 Hee-Seong Jang
Hee-Seong Jang Mi Ra Noh
Mi Ra Noh Jinu Kim
Jinu Kim Babu J. Padanilam
Babu J. Padanilam
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 12 March 2020
Sec. Nephrology
Volume 7 - 2020 | https://doi.org/10.3389/fmed.2020.00065
This article is part of the Research Topic Renal Function in Acute and Chronic Kidney Diseases View all 31 articles
The kidney is a highly metabolic organ and uses high levels of ATP to maintain electrolyte and acid-base homeostasis and reabsorb nutrients. Energy depletion is a critical factor in development and progression of various kidney diseases including acute kidney injury (AKI), chronic kidney disease (CKD), and diabetic and glomerular nephropathy. Mitochondrial fatty acid β-oxidation (FAO) serves as the preferred source of ATP in the kidney and its dysfunction results in ATP depletion and lipotoxicity to elicit tubular injury and inflammation and subsequent fibrosis progression. This review explores the current state of knowledge on the role of mitochondrial FAO dysfunction in the pathophysiology of kidney diseases including AKI and CKD and prospective views on developing therapeutic interventions based on mitochondrial energy metabolism.
The kidney demands a high energy supply to generate energy-required transport of glucose, ions, and nutrients from blood filtrate (1). Mitochondria is an essential organelle generating ATP through oxidative metabolism, as well as regulation of redox homeostasis and cell death signaling. Loss or depletion of ATP by renal tubular mitochondrial disturbance causes acute and chronic kidney diseases (2–5). Renal tubules, particularly proximal tubules that have abundant mitochondria, are metabolically active due to reabsorption of most glomerular filtrate. Because medullary region with pars recta adjunct proximal tubule has only 5–10% of total renal blood flow with tissue oxygen tension 10–20 mm Hg, the medullary proximal tubules are highly vulnerable to hypoxic condition such as ischemia/reperfusion injury (6, 7). Mitochondrial fatty acid β-oxidation (FAO) in proximal tubule is a major source of ATP generation, and its impairment is linked to ATP depletion-induced acute kidney injury (AKI) (1), lipotoxicity (8, 9), and its long-term sequelae leading to CKD (10). Several reports demonstrate that AKI is an independent risk factor for CKD (11–15), and thus promoting mitochondrial FAO is a first-rate option for preventing AKI and CKD. Recent reports indicate that podocytes are also highly sensitive to acute and chronic stimuli, because podocytes have a limited ability for adaptation to mitochondrial energy crisis (16). Here, we summarize the recent findings associated with mitochondrial dysregulation, particularly defective fatty acid (FA) metabolism and lipotoxicity in kidney diseases, which includes tubular and glomerular injury. We also discuss therapeutic strategies targeting mitochondrial energy metabolism in kidney diseases.
The mitochondria are highly dynamic intracellular organelles that generate most of the ATP in tissues, including the kidney (17). The kidney has abundant mitochondria to produce high levels of ATP through oxidative phosphorylation, to accomplish the substantial passive or active reabsorption of components of the glomerular filtrate, including various ions, glucose, and nutrients. Oxidative phosphorylation yields 36 ATP per glucose, which is highly efficient compared to that of glycolysis generating only 2 ATP (18). The most efficient ATP-generating system in cell energy metabolism is FAO, which can generate 106–129 ATP, depending on the number of carbons in the FA chain. The proximal tubule transports ~67% of glomerular filtrate and thus requires high levels of ATP for its function (19). The proximal tubule prefers FAO for ATP production and has low metabolic flexibility to glycolysis (6, 20). Moreover, it should be noted that the outer medullary proximal tubule has lower oxygen tension under normal conditions and thus less capacity to cope with hypoxic condition (6), which makes them highly sensitive to acute and chronic stimuli. On the other hand, distal tubule is less susceptible to acute stimuli such as ischemic injury and nephrotoxins, because it has better capacity for glycolytic adaptation during hypoxic/ischemic condition, despite its high energy requirement (6, 21, 22). The glomerular podocyte, which has less mitochondria than proximal tubule and depends on mitochondrial respiration for ~75% of energy, also has high vulnerability to stimuli such as glycemic condition (16), but the mechanism of its susceptibility remains to be fully defined. In diseased kidneys with impaired FAO, glycolysis and glutaminolysis can serve as a significant energy source. For example, in polycystic kidney disease (PKD), metabolic reprogramming by increased glutaminolysis, as well as glycolysis, occurs to cope with impaired FAO (23). However, in the ischemic kidney, it has been reported that poly (ADP-ribose) Polymerase 1 and Tp53 induced glycolysis and apoptosis regulator are selectively activated in the injured proximal tubules and inhibit glycolysis during ischemic injury (24, 25). This will prevent compensation of ATP production by glycolysis and makes the proximal tubules extremely vulnerable. Intriguingly, recent reports suggest upregulation of glycolysis as a compensatory mechanism to adapt to reduced FAO during persistent acute tubular injury, which may be related with tubular repair mechanism, resulting in chronic inflammation and fibrosis progression (26–28). These studies indicate that adaptation of energy metabolism for loss of mitochondrial ATP could be compensated by other metabolic processes such as glycolysis or glutaminolysis, suggesting that regulatory mechanism of metabolic pathways can be a key to develop a valuable target for treatment of kidney diseases.
Defective FA uptake, synthesis, and oxidation are tightly linked to development and progression of kidney diseases. In proximal tubular cells, FA can be taken up by membrane FA transport proteins, such as CD36 and FA-binding protein (FABP), as well as by reabsorption from glomerular filtrate by endocytosis of receptor-mediated FA-bound albumin (20, 29, 30). The kidney with AKI accumulates FAs in cytoplasm, which is a result of dysregulated FAO, leading to ATP depletion (1). Fatty acid is activated to acyl-CoA to make it permeable to the outer mitochondrial membrane (OMM) by acyl-CoA synthetases in the cytosol. Carnitine palmitoyltransferase-1 (Cpt-1), located on the OMM, catalyzes transesterification of the acyl-CoA to acylcarnitine (20, 21, 31). Acylcarnitine is shuttled across the inner mitochondrial membrane (IMM) through carnitine–acylcarnitine translocase. Acylcarnitine is reconverted to acyl-CoA by Cpt-2, an IMM protein. In the mitochondrial matrix, through β-oxidation, a serial cyclic process is trimmed the acyl-CoA to form molecules of acetyl-CoA (21). Finally, acetyl-CoA is fed into the tricarboxylic acid cycle, to generate NADH and FADH2 that serve as electron donors to the electron transport chain for ATP production (20, 29, 31) (Figure 1). It is well-known that downregulated or deficient CPT-1 or CPT-2 is critical to impaired FAO in diverse kidney diseases, such as ischemic and cisplatin AKI and diabetic nephropathy (32–34).
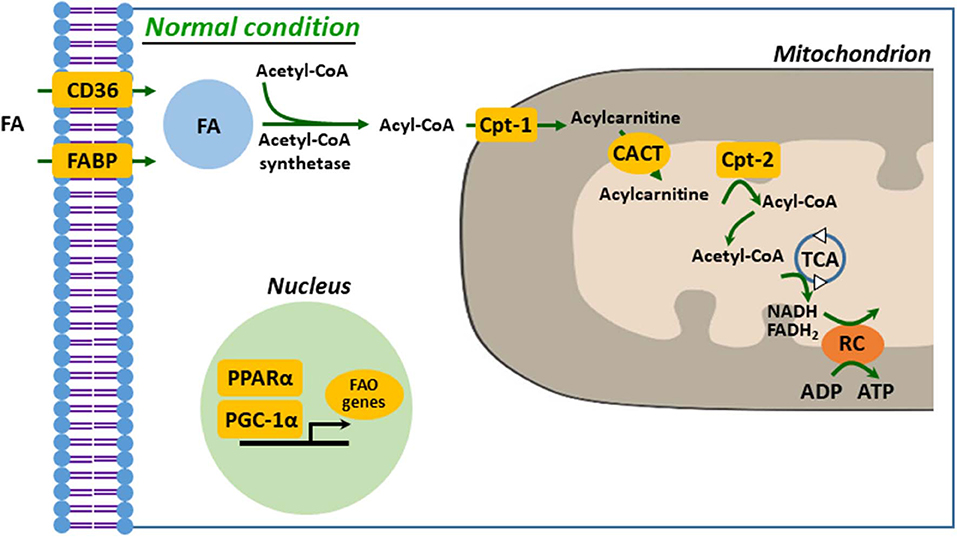
Figure 1. Mitochondrial fatty acid oxidation in kidney tubule. FA enters into cytosol of renal proximal tubule cell (PTC) via FABP or CD36. In the cytosol, FA are converted from acetyl-CoA to acyl-CoA by acetyl-CoA synthetase and then transferred to mitochondrial matrix by carnitine shuttle, Cpt-1, CACT, and Cpt-2, step by step. Acyl-CoA undergoes β-oxidation to produce acetyl-CoA for TCA. NADH and FADH2 generated by TCA are used as electron donors for RC. FA, fatty acid; FAO, fatty acid β-oxidation; Cpt, carnitine O-palmitoyltransferase; CACT, carnitine-acylcarnitine translocase; TCA, tricarboxylic acid cycle; RC, respiratory chain.
It is well-recognized that peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α)–peroxisome proliferator-activated receptor α (PPARα) axis governs transcription and regulation of FAO genes in diverse tissues, including the kidney, and its regulation has been suggested as a therapeutic target of AKI and CKD (10, 20, 35–37). We (38) and others (39–42) suggested that defective mitochondrial FAO is critical to ischemic and cisplatin-induced AKI (Figure 2). Downregulation of activity and expression of PPARα and/or PGC-1α resulted in inhibited transcriptional regulation of FAO genes, such as Cpt-1 and medium chain–specific acyl-CoA dehydrogenase, leading to decreased mitochondrial FAO (38, 40, 45). Enhanced PPARα activation by fenofibrate protects histological and functional impairment in cisplatin AKI (46). However, the upstream signaling pathway that inhibits PPARα-regulated FAO in AKI is under investigation.
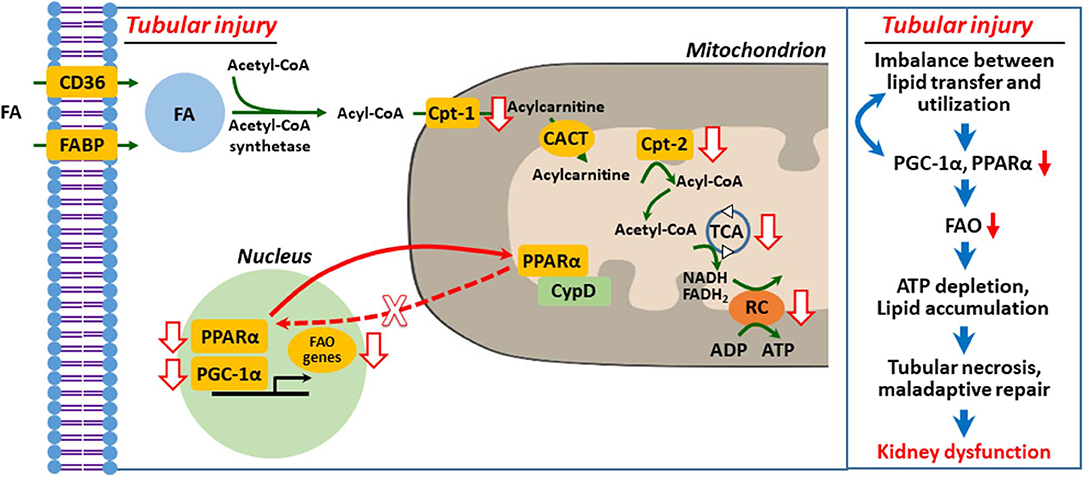
Figure 2. Defective mitochondrial fatty acid oxidation and lipid accumulation in injured kidney tubular cell. Upon tubular injury, PPARα translocates to mitochondria and binds with cyclophilin D (CypD), resulting in mitochondrial sequestration and decreased transcriptional activity of PPARα for FAO genes (38). Inhibition of FAO genes depletes ATP by impaired FAO and that in turn induces PTC necrosis, maladaptive repair, and kidney dysfunction (1, 9, 20, 29, 36, 38, 39, 43, 44). FA, fatty acid; FAO, fatty acid β-oxidation; Cpt, carnitine O-palmitoyltransferase; CACT, carnitine-acylcarnitine translocase; TCA, tricarboxylic acid cycle; RC, respiratory chain.
In our recent report (38), we hypothesized that mitochondrial interaction of proximal tubule cyclophilin D (CypD) and PPARα modulates FAO in cisplatin AKI. We demonstrated using genetic and pharmacological intervention, protein–protein interaction studies, and bioinformatics that mitochondrial CypD-PPARα binding, which modulates FAO, occurs in proximal tubule during cisplatin AKI. Mitochondrial translocation of PPARα, its binding to CypD, and sequestration led to inhibition of its nuclear translocation and transcription of PPARα-regulated FAO genes during cisplatin AKI, leading to reduced FAO, lipid accumulation, and lipotoxicity. Pharmacological or genetic inhibition of CypD promoted nuclear translocation of PPARα and enhanced the transcription of FAO genes and prevented cisplatin AKI (Figure 2).
In CKD, the PGC1α-PPARα axis and FAO key enzymes such as Cpt-1 are persistently decreased. In hypertensive and folic acid–induced CKD, tubular lipid accumulation related to defective FAO, along with tubular and functional impairment, is reported (10, 47). In proximal tubule exposed to FA, PPARα activation can eliminate ceramides, which are toxic metabolites contributing to lipotoxicity (20, 43, 48). On the other hand, in addition to its role in FAO, PGC-1α can act as a master regulator of mitochondrial biogenesis and NAD biosynthesis. This topic is reviewed in detail elsewhere (1, 8, 36, 37, 49).
Glomerular podocytes are specialized epithelial cells, integrating structural and functional maintenance of glomerular filtration barrier. Podocytes consume high energy for their function, which makes the cells highly susceptible to ATP depletion and to acute and chronic injury (16, 50, 51). Podocyte injury and loss contribute to initiation and progression of proteinuric glomerular diseases, including diabetic nephropathy, glomerular sclerosis, and membranous nephropathy (51–53). In hyperglycemic condition, mitochondrial FAO is enhanced in podocytes, but when both FAO and glycolysis were inhibited, it has a limited capacity to adapt to the changed condition and sensitize podocytes to apoptosis (16, 54). In addition, because mitochondrial respiration accounts for 75% of energy, podocytes have less glycolytic flexibility when mitochondrial function is impaired, resulting in energy deficit (16, 55). However, because glomerulus consists of other cells, including mesangial cell and endothelial cells, all of which could interact with podocytes, in diseased conditions, data from in vitro studies using only podocytes should be interpret cautiously (16). On the other hand, it seems that PPARα activation impedes the progression of diabetic nephropathy. PPARα is increased in the kidneys of streptozotocin-induced diabetic mouse model, but when PPARα was genetically deleted, the mice showed adverse effects, including glomerulosclerosis (56). Similar findings from types 1 and 2 diabetic animal models demonstrated that activation of PPARα by fibrates improved hyperglycemic and/or dislipidemic condition–induced glomerular injury and function along with lipid lowering effect (56–60). Although human data (61) revealed that fibrates improve diabetic nephropathy such as albuminuria, data from rodent studies demonstrate more effectiveness than those of clinical studies, suggesting that rodents are more sensitive to PPARα signaling. These data suggest that caution is needed to interpreting the effectiveness of fibrate treatment from animals to humans (61, 62).
Many genetic disorders are involved in initiation and progression of kidney diseases. Polycystic kidney disease due to mutations in PKD1 and PKD2, which produces polycystin 1 and 2, respectively, are the most common monogenic human kidney diseases, showing 100–1,000 fluid-filled renal cysts (63). A number of signaling pathways, including cAMP, calcium, cell cycle, mTORC1, and WNT signaling, are involved in PKD pathogenesis (63, 64). Recent reports demonstrated defective FAO, as well as glucose metabolism, can contribute to the pathogenesis of both human and animal autosomal dominant PKD (ADPKD) (64, 65). Polycystin proteins seem to be involved in mitochondrial function, because epithelial Pkd1 inactivation from proximal or distal tubule resulted in lower FAO with unchanged glycolysis (66, 67). It is reported that loss of Pkd1 drives cyst growth with declined FAO via direct repression of PPARα (23, 66). Upregulation of PPARα by fenofibrate enhanced FAO and showed beneficial effect in slowing PKD progression by suppressed renal cyst growth, fibrosis, and improved function in a slowly progressing orthologous model of ADPKD (68). On the other hand, the role of FAO in autosomal recessive PKD, a recessive form of PKD that is a rare genetic disorder characterized by enlarged kidney and biliary dysgenesis (63, 69), remains largely unknown.
Although the causal relationship is unclear, a number of reports suggest that lipid accumulation in certain tissue and cell could be harmful and is referred to as lipotoxicity (43, 70, 71). The initial hypothesis regarding lipotoxicity was that intrarenal lipid accumulation can affect structure and function in renal cells, including proximal tubule cell (71, 72). Accumulation of triglyceride, which is produced by dysregulated glycerol and non-esterified FA (NEFA) presumably derived from impaired FA transport and/or FAO in cytoplasm causes lipotoxicity, contributing to decreased production of ATP and mitochondrial energy metabolism (43, 44). NEFA triggers mitochondrial dysfunction as a cause of energetic failure of proximal tubules during hypoxia/reoxygenation, and intracellular accumulation of NEFA and triglycerides with downregulation of mitochondrial FAO (43, 73). Accumulation of triglycerides is observed in tubule injured by ischemic, cisplatin, glycerol-induced, and septic AKI, as well as in kidneys with metabolic syndrome or fibrosis progression (10, 44, 71, 74). Lipid accumulation in ischemic proximal tubule may result in persistent energy depletion with NEFA-induced mitochondrial dysfunction (43). In parallel, high-fat diet or palmitic acid overload resulted in upregulation of inflammation, fibrosis, or cell death in kidneys (75–77). However, it is still under debate whether FA or triglyceride per se is toxic, but it is clear that intrarenal lipid accumulation, by as of yet undefined mechanisms, can represent characteristics of diseased status (43, 70, 78). Recent data show that in two CKD mouse models (diabetic nephropathy and folic acid nephropathy) lipid accumulation by kidney cell–specific overexpression of CD36, a key membrane protein for FA uptake in proximal tubule (79, 80), did not generate renal fibrosis (10). It is proposed that mitochondrial defects in energy production are more detrimental than the lipid accumulation in the cytoplasm. Further studies to define the causal relationship between lipid accumulation and energy depletion and the effect of lipotoxicity during AKI and CKD are warranted.
A number of studies targeting mitochondrial dysfunction in kidney diseases have been investigated in both animals and human (29). The most treatable option targeting defective FAO in AKI and CKD to date is using agonists of PPARα, fibrates–fenofibrate, clofibrate, and others, despite its adverse effects (81, 82). Fibrates showed a preventive effect to tubular cell death and dysregulated intracellular lipid accumulation, in ischemic and cisplatin AKI models, and in high-fat diet or folic acid–induced CKD models (46, 83–86). However, fenofibrate treatment has adverse effects in kidney function as demonstrated by decreased glomerular filtrate rate and/or increased serum creatinine independent of its lipid-lowering effect (82, 87–89). These data suggest that a better understanding of the molecular mechanism of PPARα agonists and its tissue specificity is required to assess the effectiveness of fenofibrate therapy. Another promising option to modulate FAO is to target FA synthase or transporter. Administration of 5A peptide, which targets CD36 to inhibit FA transport into cell, showed promising results by lowering intrarenal lipid level in subtotal nephrectomized mice kidneys (90). Like CD36 antagonist, a blocker of FA synthase, C75, showed beneficial effect in suppression of folic acid–induced kidney fibrosis progression (10).
Other treatment strategies targeting mitochondria, but not targeting FAO per se, include the use of SS-31 (Szeto-Schiller 31) and MitoQ or MitoT. SS-31, mitochondria-targeting tetrapeptides, preserved mitochondrial structure in both proximal tubules and podocytes and thus enhanced functional recovery from ischemic AKI and prevents its long-term consequences, including interstitial fibrosis and glomerulosclerosis (91, 92). In high-fat diet–mediated proximal tubule injury, SS-31 lowered intracellular lipid accumulation by suppressing disruption of mitochondrial function (93). Mitochondria-targeted lipophilic antioxidants, MitoQ and MitoT, protected tubular injury and kidney dysfunction through suppression of mitochondrial damage and oxidative stress and improvement of mitochondrial NADPH level in septic or cisplatin AKI (94, 95). One of the major barriers to develop treatment strategies targeting mitochondrial dysfunction in AKI and CKD is to take into consideration that mitochondria is an organelle regulating redox homeostasis by reactive oxygen species production and detoxification, and its dysregulation could increase oxidative stress (8, 96). Thus, an integrated understanding for mitochondrial biology, including mitochondrial energy metabolism and redox signaling, in particular in susceptible kidney segments, should be preceded to minimize the side effects of mitochondrial targeting in kidney diseases.
Mitochondrial dysregulation, resulting in loss of ATP, is critical to energy homeostasis and pathogenesis of kidney diseases. Acute and chronic disturbance of mitochondrial FA metabolism depletes ATP, leading to tubular and glomerular injury. Lipotoxicity via impaired FA metabolism could induce cell death and inflammation and promote the chronic progression of AKI to CKD. Unveiling the role and the related molecular mechanism of mitochondrial energy metabolism is required for the development of effective therapeutics in targeting tubular and glomerular injury in acute and chronic kidney diseases.
H-SJ and BP made substantial contributions to the conception and drafting the work and revising it critically for important intellectual content, provided approval for publication of the content, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. MN and JK made contributions to drafting the work or revising it critically for important intellectual content and provided approval for publication of the content.
This work was supported by NIH grants DK-116987, DK-120533, DK120846, and American Heart Association (AHA) Grant in Aid 15GRNT25080031 (BP), AHA postdoctoral fellowship Grant 15POST25130003 (H-SJ), and grants (NRF-2016R1C1B2012080 and NRF-2019R1F1A1041410) from the National Research Foundation of Korea (JK).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. (2017) 13:629–46. doi: 10.1038/nrneph.2017.107
2. Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. (2012) 2:1303–53. doi: 10.1002/cphy.c110041
3. Maekawa H, Inagi R. Pathophysiological role of organelle stress/crosstalk in AKI-to-CKD transition. Semin Nephrol. (2019) 39:581–8. doi: 10.1016/j.semnephrol.2019.10.007
4. Weinberg JM, Venkatachalam MA, Roeser NF, Saikumar P, Dong Z, Senter RA, et al. Anaerobic and aerobic pathways for salvage of proximal tubules from hypoxia-induced mitochondrial injury. Am J Physiol Renal Physiol. (2000) 279:F927–43. doi: 10.1152/ajprenal.2000.279.5.F927
5. Ying Y, Padanilam BJ. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell Mol Life Sci. (2016) 73:2309–24. doi: 10.1007/s00018-016-2202-5
6. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. (2011) 121:4210–21. doi: 10.1172/JCI45161
7. Nourbakhsh N, Singh P. Role of renal oxygenation and mitochondrial function in the pathophysiology of acute kidney injury. Nephron Clin Pract. (2014) 127:149–52. doi: 10.1159/000363545
8. Hall AM, Schuh CD. Mitochondria as therapeutic targets in acute kidney injury. Curr Opin Nephrol Hypertens. (2016) 25:355–62. doi: 10.1097/MNH.0000000000000228
9. Stallons LJ, Funk JA, Schnellmann RG. Mitochondrial homeostasis in acute organ failure. Curr Pathobiol Rep. (2013) 1:169–77. doi: 10.1007/s40139-013-0023-x
10. Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. (2015) 21:37–46. doi: 10.1038/nm.3762
11. Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, et al. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. (2016) 27:687–97. doi: 10.1681/ASN.2015030309
12. Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. (2014) 371:58–66. doi: 10.1056/NEJMra1214243
13. Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. (2012) 81:442–8. doi: 10.1038/ki.2011.379
14. Jang HS, Kim JI, Han SJ, Park KM. Recruitment and subsequent proliferation of bone marrow-derived cells in the postischemic kidney are important to the progression of fibrosis. Am J Physiol Renal Physiol. (2014) 306:F1451–61. doi: 10.1152/ajprenal.00017.2014
15. Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. (2015) 26:1765–76. doi: 10.1681/ASN.2015010006
16. Muller-Deile J, Schiffer M. The podocyte power-plant disaster and its contribution to glomerulopathy. Front Endocrinol. (2014) 5:209. doi: 10.3389/fendo.2014.00209
17. Chandel NS. Evolution of mitochondria as signaling organelles. Cell Metab. (2015) 22:204–6. doi: 10.1016/j.cmet.2015.05.013
18. Mandel LJ. Primary active sodium transport, oxygen consumption, and ATP: coupling and regulation. Kidney Int. (1986) 29:3–9. doi: 10.1038/ki.1986.2
20. Simon N, Hertig A. Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front Med. (2015) 2:52. doi: 10.3389/fmed.2015.00052
21. Houten SM, Violante S, Ventura FV, Wanders RJ. The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu Rev Physiol. (2016) 78:23–44. doi: 10.1146/annurev-physiol-021115-105045
22. Weinberg JM, Molitoris BA. Illuminating mitochondrial function and dysfunction using multiphoton technology. J Am Soc Nephrol. (2009) 20:1164–6. doi: 10.1681/ASN.2009040419
23. Podrini C, Rowe I, Pagliarini R, Costa ASH, Chiaravalli M, Di Meo I, et al. Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun Biol. (2018) 1:194. doi: 10.1038/s42003-018-0200-x
24. Devalaraja-Narashimha K, Padanilam BJ. PARP-1 inhibits glycolysis in ischemic kidneys. J Am Soc Nephrol. (2009) 20:95–103. doi: 10.1681/ASN.2008030325
25. Kim J, Devalaraja-Narashimha K, Padanilam BJ. TIGAR regulates glycolysis in ischemic kidney proximal tubules. Am J Physiol Renal Physiol. (2015) 308:F298–308. doi: 10.1152/ajprenal.00459.2014
26. Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, et al. Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J Am Soc Nephrol. (2016) 27:3356–67. doi: 10.1681/ASN.2015020177
27. Lemos DR, McMurdo M, Karaca G, Wilflingseder J, Leaf IA, Gupta N, et al. Interleukin-1beta activates a MYC-dependent metabolic switch in kidney stromal cells necessary for progressive tubulointerstitial fibrosis. J Am Soc Nephrol. (2018) 29:1690–705. doi: 10.1681/ASN.2017121283
28. Wei Q, Su J, Dong G, Zhang M, Huo Y, Dong Z. Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am J Physiol Renal Physiol. (2019) 316:F1162–72. doi: 10.1152/ajprenal.00422.2018
29. Szeto HH. Pharmacologic approaches to improve mitochondrial function in AKI and CKD. J Am Soc Nephrol. (2017) 28:2856–65. doi: 10.1681/ASN.2017030247
30. Yang X, Okamura DM, Lu X, Chen Y, Moorhead J, Varghese Z, et al. CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat Rev Nephrol. (2017) 13:769–81. doi: 10.1038/nrneph.2017.126
31. Aon MA, Bhatt N, Cortassa SC. Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol. (2014) 5:282. doi: 10.3389/fphys.2014.00282
32. Feingold KR, Wang Y, Moser A, Shigenaga JK, Grunfeld C. LPS decreases fatty acid oxidation and nuclear hormone receptors in the kidney. J Lipid Res. (2008) 49:2179–87. doi: 10.1194/jlr.M800233-JLR200
33. Idrovo JP, Yang WL, Nicastro J, Coppa GF, Wang P. Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J Surg Res. (2012) 177:157–64. doi: 10.1016/j.jss.2012.05.053
34. Portilla D. Role of fatty acid beta-oxidation and calcium-independent phospholipase A2 in ischemic acute renal failure. Curr Opin Nephrol Hypertens. (1999) 8:473–7. doi: 10.1097/00041552-199907000-00012
35. Han SH, Wu MY, Nam BY, Park JT, Yoo TH, Kang SW, et al. PGC-1alpha protects from notch-induced kidney fibrosis development. J Am Soc Nephrol. (2017) 28:3312–22. doi: 10.1681/ASN.2017020130
36. Tran M, Parikh SM. Mitochondrial biogenesis in the acutely injured kidney. Nephron Clin Pract. (2014) 127:42–5. doi: 10.1159/000363715
37. Weinberg JM. Mitochondrial biogenesis in kidney disease. J Am Soc Nephrol. (2011) 22:431–6. doi: 10.1681/ASN.2010060643
38. Jang HS, Noh MR, Jung EM, Kim WY, Southekal S, Guda C, et al. Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int. (2020) 97:327–39. doi: 10.1016/j.kint.2019.08.019
39. Portilla D. Energy metabolism and cytotoxicity. Semin Nephrol. (2003) 23:432–8. doi: 10.1016/S0270-9295(03)00088-3
40. Portilla D, Dai G, McClure T, Bates L, Kurten R, Megyesi J, et al. Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. (2002) 62:1208–18. doi: 10.1111/j.1523-1755.2002.kid553.x
41. Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARalpha-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol. (2000) 278:F667–75. doi: 10.1152/ajprenal.2000.278.4.F667
42. Portilla D, Li S, Nagothu KK, Megyesi J, Kaissling B, Schnackenberg L, et al. Metabolomic study of cisplatin-induced nephrotoxicity. Kidney Int. (2006) 69:2194–204. doi: 10.1038/sj.ki.5000433
44. Zager RA, Johnson AC, Hanson SY. Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int. (2005) 67:111–21. doi: 10.1111/j.1523-1755.2005.00061.x
45. Tran MT, Zsengeller ZK, Berg AH, Khankin EV, Bhasin MK, Kim W, et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. (2016) 531:528–32. doi: 10.1038/nature17184
46. Nagothu KK, Bhatt R, Kaushal GP, Portilla D. Fibrate prevents cisplatin-induced proximal tubule cell death. Kidney Int. (2005) 68:2680–93. doi: 10.1111/j.1523-1755.2005.00739.x
47. Kim HJ, Moradi H, Yuan J, Norris K, Vaziri ND. Renal mass reduction results in accumulation of lipids and dysregulation of lipid regulatory proteins in the remnant kidney. Am J Physiol Renal Physiol. (2009) 296:F1297–306. doi: 10.1152/ajprenal.90761.2008
48. Kamijo Y, Hora K, Kono K, Takahashi K, Higuchi M, Ehara T, et al. PPARalpha protects proximal tubular cells from acute fatty acid toxicity. J Am Soc Nephrol. (2007) 18:3089–100. doi: 10.1681/ASN.2007020238
49. Ralto KM, Rhee EP, Parikh SM. NAD(+) homeostasis in renal health and disease. Nat Rev Nephrol. (2020) 16:99–111. doi: 10.1038/s41581-019-0216-6
50. Imasawa T, Rossignol R. Podocyte energy metabolism and glomerular diseases. Int J Biochem Cell Biol. (2013) 45:2109–18. doi: 10.1016/j.biocel.2013.06.013
51. Sieber J, Jehle AW. Free Fatty acids and their metabolism affect function and survival of podocytes. Front Endocrinol. (2014) 5:186. doi: 10.3389/fendo.2014.00186
52. Mallipattu SK, He JC. The podocyte as a direct target for treatment of glomerular disease? Am J Physiol Renal Physiol. (2016) 311:F46–51. doi: 10.1152/ajprenal.00184.2016
54. Stieger N, Worthmann K, Teng B, Engeli S, Das AM, Haller H, et al. Impact of high glucose and transforming growth factor-beta on bioenergetic profiles in podocytes. Metabolism. (2012) 61:1073–86. doi: 10.1016/j.metabol.2011.12.003
55. Abe Y, Sakairi T, Kajiyama H, Shrivastav S, Beeson C, Kopp JB. Bioenergetic characterization of mouse podocytes. Am J Physiol Cell Physiol. (2010) 299:C464–76. doi: 10.1152/ajpcell.00563.2009
56. Park CW, Kim HW, Ko SH, Chung HW, Lim SW, Yang CW, et al. Accelerated diabetic nephropathy in mice lacking the peroxisome proliferator-activated receptor alpha. Diabetes. (2006) 55:885–93. doi: 10.2337/diabetes.55.04.06.db05-1329
57. Cernuda-Morollon E, Rodriguez-Pascual F, Klatt P, Lamas S, Perez-Sala D. PPAR agonists amplify iNOS expression while inhibiting NF-kappaB: implications for mesangial cell activation by cytokines. J Am Soc Nephrol. (2002) 13:2223–31. doi: 10.1097/01.ASN.0000025786.87646.B1
58. Chen L, Zhang J, Zhang Y, Wang Y, Wang B. Improvement of inflammatory responses associated with NF-kappa B pathway in kidneys from diabetic rats. Inflamm Res. (2008) 57:199–204. doi: 10.1007/s00011-006-6190-z
59. Chen LL, Zhang JY, Wang BP. Renoprotective effects of fenofibrate in diabetic rats are achieved by suppressing kidney plasminogen activator inhibitor-1. Vascul Pharmacol. (2006) 44:309–15. doi: 10.1016/j.vph.2006.01.004
60. Muller DN, Theuer J, Shagdarsuren E, Kaergel E, Honeck H, Park JK, et al. A peroxisome proliferator-activated receptor-alpha activator induces renal CYP2C23 activity and protects from angiotensin II-induced renal injury. Am J Pathol. (2004) 164:521–32. doi: 10.1016/S0002-9440(10)63142-2
61. Kouroumichakis I, Papanas N, Zarogoulidis P, Liakopoulos V, Maltezos E, Mikhailidis DP. Fibrates: therapeutic potential for diabetic nephropathy? Eur J Intern Med. (2012) 23:309–16. doi: 10.1016/j.ejim.2011.12.007
62. Hennuyer N, Poulain P, Madsen L, Berge RK, Houdebine LM, Branellec D, et al. Beneficial effects of fibrates on apolipoprotein A-I metabolism occur independently of any peroxisome proliferative response. Circulation. (1999) 99:2445–51. doi: 10.1161/01.CIR.99.18.2445
63. Wilson PD. Polycystic kidney disease. N Engl J Med. (2004) 350:151–64. doi: 10.1056/NEJMra022161
64. Padovano V, Podrini C, Boletta A, Caplan MJ. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat Rev Nephrol. (2018) 14:678–87. doi: 10.1038/s41581-018-0051-1
65. Podrini C, Cassina L, Boletta A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell Signal. (2019) 67:109495. doi: 10.1016/j.cellsig.2019.109495
66. Hajarnis S, Lakhia R, Yheskel M, Williams D, Sorourian M, Liu X, et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun. (2017) 8:14395. doi: 10.1038/ncomms14395
67. Menezes LF, Lin CC, Zhou F, Germino GG. Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine. (2016) 5:183–92. doi: 10.1016/j.ebiom.2016.01.027
68. Lakhia R, Yheskel M, Flaten A, Quittner-Strom EB, Holland WL, Patel V. PPARalpha agonist fenofibrate enhances fatty acid beta-oxidation and attenuates polycystic kidney and liver disease in mice. Am J Physiol Renal Physiol. (2018) 314:F122–31. doi: 10.1152/ajprenal.00352.2017
69. Bergmann C. Genetics of autosomal recessive polycystic kidney disease and its differential diagnoses. Front Pediatr. (2017) 5:221. doi: 10.3389/fped.2017.00221
70. Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens. (2010) 19:393–402. doi: 10.1097/MNH.0b013e32833aa4ac
71. Moorhead JF, Chan MK, El-Nahas M, Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet. (1982) 2:1309–11. doi: 10.1016/S0140-6736(82)91513-6
72. Ruan XZ, Varghese Z, Moorhead JF. An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol. (2009) 5:713–21. doi: 10.1038/nrneph.2009.184
73. Feldkamp T, Kribben A, Roeser NF, Senter RA, Weinberg JM. Accumulation of nonesterified fatty acids causes the sustained energetic deficit in kidney proximal tubules after hypoxia-reoxygenation. Am J Physiol Renal Physiol. (2006) 290:F465–77. doi: 10.1152/ajprenal.00305.2005
74. Wahba IM, Mak RH. Obesity and obesity-initiated metabolic syndrome: mechanistic links to chronic kidney disease. Clin J Am Soc Nephrol. (2007) 2:550–62. doi: 10.2215/CJN.04071206
75. Jiang T, Wang Z, Proctor G, Moskowitz S, Liebman SE, Rogers T, et al. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J Biol Chem. (2005) 280:32317–25. doi: 10.1074/jbc.M500801200
76. Katsoulieris E, Mabley JG, Samai M, Sharpe MA, Green IC, Chatterjee PK. Lipotoxicity in renal proximal tubular cells: relationship between endoplasmic reticulum stress and oxidative stress pathways. Free Radic Biol Med. (2010) 48:1654–62. doi: 10.1016/j.freeradbiomed.2010.03.021
77. Soumura M, Kume S, Isshiki K, Takeda N, Araki S, Tanaka Y, et al. Oleate and eicosapentaenoic acid attenuate palmitate-induced inflammation and apoptosis in renal proximal tubular cell. Biochem Biophys Res Commun. (2010) 402:265–71. doi: 10.1016/j.bbrc.2010.10.012
78. Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. (2003) 100:3077–82. doi: 10.1073/pnas.0630588100
79. Baines RJ, Chana RS, Hall M, Febbraio M, Kennedy D, Brunskill NJ. CD36 mediates proximal tubular binding and uptake of albumin and is upregulated in proteinuric nephropathies. Am J Physiol Renal Physiol. (2012) 303:F1006–14. doi: 10.1152/ajprenal.00021.2012
80. Kennedy DJ, Chen Y, Huang W, Viterna J, Liu J, Westfall K, et al. CD36 and Na/K-ATPase-alpha1 form a proinflammatory signaling loop in kidney. Hypertension. (2013) 61:216–24. doi: 10.1161/HYPERTENSIONAHA.112.198770
81. Cases A, Coll E. Dyslipidemia and the progression of renal disease in chronic renal failure patients. Kidney Int Suppl. (2005) S87–93. doi: 10.1111/j.1523-1755.2005.09916.x
82. Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. (2005) 366:1849–61. doi: 10.1016/S0140-6736(05)67667-2
83. Chung HW, Lim JH, Kim MY, Shin SJ, Chung S, Choi BS, et al. High-fat diet-induced renal cell apoptosis and oxidative stress in spontaneously hypertensive rat are ameliorated by fenofibrate through the PPARalpha-FoxO3a-PGC-1alpha pathway. Nephrol Dial Transplant. (2012) 27:2213–25. doi: 10.1093/ndt/gfr613
84. Sivarajah A, Chatterjee PK, Hattori Y, Brown PA, Stewart KN, Todorovic Z, et al. Agonists of peroxisome-proliferator activated receptor-alpha (clofibrate and WY14643) reduce renal ischemia/reperfusion injury in the rat. Med Sci Monit. (2002) 8:BR532–9.
85. Takahashi K, Kamijo Y, Hora K, Hashimoto K, Higuchi M, Nakajima T, et al. Pretreatment by low-dose fibrates protects against acute free fatty acid-induced renal tubule toxicity by counteracting PPARalpha deterioration. Toxicol Appl Pharmacol. (2011) 252:237–49. doi: 10.1016/j.taap.2011.02.012
86. Tanaka Y, Kume S, Araki S, Isshiki K, Chin-Kanasaki M, Sakaguchi M, et al. Fenofibrate, a PPARalpha agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. (2011) 79:871–82. doi: 10.1038/ki.2010.530
87. Frazier R, Mehta R, Cai X, Lee J, Napoli S, Craven T, et al. Associations of fenofibrate therapy with incidence and progression of CKD in patients with type 2 diabetes. Kidney Int Rep. (2019) 4:94–102. doi: 10.1016/j.ekir.2018.09.006
88. Group AS, Ginsberg HN, Elam MB, Lovato LC, Crouse JR 3rd, Leiter LA, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. (2010) 362:1563–74. doi: 10.1056/NEJMoa1001282
89. Kim S, Ko K, Park S, Lee DR, Lee J. Effect of fenofibrate medication on renal function. Korean J Fam Med. (2017) 38:192–8. doi: 10.4082/kjfm.2017.38.4.192
90. Souza AC, Bocharov AV, Baranova IN, Vishnyakova TG, Huang YG, Wilkins KJ, et al. Antagonism of scavenger receptor CD36 by 5A peptide prevents chronic kidney disease progression in mice independent of blood pressure regulation. Kidney Int. (2016) 89:809–22. doi: 10.1016/j.kint.2015.12.043
91. Szeto HH, Liu S, Soong Y, Seshan SV, Cohen-Gould L, Manichev V, et al. Mitochondria protection after acute ischemia prevents prolonged upregulation of IL-1beta and IL-18 and arrests CKD. J Am Soc Nephrol. (2017) 28:1437–49. doi: 10.1681/ASN.2016070761
92. Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, et al. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol. (2011) 22:1041–52. doi: 10.1681/ASN.2010080808
93. Szeto HH, Liu S, Soong Y, Alam N, Prusky GT, Seshan SV. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. (2016) 90:997–1011. doi: 10.1016/j.kint.2016.06.013
94. Kong MJ, Han SJ, Kim JI, Park JW, Park KM. Mitochondrial NADP(+)-dependent isocitrate dehydrogenase deficiency increases cisplatin-induced oxidative damage in the kidney tubule cells. Cell Death Dis. (2018) 9:488. doi: 10.1038/s41419-018-0537-6
95. Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. (2008) 45:1559–65. doi: 10.1016/j.freeradbiomed.2008.09.003
Keywords: mitochondria, fatty acid β-oxidation, lipotoxicity, acute kidney injury, chronic kidney disease, diabetic nephropathy, polycystic kidney disease, glomerular nephropathy
Citation: Jang HS, Noh MR, Kim J and Padanilam BJ (2020) Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 7:65. doi: 10.3389/fmed.2020.00065
Received: 01 December 2019; Accepted: 13 February 2020;
Published: 12 March 2020.
Edited by:
John D. Imig, Medical College of Wisconsin, United StatesReviewed by:
Jonatan Barrera-Chimal, National Autonomous University of Mexico, MexicoCopyright © 2020 Jang, Noh, Kim and Padanilam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hee-Seong Jang, a2lhcm9ob2xpY0BnbWFpbC5jb20=; Babu J. Padanilam, YnBhZGFuaWxhbUB1bm1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.