 Sukanya Roy
Sukanya Roy Shannon Glaser
Shannon Glaser Sanjukta Chakraborty
Sanjukta Chakraborty
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 18 December 2019
Sec. Gastroenterology
Volume 6 - 2019 | https://doi.org/10.3389/fmed.2019.00293
This article is part of the Research Topic Mechanisms of Chronic Liver Diseases: Identifying New Therapeutic Targets View all 25 articles
Cholangiocarcinoma (CCA), or cancer of the biliary epithelium is a relatively rare but aggressive form of biliary duct cancer which has a 5-year survival rate post metastasis of 2%. Although a number of risk factors are established for CCA growth and progression, a careful evaluation of the existing literature on CCA reveals that an inflammatory environment near the biliary tree is the most common causal link between the risk factors and the development of CCA. The fact that inflammation predisposes affected individuals to CCA is further bolstered by multiple observations where the presence and maintenance of an inflammatory microenvironment at the site of the primary tumor plays a significant role in the development and metastasis of CCA. In addition, mechanisms activating the tumor vasculature and enhancing angiogenesis and lymphangiogenesis significantly contribute to CCA aggressiveness and metastasis. This review aims to address the role of an inflammatory microenvironment-CCA crosstalk and will present the basic concepts, observations, and current perspectives from recent research studies in the field of tumor stroma of CCA.
Cholangiocarcinoma (CCA) is a term used to define a group of different biliary epithelial cancers and is the second most common type of liver cancer. This group of primary biliary malignancy represents three different classically recognized kinds of biliary tree cancers, classified on the basis of anatomical point of origin in the bile duct, intrahepatic CCA (iCCA), perihilar CCA (pCCA), and distal CCA (dCCA) (1). Among these three types, iCCA originates in the intrahepatic ducts and represents the second most prevalent type of primary liver malignancy (about 10% of primary liver malignancies are iCCA). Duration of survival post-resection in intrahepatic CCA is 12.4 months (2). The most common type of CCA, is the pCCA, constituting ~50–60% of all recorded cases. pCCA comprises tumor arising from the emergence of left/right hepatic ducts at liver hilum to the confluence of cystic duct with common hepatic duct (choledocus formation) while distal CCA representing 20–30% of CCA occurs in the epithelial cells of the extra hepatic bile ducts (3). Although iCCA represents only about 5–10% of all CCA cases there is an increase in the number of iCCA among the three CCA types being observed recently (4). Internationally, CCA cases have increased since the past decade, in United States ~5,000 new cases are diagnosed each year (5). The incidence of CCA is highest among Hispanics and Asians (2.8–3.3/100,000) and lowest (2.1/100,000) among non-hispanics and African Americans (6). With a 5 year mortality rate post metastasis of 2%, CCA, originally described as a rare form of cancer is receiving more attention compared to the past decades due to its high mortality rate (1).
The number of people afflicted with CCA differs geographically primarily because of the difference in the presence of the risk factors that predispose an individual toward CCA. The number of CCA cases is higher in Asian countries (7). CCA also shows a slight bias toward the male gender (7). The number of risk factors and their extent of influence on CCA predisposition, is not as high as in other malignancies. This could be partially due to the limited number of studies focused on identifying risk factors of CCA. Presence of bile duct cysts, primary sclerosing cholangitis, liver cirrhosis, hepatobiliary parasitic infections such as with liver fluke, hepatolithiasis, and thorotrast exposure, are the most common risk factors. Further complicating this scenario is the fact that a majority of CCA cases develop without the presence of any of the above-mentioned risk factors (8). Hence, there is a need to look at new prognostic factors that will aid in predicting the surgical eligibility, outcome and survival of CCA patients. This has opened up new avenues in research and has identified the critical role of different inflammatory cytokines, increased lymphangiogenesis, relatively low angiogenesis, cancer associated fibroblasts (CAFs), mesenchymal stem cells (MSCs), and other factors, in the growth and progression of the different types of CCA. In the next section we will review some of these risk factors.
Inflammation and inflammatory mediators form a key underlying basis for several risk factors significantly associated with CCA (9). An inflammatory and obstructive autoimmune disease of the bile ducts, PSC is one of the most important risk factors of CCA. Patients with PSC have a 400-fold higher chance of developing CCA than those without PSC. Interestingly, majority of PSC patients are between the ages of 30 and 40 (in general CCA has a reported age specificity of 60–70 years in age) at the time of diagnosis (9). Up to 50% of these cases are recorded in the first year of diagnosis of PSC (10). The presence of chronic inflammation of bile ducts typically associated with PSC is thought to be one of the reasons for this heightened risk (11–13). Other factors that serve as the link between PSC and CCA include increased proliferation of the epithelial cells of the biliary tree, cholestasis in the ducts leading to the liver and presence of mutagens produced in the bile (10, 11). The role of inflammation in the growth and rapid development of CCA is also underscored by studies that identify inflammatory bowel disease (IBD) as one of the risk factors of CCA (12, 13).
Liver cirrhosis develops as a consequence of liver diseases and/or conditions such as alcoholism and hepatitis. As a result, the liver parenchyma is dominated by fibrosis/scarring of liver tissue resulting in disruption and eventual loss of normal liver function. Cirrhosis is an important risk factor for iCCA (14) and shows high degree of association, especially, in Asian populations (15). Similar to PSC, this too has an inflammatory stimulus and a sudden rise in epithelial proliferation, presence of pro-inflammatory cytokines and chemokines and the generation of fibrotic nodules in liver, mediates a link between cirrhosis and CCA (3).
Liver fluke (Clonorchis sinensis, Opisthorchis viverrini) infections have been identified as critical risk factors for CCA especially in eastern Asian countries where these infections are deemed to be endemic (16). In fact, the recognition of O. viverrini as a cancer-inducing parasite by IARC (International Agency for research on Cancer) is due to its role in the development of CCA in affected individuals. These infections are associated with a rise in inflammation, generation of fibrotic nodules, obstruction of bile ducts and/or cholestasis. Chronic inflammation in the biliary tree in O. viverrini infected patients (especially in the background of gene polymorphisms and exposure to other environmental factors) leads to CCA development (17, 18).
Viral infections such as hepatitis B and C (HBV and HCV, respectively), serve as important risk factors for CCA (19). While HBV infection is endemic to Asian countries and thus serves as the stronger risk factor for iCCA (20), HCV is the primary causative agent for iCCA in western countries (14). Cirrhosis is a common manifestation of hepatitis and leads to the development of the chronic inflammatory background that predisposes to CCA. However, the role of hepatitis viruses in causing proliferation of the hepatic epithelium is also considered to be a reason for CCA incidence in hepatitis patients (21).
Choledolithiasis and cholelithiasis are conditions that involve the presence of stones in the gall bladder and common bile ducts. The presence of these gall stones causes biliary obstruction resulting in cholestasis and serve especially as a risk factor for extrahepatic CCA (22). The presence of stones or calcium deposition inside the intrahepatic bile ducts also leads to cholestasis and chronic inflammation, ultimately serving as a risk factor for CCA. In the Asian population, 5–13% of patients with hepatolithiasis develop iCCA (15, 23).
Chronic pancreatitis is a strong risk factor for extrahepatic CCA with an odds ratio of 6.61 (95% CI 5.21–8.40) in comparison to the 2.66 odds ratio of iCCA (95% CI, 1.72–4.10). In chronic pancreatitis too, cholestasis and inflammation may arise leading to CCA (24). Also, the presence of cysts in the bile ducts (intrahepatic and extrahepatic) when left untreated leads to the development of iCCA and eCCA tumors, because of biliary duct obstruction and dilatation leading to cholestasis and inflammation (24, 25). Thus, it is evident that inflammation forms an underlying theme in the predisposition and development of CCA.
As CCAs originate from cholangiocytes from different anatomical locations of the biliary tree, they also exhibit considerable tumor heterogeneity that points to the possibility of diverse cellular origins (1, 26). In general, CCA originates from the peribiliary gland (PBG) lining epithelium of intra- and extra- hepatic ducts (IH and EH, respectively) of the biliary tree (27). Additionally, cholangiocytes and hepatocytes originating from canals of Herring can undergo mutation to give rise to tumors having varying phenotypes (28). Based on the wide range of these phenotypes, pCCA and dCCA have been characterized as adenocarcinomas mucinous in nature, while iCCA has two subtypes: iCCA arising from small bile ducts, mixed in histological phenotype and those arising directly from large intrahepatic bile duct, mucinous in histology (29, 30). While bile ductular type iCCA has been recognized to be associated with solid tumor formation not having preneoplastic lesions, iCCA arising from large intrahepatic bile duct is one which is distinctly preceded by preneoplastic lesions (biliary intraepithelial/intraductal papillary neoplasm). Additionally, bile ductular type iCCA has been correlated with chronic liver disease cases such as cirrhosis in contrast to bile duct type iCCA which is mostly correlated with PSC. These differences in histology point to the role of different cells of origin of CCA (29, 30).
The undeniable role of stem cells in CCA development and origin is proven by the fact that human hepatic stem cells (hHPSCs) are the progenitor cells giving rise to cholangiocytes and hepatocytes that mutate to give rise to CCA (28). The PBG niche starts at septal-segmental bile ducts and ends near duodenal area at hepatic pancreatic common duct. PBG niche thus distributed all across the biliary tree has a significant role in harboring a multipotent stem cell niche which forms the source of the endodermal hepatic mucinous cells that ultimately give rise to the mucinous CCA subtypes of dCCA, pCCA and bile duct type iCCA (27, 30–32). Cancer stem cells (CSCs) are more generally characterized as cellular subset that maintains tumor growth, such CSCs are recognized by the expression of extracellular markers like CD 24, CD44, CD133, epithelial cell adhesion molecule (EpCAM) etc. in liver malignancies (33). In CCA, more of these studies identifying the specific roles of CSCs are needed. As such two distinct stem cell niches are recognized for CCA development: BTSCs (biliary tree stem cell niche within PBG) and hHPSCs within canals of Herring (26, 27). These findings suggest that CCA has more than one type of cell-of-origin and the differences can be looked at to develop a treatment strategy(s) that is personified from an anatomical point of view (34).
CCA is one of the most desmoplastic tumors and the tumor microenvironment of CCA is characterized by a dense bed of connective tissue intertwining the tumor cells. This dense stroma is composed of a contiguously activated subset of fibroblasts called CAFs that play key roles in modulating several aspects of CCA progression (35). Further during tumor development and progression and resulting increase in cellular and metabolic demands there is often restricted access to nutrients and oxygen supply. This results in regions of the solid tumor having permanent or transient hypoxia, due to alterations in the tumor associated vasculature (36). The expanding vascular network is unable to meet up with the growing demands of the tumor and hypoxic regions persist and induce cellular pathways that promote more malignant phenotypes. In addition, there are immune cells, blood vessels, and lymphatic vessels that contribute to tumor progression which will be discussed in the following sections.
CAFs release a number of molecules functioning as extracellular matrix proteins (ECM) such as collagen I and fibronectin (35). In CCA, CAFs typically infiltrate the tumor stroma, and are differentially stimulated by a variety of molecular factors released by CCA tumor cells as well as hypoxia. The CAFs population in CCA thus is heterogenous in origin (37). Two of the main sources of these CAFs are liver (hepatic stellate cells, HSCs) and portal vein (portal fibroblasts), while bone marrow derived MSCs also serve as a source of CAFs to a minor extent (37). CCA tumor cells and other immune cells such as macrophages secrete inflammatory chemokines, cytokines and growth factors that not only signal fibroblasts from liver and portal vein to infiltrate the tumor microenvironment but also result in constitutive activation of fibroblasts (35). Platelet derived growth factor (PDGF-DD) overexpressed by CCA cells under hypoxic condition has been shown to be an important CAF infiltrating factor. Binding of PDGF-DD to its receptor PDGFRβ activates Cdc42, Rac1, and Rho GTPases and JNK pathways (38). PDGF-DD binding Cdc42 induces the formation of filopodia and Rac1 induces the formation of lamellipoda, thus ensuring the migration of CAFs to CCA tumor stroma. In addition to PDGF-DD, a number of other growth factors such as FGF (fibroblast growth factor), numerous factors belonging to PDGF family and TGF-β also aid CAF infiltration (39).
Alpha-smooth muscle actin-positive (α-SMA) fibroblasts promote biliary cell proliferation and correlate with poor survival in CCA. CCA fibroblasts have proliferative effects that enhance tumor promotion and progression of CCA (40). CCA patients with a high population of CAFs have poorer prognosis than patients with low number of CAFs (41). Consequently, CAF-specific α-SMA is a prognostic factor of CCA patient survival (42). The tumor boosting ability of stromal CAFs was also shown using a 3D collagen matrix-based co-culturing system, in which CCA cells and CAFs isolated from a syngeneic orthotopic rat model of CCA showed a corresponding increase in the formation of structures resembling ducts from CCA cells with the increase in CAF plating density (43). Interestingly, hepatic stellate cells (HSCs) under the influence of CCA cells can also transform into CAFs and support CCA growth (44, 45). These findings were further corroborated by studies in a syngeneic rat CCA model with selective stromal CAF depletion that exhibited improved host survival and decreased tumor growth (46).
CAFs in CCA show unique characteristics and gene signatures (47). Gene expression studies with human CCA sample derived CAFs showed significant differences between normal liver fibroblasts and CAFs. Most of the genes that were induced in CAFs were involved in controlling cellular metabolism, a prerequisite for the active production of cellular proteins to support the tumor microenvironment and promote tumorigenesis (47). In addition, exosomes also serve as important vessels for transporting regulatory molecular factors (between CAFs and CCA cells) thus supporting cross-talk between CCA cells and CAFs. While studies characterizing the exosomal cargo involved in CAF-CCA crosstalk has been relatively limited (48, 49), it has been shown that exosomes shuttle miR-195 between CAFs and CCA (50). Stimulation of MSCs to CCA cell-derived exosomes lead to increased migration and production of inflammatory tumor promoting cytokines as CXCL1, CCL2, and IL-6 (51). In addition, several growth factors contribute to the inflammatory microenvironment.
EGFA/EGFR binding has been shown to promote tumorigenesis and metastasis in CCA, another important EGFR ligand, HB-EGF was found to be highly expressed in myofibroblasts. HB-EGF activated EGF signaling promotes proliferation of CCA cells and also induces epithelial-mesenchymal (EMT) changes as well as invasion. HB-EGF secretion from fibroblasts is also activated by the pro-tumorigenic growth factor TGF-β secreted by tumor cells that in turn favors CCA growth (52).
Stromal cell derived factor 1 or SDF-1 has previously been reported to be involved in promoting cancer growth as a ligand for CXCR4/CXCR7 (53). In CCA, SDF-1 expression is only produced by the stromal CAF, possibly as a result of the HSC infiltration under stimulatory signals derived from angiotensin-II secreted by cancer cells (54). In vitro studies indicate that when SDF-1 is expressed by HSCs, a number of pro-tumorigenic responses are induced such Bcl-2, and activation of PI3K/Akt pathway. These responses initiate increased CCA cell invasion and prolonged survival in addition to inducing epithelial-mesenchymal transition (45, 55). Tumor associated macrophages were shown to produce TNF-α that induces CXCR4 expression, thus promoting SDF-1 mediated pro-tumorigenic effects (54). CAFs are also shown to release high levels of HGF (hepatocyte growth factor) that might mediate high expression of CXCR4 (43).
One of the most important cellular components of CCA stroma are MSCs. MSCs may activate a series of tumor signaling pathways through the release of cytokines and that may either promote or inhibit tumor development and progression (56). The function of MSCs in tissue repair is similar to the homing of MSCs to sites of tissue damage and to sites of tumor microenvironment (51). Injured tissues secrete a wide variety of inflammatory chemokines that sends signals to MSCs for repair. It has been seen in a number of studies that tumor cells too, while modulating several other factors in their microenvironment that foster a metastatic condition, secrete inflammatory chemokines that result in MSC infiltration (51). CCA cells also secrete exosomal vesicles that are shown to enhance expression of IL-6, CXCL-1, and CCL2 by MSCs. Further, conditioned medium from MSCs exposed to tumor cell-derived extracellular vesicles (EVs) caused an upregulation in STAT3 phosphorylation and proliferation of CCA cells, possibly by secretion of CCL2/MCP1, CXCL1/GRO-α, CXC3CL1/Fractalkine, IL-6, and PDGF-AA (51). Conditioned media from MSCs also has been found to upregulate the Wnt signaling pathway in CCA cells and increased nuclear translocation of ß-catenin (57). Further, coculture studies of CCA and MSCs have shown that increased CCR5 expression by tumor cells upregulates metalloproteinases MMP-2 and MMP-9 in CCA cells and thereby promoted angiogenesis and CCA metastasis (58).
The CCA stroma is densely populated by different infiltrating immune cells among which tumor associated macrophages (TAMs) play an important role by regulating angiogenesis, lymphangiogenesis, tumor proliferation and also modulating matrix related changes (59, 60). In a study by Wongkham et al. more than half of CCA tumor samples showed high macrophage infiltration in CCA (61). It has also been seen that CD14+/CD16+ monocyte cells which are precursors of tissue resident macrophages are present in an increased number in CCA patients. It is significant that these circulating CD14+/CD16+ monocytes have high VEGF and CXCL3 expression that promote tumor angiogenesis (62). In a correlation study it was seen that CD163+ M2 macrophages were associated with FOXP3+ regulatory T cell-related infiltration. Additionally, this study also showed that CCA conditioned media treatment of macrophages led to polarization bias toward M2 macrophages along with secretion of TGFβ, IL10, and VEGF-A (63). A high density of the M2-TAMs in patients is significantly associated with increased extrahepatic metastases possibly due to the effects on EMT pathways (41).
The association between chronic inflammation and the development and progression of malignancy is significantly pronounced in onset and development of CCA (64). Inflammation in the tumor microenvironment of CCA is promoted by a number of cytokines and chemokines that further enhance tumor progression and aid pathways involved in distant metastasis (47). Below, we discuss several inflammatory cytokines that contribute to an inflammatory tumor microenvironment and enhance CCA progression.
TNF-α is one of the most well-known mediators of inflammatory stimuli in the tumor microenvironment (65). Although TNF-α is involved in cancer progression, its more prominent pro-tumoral effects have been seen in angiogenesis and invasion of cancer cells (66, 67). During pathogenesis, TNF-α elicits an immune response at tissue injury locations. TNF-α also induces hepatic stellate cells (HSCs) so that they secrete oxidative radicals such as hydroxyl radical, nitic oxide (NO), and superoxide anion and is associated with aggressive development of CCA (68). Suksawat et al. showed that CCA cells express very high levels of eNOS and phosphorylated eNOS that correlate with poor prognosis in CCA patients. This phosphorylation mediated activation of eNOS by VEGF-C is through activation of PI3K/AKT pathway. The downstream effects of eNOS/peNOS/iNOS is thought to originate from VEGF-C pathway activation (69). TNF-α has been shown to promote migration of CCA cells by upregulating expression of S100A4, vimentin and ZEB2, molecules involved in EMT transition. In neoplastic bile ducts, these molecules have been seen to be associated with upregulation of TGF-β and downregulation of E-cadherin expression, an observation that has been correlated to poor prognosis in CCA patients (70).
Classified as one of the most important pro-inflammatory cytokines, IL-1β has been shown to be highly expressed from HSCs. The autocrine signaling mediated by CCA cells also becomes prominent in this regard as CCA cells have been shown to produce high levels of IL-1β that further enhances the CXCL5/CXCR2 pathway that in turn activates AKT/PI3K or ERK1/2 pathways. In fact, heightened CXCL5 expression has been seen to indicate poor rates of survival in CCA patients (71, 72).
Bone marrow derived MSCs (BM-MSC) when exposed to tumor conditioned medium can transform into CAFs and stimulate tumor growth via secretion of inflammatory cytokine IL-6 in the tumor stroma. In CCA, this IL-6 overexpression was found to decrease the methylation of the EGFR promoter and enhance EGFR expression that in turn is associated with poor prognosis and overall survival (64, 73). IL-6 also mediates its tumorigenic effects by causing hypermethylation based silencing of tumor suppressor genes (74). In CCA, IL-6 has been shown to activate the p38 pathway and consequently downregulate p21WAF/CIP1 a cyclin dependent kinase inhibitor, involved in cell cycle regulation (75). IL-6 also induces upregulation of STAT3 and Mcl-1 (myeloid cell leukemia-1) genes that mediate an anti-apoptotic response in neoplastic cholangiocytes (76). In addition, IL-6 also induces EMT by increasing expression of Snail and JAK/STAT and a resulting downregulation of E-cadherin and promotes CCA progression (77).
TGF-β plays dual roles in cancer progression and inhibits cell proliferation, regulates anti-inflammatory, and pro-apoptotic effects in cells under normal physiological conditions (78). It also actively promotes tumor progression and most cancer cells are resistant to its anti-proliferative effects. TGF-β activates the expression of its downstream genes (such as Bim) through differential phosphorylation and nuclear translocation of SMAD transcription factors (79). Mutational changes in the TGF-β receptor resulting in changes in Smad4 phosphorylation, increased cyclin D1 levels activate pathways that make CCA cells resistant to the tumor suppressive effects of TGF-β (80). Mouse model-based studies have shown that loss of expression of PTEN and SMAD4 gives rise to CCA (81). Correlation studies have shown that high levels of TGF- β is related to CCA metastasis to lymph nodes and distant sites as well as CCA recurrence (82). Consequently, inhibition of TGF-β resulted in significant reduction of CCA cell invasion (83). Further, altered TGF-β signaling in CCA cells also causes EMT-driven changes in cytoskeletal structure and CCA cell motility thus influencing cancer cell invasion through upregulation of EMT genes (84).
Overall, inflammatory cytokines set the stage for CCA growth by enhancing proliferation, activation of tumor promoting mechanisms such as EMT, activation of signaling pathways that promote tumor growth and loss of cell cycle checkpoints. However, the major cause for the high mortality associated with these cancers is its ability to metastasize, that is aided by the activation of lymphangiogenic (growth of new lymphatic vessels) and angiogenic (growth of new blood vessels). The various growth factors secreted by CCA cells into their stroma and other components of the tumor microenvironment foster the development of new lymphatic and blood vessels that in turn promote tumor growth and dissemination to distant organs.
Tumor cells employ several mechanisms to establish a functional and integrated vascular system comprised of both blood and lymphatic vessels to promote cellular growth and metabolism. Expansion of these vascular networks is key to migration of the tumor cells to distant sites where they establish tumor niches. A surge of recent data has implicated the roles of both lymphatic and the blood vascular in promoting CCA metastasis.
Tumor-associated lymphangiogenesis, or the sprouting of new lymphatic vessels in the tumor microenvironment is a form of tumor-associated neovascularization that has been the focus of studies concerning the metastatic spread of highly aggressive form of cancers (85). Lymphatic involvement has emerged as a hallmark of CAA with significant lymphatic invasion or lymph node metastasis implicated with poor disease prognosis (86, 87). Early metastatic CCA is characterized by a striking expansion of the intratumoral and peritumoral lymphatic vessels, which represents a key determinant of the early metastasis to the regional lymph nodes in patients rendering patients unable to opt for surgical resection. Post-surgical resection period is characterized by an increase in lymphangiogenesis and lymphatic vessel remodeling that correlates with poor post-surgical survival (86). Hence, it is critical to look at the elements in the tumor microenvironment of CCA that cause lymphangiogenesis and lymph node remodeling.
As discussed above, the tumor microenvironment of CCA is enriched with abundant cytokines and chemokines necessary for paracrine signaling that promotes development of a lymphatic bed dedicated to sustaining the growth of tumor. CAFs actively crosstalk with CCA cells in driving the development of a rich lymphatic vasculature within a pro-lymphangiogenic tumor stroma (35). High expression of VEGF-C and VEGFR-3 has been observed in the tumor microenvironment of intrahepatic CCA patients (iCCA), that also correlated with poor prognosis in patients (88–90). VEGF-C is required for the growth of small (or intial) lymphatic vessels whereas angiopoietin 1 & 2 are need by VEGF-C to form terminal lymphatic vessels in the adult body (91, 92). The dense network of lymphatic vessels and a reduced number of blood vessels, in the CCA tumor stroma also creates a hypoxic microenvironment (93). Hypoxia inducible factor-1 (HIF-1α) is known to induce lymphangiogenesis in several cancers (94). In CCA, high expression of HIF-1α promotes tumor progression and metastasis and is associated with poor patient survival (95). Interestingly, HIF-1α has also been shown to support cancer related lymphangiogenesis by upregulating the expression and subsequent secretion of Ang1/2, VEGF-C/D and PDGF-B from neoplastic cells into the tumor stroma, in several cancers as breast cancer, esophageal cancer, and oral squamous carcinoma (96–98). PDGF-D secreted from neoplastic CCA cells binds PDGFRβ on CAFs resulting in activation of ERK/NF-kB and JNK signaling networks that in turn secretes VEGF-C and promotes expansion of the lymphatic vasculature and tumor cell intravasation. Pharmacological depletion of CAFs in a CCA in vivo however, significantly reduced lymphatic vascularization and reduced lymph node metastases (99). VEGF-C expression in CCA is also mediated by M2 macrophages (63). Further, overexpression of Nerve Growth Factor Beta (NGF-B) overexpression correlated with VEGF-C overexpression, lymphatic vessel density and lymph node metastasis along with nerve cell invasion in patients of hilar CCA (100). Different correlation studies have established lymphatic vessel density (LVD) and expression of several lymphatic specific markers such as podoplanin and VEGFR-3 as prognostic biomarkers of CCA (101, 102). Podoplanin is highly expressed on the surface of CAFs as well as LECs and emerged to be a prognostic biomarker in human perihilar CCA (101). Lymph node metastasis has also been correlated with a high podoplanin expression on activated CAFs in intrahepatic CCA (90). Further studies are needed to determine the role of podoplanin in tumor lymphangiogenesis in CCA. However, podoplanin mediated regulation of small GTPases as Cdc42 induces capillary morphogenesis, polarized migration, and invasiveness of LECs (103, 104).
Thelen et al. have demonstrated that a high lymphatic vessel density or existence of lymphangiogenesis significantly correlates with poor prognosis in patients with hilar CCA. This observation adds to the role of lymphatic vessel remodeling in cancer progression, specifically the migration of cancer cells via lymphatic vessels (104, 105). In CCA, a “high” LVD is associated with increased nodal spread, and “high” LVD tumors more frequently develop recurrence (105). Indeed recent studies have shown that both peritumoral as well as intratumoral lymphatic bed is composed of capillaries that lack organization and/or drainage function thus favoring neoplastic cell infiltration because of the differential permeability or leaky nature of these vessels (106). In this regard, LECs lining these vessels also interact with tumor cells to transport them through endothelium, an event mediated by the CCL1-CCR8 chemokine axis. CC-type chemokine ligand 1 (CCL1) is expressed on the surface of LECs which bind CC-type chemokine receptor 8 (CCR8) on the surface of tumor cells and thus help in their trans-endothelial migration (107). Tumor lymphangiogenesis, which results in proliferation of LECs also functions in immune-evasion of the cancer cells. LECs in draining lymph nodes express on their surface the well-known antigen PD-L1 which binds to PD-1 on the surface of cancer specific CD 8+ cells and induces their apoptosis (108). However, there is a need to study the mechanisms of lymph node remodeling and lymphatic metastasis in CCA, that would further establish the link between lymphatic vessel remodeling, tumor stroma, tumor lymphangiogenesis and CCA metastasis.
Tumor related angiogenesis, or the sprouting of new blood vessels is one of the key mechanisms for tumor metastasis that is promoted by angiogenic factors actively secreted by tumor cells (109). Tumor angiogenesis is pronounced in CCA, one of the most aggressive and metastatic cancers. Cholangiocytes promote neo-vascularization by enhanced expression of pro-angiogenic growth factors both at the site of primary tumor as well as in the tumor stroma of distant sites where these cholangiocytes have metastasized. Thus, a sprawling network of blood vessels created by secreted factors from cholangiocytes supports the growth and spread of cholangiocytes (110). Critical mediators and activators of angiogenesis include the growth hormones VEGF, EGF, and NGF, FGF, placental growth factor, the angiopoietins and their receptors, Tie1 and Tie2. Further, neuropilin, ephrin, and leptin are being recognized as key mediators of angiogenesis and tumor growth (111). These pro-angiogenic factors play important roles both in maintenance and growth of the primary tumor as well as neo-vascularization during CCA metastasis (111). In normal tissues, following induction of angiogenesis by pro-angiogenic factors such as the VEGF factor family proteins (VEGF-A, VEGF-B, VEGF-C), remodeling of the newly formed vessel wall takes place where intercellular tight junctions and adherens junctions are created between vascular endothelial cells (BECs), that brings about permeability and elasticity in the vessel (110). After vessel remodeling is completed in normal tissues the ensuing blood flow and establishment of normoxia (normal/physiological O2 concentration) results in the inhibition of angiogenesis inhibition. In tumor cells however, hypoxia or a low oxygen environment in the region of the tumor induces expression of VEGF hormones. Cholangiocytes, accordingly, have been found to secrete high levels of both VEGF-A in the tumor stroma and VEGFR-2 during cholangiocyte hyperplasia (112). This suggests an autocrine mechanism by which cholangiocytes regulate their own growth. Similar to the studies in CCA lymphangiogenesis where high VEGFR-3 expression is enhanced under the influence of CAFs and tumor cells on the surface of LECs, it has been shown that a similar paracrine signaling mechanism exists in BECs where high levels of VEGFR-2 are expressed on its surface (112). Enhanced expression of VEGF-A and other members of the VEGF family such as VEGF-C cause BECs to secrete MMP-9 and MMP-7 which help in remodeling of the basement membrane and surrounding ECM and promotes tumor metastasis. Interestingly, it has been shown that TGF-β and VEGF are co-expressed in human CCA and that overexpression and functional interaction of TGF-β and VEGF could potentially contribute to the “angiogenic switch” and the malignant phenotype in human CCC (113). In addition to hypoxia stimulating production of VEGF, additional factors such as estrogen along with IGF1 (insulin like growth factor 1) and IGFR (IGF1 receptor) synergistically increases the expression of VEGFs such as VEGF-A, VEGF-C and their corresponding receptors in cultured CCA cells (114). In addition, metastasis-associated in colon cancer-1 (MACC1) protein upregulates VEGF-A thus favoring the growth of CCA (115). Overexpression of histidine decarboxylase (HDC) enzyme correlated with that of VEGF-A/C expression. HDC knockdown/inhibition significantly reduced tumor growth by reducing tumor cell proliferation and VEGF expression (109).
Growing evidence from literature suggests that microRNAs (miRNA), endogenous small non-coding RNAs (19–24 nucleotides) regulate various aspects of cholangiopathies including CCA and has been extensively reviewed elsewhere (116, 117). However, studies evaluating their role in regulation of lymphangiogenesis associated with CCA is very limited. The miRNAs involved in CCA associated angiogenesis have been more extensively investigated and miR-92a, miR-126, miR-132, and miR-296 regulate several key pathways that enhance CCA associated angiogenesis (118). Overexpression of miR 16 and miR-424 has been shown to regulate the VEGF-A/FGF signaling cascades and reduce tumor cell proliferation and migration (119). miR-101, an miRNA highly expressed in liver was found to inhibit the growth of CCA by inhibiting VEGF expression (120). Understanding how miRNA regulate different molecular players involved at different levels of CCA progression also will help design better therapeutic interventions for arresting tumor progression. Further these miRNAs have the potential of being diagnostic biomarkers for CCA metastasis.
The epidemiology of CCA varies across different regions owing to the differences in the number and intensity of the risk factors present in each place, the malignancy also varies in terms of the epidemiology of its types (iCCA, pCCA, dCCA), however based on the data above it can be postulated that inflammation of the tumor microenvironment and its associated players have a crucial role in shaping the response of the CCA cells to therapeutic strategies, their growth and progression. To this end, the early metastatic events of CCA is an area that can be pursued in the future to look for new therapeutic targets as well as to unravel the intricacies of the inflammatory tumor microenvironment-CCA crosstalk. While therapies targeting specific molecules and signaling pathways have shown promise, combinatorial therapies as a whole have come up to be effective in different cancer types. Hence, a better understanding of the different components of the tumor stroma that the CCA cells modulate and exploit in order to give rise to a pro-inflammatory and pro-tumorigenic environment can lead to a holistic understanding and approach toward treating CCA. Some of these key mechanisms that interact and promote the onset and progression and subsequent metastasis of CCA is shown in Figure 1. It is also evident that the aggressiveness of this cancer is directly related to its ability to metastasize and hence understanding key events that promote lymphatic metastasis in the early stages of the cancer will be critical for development of targeted therapies. Specific traits of CCA such as the high rate of lymphangiogenesis vs. the low rate of angiogenesis, deserve special research focus to unravel some of the underlying molecular pathways that mediate disease progression.
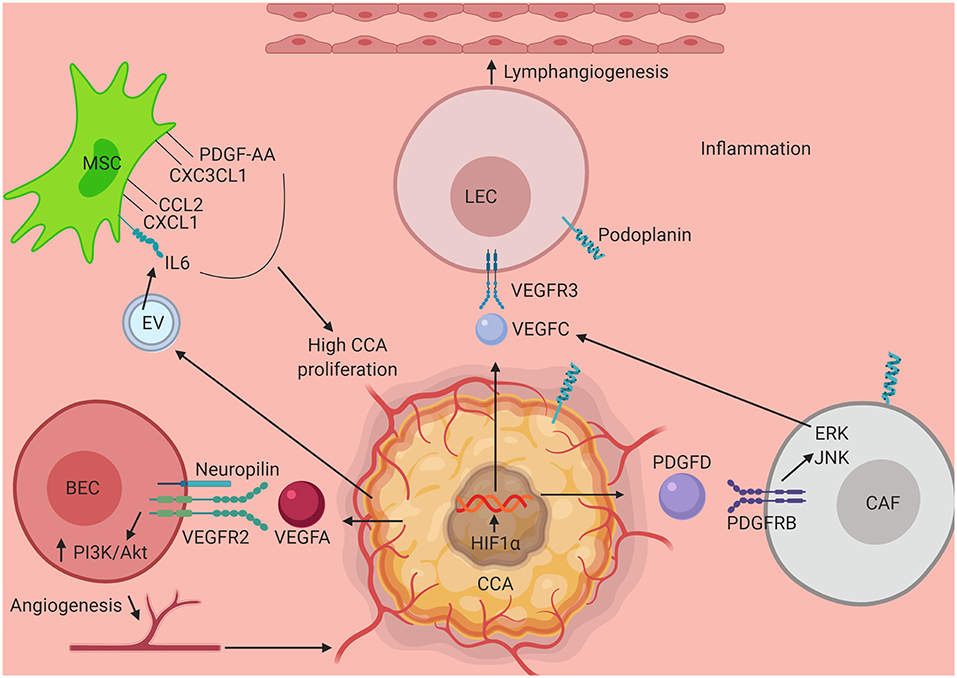
Figure 1. Schematic illustrating interaction of key elements in the tumor stroma in CCA progression. Several components of the CCA tumor microenvironment activate mechanisms that promote tumor growth, migration and activation of tumor associated angiogenesis and lymphangiogenesis. Interaction between VEC and CCA cell via VEGFR2-VEGFA assisted by neuropilin leads to tumor angiogenesis via upregulation of PI3K/Akt pathway. HIF1α activated by an inflamed tumor microenvironment stimulates CCA progression. VEGFC secreted by CCA as well by CAF (via PDGFD stimulation of tumor cells) aids in the process of lymphangiogenesis by stimulating LECs to divide via VEGFR3 engagement and upregulation of ERK/JNK pathway. Further contributing to the surrounding milieu, exosomal vesicles secreted by CCA triggers production of IL6, CCL2, CXCL1, CXC3CL1, and PDGF-AA which in turn when secreted in the tumor stroma induces CCA proliferation and growth pathways. CCA, Cholangiocarcinoma; MSC, Mesenchymal Stem Cell; LEC, Lymphatic Endothelial Cell; CAF, Cancer Associated Fibroblast; VEC, Vascular Endothelial Cell; PDGF-AA, Platelet Derived Growth Factor-AA; CXC3CL1, Chemokine Ligand 1 (Fractalkine); CCL2, C-C Motif Chemokine Ligand 2; CXCL1, C-X-C Motif Chemokine Ligand 1; IL6, Interleukin 6; VEGFR3, Vascular Endothelial Growth Factor Receptor 3; VEGFC, Vascular Endothelial Growth Factor C; VEGFA, Vascular Endothelial Growth Factor A; VEGFR2, Vascular Endothelial Growth Factor 2; PI3K, Phosphoinositide 3-kinase; Akt, Protein Kinase B; HIF1α, Hypoxia Inducible Factor 1 α; PDGFD, Platelet Derived Growth Factor D; PDGFRB, Platelet Derived Growth Factor Receptor B; ERK, Extracellular Receptor Kinase; JNK, c-Jun N-terminal Kinase.
SR, SG, and SC planned and wrote the manuscript and approved the final submitted version. SR made the figure.
This work was supported by Auf-X-Grant Award from Texas A&M University Health Science Center and Department of Medical Physiology, to SC, faculty start-up funds from Texas A&M University Health Science Center College of Medicine to SC and NIH grant DK110035 to SG, and American Heart Association Scientist Development grant 17SDG33670306 to SC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol. (2016) 13:261–80. doi: 10.1038/nrgastro.2016.51
2. Sriputtha S, Khuntikeo N, Promthet S, Kamsa-Ard S. Survival rate of intrahepatic cholangiocarcinoma patients after surgical treatment in Thailand. Asian Pac J Cancer Prev. (2013) 14:1107–10. doi: 10.7314/APJCP.2013.14.2.1107
3. Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. (2013) 145:1215–29. doi: 10.1053/j.gastro.2013.10.013
4. Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol. (2018) 15:95–111. doi: 10.1038/nrclinonc.2017.157
5. Ghouri YA, Mian I, Blechacz B. Cancer review: cholangiocarcinoma. J Carcinog. (2015) 14:1. doi: 10.4103/1477-3163.151940
6. Everhart JE, Ruhl CE. Burden of digestive diseases in the United States Part III: Liver, biliary tract, and pancreas. Gastroenterology. (2009) 136:1134–44. doi: 10.1053/j.gastro.2009.02.038
7. Khan SA, Taylor-Robinson SD, Toledano MB, Beck A, Elliott P, Thomas HC. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J Hepatol. (2002) 37:806–13. doi: 10.1016/S0168-8278(02)00297-0
8. Endo I, Gonen M, Yopp AC, Dalal KM, Zhou Q, Klimstra D, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg. (2008) 248:84–96. doi: 10.1097/SLA.0b013e318176c4d3
9. Boonstra K, Weersma RK, van Erpecum KJ, Rauws EA, Spanier BW, Poen AC, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology. (2013) 58:2045–55. doi: 10.1002/hep.26565
10. Tyson GL, El-Serag HB. Risk factors for cholangiocarcinoma. Hepatology. (2011) 54:173–84. doi: 10.1002/hep.24351
11. Patel T. Cholangiocarcinoma. Nat Clin Pract Gastroenterol Hepatol. (2006) 3:33–42. doi: 10.1038/ncpgasthep0389
12. Erichsen R, Jepsen P, Vilstrup H, Ekbom A, Sørensen HT Incidence and prognosis of cholangiocarcinoma in Danish patients with and without inflammatory bowel disease: a national cohort study 1978-2003. Eur J Epidemiol. (2009) 24:513–20. doi: 10.1007/s10654-009-9365-4
13. Sørensen JØ, Nielsen OH, Andersson M, Ainsworth MA, Ytting H, Bélard E, et al. Inflammatory bowel disease with primary sclerosing cholangitis: a Danish population-based cohort study 1977-2011. Liver Int. (2018) 38:532–541. doi: 10.1111/liv.13548
14. Palmer WC, Patel T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J Hepatol. (2012) 57:69–76. doi: 10.1016/j.jhep.2012.02.022
15. Chang JS, Tsai CR, Chen LT. Medical risk factors associated with cholangiocarcinoma in Taiwan: a population-based case-control study. PLoS ONE. (2013) 8:e69981. doi: 10.1371/journal.pone.0069981
16. Sithithaworn P, Yongvanit P, Duenngai K, Kiatsopit N, Pairojkul C. Roles of liver fluke infection as risk factor for cholangiocarcinoma. J Hepatobil Pancreat Sci. (2014) 21:301–8. doi: 10.1002/jhbp.62
17. Honjo S, Srivatanakul P, Sriplung H, Kikukawa H, Hanai S, Uchida K, et al. Genetic and environmental determinants of risk for cholangiocarcinoma via Opisthorchis viverrini in a densely infested area in Nakhon Phanom, northeast Thailand. Int J Cancer. (2005) 117:854–60. doi: 10.1002/ijc.21146
18. Brindley PJ, Loukas A. Helminth infection-induced malignancy. PLoS Pathog. (2017) 13:e1006393. doi: 10.1371/journal.ppat.1006393
19. Li H, Hu B, Zhou Z-Q, Guan J, Zhang Z-Y, Zhou G-W. Hepatitis C virus infection and the risk of intrahepatic cholangiocarcinoma and extrahepatic cholangiocarcinoma: evidence from a systematic review and meta-analysis of 16 case-control studies. World J Surg Oncol. (2015) 13:161. doi: 10.1186/s12957-015-0583-9
20. Matsumoto K, Onoyama T, Kawata S, Takeda Y, Harada K, Ikebuchi Y, et al. Hepatitis B and C virus infection is a risk factor for the development of cholangiocarcinoma. Intern Med. (2014) 53:651–4. doi: 10.2169/internalmedicine.53.1410
21. Ralphs S, Khan SA. The role of the hepatitis viruses in cholangiocarcinoma. J Viral Hepat. (2013) 20:297–305. doi: 10.1111/jvh.12093
22. Schottenfeld D, Beebe-Dimmer J. Chronic inflammation: a common and important factor in the pathogenesis of neoplasia. CA Cancer J Clin. (2006) 56:69–83. doi: 10.3322/canjclin.56.2.69
23. Kim HJ, Kim JS, Joo MK, Lee BJ, Kim JH, Yeon JE, et al. Hepatolithiasis and intrahepatic cholangiocarcinoma: a review. World J Gastroenterol. (2015) 21:13418–31. doi: 10.3748/wjg.v21.i48.13418
24. Petrick JL, Yang B, Altekruse SF, Van Dyke AL, Koshiol J, Graubard BI, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based study in SEER-Medicare. PLoS ONE. (2017) 12:e0186643. doi: 10.1371/journal.pone.0186643
25. Abdalla EK, Forsmark CE, Lauwers GY, Vauthey JN, Monolobar Caroli's Disease and cholangiocarcinoma. HPB Surg. (1999) 11:271–6; discussion: 276–7. doi: 10.1155/1999/70985
26. Komuta M, Govaere O, Vandecaveye V, Akiba J, Van Steenbergen W, Verslype C, et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology. (2012) 55:1876–88. doi: 10.1002/hep.25595
27. Nakanuma Y, Sato Y, Harada K, Sasaki M, Xu J, Ikeda H. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J Hepatol. (2010) 2:419–27. doi: 10.4254/wjh.v2.i12.419
28. Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. (2006) 25:3818–22. doi: 10.1038/sj.onc.1209558
29. Aishima S, Oda Y. Pathogenesis and classification of intrahepatic cholangiocarcinoma: different characters of perihilar large duct type versus peripheral small duct type. J Hepatobiliary Pancreat Sci. (2015) 22:94–100. doi: 10.1002/jhbp.154
30. Liau JY, Tsai JH, Yuan RH, Chang CN, Lee HJ, Jeng YM. Morphological subclassification of intrahepatic cholangiocarcinoma: etiological, clinicopathological, and molecular features. Mod Pathol. (2014) 27:1163–73. doi: 10.1038/modpathol.2013.241
31. Cardinale V, Wang Y, Carpino G, Cui CB, Gatto M, Rossi M, et al. Multipotent stem/progenitor cells in human biliary tree give rise to hepatocytes, cholangiocytes, and pancreatic islets. Hepatology. (2011) 54:2159–72. doi: 10.1002/hep.24590
32. Carpino G, Cardinale V, Onori P, Franchitto A, Berloco PB, Rossi M, et al. Biliary tree stem/progenitor cells in glands of extrahepatic and intraheptic bile ducts: an anatomical in situ study yielding evidence of maturational lineages. J Anat. (2012) 220:186–99. doi: 10.1111/j.1469-7580.2011.01462.x
33. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. (2008) 8:755–68. doi: 10.1038/nrc2499
34. Cardinale V, Carpino G, Reid L, Gaudio E, Alvaro D. Multiple cells of origin in cholangiocarcinoma underlie biological, epidemiological and clinical heterogeneity. World J Gastrointest Oncol. (2012) 4:94–102. doi: 10.4251/wjgo.v4.i5.94
35. Cadamuro M, Morton SD, Strazzabosco M, Fabris L. Unveiling the role of tumor reactive stroma in cholangiocarcinoma: an opportunity for new therapeutic strategies. Transl Gastrointest Cancer. (2013) 2:130–44. doi: 10.3978/j.issn.2224-4778.2013.04.02
36. Brivio S, Cadamuro M, Fabris L, Strazzabosco M. Epithelial-to-mesenchymal transition and cancer invasiveness: what can we learn from cholangiocarcinoma? J Clin Med. (2015) 4:2028–41. doi: 10.3390/jcm4121958
37. Leyva-Illades D, McMillin M, Quinn M, Demorrow S. Cholangiocarcinoma pathogenesis: role of the tumor microenvironment. Transl Gastrointest Cancer. (2012) 1:71–80. doi: 10.3978/j.issn.2224-4778.2011.11.05
38. Cadamuro M, Nardo G, Indraccolo S, Dall'olmo L, Sambado L, Moserle L, et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology. (2013) 58:1042–53. doi: 10.1002/hep.26384
39. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. (2013) 19:1423–37. doi: 10.1038/nm.3394
40. Chuaysri C, Thuwajit P, Paupairoj A, Chau-In S, Suthiphongchai T, Thuwajit C. Alpha-smooth muscle actin-positive fibroblasts promote biliary cell proliferation and correlate with poor survival in cholangiocarcinoma. Oncol Rep. (2009) 21:957–69. doi: 10.3892/or_00000309
41. Thanee M, Loilome W, Techasen A, Namwat N, Boonmars T, Pairojkul C, et al. Quantitative changes in tumor-associated M2 macrophages characterize cholangiocarcinoma and their association with metastasis. Asian Pac J Cancer Prev. (2015) 16:3043–50. doi: 10.7314/APJCP.2015.16.7.3043
42. Okabe H, Beppu T, Hayashi H, Horino K, Masuda T, Komori H, et al. Hepatic stellate cells may relate to progression of intrahepatic cholangiocarcinoma. Ann Surg Oncol. (2009) 16:2555–64. doi: 10.1245/s10434-009-0568-4
43. Campbell DJ, Dumur CI, Lamour NF, Dewitt JL, Sirica AE. Novel organotypic culture model of cholangiocarcinoma progression. Hepatol Res. (2012) 42:1119–30. doi: 10.1111/j.1872-034X.2012.01026.x
44. Okabe H, Beppu T, Hayashi H, Ishiko T, Masuda T, Otao R, et al. Hepatic stellate cells accelerate the malignant behavior of cholangiocarcinoma cells. Ann Surg Oncol. (2011) 18:1175–84. doi: 10.1245/s10434-010-1391-7
45. Gentilini A, Rombouts K, Galastri S, Caligiuri A, Mingarelli E, Mello T, et al. Role of the stromal-derived factor-1 (SDF-1)-CXCR4 axis in the interaction between hepatic stellate cells and cholangiocarcinoma. J Hepatol. (2012) 57:813–20. doi: 10.1016/j.jhep.2012.06.012
46. Mertens JC, Fingas CD, Christensen JD, Smoot RL, Bronk SF, Werneburg NW, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer Res. (2013) 73:897–907. doi: 10.1158/0008-5472.CAN-12-2130
47. Utispan K, Thuwajit P, Abiko Y, Charngkaew K, Paupairoj A, Chau-in S, et al. Gene expression profiling of cholangiocarcinoma-derived fibroblast reveals alterations related to tumor progression and indicates periostin as a poor prognostic marker. Mol Cancer. (2010) 9:13. doi: 10.1186/1476-4598-9-13
48. Dutta S, Reamtong O, Panvongsa W, Kitdumrongthum S, Janpipatkul K, Sangvanich P, et al. Proteomics profiling of cholangiocarcinoma exosomes: a potential role of oncogenic protein transferring in cancer progression. Biochim Biophys Acta. (2015) 1852:1989–99. doi: 10.1016/j.bbadis.2015.06.024
49. Kitdumrongthum S, Metheetrairut C, Charoensawan V, Ounjai P, Janpipatkul K, Panvongsa W, et al. Dysregulated microRNA expression profiles in cholangiocarcinoma cell-derived exosomes. Life Sci. (2018) 210:65–75. doi: 10.1016/j.lfs.2018.08.058
50. Li L, Piontek K, Ishida M, Fausther M, Dranoff JA, Fu R, et al. Extracellular vesicles carry microRNA-195 to intrahepatic cholangiocarcinoma and improve survival in a rat model. Hepatology. (2017) 65:501–14. doi: 10.1002/hep.28735
51. Haga H, Yan IK, Takahashi K, Wood J, Zubair A, Patel T. Tumour cell-derived extracellular vesicles interact with mesenchymal stem cells to modulate the microenvironment and enhance cholangiocarcinoma growth. J Extracell Vesicles. (2015) 4:24900. doi: 10.3402/jev.v4.24900
52. Clapéron A, Mergey M, Aoudjehane L, Ho-Bouldoires TH, Wendum D, Prignon A, et al. Hepatic myofibroblasts promote the progression of human cholangiocarcinoma through activation of epidermal growth factor receptor. Hepatology. (2013) 58:2001–11. doi: 10.1002/hep.26585
53. Chatterjee S, Behnam Azad B, Nimmagadda S. The intricate role of CXCR4 in cancer. Adv Cancer Res. (2014) 124:31–82. doi: 10.1016/B978-0-12-411638-2.00002-1
54. Ohira S, Sasaki M, Harada K, Sato Y, Zen Y, Isse K, et al. Possible regulation of migration of intrahepatic cholangiocarcinoma cells by interaction of CXCR4 expressed in carcinoma cells with tumor necrosis factor-alpha and stromal-derived factor-1 released in stroma. Am J Pathol. (2006) 168:1155–68. doi: 10.2353/ajpath.2006.050204
55. Okamoto K, Tajima H, Nakanuma S, Sakai S, Makino I, Kinoshita J, et al. Angiotensin II enhances epithelial-to-mesenchymal transition through the interaction between activated hepatic stellate cells and the stromal cell-derived factor-1/CXCR4 axis in intrahepatic cholangiocarcinoma. Int J Oncol. (2012) 41:573–82. doi: 10.3892/ijo.2012.1499
56. Chang AI, Schwertschkow AH, Nolta JA, Wu J Involvement of mesenchymal stem cells in cancer progression and metastases. Curr Cancer Drug Targets. (2015) 15:88–98. doi: 10.2174/1568009615666150126154151
57. Wang W, Zhong W, Yuan J, Yan C, Hu S, Tong Y, et al. Involvement of Wnt/beta-catenin signaling in the mesenchymal stem cells promote metastatic growth and chemoresistance of cholangiocarcinoma. Oncotarget. (2015) 6:42276–89. doi: 10.18632/oncotarget.5514
58. Zhong W, Tong Y, Li Y, Yuan J, Hu S, Hu T, et al. Mesenchymal stem cells in inflammatory microenvironment potently promote metastatic growth of cholangiocarcinoma via activating Akt/NF-kappaB signaling by paracrine CCL5. Oncotarget. (2017) 8:73693–704. doi: 10.18632/oncotarget.17793
59. Gentilini A, Pastore M, Marra F, Raggi C. The role of stroma in cholangiocarcinoma: the intriguing interplay between fibroblastic component, immune cell subsets and tumor epithelium. Int J Mol Sci. (2018) 19:2885. doi: 10.3390/ijms19102885
60. Brivio S, Cadamuro M, Strazzabosco M, Fabris L. Tumor reactive stroma in cholangiocarcinoma: the fuel behind cancer aggressiveness. World J Hepatol. (2017) 9:455–68. doi: 10.4254/wjh.v9.i9.455
61. Subimerb C, Pinlaor S, Khuntikeo N, Leelayuwat C, Morris A, McGrath MS, et al. Tissue invasive macrophage density is correlated with prognosis in cholangiocarcinoma. Mol Med Rep. (2010) 3:597–605. doi: 10.3892/mmr_00000303
62. Subimerb C, Pinlaor S, Lulitanond V, Khuntikeo N, Okada S, McGrath MS, et al. Circulating CD14(+) CD16(+) monocyte levels predict tissue invasive character of cholangiocarcinoma. Clin Exp Immunol. (2010) 161:471–9. doi: 10.1111/j.1365-2249.2010.04200.x
63. Hasita H, Komohara Y, Okabe H, Masuda T, Ohnishi K, Lei XF, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. (2010) 101:1913–9. doi: 10.1111/j.1349-7006.2010.01614.x
64. Wehbe H, Henson R, Meng F, Mize-Berge J, Patel T. Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. (2006) 66:10517–24. doi: 10.1158/0008-5472.CAN-06-2130
65. Popa C, Netea MG, van Riel PL, van der Meer JW, Stalenhoef AF. The role of TNF-alpha in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J Lipid Res. (2007) 48:751–62. doi: 10.1194/jlr.R600021-JLR200
66. Moore RJ, Owens DM, Stamp G, Arnott C, Burke F, East N, et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat Med. (1999) 5:828–31. doi: 10.1038/10552
67. Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. (2006) 25:409–16. doi: 10.1007/s10555-006-9005-3
68. Jaiswal M, LaRusso NF, Burgart LJ, Gores GJ. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. (2000) 60:184–90. Available online at: https://cancerres.aacrjournals.org
69. Suksawat M, Techasen A, Namwat N, Boonsong T, Titapun A, Ungarreevittaya P, et al. Inhibition of endothelial nitric oxide synthase in cholangiocarcinoma cell lines - a new strategy for therapy. FEBS Open Bio. (2018) 8:513–22. doi: 10.1002/2211-5463.12388
70. Techasen A, Loilome W, Namwat N, Khuntikeo N, Puapairoj A, Jearanaikoon P, et al. Loss of E-cadherin promotes migration and invasion of cholangiocarcinoma cells and serves as a potential marker of metastasis. Tumour Biol. (2014) 35:8645–52. doi: 10.1007/s13277-014-2087-6
71. Okabe H, Beppu T, Ueda M, Hayashi H, Ishiko T, Masuda T, et al. Identification of CXCL5/ENA-78 as a factor involved in the interaction between cholangiocarcinoma cells and cancer-associated fibroblasts. Int J Cancer. (2012) 131:2234–41. doi: 10.1002/ijc.27496
72. Zhou SL, Dai Z, Zhou ZJ, Chen Q, Wang Z, Xiao YS, et al. CXCL5 contributes to tumor metastasis and recurrence of intrahepatic cholangiocarcinoma by recruiting infiltrative intratumoral neutrophils. Carcinogenesis. (2014) 35:597–605. doi: 10.1093/carcin/bgt397
73. Mishra PJ, Mishra PJ, Humeniuk R, Medina DJ, Alexe G, Mesirov JP, et al. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. (2008) 68:4331–9. doi: 10.1158/0008-5472.CAN-08-0943
74. Gasche JA, Hoffmann J, Boland CR, Goel A Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int J Cancer. (2011) 129:1053–63. doi: 10.1002/ijc.25764
75. Tadlock L, Patel T. Involvement of p38 mitogen-activated protein kinase signaling in transformed growth of a cholangiocarcinoma cell line. Hepatology. (2001) 33:43–51. doi: 10.1053/jhep.2001.20676
76. Yu C, Bruzek LM, Meng XW, Gores GJ, Carter CA, Kaufmann SH, et al. The role of Mcl-1 downregulation in the proapoptotic activity of the multikinase inhibitor BAY 43-9006. Oncogene. (2005) 24:6861–9. doi: 10.1038/sj.onc.1208841
77. Yadav A, Kumar B, Datta J, Teknos TN, Kumar P IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. (2011) 9:1658–67. doi: 10.1158/1541-7786.MCR-11-0271
78. Santibanez JF, Quintanilla M, Bernabeu C. TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin Sci. (2011) 121:233–51. doi: 10.1042/CS20110086
80. Zen Y, Harada K, Sasaki M, Chen TC, Chen MF, Yeh TS, et al. Intrahepatic cholangiocarcinoma escapes from growth inhibitory effect of transforming growth factor-beta1 by overexpression of cyclin D1. Lab Invest. (2005) 85:572–81. doi: 10.1038/labinvest.3700236
81. Xu X, Ehdaie B, Ohara N, Yoshino T, Deng CX. Synergistic action of Smad4 and Pten in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene. (2010) 29:674–86. doi: 10.1038/onc.2009.375
82. Chen Y, Ma L, He Q, Zhang S, Zhang C, Jia W TGF-beta1 expression is associated with invasion and metastasis of intrahepatic cholangiocarcinoma. Biol Res. (2015) 48:26. doi: 10.1186/s40659-015-0016-9
83. Sato Y, Harada K, Itatsu K, Ikeda H, Kakuda Y, Shimomura S, et al. Epithelial-mesenchymal transition induced by transforming growth factor-{beta}1/Snail activation aggravates invasive growth of cholangiocarcinoma. Am J Pathol. (2010) 177:141–52. doi: 10.2353/ajpath.2010.090747
84. Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T, et al. E/N-cadherin switch mediates cancer progression via TGF-beta-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer. (2011) 105:1885–93. doi: 10.1038/bjc.2011.452
85. Inoue K, Makuuchi M, Takayama T, Torzilli G, Yamamoto J, Shimada K, et al. Long-term survival and prognostic factors in the surgical treatment of mass-forming type cholangiocarcinoma. Surgery. (2000) 127:498–505. doi: 10.1067/msy.2000.104673
86. Fabris L, Alvaro D. The prognosis of perihilar cholangiocarcinoma after radical treatments. Hepatology. (2012) 56:800–2. doi: 10.1002/hep.25808
87. Sha M, Jeong S, Wang X, Tong Y, Cao J, Sun HY, et al. Tumor-associated lymphangiogenesis predicts unfavorable prognosis of intrahepatic cholangiocarcinoma. BMC Cancer. (2019) 19:208. doi: 10.1186/s12885-019-5420-z
88. Fava G, Demorrow S, Gaudio E, Franchitto A, Onori P, Carpino G, et al. Endothelin inhibits cholangiocarcinoma growth by a decrease in the vascular endothelial growth factor expression. Liver Int. (2009) 29:1031–42. doi: 10.1111/j.1478-3231.2009.01997.x
89. Augustin HG, Koh GY, Thurston G, Alitalo K Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. (2009) 10:165–77. doi: 10.1038/nrm2639
90. Aishima S, Nishihara Y, Iguchi T, Taguchi K, Taketomi A, Maehara Y, et al. Lymphatic spread is related to VEGF-C expression and D2-40-positive myofibroblasts in intrahepatic cholangiocarcinoma. Mod Pathol. (2008) 21:256–64. doi: 10.1038/modpathol.3800985
91. Xu Y, Yuan L, Mak J, Pardanaud L, Caunt M, Kasman I, et al. Neuropilin-2 mediates VEGF-C-induced lymphatic sprouting together with VEGFR3. J Cell Biol. (2010) 188:115–30. doi: 10.1083/jcb.200903137
92. Okada K, Osaki M, Araki K, Ishiguro K, Ito H, Ohgi S. Expression of hypoxia-inducible factor (HIF-1alpha), VEGF-C and VEGF-D in non-invasive and invasive breast ductal carcinomas. Anticancer Res. (2005) 25:3003–9. Available online at: http://ar.iiarjournals.org
93. Park BK, Paik YH, Park JY, Park KH, Bang S, Park SW, et al. The clinicopathologic significance of the expression of vascular endothelial growth factor-C in intrahepatic cholangiocarcinoma. Am J Clin Oncol. (2006) 29:138–42. doi: 10.1097/01.coc.0000204402.29830.08
94. Pezzuto A, Carico E. Role of HIF-1 in cancer progression: novel insights. A review. Curr Mol Med. (2018) 18:343–51. doi: 10.2174/1566524018666181109121849
95. Thongchot S, Yongvanit P, Loilome W, Seubwai W, Phunicom K, Tassaneeyakul W, et al. High expression of HIF-1alpha, BNIP3 and PI3KC3: hypoxia-induced autophagy predicts cholangiocarcinoma survival and metastasis. Asian Pac J Cancer Prev. (2014) 15:5873–8. doi: 10.7314/APJCP.2014.15.14.5873
96. Mills CN, Joshi SS, Niles RM. Expression and function of hypoxia inducible factor-1 alpha in human melanoma under non-hypoxic conditions. Mol Cancer. (2009) 8:104. doi: 10.1186/1476-4598-8-104
97. Currie MJ, Hanrahan V, Gunningham SP, Morrin HR, Frampton C, Han C, et al. Expression of vascular endothelial growth factor D is associated with hypoxia inducible factor (HIF-1alpha) and the HIF-1alpha target gene DEC1, but not lymph node metastasis in primary human breast carcinomas. J Clin Pathol. (2004) 57:829–34. doi: 10.1136/jcp.2003.015644
98. Liang X, Yang D, Hu J, Hao X, Gao J, Mao Z. Hypoxia inducible factor-alpha expression correlates with vascular endothelial growth factor-C expression and lymphangiogenesis/angiogenesis in oral squamous cell carcinoma. Anticancer Res. (2008) 28:1659–66. Available online at: http://ar.iiarjournals.org
99. Cadamuro M, Vismara M, Brivio S, Furlanetto A, Strazzabosco M, Fabris L. JNK signaling activated by Platelet-Derived Growth Factor D (PDGF-D) stimulates secretion of Vascular Endothelial Growth Factor-C (VEGF-C) by cancer-associated fibroblasts to promote lymphangiogenesis and early metastatization in cholangiocarcinoma. Digest Liver Dis. (2015) 47:e22–3. doi: 10.1016/j.dld.2015.01.052
100. Xu LB, Liu C, Gao GQ, Yu XH, Zhang R, Wang J. Nerve growth factor-beta expression is associated with lymph node metastasis and nerve infiltration in human hilar cholangiocarcinoma. World J Surg. (2010) 34:1039–45. doi: 10.1007/s00268-010-0417-4
101. Obulkasim H, Shi X, Wang J, Li J, Dai B, Wu P, et al. Podoplanin is an important stromal prognostic marker in perihilar cholangiocarcinoma. Oncol Lett. (2018) 15:137–46. doi: 10.3892/ol.2017.7335
102. Sha M, Jeong S, Chen XS, Tong Y, Cao J, Sun HY, et al. Expression of VEGFR-3 in intrahepatic cholangiocarcinoma correlates with unfavorable prognosis through lymphangiogenesis. Int J Biol Sci. (2018) 14:1333–42. doi: 10.7150/ijbs.26045
103. Cueni LN, Hegyi I, Shin JW, Albinger-Hegyi A, Gruber S, Kunstfeld R, et al. Tumor lymphangiogenesis and metastasis to lymph nodes induced by cancer cell expression of podoplanin. Am J Pathol. (2010) 177:1004–16. doi: 10.2353/ajpath.2010.090703
104. Navarro A, Perez RE, Rezaiekhaligh M, Mabry SM, Ekekezie II, et al. T1alpha/podoplanin is essential for capillary morphogenesis in lymphatic endothelial cells. Am J Physiol Lung Cell Mol Physiol. (2008) 295:L543–51. doi: 10.1152/ajplung.90262.2008
105. Thelen A, Scholz A, Benckert C, Schröder M, Weichert W, Wiedenmann B, et al. Microvessel density correlates with lymph node metastases and prognosis in hilar cholangiocarcinoma. J Gastroenterol. (2008) 43:959–66. doi: 10.1007/s00535-008-2255-9
106. Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, et al. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. (2002) 296:1883–6. doi: 10.1126/science.1071420
107. Alitalo K, Mohla S, Ruoslahti E. Lymphangiogenesis and cancer: meeting report. Cancer Res. (2004) 64:9225–9. doi: 10.1158/0008-5472.CAN-04-2475
108. Tewalt EF, Cohen JN, Rouhani SJ, Guidi CJ, Qiao H, Fahl SP, et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood. (2012) 120:4772–82. doi: 10.1182/blood-2012-04-427013
109. Francis H, DeMorrow S, Venter J, Onori P, White M, Gaudio E, et al. Inhibition of histidine decarboxylase ablates the autocrine tumorigenic effects of histamine in human cholangiocarcinoma. Gut. (2012) 61:753–64. doi: 10.1136/gutjnl-2011-300007
110. Leite de Oliveira R, Hamm A, Mazzone M. Growing tumor vessels: more than one way to skin a cat - implications for angiogenesis targeted cancer therapies. Mol Aspects Med. (2011) 32:71–87. doi: 10.1016/j.mam.2011.04.001
111. Cherry-Bohannan J, Baker K, Francis HJ. VEGF and cholangiocarcinoma: feeding the tumor. Transl Gastrointest Cancer. (2012) 1:95–102. doi: 10.3978/j.issn.2224-4778.2011.12.06
112. Gaudio E, Barbaro B, Alvaro D, Glaser S, Francis H, Ueno Y, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. (2006) 130:1270–82. doi: 10.1053/j.gastro.2005.12.034
113. Benckert C, Jonas S, Cramer T, Von Marschall Z, Schäfer G, Peters M, et al. Transforming growth factor beta 1 stimulates vascular endothelial growth factor gene transcription in human cholangiocellular carcinoma cells. Cancer Res. (2003) 63:1083–92. Available online at: https://cancerres.aacrjournals.org
114. Mancino A, Mancino MG, Glaser SS, Alpini G, Bolognese A, Izzo L, et al. Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig Liver Dis. (2009) 41:156–63. doi: 10.1016/j.dld.2008.02.015
115. Peng T, Li Z, Li D, Wang S. MACC1 promotes angiogenesis in cholangiocarcinoma by upregulating VEGFA. Onco Targets Ther. (2019) 12:1893–903. doi: 10.2147/OTT.S197319
116. Zheng B, Jeong S, Zhu Y, Chen L, Xia Q miRNA and lncRNA as biomarkers in cholangiocarcinoma(CCA). Oncotarget. (2017) 8:100819–30. doi: 10.18632/oncotarget.19044
117. Meadows V, Francis H. MicroRNAs and cholangiocarcinoma: elucidating the effects of tiny giants. AME Med J. (2018) 3:98. doi: 10.21037/amj.2018.09.10
118. Anand S, Cheresh DA. MicroRNA-mediated regulation of the angiogenic switch. Curr Opin Hematol. (2011) 18:171–6. doi: 10.1097/MOH.0b013e328345a180
119. Qu KZ, Zhang K, Li H, Afdhal NH, Albitar M Circulating microRNAs as biomarkers for hepatocellular carcinoma. J Clin Gastroenterol. (2011) 45:355–60. doi: 10.1097/MCG.0b013e3181f18ac2
Keywords: microenvironment, inflammatory signaling, cholangiocarcinoma, tumor lymphangiogenesis, angiogenesis
Citation: Roy S, Glaser S and Chakraborty S (2019) Inflammation and Progression of Cholangiocarcinoma: Role of Angiogenic and Lymphangiogenic Mechanisms. Front. Med. 6:293. doi: 10.3389/fmed.2019.00293
Received: 22 October 2019; Accepted: 29 November 2019;
Published: 18 December 2019.
Edited by:
Zongxin Ling, Zhejiang University, ChinaReviewed by:
Guido Carpino, Foro Italico University of Rome, ItalyCopyright © 2019 Roy, Glaser and Chakraborty. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanjukta Chakraborty, c2NoYWtyYWJvcnR5QHRhbXUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.