculosis (258). Synovial ADA fluid measurement, in association with CRP and ESR levels, can distinguish OA from other rheumatic diseases, like RA (186). There is a study in the Indian population that links increased ADA activity with being overweight and obesity (150). This link should be studied in obese patients with OA to understand better the role of ADA as an adenosine level regulator.
As mentioned above, several purinergic receptors are involved in inflammasome activation in different diseases. The study of these mechanisms in the inflammation of OA could be an interesting field of research.
In conclusion, the purinergic system is a key modulator of metainflammation, and its contribution to the pathogenesis of OA opens future therapeutic approaches for the treatment of the disease.
Author Contributions
AM, GH-B, and RL conceptualized, wrote, and revised the manuscript. PG, AL, JM, and IB-Á were primarily responsible for writing, editing, and revising the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Instituto de Salud Carlos III through the Miguel Servet program (CP15/00053), co-funded by Fondo Europeo de Desarrollo Regional (FEDER) and research grants from the Spanish Instituto de Salud Carlos III (PI16/00991 and PI19/00744). PG and AL are recipients of a research fellowship from the Fundación Conchita Rábago. JM and IB-Á are funded by the Instituto de Salud Carlos III through PIE15/00048 and FI19/00060, respectively.
Conflict of Interest
AM has filed a patent for the use of adenosine A2AR agonists to prevent prosthesis loosening (pending) and a separate patent concerning the use of A2AR agonists and agents that increase adenosine levels to promote bone formation/regeneration. RL and GH-B have filed a patent on the use of 6-shogaol in osteoarthritis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Burnstock G. Purinergic receptors. J Theor Biol. (1976) 62:491–503. doi: 10.1016/0022-5193(76)90133-8
3. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. (2014) 509:310–7. doi: 10.1038/nature13085
4. Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, et al. Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature. (2010) 467:863–7. doi: 10.1038/nature09413
5. Eltzschig HK, Eckle T, Mager A, Küper N, Karcher C, Weissmüller T, et al. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. (2006) 99:1100–8. doi: 10.1161/01.RES.0000250174.31269.70
6. Velasquez S, Eugenin EA. Role of Pannexin-1 hemichannels and purinergic receptors in the pathogenesis of human diseases. Front Physiol. (2014) 5:96. doi: 10.3389/fphys.2014.00096
7. Haskó G, Antonioli L, Cronstein BN. Adenosine metabolism, immunity and joint health. Biochem Pharmacol. (2018) 151:307–13. doi: 10.1016/j.bcp.2018.02.002
8. Taruno A, Vingtdeux V, Ohmoto M, Ma Z, Dvoryanchikov G, Li A, et al. CALHM1 ion channel mediates purinergic neurotransmission of sweet, bitter and umami tastes. Nature. (2013) 495:223–6. doi: 10.1038/nature11906
10. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci. (2018) 19:1–16. doi: 10.3390/ijms19041222
11. Wang Y, Martins I, Ma Y, Kepp O, Galluzzi L, Kroemer G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy. (2013) 9:1624–5. doi: 10.4161/auto.25873
12. Fader CM, Aguilera MO, Colombo MI. ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy. (2012) 8:1741–56. doi: 10.4161/auto.21858
13. Knowles AF. The GDA1_CD39 superfamily: NTPDases with diverse functions. Purinergic Signal. (2011) 7:21–45. doi: 10.1007/s11302-010-9214-7
14. Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5′-nucleotidase (CD73). Purinergic Signal. (2006) 2:351–60. doi: 10.1007/s11302-005-5302-5
15. Zimmermann H, Zebisch M, Sträter N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. (2012) 8:437–502. doi: 10.1007/s11302-012-9309-4
16. Zimmermann H. 5'-Nucleotidase: molecular structure and functional aspects. Biochem J. (1992) 285:345–65. doi: 10.1042/bj2850345
17. Moreno E, Canet J, Gracia E, Lluís C, Mallol J, Canela EI, et al. Molecular evidence of adenosine deaminase linking adenosine A2A receptor and CD26 proteins. Front Pharmacol. (2018) 9:106. doi: 10.3389/fphar.2018.00106
18. Corciulo C, Cronstein BN. Signaling of the purinergic system in the joint. Front Pharmacol. (2020) 10:1591. doi: 10.3389/fphar.2019.01591
19. Jacobson KA, Merighi S, Varani K, Borea PA, Baraldi S, Aghazadeh Tabrizi M, et al. A3 adenosine receptors as modulators of inflammation: from medicinal chemistry to therapy. Med Res Rev. (2018) 38:1031–72. doi: 10.1002/med.21456
20. Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors - an update. Pharmacol Rev. (2011) 63:1–34. doi: 10.1124/pr.110.003285
21. Jacobson KA, Delicado EG, Gachet C, Kennedy C, von Kügelgen I, Li B, et al. Update of P2Y receptor pharmacology: IUPHAR review 27. Br J Pharmacol. (2020) 177:2413–33. doi: 10.1111/bph.15005
22. Strazzulla LC, Cronstein BN. Regulation of bone and cartilage by adenosine signaling. Purinergic Signal. (2016) 12:583–93. doi: 10.1007/s11302-016-9527-2
23. Burnstock G. Purine and purinergic receptors. Brain Neurosci Adv. (2018) 2:239821281881749. doi: 10.1177/2398212818817494
24. Burnstock G. Introduction to the special issue on purinergic receptors. Adv Exp Med Biol. (2017) 1051:1–6. doi: 10.1007/5584_2017_12
25. Verkhratsky A, Burnstock G. Biology of purinergic signalling: its ancient evolutionary roots, its omnipresence and its multiple functional significance. BioEssays. (2014) 36:697–705. doi: 10.1002/bies.201400024
26. Johnson VL, Hunter DJ. The epidemiology of osteoarthritis. Best Pract Res Clin Rheumatol. (2014) 28:5–15. doi: 10.1016/j.berh.2014.01.004
27. Vina ER, Kwoh CK. Epidemiology of osteoarthritis: literature update. Curr Opin Rheumatol. (2018) 30:160–7. doi: 10.1097/BOR.0000000000000479
28. Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, et al. Osteoarthritis. Nat Rev Dis Prim. (2016) 2:16072. doi: 10.1038/nrdp.2016.72
29. O'Neill TW, McCabe PS, McBeth J. Update on the epidemiology, risk factors and disease outcomes of osteoarthritis. Best Pract Res Clin Rheumatol. (2018) 32:312–26. doi: 10.1016/j.berh.2018.10.007
30. Malemud CJ. The biological basis of osteoarthritis: state of the evidence HHS public access. Curr Opin Rheumatol. (2015) 27:289–94. doi: 10.1097/BOR.0000000000000162
31. Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthr Cartil. (2013) 21:16–21. doi: 10.1016/j.joca.2012.11.012
32. Rezuş E, Cardoneanu A, Burlui A, Luca A, Codreanu C, Tamba BI, et al. The link between inflammaging and degenerative joint diseases. Int J Mol Sci. (2019) 20:614. doi: 10.3390/ijms20030614
33. van Namen M, Prendergast L, Peiris C. Supervised lifestyle intervention for people with metabolic syndrome improves outcomes and reduces individual risk factors of metabolic syndrome: a systematic review and meta-analysis. Metabolism. (2019) 101:153988. doi: 10.1016/j.metabol.2019.153988
34. Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, Nissen M, et al. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. (2001) 24:683–9. doi: 10.2337/diacare.24.4.683
35. Niu J, Clancy M, Aliabadi P, Vasan R, Felson DT. Metabolic syndrome, its components, and knee osteoarthritis: the framingham osteoarthritis study. Arthritis Rheumatol. (2017) 69:1194–203. doi: 10.1002/art.40087
36. Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. (2011) 29:415–45. doi: 10.1146/annurev-immunol-031210-101322
37. Wang X, Hunter D, Xu J, Ding C. Metabolic triggered inflammation in osteoarthritis. Osteoarthr Cartil. (2015) 23:22–30. doi: 10.1016/j.joca.2014.10.002
38. Gao Y-H, Zhao C-W, Liu B, Dong N, Ding L, Li Y-R, et al. An update on the association between metabolic syndrome and osteoarthritis and on the potential role of leptin in osteoarthritis. Cytokine. (2020) 129:155043. doi: 10.1016/j.cyto.2020.155043
39. Courties A, Sellam J, Berenbaum F. Metabolic syndrome-associated osteoarthritis. Curr Opin Rheumatol. (2017) 29:214–22. doi: 10.1097/BOR.0000000000000373
40. Xie C, Chen Q. Adipokines: new therapeutic target for osteoarthritis? Curr Rheumatol Rep. (2019) 21:71. doi: 10.1007/s11926-019-0868-z
41. Herrero-Beaumont G, Pérez-Baos S, Sánchez-Pernaute O, Roman-Blas JA, Lamuedra A, Largo R. Targeting chronic innate inflammatory pathways, the main road to prevention of osteoarthritis progression. Biochem Pharmacol. (2019) 165:24–32. doi: 10.1016/j.bcp.2019.02.030
42. Wluka AE, Lombard CB, Cicuttini FM. Tackling obesity in knee osteoarthritis. Nat Rev Rheumatol. (2013) 9:225–35. doi: 10.1038/nrrheum.2012.224
43. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
44. Sowers MR, Karvonen-Gutierrez CA. The evolving role of obesity in knee osteoarthritis. Curr Opin Rheumatol. (2010) 22:533–7. doi: 10.1097/BOR.0b013e32833b4682
45. Robinson WH, Lepus CM, Wang Q, Raghu H, Mao R, Lindstrom TM, et al. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat Rev Rheumatol. (2016) 12:580–92. doi: 10.1038/nrrheum.2016.136
46. Dumond H, Presle N, Terlain B, Mainard D, Loeuille D, Netter P, et al. Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum. (2003) 48:3118–29. doi: 10.1002/art.11303
47. Yan M, Zhang J, Yang H, Sun Y. The role of leptin in osteoarthritis. Medicine. (2018) 97:e0257. doi: 10.1097/MD.0000000000010257
48. Vuolteenaho K, Koskinen A, Kukkonen M, Nieminen R, Päivärinta U, Moilanen T, et al. Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage—mediator role of no in leptin-induced PGE2, IL-6, and IL-8 production. Mediat Inflamm. (2009) 2009:1–10. doi: 10.1155/2009/345838
49. Koskinen A, Vuolteenaho K, Nieminen R, Moilanen T, Moilanen E. Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from oa patients. Clin Exp Rheumatol. (2011) 29:57–64. doi: 10.1016/S1063-4584(10)60038-6
50. Lee JH, Ort T, Ma K, Picha K, Carton J, Marsters PA, et al. Resistin is elevated following traumatic joint injury and causes matrix degradation and release of inflammatory cytokines from articular cartilage in vitro. Osteoarthr Cartil. (2009) 17:613–20. doi: 10.1016/j.joca.2008.08.007
51. Thommesen L, Stunes AK, Monjo M, Grøsvik K, Tamburstuen M V., Kjøbli E, et al. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem. (2006) 99:824–34. doi: 10.1002/jcb.20915
52. Patel L, Buckels AC, Kinghorn IJ, Murdock PR, Holbrook JD, Plumpton C, et al. Resistin is expressed in human macrophages and directly regulated by PPARγ activators. Biochem Biophys Res Commun. (2003) 300:472–6. doi: 10.1016/S0006-291X(02)02841-3
53. Filková M, Haluzík M, Gay S, Šenolt L. The role of resistin as a regulator of inflammation: implications for various human pathologies. Clin Immunol. (2009) 133:157–70. doi: 10.1016/j.clim.2009.07.013
54. Mu H, Ohashi R, Yan S, Chai H, Yang H, Lin P, et al. Adipokine resistin promotes in vitro angiogenesis of human endothelial cells. Cardiovasc Res. (2006) 70:146–57. doi: 10.1016/j.cardiores.2006.01.015
55. Chen T-H, Chen L, Hsieh M-S, Chang C-P, Chou D-T, Tsai S-H. Evidence for a protective role for adiponectin in osteoarthritis. Biochim Biophys Acta Mol Basis Dis. (2006) 1762:711–8. doi: 10.1016/j.bbadis.2006.06.008
56. Kroon FPB, Veenbrink AI, de Mutsert R, Visser AW, van Dijk KW, le Cessie S, et al. The role of leptin and adiponectin as mediators in the relationship between adiposity and hand and knee osteoarthritis. Osteoarthr Cartil. (2019) 27:1761–7. doi: 10.1016/j.joca.2019.08.003
57. Sun AR, Panchal SK, Friis T, Sekar S, Crawford R, Brown L, et al. Obesity-associated metabolic syndrome spontaneously induces infiltration of pro-inflammatory macrophage in synovium and promotes osteoarthritis. Gualillo O, editor. PLoS ONE. (2017) 12:e0183693. doi: 10.1371/journal.pone.0183693
58. Larrañaga-Vera A, Lamuedra A, Pérez-Baos S, Prieto-Potin I, Peña L, Herrero-Beaumont G, et al. Increased synovial lipodystrophy induced by high fat diet aggravates synovitis in experimental osteoarthritis. Arthritis Res Ther. (2017) 19:264. doi: 10.1186/s13075-017-1473-z
59. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. (2011) 11:98–107. doi: 10.1038/nri2925
60. Ramage L, Nuki G, Salter DM. Signalling cascades in mechanotransduction: cell-matrix interactions and mechanical loading. Scand J Med Sci Sports. (2009) 19:457–69. doi: 10.1111/j.1600-0838.2009.00912.x
61. Mohtai M, Gupta M, Donlon B, Ellison B, Cooke J, Gibbons G, et al. Interleukin-6 in osteoarthritic chondrocytes and effects. J Orthop Res. (1996) 14:67–73. doi: 10.1002/jor.1100140112
62. Millward-Sadler SJ, Wright MO, Davies LW, Nuki G, Salter DM. Mechanotransduction via integrins and interleukinâ results in altered aggrecan and matrix metalloproteinase 3 gene expression in normal, but not osteoarthritic, human articular chondrocytes. Arthritis Rheum. (2000) 43:2091–9 doi: 10.1002/1529-0131(200009)43:9<2091::AID-ANR21>3.0.CO;2-C
63. Salter DM, Millward-Sadler SJ, Nuki G, Wright MO. Differential Responses of Chondrocytes From Normal and Osteoarthritic Human Articular Cartilage to Mechanical Stimulation. Amsterdam: IOS Press. (2001).
64. Goldring M, Otero M, Plumb D, Dragomir C, Favero M, EI Hachem K, et al. Roles of inflammatory and anabolic cytokines in cartilage metabolism: signals and multiple effectors converge upon MMP-13 regulation in osteoarthritis. Eur Cells Mater. (2011) 21:202–20. doi: 10.22203/eCM.v021a16
65. Egloff C, Hügle T, Valderrabano V. Biomechanics and pathomechanisms of osteoarthritis. Swiss Med Wkly. (2012) 142:1–14. doi: 10.4414/smw.2012.13583
66. World Health Organization. Definition, Diagnosis and Classification of Diabetes Mellitus and Its Complications. Part 1: Diagnosis and Classification of Diabetes mellitus. Geneva: World Health Organization; Department of Noncommunicable Disease Surveillance. (1999).
67. Neumann J, Hofmann FC, Heilmeier U, Ashmeik W, Tang K, Gersing AS, et al. Type 2 diabetes patients have accelerated cartilage matrix degeneration compared to diabetes free controls: data from the osteoarthritis initiative. Osteoarthr Cartil. (2018) 26:751–61. doi: 10.1016/j.joca.2018.03.010
68. Louati K, Vidal C, Berenbaum F, Sellam J. Association between diabetes mellitus and osteoarthritis: systematic literature review and meta-analysis. RMD Open. (2015) 1:e000077. doi: 10.1136/rmdopen-2015-000077
69. Wang H, Cheng Y, Shao D, Chen J, Sang Y, Gui T, et al. Metabolic syndrome increases the risk for knee osteoarthritis: a meta-analysis. Evidence-Based Comple Altern Med. (2016) 2016:1–7. doi: 10.1155/2016/7242478
70. Courties A, Berenbaum F, Sellam J. The phenotypic approach to osteoarthritis: a look at metabolic syndrome-associated osteoarthritis. Jt Bone Spine. (2019) 86:725–30. doi: 10.1016/j.jbspin.2018.12.005
71. Berenbaum F. Diabetes-induced osteoarthritis: from a new paradigm to a new phenotype. Ann Rheum Dis. (2011) 70:1354–6. doi: 10.1136/ard.2010.146399
72. Laiguillon M-C, Courties A, Houard X, Auclair M, Sautet A, Capeau J, et al. Characterization of diabetic osteoarthritic cartilage and role of high glucose environment on chondrocyte activation: toward pathophysiological delineation of diabetes mellitus-related osteoarthritis. Osteoarthr Cartil. (2015) 23:1513–22. doi: 10.1016/j.joca.2015.04.026
73. Schett G, Kleyer A, Perricone C, Sahinbegovic E, Iagnocco A, Zwerina J, et al. Diabetes is an independent predictor for severe osteoarthritis: results from a longitudinal cohort study. Diabetes Care. (2013) 36:403–9. doi: 10.2337/dc12-0924
74. Arellano Perez Vertti RD, Aguilar Muñiz LS, Morán Martínez J, González Galarza FF, Arguello Astorga R. Cartilage oligomeric matrix protein levels in type 2 diabetes associated with primary knee osteoarthritis patients. Genet Test Mol Biomark. (2019) 23:16–22. doi: 10.1089/gtmb.2018.0184
75. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. (2012) 18:363–74. doi: 10.1038/nm.2627
76. Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. (2014) 105:141–50. doi: 10.1016/j.diabres.2014.04.006
77. Largo R, Alvarez-Soria M., Diez-Ortego I, Calvo E, Sánchez-Pernaute O, Egido J, et al. Glucosamine inhibits IL-1β-induced NFκB activation in human osteoarthritic chondrocytes. Osteoarthr Cartil. (2003) 11:290–8. doi: 10.1016/S1063-4584(03)00028-1
78. Gui T, Lin Y, Huan S, Li Y, Wang B, Yang J, et al. Elevated expression of ICAM-1 in synovium is associated with early inflammatory response for cartilage degeneration in type 2 diabetes mellitus. J Cell Biochem. (2019) 120:13177–86. doi: 10.1002/jcb.28592
79. Fulop T, Witkowski JM, Olivieri F, Larbi A. The integration of inflammaging in age-related diseases. Semin Immunol. (2018) 40:17–35. doi: 10.1016/j.smim.2018.09.003
80. Bektas A, Schurman SH, Sen R, Ferrucci L. Aging, inflammation and the environment. Exp Gerontol. (2018) 105:10–8. doi: 10.1016/j.exger.2017.12.015
81. Sellam J, Berenbaum F. Is osteoarthritis a metabolic disease? Jt Bone Spine. (2013) 80:568–73. doi: 10.1016/j.jbspin.2013.09.007
82. Frasca D, Blomberg BB, Paganelli R. Aging, obesity, and inflammatory age-related diseases. Front Immunol. (2017) 8:1745. doi: 10.3389/fimmu.2017.01745
83. Millerand M, Berenbaum F, Jacques C. Danger signals and inflammaging in osteoarthritis. Clin Exp Rheumatol. (2019) 37:48–56. Available online at: https://www.clinexprheumatol.org/article.asp?a=14559
84. Martin JA, Brown TD, Heiner AD, Buckwalter JA. Chondrocyte senescence, joint loading and osteoarthritis. Clin Orthop Relat Res. (2004) 427:96–103. doi: 10.1097/01.blo.0000143818.74887.b1
85. Gratal P, Mediero A, Sánchez-Pernaute O, Prieto-Potin I, Lamuedra A, Herrero-Beaumont G, et al. Chondrocyte enlargement is a marker of osteoarthritis severity. Osteoarthr Cartil. (2019) 27:1229–34. doi: 10.1016/j.joca.2019.04.013
86. Tardio L, Andrés-Bergós J, Zachara NE, Larrañaga-Vera A, Rodriguez-Villar C, Herrero-Beaumont G, et al. O-linked N-acetylglucosamine (O-GlcNAc) protein modification is increased in the cartilage of patients with knee osteoarthritis. Osteoarthr Cartil. (2014) 22:259–63. doi: 10.1016/j.joca.2013.12.001
87. Caramés B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. (2012) 71:575–81. doi: 10.1136/annrheumdis-2011-200557
88. Zhi LQ, Yao SX, Liu HL, Li M, Duan N, Ma JB. Hydroxytyrosol inhibits the inflammatory response of osteoarthritis chondrocytes via SIRT6-mediated autophagy. Mol Med Rep. (2018) 17:4035–42. doi: 10.3892/mmr.2017.8353
89. Zhong G, Long H, Ma S, Shunhan Y, Li J, Yao J. miRNA-335-5p relieves chondrocyte inflammation by activating autophagy in osteoarthritis. Life Sci. (2019) 226:164–72. doi: 10.1016/j.lfs.2019.03.071
90. Caramés B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. (2010) 62:791–801. doi: 10.1002/art.27305
91. Guilak F. Biochemical factors in osteoarthritis. Best Pract Res Clin Rheumatol. (2011) 25:815–23. doi: 10.1016/j.berh.2011.11.013
92. DeFrate L, Kim-Wang S, Englander Z, McNulty A. Osteoarthritis year in review 2018: mechanics. Osteoarthr Cartil. (2019) 27:392–400. doi: 10.1016/j.joca.2018.12.011
93. Gómez R, Villalvilla A, Largo R, Gualillo O, Herrero-Beaumont G. TLR4 signalling in osteoarthritis-finding targets for candidate DMOADs. Nat Rev Rheumatol. (2015) 11:159–70. doi: 10.1038/nrrheum.2014.209
94. Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. (2014) 5:1–7. doi: 10.1038/cddis.2014.70
95. Haskó G, Cronstein B. Regulation of inflammation by adenosine. Front Immunol. (2013) 4:1–8. doi: 10.3389/fimmu.2013.00085
96. Antonioli L, Csóka B, Fornai M, Colucci R, Kókai E, Blandizzi C, et al. Adenosine and inflammation: What's new on the horizon? Drug Discov Today. (2014) 19:1051–68. doi: 10.1016/j.drudis.2014.02.010
97. Eltzschig HK, Sitkovsky M V., Robson SC. Purinergic signaling during inflammation. N Engl J Med. (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
98. Cronstein BN, Sitkovsky M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat Rev Rheumatol. (2017) 13:41–51. doi: 10.1038/nrrheum.2016.178
99. Haskó G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. (2004). 25:33–9. doi: 10.1016/j.it.2003.11.003
100. Trautmann A. Extracellular ATP in the immune system: more than just a “danger signal.” Sci Signal. (2009) 2:pe6. doi: 10.1126/scisignal.256pe6
101. Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun. (2019) 10:1916. doi: 10.1038/s41467-019-09801-x
102. Wakai A, Wang JH, Winter DC, Street JT, O'Sullivan RG, Redmond HP. Adenosine inhibits neutrophil vascular endothelial growth factor release and transendothelial migration via A2B receptor activation. Shock. (2001) 15:297–301. doi: 10.1097/00024382-200115040-00008
103. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Pub Group. (2016) 14:177–92. doi: 10.1038/nri.2016.4
104. Gorini S, Gatta L, Pontecorvo L, Vitiello L, la Sala A. Regulation of innate immunity by extracellular nucleotides. Am J Blood Res. (2013) 3:14–28. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3555188/pdf/ajbr0003-0014.pdf
105. Antonioli L, Fornai M, Blandizzi C, Pacher P, Haskó G. Adenosine signaling and the immune system: when a lot could be too much. Immunol Lett. (2019) 205:9–15. doi: 10.1016/j.imlet.2018.04.006
106. Kumar V, Sharma A. Adenosine: an endogenous modulator of innate immune system with therapeutic potential. Euro J Pharmacol. (2009) 15:7–15. doi: 10.1016/j.ejphar.2009.05.005
107. Csóka B, Németh ZH, Virág L, Gergely P, Leibovich SJ, Pacher P, et al. A2A adenosine receptors and C/EBPβ are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. (2007) 110:2685–95. doi: 10.1182/blood-2007-01-065870
108. Bowser JL, Lee JW, Yuan X, Eltzschig HK. The hypoxia-adenosine link during inflammation. J Appl Physiol. (2017) 123:1303–20. doi: 10.1152/japplphysiol.00101.2017
109. Linden J, Koch-Nolte F, Dahl G. Purine release, metabolism, and signaling in the inflammatory response. Annu Rev Immunol. (2019) 37:325–47. doi: 10.1146/annurev-immunol-051116-052406
110. Di Virgilio F, Sarti AC, Grassi F. Modulation of innate and adaptive immunity by P2X ion channels. Curr Opin Immunol. (2018) 52:51–9. doi: 10.1016/j.coi.2018.03.026
111. Ritchie PK, Spangelo BL, Krzymowski DK, Rossiter TB, Kurth E, Judd AM. Adenosine increases interleukin 6 release and decreases tumour necrosis factor release from rat adrenal zona glomerulosa cells, ovarian cells, anterior pituitary cells and peritoneal macrophages. Cytokine. (1997) 9:187–98. doi: 10.1006/cyto.1996.0153
112. Link A, Kino T, Worth JA, McGuire JL, Crane ML, Chrousos GP, et al. Ligand-activation of the adenosine A2a receptors inhibits IL-12 production by human monocytes. J Immunol. (2000) 164:436–42. doi: 10.4049/jimmunol.164.1.436
113. Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. (2005) 175:3463–8. doi: 10.4049/jimmunol.175.6.3463
114. Feng WG, Wang YB, Zhang JS, Wang XY, Li CL, Chang ZL. cAMP elevators inhibit LPS-induced IL-12 p40 expression by interfering with phosphorylation of p38 MAPK in murine peritoneal macrophages. Cell Res. (2002) 12:331–7. doi: 10.1038/sj.cr.7290135
115. Shames BD, McIntyre RC, Bensard DD, Pulido EJ, Selzman CH, Reznikov LL, et al. Suppression of tumor necrosis factor α production by cAMP in human monocytes: dissociation with mRNA level and independent of interleukin-10. J Surg Res. (2001) 99:187–93. doi: 10.1006/jsre.2001.6178
116. Karsdal MA, Sumer EU, Wulf H, Madsen SH, Christiansen C, Fosang AJ, et al. Induction of increased cAMP levels in articular chondrocytes blocks matrix metalloproteinase–mediated cartilage degradation, but not aggrecanase-mediated cartilage degradation. Arthritis Rheum. (2007) 56:1549–58. doi: 10.1002/art.22599
117. Campo GM, Avenoso A, D'Ascola A, Prestipino V, Scuruchi M, Nastasi G, et al. The stimulation of adenosine 2A receptor reduces inflammatory response in mouse articular chondrocytes treated with hyaluronan oligosaccharides. Matrix Biol. (2012) 31:338–51. doi: 10.1016/j.matbio.2012.07.001
118. Campo GM, Avenoso A, D'Ascola A, Prestipino V, Scuruchi M, Nastasi G, et al. Protein kinase a mediated anti-inflammatory effects exerted by adenosine treatment in mouse chondrocytes stimulated with IL-1β. BioFactors. (2012) 38:429–39. doi: 10.1002/biof.1040
119. Faas MM, Sáez T, de Vos P. Extracellular ATP and adenosine: the yin and yang in immune responses? Mol Aspects Med. (2017) 55:9–19. doi: 10.1016/j.mam.2017.01.002
120. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and inflammation. Immunity. (2017) 47:15–31. doi: 10.1016/j.immuni.2017.06.020
121. Di Virgilio F, Boeynaems J-M, Robson SC. Extracellular nucleotides as negative modulators of immunity. Curr Opin Pharmacol. (2009) 9:507–13. doi: 10.1016/j.coph.2009.06.021
122. Boeynaems J-M, Communi D. Modulation of inflammation by extracellular nucleotides. J Invest Dermatol. (2006) 126:943–4. doi: 10.1038/sj.jid.5700233
123. Gaikwad S, Patel D, Agrawal-Rajput R. CD40 negatively regulates ATP-TLR4-activated inflammasome in microglia. Cell Mol Neurobiol. (2017) 37:351–9. doi: 10.1007/s10571-016-0358-z
124. Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. (2012) 51:249–57. doi: 10.1016/j.bone.2012.02.012
125. Orriss IR. The role of purinergic signalling in the musculoskeletal system. Auton Neurosci Basic Clin. (2015) 191:124–34. doi: 10.1016/j.autneu.2015.04.013
126. Koolpe M, Pearson D, Benton HP. Expression of both P1 and P2 purine receptor genes by human articular chondrocytes and profile of ligand-mediated prostaglandin E2 release. Arthritis Rheum. (1999) 42:258–67. doi: 10.1002/1529-0131(199902)42:2<258::AID-ANR7>3.0.CO;2-O
127. Varani K, De Mattei M, Vincenzi F, Gessi S, Merighi S, Pellati A, et al. Characterization of adenosine receptors in bovine chondrocytes and fibroblast-like synoviocytes exposed to low frequency low energy pulsed electromagnetic fields. Osteoarthr Cartil. (2008) 16:292–304. doi: 10.1016/j.joca.2007.07.004
128. Pinto-Cardoso R, Pereira-Costa F, Pedro Faria J, Bandarrinha P, Bessa-Andrês C, Correia-de-Sá P, et al. Adenosinergic signalling in chondrogenesis and cartilage homeostasis: friend or foe? Biochem Pharmacol. (2020) 174:113784. doi: 10.1016/j.bcp.2019.113784
129. Bekisz JM, Lopez CD, Corciulo C, Mediero A, Coelho PG, Witek L, et al. The role of adenosine receptor activation in attenuating cartilaginous inflammation. Inflammation. (2018) 41:1135–41. doi: 10.1007/s10753-018-0781-z
130. Corciulo C, Lendhey M, Wilder T, Schoen H, Cornelissen AS, Chang G, et al. Endogenous adenosine maintains cartilage homeostasis and exogenous adenosine inhibits osteoarthritis progression. Nat Commun. (2017) 8:15019. doi: 10.1038/ncomms15019
131. Massari L, Benazzo F, Falez F, Perugia D, Pietrogrande L, Setti S, et al. Biophysical stimulation of bone and cartilage: state of the art and future perspectives. Int Orthop. (2019) 43:539–51. doi: 10.1007/s00264-018-4274-3
132. Iwasa K, Reddi AH. Pulsed electromagnetic fields and tissue engineering of the joints. Tissue Eng - Part B Rev. (2018) 24:144–54. doi: 10.1089/ten.TEB.2017.0294
133. Liu X, Corciulo C, Arabagian S, Ulman A, Cronstein BN. Adenosine-functionalized biodegradable PLA-b-PEG nanoparticles ameliorate osteoarthritis in rats. Sci Rep. (2019) 9:7430. doi: 10.1038/s41598-019-43834-y
134. Tesch AM, MacDonald MH, Kollias-Baker C, Benton HP. Endogenously produced adenosine regulates articular cartilage matrix homeostasis: enzymatic depletion of adenosine stimulates matrix degradation. Osteoarthr Cartil. (2004) 12:349–59. doi: 10.1016/j.joca.2004.01.002
135. Campo GM, Avenoso A, D'Ascola A, Scuruchi M, Prestipino V, Nastasi G, et al. Adenosine A2A receptor activation and hyaluronan fragment inhibition reduce inflammation in mouse articular chondrocytes stimulated with interleukin-1β. FEBS J. (2012) 279:2120–33. doi: 10.1111/j.1742-4658.2012.08598.x
136. Tesch AM, MacDonald MH, Kollias-Baker C, Benton HP. Effects of an adenosine kinase inhibitor and an adenosine deaminase inhibitor on accumulation of extracellular adenosine by equine articular chondrocytes. Am J Vet Res. (2002) 63:1512–9. doi: 10.2460/ajvr.2002.63.1512
137. Lotz M. The role of nitric oxide in articular cartilage damage. Rheum Dis Clin North Am. (1999) 25:269–82. doi: 10.1016/S0889-857X(05)70067-3
138. Nc W, Sf H, Rm R. The role of extra-cellular adenosine receptors in modulating pain progression during osteoarthritis (OA): a systematic review of the literature. Rheumatol Orthop Med. (2018) 3:1–5. doi: 10.15761/ROM.1000157
139. Johansson SM, Lindgren E, Yang J-N, Herling AW, Fredholm BB. Adenosine A1 receptors regulate lipolysis and lipogenesis in mouse adipose tissue — Interactions with insulin. Eur J Pharmacol. (2008) 597:92–101. doi: 10.1016/j.ejphar.2008.08.022
140. Szkudelski T, Szkudelska K, Nogowski L. Effects of adenosine A1 receptor antagonism on lipogenesis and lipolysis in isolated rat adipocytes. Physiol Res. (2009) 58:863–71. Available online at: http://www.biomed.cas.cz/physiolres/pdf/58/58_863.pdf
141. Gharibi B, Abraham AA, Ham J, Evans BAJ. Contrasting effects of A1 and A2b adenosine receptors on adipogenesis. Int J Obes. (2012) 36:397–406. doi: 10.1038/ijo.2011.129
142. Cheng JT, Liu IM, Chi TC, Shinozuka K, Lu FH, Wu TJ, et al. Role of adenosine in insulin-stimulated release of leptin from isolated white adipocytes of Wistar rats. Diabetes. (2000) 49:20–4. doi: 10.2337/diabetes.49.1.20
143. Rice AM, Fain JN, Rivkees SA. A1 adenosine receptor activation increases adipocyte leptin secretion 1. Endocrinology. (2000) 141:1442–5. doi: 10.1210/endo.141.4.7423
144. Dong Q, Ginsberg HN, Erlanger BF. Overexpression of the A 1 adenosine receptor in adipose tissue protects mice from obesity-related insulin resistance. Diabetes Obes Metab. (2001) 3:360–6. doi: 10.1046/j.1463-1326.2001.00158.x
145. Faulhaber-Walter R, Jou W, Mizel D, Li L, Zhang J, Kim SM, et al. Impaired glucose tolerance in the absence of adenosine A1 receptor signaling. Diabetes. (2011) 60:2578–87. doi: 10.2337/db11-0058
146. Yang T, Gao X, Sandberg M, Zollbrecht C, Zhang X-M, Hezel M, et al. Abrogation of adenosine A1 receptor signalling improves metabolic regulation in mice by modulating oxidative stress and inflammatory responses. Diabetologia. (2015) 58:1610–20. doi: 10.1007/s00125-015-3570-3
147. Töpfer M, Burbiel CE, Müller CE, Knittel J, Verspohl EJ. Modulation of insulin release by adenosine A 1 receptor agonists and antagonists in INS-1 cells: the possible contribution of 86 Rb + efflux and 45 Ca 2+ uptake. Cell Biochem Funct. (2008) 26:833–43. doi: 10.1002/cbf.1514
148. Tozzi M, Novak I. Purinergic receptors in adipose tissue as potential targets in metabolic disorders. Front Pharmacol. (2017) 8:878. doi: 10.3389/fphar.2017.00878
149. Antonioli L, Blandizzi C, Csóka B, Pacher P, Haskó G. Adenosine signalling in diabetes mellitus—pathophysiology and therapeutic considerations. Nat Rev Endocrinol. (2015) 11:228–41. doi: 10.1038/nrendo.2015.10
150. Jadhav AA, Jain A. Elevated adenosine deaminase activity in overweightand obese Indian subjects. Arch Physiol Biochem. (2012) 118:1–5. doi: 10.3109/13813455.2011.603341
151. Shkhyan R, Lee S, Gullo F, Li L, Peleli M, Carlstrom M, et al. Genetic ablation of adenosine receptor A3 results in articular cartilage degeneration. J Mol Med. (2018) 96:1049–60. doi: 10.1007/s00109-018-1680-3
152. Johnston-Cox H, Koupenova M, Yang D, Corkey B, Gokce N, Farb MG, et al. The A2b adenosine receptor modulates glucose homeostasis and obesity. Eltzschig HK, editor. PLoS ONE. (2012) 7:e40584. doi: 10.1371/journal.pone.0040584
153. Cieślak M, Cieślak M. Role of purinergic signalling and proinflammatory cytokines in diabetes. Clin Diabetol. (2017) 6:90–100. doi: 10.5603/DK.2017.0015
154. Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy S V., Dikov MM, Blackburn MR, et al. Effect of A 2B adenosine receptor gene ablation on proinflammatory adenosine signaling in mast cells. J Immunol. (2008) 180:7212–20. doi: 10.4049/jimmunol.180.11.7212
155. Bar-Yehuda S, Rath-Wolfson L, Del Valle L, Ochaion A, Cohen S, Patoka R, et al. Induction of an antiinflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum. (2009) 60:3061–71. doi: 10.1002/art.24817
156. Varani K, De Mattei M, Vincenzi F, Tosi A, Gessi S, Merighi S, et al. Pharmacological characterization of P2X1 and P2X3 purinergic receptors in bovine chondrocytes. Osteoarthr Cartil. (2008) 16:1421–9. doi: 10.1016/j.joca.2008.03.016
157. Ruiz-Rodríguez VM, Cortes-García JD, de Jesús Briones-Espinoza M, Rodríguez-Varela E, Vega-Cárdenas M, Gómez-Otero A, et al. P2X4 receptor as a modulator in the function of P2X receptor in CD4+ T cells from peripheral blood and adipose tissue. Mol Immunol. (2019) 112:369–77. doi: 10.1016/j.molimm.2019.06.009
158. Jacques-Silva MC, Correa-Medina M, Cabrera O, Rodriguez-Diaz R, Makeeva N, Fachado A, et al. ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic cell. Proc Natl Acad Sci USA. (2010) 107:6465–70. doi: 10.1073/pnas.0908935107
159. Thaning P, Bune LT, Hellsten Y, Pilegaard H, Saltin B, Rosenmeier JB. Attenuated purinergic receptor function in patients with type 2 diabetes. Diabetes. (2010) 59:182–9. doi: 10.2337/db09-1068
160. Burnstock G, Gentile D. The involvement of purinergic signalling in obesity. Purinergic Signal. (2018) 14:97–108. doi: 10.1007/s11302-018-9605-8
161. Mancini JE, Ortiz G, Potilinstki C, Salica JP, Lopez ES, Croxatto JO, et al. Possible neuroprotective role of P2X2 in the retina of diabetic rats. Diabetol Metab Syndr. (2018) 10:31. doi: 10.1186/s13098-018-0332-7
162. Teixeira JM, Bobinski F, Parada CA, Sluka KA, Tambeli CH. P2X3 and P2X2/3 receptors play a crucial role in articular hyperalgesia development through inflammatory mechanisms in the knee joint experimental synovitis. Mol Neurobiol. (2017) 54:6174–86. doi: 10.1007/s12035-016-0146-2
163. Johnson K, Terkeltaub R. Upregulated ank expression in osteoarthritis can promote both chondrocyte MMP-13 expression and calcification via chondrocyte extracellular PPi excess. Osteoarthr Cartil. (2004) 12:321–35. doi: 10.1016/j.joca.2003.12.004
164. Rosenthal AK, Gohr CM, Mitton-Fitzgerald E, Lutz MK, Dubyak GR, Ryan LM. The progressive ankylosis gene product ANK regulates extracellular ATP levels in primary articular chondrocytes. Arthritis Res Ther. (2013) 15:R154. doi: 10.1186/ar4337
165. Burnstock G, Novak I. Purinergic signalling and diabetes. Purinergic Signal. (2013) 9:307–24. doi: 10.1007/s11302-013-9359-2
166. Kim H, Kajikawa T, Walsh MC, Takegahara N, Jeong YH, Hajishengallis G, et al. The purinergic receptor P2X5 contributes to bone loss in experimental periodontitis. BMB Rep. (2018) 51:468–73. doi: 10.5483/BMBRep.2018.51.9.126
167. Ussar S, Lee KY, Dankel SN, Boucher J, Haering M-F, Kleinridders A, et al. ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Sci Transl Med. (2014) 6:247ra103. doi: 10.1126/scitranslmed.3008490
168. Tanigawa H, Toyoda F, Kumagai K, Okumura N, Maeda T, Matsuura H, et al. P2X7 ionotropic receptor is functionally expressed in rabbit articular chondrocytes and mediates extracellular ATP cytotoxicity. Purinergic Signal. (2018) 14:245–58. doi: 10.1007/s11302-018-9611-x
169. Hu H, Yang B, Li Y, Zhang S, Li Z. Blocking of the P2X7 receptor inhibits the activation of the MMP-13 and NF-κB pathways in the cartilage tissue of rats with osteoarthritis. Int J Mol Med. (2016) 38:1922–32. doi: 10.3892/ijmm.2016.2770
170. Zippel N, Limbach CA, Ratajski N, Urban C, Luparello C, Pansky A, et al. Purinergic receptors influence the differentiation of human mesenchymal stem cells. Stem Cells Dev. (2012) 21:884–900. doi: 10.1089/scd.2010.0576
171. Beaucage KL, Xiao A, Pollmann SI, Grol MW, Beach RJ, Holdsworth DW, et al. Loss of P2X7 nucleotide receptor function leads to abnormal fat distribution in mice. Purinergic Signal. (2014) 10:291–304. doi: 10.1007/s11302-013-9388-x
172. Glas R, Sauter NS, Schulthess FT, Shu L, Oberholzer J, Maedler K. Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia. (2009) 52:1579–88. doi: 10.1007/s00125-009-1349-0
173. Burnstock G, Arnett TR, Orriss IR. Purinergic signalling in the musculoskeletal system. Purinergic Signal. (2013) 9:541–72. doi: 10.1007/s11302-013-9381-4
174. Ciciarello M, Zini R, Rossi L, Salvestrini V, Ferrari D, Manfredini R, et al. Extracellular purines promote the differentiation of human bone marrow-derived mesenchymal stem cells to the osteogenic and adipogenic lineages. Stem Cells Dev. (2013) 22:1097–111. doi: 10.1089/scd.2012.0432
175. Laplante M-A, Monassier L, Freund M, Bousquet P, Gachet C. The purinergic P2Y1 receptor supports leptin secretion in adipose tissue. Endocrinology. (2010) 151:2060–70. doi: 10.1210/en.2009-1134
176. Millward-Sadler SJ, Wright MO, Flatman PW, Salter DM. ATP in the mechanotransduction pathway of normal human chondrocytes. Biorheology. (2004) 41:567–75. Available online at: https://content.iospress.com/articles/biorheology/bir324
177. Albrecht P, Merz J, Stachon P, Bode C, Zirlik A. The deficiency of the purinergic P2Y2 receptor protects hyperlipidemic mice from suffering metabolic syndrome. Basic Res Cardiol. (2018) 113:45. doi: 10.1007/s00395-018-0702-1
178. Lemaire A, Vanorlé M, Horckmans M, di Pietrantonio L, Clouet S, Robaye B, et al. Mouse P2Y 4 nucleotide receptor is a negative regulator of cardiac adipose-derived stem cell differentiation and cardiac fat formation. Stem Cells Dev. (2017) 26:363–73. doi: 10.1089/scd.2016.0166
179. Coutinho-Silva R, Parsons M, Robson T, Lincoln J, Burnstock G. P2X and P2Y purinoceptor expression in pancreas from streptozotocin-diabetic rats. Mol Cell Endocrinol. (2003) 204:141–54. doi: 10.1016/S0303-7207(03)00003-0
180. Steculorum SM, Timper K, Engström Ruud L, Evers N, Paeger L, Bremser S, et al. Inhibition of P2Y6 signaling in AgRP neurons reduces food intake and improves systemic insulin sensitivity in obesity. Cell Rep. (2017) 18:1587–97. doi: 10.1016/j.celrep.2017.01.047
181. Ohtani M, Suzuki J-I, Jacobson KA, Oka T. Evidence for the possible involvement of the P2Y6 receptor in Ca2+ mobilization and insulin secretion in mouse pancreatic islets. Purinergic Signal. (2008) 4:365–75. doi: 10.1007/s11302-008-9122-2
182. Prada MP, Syed AU, Buonarati OR, Reddy GR, Nystoriak MA, Ghosh D, et al. A Gs-coupled purinergic receptor boosts Ca2+ influx and vascular contractility during diabetic hyperglycemia. Elife. (2019) 8:e42214. doi: 10.7554/eLife.42214
183. Merz J, Rofa K, Dimanski D, Ahmed I, Hoppe N, Bode C, et al. Knockout of purinergic receptor Y13 (P2Y13) results in an improved outcome in metabolic syndrome in mice. Eur Heart J. (2019) 40 (Suppl_1):ehz746.0070. doi: 10.1093/eurheartj/ehz746.0070
184. Tan C, Voss U, Svensson S, Erlinge D, Olde B. High glucose and free fatty acids induce beta cell apoptosis via autocrine effects of ADP acting on the P2Y13 receptor. Purinergic Signal. (2013) 9:67–79. doi: 10.1007/s11302-012-9331-6
185. Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A 3 adenosine receptor agonists. Drug Disc Today. (2012) 17:359–66. doi: 10.1016/j.drudis.2011.10.007
186. Zakeri Z, Izadi S, Niazi A, Bari Z, Zendeboodi S, Shakiba M, et al. Comparison of adenosine deaminase levels in serum and synovial fluid between patients with rheumatoid arthritis and osteoarthritis. Int J Clin Exp Med. (2012) 5:195–200. Available online at: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3342715/pdf/ijcem0005-0195.pdf
187. Mistry D, Chambers MG, Mason RM. The role of adenosine in chondrocyte death in murine osteoarthritis and in a murine chondrocyte cell line. Osteoarthr Cartil. (2006) 14:486–95. doi: 10.1016/j.joca.2005.11.015
188. Nakamachi Y, Koshiba M, Nakazawa T, Hatachi S, Saura R, Kurosaka M, et al. Specific increase in enzymatic activity of adenosine deaminase 1 in rheumatoid synovial fibroblasts. Arthritis Rheum. (2003) 48:668–74. doi: 10.1002/art.10956
189. Fredholm BB. Adenosine—a physiological or pathophysiological agent? J Mol Med. (2014) 92:201–6. doi: 10.1007/s00109-013-1101-6
190. Pardo F, Villalobos-Labra R, Chiarello DI, Salsoso R, Toledo F, Gutierrez J, et al. Molecular implications of adenosine in obesity. Mol Aspects Med. (2017) 55:90–101. doi: 10.1016/j.mam.2017.01.003
191. Nieto-Vazquez I, Fernandez-Veledo S, de Alvaro C, Lorenzo M. Dual role of interleukin-6 in regulating insulin sensitivity in murine skeletal muscle. Diabetes. (2008) 57:3211–21. doi: 10.2337/db07-1062
192. Corciulo C, Castro C, Coughlin T, Wilder T, Kennedy O, Cronstein B. Regenerating cartilage and reversing osteoarthritis (OA) stimulation of adenosine A2A receptors (A2AR) increases cartilage volume and matrix in vitro and in vivo. Arhtritis Rheumatol. (2017) 69. Available online at: https://acrabstracts.org/abstract/regenerating-cartilage-and-reversing-osteoarthritis-oa-stimulation-of-adenosine-a2a-receptors-a2ar-increases-cartilage-volume-and-matrix-in-vitro-and-in-vivo/
193. Wang Y, Hussain SM, Wluka AE, Lim YZ, Abram F, Pelletier J-P, et al. Association between metformin use and disease progression in obese people with knee osteoarthritis: data from the osteoarthritis initiative—a prospective cohort study. Arthritis Res Ther. (2019) 21:127. doi: 10.1186/s13075-019-1915-x
194. Jacobson KA, Reitman ML. Adenosine-related mechanisms in non-adenosine receptor drugs. Cells. (2020) 9:956. doi: 10.3390/cells9040956
195. Burnstock G. The therapeutic potential of purinergic signalling. Biochem Pharmacol. (2018) 151:157–65. doi: 10.1016/j.bcp.2017.07.016
196. Caswell AM, Leong WS, Russell RGG. Evidence for the presence of P2-purinoceptors at the surface of human articular chondrocytes in monolayer culture. BBA - Gen Subj. (1991) 1074:151–8. doi: 10.1016/0304-4165(91)90054-K
197. Kwon HJ. Extracellular ATP signaling via P2X4 receptor and camp/PKA signaling mediate ATP oscillations essential for prechondrogenic condensation. J Endocrinol. (2012) 214:337–48. doi: 10.1530/JOE-12-0131
198. Chowdhury TT, Knight MM. Purinergic pathway suppresses the release of NO and stimulates proteoglycan synthesis in chondrocyte/agarose constructs subjected to dynamic compression. J Cell Physiol. (2006) 209:845–53. doi: 10.1002/jcp.20768
199. Burnstock G. Purinergic signalling: therapeutic developments. Front Pharmacol. (2017) 8:661. doi: 10.3389/fphar.2017.00661
200. Bravo D, Maturana CJ, Pelissier T, Hernández A, Constandil L. Interactions of pannexin 1 with NMDA and P2X7 receptors in central nervous system pathologies: possible role on chronic pain. Pharmacol Res. (2015) 101:86–93. doi: 10.1016/j.phrs.2015.07.016
201. Kvist TM, Schwarz P, Jørgensen NR. The P2X7 receptor: a key player in immune-mediated bone loss? Sci World J. (2014) 2014:10. doi: 10.1155/2014/954530
202. Rosenthal AK, Hempel D, Kurup I V., Masuda I, Ryan LM. Purine receptors modulate chondrocyte extracellular inorganic pyrophosphate production. Osteoarthr Cartil. (2010) 18:1496–501. doi: 10.1016/j.joca.2010.08.004
203. Kudirka JC, Panupinthu N, Tesseyman MA, Dixon SJ, Bernier SM. P2Y nucleotide receptor signaling through MAPK/ERK is regulated by extracellular matrix: involvement of β3 integrins. J Cell Physiol. (2007) 213:54–64. doi: 10.1002/jcp.21087
204. Wann AKT, Zuo N, Haycraft CJ, Jensen CG, Poole CA, McGlashan SR, et al. Primary cilia mediate mechanotransduction through control of ATP-induced Ca 2+ signaling in compressed chondrocytes. FASEB J. (2012) 26:1663–71. doi: 10.1096/fj.11-193649
205. He Z, Leong DJ, Zhuo Z, Majeska RJ, Cardoso L, Spray DC, et al. Strain-induced mechanotransduction through primary cilia, extracellular ATP, purinergic calcium signaling, and ERK1/2 transactivates CITED2 and downregulates MMP-1 and MMP-13 gene expression in chondrocytes. Osteoarthr Cartil. (2016) 24:892–901. doi: 10.1016/j.joca.2015.11.015
206. Coleman MC, Ramakrishnan PS, Brouillette MJ, Martin JA. Injurious loading of articular cartilage compromises chondrocyte respiratory function. Arthritis Rheumatol. (2016) 68:662–71. doi: 10.1002/art.39460
207. Zhai G. Alteration of metabolic pathways in osteoarthritis. Metabolites. (2019) 9:11. doi: 10.3390/metabo9010011
208. Lane RS, Fu Y, Matsuzaki S, Kinter M, Humphries KM, Griffin TM. Mitochondrial respiration and redox coupling in articular chondrocytes. Arthritis Res Ther. (2015) 17:54. doi: 10.1186/s13075-015-0566-9
209. Mobasheri A, Rayman MP, Gualillo O, Sellam J, Van Der Kraan P, Fearon U. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. (2017) 13:302–11. doi: 10.1038/nrrheum.2017.50
210. Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. (2016) 12:412–20. doi: 10.1038/nrrheum.2016.65
211. Zhao X, Petursson F, Viollet B, Lotz M, Terkeltaub R, Liu-Bryan R. Peroxisome proliferator-activated receptor γ coactivator 1α and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheumatol. (2014) 66:3073–82. doi: 10.1002/art.38791
212. Chen LY, Wang Y, Terkeltaub R, Liu-Bryan R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthr Cartil. (2018) 26:1539–50. doi: 10.1016/j.joca.2018.07.004
213. Qiu L, Luo Y, Chen X. Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. Biomed Pharmacother. (2018) 103:1585–91. doi: 10.1016/j.biopha.2018.05.003
214. Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu-Bryan R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. (2015) 67:2141–53. doi: 10.1002/art.39182
215. Aymerich I, Foufelle F, Ferré P, Casado FJ, Pastor-Anglada M. Extracellular adenosine activates AMP-dependent protein kinase (AMPK). J Cell Sci. (2006) 119:1612–21. doi: 10.1242/jcs.02865
216. Friedman B, Cronstein B. Adenosine A2A Receptor Signals Through AMPK and SIRT1 to Increase Chondrocyte Homeostasis - ACR Meeting Abstracts. ACR/ARP Annu Meet. (2019).
217. Habiballa L, Salmonowicz H, Passos JF. Mitochondria and cellular senescence: implications for musculoskeletal ageing. Free Radic Biol Med. (2019) 132:3–10. doi: 10.1016/j.freeradbiomed.2018.10.417
218. Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Rad Biol Med. (2019) 132:73–82. doi: 10.1016/j.freeradbiomed.2018.08.03
219. Cillero-Pastor B, Caramés B, Lires-Deán M, Vaamonde-García C, Blanco FJ, López-Armada MJ. Mitochondrial dysfunction activates cyclooxygenase 2 expression in cultured normal human chondrocytes. Arthritis Rheum. (2008) 58:2409–19. doi: 10.1002/art.23644
220. Martel-Pelletier J, Pelletier JP, Fahmi H. Cyclooxygenase-2 and prostaglandins in articular tissues. Semin Arthritis Rheum. (2003) 33:155–67. doi: 10.1016/S0049-0172(03)00134-3
221. Caiazzo E, Maione F, Morello S, Lapucci A, Paccosi S, Steckel B, et al. Adenosine signalling mediates the anti-inflammatory effects of the COX-2 inhibitor nimesulide. Biochem Pharmacol. (2016) 112:72–81. doi: 10.1016/j.bcp.2016.05.006
222. Castro CM, Corciulo C, Solesio ME, Liang F, Pavlov EV., Cronstein BN. Adenosine A2A receptor (A2AR) stimulation enhances mitochondrial metabolism and mitigates reactive oxygen species-mediated mitochondrial injury. FASEB J. (2020) 34:5027–45. doi: 10.1096/fj.201902459R
223. So A, Busso N. Osteoarthritis: crystal-gazing into the pathogenesis of osteoarthritis. Nat Rev Rheumatol. (2011) 7:688–9. doi: 10.1038/nrrheum.2011.165
224. Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. (2003) 4:95–104. doi: 10.1038/nrm1019
225. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
226. Jin C, Frayssinet P, Pelker R, Cwirka D, Hu B, Vignery A, et al. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc Natl Acad Sci USA. (2011) 108:14867–72. doi: 10.1073/pnas.1111101108
227. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-κb activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
228. McAllister MJ, Chemaly M, Eakin AJ, Gibson DS, McGilligan VE. NLRP3 as a Potentially novel biomarker for the management of osteoarthritis. Osteoarthri Cartil. (2018) 26:612–9. doi: 10.1016/j.joca.2018.02.901
229. Schett G, Dayer JM, Manger B. Interleukin-1 function and role in rheumatic disease. Nat Rev Rheumatol. (2016) 12:14–24. doi: 10.1038/nrrheum.2016.166
230. Ruan G, Xu J, Wang K, Wu J, Zhu Q, Ren J, et al. Associations between knee structural measures, circulating inflammatory factors and MMP13 in patients with knee osteoarthritis. Osteoarthr Cartil. (2018) 26:1063–9. doi: 10.1016/j.joca.2018.05.003
231. Koh SM, Chan CK, Teo SH, Singh S, Merican A, Ng WM, et al. Elevated plasma and synovial fluid interleukin-8 and interleukin-18 may be associated with the pathogenesis of knee osteoarthritis. Knee. (2020) 27:26–35. doi: 10.1016/j.knee.2019.10.028
232. Bao J, Chen Z, Xu L, Wu L, Xiong Y. Rapamycin protects chondrocytes against IL-18-induced apoptosis and ameliorates rat osteoarthritis. Aging. (2020) 12:5152–67. doi: 10.18632/aging.102937
233. Bougault C, Gosset M, Houard X, Salvat C, Godmann L, Pap T, et al. Stress-induced cartilage degradation does not depend on the NLRP3 inflammasome in human osteoarthritis and mouse models. Arthritis Rheum. (2012) 64:3972–81. doi: 10.1002/art.34678
234. Zu Y, Mu Y, Li Q, Zhang S-T, Yan H-J. Icariin alleviates osteoarthritis by inhibiting NLRP3-mediated pyroptosis. J Orthop Surg Res. (2019) 14:307. doi: 10.1186/s13018-019-1307-6
235. Zhao L-R, Xing R-L, Wang P-M, Zhang N-S, Yin S-J, Li X-C, et al. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP-induced pyroptosis in knee osteoarthritis. Mol Med Rep. (2018) 17:5463–9. doi: 10.3892/mmr.2018.8520
236. Chen Z, Zhong H, Wei J, Lin S, Zong Z, Gong F, et al. Inhibition of Nrf2/HO-1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res Ther. (2019) 21:300. doi: 10.1186/s13075-019-2085-6
237. Conway R, McCarthy GM. Calcium-containing crystals and osteoarthritis: an unhealthy alliance. Curr Rheumatol Rep. (2018) 20:13. doi: 10.1007/s11926-018-0721-9
238. Stack J, McCarthy G. Basic Calcium phosphate crystals and osteoarthritis pathogenesis: novel pathways and potential targets. Curr Opin Rheumatol. (2016) 28:122–6. doi: 10.1097/BOR.0000000000000245
239. Ea HK, Chobaz V, Nguyen C, Nasi S, van Lent P, Daudon M, et al. Pathogenic role of basic calcium phosphate crystals in destructive arthropathies. PLoS ONE. (2013) 8:e57352. doi: 10.1371/journal.pone.0057352
240. Nasi S, Ea H-K, So A, Busso N. Revisiting the role of interleukin-1 pathway in osteoarthritis: interleukin-1α and−1β, and NLRP3 inflammasome are not involved in the pathological features of the murine menisectomy model of osteoarthritis. Front Pharmacol. (2017) 8:282. doi: 10.3389/fphar.2017.00282
241. Mahon OR, Dunne A. Disease-associated particulates and joint inflammation; mechanistic insights and potential therapeutic targets. Front Immunol. (2018) 9:1145. doi: 10.3389/fimmu.2018.01145
242. Pazár B, Ea H-K, Narayan S, Kolly L, Bagnoud N, Chobaz V, et al. Basic calcium phosphate crystals induce monocyte/macrophage IL-1β secretion through the NLRP3 inflammasome in vitro. J Immunol. (2011) 186:2495–502. doi: 10.4049/jimmunol.1001284
243. Baron L, Gombault A, Fanny M, Villeret B, Savigny F, Guillou N, et al. The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis. (2015) 6:e1629. doi: 10.1038/cddis.2014.576
244. Ouyang X, Ghani A, Malik A, Wilder T, Colegio OR, Flavell RA, et al. Adenosine is required for sustained inflammasome activation via the A2A receptor and the HIF-1α pathway. Nat Commun. (2013) 4:2909. doi: 10.1038/ncomms3909
245. North RA. Molecular physiology of P2X receptors. Physiol Rev. (2002) 82:1013–67. doi: 10.1152/physrev.00015.2002
246. Kopp R, Krautloher A, Ramírez-Fernández A, Nicke A. P2X7 interactions and signaling – making head or tail of it. Front Mol Neurosci. (2019) 12:183. doi: 10.3389/fnmol.2019.00183
247. Ribeiro DE, Roncalho AL, Glaser T, Ulrich H, Wegener G, Joca S. P2X7 Receptor Signaling in Stress and Depression. Int J Mol Sci. (2019) 20:2778. doi: 10.3390/ijms20112778
248. Guerra Martinez C. P2X7 receptor in cardiovascular disease: The heart side. Clin Experi Pharmacol Physiol. (2019) 46:513–26. doi: 10.1111/1440-1681.13079
249. Pelegrin P, Surprenant A. Pannexin-1 mediates large pore formation and interleukin-1beta release by the ATP-gated P2X7 receptor. EMBO J. (2006) 25:5071–82. doi: 10.1038/sj.emboj.7601378
250. Parzych K, Zetterqvist A V., Wright WR, Kirkby NS, Mitchell JA, Paul-Clark MJ. Differential role of pannexin-1/ATP/P2X7 axis in IL-1β release by human monocytes. FASEB J. (2017) 31:2439–45. doi: 10.1096/fj.201600256
251. Zeng D, Yao P, Zhao H. P2X7, a critical regulator and potential target for bone and joint diseases. J Cell Physiol. (2019) 234:2095–103. doi: 10.1002/jcp.27544
252. Riteau N, Baron L, Villeret B, Guillou N, Savigny F, Ryffel B, et al. ATP release and purinergic signaling: a common pathway for particle-mediated inflammasome activation. Cell Death Dis. (2012) 3:e403. doi: 10.1038/cddis.2012.144
253. Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The TWIK2 Potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity. (2018) 49:56–65.e4. doi: 10.1016/j.immuni.2018.04.032
254. Dunton CL, Purves JT, Hughes FM, Jin H, Nagatomi J. Elevated hydrostatic pressure stimulates ATP release which mediates activation of the NLRP3 inflammasome via P2X4 in rat urothelial cells. Int Urol Nephrol. (2018) 50:1607–17. doi: 10.1007/s11255-018-1948-0
255. Lyons C, Kennedy E, Roche H. Metabolic inflammation-differential modulation by dietary constituents. Nutrients. (2016) 8:247. doi: 10.3390/nu8050247
256. Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis factor α. Arthritis Rheum. (2011) 63:1928–37. doi: 10.1002/art.30333
257. Zhou S, Lu W, Chen L, Ge Q, Chen D, Xu Z, et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep. (2017) 7:43245. doi: 10.1038/srep43245
Keywords: purinergic system, A2AR, P2X7 receptor, osteoarthritis, metainflammation, mitochondrial metabolism, Inflammasome, rheumatic diseases
Citation: Gratal P, Lamuedra A, Medina JP, Bermejo-Álvarez I, Largo R, Herrero-Beaumont G and Mediero A (2020) Purinergic System Signaling in Metainflammation-Associated Osteoarthritis. Front. Med. 7:506. doi: 10.3389/fmed.2020.00506
Received: 15 May 2020; Accepted: 22 July 2020;
Published: 28 August 2020.
Edited by:
Ying Ying Leung, Duke-NUS Medical School, SingaporeReviewed by:
Zoltan Nemeth, Morristown Medical Center, United StatesLuiz Eduardo Baggio Savio, Federal University of Rio de Janeiro, Brazil
Patricia Teixeira Santana, Federal University of Rio de Janeiro, Brazil
Copyright © 2020 Gratal, Lamuedra, Medina, Bermejo-Álvarez, Largo, Herrero-Beaumont and Mediero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aránzazu Mediero, YXJhbnphenUubWVkaWVybyYjeDAwMDQwO3F1aXJvbnNhbHVkLmVz
†These authors have contributed equally to this work