 Simon Sutcliffe1
Simon Sutcliffe1 Shirin Kalyan1,2
Shirin Kalyan1,2 Jim Pankovich1Jenny M. H. Chen1Rashieda Gluck1
Jim Pankovich1Jenny M. H. Chen1Rashieda Gluck1 Darby Thompson3,4
Darby Thompson3,4 Momir Bosiljcic1
Momir Bosiljcic1 Mark Bazett1Richard N. Fedorak5†
Mark Bazett1Richard N. Fedorak5† Remo Panaccione6
Remo Panaccione6 Jeffrey Axler7John K. Marshall8
Jeffrey Axler7John K. Marshall8 David W. Mullins9Boyko Kabakchiev10Dermot P. B. McGovern11Julie Jang1Andrew Coldman12
David W. Mullins9Boyko Kabakchiev10Dermot P. B. McGovern11Julie Jang1Andrew Coldman12 Gillian Vandermeirsch1Brian Bressler13Hal Gunn1*
Gillian Vandermeirsch1Brian Bressler13Hal Gunn1*- 1Qu Biologics Inc., Vancouver, BC, Canada
- 2Department of Medicine, University of British Columbia, Vancouver, BC, Canada
- 3Emmes Canada, Burnaby, BC, Canada
- 4Department of Statistics and Actuarial Sciences, Simon Fraser University, Burnaby, BC, Canada
- 5Division of Gastroenterology, University of Alberta, Edmonton, AB, Canada
- 6Inflammatory Bowel Disease Unit, University of Calgary, Calgary, AB, Canada
- 7Toronto Digestive Disease Associates Inc., Vaughan, ON, Canada
- 8Department of Medicine and Farncombe Family Digestive Health Research Institute, McMaster University, Hamilton, ON, Canada
- 9Department of Microbiology, Immunology and Medical Education, Geisel School of Medicine at Dartmouth, Hanover, NH, United States
- 10Zane Cohen Centre for Digestive Diseases, Samuel Lunenfeld Research Institute, Mount Sinai Hospital, Toronto, ON, Canada
- 11Cedars-Sinai Medical Center, Los Angeles, CA, United States
- 12Cancer Control Research, British Columbia Cancer Agency, Vancouver, BC, Canada
- 13Gastrointestinal Research Institute, Vancouver, BC, Canada
Background: Current Crohn's disease (CD) therapies focus on suppressing immune function and come with consequent risk, such as infection and cancer. Notwithstanding, most CD patients still experience disease progression. There is a need for new CD treatment strategies that offer better health outcomes for patients.
Aims: To assess safety, efficacy, and tolerability of a novel microbial-derived immunotherapy, QBECO, that aims to restore rather than suppress immune function in CD.
Methods: A randomized, double-blind, placebo-controlled trial was conducted in 68 patients with moderate-to-severe CD. Primary endpoints: safety and Week 8 clinical improvement. Secondary endpoints: Week 8 clinical response and remission. Week 8 responders continued blinded treatment through Week 16; non-responders received open-label QBECO from Weeks 9–16. Exploratory analyses included immune biomarker and genotype assessments.
Results: QBECO was well-tolerated. Mean reduction in Crohn's Disease Activity Index (CDAI) score was −68 for QBECO vs. −31 for placebo at Week 8. Improvement with QBECO continued through Week 16 (-130 CDAI reduction). Week 8 QBECO clinical response, improvement and remission rates were 41.2%, 32.4%, 29.4% vs. 26.5%, 23.5%, 23.5% for placebo. TNFα inhibitor-naïve subjects achieved higher response rates at Week 8 with QBECO (64%) vs. placebo (26%). Specific immune biomarkers were identified that linked to QBECO response.
Conclusion: This proof-of-concept study supports further investigation for the use of QBECO as a novel immunotherapy approach for CD. Biomarker analyses suggests it may be feasible to personalize CD treatment with QBECO. Larger trials are now needed to confirm clinical improvement and the unique biological findings.
Clinical Trial Number: NCT01809275 (https://clinicaltrials.gov/ct2/show/NCT01809275)
Introduction
Crohn's disease (CD) is a chronic inflammatory disease of the gastrointestinal (GI) tract with a relapsing and remitting course. Chronic uncontrolled inflammation can lead to progressive bowel damage and complications such as stenosis and fistula, often leading to surgery (1, 2). Front-line therapies for CD target adaptive immune pathways, and many patients treated with these immunosuppressive therapeutics still develop progressive disabling disease (2) as well as increased risk of infections, malignancy, lupus-like autoimmunity, demyelinating central nervous system disease, and hypersensitivity reactions (3, 4). Thus, new treatment approaches for CD that are safe, cost-effective, and able to achieve durable remission are required.
Accumulating clinical and genetic evidence suggests that a defective innate immune response may be fundamental to the pathogenesis of CD (5–8) and precedes the consequent over-reactive adaptive immune response that is characteristic of the disease and the target of current treatments (9, 10). Segal and Lowei first demonstrated that patients with CD exhibited an impaired systemic acute inflammatory response (11). Subsequent genetic studies provided further support for the hypothesis that defective or inefficient innate immune function, particularly that of macrophages, is linked to CD (12, 13). In this work, we present the results from a Phase 1/2 randomized, double-blind, placebo-controlled trial of a novel immunotherapy approach to optimize innate immune function in CD. QBECO, an investigational immunotherapy derived from an inactivated GI pathogen, aims to elicit an acute innate immune response targeting the GI tract to re-establish competent barrier function and immune competency (14). Treatment is self-administered by subcutaneous injection. Promising early clinical experience with QBECO for the treatment of CD in a compassionate use program (15) and a translational study in ulcerative colitis showing improved GI barrier function with QBECO treatment (14) motivated this proof-of-concept clinical trial to explore safety, efficacy, and tolerability of this novel immunotherapy in subjects with moderate-to-severe CD.
Methods
Study Design, Randomization, and Treatment Strategy
This was a Phase 1/2 randomized, double-blind, placebo-controlled study (NCT01809275; Health Canada approval 27-02-2013) for the treatment of moderate-to-severe CD. All study subjects provided written informed consent and the trial was conducted in compliance with the Declaration of Helsinki and Good Clinical Practice guidelines. The study protocol was approved by the institutional research ethics boards at the four study sites.
Eligible subjects were randomized 1:1 to receive self-administered subcutaneous injections of blinded placebo or blinded QBECO every second day for 8 weeks. At the time the study was conducted, the Crohn's Disease Activity Index (CDAI) was the instrument of choice to assess disease severity (16–18). The CDAI is composed of 8 weighted components present over 7 days which include: number of liquid/soft stools per day, abdominal pain, general feeling of well-being, presence of complications, taking opioid-based medication for diarrhea, presence of an abdominal mass, hematocrit <0.47 for men or <0.42 in women, and deviation from one's normal weight (by percentage). Subjects with a clinical response (defined as a decrease in CDAI ≥ 70 points) at Week 8 continued their randomized assignment in a blinded manner through 16 weeks. Subjects without a clinical response at Week 8 received open-label QBECO from Week 9–16. Subcutaneous injection procedures were identical for the randomized groups and included an initial 5-day training at the study site. Subjects titrated their dose beginning at 0.05 mL, increasing by 0.02 mL every other day until experiencing a 2.5–5 cm erythema at the injection site on the day following injection, or until a maximum 0.2 mL dose was reached. The dose was reduced by 0.01 mL if erythema exceeded 5 cm until the targeted 2.5–5 cm erythema was reached. Subjects switching to open-label QBECO at 8 weeks were re-titrated irrespective of their randomized group. Randomization was based on a pre-defined list using a permuted block design (size 4). Product was dispensed in randomly numbered kits. Pharmacy, clinical site staff and subjects were blinded to assignment.
Patient Population
Subjects ≥ 18 years with a diagnosis of CD of >6 months duration established by clinical, endoscopic or radiological, and histopathological assessment were enrolled following informed consent. All subjects had moderate-to-severe disease, defined by CDAI > 220 but <450 points, AND either a CRP (C-reactive protein) level > 2.87 mg/L or a fecal calprotectin (FCP) level > 250 μg/g, or an ileocolonoscopy or radiographic tests showing active CD within the last 6 months. CRP, FCP, and standard laboratory measures were taken at each study visit. Continued stable doses of the following medications were allowed: oral 5-ASA compounds, oral corticosteroid prednisone equivalent dose <30 mg/day or budesonide <9 mg/day if there was dose stability >2 weeks before trial screening visit; probiotics, anti-diarrheal medications, azathioprine or 6-mercaptopurine, or methotrexate provided the dose had been stable for 8 weeks preceding first study dose; and antibiotics providing dose stability was present for 2 weeks prior to first study dose. Male and pre-menopausal female subjects were required to agree to practice effective birth control.
QBECO and Placebo Composition, Formulation, and Administration
QBECO is an investigational immune modulator consisting of all major macromolecules of an inactivated pathogenic strain of E. coli, isolated from a patient with an E. coli GI infection, suspended in physiological saline with 0.4% phenol as a preservative. The placebo used in this clinical trial was physiological saline with 0.4% phenol. Study ampules were tinted to prevent comparison of turbidity between QBECO and placebo.
Following three in-clinic supervised doses, subjects self-administered QBECO at home, recording the date of the injection, dose (volume) injected, location of the injection site, local skin response diameter (if present) on the day following administration, and any other relevant observations. Compliance was evaluated by diary review and clinic visit assessment.
Safety Assessment
Safety variables included adverse events, concomitant therapies, physical exams, and laboratory tests to assess hematologic, hepatic, and renal function at scheduled time points. Adverse events were graded according to National Cancer Institute Common Terminology Criteria, Version 4.0 (http://avs.nci.nih.gov). Subjects experiencing serious adverse events discontinued study treatment.
Study Outcome Variables
Primary endpoints were safety (adverse events, clinical laboratory findings, concomitant therapies) and clinical improvement defined as a decrease in CDAI from baseline of ≥100 points or a CDAI score of ≤ 150 points at Week 8. Secondary endpoints were clinical remission at Week 8 (CDAI ≤ 150 points); clinical response at Week 8 (decrease in CDAI score from baseline ≥ 70 points); and CDAI score change from baseline to Week 8. Treatment failure was defined by the need for rescue medications, major surgical intervention for the treatment of CD, or QBECO-related adverse events leading to discontinuation of QBECO.
Exploratory end-points included the relationship between QBECO response and baseline demographic and disease characteristics, concomitant therapies, serum immune biomarkers, and CD-associated genotypes.
Immune Biomarker Analysis
Serum immune cytokines and chemokines were analyzed by multiplex technology (assay performed by Eve Technologies, Calgary, AB, Canada) using the Human Cytokine/Chemokine Array 42-Plex with IL-18 (HD42; Millipore).
Genetic Analysis
Genetic analysis with respect to QBECO response/non-response was performed on 30 CD subjects treated with QBECO, including 27 subjects from this trial and 3 subjects treated in a compassionate use program (15). Genetic analysis was not included in the original trial design and thus, consent for genetic testing sought retrospectively in subjects treated with QBECO. The analysis was done on 16 trial subjects who provided consent, 11 trial subjects for whom ethics approval was obtained for genome analysis on anonymized samples identified only as “QBECO responder” or “QBECO non-responder,” and 3 subjects treated in a compassionate use program who provided consent.
Genotyping was performed using the InfiniumOmni2-5-8 v1.3 array (Illumina, San Diego, CA). Preprocessing and quality control were completed in Genome Studio v2011.1 (Illumina). Only single-nucleotide polymorphisms (SNPs) with GenCall scores >0.2 in 90% of samples, call rates > 95%, minor allele frequency >5%, and Hardy–Weinberg equilibrium p-values > 10−6 were considered for further analysis. Two-hundred forty IBD susceptibility SNPs (19, 20) were selected in a hypothesis-driven approach; 113 passed quality control for analysis.
Statistical Analysis
An initial sample size of 60 subjects was chosen to achieve 80% power with a two-sided 0.05-level chi-square test assuming a 42% (33 vs. 75%) difference in clinical improvement rate between QBECO and placebo at Week 8. An interim sample-size re-estimation was performed based on aggregate, blinded, clinical improvement rates, and the sample size was increased to 68 subjects.
Week 8 and 16 clinical response, improvement and remission rates were compared between placebo and QBECO groups using a 2-sided Fisher's Exact test. Change in CDAI from baseline was compared using a 2-sided, 2-sample paired t-test.
The intention to treat (ITT) analysis evaluated all 68 subjects by randomized group. Subjects considered as treatment failures or who did not provide Week 8 CDAI scores were considered non-responders in the ITT analysis. The Week 8 per protocol (PP) analysis excludes 10 subjects who did not complete 8 weeks of treatment (n = 8) or failed to meet all eligibility criteria (n = 2), comprised of 7 from the QBECO arm and 3 from the placebo arm (Figure 1).
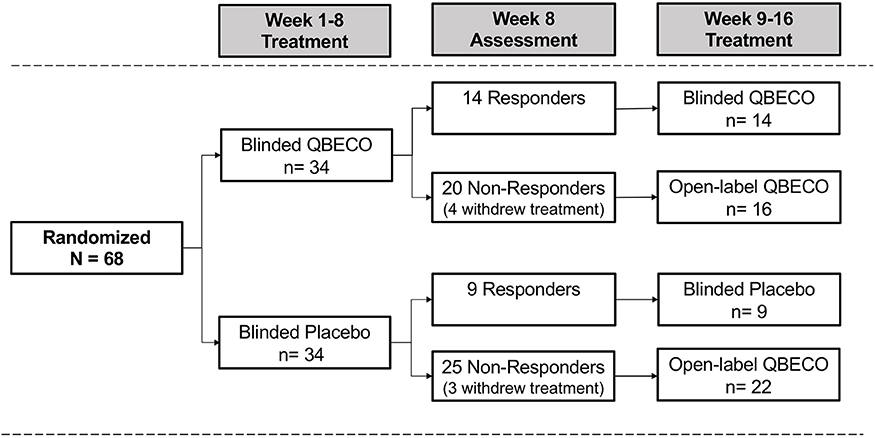
Figure 1. Patient flow by treatment assignment. Subjects with moderate-to-severe Crohn's disease were randomized 1:1 to QBECO or Placebo for 8 weeks. Those responding to allocated blinded treatment continued blinded treatment for another 8 weeks; all 8 Week non-responders commenced open-label QBECO for 8 weeks.
To account for imbalances in important baseline variables, change in CDAI score from baseline through Week 8 was modeled using a linear regression model that included the following criteria: CDAI, disease severity at baseline (≥250 vs. <250), use of concomitant immunosuppressive medication (Y/N), disease duration (years) and prior exposure to anti-TNF agents (Y/N). These variables were selected prior to the regression analysis based on scientific and clinical judgement; no model selection was performed. Population marginal means for QBECO and placebo (i.e., assuming a balanced population) were generated using least-square means analysis and statistically compared using a t-test.
Cytokines were evaluated by change in concentration over time with exposure to study treatment (using one-sample Mann-Whitney test of the paired differences) and by differential change over time by QBECO clinical response status (in QBECO treated subjects only). Linear mixed effect model of cytokine concentration was computed with p-values estimated from a Wald test of the interaction between clinical response and time. Finally, associations between baseline concentration of cytokines and Week 8 clinical response and remission status (two-sample Mann-Whitney) were assessed. P-values were adjusted for multiple comparisons across 42 cytokines using a Benjamini-Hochberg procedure.
Genetic analysis used generalized linear models with an additive genetic model for categorical response and predictor variables. Kruskal-Wallis analysis of variance was used for continuous and ordinal response variables. Benjamini-Hochberg procedure was used to adjust for multiple comparisons across the 113 SNPs that passed quality control. An algorithm combining the top three SNPs linked to QBECO response was formulated using methodology previously described in a genetic association study of CD and ulcerative colitis phenotypes (21).
All authors had access to the study data and reviewed and approved the final manuscript.
Results
Study Subjects
Sixty-eight subjects were randomly assigned (1:1) to receive blinded QBECO or placebo for 8 weeks. Demographic and baseline characteristics are shown for both treatment groups in Table 1. The two arms markedly differed with respect to prior anti-TNFα exposure (59% QBECO vs. 21% placebo). Patients randomized to QBECO were older (average age 42.4 vs. 31.8 years for placebo), and more likely to have baseline CDAI > 250 compared to the placebo group (24 vs. 19).

Table 1. Baseline and demographic variables.
Overall, 56 of the 68 subjects (82.4%) completed the study through Week 24, including 27 (79.4%) subjects initially randomized to QBECO and 29 (85.3%) to placebo. Figure 1 shows the flow of subjects over 16 weeks by treatment. Compliance with treatment administration was high in both groups (>90% of expected injections).
Change in Crohn's Disease Status by Treatment Group
Mean reduction in CDAI score from baseline to Week 8 was greater for QBECO vs. placebo subjects in both the ITT (p = 0.068) and PP (p = 0.027) analyses (Figure 2). Table 2 presents the rates of clinical response (41.2% QBECO vs. 26.5% placebo), improvement (32.4% QBECO vs. 23.5% placebo) and remission (29.4% QBECO vs. 23.5% placebo), which did not achieve statistical significance at Week 8. Subjects treated with QBECO continued to improve through Week 16, experiencing a further mean CDAI reduction of 50 points from Week 8, reaching a 130-point reduction by Week 16 (Figure 3; Supplement Table 1 provides this data in tabular form, which includes the number of patients moving through each arm of the study to Week 16).
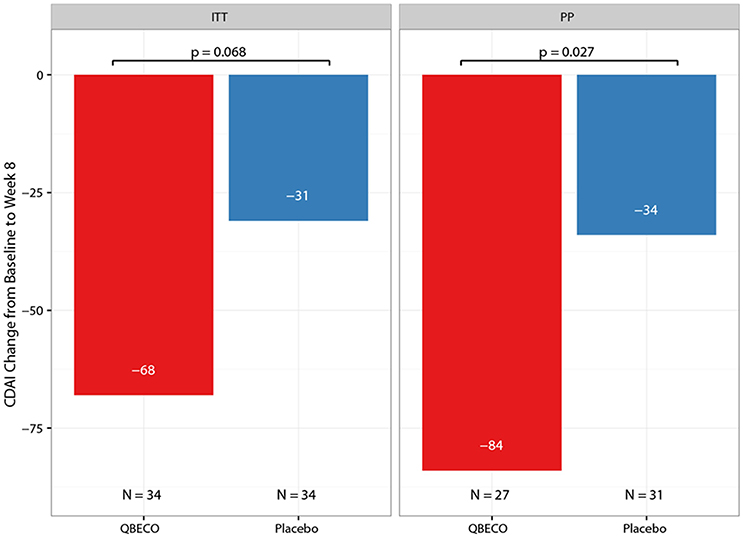
Figure 2. Mean change in Crohn's Disease Activity Index (CDAI) score from baseline to Week 8 by treatment group. The mean reduction in CDAI from baseline to Week 8 in patients with moderate-to-severe Crohn's disease blinded to QBECO (red bar) or Placebo (blue bar) treatment. ITT, Intention to Treat analysis; PP, Per Protocol analysis.
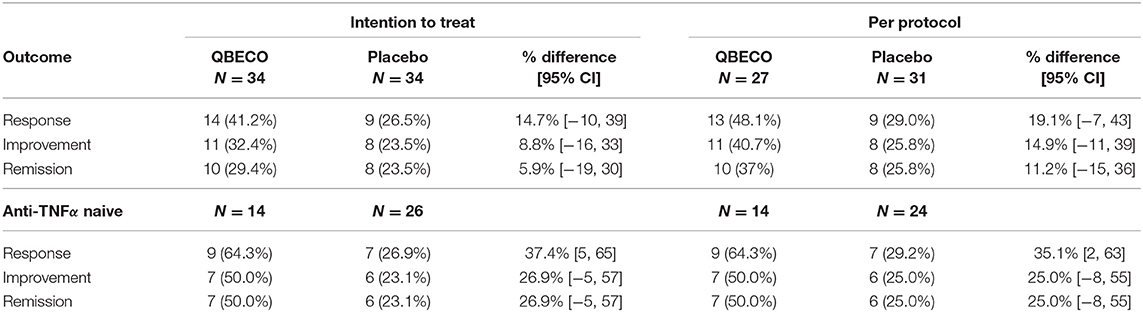
Table 2. Clinical response, improvement, and remission rates at Week 8 by treatment.
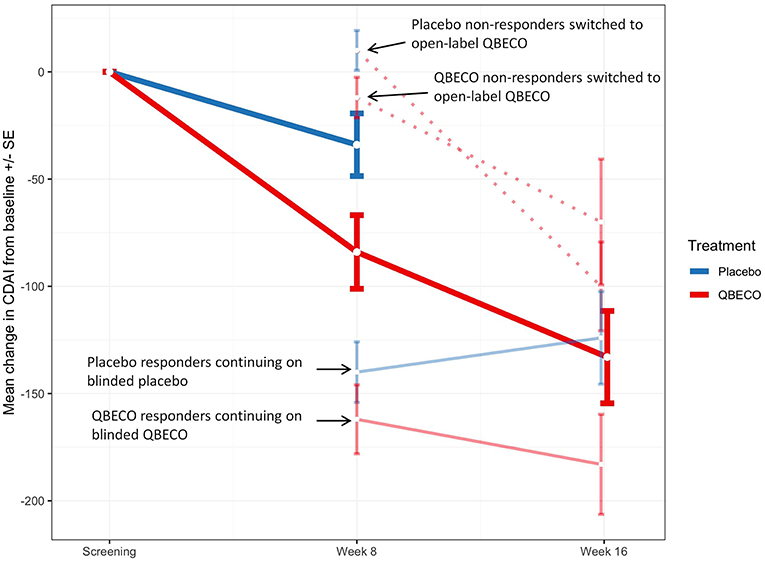
Figure 3. Mean change in Crohn's Disease Activity Index (CDAI) score in study groups Week 8 and Week 16. Mean change in CDAI score for subjects randomized to QBECO (solid red line) and placebo (solid blue line) from baseline to Week 8 and Weeks 16 of study treatment. Those responding to allocated blinded treatment at week 8 continued blinded treatment for another 8 weeks [light blue solid line for responders originally blinded to placebo treatment (n = 9); light red solid line for QBECO responders (n = 13)]. All placebo (n = 22) and QBECO (n = 14) non-responders at week 8 received open-label QBECO (dashed red lines) for weeks 9–16. Dark solid red line represents the average of all subjects on QBECO for weeks 9–16.
Patients naïve to anti-TNFα agents achieved a 64% response rate at Week 8 with QBECO vs. 26% placebo (p = 0.041; ITT analysis), and more than double the improvement and remission rates (Table 2). Anti-TNFα naïve Week 8 placebo non-responders (n = 17) treated with 8 weeks open-label QBECO from weeks 9 to 16 achieved response, improvement and remission rates of 71, 47, and 47%, respectively. This was similar to the response, improvement and remission rates observed after 8 weeks of treatment in the anti-TNFα naïve group initially randomized to QBECO. A longitudinal analysis including previous TNFα inhibitor exposure interaction with treatment following week 8 shows that subjects previously treated with anti-TNFα agents may respond to QBECO treatment with longer course of treatment (Supplement Figure 1).
In subjects with baseline CDAI ≥ 250 (n = 24 [70.6%] QBECO, n = 19 [55.9%] placebo), the response, improvement and remission rates in subjects treated with QBECO vs. placebo were: 42%, 29%, 25% vs. 16%, 11%, 11%, respectively. To account for differences in baseline characteristics that may be important in the change in CDAI score by treatment, a regression analysis was performed taking into account baseline CDAI, disease severity at baseline (>250), use of concomitant immunosuppressive medication, disease duration and prior exposure to anti-TNFα agents (Supplement Figure 2). Following adjustment for imbalanced prognostic variables, the reduction in CDAI at Week 8 was 48 points greater in the QBECO than placebo treated cohort (p = 0.024; ITT analysis).
Safety Evaluation
QBECO treatment was well-tolerated. Adverse events experienced by >5% of blinded participants for weeks 1–8 of the study (Table 3) and subjects who received open-label QBECO for weeks 9–16 (Table 4). The majority of adverse events reported during the blinded period (weeks 1–8) were Grade 1 (82.1 and 88.1% of all events for subjects receiving QBECO-01 and placebo, respectively) and transient. No significant difference between placebo and QBECO groups was identified. Those receiving QBECO experienced more transient flu-like symptoms, which are considered to be related to the mechanism of action of QBECO.
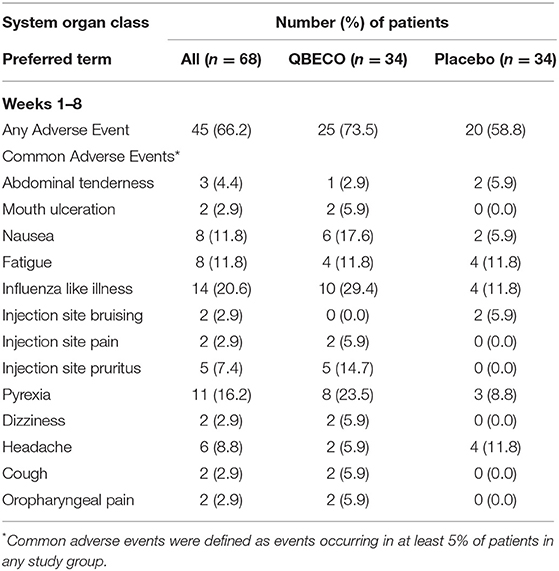
Table 3. Adverse events affecting at least 5% of subjects receiving study drug.
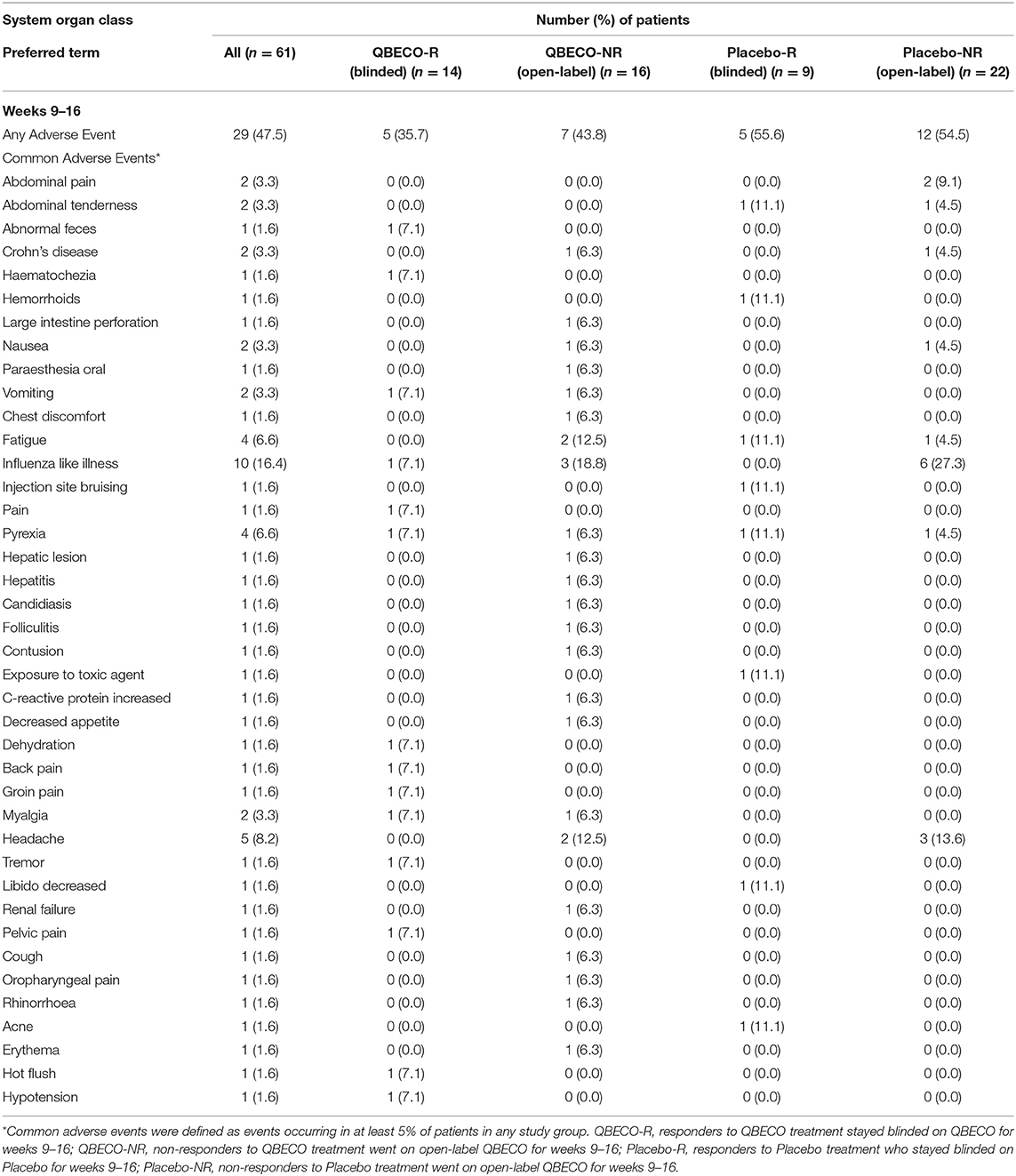
Table 4. Adverse events affecting at least 5% of subjects receiving study drug.
Severe Adverse Events (SAE) were uncommon with 6.4 and 2.3% of all reported events reported as Grade 3 for those receiving QBECO and placebo, respectively. Seven of these eight (87.5%) individuals had past exposure to anti-TNFα therapies. Five of eight SAEs were considered to be unlikely related or unrelated to treatment, with 3 (fever/chills, liver/kidney issues, exacerbations of benign lung nodules) possibly related to treatment. The only SAE in an anti-TNFα-naïve subject was a C. difficile infection in an individual randomized to placebo. SAEs experienced during the course of the study and reasons for not completing the study are listed in Table 5.
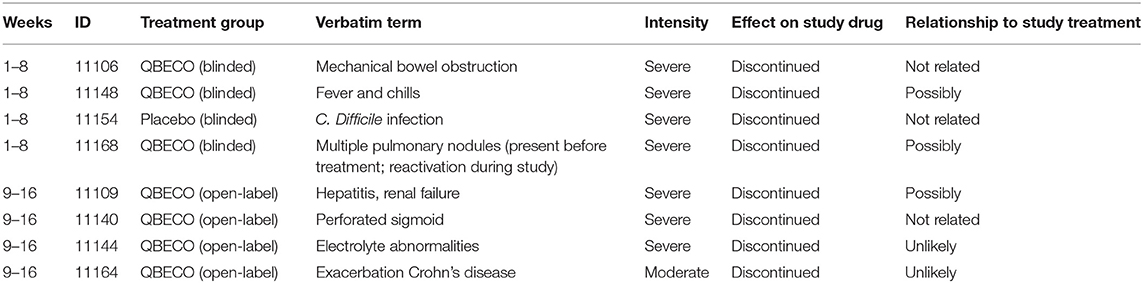
Table 5. Serious adverse events experienced over the course of study.
Immune Biomarker Analysis
Forty-two serum immune factors were assessed at baseline, Week 8, 16, and 24. Interleukin-18 (IL-18) increased from baseline to Week 8 and 16 with QBECO treatment (median change 24 pg/mL, adjusted p = 0.066 at Week 8 and 56 pg/mL, adjusted p = 0.067 at Week 16), but not with placebo treatment. None of the serum cytokine biomarkers remained elevated at Week 24 (i.e., 8 weeks after stopping study treatment).
Among QBECO treated subjects, IFNγ, IL-12p70, IL-17A, and TGFα were significantly elevated in QBECO responders vs. QBECO non-responders (adjusted p = 0.037 for all) (Figure 4).
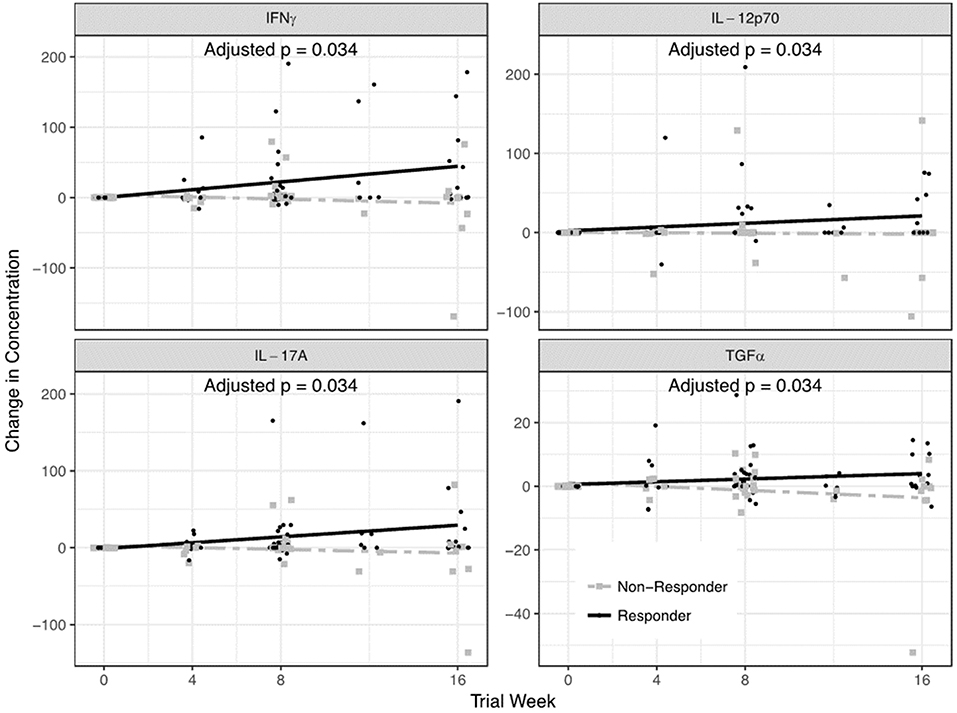
Figure 4. Serum cytokines that differed in QBECO responders and non-responders. A 42-plex cytokine/chemokine analysis was performed on serum. Four cytokines- IFNγ, IL-12p70, IL-17A, and TGFα- differentiated QBECO responders from non-responders over the study period after adjusting for multiple comparisons.
Week 8 clinical remission with QBECO treatment occurred more frequently in patients with lower baseline serum Eotaxin-1 levels (adjusted p = 0.0062) and IL-10 and IL-12p40 (p > 0.05 after correcting for multiple comparisons). This relationship between lower Eotaxin-1 levels and increased Week 8 remission was not found in placebo treated patients. These 3 cytokines tended to be higher in patients previously treated with TNFα inhibitors (Supplement Table 2), and in a longitudinal analysis, as performed with previous anti-TNFα agent use, patients with high baseline Eotaxin-1 levels responded equally well to QBECO treatment with a longer course of treatment (Supplement Figure 3).
CRP, an acute phase response protein upregulated in response to bacterial infections, and FCP, a cation-binding protein released by granulocytes in response to infection, were not anticipated to be reduced during active QBECO treatment given its mechanism of action. We assessed levels of these immune biomarkers leading up to Week 24 (when subjects were off study treatments). At Week 24, 44% of those who had been on QBECO from the beginning of the study, 42% of those who had switched to QBECO from placebo at Week 8, and 0% of those who were on placebo since the beginning had CRP levels < 5 mg/L (Supplement Table 3). Similarly, 35% of those who had been on QBECO from the beginning of the study, 18% of those who had switched to QBECO from placebo at Week 8, and 0% of those who were on placebo since the beginning had FCP levels of <250 ug/g (Supplement Table 4).
Genetic Associations With Response to QBECO
One hundred and thirteen SNPs reported to be linked to IBD were analyzed for response to QBECO treatment. A gene risk score, which is a weighted value based on variation in multiple genetic loci, was computed to assess the ability to stratify subjects' response to QBECO. Figure 5 shows that the gene risk score could differentiate QBECO responders from non-responders in this cohort, p = 0.0000243. Supplement Table 5 lists the IBD-linked SNPs and their weighted contribution to the construction of the gene risk score.
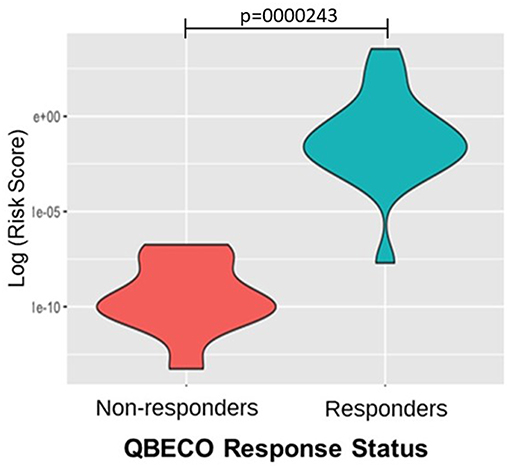
Figure 5. Gene risk score separates QBECO responders from non-responders. One hundred and thirteen inflammatory bowel disease (IBD)-related SNPs were included in computing the gene risk score for response to QBECO to assess the potential contribution of subjects' genetics to treatment outcome. The derived gene risk score could successfully distinguish QBECO responders from non-responders, p = 0.0000243.
Discussion
In this proof-of-concept study assessing QBECO, a first-in-class microbial-based immunotherapy, for the treatment of CD, a greater reduction in disease was observed by Week 8 in subjects randomized to QBECO compared to placebo. For the pre-specified Week 8 primary analysis in this 68-patient study, the difference did not reach statistical significance, but secondary analyses suggest that the biological effect induced by QBECO may be of benefit to patients with moderate-to-severe CD and warrants further study.
Notably, patients with prior exposure to anti-TNFα agents, who are known to generally be more difficult to treat (22, 23), were less likely to respond to QBECO by Week 8. Due to unequal randomization, these patients were significantly more prevalent in the QBECO arm than in the placebo arm. However, subjects previously treated with TNFα inhibitors did experience symptom improvement with QBECO as treatment continued to Week 16, suggesting that a longer course of treatment may be required to achieve optimal results in these subjects. QBECO Week 8 clinical response, improvement and remission rates in anti-TNFα naïve patients compare favorably to those reported at similar time-points in recent Phase 3 trials with biologics such as vedolizumab and ustekinumab (22, 23).
The current treatment approach for CD largely targets the overzealous adaptive immune response to invading bacteria in the GI tract, but accumulating evidence suggests patients with CD have impaired or deficient innate immunity that predisposes to defective barrier function (3, 5, 7, 8). Identification of genetic variants linked to CD that associate with innate immune function lends support to the idea that innate immune insufficiency plays a role in disease pathophysiology, at least for a significant segment of those suffering from CD (5, 7). Serum cytokine analysis in this study demonstrated a QBECO-induced increase in IL-18, a cytokine known to promote phagocytosis and bacterial clearance (24). This corroborates our findings in experimental models of colitis in which colonic expression of IL-18 increased in response to QBECO treatment—resulting in marked improvements in gastrointestinal histopathology and barrier function (14). Other studies of colitis have shown a lack of IL-18 results in more severe disease (25), and the administration of IL-18 can reverse the phenotype (26). IL-18 with IL-12 acts on natural killer (NK) cells, γδ T cells and other “Th1” cells to stimulate the production of IFNγ (27, 28), which in turn acts on macrophages to further enhance phagocytosis, bacterial clearance and antigen presentation (24). CD patients who improved with QBECO treatment produced IL-18 and had increases in serum IFNγ, IL-12p70, and IL-17A levels, whereas, subjects deemed as QBECO non-responders did not show the same increases in these three cytokines. This may reflect an inability to launch a productive immune response to bacterial stimulation. The observed higher incidence of transient flu-like symptoms in QBECO treated subjects likely reflects this immune mobilization, and we believe it is part of QBECO's mechanism of action. Of note, CD patients who improved with QBECO treatment also had increases in their levels of TGFα, which has been reported to be reduced in diseased regions of the colon of patients with inflammatory bowel disease and increased in healthy regions (29).
Patients with lower baseline levels of Eotaxin-1, IL-10, and IL-12p40 were more likely to achieve response and remission with 8 weeks of QBECO treatment. Of note, these cytokines tended to be higher in those who had previously been treated with anti-TNFα therapy and may reflect greater immune dysregulation in patients (30–33). For such individuals, a longer course of QBECO treatment may be required to overcome the presence of greater immune dysfunction, as is suggested by the study's 16-week data showing that patients with higher baseline levels of these cytokines achieved more optimal responses with a longer duration of treatment.
A personalized approach to CD treatment has been elusive to date, possibly because current treatments focus on symptom management (3, 34, 35), rather than upstream biological processes predisposing CD symptoms. Subject genotype was found to differentiate QBECO responders from non-responders. Collectively, the genetic and cytokine findings of this study provide promise for personalized medicine in CD with QBECO, and they now need replication in larger cohorts.
This proof-of-concept study is limited by its small size, short treatment duration, lack of stratification for previous TNFα inhibitor use, and lack of endoscopic and histological assessment. The therapeutic paradigm has now moved from symptom-based assessment to objective measures of disease activity (36). QBECO treatment has shown endoscopic and histological improvement in moderate-to-severe ulcerative colitis (14) and now needs to be demonstrated in patients with CD.
In conclusion, QBECO warrants further study as a novel immunotherapy approach for the management of CD. This approach not only provides a new way of thinking about the treatment of the disease, but also sheds more light on the heterogeneity of CD pathogenesis. QBECO may be the optimal choice for those patients with disease characterized by innate immune dysfunction rather than other underlying etiologies. The data from this trial will inform the design of larger definitive Phase II trials, which will include evaluation of endoscopic and histological endpoints, assessment of the impact of prior TNFα inhibitor exposure on QBECO response, evaluation of patients over a longer treatment period, microbiome assessment, and confirmation of the genetic and immune biomarker findings.
Ethics Statement
This was a Phase 1/2 randomized, double-blind, placebo-controlled study (NCT01809275; Health Canada approval 27-02-2013) for the treatment of moderate-to-severe CD. All study subjects provided written informed consent and the trial was conducted in compliance with the Declaration of Helsinki and Good Clinical Practice guidelines. The study protocol was approved by the institutional research ethics boards at the four study sites.
Author Contributions
HG, SS, BB, and AC: study concept and design, analysis and interpretation of data, drafting and review of manuscript. SK, JP, MBa, GV, RF, RP, JA, JM, DMu, and DMc: analysis and interpretation of data, drafting manuscript, critical revision of manuscript, and acquisition of data. DT: statistical analysis of the trial. BK: statistical analysis of the genetic study. RG, JC, MBo, and JJ: acquisition of data, conduct of study, and patient engagement. All authors read and approved the final draft of the manuscript.
Funding
This study was funded by Qu Biologics and Genome BC (SoFI Program).
Conflict of Interest Statement
SS, DT, RF, RP, DMu, BK, DMc, AC, and BB have served as consultants and/or advisors to Qu Biologics. SK, JP, JC, RG, MBo, MBa, JJ, and GV are (or were) employees of Qu Biologics. HG is the CEO and major shareholder of Qu Biologics. Qu Biologics owns patents across all the major markets (including U.S. Patent No. 8,980,279) relating to the use of Site Specific Immunomodulators derived from components of E. coli (QBECO) to treat inflammatory bowel disease. Qu Biologics has also filed patents for the use of immune and genetic biomarkers for the use of QBECO in patients with inflammatory bowel disease. In addition to the above, the following author affiliations are non-academic incorporated for-profit entities: Emmes Canada (DT) and Toronto Digestive Disease Associates, Inc. (JA).
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2019.00170/full#supplementary-material
References
1. Beaugerie L, Seksik P, Nion-Larmurier I, Gendre JP, Cosnes J. Predictors of Crohn's disease. Gastroenterology. (2006) 130:650–6. doi: 10.1053/j.gastro.2005.12.019
2. Magro F, Dias CC, Coelho R, Santos PM, Fernandes S, Caetano C, et al. Impact of early surgery and immunosuppression on Crohn's disease disabling outcomes. Inflamm Bowel Dis. (2017) 23:289–97. doi: 10.1097/MIB.0000000000001007
3. Ahluwalia JP. Immunotherapy in inflammatory bowel disease. Med Clin North Am. (2012) 96:525–44. doi: 10.1016/j.mcna.2012.04.009
4. McLean MH, Neurath MF, Durum SK. Targeting interleukins for the treatment of inflammatory bowel disease-what lies beyond anti-TNF therapy? Inflamm Bowel Dis. (2014) 20:389–97. doi: 10.1097/01.MIB.0000437616.37000.41
5. Vinh DC, Behr MA. Crohn's as an immune deficiency: from apparent paradox to evolving paradigm. Expert Rev Clin Immunol. (2013) 9:17–30. doi: 10.1586/eci.12.87
6. Hayee B, Rahman FZ, Sewell G, Smith AM, Segal AW. Crohn's disease as an immunodeficiency. Expert Rev Clin Immunol. (2010) 6:585–96. doi: 10.1586/eci.10.32
7. Marks DJ, Rahman FZ, Sewell GW, Segal AW. Crohn's disease: an immune deficiency state. Clin Rev Allergy Immunol. (2010) 38:20–31. doi: 10.1007/s12016-009-8133-2
8. Sewell GW, Rahman FZ, Levine AP, Jostins L, Smith PJ, Walker AP, et al. Defective tumor necrosis factor release from Crohn's disease macrophages in response to Toll-like receptor activation: relationship to phenotype and genome-wide association susceptibility loci. Inflamm Bowel Dis. (2012) 18:2120–7. doi: 10.1002/ibd.22952
9. Funderburg NT, Stubblefield Park SR, Sung HC, Hardy G, Clagett B, Ignatz-Hoover J, et al. Circulating CD4(+) and CD8(+) T cells are activated in inflammatory bowel disease and are associated with plasma markers of inflammation. Immunology. (2013) 140:87–97. doi: 10.1111/imm.12114
10. Elson CO, Alexander KL. Host-microbiota interactions in the intestine. Dig Dis. (2015) 33:131–6. doi: 10.1159/000369534
11. Segal AW, Loewi G. Neutrophil dysfunction in Crohn's disease. Lancet. (1976) 2:219–21. doi: 10.1016/S0140-6736(76)91024-2
12. Casanova JL, Abel L. Revisiting Crohn's disease as a primary immunodeficiency of macrophages. J Exp Med. (2009) 206:1839–43. doi: 10.1084/jem.20091683
13. Uniken Venema WT, Voskuil MD, Dijkstra G, Weersma RK, Festen EA. The genetic background of inflammatory bowel disease: from correlation to causality. J Pathol. (2017) 241:146–58. doi: 10.1002/path.4817
14. Sham HP, Bazett M, Bosiljcic M, Yang H, Luk B, Law HT, et al. Immune stimulation using a gut microbe-based immunotherapy reduces disease pathology and improves barrier function in ulcerative colitis. Front Immunol. (2018) 9:2211. doi: 10.3389/fimmu.2018.02211
15. Bressler B, Bethel KP, Kleef R, Reynolds SL, Sutcliffe S, Mullins DW, et al. Site-specific immunomodulator: a novel treatment for Crohn's disease. Gastroenterol Res Pract. (2015) 2015:231243. doi: 10.1155/2015/231243
16. Harvey RF, Bradshaw JM. A simple index of Crohn's-disease activity. Lancet. (1980) 1:514. doi: 10.1016/S0140-6736(80)92767-1
17. Best WR, Becktel JM, Singleton JW, Kern F Jr. Development of a Crohn's disease activity index. National Cooperative Crohn's Disease Study. Gastroenterology. (1976) 70:439–44. doi: 10.1016/S0016-5085(76)80163-1
18. Best WR, Becktel JM, Singleton JW. Rederived values of the eight coefficients of the Crohn's Disease Activity Index (CDAI). Gastroenterology. (1979) 77:843–6. doi: 10.1016/0016-5085(79)90384-6
19. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
20. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
21. Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, Ahmad T, et al. Inherited determinants of Crohn's disease and ulcerative colitis phenotypes: a genetic association study. Lancet. (2016) 387:156–67. doi: 10.1016/S0140-6736(15)00465-1
22. Sandborn WJ, Gasink C, Gao LL, Blank MA, Johanns J, Guzzo C, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med. (2012) 367:1519–28. doi: 10.1056/NEJMoa1203572
23. Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. (2013) 369:711–21. doi: 10.1056/NEJMoa1215739
24. Kinoshita M, Miyazaki H, Ono S, Seki S. Immunoenhancing therapy with interleukin-18 against bacterial infection in immunocompromised hosts after severe surgical stress. J Leukoc Biol. (2013) 93:689–98. doi: 10.1189/jlb.1012502
25. Takagi H, Kanai T, Okazawa A, Kishi Y, Sato T, Takaishi H, et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand J Gastroenterol. (2003) 38:837–44. doi: 10.1080/00365520310004047
26. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. (2010) 32:367–78. doi: 10.1016/j.immuni.2010.02.012
27. Balasubramani A, Shibata Y, Crawford GE, Baldwin AS, Hatton RD, Weaver CT. Modular utilization of distal cis-regulatory elements controls Ifng gene expression in T cells activated by distinct stimuli. Immunity. (2010) 33:35–47. doi: 10.1016/j.immuni.2010.07.004
28. Tsai CY, Liong KH, Gunalan MG, Li N, Lim DS, Fisher DA, et al. Type I IFNs and IL-18 regulate the antiviral response of primary human gammadelta T cells against dendritic cells infected with Dengue virus. J Immunol. (2015) 194:3890–900. doi: 10.4049/jimmunol.1303343
29. Hormi K, Cadiot G, Kermorgant S, Dessirier V, Le Romancer M, Lewin MJ, et al. Transforming growth factor-alpha and epidermal growth factor receptor in colonic mucosa in active and inactive inflammatory bowel disease. Growth Factors. (2000) 18:79–91. doi: 10.3109/08977190009003235
30. Adar T, Shteingart S, Ben Ya'acov A, Bar-Gil Shitrit A, Goldin E. From airway inflammation to inflammatory bowel disease: eotaxin-1, a key regulator of intestinal inflammation. Clin Immunol. (2014) 153:199–208. doi: 10.1016/j.clim.2014.04.012
31. Blackburn SD, Wherry EJ. IL-10, T cell exhaustion and viral persistence. Trends Microbiol. (2007) 15:143–6. doi: 10.1016/j.tim.2007.02.006
32. Croxford AL, Kulig P, Becher B. IL-12-and IL-23 in health and disease. Cytokine Growth Factor Rev. (2014) 25:415–21. doi: 10.1016/j.cytogfr.2014.07.017
33. Murdoch JR, Lloyd CM. Chronic inflammation and asthma. Mutat Res. (2010) 690:24–39. doi: 10.1016/j.mrfmmm.2009.09.005
34. Suares NC, Hamlin PJ, Greer DP, Warren L, Clark T, Ford AC. Efficacy and tolerability of methotrexate therapy for refractory Crohn's disease: a large single-centre experience. Aliment Pharmacol Ther. (2012) 35:284–91. doi: 10.1111/j.1365–2036.2011.04925.x
35. Cohen BL, Torres J, Colombel JF. Immunosuppression in inflammatory bowel disease: how much is too much? Curr Opin Gastroenterol. (2012) 28:341–8. doi: 10.1097/MOG.0b013e328354567f
Keywords: Crohn's disease, randomized placebo-controlled trial, immunotherapy, innate immunity, biologic, biomarkers, microbial-based therapy
Citation: Sutcliffe S, Kalyan S, Pankovich J, Chen JMH, Gluck R, Thompson D, Bosiljcic M, Bazett M, Fedorak RN, Panaccione R, Axler J, Marshall JK, Mullins DW, Kabakchiev B, McGovern DPB, Jang J, Coldman A, Vandermeirsch G, Bressler B and Gunn H (2019) Novel Microbial-Based Immunotherapy Approach for Crohn's Disease. Front. Med. 6:170. doi: 10.3389/fmed.2019.00170
Received: 19 December 2018; Accepted: 08 July 2019;
Published: 19 July 2019.
Edited by:
Yeong Yeh Lee, University of Science, MalaysiaReviewed by:
Nazri Mustaffa, University of Science, MalaysiaAngel Lanas, University of Zaragoza, Spain
Copyright © 2019 Sutcliffe, Kalyan, Pankovich, Chen, Gluck, Thompson, Bosiljcic, Bazett, Fedorak, Panaccione, Axler, Marshall, Mullins, Kabakchiev, McGovern, Jang, Coldman, Vandermeirsch, Bressler and Gunn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hal Gunn, aGFsQHF1YmlvbG9naWNzLmNvbQ==
†Passed away on November 8th, 2018