 Reiner K. W. Mailer
Reiner K. W. Mailer Lorena Hänel
Lorena Hänel Mikel Allende
Mikel Allende Thomas Renné
Thomas Renné- Institute of Clinical Chemistry and Laboratory Medicine, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Activated platelets and mast cells expose the inorganic polymer, polyphosphate (polyP) on their surfaces. PolyP initiates procoagulant and proinflammatory reactions and the polymer has been recognized as a therapeutic target for interference with blood coagulation and vascular hyperpermeability. PolyP content and chain length depend on the specific cell type and energy status, which may affect cellular functions. PolyP metabolism has mainly been studied in bacteria and yeast, but its roles in eukaryotic cells and mammalian systems have remained enigmatic. In this review, we will present an overview of polyP functions, focusing on intra- and extracellular roles of the polymer and discuss open questions that emerge from the current knowledge on polyP regulation.
Polyphosphate Metabolism
Inorganic polyphosphate (polyP) is abundantly found in every cell in nature, however previous studies had mainly focused on prokaryotes to investigate polyP metabolism. PolyP is a polyanion consisting of up to several hundreds of phosphate units (Pi) linked by energy-rich phosphoanhydride bonds. PolyP is formed in bacteria by polyP kinase through transfer of ATP/GTP's γ-phosphate residues onto the nascent polymer chain (1, 2), while depolymerization of polyP is catalyzed by exopolyphosphatase (ppx) (3). Dependency of polyP for bacterial growth and survival initiated efforts to develop drugs that target polyP metabolism [reviewed in (4–6)].
Eukaryotic polyP metabolism is poorly understood with exception for S. cerevisiae cells. Yeast expresses a polyP polymerase, vacuolar transporter chaperone, and polyP phosphatases including exopolyphosphatase (Ppx1), endopolyphosphatases (Ppn1) and diadenosine and diphosphoinositol phosphohydrolase (Ddp1) (7–10). Despite intensive investigations, mammalian homologs for these polyP-related enzymes have not been identified, however diphosphoinositol polyP phosphohydrolases (DIPPs) have been shown to degrade polyP in alkaline conditions (10). The same report also demonstrated that intracellular polyP concentrations were dependent on enzymes regulating inositol phosphorylation, such as phospholipase C, inositol polyP multikinase and inositol hexakisphosphate kinase (Ip6k1) in yeast (10). Notably, genetic ablation of Ip6k1, a gene coding for the kinase that generates diphospho-moieties through phosphorylation of the 5-position of inositol penta- and hexakisphosphate, has been shown to reduce platelet polyP levels in mice (11). Together, the data suggest that polyP is intertwined with polyphosphorylated inositol metabolism. Multiple cellular processes are regulated by the (poly)phosphorylated inositol, including signal transduction, Ca2+ channel permeability and gene expression [reviewed in (12)]. Particularly, immune cell activation depends on membrane-bound phosphoinositides and soluble inositol phosphates [reviewed in (13)].
In yeast, intracellular concentrations of Pi are sensed through Pi-responsive signaling (Pho-regulon) mediated by both, polyP and diphosphoinositol pentakisphosphate (14, 15). Under phosphate-limiting growth conditions, the transcription factor Pho4 controls expression of high affinity Pi transporters and secreted acid phosphatases to replenish intracellular Pi. Orthologs of the yeast Pho-regulon seem not to exist in multicellular eukaryotes. However, Pi sensor domains have been identified as part of Pi transporters that are conserved among various organisms and share a SPX domain (16). This common SPX domain, named after yeast Syg1 and Pho81 and human XPR1 (xenotropic and polytropic retrovirus receptor 1), binds to diphosphoinositol pentakisphosphate and increases polyP synthesis in yeast and plants. Mice with conditional deficiency of Xpr1 in renal tubular cells develop proximal tubular dysfunction (17). In humans, impaired XPR1 function due to mutations in the Pi exporter is associated with primary familial brain calcification (18). These findings indicate a link between Pi sensing, intracellular Pi levels and polyP, although further studies in eukaryotes are required to elucidate polyP regulation in vivo.
Intracellular Roles of Polyphosphate
High cytoplasmic polyP levels have been found in various cell lines, including NIH3T3 fibroblasts, Vero epithelial kidney cells and Jurkat CD4+ T cells (19). Tissues with high energy demand and high regeneration or proliferation capacity (e.g., brain, heart, liver, and cancer cells) are rich in polyP (19–21). In line with polyP's function as energy storage pool, defective polyP synthesis confers disadvantages for growth and survival of bacteria, fungi and protozoa (9, 22, 23). In bacteria, polyP is bound to Ca2+ ions and complexed with poly-β-hydroxybutyrate forming membrane channels utilized for DNA uptake (24, 25).
In eukaryotes, polyP has functions in mitochondria and the polymer is enriched within the intermembrane compartment and uncouplers of oxidative phosphorylation decrease mitochondrial polyP levels in S. cerevisiae (26, 27). Degradation of polyP using Ppx1 polyphosphatase, that is localized to the mitochondrial intermembrane space by a cytochrome c-derived targeting sequence, decreases the membrane potential and increases NADH levels in mitochondria (28, 29). Moreover, polyP appears to contribute to opening of the mitochondrial permeability transition pore in cardiac myocytes. In these cells in culture, Ppx1-mediated polyP degradation inhibits mitochondrial Ca2+ ion accumulation and interferes with Ca2+-induced cell death that is associated with myocardial infarction and ischemia reperfusion injury (30).
Based on the function of the polymer for growth and survival, polyP is believed to have a role in malignant diseases. In cancer cells, polyP has been detected in epithelial tumor cell lines derived from prostate and mammary gland as well as in primary myeloma B cells (21, 31, 32). In addition to cancer-associated pro-coagulant activities of extracellular polyP (see below), the polymer facilitates various intracellular functions in cancer cells. PolyP increases the kinase activity of mammalian target of rapamycin (mTOR) in MCF-7 tumor cells and accumulates at nucleolar transcription sites in myeloma cells. In contrast, Ppx1-mediated polyP degradation and actinomycin D-induced transcription inhibition abrogate these polyP effects (31, 32). Together, the data suggest, that polyP may act as metabolic driving force in different cellular compartments, thereby promoting tumorigenesis.
As a negatively charged polymer, polyP exerts chaperone activity in bacteria such as E. coli (33, 34). Short and long chain polyP regulate ribosomal translation efficiency and ribosomal protein degradation, indicating a differential role of polyP dependent on chain lengths in bacterial protein biosynthesis (35, 36). Similarly to the polymer starch and its monomeric form glucose, polyP is condensed phosphate and reduces the concentration of Pi and thus the intracellular osmotic pressure conferred by the ion. The polymer acts as Pi storage pool and serves as non-enzymatic protein (pyro-)phosphorylation mediator (37, 38) [as does inositol pyrophosphate (39, 40)] in eukaryotes. PolyP also activates the cytoplasmic portion of transient receptor potential (TRP) A1 and melastatin 8 in neurons (41, 42). Moreover, a recent report identified a glucokinase that relies on polyP as phosphoryl donor in the liver (43), indicating that future research on tissue-specific polyP functions may broaden our view of its physiological roles in various cell types. In this context, Ca2+-dependent, mTOR- and TRP-regulated immune responses could be a potential target for polyP.
Besides its established mitochondrial and nuclear distribution, polyP accumulates in intracellular vesicles. Yeast, parasites, mast cells and platelets store polyP in vacuoles, acidocalcisomes, heparin-containing granules and dense granules, respectively (44–48). Ca2+-complexed polyP derived from these acidic organelles is insoluble, retained on the plasma membrane in nanoparticle form upon release and provides the pro-coagulant surface for factor XII (FXII) contact activation (49). PolyP-containing vesicles are also found in lysosome-related organelles derived from fibroblasts and astrocytes (50, 51). The latter cell type releases polyP as a neurotransmitter from lysosomes expressing vesicular nucleotide transporter in response to pH changes, exogenous polyP and Ca2+ ion signaling (52). Taken together, these studies indicate that polyP have fundamental functions for secretion and possibly stability of lysosome-related vesicles in other cell types, such as immune cells. However, quantification of Pi via malachite green assay and 32Pi via radiodetection obtained from hydrolysed polyP has to be interpreted with caution, since microbial contaminations interfered with these assays to detect polyP in human granulocytes in a previous report [update to (53)]. Analysis of in vivo polyP functions is an area of ongoing research and recently a flow cytometry-based assay has been established to quantify the polymer on cell surfaces (54). Table 1 provides a summary of polyP functions in different organisms, cell populations and intracellular compartments.
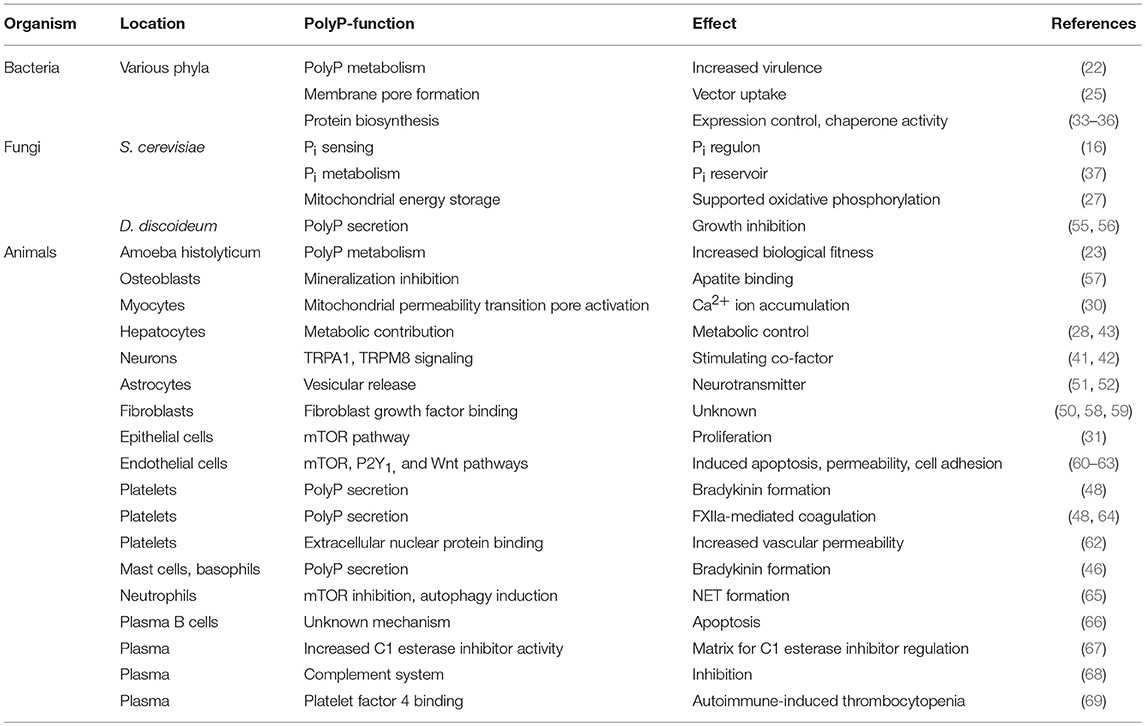
Table 1. Overview on polyP-mediated activities in various cell types.
Extracellular Polyphosphates
While intracellular polyP activities are established only recent data have revealed a role of extracellular polyP in mammalian and human cardiovascular biology. The in vivo activation of FXII in the initiating steps of the intrinsic blood coagulation system has been an enigma for many years [reviewed in (70)]. FXII activation by binding to negatively charged kaolin or ellagic acid (“contact activation”) provides the mechanistic basis for activated partial thromboplastin time (aPTT), the clinical coagulation test (with an estimated 4–5 billion tests annually). However, the natural surface that induces FXII contact activation in vivo had been unknown. Activated platelets induce plasma clotting in a FXII-dependent manner, indicating that platelets release FXII-activating structures. Studies in mice and human plasma revealed that polyP serves as the long sought FXII-activating surface on activated platelets linking primary and secondary hemostasis. Vice versa, humans with polyP deficiency (Hermansky Pudlak Syndrome) have defective platelet-driven FXII activation and clotting [(48) and reviewed in (71) and (72)]. In addition to FXII activation, polyP has been reported to modulate other coagulation reactions in vitro, however a potential role of these pathways in vivo remains to be demonstrated (64, 73–75). The chain length of synthetic polyP determines its solubility and FXII activation capacity in plasma (76). However, natural platelet polyP forms insoluble Ca2+ ion-rich nanoparticles independently of the chain length of the polyP molecules that are maintained on the surfaces of pro-coagulant platelets and thus activate FXII (49). Hence size of polyP does not matter for FXII-activating potency of the physiologically occurring Ca2+-saturated polymer. Similarly to polyP, also synthetic polyI:C, a dsRNA analog that activates Toll-like receptor 3, has been shown to have pro-coagulant activity (77).
PolyP is a chelator for positive metal ions and dense granules of platelets contain polyP bound to Ca2+ and possible Zn2+ ions at high concentrations (47). In plasma, Ca2+-saturated polyP has a half-life of about 90 min (76), due to degradation by polyphosphatases, such as alkaline phosphatase that has exopolyphosphatase activity (48, 78). Synthetic polyP derived from melted phosphoric acid is mostly complexed to Na+ ions. Counterions regulate structure, biophysical properties and biological activities of polyP (49). Most of the in vitro studies have been performed with synthetic (often Ca2+-free) polyP in the absence of phosphatases. Physiologically occurring Ca2+-polyP is not soluble and operates on the cell surface. The low solubility in plasma challenges suggested functions for the extracellular polymer in solution. In contrast Ca2+-free synthetic polyP of short and medium sized chain length (50–150 Pi moieties) depletes free Ca2+ ions leading to anti-coagulant effects similarly to EDTA (76).
Most cell culture experiments have been performed with medium sized synthetic polyP (10–50 μM, based on monophosphate units). In buffer or plasma environments in vitro, synthetic polyP is associated with an array of binding partners, including von Willebrand factor, coagulation factors XII, V and XI, complement factors C1s, C5b6, C6, and C7 (64, 68, 73, 74). Medium chain polyP and heparin have been reported to bind C1 esterase inhibitor (C1INH), thereby increasing function of the inhibitor (67, 79). However, FXII is readily activated by platelet-derived polyP to initiate the intrinsic coagulation cascade in vivo, indicating that increased activity of physiologic C1INH levels following polyanion binding is not sufficient to prevent FXIIa-driven coagulation. In contrast, high dosages of C1INH (15 IU/kg) but not low dosages (7.5 IU/kg) decrease ischemia/reperfusion injuries (80), suggesting that polyP binding to supra-physiological C1INH levels has the capacity for regulating thrombosis. The amount of polyP exposed on activated platelet surface is not precisely known, and it remains to be analyzed whether low amounts of platelet-derived C1INH that is retained on the surface of activated platelets and is possibly bound to polyP have a role in regulating serine proteases in vivo (81).
PolyP increases endothelial cell permeability in vivo, stimulates expression of cell adhesion molecules and induces apoptosis in culture (60). Follow-up studies, have elucidated the pathways involved in these endothelial reactions. PolyP application activates NF-κb and β-catenin pathways via receptor for advanced glycation end products, P2Y1 receptor signaling and Wnt signaling (61, 62). Vice versa, the anti-thyroid drug methylthiouracil interferes with polyP-mediated inflammation of the endothelium in vivo (82). On the other hand, probiotic-derived polyP improves epithelial barrier function and may contribute to immunomodulatory mechanisms against commensal bacteria in the gut via NF-κb inhibition (83).
High extracellular polyP levels (>500 μM) elicit scaffolding effects that control amyloidogenic processes (84). In addition, polyP binds to fibroblast growth factor (FGF) (58). However, possible effects of polyP-FGF complexes on FGF-receptor signaling have remained controversial, as both increased and inhibited mitogenic activities have been reported (58, 59). A possible explanation for the dual activities might be that FXII, which initially promotes angiogenesis via urokinase plasminogen activator receptor (uPAR) signaling (85), gets rapidly depleted following polyP application via binding of FXIIa to C1INH. In the presence of serum-derived C1INH, FXIIa gets rapidly cleared and might limit FXII(a)/uPAR-driven pathways. Supporting this idea, tumor cells secrete exosomes that expose polyP and are associated with worse outcome involving immune evasion possibly through reduced FXII(a)/uPAR-driven differentiation of tumor-reactive T cell subsets (21, 86, 87). Moreover, human plasma cells induce apoptosis following polyP treatment, whereas other lymphocyte populations do not (66).
Presentation of polyP on activated platelets allows for contact of polyP with immune cells. Activation of platelets increases the intracellular Ca2+ ion concentration via stromal interaction molecule 1 to release Ca2+ ions from the sarcoplasmatic reticulum (88), which opens store-operated calcium entry channels in the plasma membrane (89). Ca2+-mediated platelet activation mirrors the process of T cell stimulation (90). Moreover, conjugates of platelet-CD4+ T cells have been reported (91) and T-cell proliferation and differentiation is modulated by platelet-released molecules, such as platelet factor 4 (92), soluble CD40 ligand (93), ADP/ATP (94) and transforming growth factor-β (95). Recent findings also suggest that platelets inhibit pro-inflammatory interleukin (IL)-17 secreting effector T helper (Th17) cells in a tumor model (96). Taken together, these studies suggest that polyP modulates immune responses via direct or indirect pathways.
Neutrophils express FXII that is translocated to the plasma membrane upon activation and facilitates uPAR signaling via an autocrine mechanism (97). FXII signaling in neutrophils is independent of FXII cleavage and promotes adhesion, migration and formation of neutrophil extracellular traps (NET). In transfer experiments, neutrophils derived from FXII-deficient bone marrow show reduced tissue infiltration and better wound healing in the host, compared to neutrophils derived from wild-type bone marrow. PolyP treatment of neutrophils in vitro is independent on uPAR signaling and promotes the generation of NET through decreased mTOR acitivity and autophagy induction (65). Supporting a link between polyP and neutrophils, neutrophil-mediated inflammation is reduced in polyP-deficient mice in two independent mouse models with (i) genetic IP6K1 ablation or (ii) pharmacological IP6K1 inhibition (98). The data suggest that both neutrophil FXII and platelet-derived polyP contribute to NET formation.
Both polyP and heparin are negatively charged polymers and polyP has been shown to substitute for heparin. Both heparin and polyP derived from mast cells drive the activation of the kallikrein-kinin system (46, 99). Antibodies that recognize complexes of platelet factor 4 (PF4) bound to heparin induce platelet aggregation in heparin-induced thrombocytopenia (HIT). These antibodies also cross-react with PF4/polyP complexes (69), indicating that polyP might contribute to HIT offering a rational for the recently described heparin-independent HIT forms (100).
Polyphosphate-Mediated Inflammation
PolyP promotes the autocatalytic activation of FXII to the serine protease FXIIa and FXIIa production is amplified by the protease plasma kallikrein (PKa) through a feed-forward mechanism of FXIIa-cleaved plasma prekallikrein (PK, fluid phase activation). Deficiency in FXII is not associated with obvious abnormalities in humans and mice. However, FXII-deficient plasma (Hageman trait) has a prolonged aPTT. Mice lacking FXII (F12−/−) have largely defective arterial and venous thrombus formation (101) and are protected from thromboembolic diseases without showing hemostatic abnormalities (48, 102). The impact of FXII on thrombosis recently steered the World Health Organization to establish an international standard for human plasma-derived FXII (103). The fact that FXII deficiency is not associated with bleeding, suggests that FXII-mediated intrinsic blood coagulation contributes to other vascular pathways rather than hemostasis [reviewed in (104)].
FXII and FXIIa are both ligands of uPAR that is expressed on antigen-presenting cells and promotes differentiation of pro-inflammatory Th17 cells (85, 87). Consistently, F12−/− mice were shown to be protected from Th17-driven experimental autoimmune encephalomyelitis (EAE) development (87). Autoimmune diseases, such as EAE, require the extravasation of auto-reactive Th1 and Th17 cells to infiltrate and attack target cell structures. Furthermore, FXIIa increases vascular permeability via the kallikrein-kinin system, in which bradykinin (BK) is produced by PKa-mediated high molecular weight kininogen cleavage. BK is a peptide hormone and BK signal transduction is mediated by G-coupled kinin B1 and B2 receptors on endothelial cells as well as leukocytes [reviewed in (105)]. Kinin B1 receptor expression is induced by pro-inflammatory cytokines (e.g., IL-1β) and stimulated by BK and the C-terminal truncated BK derivate des-Arg(9)BK, whereas constitutively expressed kinin B2 receptors recognize BK [reviewed in (106)]. Deficiency in kinin B1 receptors affects Th-cell migration and outcome in murine EAE models and possibly humans with multiple sclerosis (107), suggesting that several FXII-dependent pathways can interact with T-cell homeostasis. In line with this notion, increased populations of Th17 cells have been reported in patients with defective FXIIa regulation (108).
Proteolytic activity of FXIIa is regulated by C1INH, a serpin that irreversibly inhibits FXIIa through covalent binding into its reactive center. C1INH also has the capacity to inhibit several other serine proteases, including PKa, active factor XI (FXIa) and complement factors C1r and C1s. Impaired FXIIa inhibition augments BK formation by the kallikrein-kinin system and is associated with a BK-mediated life-threatening inherited swelling disorder, hereditary angioedema (HAE) [reviewed in (109)]. C1INH deficiency has no impact on thrombosis and HAE patients have a normal thromboembolic risk [reviewed in (110)]. HAE is a rare disease that is mainly autosomal dominant inherited and characterized by reduced C1INH levels (HAE type I) or function (HAE type II). In addition HAE has been reported in patients with normal C1 esterase inhibitor activity (HAE type III). Disease-causing HAE type III mutations have been mapped to the F12 gene. Analysis of mutant FXII T309K and T309R (positions refer to the mature protein) revealed that mutant FXII is defective in a single O-linked glycosylation, which promotes contact-driven FXIIa formation (111). Additionally, the mutations create new serine protease cleavage sites and thus increase FXIIa- and plasmin-mediated mutant FXII zymogen activation (112). Other HAE type III associated mutations have been linked to plasminogen and angiopoeitin (113, 114). While BK is short-lived and degraded within seconds, the activity of FXIIa or activation of FXII is increased in HAE leading to sustained and prolonged BK formation (115, 116). A schematic overview depicting PolyP/FXII-mediated pathways is shown in Figure 1.
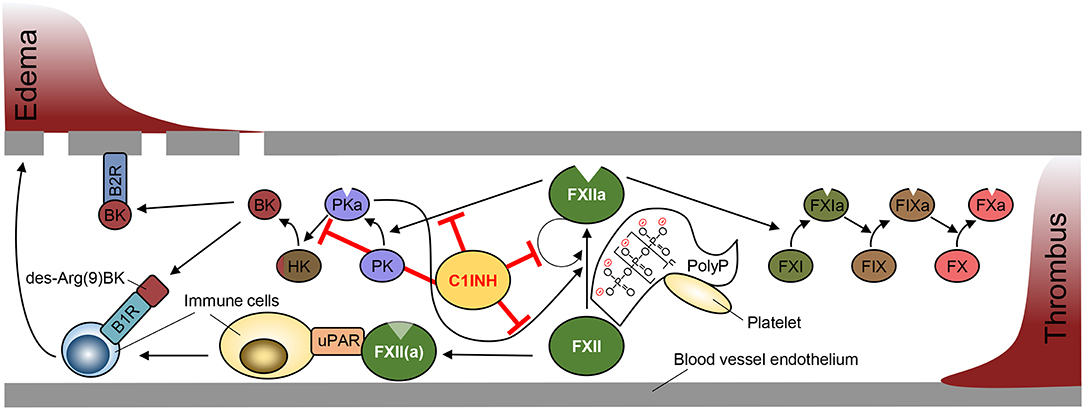
Figure 1. Schematic overview of Factor XII-mediated pathways in edema and thrombosis formation. Factor XII (FXII) activation is driven by autocatalytic activation promoted by binding to negatively charged surfaces (contact activation), such as polyphosphate (PolyP) retained on platelet surface. FXIIa production is amplified by plasma kallikrein (PKa) that is produced from FXIIa-cleaved plasma prekallikrein (PK). C1 esterase inhibitor (C1INH) blocks the activity of FXIIa and PKa to prevent PKa-driven edema formation via cleavage of high molecular weight kininogen (HK) to release bradykinin (BK). Binding of BK and its metabolite des-Arg(9)BK to bradykinin receptors (B1R, B2R) promotes vascular permeability and immune cell activation. FXII and FXIIa bind urokinase plasminogen activator receptor (uPAR) to stimulate immune responses (left). FXIIa initiates the activation of the intrinsic coagulation cascade via activation of factor XI (FXI), factor IX (FIX) and factor X (FX) to form a thrombus (right).
In addition to the kallikrein-kinin system, FXIIa triggers the intrinsic blood coagulation pathway that has a role in thrombus formation [reviewed in (117)]. Thrombi occlude vessels causing tissue ischemia but also function in host defense in a concept termed “immunothrombosis.” A recent report on bacterial sepsis-associated pro-coagulant mechanisms showed that inflammation-driven thrombosis occurs earlier in the spleen compared to liver despite similar bacterial burden in both organs, suggesting that organ-specific environment rather than bacterial components contribute to microthrombus formation (118). Bacteria-derived polyP is highly pro-coagulant in vitro, however, it is unknown whether bacteria have the capacity to expose or secrete their long chain polyP (>1,000 Pi moieties).
Intracellular polyP gets released during necrosis and may act as a damage associated molecular pattern inducing immune responses through P2Y signaling (62). Indeed, long chain polyP induces thrombosis via FXII activation but also leads to platelet activation and consumptive coagulopathy (119). Moreover, polyP serves as phosphoryl donor for adenylate kinase in the extracellular space (120). Thus, polyP may regulate ADP/ATP ratio and signaling via purinergic receptors in immune cells. An overview of the complex network by which polyP impacts on cellular mechanisms in different cell types to facilitate coagulation and inflammation is depicted in Figure 2.
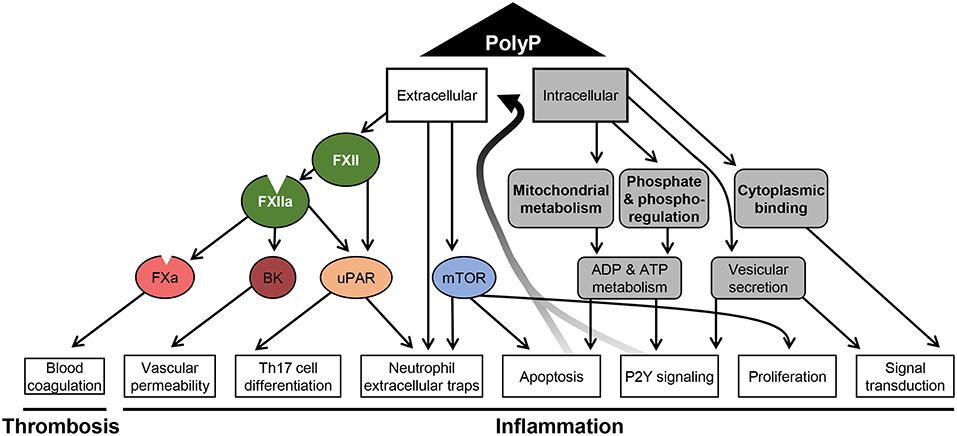
Figure 2. Schematic overview of polyphosphate-mediated pathways. A summary of the diverse mechanisms that are affected by polyP and have been reported for distinct cell types is shown. Polyphosphate (polyP) regulates intracellular mechanisms (gray) related to metabolism, signaling and apoptosis. The effect of polyP on phoshoregulation has been shown in yeast, the impact on mitochondrial activity in yeast and cardiomyocytes. Protein binding and vesicular secretion of polyP was reported for neurons and polyP-driven proliferation has been shown for tumor cells. Active and passive release of polyP promotes extracellular pathways to activate coagulation factors (FXIIa and FXa), to generate bradykinin (BK) and to stimulate cells via urokinase plasminogen activator receptor (uPAR)- and mammalian target of rapamycin (mTOR)-driven mechanisms. Signaling via these mediators were reported to affect endothelial cells and immune cells.
Interference with Polyphosphate Functions
Intracellular polyP levels have been modulated in S. cerevisiae through phosphate starvation and abrogation of the mitochondrial membrane potential by uncouplers (27), indicating that intracellular polyP broadly influences cell metabolism. Moreover, Ppx1-mediated degradation of polyP regulates mitochondrial Ca2+ ion efflux (30).
Mammalian enzymes that control formation and degradation of polyP have remained unknown. To interfere with polyP/FXIIa-driven inflammation plasma derived C1INH (Berinert™) and B2R antagonist (Icatibant™) dampen bradykinin formation and signaling and have been used for treatment of excessive bradykinin-mediated swellings in HAE patients (121, 122). Neutralizing FXIIa antibody 3F7 prevents thromboembolism and bradykinin generation with minimal therapy-associated adverse effects such as increased bleeding (123). Interference with the extracellular polyP/FXII pathway is a promising strategy to dampen coagulation and inflammation [reviewed in (124)]. To directly target extracellular polyP, neutralization and degradation approaches have been tested. Structural homology of polyP and heparin suggests that the polycation protamine sulfat, the heparin antidote, can also neutralize polyP. Indeed, polyP inhibitor screening identified various polycation substrates, such as spermidine, histone H1, polymyxin B and cationic polymers as possible agents for interference with polyP-mediated coagulation and inflammation (125). Moreover, the recombinant polyP-binding domains of E. coli ppx (PPX_Δ12) bind to polyP with high affinity and block polyP-mediated FXII activation. Similarly, degradation of polyP with recombinant ppx inhibits arterial and venous thrombus formation without interfering with hemostasis, indicating that polyP operates via FXII in in vivo coagulation (126).
Interference with polyP developed evolutionary in blood-sucking insects. Sand flies express salivary proteins that enable the insects to feed on mammalian blood without triggering BK-mediated itching or FXIIa-mediated clotting. These proteins, termed PdSP15a and PdSP15b, provide a positively charged helix that efficiently binds to and neutralizes polyP (127). Taken together, polyP inhibition has been shown to block thrombosis and inflammation and provides an opportunity for efficient and safe future treatment.
Outlook
Recent advantages to visualize, measure and detect polyP and to modulate polyP metabolism offers the possibility to analyze cell-specific roles for polyP (11, 28, 126, 128). Open research questions include regulation of polyP in mammalians and eukaryotic cells, and possibilities to target polyP-mediated activities in therapeutic settings.
Author Contributions
RM wrote the manuscript and created the figures. LH, MA, and TR reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Projektnummer A11—SFB 877 and Projektnummer B8—SFB 841 and an European Research Council grant (ERC-StG-2012-311575_F-12).
References
1. Ahn K, Kornberg A. Polyphosphate kinase from Escherichia coli. Purification and demonstration of a phosphoenzyme intermediate. J Biol Chem. (1990) 265:11734–9.
2. Zhang H, Ishige K, Kornberg A. A polyphosphate kinase (PPK2) widely conserved in bacteria. Proc Natl Acad Sci USA. (2002) 99:16678–83. doi: 10.1073/pnas.262655199
3. Akiyama M, Crooke E, Kornberg A. An exopolyphosphatase of Escherichia coli. The enzyme and its ppx gene in a polyphosphate operon. J Biol Chem. (1993) 268:633–9.
4. Xie L, Jakob U. Inorganic polyphosphate, a multifunctional polyanionic protein scaffold. J Biol Chem. (2018) 294:2180–90. doi: 10.1074/jbc.REV118.002808
5. Gautam LK, Sharma P, Capalash N. Bacterial polyphosphate kinases revisited: role in pathogenesis and therapeutic potential. Curr Drug Targets. (2018) 20:292–301. doi: 10.2174/1389450119666180801120231
6. Rao NN, Gómez-García MR, Kornberg A. Inorganic polyphosphate: essential for growth and survival. Annu Rev Biochem. (2009) 78:605–47. doi: 10.1146/annurev.biochem.77.083007.093039
7. Hothorn M, Neumann H, Lenherr ED, Wehner M, Rybin V, Hassa PO, et al. Catalytic core of a membrane-associated eukaryotic polyphosphate polymerase. Science. (2009) 324:513–6. doi: 10.1126/science.1168120
8. Wurst H, Kornberg A. A soluble exopolyphosphatase of Saccharomyces cerevisiae. Purification and characterization. J Biol Chem. (1994) 269:10996–1001.
9. Sethuraman A, Rao NN, Kornberg A. The endopolyphosphatase gene: essential in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. (2001) 98:8542–7. doi: 10.1073/pnas.151269398
10. Lonetti A, Szijgyarto Z, Bosch D, Loss O, Azevedo C, Saiardi A. Identification of an evolutionarily conserved family of inorganic polyphosphate endopolyphosphatases. J Biol Chem. (2011) 286:31966–74. doi: 10.1074/jbc.M111.266320
11. Ghosh S, Shukla D, Suman K, Lakshmi BJ, Manorama R, Kumar S, et al. Inositol hexakisphosphate kinase 1 maintains hemostasis in mice by regulating platelet polyphosphate levels. Blood. (2013) 122:1478–86. doi: 10.1182/blood-2013-01-481549
12. Hatch AJ, York JD. SnapShot: inositol phosphates. Cell. (2010) 143:1030–1030 e1. doi: 10.1016/j.cell.2010.11.045
13. Sauer K, Cooke MP. Regulation of immune cell development through soluble inositol−1,3,4,5-tetrakisphosphate. Nat Rev Immunol. (2010) 10:257–71. doi: 10.1038/nri2745
14. Auesukaree C, Homma T, Tochio H, Shirakawa M, Kaneko Y, Harashima S. Intracellular phosphate serves as a signal for the regulation of the PHO pathway in Saccharomyces cerevisiae. J Biol Chem. (2004) 279:17289–94. doi: 10.1074/jbc.M312202200
15. Lee YS, Mulugu S, York JD, O'Shea EK. Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science. (2007) 316:109–12. doi: 10.1126/science.1139080
16. Wild R, Gerasimaite R, Jung JY, Truffault V, Pavlovic I, Schmidt A, et al. Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Science. (2016) 352:986–90. doi: 10.1126/science.aad9858
17. Ansermet C, Moor MB, Centeno G, Auberson M, Hu DZ, Baron R, et al. Renal fanconi syndrome and hypophosphatemic rickets in the absence of xenotropic and polytropic retroviral receptor in the nephron. J Am Soc Nephrol. (2017) 28:1073–8. doi: 10.1681/ASN.2016070726
18. Legati A, Giovannini D, Nicolas G, López-Sánchez U, Quintáns B, Oliveira JR, et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet. (2015) 47:579–81. doi: 10.1038/ng.3289
19. Kumble KD, Kornberg A. Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem. (1995) 270:5818–22. doi: 10.1074/jbc.270.11.5818
20. Lynn WS, Brown RH. Synthesis of polyphosphate by rat liver mitochondria. Biochem Biophys Res Commun. (1963) 11:367–71.
21. Nickel KF, Ronquist G, Langer F, Labberton L, Fuchs TA, Bokemeyer C, et al. The polyphosphate-factor XII pathway drives coagulation in prostate cancer-associated thrombosis. Blood. (2015) 126:1379–89. doi: 10.1182/blood-2015-01-622811
22. Wang L, Yan J, Wise MJ, Liu Q, Asenso J, Huang Y, et al. Distribution patterns of polyphosphate metabolism pathway and its relationships with bacterial durability and virulence. Front Microbiol. (2018) 9:782. doi: 10.3389/fmicb.2018.00782
23. Zhang H, Gómez-García MR, Brown MR, Kornberg A. Inorganic polyphosphate in Dictyostelium discoideum: influence on development, sporulation, and predation. Proc Natl Acad Sci USA. (2005) 102:2731–5. doi: 10.1073/pnas.0500023102
24. Reusch RN, Sadoff HL. Putative structure and functions of a poly-beta-hydroxybutyrate/calcium polyphosphate channel in bacterial plasma membranes. Proc Natl Acad Sci USA. (1988) 85:4176–80.
25. Castuma CE, Huang R, Kornberg A, Reusch RN. Inorganic polyphosphates in the acquisition of competence in Escherichia coli. J Biol Chem. (1995) 270:12980–3. doi: 10.1074/jbc.270.22.12980
26. Kulakovskaya TV, Lichko LP, Vagabov VM, Kulaev IS. Inorganic polyphosphates in mitochondria. Biochemistry. (2010) 75:825–31. doi: 10.1134/S0006297910070035
27. Pestov NA, Kulakovskaya TV, Kulaev IS. Inorganic polyphosphate in mitochondria of Saccharomyces cerevisiae at phosphate limitation and phosphate excess. FEMS Yeast Res. (2004) 4:643–8. doi: 10.1016/j.femsyr.2003.12.008
28. Abramov AY, Fraley C, Diao CT, Winkfein R, Colicos MA, Duchen MR, et al. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc Natl Acad Sci USA. (2007) 104:18091–6. doi: 10.1073/pnas.0708959104
29. Pavlov E, Aschar-Sobbi R, Campanella M, Turner RJ, Gómez-García MR, Abramov AY. Inorganic polyphosphate and energy metabolism in mammalian cells. J Biol Chem. (2010) 285:9420–8. doi: 10.1074/jbc.M109.013011
30. Seidlmayer LK, Gomez-Garcia MR, Blatter LA, Pavlov E, Dedkova EN. Inorganic polyphosphate is a potent activator of the mitochondrial permeability transition pore in cardiac myocytes. J Gen Physiol. (2012) 139:321–31. doi: 10.1085/jgp.201210788
31. Wang L, Fraley CD, Faridi J, Kornberg A, Roth RA. Inorganic polyphosphate stimulates mammalian TOR, a kinase involved in the proliferation of mammary cancer cells. Proc Natl Acad Sci USA. (2003) 100:11249–54. doi: 10.1073/pnas.1534805100
32. Jimenez-Nuñez MD, Moreno-Sanchez D, Hernandez-Ruiz L, Benítez-Rondán A, Ramos-Amaya A, Rodríguez-Bayona B, et al. Myeloma cells contain high levels of inorganic polyphosphate which is associated with nucleolar transcription. Haematologica. (2012) 97:1264–71. doi: 10.3324/haematol.2011.051409
33. Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, et al. Polyphosphate is a primordial chaperone. Mol Cell. (2014) 53:689–99. doi: 10.1016/j.molcel.2014.01.012
34. Yoo NG, Dogra S, Meinen BA, Tse E, Haefliger J, Southworth DR, et al. Polyphosphate stabilizes protein unfolding intermediates as soluble amyloid-like oligomers. J Mol Biol. (2018) 430:4195–208. doi: 10.1016/j.jmb.2018.08.016
35. McInerney P, Mizutani T, Shiba T. Inorganic polyphosphate interacts with ribosomes and promotes translation fidelity in vitro and in vivo. Mol Microbiol. (2006) 60:438–47. doi: 10.1111/j.1365-2958.2006.05103.x
36. Kuroda A, Nomura K, Ohtomo R, Kato J, Ikeda T, Takiguchi N, et al. Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science. (2001) 293:705–8. doi: 10.1126/science.1061315
37. Vagabov VM, Trilisenko LV, Kulaev IS. Dependence of inorganic polyphosphate chain length on the orthophosphate content in the culture medium of the yeast Saccharomyces cerevisiae. Biochemistry. (2000) 65:349–54.
38. Azevedo C, Livermore T, Saiardi A. Protein polyphosphorylation of lysine residues by inorganic polyphosphate. Mol Cell. (2015) 58:71–82. doi: 10.1016/j.molcel.2015.02.010
39. Saiardi A, Bhandari R, Resnick AC, Snowman AM, Snyder SH. Phosphorylation of proteins by inositol pyrophosphates. Science. (2004) 306:2101–5. doi: 10.1126/science.1103344
40. Bhandari R, Saiardi A, Ahmadibeni Y, Snowman AM, Resnick AC, Kristiansen TZ, et al. Protein pyrophosphorylation by inositol pyrophosphates is a posttranslational event. Proc Natl Acad Sci USA. (2007) 104:15305–10. doi: 10.1073/pnas.0707338104
41. Kim D, Cavanaugh EJ. Requirement of a soluble intracellular factor for activation of transient receptor potential A1 by pungent chemicals: role of inorganic polyphosphates. J Neurosci. (2007) 27:6500–9. doi: 10.1523/JNEUROSCI.0623-07.2007
42. Zakharian E, Thyagarajan B, French RJ, Pavlov E, Rohacs T. Inorganic polyphosphate modulates TRPM8 channels. PLoS ONE. (2009) 4:e5404. doi: 10.1371/journal.pone.0005404
43. Ali A, Wathes DC, Swali A, Burns H, Burns S. A novel mammalian glucokinase exhibiting exclusive inorganic polyphosphate dependence in the cell nucleus. Biochem Biophys Rep. (2017) 12:151–7. doi: 10.1016/j.bbrep.2017.09.004
44. Lander N, Cordeiro C, Huang G, Docampo R. Polyphosphate and acidocalcisomes. Biochem Soc Trans. (2016) 44:1–6. doi: 10.1042/BST20150193
45. Hernández-Ruiz L, Sáez-Benito A, Pujol-Moix N, Rodríguez-Martorell J, Ruiz FA. Platelet inorganic polyphosphate decreases in patients with delta storage pool disease. J Thromb Haemost. (2009) 7:361–3. doi: 10.1111/j.1538-7836.2008.03238.x
46. Moreno-Sanchez D, Hernandez-Ruiz L, Ruiz FA, Docampo R. Polyphosphate is a novel pro-inflammatory regulator of mast cells and is located in acidocalcisomes. J Biol Chem. (2012) 287:28435–44. doi: 10.1074/jbc.M112.385823
47. Ruiz FA, Lea CR, Oldfield E, Docampo R. Human platelet dense granules contain polyphosphate and are similar to acidocalcisomes of bacteria and unicellular eukaryotes. J Biol Chem. (2004) 279:44250–7. doi: 10.1074/jbc.M406261200
48. Müller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, et al. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell. (2009) 139:1143–56. doi: 10.1016/j.cell.2009.11.001
49. Verhoef JJ, Barendrecht AD, Nickel KF, Dijkxhoorn K, Kenne E, Labberton L, et al. Polyphosphate nanoparticles on the platelet surface trigger contact system activation. Blood. (2017) 129:1707–17. doi: 10.1182/blood-2016-08-734988
50. Pisoni RL, Lindley ER. Incorporation of [32P]orthophosphate into long chains of inorganic polyphosphate within lysosomes of human fibroblasts. J Biol Chem. (1992) 267:3626–31.
51. Holmström KM, Marina N, Baev AY, Wood NW, Gourine AV, Abramov AY. Signalling properties of inorganic polyphosphate in the mammalian brain. Nat Commun. (2013) 4: 1362. doi: 10.1038/ncomms2364
52. Angelova PR, Iversen KZ, Teschemacher AG, Kasparov S, Gourine AV, Abramov AY. Signal transduction in astrocytes: localization and release of inorganic polyphosphate. Glia. (2018) 66:2126–36. doi: 10.1002/glia.23466
53. Cowling RT, Birnboim HC. Incorporation of [32P]orthophosphate into inorganic polyphosphates by human granulocytes and other human cell types. J Biol Chem. (1994) 269:9480–5.
54. Labberton L, Long AT, Gendler SJ, Snozek CL, Stavrou EX, Nickel KF, et al. A flow cytometry-based assay for procoagulant platelet polyphosphate. Cytometry B Clin Cytom. (2018) 94:369–73. doi: 10.1002/cyto.b.21492
55. Suess PM, Gomer RH. Extracellular polyphosphate inhibits proliferation in an autocrine negative feedback loop in dictyostelium discoideum. J Biol Chem. (2016) 291:20260–9. doi: 10.1074/jbc.M116.737825
56. Suess PM, Watson J, Chen W, Gomer RH. Extracellular polyphosphate signals through Ras and Akt to prime Dictyostelium discoideum cells for development. J Cell Sci. (2017) 130:2394–404. doi: 10.1242/jcs.203372
57. Hoac B, Kiffer-Moreira T, Millán JL, McKee MD. Polyphosphates inhibit extracellular matrix mineralization in MC3T3-E1 osteoblast cultures. Bone. (2013) 53:478–86. doi: 10.1016/j.bone.2013.01.020
58. Shiba T, Nishimura D, Kawazoe Y, Onodera Y, Tsutsumi K, Nakamura R, et al. Modulation of mitogenic activity of fibroblast growth factors by inorganic polyphosphate. J Biol Chem. (2003) 278:26788–92. doi: 10.1074/jbc.M303468200
59. Han KY, Hong BS, Yoon YJ, Yoon CM, Kim YK, Kwon YG, et al. Polyphosphate blocks tumour metastasis via anti-angiogenic activity. Biochem J. (2007) 406:49–55. doi: 10.1042/BJ20061542
60. Bae JS, Lee W, Rezaie AR. Polyphosphate elicits pro-inflammatory responses that are counteracted by activated protein C in both cellular and animal models. J Thromb Haemost. (2012) 10:1145–51. doi: 10.1111/j.1538-7836.2012.04671.x
61. Hassanian SM, Ardeshirylajimi A, Dinarvand P, Rezaie AR. Inorganic polyphosphate promotes cyclin D1 synthesis through activation of mTOR/Wnt/beta-catenin signaling in endothelial cells. J Thromb Haemost. (2016) 14:2261–73. doi: 10.1111/jth.13477
62. Dinarvand P, Hassanian SM, Qureshi SH, Manithody C, Eissenberg JC, Yang L, et al. Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor for advanced glycation end products and P2Y1 purinergic receptor. Blood. (2014) 123:935–45. doi: 10.1182/blood-2013-09-529602
63. Hassanian SM, Dinarvand P, Smith SA, Rezaie AR. Inorganic polyphosphate elicits pro-inflammatory responses through activation of the mammalian target of rapamycin complexes 1 and 2 in vascular endothelial cells. J Thromb Haemost. (2015) 13:860–71. doi: 10.1111/jth.12899
64. Smith SA, Mutch NJ, Baskar D, Rohloff P, Docampo R, Morrissey JH. Polyphosphate modulates blood coagulation and fibrinolysis. Proc Natl Acad Sci USA. (2006) 103:903–8. doi: 10.1073/pnas.0507195103
65. Chrysanthopoulou A, Kambas K, Stakos D, Mitroulis I, Mitsios A, Vidali V, et al. Interferon lambda1/IL−29 and inorganic polyphosphate are novel regulators of neutrophil-driven thromboinflammation. J Pathol. (2017) 243:111–22. doi: 10.1002/path.4935
66. Hernandez-Ruiz L, González-García I, Castro C, Brieva JA, Ruiz FA. Inorganic polyphosphate and specific induction of apoptosis in human plasma cells. Haematologica. (2006) 91:1180–6.
67. Wijeyewickrema LC, Lameignere E, Hor L, Duncan RC, Shiba T, Travers RJ, et al. Polyphosphate is a novel cofactor for regulation of complement by a serpin, C1 inhibitor. Blood. (2016) 128:1766–76. doi: 10.1182/blood-2016-02-699561
68. Wat JM, Foley JH, Krisinger MJ, Ocariza LM, Lei V, Wasney GA, et al. Polyphosphate suppresses complement via the terminal pathway. Blood. (2014) 123:768–76. doi: 10.1182/blood-2013-07-515726
69. Cines DB, Yarovoi SV, Zaitsev SV, Lebedeva T, Rauova L, Poncz M, et al. Polyphosphate/platelet factor 4 complexes can mediate heparin-independent platelet activation in heparin-induced thrombocytopenia. Blood Adv. (2016) 1:62–74. doi: 10.1182/bloodadvances.2016000877
70. Long AT, Kenne E, Jung R, Fuchs TA, Renné T. Contact system revisited: an interface between inflammation, coagulation, and innate immunity. J Thromb Haemost. (2016) 14:427–37. doi: 10.1111/jth.13235
71. Nickel KF, Long AT, Fuchs TA, Butler LM, Renné T. Factor XII as a Therapeutic target in thromboembolic and inflammatory diseases. Arterioscler Thromb Vasc Biol. (2017) 37:13–20. doi: 10.1161/ATVBAHA.116.308595
72. Maas C, Renné T. Coagulation factor XII in thrombosis and inflammation. Blood. (2018) 131:1903–909. doi: 10.1182/blood-2017-04-569111
73. Choi SH, Smith SA, Morrissey JH. Polyphosphate is a cofactor for the activation of factor XI by thrombin. Blood. (2011) 118:6963–70. doi: 10.1182/blood-2011-07-368811
74. Mutch NJ, Engel R, Uitte de Willige S, Philippou H, Ariëns RA. Polyphosphate modifies the fibrin network and down-regulates fibrinolysis by attenuating binding of tPA and plasminogen to fibrin. Blood. (2010) 115:3980–8. doi: 10.1182/blood-2009-11-254029
75. Biswas I, Panicker SR, Cai X, Mehta-D'souza P, Rezaie AR. Inorganic polyphosphate amplifies high mobility group box 1-mediated von willebrand factor release and platelet string formation on endothelial cells. Arterioscler Thromb Vasc Biol. (2018) 38:1868–77. doi: 10.1161/ATVBAHA.118.311165
76. Smith SA, Choi SH, Davis-Harrison R, Huyck J, Boettcher J, Rienstra CM, et al. Polyphosphate exerts differential effects on blood clotting, depending on polymer size. Blood. (2010) 116:4353–9. doi: 10.1182/blood-2010-01-266791
77. Smith SA, Gajsiewicz JM, Morrissey JH. Ability of polyphosphate and nucleic acids to trigger blood clotting: some observations and caveats. Front Med. (2018) 5:107. doi: 10.3389/fmed.2018.00107
78. Lorenz B, Schröder HC. Mammalian intestinal alkaline phosphatase acts as highly active exopolyphosphatase. Biochim Biophys Acta. (2001) 1547:254–61. doi: 10.1016/S0167-4838(01)00193-5
79. Rajabi M, Struble E, Zhou Z, Karnaukhova E. Potentiation of C1-esterase inhibitor by heparin and interactions with C1s protease as assessed by surface plasmon resonance. Biochim Biophys Acta. (2012) 1820:56–63. doi: 10.1016/j.bbagen.2011.10.008
80. Heydenreich N, Nolte MW, Göb E, Langhauser F, Hofmeister M, Kraft P, et al. C1-inhibitor protects from brain ischemia-reperfusion injury by combined antiinflammatory and antithrombotic mechanisms. Stroke. (2012) 43:2457–67. doi: 10.1161/STROKEAHA.112.660340
81. Schmaier AH, Amenta S, Xiong T, Heda GD, Gewirtz AM. Expression of platelet C1 inhibitor. Blood. (1993) 82:465–74.
82. Min G, Ku SK, Jeong S, Baek MC, Bae JS. Suppressive effects of methylthiouracil on polyphosphate-mediated vascular inflammatory responses. J Cell Mol Med. (2016) 20:2333–40. doi: 10.1111/jcmm.12925
83. Segawa S, Fujiya M, Konishi H, Ueno N, Kobayashi N, Shigyo T, et al. Probiotic-derived polyphosphate enhances the epithelial barrier function and maintains intestinal homeostasis through integrin-p38 MAPK pathway. PLoS ONE. (2011) 6:e23278. doi: 10.1371/journal.pone.0023278
84. Cremers CM, Knoefler D, Gates S, Martin N, Dahl JU, Lempart J, et al. Polyphosphate: a conserved modifier of amyloidogenic processes. Mol Cell. (2016) 63:768–80. doi: 10.1016/j.molcel.2016.07.016
85. LaRusch GA, Mahdi F, Shariat-Madar Z, Adams G, Sitrin RG, Zhang WM, et al. Factor XII stimulates ERK1/2 and Akt through uPAR, integrins, and the EGFR to initiate angiogenesis. Blood. (2010) 115:5111–20. doi: 10.1182/blood-2009-08-236430
86. Nickel KF, Labberton L, Long AT, Langer F, Fuchs TA, Stavrou EX, et al. The polyphosphate/factor XII pathway in cancer-associated thrombosis: novel perspectives for safe anticoagulation in patients with malignancies. Thromb Res. (2016) 141(Suppl. 2):S4–7. doi: 10.1016/S0049-3848(16)30353-X
87. Göbel K, Pankratz S, Asaridou CM, Herrmann AM, Bittner S, Merker M, et al. Blood coagulation factor XII drives adaptive immunity during neuroinflammation via CD87-mediated modulation of dendritic cells. Nat Commun. (2016) 7:11626. doi: 10.1038/ncomms11626
88. Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, et al. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med. (2008) 205:1583–91. doi: 10.1084/jem.20080302
89. Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. (2009) 113:2056–63. doi: 10.1182/blood-2008-07-171611
90. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. (2015) 33:291–353. doi: 10.1146/annurev-immunol-032414-112212
91. Zamora C, Cantó E, Nieto JC, Ortiz MA, Diaz-Torné C, Diaz-Lopez C, et al. Functional consequences of platelet binding to T lymphocytes in inflammation. J Leukoc Biol. (2013) 94:521–9. doi: 10.1189/jlb.0213074
92. Liu CY, Battaglia M, Lee SH, Sun QH, Aster RH, Visentin GP. Platelet factor 4 differentially modulates CD4+CD25+ (regulatory) versus CD4+CD25- (nonregulatory) T cells. J Immunol. (2005) 174:2680–6. doi: 10.4049/jimmunol.174.5.2680
93. Huang J, Jochems C, Talaie T, Anderson A, Jales A, Tsang KY, et al. Elevated serum soluble CD40 ligand in cancer patients may play an immunosuppressive role. Blood. (2012) 120:3030–8. doi: 10.1182/blood-2012-05-427799
94. Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. (1997) 90:1600–10.
95. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci USA. (2009) 106:13445–50. doi: 10.1073/pnas.0901944106
96. Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, et al. Platelets subvert T cell immunity against cancer via GARP-TGFbeta axis. Sci Immunol. (2017) 2:eaai7911. doi: 10.1126/sciimmunol.aai7911
97. Stavrou EX, Fang C, Bane KL, Long AT, Naudin C, Kucukal E, et al. Factor XII and uPAR upregulate neutrophil functions to influence wound healing. J Clin Invest. (2018) 128:944–59. doi: 10.1172/JCI92880
98. Hou Q, Liu F, Chakraborty A, Jia Y, Prasad A, Yu H, et al. Inhibition of IP6K1 suppresses neutrophil-mediated pulmonary damage in bacterial pneumonia. Sci Transl Med. (2018) 10:eaal4045. doi: 10.1126/scitranslmed.aal4045
99. Oschatz C, Maas C, Lecher B, Jansen T, Björkqvist J, Tradler T, et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity. (2011) 34:258–68. doi: 10.1016/j.immuni.2011.02.008
100. Krauel K, Preuße P, Warkentin TE, Trabhardt C, Brandt S, Jensch I, et al. Fibronectin modulates formation of PF4/heparin complexes and is a potential factor for reducing risk of developing HIT. Blood. (2018) 133:978–89. doi: 10.1182/blood-2018-05-850370
101. Renné T, Pozgajová M, Grüner S, Schuh K, Pauer HU, Burfeind P, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. (2005) 202:271–81. doi: 10.1084/jem.20050664
102. Kleinschnitz C, Stoll G, Bendszus M, Schuh K, Pauer HU, Burfeind P, et al. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J Exp Med. (2006) 203:513–8. doi: 10.1084/jem.20052458
103. Wilmot HV, Hockley J, Rigsby P, Gray E. Establishment of the World Health Organization first international standard for factor XII, Plasma, Human. Front Med. (2018) 5:46. doi: 10.3389/fmed.2018.00046
104. Bickmann JK, Baglin T, Meijers JCM, Renné T. Novel targets for anticoagulants lacking bleeding risk. Curr Opin Hematol. (2017) 24:419–26. doi: 10.1097/MOH.0000000000000367
105. Böckmann S, Paegelow I. Kinins and kinin receptors: importance for the activation of leukocytes. J Leukoc Biol. (2000) 68:587–92. doi: 10.1189/jlb.68.5.587
106. McLean PG, Perretti M, Ahluwalia A. Kinin B(1) receptors and the cardiovascular system: regulation of expression and function. Cardiovasc Res. (2000) 48:194–210. doi: 10.1016/S0008-6363(00)00184-X
107. Göbel K, Pankratz S, Schneider-Hohendorf T, Bittner S, Schuhmann MK, Langer HF, et al. Blockade of the kinin receptor B1 protects from autoimmune CNS disease by reducing leukocyte trafficking. J Autoimmun. (2011) 36:106–14. doi: 10.1016/j.jaut.2010.11.004
108. Arcoleo F, Lo Pizzo M, Misiano G, Milano S, Romano GC, Muggeo V, et al. The complex alteration in the network of IL−17-type cytokines in patients with hereditary angioedema. Clin Exp Med. (2018) 18:355–61. doi: 10.1007/s10238-018-0499-0
109. De Maat S, Hofman LM, Maas C. Hereditary angioedema: the plasma contact system out of control. J Thromb Haemost. (2018) 16:1674–85. doi: 10.1111/jth.14209
110. Bender L, Weidmann H, Rose-John S, Renné T, Long AT. Factor XII-driven inflammatory reactions with implications for anaphylaxis. Front Immunol. (2017) 8: 1115. doi: 10.3389/fimmu.2017.01115
111. Björkqvist J, de Maat S, Lewandrowski U, Di Gennaro A, Oschatz C, Schönig K, et al. Defective glycosylation of coagulation factor XII underlies hereditary angioedema type III. J Clin Invest. (2015) 125:3132–46. doi: 10.1172/JCI77139
112. de Maat S, Björkqvist J, Suffritti C, Wiesenekker CP, Nagtegaal W, Koekman A, et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J Allergy Clin Immunol. (2016) 138:1414–23 e9. doi: 10.1016/j.jaci.2016.02.021
113. Dewald G. A missense mutation in the plasminogen gene, within the plasminogen kringle 3 domain, in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. (2018) 498:193–8. doi: 10.1016/j.bbrc.2017.12.060
114. Bafunno V, Firinu D, D'Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin−1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. (2018) 141:1009–17. doi: 10.1016/j.jaci.2017.05.020
115. Csuka D, Veszeli N, Imreh É, Zotter Z, Skopál J, Prohászka Z, et al. Comprehensive study into the activation of the plasma enzyme systems during attacks of hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis. (2015) 10:132. doi: 10.1186/s13023-015-0351-5
116. Björkqvist J, Nickel KF, Stavrou E, Renné T. In vivo activation and functions of the protease factor XII. Thromb Haemost. (2014) 112:868–75. doi: 10.1160/TH14-04-0311
117. Naudin C, Burillo E, Blankenberg S, Butler L, Renné T. Factor XII contact activation. Semin Thromb Hemost. (2017) 43:814–26. doi: 10.1055/s-0036-1598003
118. Beristain-Covarrubias N, Perez-Toledo M, Flores-Langarica A, Zuidscherwoude M, Hitchcock JR, Channell WM, et al. Salmonella-induced thrombi in mice develop asynchronously in the spleen and liver and are not effective bacterial traps. Blood. (2019) 133:600–4. doi: 10.1182/blood-2018-08-867267
119. Zilberman-Rudenko J, Reitsma SE, Puy C, Rigg RA, Smith SA, Tucker EI, et al. Factor XII activation promotes platelet consumption in the presence of bacterial-type long-chain polyphosphate in vitro and in vivo. Arterioscler Thromb Vasc Biol. (2018) 38:1748–60. doi: 10.1161/ATVBAHA.118.311193
120. Müller WEG, Wang S, Neufurth M, Kokkinopoulou M, Feng Q, Schröder HC, et al. Polyphosphate as a donor of high-energy phosphate for the synthesis of ADP and ATP. J Cell Sci. (2017) 130:2747–56. doi: 10.1242/jcs.204941
121. Craig TJ, Bewtra AK, Bahna SL, Hurewitz D, Schneider LC, Levy RJ, et al. C1 esterase inhibitor concentrate in 1085 Hereditary Angioedema attacks–final results of the I.M.P.A.C.T.2 study. Allergy. (2011) 66:1604–11. doi: 10.1111/j.1398-9995.2011.02702.x
122. Lumry WR, Li HH, Levy RJ, Potter PC, Farkas H, Moldovan D, et al. Randomized placebo-controlled trial of the bradykinin B(2) receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST−3 trial. Ann Allergy Asthma Immunol. (2011) 107:529–37. doi: 10.1016/j.anai.2011.08.015
123. Worm M, Köhler EC, Panda R, Long A, Butler LM, Stavrou EX, et al. The factor XIIa blocking antibody 3F7: a safe anticoagulant with anti-inflammatory activities. Ann Transl Med. (2015) 3:247. doi: 10.3978/j.issn.2305-5839.2015.09.07
124. Weidmann H, Heikaus L, Long AT, Naudin C, Schlüter H, Renné T. The plasma contact system, a protease cascade at the nexus of inflammation, coagulation and immunity. Biochim Biophys Acta Mol Cell Res. (2017) 1864(11 Pt B):2118–27. doi: 10.1016/j.bbamcr.2017.07.009
125. Smith SA, Choi SH, Collins JN, Travers RJ, Cooley BC, Morrissey JH. Inhibition of polyphosphate as a novel strategy for preventing thrombosis and inflammation. Blood. (2012) 120:5103–10. doi: 10.1182/blood-2012-07-444935
126. Labberton L, Kenne E, Long AT, Nickel KF, Di Gennaro A, Rigg RA, et al. Neutralizing blood-borne polyphosphate in vivo provides safe thromboprotection. Nat Commun. (2016) 7:12616. doi: 10.1038/ncomms12616
127. Alvarenga PH, Xu X, Oliveira F, Chagas AC, Nascimento CR, Francischetti IM, et al. Novel family of insect salivary inhibitors blocks contact pathway activation by binding to polyphosphate, heparin, and dextran sulfate. Arterioscler Thromb Vasc Biol. (2013) 33:2759–70. doi: 10.1161/ATVBAHA.113.302482
Keywords: polyphosphate, factor XII, hereditary angioedema, thrombosis, inflammation, coagulation, immune activation, vascular permeability
Citation: Mailer RKW, Hänel L, Allende M and Renné T (2019) Polyphosphate as a Target for Interference With Inflammation and Thrombosis. Front. Med. 6:76. doi: 10.3389/fmed.2019.00076
Received: 07 January 2019; Accepted: 28 March 2019;
Published: 12 April 2019.
Edited by:
Keith McCrae, Cleveland Clinic, United StatesReviewed by:
Wei Li, Marshall University, United StatesBrian R. Branchford, University of Colorado Denver, United States
Copyright © 2019 Mailer, Hänel, Allende and Renné. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reiner K. W. Mailer, ci5tYWlsZXJAdWtlLmRl