 Yang-Hsiang Lin
Yang-Hsiang Lin Kwang-Huei Lin
Kwang-Huei Lin Chau-Ting Yeh
Chau-Ting Yeh- 1Liver Research Center, Chang Gung Memorial Hospital, Taoyuan, Taiwan
- 2Department of Biochemistry, College of Medicine, Chang Gung University, Taoyuan, Taiwan
Thyroid hormone (TH) and its receptor (TR) are involved in differentiation, metabolic process, and growth regulation in hepatocellular carcinoma (HCC). The TH/TR complexes are ligand-dependent transcriptional factors, functioning through binding to thyroid hormone response elements (TREs) upstream of the target genes. To date, deciphering the biological effects of TH in cancer progression remains challenging. Several lines of evidence suggest a growth inhibitory effect of TH in liver cancer. Mutation and aberrant expression of TRs are highly correlated with several types of cancers including HCC. Several reports show that TH inhibits cell growth in liver cancer through regulation of cell-cycle-related genes and non-coding RNAs. A case–control study indicates that hypothyroidism is associated with an increased risk of HCC. Moreover, TH/TR suppresses hepatocarcinogenesis via selective autophagy. Conversely, other groups have indicated that TH promotes cancer cell proliferation. In vitro and in vivo experiments show that TH/TR enhances cancer cell migration and invasion, anticancer drug resistance, angiogenesis, and cancer stem cell self-renewal. Adding to the complexity of this issue, non-genomic effects of TH mediated by integrin receptor on cell surface can also modulate several biological functions. Accumulating evidence indicate that regulations by genomic and non-genomic effects of TH overlap. Taken together, these observations suggest that the functions of TH depend largely on cell context, and TH/TR plays a duel role in cancer progression. Therefore, understanding the maze of biological effects of TH has become a necessity when attempting to develop effective therapeutic and preventive strategies in liver cancer.
Introduction
Thyroid hormone (TH) plays an important role in countless cellular processes, including cell differentiation, proliferation, autophagy, and metabolism (1). The effects of TH on cancer cells are mediated by either genomic or non-genomic signal transduction (2). Deregulation of TH-mediated pathway results in disturbances of cellular functions such as those observed in tumorigenesis (1). An important issue of TH involved in cancer progression is the “TH concentration” in patients (3). Increasing evidence has shown that TH status (hypothyroidism and hyperthyroidism) is involved in the development of several cancers, including ovarian cancer (4), prostate cancer (5), breast cancer (6), and hepatocellular carcinoma (HCC) (7), evidenced by population-based case–control study.
HCC is one of the most common and aggressive human malignancy in the world. Most patients with HCC have an established background of chronic liver disease and cirrhosis. The major risk factors for liver cirrhosis include chronic infection with hepatitis B virus (HBV) and hepatitis C virus (HCV), and alcoholism (8). Cirrhosis develops following long periods of chronic necroinflammation of hepatocytes and is characterized by a decreased capability of hepatocyte proliferation, indicating an exhaustion of the regenerative potential of the liver. Several preclinical investigations have linked TH-mediated response to tumor-suppressing functions or tumor-promoting actions in hepatocarcinogenesis (1).
The main objective of this review is to summarize the basic properties and functional roles of the TH in HCC. In particular, we have encapsulated current knowledge on the contribution of TH-regulated genes that alter cell growth, cell cycle distribution, drug resistance, and metastasis. Hopefully, these pieces of information can provide clues for future anticancer discoveries.
The Genomic and Non-genomic Actions of TH
The effects of TH on cancer cells can be divided into two categories: genomic and non-genomic pathways (2). Dysregulation of any of these mechanisms has significant effects on cancer formation and progression. For genomic effect of TH (Figure 1A), T3 interacts with thyroid hormone receptor (TR) and other nuclear receptors, such as vitamin D receptor (VDR) and retinoid X receptor (RXR), which in turn regulates transcriptional activities of the target genes (9). TRs are encoded by two separate genes, THRA (TRα) and THRB (TRβ), located on human chromosomes 17 and 3, respectively. The functional domains of TRs are composed of amino terminal A/B domain that may recruit regulatory proteins, DNA-binding domain (DBD), a hinge region that contains the nuclear localization signal, and carboxy-terminal ligand-binding domain that binds to T3. TRs function in a ligand-dependent manner via binding to thyroid hormone response elements (TREs) of target gene promoter regions (9). So far, direct repeat (DR4), inverted palindrome (F2), and palindrome (pal) types of TRE have been identified (10). T3 binding induces TR conformational changes, causing coactivator [e.g., steroid receptor coactivator 1 (SRC-1)] binding and corepressor [e.g., nuclear receptor corepressor (NCoR) and silencing mediator of retinoic and thyroid receptor (SMRT)] release that lead to chromatin modifications and transcriptional activation (Figure 1A).
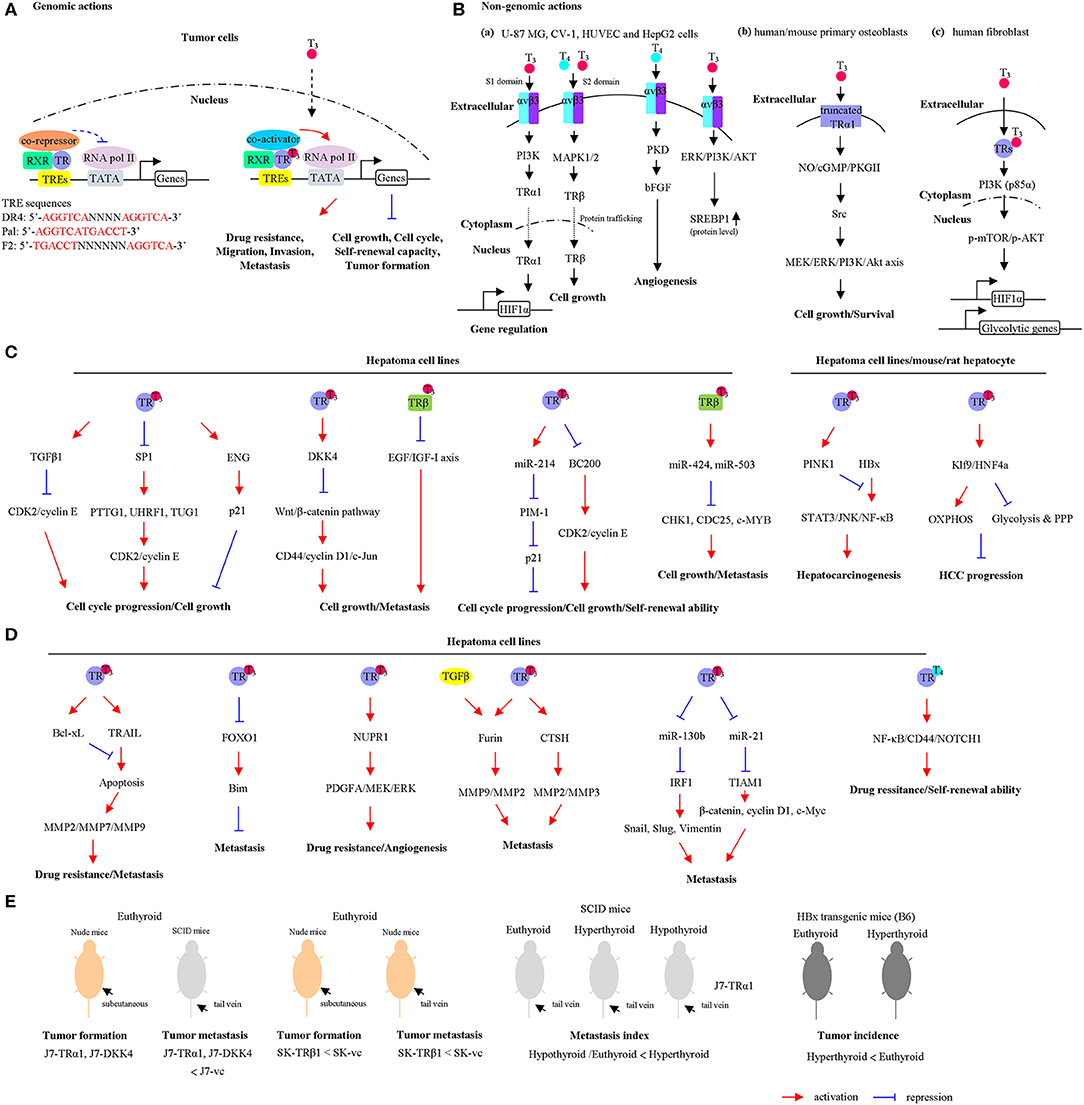
Figure 1. The actions of thyroid hormones (THs) and their corresponding receptors. (A) Genomic actions of the TH/thyroid hormone receptor (TR). In the absence or presence of TH, TR/retinoid X receptor (RXR) heterodimers bind to thyroid hormone response elements (TREs). Unliganded TRs associate with corepressors that repress gene transcription. Binding of T3 causes corepressor release, coactivator recruitment, and transcriptional activation. (B) Non-genomic actions of the TH. (a) The TH interacts with the integrin receptor and activates extracellular signal-regulated kinase (ERK)/phospholipase C (PLC)/protein kinase Cα (PKCα) cascade, which upregulates cell proliferation. U-87 MG cells, glioma cells; CV-1 cells, fibroblast cell lines that lacks functional TRs; HUVEC cells, endothelial cells; HepG2 cells, hepatoma cell line. (b) The T3 interacts with the αvβ3 and enhances the phosphatidylinositol 3-kinase (PI3K) pathway, leading to direct trafficking of TRα1 from the cytoplasm to the nucleus and promotes hypoxia-inducible factor-1α (HIF-1α) expression. (c) In the cytoplasm, the liganded TRβ1 associates with the PI3K regulatory subunit, p85α, and activates Akt/mammalian target of rapamycin (mTOR) axis. Consequently, these events regulate gene expression, including HIF1α and glycolytic genes. (d) T3 interacts with plasma membrane-associated truncated TRα1 and induces the NO-cGMP-PKGII cascade. Subsequently, this cascade initiates Src/MEK/ERK/PI3K/Akt signaling, which results in induced cell proliferation and survival. The cell lines used in each pathway were shown. The schematic diagram summarizes the findings mentioned in the text including the types of cell lines and animal models. (C) TH/TR-mediated cell growth and cell cycle progression via specific signaling pathway are shown. OXPHOS, oxidative phosphorylation; PPP, pentose phosphate pathway. (D) TH/TR-mediated cell metastasis, angiogenesis, self-renewal ability, and drug resistance via specific signaling pathway are shown. (E) TH/TR-mediated tumor growth and tumor metastasis in murine model are shown. Euthyroid, control; hypothyroid, low T3 levels; hyperthyroid, high T3 levels. For a detailed description, please see in the corresponding section.
For non-genomic effects of TH (Figure 1B), previous studies have shown that the regulatory mechanisms of TH's functions are not totally mediated by nuclear TRs. Other nonnuclear events are involved. It is referred to as non-genomic effects. These reactions are rapid in contrast to genomic effects that occur over minutes or hours and are not involved in gene transcription or protein synthesis. Based on earlier findings, TH (T4 and T3) interacts with cell surface protein, integrin αvβ3, or plasma-membrane-associated truncated TR or cytoplasmic TR and contributes to regulation of multiple physiological functions (11). In fact, TH interacts with integrin αvβ3 near its Arg–Gly–Asp (RGD)-binding site. Two TH-binding domains of integrin αvβ3 are identified (12). Notably, T3 could bind to the S1 domain and activate the phosphatidylinositol 3-kinase (PI3K)/Akt/protein kinase B (PKB) pathway through Src kinase. S1 domain regulates Src and PI3K-mediated TRα translocation from the cytoplasm to the nucleus and enhances target gene expressions such as hypoxia-inducible factor-1α (HIF-1α) (Figure 1Ba) (13). Furthermore, S2 domain mainly recognizes T4 and T3, consequently activating the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK1/2) pathway via phospholipase C (PLC) and protein kinase Cα (PKCα). Specifically, S2 domain promotes tumor cell proliferation via regulation of nuclear trafficking of TRβ1 from the cytoplasm (Figure 1Ba) (14). Gnoni et al. (15) indicated that translational efficiency of the precursor form of sterol regulatory element-binding transcription factor 1 (SREBP1), which functioned as master regulator of lipid metabolism, was upregulated by T3 through activation of MAPK/ERK and PI3K/AKT/mTOR-C1 pathway in HepG2 cells (Figure 1Ba). Notably, these effects were partially abrogated by tetraiodothyroacetic acid (tetrac), which repressed the binding of T4 and T3 to the integrin αvβ3. These findings suggested that SREBP1-mediated lipid metabolism was regulated by T3 via non-genomic effects in hepatoma cell lines (15). In addition, T3 interacts with plasma membrane-associated truncated TRα1 and induces the NO-cGMP-PKGII cascade (16). In this TRα-depleted osteoblasts model, re-overepxression of truncated TRα1 partly restored T3-mediated effects. Subsequently, this cascade initiates Src/MEK/ERK/PI3K/Akt signaling, which results in induced cell proliferation and survival (Figure 1Bb). Using the human fibroblast-harboring wild-type or mutant TRβ (resistance to thyroid hormones) models, liganded TRβ1 interacts with p85α in the cytoplasm, leading to activating phosphorylation of Akt (Figure 1Bc). Activated Akt promotes the nuclear mammalian target of rapamycin (mTOR)/p70S6K cascade and sequential regulation of several HIF-1α target genes, including glucose transporter 1 (GLUT1), platelet-type phosphofructokinase (PFKP), and monocarboxylate transporter 4 (MCT4) (17–20). Although several non-genomic effects of TH were not observed in hepatoma cell lines, these models have several advantages. For instance, the CV-1, a fibroblast cell line lacking endogenous TR expression, was a good cell line model for observing non-genomic actions of TR (21). Additionally, T4 induced angiogenesis through αvβ3/protein kinase D (PKD)/basic fibroblast growth factor (bFGF) in human umbilical vein endothelial cells (HUVECs) (Figure 1Ba) (22), suggesting TH crosstalk with tumor microenvironment via non-genomic actions.
Crosstalk between genomic and non-genomic actions of TH via TR and integrins has been explored. Cao et al. have demonstrated that TH binds to αvβ3 at plasma membrane and controls TRβ1 localization (23). In addition, posttranslational modifications of TRβ1 are regulated by TH/αvβ3. TH treatment triggers MAPK-dependent phosphorylation of TRβ1 at Ser142 (24). This event causes shedding by the receptor of corepressor (SMRT) and recruitment of coactivators that contribute to the transcriptional activity. Understanding how crosstalk occurs between genomic and non-genomic actions to regulate important biological functions will help us design novel drugs or inhibitors to improve treatment of TH-dependent cancers.
The Impacts of Thyroid Hormone on Cell Growth and Cell Cycle Progression in HCC
Previously, a transplantation model for investigating the effects of TH on hepatocytes was established (25). Small pieces of thyroid tissues were transplanted through the portal vein in to the liver of thyroidectomized rats. After short- and long-term transplantation, the proliferative, synthesize thyroglobulin, and T3 release ability were increased. In addition, Columbano et al. (26) group demonstrated that thyromimetic agent GC-1, which was highly selective for interacting and activation of TRβ1 functions, promoted rat hepatocyte and pancreatic cell proliferation. Mechanically, cyclin D1 messenger RNA (mRNA) was upregulated by GC-1 treatment. These findings indicated that TRβ-selective agent GC-1 is an important mitogen for both hepatocyte and pancreatic cells. Work by the same group also showed that T3 treatment result in increased incorporation of the DNA replication marker, bromodeoxyuridine (BrdU), in mouse hepatocytes. In addition, treatment with T3 and GC-1 has been shown to prevent a spectrum of liver-related diseases such as non-alcoholic fatty liver in rats (27). GC-1 treatment reduced hepatoma cell proliferation and tumor burden through inhibition of p-Met (Y1234/1235), p-ERK1/2, and p-STAT3 pathway (28). Meanwhile, GC-1 treatment did not activate β-catenin and its downstream targets. These findings suggested that GC-1 exerted an antitumor effect in HCC. Transforming growth factor (TGF-β) is induced by T3 in HepG2-TR overexpressing cells (29). Moreover, treatment by TGF-β neutralizing antibody but not the control antibody, restored the cell proliferation suppression effect of T3. Mechanically, several cell-cycle-related genes such as CDK2, clyclin E, and ppRb were downregulated by T3/TR, while p21 was induced by T3/TR. Notably, suppression of cyclin E, cdk2, and ppRb expression by T3 was blocked by treating with TGF-β neutralizing antibody. Pituitary tumor transforming gene 1 (PTTG1) is cell cycle modulator and acts as a securin that suppresses sister chromatid separation and acts as an oncogene implicated in cancer progression (30, 31). Chen et al. demonstrated that PTTG1 was repressed by T3/TR through suppression of transcription factor, SP1 (32). Overexpression of PTTG1 enhanced cell growth and cell cycle progression in Hep3B cells. Notably, the expression levels of PTTG1 and SP1 were inversely correlated with the expressions of TRs in HCC specimens. Endoglin (ENG) is a component of the TGFβ receptor complex that binds to TGF-β1, TGF-β3, activin-A, and bone morphogenetic protein-2 and regulates TGFβ1-mediated responses. ENG is directly upregulated by T3/TRs in HepG2-overexpressing TR cells (33). T3/TR dramatically induced G0/G1 phase cell cycle arrest. Knockdown of ENG in HepG2-TRα1 cells facilitated p21 polyubquitination and promoted cell growth and cell cycle progression in the presence of T3. In addition, antagonization of Wnt signal pathway by Dickkopf 4 (DKK4) was induced by T3 in HepG2-TR-overexpressing cells and J7-TRα1 cells (34). Functionally, ectopic expression of DKK4 in J7 cells reduced cell growth and invasion. The effect of DKK4 and TRα1 on tumor growth and metastasis of J7 in animal models was observed without treating T3. The tumor growth and metastatic index were repressed by overexpression of DKK4 and TRα1. These observations suggest that TR/DKK4/Wnt/β-catenin axis modulates proliferation and migration of hepatoma cells during metastasis. Dysregulation of epigenetic regulator may contribute to cancer progression (35). Expression of an epigenetic regulator, ubiquitin-like with PHD and ring finger domains 1 (UHRF1), is regulated by TH. The regulatory effect is indirect and mediated by SP1 (36). Inhibition of UHRF1 expression reduced cell growth in hepatoma cell lines via cyclin E/CDK2/p21 axis. Furthermore, overexpression of UHRF1 abolished T3-induced cell cycle arrest. Quantitative assessment using clinical samples revealed that expression levels of UHRF1 and Sp1 were elevated in HCC and inversely correlated with TRα1 expression. Another oncoprotein, Stathmin (STMN1), highly expressed in HCC, was directly repressed by T3 (37). Depletion of STMN1 inhibited tumor growth in vitro and in vivo. Additionally, T3-mediated cell cycle distributions were attenuated by STMN1. Notably, cyclin A, B, and D levels were reduced in STMN1 knockdown cells. High-throughput screening experiments revealed more specific genes that were directly regulated by T3/TR, when chromatin immunoprecipitation coupled with microarray (ChIP-on-ChIP) analysis was performed (38). Among these potential target genes, 304 upregulated and 176 downregulated genes were simultaneously bound by T3/TR. A transcription factor, E74-like factor 2 (ELF2), directly downregulated by T3/TR, was involved in T3-mediated cell growth and cell cycle progression effect. Knockdown of ELF2 in SK-Hep1 cells promoted p21 and p27 expression. Consistent with our results, TH/TR-induced antitumor effects through induction of a differentiation program in the preneoplastic hepatocytes. This view has been supported by experiments using a carcinogen-induced HCC animal model (39, 40). Taken together, T3/TR plays tumor suppressor role in regulating cell growth and cell cycle progression (Figure 1C).
Non-coding RNAs (ncRNA) are a class of non-protein coding transcripts frequently dysregulated in various cancers, which also play important roles in biological processes, such as cell proliferation, metabolism, drug resistance, and cell metastasis (41). Expression of a microRNA (miRNA), miR-214, was stimulated upon T3 treatment (42). PIM-1 is a miR-214 target gene. Furthermore, the effect of T3-repressed cell growth was partially restored upon miR-214 knockdown. MiR-424 and miR-503 were positively regulated by TH in SK-overexpressing TRβ cell lines (43). Knockdown of those miRNAs reversed the inhibitory effect of TH on CHK1, WEE1, CDC25, c-MYB, and E2F3, in turn impairing the effect of TH on cell growth. A long ncRNA, BC200, directly downregulated by T3 was identified in hepatoma (44). BC200 enhanced cell proliferation through regulation of cell-cycle-related genes such as cyclin E, CDK2, and p21. Meanwhile, depletion of BC200 repressed the tumor sphere formation frequencies of CD133+ subsets of HepG2 and Huh7 cells, compared with control cells. Moreover, the BC200/cyclin E2/CDK2 axis is correlated with poor prognosis in HCC patients (Figure 1C). In contrast, Wang and coworkers demonstrated that T4/TRα promoted hepatoma cell, including CSQT-2 and MHCC97-H cells, self-renewal ability through activation of nuclear factor kappa B (NF-κB) and stem cell genes such as CD44, NOTCH1, BMI1, and HIF1α (45). In addition, T4 enhanced the percentage of CD90-positive HCC cells and drug resistance. In this model, T4 acted as promoter of CSC self-renewal (Figure 1D). Although T4 is predominantly secreted from the thyroid gland, the affinity of the TR for T4 is lower than that for T3. Different treatment (T4 or T3) may trigger different signaling pathways via genomic or non-genomic actions and results in controversial results. Other studies reported that TH/TRs promoted cell growth in other cancer cell types, including glioma (46), breast (47), and prostate cancer (48). Kress et al. (49) demonstrated that overexpression of TRα1 and activation of Wnt/β-catenin pathway promoted colorectal cancer formation. TRα1 enhanced tumorigenesis in APC+/1638N mice with a Wnt-activated genetic background. Notably, a study reported that thyroid status regulated tumor formation and metastasis in experimental animal models and humans (50). Lin et al. (51) reported that mutated TRs in human HCCs and cultured cells lose its T3 binding and transcriptional activities and subsequently exhibit dominant-negative activity. Another evidence demonstrated that mutation form of TRs isolated from HCC patients (HCC-TR) were transfected into HepG2 cell lines (52). Moreover, differential expressed genes in those cell lines were measured using microarray analysis. Interestingly, only a subset of genes regulated by HCC-derived TR mutants was also regulated by the corresponding wild-type TRs. Furthermore, the mutation form of TR could regulate additional target genes not modulated by the wild-type receptors. Due to the complexity of these effects, TH/TR could play a tumor-suppressive and/or protumorigenic role in a cellular context-dependent manner.
The Impacts of Thyroid Hormone on Autophagy in HCC
Hepatitis B virus X protein (HBx) encoded by the X gene of the HBV is involved in tumor formation, apoptosis, drug resistance, and metastasis through various pathway (53). By treating with or without T3 in HBx-transgenic mice, T3 could protect hepatocytes from HBx-triggered cell damage through regulation of mitochondrial quality control in vivo (54). PTEN-induced kinase 1 (PINK1) is induced by T3 in vitro and in vivo. PINK1 can activate and recruit Parkin, an E3 ligase, to enhance HBx protein ubiquitination and trigger selective mitophagy (Figure 1C). Furthermore, Chi et al. demonstrated that TH treatment repressed liver carcinogenesis through activation of autophagy (55). Death-associated protein kinase 2 (DAPK2) expression was positively regulated by T3. In a murine model, overexpression of DAPK2 attenuated diethylnitrosamine-induced toxicity and DNA damage via promotion of autophagy. These findings suggest that TH treatment can be an effective therapeutic option for liver cancer.
The Impacts of Thyroid Hormone on Drug Resistance in HCC
Drug resistance is a complex process, with many intrinsic and extrinsic factors regulating the resistance potential of HCC cells (56). In addition, drug resistance is a well-known nightmare when cancers and diseases become tolerant to drug treatments. Although several types of cancers are initially susceptible to anticancer treatment, over time, they can develop resistance through mechanisms such as increased tumor heterogeneity, alteration of tumor microenvironment, and renewal of cancer stem cells. Several studies demonstrated that T3 leads to promotion of anticancer drug resistance. These associations between TH/TR and drug resistance are shown in Figure 1D. Chi and coworkers demonstrated that tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) was directly upregulated by T3 in TR-overexpressing HepG2 and J7 cells (57). In those cell lines, T3 suppresses r-TRAIL and chemotherapeutic drugs-induced apoptosis. Several experimental results demonstrated that T3-induced Bcl-xL, an antiapoptotic gene, prevented hepatoma cells from TRAIL-induced apoptosis, in turn promoting hepatoma cell metastasis. Another report indicated that Bim, a proapoptotic gene, was indirectly regulated by T3 (58). Furthermore, Bim was directly regulated by Forkhead box protein O1 (FoxO1), which was repressed by T3 through genomic and non-genomic effects. Of note, T3-repressed Bim prevented hepatoma cells from cisplatin, doxorubicin, and TRAIL-induced apoptosis. Nuclear protein-1 (NUPR1) was positive regulated by T3/TR through direct binding to the−2066/-1910 region of the NUPR1 promoter (59). Ectopic expression of NUPR1 promoted angiogenesis through the regulation of PDGFA/MEK/ERK signaling cascade. Furthermore, TR/NUPR1 axis enhanced sorafenib resistance in Mahlavu and Huh7 cell lines in vitro and in vivo. In addition, Wang et al. reported that T4 enhanced drug resistance of CSQT-2 and MHCC97-H cells in a TRα-dependent manner (45). Previous studies reported that TH, including T4 and T3, induced P-glycoprotein (P-gp) gene expression, which is also known as multidrug resistance protein 1, and plays a significant role in multidrug-resistant phenotype (60–62). Accordingly, TH/TR manifests an oncogene-like effect via increasing angiogenesis and drug resistance in hepatic tumor microenvironment.
The Impacts of Thyroid Hormone on Cancer Cell Metastasis in HCC
The process of cancer metastasis remains an important issue when it comes to successful management of malignant diseases (63). Martinez-Iglesias et al. (64) group indicated that TRβ1 acts as a suppressor of tumor metastasis in hepatoma (SK-Hep1) and breast cancer (MDA-MB-468) cells. Ectopic expression of TRβ1 in those cells inhibited tumor metastasis, both in vitro and in vivo, validating a suppressor role of TRβ (Figure 1C). This study showed that TRβ1 had major suppressive effect on metastasis. Furthermore, the authors showed that double-knockout thra thrb mice are induced to generate epithelial tumors, suggesting that TR deficiency could inhibit tumor formation at early stages while enhancing cancer progression at later stages in the skin carcinogenesis protocol. The same group has also demonstrated that hypothyroidism could retard tumor formation and promote metastasis independent of TRβ1 expression (65), suggesting that the influence of thyroid status in HCC progression is more significant than that of the TRβ1-mediated effects. In addition, T3-mediated Furin upregulation cooperated with TGF-β1 signaling pathway to regulate cellular function (66). Ectopic expression of Furin promoted cell migration, invasion, and metastasis in vitro and in vivo (Figure 1D). In addition, an oncomiR, miR-21, was upregulated by T3 in HepG2-TR cell lines (67). Inhibition of miR-21-induced Hep3B-TRα1 cell migration was rescued by T3 treatment. Meanwhile, T-cell lymphoma invasion and metastasis 1 (TIAM1) was a potential target of miR-21 and downregulated by T3. Quantitative assessment using clinical samples showed that miR-21 was positively correlated with TRα1 in HCC specimens (Figure 1D). On the other hand, miR-130b was negatively regulated by T3/TR (68). Cell migration was enhanced by T3 but partially repressed upon miR-130b overexpression. Notably, miR-130b regulated cell migration, invasion, and metastasis, which was mediated by interferon regulatory factor 1 (IRF1) (Figure 1D). Hyperthyroidism in J7-TRα1 xenograft models promotes cell metastasis compared to eu- and hypothyroid. T3/TR, miR-130b, IRF1, and EMT-related genes axis regulated the motility and invasion of hepatoma cells. Thus, these studies suggest that T3/TR exerts an oncogene-like effect in metastasis of liver cancer. Due to different experimental approaches, overexpression of TRs in J7 and SK-Hep1 cells under normal physiological concentration of TH (euthyroid) suppressed tumor metastasis in nude and severe combined immunodeficient (SCID) mice (Figure 1E). However, it has the opposite effects by treating T3 (hyperthyroid/hypothyroid vs. euthyroid group) in xenograft model by tail vein injection method (Figure 1E). In addition, TH suppressed HBx-induced hepatocarcinogenesis in a transgenic mice model. The different genetic background (immune vs. immunodeficient mouse model) maybe responsible for the conflicting effects of TH/TR in HCC progression. On the other hand, tumor microenvironment can modulate tumor formation, metastasis, drug resistance, and therapeutic response. Stromal cells orchestrate the microenvironment and provide specific molecules such as growth factors and proangiogenic factors to facilitate tumor cell survival and metastasis (69). T3 enhanced phosphorylation of GSK-3β and Akt in TRα1-overexpressing endothelial cells through non-genomic actions (70). The crosstalk between cancer cell and stromal cells may modulate the function of tumor suppressors, resulting in a switch to promote oncogenesis.
The Impacts of Thyroid Hormone on Metabolism in HCC
Previous experiments by our group showed that metabolism-related genes, including aldo-keto reductase family 1, member b1 (AKR1B1) (71), methionine adenosyltransferase 1a (MAT1A) (72), and chromosome 19 open reading frame 80 (C19orf80) (73), upregulated by T3 in TR-overexpressing HepG2 cells, modulated HCC progression. In particular, Tseng and coworkers demonstrated that TH/TR regulates lipid metabolism via C19orf80-triggered autophagy pathway (73). Multiple lines of evidence, including ours, indicated that hepatic lipid turnover rate was induced by TH treatment. Major risk factors such as non-alcoholic fatty liver disease (NAFLD) and metabolic syndrome for HCC, especially for non-hepatitis B and non-hepatitis C HCC (NBNC-HCC), have been reported (74). Recently, it was found that TUG1, responsible for regulating glycolysis and metastasis, was downregulated by T3/TR in HepG2-TR cells (75). Depletion of TUG1 reduced cell cycle progression and soft agar colony formation (Figure 1C). Notably, α-fetoprotein was partially downregulated by T3/TR through suppression of TUG1. By analyzing clinical samples, mRNA expression levels of AFP were strongly and positively correlated with TUG1 and associated with unfavorable prognosis in patients with NBNC-HCC. Kowalik et al. (76) demonstrated that T3 treatment in HCC-bearing rats dramatically repressed the number and burden of HCC. Notably, T3-induced metabolic reprogramming (switch from glycolysis to oxidative metabolism) precedes preneoplastic nodules regression (Figure 1C). The identified metabolites are the intermediate products of the responsible metabolic processes. Further study is mandatory to determine the oncometabolites regulated by TH/TR to overcome the current limitations of HCC therapy.
Clinical Significance of Thyroid Hormone and Its Receptor in Liver Cancer
A case–control study reported that long-term hypothyroidism was associated with significantly elevated risk of HCC, independent of other major HCC risk factors (7). In particular, this finding was statistically significant only among women. Subsequently, the correlations between thyroid functions, clinical parameters, and survival functions were investigated by Pinter et al. (77). They found that thyroid-stimulating hormone (TSH) and free T4 (fT4) levels were highly correlated with prognostic factors such as tumor size and C-reactive protein. Notably, high serum fT4 levels were independently correlated with poor prognosis in HCC. Chu et al. demonstrated that higher levels of TSH and fT4 were independently associated with favorable progression-free and overall survival in HCC patients (78). However, the outcome associations were opposite between advanced HCC patients treated by sorafenib and systemic chemotherapy. In addition, truncating and missense mutations of TRs were identified in HCC specimens and HCC cell lines. Of note, v-erbA is a mutant form of TRα with disrupted T3-mediated regulation functions for genes such as follistatin, activin βC, thrombomodulin, N-myc downstream-regulated gene 2, Sine oculis-related homeobox-1 homolog, and Ras-GTP-releasing protein 3, in turn leading to liver carcinogenesis in a transgene mouse model (79). Our previous study has shown that thyroid-stimulating hormone receptor (TSHR) is highly expressed in HCC and associated with unfavorable survival outcomes (80). Meanwhile, mutations of TSHR in exon 10 were identified in HCC specimens, but not in the adjacent non-cancerous tissues, suggesting a growth advantage of tumor cells harboring such mutations.
Conclusions
Based on these findings, the distinct functions of TH/TR in HCC depend on TH (T4 vs. T3), TRs expression status (mutation/different isoforms), integrin αvβ3, coregulators, cell lines, cell context, and cancer stage. Liver cancer is a complex experimental model when it comes to exploring the relationship between the TH/TR and tumor progression. In the current article, the T3/TR-regulated genes in hepatoma cell lines, involved in regulations of tumor growth, cell cycle progression, drug resistance, and metastasis, are comprehensively shown in Figure 1 and listed in Table 1. Although several protein-coding genes and ncRNAs are known to be regulated by T3/TR, existing knowledge regarding the key signaling pathways responsible for hypothyroidism-related liver carcinogenesis is incomplete. The collective results support distinct associations between the target genes and TH/TR networks that regulate cancer cell growth, anticancer drug resistance and metastasis. In addition, these associations could be different depending on cancer types. Understanding the details of these cross-talks is therefore essential to successfully identify novel therapeutic targets.
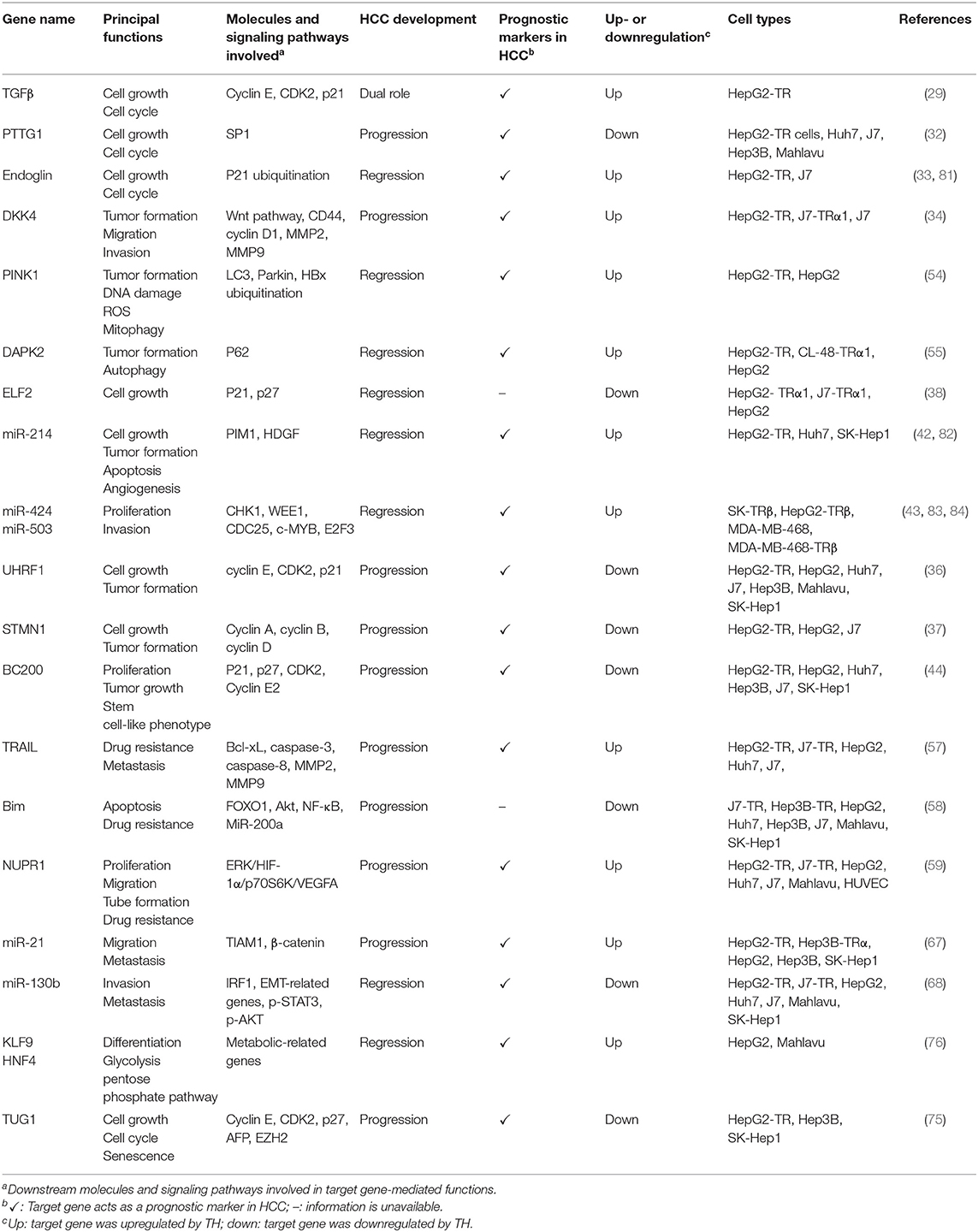
Table 1. Thyroid hormone (TH)/thyroid hormone receptor (TR)-regulated genes and their potential mechanisms in hepatocellular carcinoma (HCC).
Author Contributions
Y-HL: writing—original draft preparation. K-HL and C-TY: writing—review and editing. C-TY: supervision.
Funding
This work was supported by grants from Chang Gung Memorial Hospital, Taoyuan, Taiwan (CMRPG3H0721, CMRPG3H0722, CMRPG3H0723, NZRPG3G0171, NZRPG3G0172, NZRPG3G0173, NMRPG3H0561, and NRRPG3J0141 to Y-HL) and the Ministry of Science and Technology of the Republic of China (MOST 106-2321-B-182A-004-MY3, MOST 107-2320-B-182A-028-, and MOST 108-2320-B-182A-004- to Y-HL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Krashin E, Piekielko-Witkowska A, Ellis M, Ashur-Fabian O. Thyroid hormones and cancer: a comprehensive review of preclinical and clinical studies. Front Endocrinol. (2019) 10:59. doi: 10.3389/fendo.2019.00059
2. Hammes SR, Davis PJ. Overlapping nongenomic and genomic actions of thyroid hormone and steroids. Best Pract Res Clin Endocrinol Metab. (2015) 29:581–93. doi: 10.1016/j.beem.2015.04.001
3. Khan SR, Chaker L, Ruiter R, Aerts JG, Hofman A, Dehghan A, et al. Thyroid function and cancer risk: the rotterdam study. J Clin Endocrinol Metab. (2016) 101:5030–6. doi: 10.1210/jc.2016-2104
4. Journy NMY, Bernier MO, Doody MM, Alexander BH, Linet MS, Kitahara CM. Hyperthyroidism, hypothyroidism, and cause-specific mortality in a large cohort of women. Thyroid. (2017) 27:1001–10. doi: 10.1089/thy.2017.0063
5. Mondul AM, Weinstein SJ, Bosworth T, Remaley AT, Virtamo J, Albanes D. Circulating thyroxine, thyroid-stimulating hormone, and hypothyroid status and the risk of prostate cancer. PLoS ONE. (2012) 7:e47730. doi: 10.1371/journal.pone.0047730
6. Ortega-Olvera C, Ulloa-Aguirre A, Angeles-Llerenas A, Mainero-Ratchelous FE, Gonzalez-Acevedo CE, Hernandez-Blanco ML, et al. Thyroid hormones and breast cancer association according to menopausal status and body mass index. Breast Cancer Res. (2018) 20:94. doi: 10.1186/s13058-018-1017-8
7. Hassan MM, Kaseb A, Li D, Patt YZ, Vauthey JN, Thomas MB, et al. Association between hypothyroidism and hepatocellular carcinoma: a case-control study in the United States. Hepatology. (2009) 49:1563–70. doi: 10.1002/hep.22793
8. Zamor PJ, deLemos AS, Russo MW. Viral hepatitis and hepatocellular carcinoma: etiology and management. J Gastrointest Oncol. (2017) 8:229–42. doi: 10.21037/jgo.2017.03.14
9. Brent GA. Mechanisms of thyroid hormone action. J Clin Invest. (2012) 122:3035–43. doi: 10.1172/JCI60047
10. Velasco LF, Togashi M, Walfish PG, Pessanha RP, Moura FN, Barra GB, et al. Thyroid hormone response element organization dictates the composition of active receptor. J Biol Chem. (2007) 282:12458–66. doi: 10.1074/jbc.M610700200
11. Cody V, Davis PJ, Davis FB. Molecular modeling of the thyroid hormone interactions with alpha v beta 3 integrin. Steroids. (2007) 72:165–70. doi: 10.1016/j.steroids.2006.11.008
12. Lin HY, Cody V, Davis FB, Hercbergs AA, Luidens MK, Mousa SA, et al. Identification and functions of the plasma membrane receptor for thyroid hormone analogues. Discov Med. (2011) 11:337–47.
13. Lin HY, Sun M, Tang HY, Lin C, Luidens MK, Mousa SA, et al. L-Thyroxine vs. 3,5,3'-triiodo-L-thyronine and cell proliferation: activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol. (2009) 296:C980–91. doi: 10.1152/ajpcell.00305.2008
14. Davis PJ, Goglia F, Leonard JL. Nongenomic actions of thyroid hormone. Nat Rev Endocrinol. (2016) 12:111–21. doi: 10.1038/nrendo.2015.205
15. Gnoni GV, Rochira A, Leone A, Damiano F, Marsigliante S, Siculella L. 3,5,3'triiodo-L-thyronine induces SREBP-1 expression by non-genomic actions in human HEP G2 cells. J Cell Physiol. (2012) 227:2388–97. doi: 10.1002/jcp.22974
16. Kalyanaraman H, Schwappacher R, Joshua J, Zhuang S, Scott BT, Klos M, et al. Nongenomic thyroid hormone signaling occurs through a plasma membrane-localized receptor. Sci Signal. (2014) 7:ra48. doi: 10.1126/scisignal.2004911
17. Cohen K, Ellis M, Khoury S, Davis PJ, Hercbergs A, Ashur-Fabian O. Thyroid hormone is a MAPK-dependent growth factor for human myeloma cells acting via alphavbeta3 integrin. Mol Cancer Res. (2011) 9:1385–94. doi: 10.1158/1541-7786.MCR-11-0187
18. Shinderman-Maman E, Cohen K, Weingarten C, Nabriski D, Twito O, Baraf L, et al. The thyroid hormone-alphavbeta3 integrin axis in ovarian cancer: regulation of gene transcription and MAPK-dependent proliferation. Oncogene. (2016) 35:1977–87. doi: 10.1038/onc.2015.262
19. Barbakadze T, Natsvlishvili N, Mikeladze D. Thyroid hormones differentially regulate phosphorylation of ERK and Akt via integrin alphavbeta3 receptor in undifferentiated and differentiated PC-12 cells. Cell Biochem Funct. (2014) 32:282–6. doi: 10.1002/cbf.3013
20. Otto T, Fandrey J. Thyroid hormone induces hypoxia-inducible factor 1alpha gene expression through thyroid hormone receptor beta/retinoid x receptor alpha-dependent activation of hepatic leukemia factor. Endocrinology. (2008) 149:2241–50. doi: 10.1210/en.2007-1238
21. Lin HY, Martino LJ, Wilcox BD, Davis FB, Gordinier JK, Davis PJ. Potentiation by thyroid hormone of human IFN-gamma-induced HLA-DR expression. J Immunol. (1998) 161:843–9.
22. Liu X, Zheng N, Shi YN, Yuan J, Li L. Thyroid hormone induced angiogenesis through the integrin alphavbeta3/protein kinase D/histone deacetylase 5 signaling pathway. J Mol Endocrinol. (2014) 52:245–54. doi: 10.1530/JME-13-0252
23. Cao HJ, Lin HY, Luidens MK, Davis FB, Davis PJ. Cytoplasm-to-nucleus shuttling of thyroid hormone receptor-beta1 (Trbeta1) is directed from a plasma membrane integrin receptor by thyroid hormone. Endocr Res. (2009) 34:31–42. doi: 10.1080/07435800902911810
24. Davis PJ, Shih A, Lin HY, Martino LJ, Davis FB. Thyroxine promotes association of mitogen-activated protein kinase and nuclear thyroid hormone receptor (TR) and causes serine phosphorylation of TR. J Biol Chem. (2000) 275:38032–9. doi: 10.1074/jbc.M002560200
25. Dombrowski F, Klotz L, Hacker HJ, Li Y, Klingmuller D, Brix K, et al. Hyperproliferative hepatocellular alterations after intraportal transplantation of thyroid follicles. Am J Pathol. (2000) 156:99–113. doi: 10.1016/S0002-9440(10)64710-4
26. Columbano A, Pibiri M, Deidda M, Cossu C, Scanlan TS, Chiellini G, et al. The thyroid hormone receptor-beta agonist GC-1 induces cell proliferation in rat liver and pancreas. Endocrinology. (2006) 147:3211–8. doi: 10.1210/en.2005-1561
27. Perra A, Simbula G, Simbula M, Pibiri M, Kowalik MA, Sulas P, et al. Thyroid hormone (T3) and TRbeta agonist GC-1 inhibit/reverse nonalcoholic fatty liver in rats. FASEB J. (2008) 22:2981–9. doi: 10.1096/fj.08-108464
28. Puliga E, Min Q, Tao J, Zhang R, Pradhan-Sundd T, Poddar M, et al. Thyroid hormone receptor-beta agonist GC-1 inhibits met-beta-catenin-driven hepatocellular cancer. Am J Pathol. (2017) 187:2473–85. doi: 10.1016/j.ajpath.2017.07.005
29. Yen CC, Huang YH, Liao CY, Liao CJ, Cheng WL, Chen WJ, et al. Mediation of the inhibitory effect of thyroid hormone on proliferation of hepatoma cells by transforming growth factor-beta. J Mol Endocrinol. (2006) 36:9–21. doi: 10.1677/jme.1.01911
30. Yoon CH, Kim MJ, Lee H, Kim RK, Lim EJ, Yoo KC, et al. PTTG1 oncogene promotes tumor malignancy via epithelial to mesenchymal transition and expansion of cancer stem cell population. J Biol Chem. (2012) 287:19516–27. doi: 10.1074/jbc.M111.337428
31. Chen R, Duan J, Li L, Ma Q, Sun Q, Ma J, et al. mTOR promotes pituitary tumor development through activation of PTTG1. Oncogene. (2017) 36:979–88. doi: 10.1038/onc.2016.264
32. Chen RN, Huang YH, Yeh CT, Liao CH, Lin KH. Thyroid hormone receptors suppress pituitary tumor transforming gene 1 activity in hepatoma. Cancer Res. (2008) 68:1697–706. doi: 10.1158/0008-5472.CAN-07-5492
33. Lin YH, Huang YH, Wu MH, Wu SM, Chi HC, Liao CJ, et al. Thyroid hormone suppresses cell proliferation through endoglin-mediated promotion of p21 stability. Oncogene. (2013) 32:3904–14. doi: 10.1038/onc.2013.5
34. Liao CH, Yeh CT, Huang YH, Wu SM, Chi HC, Tsai MM, et al. Dickkopf 4 positively regulated by the thyroid hormone receptor suppresses cell invasion in human hepatoma cells. Hepatology. (2012) 55:910–20. doi: 10.1002/hep.24740
35. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. (2016) 8:a019505. doi: 10.1101/cshperspect.a019505
36. Wu SM, Cheng WL, Liao CJ, Chi HC, Lin YH, Tseng YH, et al. Negative modulation of the epigenetic regulator, UHRF1, by thyroid hormone receptors suppresses liver cancer cell growth. Int J Cancer. (2015) 137:37–49. doi: 10.1002/ijc.29368
37. Tseng YH, Huang YH, Lin TK, Wu SM, Chi HC, Tsai CY, et al. Thyroid hormone suppresses expression of stathmin and associated tumor growth in hepatocellular carcinoma. Sci Rep. (2016) 6:38756. doi: 10.1038/srep38756
38. Chung IH, Liu H, Lin YH, Chi HC, Huang YH, Yang CC, et al. ChIP-on-chip analysis of thyroid hormone-regulated genes and their physiological significance. Oncotarget. (2016) 7:22448–59. doi: 10.18632/oncotarget.7988
39. Perra A, Kowalik MA, Pibiri M, Ledda-Columbano GM, Columbano A. Thyroid hormone receptor ligands induce regression of rat preneoplastic liver lesions causing their reversion to a differentiated phenotype. Hepatology. (2009) 49:1287–96. doi: 10.1002/hep.22750
40. Ledda-Columbano GM, Perra A, Concas D, Cossu C, Molotzu F, Sartori C, et al. Different effects of the liver mitogens triiodo-thyronine and ciprofibrate on the development of rat hepatocellular carcinoma. Toxicol Pathol. (2003) 31:113–20. doi: 10.1080/01926230390173851
41. Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. (2018) 18:5–18. doi: 10.1038/nrc.2017.99
42. Huang PS, Lin YH, Chi HC, Chen PY, Huang YH, Yeh CT, et al. Thyroid hormone inhibits growth of hepatoma cells through induction of miR-214. Sci Rep. (2017) 7:14868. doi: 10.1038/s41598-017-14864-1
43. Ruiz-Llorente L, Ardila-Gonzalez S, Fanjul LF, Martinez-Iglesias O, Aranda A. microRNAs 424 and 503 are mediators of the anti-proliferative and anti-invasive action of the thyroid hormone receptor beta. Oncotarget. (2014) 5:2918–33. doi: 10.18632/oncotarget.1577
44. Lin YH, Wu MH, Huang YH, Yeh CT, Chi HC, Tsai CY, et al. Thyroid hormone negatively regulates tumorigenesis through suppression of BC200. Endocr Relat Cancer. (2018) 25:967–79. doi: 10.1530/ERC-18-0176
45. Wang T, Xia L, Ma S, Qi X, Li Q, Xia Y, et al. Hepatocellular carcinoma: thyroid hormone promotes tumorigenicity through inducing cancer stem-like cell self-renewal. Sci Rep. (2016) 6:25183. doi: 10.1038/srep25183
46. Davis FB, Tang HY, Shih A, Keating T, Lansing L, Hercbergs A, et al. Acting via a cell surface receptor, thyroid hormone is a growth factor for glioma cells. Cancer Res. (2006) 66:7270–5. doi: 10.1158/0008-5472.CAN-05-4365
47. Hall LC, Salazar EP, Kane SR, Liu N. Effects of thyroid hormones on human breast cancer cell proliferation. J Steroid Biochem Mol Biol. (2008) 109:57–66. doi: 10.1016/j.jsbmb.2007.12.008
48. Tsui KH, Hsieh WC, Lin MH, Chang PL, Juang HH. Triiodothyronine modulates cell proliferation of human prostatic carcinoma cells by downregulation of the B-cell translocation gene 2. Prostate. (2008) 68:610–9. doi: 10.1002/pros.20725
49. Kress E, Skah S, Sirakov M, Nadjar J, Gadot N, Scoazec JY, et al. Cooperation between the thyroid hormone receptor TRalpha1 and the WNT pathway in the induction of intestinal tumorigenesis. Gastroenterology. (2010) 138:1863–74. doi: 10.1053/j.gastro.2010.01.041
50. Smyth PP. The thyroid and breast cancer: a significant association? Ann Med. (1997) 29:189–91. doi: 10.3109/07853899708999335
51. Lin K, Chen S, Zhu XG, Shieh H, McPhie P, Cheng S. The gene regulating activity of thyroid hormone nuclear receptors is modulated by cell-type specific factors. Biochem Biophys Res Commun. (1997) 238:280–4. doi: 10.1006/bbrc.1997.7285
52. Chan IH, Privalsky ML. Thyroid hormone receptor mutants implicated in human hepatocellular carcinoma display an altered target gene repertoire. Oncogene. (2009) 28:4162–74. doi: 10.1038/onc.2009.265
53. Wang C, Yang W, Yan HX, Luo T, Zhang J, Tang L, et al. Hepatitis B virus X (HBx) induces tumorigenicity of hepatic progenitor cells in 3,5-diethoxycarbonyl-1,4-dihydrocollidine-treated HBx transgenic mice. Hepatology. (2012) 55:108–20. doi: 10.1002/hep.24675
54. Chi HC, Chen SL, Lin SL, Tsai CY, Chuang WY, Lin YH, et al. Thyroid hormone protects hepatocytes from HBx-induced carcinogenesis by enhancing mitochondrial turnover. Oncogene. (2017) 36:5274–84. doi: 10.1038/onc.2017.136
55. Chi HC, Chen SL, Tsai CY, Chuang WY, Huang YH, Tsai MM, et al. Thyroid hormone suppresses hepatocarcinogenesis via DAPK2 and SQSTM1-dependent selective autophagy. Autophagy. (2016) 12:2271–85. doi: 10.1080/15548627.2016.1230583
56. Lohitesh K, Chowdhury R, Mukherjee S. Resistance a major hindrance to chemotherapy in hepatocellular carcinoma: an insight. Cancer Cell Int. (2018) 18:44. doi: 10.1186/s12935-018-0538-7
57. Chi HC, Chen SL, Liao CJ, Liao CH, Tsai MM, Lin YH, et al. Thyroid hormone receptors promote metastasis of human hepatoma cells via regulation of TRAIL. Cell Death Differ. (2012) 19:1802–14. doi: 10.1038/cdd.2012.58
58. Chi HC, Chen SL, Cheng YH, Lin TK, Tsai CY, Tsai MM, et al. Chemotherapy resistance and metastasis-promoting effects of thyroid hormone in hepatocarcinoma cells are mediated by suppression of FoxO1 and Bim pathway. Cell Death Dis. (2016) 7:e2324. doi: 10.1038/cddis.2016.227
59. Chen CY, Wu SM, Lin YH, Chi HC, Lin SL, Yeh CT, et al. Induction of nuclear protein-1 by thyroid hormone enhances platelet-derived growth factor A mediated angiogenesis in liver cancer. Theranostics. (2019) 9:2361–79. doi: 10.7150/thno.29628
60. Mitin T, Von Moltke LL, Court MH, Greenblatt DJ. Levothyroxine up-regulates P-glycoprotein independent of the pregnane X receptor. Drug Metab Dispos. (2004) 32:779–82. doi: 10.1124/dmd.32.8.779
61. Callaghan R, Luk F, Bebawy M. Inhibition of the multidrug resistance P-glycoprotein: time for a change of strategy? Drug Metab Dispos. (2014) 42:623–31. doi: 10.1124/dmd.113.056176
62. Davis PJ, Incerpi S, Lin HY, Tang HY, Sudha T, Mousa SA. Thyroid hormone and P-glycoprotein in tumor cells. Biomed Res Int. (2015) 2015:168427. doi: 10.1155/2015/168427
63. Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. (2013) 18:43–73. doi: 10.1615/CritRevOncog.v18.i1-2.40
64. Martinez-Iglesias O, Garcia-Silva S, Tenbaum SP, Regadera J, Larcher F, Paramio JM, et al. Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res. (2009) 69:501–9. doi: 10.1158/0008-5472.CAN-08-2198
65. Martinez-Iglesias O, Garcia-Silva S, Regadera J, Aranda A. Hypothyroidism enhances tumor invasiveness and metastasis development. PLoS ONE. (2009) 4:e6428. doi: 10.1371/journal.pone.0006428
66. Chen RN, Huang YH, Lin YC, Yeh CT, Liang Y, Chen SL, et al. Thyroid hormone promotes cell invasion through activation of furin expression in human hepatoma cell lines. Endocrinology. (2008) 149:3817–31. doi: 10.1210/en.2007-0989
67. Huang YH, Lin YH, Chi HC, Liao CH, Liao CJ, Wu SM, et al. Thyroid hormone regulation of miR-21 enhances migration and invasion of hepatoma. Cancer Res. (2013) 73:2505–17. doi: 10.1158/0008-5472.CAN-12-2218
68. Lin YH, Wu MH, Liao CJ, Huang YH, Chi HC, Wu SM, et al. Repression of microRNA-130b by thyroid hormone enhances cell motility. J Hepatol. (2015) 62:1328–40. doi: 10.1016/j.jhep.2014.12.035
69. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
70. Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, et al. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci USA. (2006) 103:14104–9. doi: 10.1073/pnas.0601600103
71. Liao CS, Tai PJ, Huang YH, Chen RN, Wu SM, Kuo LW, et al. Regulation of AKR1B1 by thyroid hormone and its receptors. Mol Cell Endocrinol. (2009) 307:109–17. doi: 10.1016/j.mce.2009.04.013
72. Wu SM, Huang YH, Lu YH, Chien LF, Yeh CT, Tsai MM, et al. Thyroid hormone receptor-mediated regulation of the methionine adenosyltransferase 1 gene is associated with cell invasion in hepatoma cell lines. Cell Mol Life Sci. (2010) 67:1831–43. doi: 10.1007/s00018-010-0281-2
73. Tseng YH, Ke PY, Liao CJ, Wu SM, Chi HC, Tsai CY, et al. Chromosome 19 open reading frame 80 is upregulated by thyroid hormone and modulates autophagy and lipid metabolism. Autophagy. (2014) 10:20–31. doi: 10.4161/auto.26126
74. Huang SF, Chang IC, Hong CC, Yen TC, Chen CL, Wu CC, et al. Metabolic risk factors are associated with non-hepatitis B non-hepatitis C hepatocellular carcinoma in Taiwan, an endemic area of chronic hepatitis B. Hepatol Commun. (2018) 2:747–59. doi: 10.1002/hep4.1182
75. Lin YH, Wu MH, Huang YH, Yeh CT, Lin KH. TUG1 is a regulator of AFP and serves as prognostic marker in non-hepatitis B non-hepatitis C hepatocellular carcinoma. Cells. (2020) 9:262. doi: 10.3390/cells9020262
76. Kowalik MA, Puliga E, Cabras L, Sulas P, Petrelli A, Perra A, et al. Thyroid hormone inhibits hepatocellular carcinoma progression via induction of differentiation and metabolic reprogramming. J Hepatol. (2020) 72:1159–69. doi: 10.1016/j.jhep.2019.12.018
77. Pinter M, Haupt L, Hucke F, Bota S, Bucsics T, Trauner M, et al. The impact of thyroid hormones on patients with hepatocellular carcinoma. PLoS ONE. (2017) 12:e0181878. doi: 10.1371/journal.pone.0181878
78. Chu YD, Lin KH, Huang YH, Lin CC, Hung CF, Yeh TS, et al. A novel thyroid function index associated with opposite therapeutic outcomes in advanced hepatocellular carcinoma patients receiving chemotherapy or sorafenib. Asia Pac J Clin Oncol. (2018) 14:e341–e51. doi: 10.1111/ajco.12983
79. Ventura-Holman T, Mamoon A, Subauste MC, Subauste JS. The effect of oncoprotein v-erbA on thyroid hormone-regulated genes in hepatocytes and their potential role in hepatocellular carcinoma. Mol Biol Rep. (2011) 38:1137–44. doi: 10.1007/s11033-010-0211-2
80. Shih YL, Huang YH, Lin KH, Chu YD, Yeh CT. Identification of functional thyroid stimulating hormone receptor and TSHR gene mutations in hepatocellular carcinoma. Anticancer Res. (2018) 38:2793–802. doi: 10.21873/anticanres.12523
81. Kasprzak A, Adamek A. Role of endoglin (cd105) in the progression of hepatocellular carcinoma and anti-angiogenic therapy. Int J Mol Sci. (2018) 19:3887. doi: 10.3390/ijms19123887
82. Shih TC, Tien YJ, Wen CJ, Yeh TS, Yu MC, Huang CH, et al. MicroRNA-214 downregulation contributes to tumor angiogenesis by inducing secretion of the hepatoma-derived growth factor in human hepatoma. J Hepatol. (2012) 57:584–91. doi: 10.1016/j.jhep.2012.04.031
83. Yang H, Zheng W, Shuai X, Chang RM, Yu L, Fang F, et al. MicroRNA-424 inhibits Akt3/E2F3 axis and tumor growth in hepatocellular carcinoma. Oncotarget. (2015) 6:27736–50. doi: 10.18632/oncotarget.4811
Keywords: thyroid hormone, thyroid hormone receptor, hypothyroidism, therapeutic target, hepatocellular carcinoma
Citation: Lin Y-H, Lin K-H and Yeh C-T (2020) Thyroid Hormone in Hepatocellular Carcinoma: Cancer Risk, Growth Regulation, and Anticancer Drug Resistance. Front. Med. 7:174. doi: 10.3389/fmed.2020.00174
Received: 26 November 2019; Accepted: 15 April 2020;
Published: 22 May 2020.
Edited by:
Andrea Perra, University of Cagliari, ItalyReviewed by:
Diego Francesco Calvisi, University of Regensburg, GermanyHai Feng, Harbin Medical University, China
Hung-Yun Lin, Taipei Medical University, Taiwan
Copyright © 2020 Lin, Lin and Yeh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chau-Ting Yeh, Y2hhdXRpbmd5JiN4MDAwNDA7Z21haWwuY29t