 Marie Robert
Marie Robert Pierre Miossec
Pierre Miossec
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med., 14 January 2019
Sec. Rheumatology
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00364
This article is part of the Research TopicSynovial Tissue: Turning the Page to Precision Medicine in Arthritis?View all 16 articles
Interleukin (IL)-17A has a direct contribution in early induction and late chronic stages of various inflammatory diseases. In vitro and in vivo experiments have first characterized its local effects on different cell types and then its systemic effects. For instance, IL-17 axis is now identified as a key driver of psoriasis through its effects on keratinocytes. Similar observations apply for rheumatoid arthritis (RA) where IL-17A triggers changes in the synovium that lead to synovitis and maintain local inflammation. These results have prompted the development of biologics to target this cytokine. However, while convincing studies are reported on the efficacy of IL-17 inhibitors in psoriasis, there are conflicting results in RA. Patient heterogeneity but also the involvement of mediators that regulate IL-17 function may explain these results. Therefore, new tools and concepts are required to identify patients that could benefit from these IL-17 targeted therapies in RA and the development of predictive biomarkers of response has started with the emergence of various bioassays. Current strategies are also focusing on synovial biopsies that may be used to stratify patients. From local to systemic levels, new approaches are developing and move the field of RA management into the era of precision medicine.
Interleukin (IL)-17A is a pro-inflammatory cytokine that contributes to the pathogenesis of several auto-immune and inflammatory diseases (1). In vitro and in vivo experiments have identified IL-17 effects on various cell types explaining its involvement in early induction and late chronic stages of many diseases. For instance, IL-17A acts on keratinocytes to induce the expression of several chemokines leading to the recruitment of immune cells that characterized psoriasis (2). Furthermore, in rheumatoid arthritis (RA), the most prevalent chronic inflammatory disease (3), IL-17A acts locally on synoviocytes and osteoblasts contributing to synovitis and joint destruction (4, 5).
These observations have prompted the development of biologics targeting IL-17A and various strategies are currently being tested (2). In psoriasis, inhibitors of IL-17A axis bring a clear benefit in patient care management. Among diseases affecting joints, IL-17 inhibitors are effective in active ankylosing spondylitis and psoriatic arthritis, whereas conflicting results are reported for RA with a high degree of heterogeneity in response (6–9). To potentiate the use of such therapies in RA, an effort is needed to precisely identify patients that would respond to IL-17A inhibition. Current strategies are focusing on the development of biomarkers (5, 10) but also on synovial biopsies (11) to explain patient heterogeneity and treatment response.
The present review discusses the effects of IL-17A on synovium, its regulation and current strategies to detect bioactive IL-17A. Regarding the role of IL-17 in RA pathogenesis, these observations emphasize that this cytokine and its inhibitors should now be considered in the development of precision medicine in RA.
The IL-17 family is composed of six members: IL-17A to IL-17F. The IL-17A was the first isoform discovered in 1993. Initially described as cytotoxic T lymphocyte-associated antigen 8, a product of T cells in rodents, the effects of human IL-17A were then characterized (12, 13). One of its earliest documented biological activities was its effects on RA synoviocytes (14). Then, it was shown that this cytokine promotes granulopoiesis and protects the host against bacterial and fungal infections (1).
Among the IL-17 family, IL-17A and IL-17F share the greatest homology with a 50% sequence identity and can be secreted as homodimer or heterodimer (15, 16). Many of the effects of IL-17A and IL-17F are found similar even if IL-17F is usually less active at inducing inflammation (1).
Conversely, IL-17E (also known as IL-25) has the lowest homology with IL-17A with only 20% sequence identity (17). IL-17E is a mediator of T-helper (Th) 2 cell responses especially in host defense against parasites (18) and allergy (19). In addition, it also regulates Th17 inflammatory response and IL-17 function (20) (Figure 1).
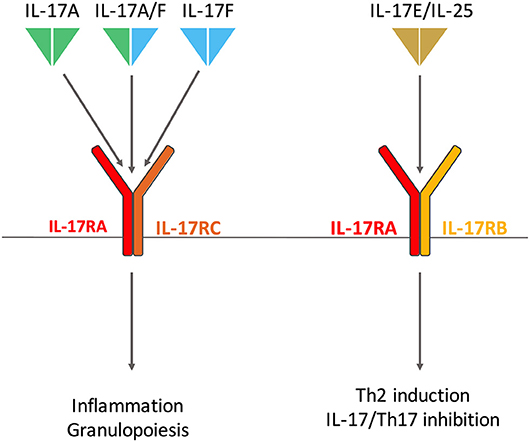
Figure 1. Interleukin (IL)-17 and Receptor family involved in rheumatoid arthritis. IL-17A and IL-17F homodimers and the IL-17A/IL-17F heterodimer bind the same receptor composed of IL-17RA and IL-17RC subunits. IL-17RA is also involved in IL-17E (also known as IL-25) receptor with IL-17RB. IL-17A/F and IL-17E have distinct biological effects, the first triggers inflammation and granulopoiesis; the latter promotes T-helper (Th) 2 responses in host defense against parasites and allergy. IL-17E also regulates Th17 inflammatory response.
The first cellular source of IL-17 was identified in 1999 as a particular subtype of CD4+ T cells (21). Th17 cells were finally described in 2005 in the mouse being different from the classical Th1 and Th2 cells (22). The differentiation of Th17 cells is a multi-step process involving transforming growth factor ß, IL-21, IL-1ß, IL-6, and IL-23 in humans (23, 24). The lineage-specific transcription factor retinoic acid receptor-related orphan receptor (RORc, RORγt in mice) is required for the differentiation (23). Other subsets of immune cells can produce IL-17 including γδ T cells, natural killer cells, invariant natural killer T cells, innate lymphoid cells and CD8+ T cells (2).
The first receptor of IL-17 to be identified was discovered in 1995 (25). The IL-17 receptor (IL-17R) family now includes 5 subunits, from IL-17RA to IL-17RE (26). IL-17A, IL-17F, and IL-17A/F bind the same receptor composed of IL-17RA and IL-17RC subunits (27). IL-25 binds a receptor made of IL-17RA and IL-17RB (28). Despite their opposite biological effects, IL-17A and IL-25 share a common receptor chain, an important point to consider when targeting IL-17RA in clinic (2) (Figure 1).
Upon ligand binding, the association of IL-17R with Act1 (also known as connection to iκB kinase and stress-activated protein kinases) induces the recruitment and the ubiquitination of tumor necrosis factor (TNF)-receptor associated factor-6 triggering nuclear factor-κ B (NF-κB), CCAAT/enhancer binding protein-ß, CCAAT/enhancer binding protein-δ and mitogen-activated protein kinase pathways. IL-17R and Act1 also activate extracellular signal-regulated kinase-5. These two signaling pathways mediated by IL-17 induce the transcription of inflammatory genes. IL-17 signaling also increases mRNA stability of IL-17 target genes (29). mRNA stabilization is one of the process by which IL-17 and other cytokines synergize, as described below for TNFα (30). Interestingly, peptide that blocks the interaction between Act1 and IL-17RA decreases both IL-17A and IL-25-induced inflammation (31).
Many changes occur in the RA synovium, which is characterized by hyperplasia, neoangiogenesis and local infiltration by immune cells (32, 33). These modifications trigger the destruction of cartilage and bone. The role of IL-17 in the synovitis pathogenesis was first characterized by observations on RA explants. Then, its effects on synovial and bone biopsies and in vitro are described.
Shortly after the description of IL-17, observations on synovial tissues and fluids of RA patients suggest that this cytokine may be involved in joint destruction. Indeed, immunostaining of the synovial tissues of RA patients demonstrates that a subset of CD4+CD45RO+ memory T cells produces IL-17; these IL-17 positive cells being not detected in synovial tissue from osteoarthritis (OA) patients. Moreover, concentration of IL-17 in synovial fluid is also higher in RA patients than in OA, trauma and gout patients (34). Interestingly, there is a spontaneous secretion of IL-17 by RA synovium compared with OA and normal synovium (35, 36). IL-17 synovial membrane mRNA level predicts damage progression (37). Double-immunofluorescence studies show that RORc co-localized with IL-17A and IL-17F staining suggesting that Th17 cells participate to the local cytokine production. IL-17A and IL-17F producing-cells are detected in the lymphocytic infiltrates and in hyperplasic lining cells of RA synovium (30). The recruitment of Th17 cells to the joint leads to interactions with local cells that perpetuate chronic inflammation (38). Specifically, cell interactions between Th17 cells and synoviocytes are crucial as they lead to a massive production of IL-17. The interaction molecule podoplanin contributes widely to this high IL-17 secretion (39, 40). In vitro and in vivo experiments show that IL-17A and IL-17F-producing cells have a plasma-cell like morphology (30, 41). This morphology has been associated with increased secretion in vitro and probably in vivo. Experiments on synovial explants from RA show that the Th2 cytokines IL-4 and IL-13 completely inhibit the production of IL-17 (35).
All together, these findings suggest a local production of IL-17 in RA synovium, mainly mediated by Th17 cells. The interactions between local mesenchymal cells and Th17 cells are crucial for a higher and more sustained production.
Having characterized the production of IL-17 in RA synovitis and the cells involved, IL-17 effects on synovial and bone explants are now described.
Structural damage in RA includes cartilage destruction and bone erosion (42). Cartilage damage is partially induced by synovial cytokines such as IL-17. Experiments on RA synovial samples show that IL-17 triggers the production of IL-6, leukemia inhibitory factor (LIF) and macrophage inflammatory protein (MIP)-3α/chemokine (C-C motif) ligand-20 by RA synovium (35, 43, 44). Moreover, the addition of an anti-IL-17 antibody to RA synovium cultures significantly decreases matrix metalloproteinase (MMP)-1 production, collagenase activity but not tissue inhibitor of MMP (TIMP)-1 production suggesting the direct contribution of IL-17 to joint destruction (45). The MMP/TIMP system plays a role in the collagen tissue turnover; a shift toward MMP production suggests degradation of the collagen framework. MMP-1 induces collagen degradation and the release of carboxy-terminal telopeptides. IL-17 increases carboxy-terminal telopeptides production in RA synovium explants, an effect that is reversed when adding an anti-IL-17 antibody (45, 46). Keeping with this, the C-pro-peptide of type I collagen, representing the production of type I collagen as part of repair efforts, is inhibited when adding IL-17 to RA synovium (46). All together, these results suggest that IL-17 promotes cartilage destruction at the expense of cartilage synthesis.
As mentioned above, RA also leads to bone erosion and particularly to early juxta-articular bone loss (42). Keeping with the results on RA synovium, IL-17 alone, and more in combination with IL-1 or TNFα, increases the production of IL-6 by RA bone explants (46, 47). In addition, IL-17 reduces bone formation and increases its destruction (46).
To go further into the comprehension of IL-17-induced destruction, effects of IL-17 on isolated cells are now described (Figure 2). IL-17A and IL-17F induce synoviocyte activation with increased cytokine and chemokine production, especially of IL-6 and IL-8 (4, 48–51). Moreover, IL-17 triggers synoviocyte migration and promotes an invasive phenotype that favors tissue destruction (33, 52, 53). Tissue destruction includes cartilage matrix destruction and bone erosion. Matrix destruction is mainly mediated by MMP. Among them, MMP-1,−2,−9, and−13 are induced by IL-17 in RA synoviocytes and chondrocytes (45, 54).
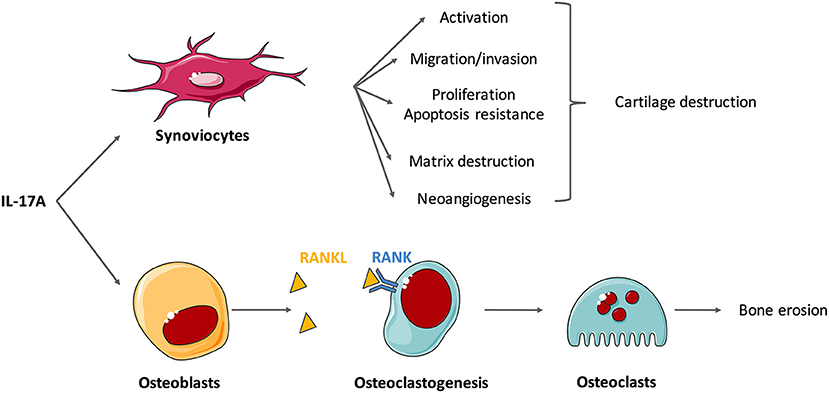
Figure 2. Effects of interleukin (IL)-17A on isolated cells involved in the synovitis. Rheumatoid synovium is characterized by hyperplasia, neoangiogenesis and excessive inflammation. IL-17A mediates cartilage and bone destruction that occurs in rheumatoid arthritis (RA) mainly through its action on synoviocytes and osteoblasts. In vitro experiments show that IL-17A induces synoviocyte activation (e.g., production of IL-6 and IL-8), migration and invasion that promote cartilage destruction. Through the induction of matrix metalloproteinases (MMP), IL-17A induces matrix destruction. It also favors proliferation and apoptosis resistance and the neoangiogenesis required for pannus development. In addition, IL-17 promotes receptor activator of NF-κB ligand (RANKL) expression on osteoblasts that binds to RANK, activates osteoclastogenesis and finally triggers bone erosion by osteoclasts.
Bone remodeling roughly depends on the balance between the activity of osteoclasts, that favor destruction, and osteoblasts, that promote bone formation. IL-17 promotes the expression of receptor activator of NF-κB ligand (RANKL) on osteoblasts and synoviocytes and then activates RANK signaling in osteoclasts (1, 55, 56). These results suggest that IL-17 plays a role in osteoclastogenesis, thereby promoting bone destruction (34, 57). Moreover, IL-17A could inhibit osteoblast and osteocyte activity in vitro but this should be confirmed (58).
Neoangiogenesis is crucial for pannus development in RA synovium. IL-17 is involved in this process inducing the production of vascular endothelial growth factor by synovial fibroblasts (59, 60). The RA synovium is also characterized by hyperplasia of synovial lining cells. IL-17 stimulates synoviocyte proliferation (61). This excessive proliferation combined with apoptosis resistance causes synovial hypertrophy. More specifically, IL-17 up-regulates anti-apoptotic genes and down-regulates pro-apoptotic genes (61, 62). IL-17 alone, and especially when combined with TNFα, increases the expression of the anti-apoptotic adhesion molecule Amigo 2 (63) and that of synoviolin, that prolong the survival of RA synoviocytes (50, 64). IL-17 also impairs apoptosis through activation of autophagy (65).
Therefore, observations on synovial and bone samples from RA patients and in vitro experiments confirm the role of IL-17 in synovitis.
Even if IL-17 effects on RA synovitis are clear, some mediators interfere with this system by regulating positively and negatively IL-17 function (Figure 3).
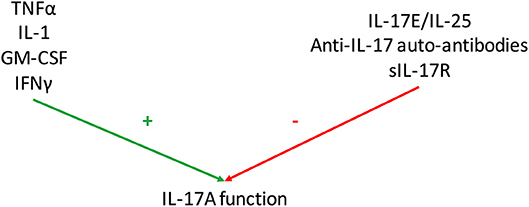
Figure 3. Regulation of Interleukin (IL)-17 function. Various mediators regulate IL-17A function. Some have additive or synergistic effects with IL-17A as tumor necrosis factor-α (TNFα), IL-1, granulocyte-macrophage colony stimulating factor (GM-CSF) and interferon (IFN) γ. Conversely, IL-25 (also known as IL-17E), anti-IL-17 autoantibodies and soluble IL-17 receptor (sIL-17R) inhibit IL-17 function.
Concomitant with the description of human IL-17A (13), experiments showed that TNFα potentiates the effect of IL-17A on IL-6 and IL-8-induced secretion by rheumatoid synoviocytes (14, 56). Similar results were obtained on RA synovium explants (37, 44). Moreover, IL-17F also synergizes with TNFα (4, 48). Mechanisms underlying this synergistic interaction were later described when IL-17A and IL-17F were shown to induce TNF receptor II expression and production. Microarrays analysis reveal that almost 90% of genes modified by the combination of IL-17A and TNFα showed a pattern of additivity and 1% of synergy (4, 30). Interestingly, IL-17 and TNFα mainly synergize through the induction of mRNA stabilization independently of TNF-receptor associated factor-6 (66, 67). Some genes synergistically induced by this combination are of importance in RA (e.g., IL-6, IL-8, chemokine (C-C motif) ligand-20, etc …). For instance, IL-17 and TNFα promote an invasive phenotype in synoviocytes (53) but also neutrophil survival (68).
IL-1 is involved in RA pathogenesis through bone and cartilage destruction (69). In vitro experiments on RA synoviocytes show a synergistic effect of IL-17 and IL-1ß on the production of IL-6 whereas an additive effect is observed for LIF production (43, 70). Interestingly, IL-17 and IL-1ß induce synergistically chemokine (C-C motif) ligand-20 production, which in turn recruits Th17 cells (44). The synergistic effect of IL-17 and IL-1ß is also demonstrated in RA bone explants where the two cytokines increase bone destruction and reduce its formation (46). Similar results are observed with the collagen-induced arthritis mouse models (71, 72). Blocking of both IL-1ß and IL-17A with a bi-specific antibody appears to reduce joint inflammation, destruction and synovial proliferation notably through the reduction of NF-κB activation (72). While IL-1 inhibitors (anti-IL-1ß antibody or soluble type I IL-1 receptor) have modest effect in RA, it would be of interest to develop biological agents that block both IL-17 and IL-1 (69).
Granulocyte-macrophage colony stimulating factor (GM-CSF) is produced by many cell types (e.g., myeloid cells, tissue-resident cells) and plays a key role in the differentiation of myeloid cells and in the production of neutrophils, eosinophils and monocytes. GM-CSF is also involved in adaptive immunity. In vivo experiments show that GM-CSF is involved in RA pathogenesis (69). GM-CSF level is also increased in synovial fluid and blood from RA patients (73). In experimental arthritis, the combination of IL-17 and GM-CSF shows complementary and local additive effects and induces a more severe phenotype (74).
Interferon (IFN) γ plays a role in anti-infectious host defense, in inflammatory and in auto-immune diseases (75). IL-17A and IFNγ have an additive effect on IL-6 secretion by RA synoviocytes (14). Experiments on other cell types show that the combination of IL-17 and IFNγ increases the production of IL-6, IL-8, intracellular adhesion molecule-1 and nitric oxide (50, 76).
As described above, IL-17A and IL-25 bear the lowest homology and their receptors share the common chain IL-17RA (1).
In a mouse model of type I diabetes, IL-25 effect is similar to that of anti-IL-17 to reduce peri-islet CD4 and CD8 T-cell infiltrates while increasing the proportion of the Treg cell population. Interestingly, only IL-25 treatment reduces the amount of autoreactive Th2 and Th17 cells in delayed recurrent autoimmunity. This study highlights the potential shift induced by IL-25 into the Th17/Treg balance (77).
Administration of IL-25 reduces collagen-induced arthritis development in mice and suppresses Th17 cell responses in an IL-13 dependent manner (78). Similar observations are made in experimental autoimmune encephalomyelitis mice where IL-13 is also required to induce Th17 suppression (20).
IL-25 level is higher in serum and synovial fluid from RA patients compared with OA patients and healthy controls (78, 79). Similarly, IL-25 level is correlated with disease activity and with inflammatory cytokines (e.g., TNFα, IL-1ß, IL-17A, IL-6) in RA patients. Moreover, when stimulated peripheral blood mononuclear cells from RA patients are treated with recombinant IL-25, Th17 cells and IL-17A expression are inhibited and that of IL-4 increased (78).
Interestingly, there is a spontaneous secretion of IL-25 by RA synoviocytes that is delayed compared with the production of IL-6. Similar results were obtained in a model known to mimic the inflammatory site of RA synovium (synoviocytes/peripheral blood mononuclear cells coculture), IL-25 production being delayed compared with that of IL-17A. In turn, IL-25 can inhibit IL-17A function acting as a receptor antagonist (79).
Considering the interaction between IL-17A and TNFα, IL-25 reduces the production of IL-6 induced by these two cytokines. Interestingly, IL-17A and TNFα decrease IL-25 production while TNFα alone increases IL-17RB in synoviocytes, being a potential way for TNFα to regulate inflammation (79). Indeed, IL-17RB is required for IL-25 signaling that in turn controls Th1 and Th17 responses and inhibits monocyte-derived inflammatory cytokines (20, 79–81).
All of these results suggest that IL-25 acts as a regulatory pathway in response to inflammation to then down-regulates excessive Th17 and IL-17 immune response.
Autoantibodies against pro-inflammatory cytokines were first described for IL-1α and constitute a marker of good prognosis in RA (82, 83). They bind their antigen and form immune complexes with the cytokine. Anti-IL-17 antibodies are absent in healthy controls while there are detected in almost 40% of RA patients. As opposed to bioactive IL-17A, anti-IL-17 antibodies are increased in non-severe RA and so linked to a better prognosis. As expected, higher titers of immune complexes are detected in non-destructive compared with destructive RA (10).
The expression of cytokines is regulated by various mechanisms. For instance, IL-1 receptor antagonist binds to IL-1 receptors, competitively antagonizes the binding of IL-1 and finally decreases its biological effects (84). Soluble type II IL-1 receptor also acts as an inhibitor of IL-1 function (85).
Although not fully demonstrated, it makes sense to consider the contribution of soluble IL-17R (sIL-17R) in the regulation of IL-17 function. Interestingly, sIL-17RB is increased in alveolar echinococcosis infected patients compared with controls and its level is correlated with disease severity. Conversely, sIL-17RA shows an opposite trend. These variations of soluble receptors may silence the IL-25 mediated response, thereby promoting disease progression (86).
This example, far from the RA, illustrates that sIL-17R is involved in the modulation of IL-17 levels. In vitro and ex vivo experiments with RA samples have shown that the combined inhibition of IL-17, IL-1 and TNFα with soluble receptors increased the degree of response (44, 87, 88).
Therefore, many mediators regulate positively or negatively IL-17 function; these results are summarized in Figure 3. Considering all these interactions, it remains a challenge to detect the specific effect of IL-17 both at local and systemic level.
It makes sense that patients with high level of IL-17 would be more sensitive to an anti-IL-17 inhibitor. This concept was developed after the emergence of TNFα inhibitors. In a majority of patients, this treatment leads to the reduction of symptoms, inflammation and bone destruction. However, around 30 % of these RA patients do not respond. To better understand this observation, a bioassay was developed to evaluate TNFα bioactivity before treatment (89). It is based on the ability of synoviocytes to produce IL-6 in response to TNFα (90). Indeed, 60% of patients have a good ability of their plasma to induce IL-6 production before infliximab therapy (a TNFα inhibitor), this production being inhibited 4 h after the first infliximab infusion. Another pattern of patients has moderate or no IL-6 production before infusion, therefore no inhibition by infliximab. The difference of IL-6 production before and 4 h after the first infliximab infusion is correlated with clinical response. This may explain the heterogeneity in treatment response to TNFα inhibitors (89). Interestingly, intra-articular administration of etanercept (a TNFα inhibitor) results in a significant improvement of the composite change index compared to placebo in RA and psoriatic arthritis patients. Serum etanercept levels were comparable between composite change index good and non-responders, thus indicating that local inhibition of TNFα would be effective (91).
Similar experiments would be of interest for IL-17 since IL-17 systemic inhibitors show heterogeneous results in RA. Interestingly, IL-17A synovial fluid levels are higher than serum levels in early RA cases, suggesting that local production may be reflected by circulating levels (92).
More recently, studies from the Pathobiology of Early Arthritis Cohort have been set up with the aim to define from synovial biopsies and blood samples the involvement of cellular/molecular signatures in determining clinical phenotypes (11, 93, 94). For instance, in early RA, synovial transcripts correlating with disease activity (disease activity score-28/C-reactive protein) are significantly enriched in TNFα-induced genes and predict poor response to first-line therapy (95). Considering the important interpatient heterogeneity, such approaches on synovial biopsies may be used to stratify patient for tailored drug delivery strategies (94), especially in the case of IL-17 inhibitors where results showed a high heterogeneity. Indeed, IL-17 and its receptor are up-regulated within synovial ectopic lymphoid structures and further contribute to the chronicity of local inflammation (64, 96). These structures have the ability to function as germinal centers and there is a significant association between their presence and erosive disease. Aggressive treatments are recommended for these patients to prevent the onset of erosions. Considering the key role of IL-17 in the formation of these ectopic lymphoid structures, IL-17 inhibition would be of interest in these patients selected with a synovial biopsy (97, 98).
Considering the results described above, there is no doubt that IL-17 is involved in RA pathogenesis. However, IL-17A circulating levels measured by ELISA vary a lot across studies, from undetectable to pg/ml or even ng/ml concentration (99, 100). Moreover, these tests do not detect the bioactive form that is crucial since there are circulating inhibitors (IL-25, anti-IL-17 autoantibodies, sIL-17R) and activators of IL-17 (TNFα, IL-1, GM-CSF, IFNγ) (Figure 3). To measure the level of bioactive IL-17A, a cell-based bioassay was developed on the ability of RA synoviocytes to produce IL-6. RA synoviocytes are exposed to plasma samples and IL-6 production is measured with or without an anti-IL-17 antibody (35). The test was then extended to human endothelial cells that are able to produce IL-8 in presence of IL-17A (5). By blocking IL-17A, it allows to quantify its specific contribution in the production of pro-inflammatory cytokines.
While many studies are performed to identify predictive biomarkers of RA development (e.g., cigarette smoking, infection), another issue is also to predict which therapy is the best suited for patients that have developed RA (101).
Using the bioassay described above, bioactive IL-17A is higher in RA patients compared with healthy controls and its level is correlated with destruction (5). As mentioned earlier, anti-IL-17 antibodies and immune complexes are elevated in non-destructive RA (10). Detection of these biomarkers represents an interesting tool to identify patients with an IL-17 driven disease that could respond better to IL-17 inhibitors.
In RA patients, a meta-analysis shows the superiority of secukinumab (anti-IL-17A) and ixekizumab (anti-IL-17A) compared with placebo based on American College of Rheumatology (ACR)-20 and ACR50 clinical response. However, it does not reach statistical significance for ACR70 response and analysis of individual response rate shows a high degree of heterogeneity. Moreover, brodalumab (anti-IL-17RA) is not effective in achieving ACR20 (2, 102). These observations rely on different explanations. First, immunohistochemical analysis reveal a high variability of IL-17A, IL-17F and their receptor expression in RA synovitis (103). IL-17 inhibition would not be sufficient in these patients with low expression of IL-17. Then, different strategies are developed to block the IL-17 pathway with anti-IL-17A, anti-IL-17A/F and anti-IL-17RA (2). These antibodies may encounter some pitfalls; for instance, the inhibition of IL-17RA could inhibit the anti-inflammatory effect mediated by IL-25 (28). The dual inhibition with bi-specific antibodies against TNFα and IL-17A would have been of interest to prevent their synergistic interaction but recent papers show no clear benefit, especially when compared to TNFα inhibition alone (104–106). The structure of the dual inhibitor and the respective location of the two binding sites have to be considered.
Therefore, as for TNF inhibitors, an effort is needed to identify RA patients that would benefit from IL-17 targeted therapies. The development of predictive biomarkers of response to IL-17 inhibitors is beginning; for instance, the cell-based bioassay detecting bioactive IL-17A is of interest but only constitutes the spearhead of more research. IL-17 expression in synovial tissue may be another way to stratify patients to potentiate the beneficial effect of these inhibitors (11, 96, 103). Even if robust evidence is still needed to confirm the use of such biomarkers in clinical routine, these strategies can move the field of RA management into the new era of precision medicine in the future.
IL-17A is involved in early induction and late chronic stages of various inflammatory diseases. The inhibition of its signaling brings a clear improvement in psoriasis, psoriatic arthritis and in ankylosing spondylitis treatment but results are less convincing in RA. However, ex vivo and in vitro studies clearly show that IL-17A is one of the culprit that perpetuates local inflammation in synovium and especially in RA. Explanations of such unexpected results may come from the many mediators that modulate IL-17 function, with either agonist or antagonist effects. The significant heterogeneity of IL-17 expression between patients also imposes a stratification of them to identify the ones that could benefit from IL-17 inhibitors. The development of predictive biomarkers as bioactive IL-17 or anti-IL-17-autoantibodies or the use of synovial biopsies still requires robust evidence but would be interesting to turn the page to precision medicine in RA.
PM holds a patent on the IL-17 bioassay.
MR: writing and figures. PM: concept and proof reading.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
MR is supported by the Ecole de l'Inserm Liliane Bettencourt. PM is a senior member of the Institut Universitaire de France. His laboratory is supported in part by the IHU OPERA.
IL, interleukin; RA, rheumatoid arthritis; Th, T-helper; ROR, retinoic acid receptor-related orphan receptor; IL-17R, IL-17 receptor; TNF, tumor necrosis factor; NF-κB, nuclear factor-κ B; OA, osteoarthritis; LIF, leukemia inhibitory factor; MIP, macrophage inflammatory protein; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of MMP; RANKL, receptor activator of NF-κB ligand; GM-CSF, granulocyte-macrophage colony stimulating factor; IFN, interferon; sIL-17R, soluble IL-17R; ACR, American College of Rheumatology.
1. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. (2012) 11:763–76. doi: 10.1038/nrd3794
2. Beringer A, Noack M, Miossec P. IL-17 in Chronic Inflammation: from discovery to targeting. Trends Mol Med. (2016) 22:230–41. doi: 10.1016/j.molmed.2016.01.001
3. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
4. Hot A, Miossec P. Effects of interleukin (IL)-17A and IL-17F in human rheumatoid arthritis synoviocytes. Ann Rheum Dis. (2011) 70:727–32. doi: 10.1136/ard.2010.143768
5. Ndongo-Thiam N, Miossec P. A cell-based bioassay for circulating bioactive IL-17: application to destruction in rheumatoid arthritis. Ann Rheum Dis. (2015) 74:1629–31. doi: 10.1136/annrheumdis-2014-207110
6. Fragoulis GE, Siebert S, McInnes IB. Therapeutic Targeting of IL-17 and IL-23 cytokines in immune-mediated diseases. Annu Rev Med. (2016) 67:337–53. doi: 10.1146/annurev-med-051914-021944
7. Pavelka K, Kivitz A, Dokoupilova E, Blanco R, Maradiaga M, Tahir H, et al. Efficacy, safety, and tolerability of secukinumab in patients with active ankylosing spondylitis: a randomized, double-blind phase 3 study, MEASURE 3. Arthritis Res Ther. (2017) 19:285. doi: 10.1186/s13075-017-1490-y
8. Sieper J, Deodhar A, Marzo-Ortega H, Aelion JA, Blanco R, Jui-Cheng T, et al. Secukinumab efficacy in anti-TNF-naive and anti-TNF-experienced subjects with active ankylosing spondylitis: results from the MEASURE 2 Study. Ann Rheum Dis. (2017) 76:571–92. doi: 10.1136/annrheumdis-2016-210023
9. Braun J, Baraliakos X, Deodhar A, Baeten D, Sieper J, Emery P, et al. Effect of secukinumab on clinical and radiographic outcomes in ankylosing spondylitis: 2-year results from the randomised phase III MEASURE 1 study. Ann Rheum Dis. (2017) 76:1070–7. doi: 10.1136/annrheumdis-2016-209730
10. Ndongo-Thiam N, Clement A, Pin JJ, Razanajaona-Doll D, Miossec P. Negative association between autoantibodies against IL-17, IL-17/anti-IL-17 antibody immune complexes and destruction in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1420–2. doi: 10.1136/annrheumdis-2016-209149
11. Astorri E, Nerviani A, Bombardieri M, Pitzalis C. Towards a stratified targeted approach with biologic treatments in rheumatoid arthritis: role of synovial pathobiology. Curr Pharm Des. (2015) 21:2216–24. doi: 10.2174/1381612821666150310145758
12. Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P. CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol. (1993) 150:5445–56.
13. Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol. (1995) 155:5483–6.
14. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. (1996) 183:2593–603.
15. Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, et al. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. (2001) 20:5332–41. doi: 10.1093/emboj/20.19.5332
16. Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem. (2007) 282:13447–55. doi: 10.1074/jbc.M700499200
17. Lee J, Ho WH, Maruoka M, Corpuz RT, Baldwin DT, Foster JS, et al. IL-17E, a novel proinflammatory ligand for the IL-17 receptor homolog IL-17Rh1. J Biol Chem. (2001) 276:1660–4. doi: 10.1074/jbc.M008289200
18. Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. (2006) 203:1105–16. doi: 10.1084/jem.20051615
19. Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, et al. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Aller Clin Immunol. (2007) 120:1324–31. doi: 10.1016/j.jaci.2007.07.051
20. Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. (2007) 204:161–70. doi: 10.1084/jem.20061738
21. Aarvak T, Chabaud M, Miossec P, Natvig JB. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol. (1999) 162:1246–51.
22. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
23. Noack M, Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun Rev. (2014) 13:668–77. doi: 10.1016/j.autrev.2013.12.004
24. Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. (2009) 361:888–98. doi: 10.1056/NEJMra0707449
25. Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, et al. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. (1995) 3:811–21.
26. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
27. Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, et al. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. (2006) 177:36–9. doi: 10.4049/jimmunol.177.1.36
28. Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, et al. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol. (2008) 181:4299–310. doi: 10.4049/jimmunol.181.6.4299
29. Amatya N, Garg AV, Gaffen SL. IL-17 Signaling: the Yin and the Yang. Trends Immunol. (2017) 38:310–22. doi: 10.1016/j.it.2017.01.006
30. Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, Miossec P. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol. (2009) 182:3112–20. doi: 10.4049/jimmunol.0801967
31. Liu C, Swaidani S, Qian W, Kang Z, Sun P, Han Y, et al. A CC' loop decoy peptide blocks the interaction between Act1 and IL-17RA to attenuate IL-17- and IL-25-induced inflammation. Sci Signal. (2011) 4:ra72. doi: 10.1126/scisignal.2001843
32. Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. Int J Biochem Cell Biol. (2004) 36:372–8. doi: 10.1016/S1357-2725(03)00259-0
33. Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. (2013) 9:24–33. doi: 10.1038/nrrheum.2012.190
34. Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest. (1999) 103:1345–52. doi: 10.1172/JCI5703
35. Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, et al. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. (1999) 42:963–70.
36. Honorati MC, Meliconi R, Pulsatelli L, Cane S, Frizziero L, Facchini A. High in vivo expression of interleukin-17 receptor in synovial endothelial cells and chondrocytes from arthritis patients. Rheumatology (Oxford). (2001) 40:522–7. doi: 10.1093/rheumatology/40.5.522
37. Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, et al. Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum. (2006) 54:1122–31. doi: 10.1002/art.21749
38. Eljaafari A, Tartelin ML, Aissaoui H, Chevrel G, Osta B, Lavocat F, et al. Bone marrow-derived and synovium-derived mesenchymal cells promote Th17 cell expansion and activation through caspase 1 activation: contribution to the chronicity of rheumatoid arthritis. Arthritis Rheum. (2012) 64:2147–57. doi: 10.1002/art.34391
39. Noack M, Ndongo-Thiam N, Miossec P. Interaction among activated lymphocytes and mesenchymal cells through podoplanin is critical for a high IL-17 secretion. Arthritis Res Ther. (2016) 18:148. doi: 10.1186/s13075-016-1046-6
40. Noack M, Ndongo-Thiam N, Miossec P. Role of podoplanin in the high interleukin-17A secretion resulting from interactions between activated lymphocytes and psoriatic skin-derived mesenchymal cells. Clin Exp Immunol. (2016) 186:64–74. doi: 10.1111/cei.12830
41. Page G, Sattler A, Kersten S, Thiel A, Radbruch A, Miossec P. Plasma cell-like morphology of Th1-cytokine-producing cells associated with the loss of CD3 expression. Am J Pathol. (2004) 164:409–17. doi: 10.1016/S0002-9440(10)63131-8
42. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
43. Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. (1998) 161:409–14.
44. Chabaud M, Page G, Miossec P. Enhancing effect of IL-1, IL-17, and TNF-alpha on macrophage inflammatory protein-3alpha production in rheumatoid arthritis: regulation by soluble receptors and Th2 cytokines. J Immunol. (2001) 167:6015–20. doi: 10.4049/jimmunol.167.10.6015
45. Chabaud M, Garnero P, Dayer JM, Guerne PA, Fossiez F, Miossec P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine. (2000) 12:1092–9. doi: 10.1006/cyto.2000.0681
46. Chabaud M, Lubberts E, Joosten L, van Den Berg W, Miossec P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. (2001) 3:168–77. doi: 10.1186/ar294
47. Osta B, Roux JP, Lavocat F, Pierre M, Ndongo-Thiam N, Boivin G, et al. Differential Effects of IL-17A and TNF-alpha on osteoblastic differentiation of isolated synoviocytes and on bone explants from arthritis patients. Front Immunol. (2015) 6:151. doi: 10.3389/fimmu.2015.00151
48. Hot A, Zrioual S, Toh ML, Lenief V, Miossec P. IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann Rheum Dis. (2011) 70:341–8. doi: 10.1136/ard.2010.132233
49. Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, et al. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. (2004) 6:R120–8. doi: 10.1186/ar1038
50. Benedetti G, Miossec P. Interleukin 17 contributes to the chronicity of inflammatory diseases such as rheumatoid arthritis. Eur J Immunol. (2014) 44:339–47. doi: 10.1002/eji.201344184
51. Zrioual S, Toh ML, Tournadre A, Zhou Y, Cazalis MA, Pachot A, et al. IL-17RA and IL-17RC receptors are essential for IL-17A-induced ELR+ CXC chemokine expression in synoviocytes and are overexpressed in rheumatoid blood. J Immunol. (2008) 180:655–63. doi: 10.4049/jimmunol.180.1.655
52. Li G, Zhang Y, Qian Y, Zhang H, Guo S, Sunagawa M, et al. Interleukin-17A promotes rheumatoid arthritis synoviocytes migration and invasion under hypoxia by increasing MMP2 and MMP9 expression through NF-kappaB/HIF-1alpha pathway. Mol Immunol. (2013) 53:227–36. doi: 10.1016/j.molimm.2012.08.018
53. Hot A, Zrioual S, Lenief V, Miossec P. IL-17 and tumour necrosis factor alpha combination induces a HIF-1alpha-dependent invasive phenotype in synoviocytes. Ann Rheum Dis. (2012) 71:1393–401. doi: 10.1136/annrheumdis-2011-200867
54. Moran EM, Mullan R, McCormick J, Connolly M, Sullivan O, Fitzgerald O, et al. Human rheumatoid arthritis tissue production of IL-17A drives matrix and cartilage degradation: synergy with tumour necrosis factor-alpha, Oncostatin M and response to biologic therapies. Arthritis Res Ther. (2009) 11:R113. doi: 10.1186/ar2772
55. Van Bezooijen RL, Papapoulos SE, Lowik CW. Effect of interleukin-17 on nitric oxide production and osteoclastic bone resorption: is there dependency on nuclear factor-kappaB and receptor activator of nuclear factor kappaB (RANK)/RANK ligand signaling? Bone. (2001) 28:378–86. doi: 10.1016/S8756-3282(00)00457-9
56. Lavocat F, Maggi L, Annunziato F, Miossec P. T-cell clones from Th1, Th17 or Th1/17 lineages and their signature cytokines have different capacity to activate endothelial cells or synoviocytes. Cytokine. (2016) 88:241–50. doi: 10.1016/j.cyto.2016.09.019
57. Lavocat F, Osta B, Miossec P. Increased sensitivity of rheumatoid synoviocytes to Schnurri-3 expression in TNF-alpha and IL-17A induced osteoblastic differentiation. Bone. (2016) 87:89–96. doi: 10.1016/j.bone.2016.04.008
58. Uluckan O, Jimenez M, Karbach S, Jeschke A, Grana O, Keller J, et al. Chronic skin inflammation leads to bone loss by IL-17-mediated inhibition of Wnt signaling in osteoblasts. Sci Transl Med. (2016) 8:330ra37. doi: 10.1126/scitranslmed.aad8996
59. Daoussis D, Andonopoulos AP, Liossis SN. Wnt pathway and IL-17: novel regulators of joint remodeling in rheumatic diseases. Looking beyond the RANK-RANKL-OPG axis. Semin Arthritis Rheum. (2010) 39:369–83. doi: 10.1016/j.semarthrit.2008.10.008
60. Honorati MC, Neri S, Cattini L, Facchini A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage. (2006) 14:345–52. doi: 10.1016/j.joca.2005.10.004
61. Zhang Q, Wu J, Cao Q, Xiao L, Wang L, He D, et al. A critical role of Cyr61 in interleukin-17-dependent proliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Rheum. (2009) 60:3602–12. doi: 10.1002/art.24999
62. Lee SY, Kwok SK, Son HJ, Ryu JG, Kim EK, Oh HJ, et al. IL-17-mediated Bcl-2 expression regulates survival of fibroblast-like synoviocytes in rheumatoid arthritis through STAT3 activation. Arthritis Res Ther. (2013) 15:R31. doi: 10.1186/ar4179
63. Benedetti G, Bonaventura P, Lavocat F, Miossec P. IL-17A and TNF-alpha Increase the Expression of the Antiapoptotic Adhesion Molecule Amigo-2 in Arthritis Synoviocytes. Front Immunol. (2016) 7:254. doi: 10.3389/fimmu.2016.00254
64. Toh ML, Gonzales G, Koenders MI, Tournadre A, Boyle D, Lubberts E, et al. Role of interleukin 17 in arthritis chronicity through survival of synoviocytes via regulation of synoviolin expression. PLoS ONE (2010) 5:e13416. doi: 10.1371/journal.pone.0013416
65. Kim EK, Kwon JE, Lee SY, Lee EJ, Kim DS, Moon SJ, et al. IL-17-mediated mitochondrial dysfunction impairs apoptosis in rheumatoid arthritis synovial fibroblasts through activation of autophagy. Cell Death Dis. (2017) 8:e2565. doi: 10.1038/cddis.2016.490
66. Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. (2007) 179:4135–41. doi: 10.4049/jimmunol.179.6.4135
67. Hartupee J, Liu C, Novotny M, Sun D, Li X, Hamilton TA. IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6. J Immunol. (2009) 182:1660–6. doi: 10.4049/jimmunol.182.3.1660
68. Parsonage G, Filer A, Bik M, Hardie D, Lax S, Howlett K, et al. Prolonged, granulocyte-macrophage colony-stimulating factor-dependent, neutrophil survival following rheumatoid synovial fibroblast activation by IL-17 and TNFalpha. Arthritis Res Ther. (2008) 10:R47. doi: 10.1186/ar2406
69. Noack M, Miossec P. Selected cytokine pathways in rheumatoid arthritis. Semin Immunopathol. (2017) 39:365–83. doi: 10.1007/s00281-017-0619-z
70. Kehlen A, Pachnio A, Thiele K, Langner J. Gene expression induced by interleukin-17 in fibroblast-like synoviocytes of patients with rheumatoid arthritis: upregulation of hyaluronan-binding protein TSG-6. Arthritis Res Ther. (2003) 5:R186–92. doi: 10.1186/ar762
71. Zhang Y, Ren G, Guo M, Ye X, Zhao J, Xu L, et al. Synergistic effects of interleukin-1beta and interleukin-17A antibodies on collagen-induced arthritis mouse model. Int Immunopharmacol. (2013) 15:199–205. doi: 10.1016/j.intimp.2012.12.010
72. Wu Q, Wang Y, Wang Q, Yu D, Wang Y, Song L, et al. The bispecific antibody aimed at the vicious circle of IL-1beta and IL-17A, is beneficial for the collagen-induced rheumatoid arthritis of mice through NF-kappaB signaling pathway. Immunol Lett. (2016) 179:68–79. doi: 10.1016/j.imlet.2016.09.001
73. Xu WD, Firestein GS, Taetle R, Kaushansky K, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. II. Granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J Clin Invest. (1989) 83:876–82. doi: 10.1172/JCI113971
74. van Nieuwenhuijze AE, van de Loo FA, Walgreen B, Bennink M, Helsen M, van den Bersselaar L, et al. Complementary action of granulocyte macrophage colony-stimulating factor and interleukin-17A induces interleukin-23, receptor activator of nuclear factor-kappaB ligand, and matrix metalloproteinases and drives bone and cartilage pathology in experimental arthritis: rationale for combination therapy in rheumatoid arthritis. Arthritis Res Ther. (2015) 17:163. doi: 10.1186/s13075-015-0683-5
75. Billiau A, Matthys P. Interferon-gamma: a historical perspective. Cytokine Growth Factor Rev. (2009) 20:97–113. doi: 10.1016/j.cytogfr.2009.02.004
76. Robert M, Miossec P. Effects of Interleukin 17 on the cardiovascular system. Autoimmun Rev. (2017) 16:984–91. doi: 10.1016/j.autrev.2017.07.009
77. Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes. (2009) 58:1302–11. doi: 10.2337/db08-1113
78. Liu D, Cao T, Wang N, Liu C, Ma N, Tu R, et al. IL-25 attenuates rheumatoid arthritis through suppression of Th17 immune responses in an IL-13-dependent manner. Sci Rep. (2016) 6:36002. doi: 10.1038/srep36002
79. Lavocat F, Ndongo-Thiam N, Miossec P. Interleukin-25 Produced by Synoviocytes Has Anti-inflammatory Effects by Acting As a Receptor Antagonist for Interleukin-17A Function. Front Immunol. (2017) 8:647. doi: 10.3389/fimmu.2017.00647
80. Caruso R, Sarra M, Stolfi C, Rizzo A, Fina D, Fantini MC, et al. Interleukin-25 inhibits interleukin-12 production and Th1 cell-driven inflammation in the gut. Gastroenterology (2009) 136:2270–9. doi: 10.1053/j.gastro.2009.02.049
81. Caruso R, Stolfi C, Sarra M, Rizzo A, Fantini MC, Pallone F, et al. Inhibition of monocyte-derived inflammatory cytokines by IL-25 occurs via p38 Map kinase-dependent induction of Socs-3. Blood (2009) 113:3512–9. doi: 10.1182/blood-2008-08-172767
82. Jouvenne P, Fossiez F, Banchereau J, Miossec P. High levels of neutralizing autoantibodies against IL-1 alpha are associated with a better prognosis in chronic polyarthritis: a follow-up study. Scand J Immunol. (1997) 46:413–8.
83. Miossec P. Anti-interleukin 1alpha autoantibodies. Ann Rheum Dis. (2002) 61:577–9. doi: 10.1136/ard.61.7.577
84. Carter DB, Deibel MR, Jr., Dunn CJ, Tomich CS, Laborde AL, Slightom JL, et al. Purification, cloning, expression and biological characterization of an interleukin-1 receptor antagonist protein. Nature. (1990) 344:633–8. doi: 10.1038/344633a0
85. Jouvenne P, Vannier E, Dinarello CA, Miossec P. Elevated levels of soluble interleukin-1 receptor type II and interleukin-1 receptor antagonist in patients with chronic arthritis: correlations with markers of inflammation and joint destruction. Arthritis Rheum. (1998) 41:1083–9.
86. Lechner CJ, Gruner B, Huang X, Hoffmann WH, Kern P, Soboslay PT. Parasite-specific IL-17-type cytokine responses and soluble IL-17 receptor levels in Alveolar Echinococcosis patients. Clin Dev Immunol. (2012) 2012:735342. doi: 10.1155/2012/735342
87. Chevrel G, Garnero P, Miossec P. Addition of interleukin 1 (IL1) and IL17 soluble receptors to a tumour necrosis factor alpha soluble receptor more effectively reduces the production of IL6 and macrophage inhibitory protein-3alpha and increases that of collagen in an in vitro model of rheumatoid synoviocyte activation. Ann Rheum Dis. (2002) 61:730–3.
88. Chabaud M, Miossec P. The combination of tumor necrosis factor alpha blockade with interleukin-1 and interleukin-17 blockade is more effective for controlling synovial inflammation and bone resorption in an ex vivo model. Arthritis Rheum. (2001) 44:1293–303. doi: 10.1002/1529-0131(200106)44:6<1293::AID-ART221>3.0.CO;2-T
89. Marotte H, Maslinski W, Miossec P. Circulating tumour necrosis factor-alpha bioactivity in rheumatoid arthritis patients treated with infliximab: link to clinical response. Arthritis Res Ther. (2005) 7:R149–55. doi: 10.1186/ar1465
90. Ziolkowska M, Kurowska M, Radzikowska A, Luszczykiewicz G, Wiland P, Dziewczopolski W, et al. High levels of osteoprotegerin and soluble receptor activator of nuclear factor kappa B ligand in serum of rheumatoid arthritis patients and their normalization after anti-tumor necrosis factor alpha treatment. Arthritis Rheum. (2002) 46:1744–53. doi: 10.1002/art.10388
91. Aalbers C, Gerlag D, Vos K, Vervoordeldonk M, Landewe R, Tak PP. Intra-articular etanercept treatment in inflammatory arthritis: a randomized double-blind placebo-controlled proof of mechanism clinical trial validating TNF as a potential therapeutic target for local treatment. Joint Bone Spine. (2015) 82:338–44. doi: 10.1016/j.jbspin.2015.03.002
92. Rosu A, Margaritescu C, Stepan A, Musetescu A, Ene M. IL-17 patterns in synovium, serum and synovial fluid from treatment-naive, early rheumatoid arthritis patients. Rom J Morphol Embryol. (2012) 53:73–80.
93. Pitzalis C, Kelly S, Humby F. New learnings on the pathophysiology of RA from synovial biopsies. Curr Opin Rheumatol. (2013) 25:334–44. doi: 10.1097/BOR.0b013e32835fd8eb
94. Ferrari M, Onuoha SC, Pitzalis C. Trojan horses and guided missiles: targeted therapies in the war on arthritis. Nat Rev Rheumatol. (2015) 11:328–37. doi: 10.1038/nrrheum.2015.17
95. De Groof A, Ducreux J, Humby F, Nzeusseu Toukap A, Badot V, Pitzalis C, et al. Higher expression of TNFalpha-induced genes in the synovium of patients with early rheumatoid arthritis correlates with disease activity, and predicts absence of response to first line therapy. Arthritis Res Ther. (2016) 18:19. doi: 10.1186/s13075-016-0919-z
96. Boutet MA, Nerviani A, Gallo Afflitto G, Pitzalis C. Role of the IL-23/IL-17 axis in psoriasis and psoriatic arthritis: the clinical importance of its divergence in skin and joints. Int J Mol Sci. (2018) 19:E530. doi: 10.3390/ijms19020530
97. Humby F, Manzo A, Kirkham B, Pitzalis C. The synovial membrane as a prognostic tool in rheumatoid arthritis. Autoimmun Rev. (2007) 6:248–52. doi: 10.1016/j.autrev.2006.08.013
98. Humby FC, Al Balushi F, Lliso G, Cauli A, Pitzalis C. Can Synovial Pathobiology Integrate with Current Clinical and Imaging Prediction Models to Achieve Personalized Health Care in Rheumatoid Arthritis? Front Med. (2017) 4:41. doi: 10.3389/fmed.2017.00041
99. DeForge LE, Loyet KM, Delarosa D, Chinn J, Zamanian F, Chuntharapai A, et al. Evaluation of heterophilic antibody blocking agents in reducing false positive interference in immunoassays for IL-17AA, IL-17FF, and IL-17AF. J Immunol Methods. (2010) 362:70–81. doi: 10.1016/j.jim.2010.09.004
100. Singh R, Aggarwal A, Misra R. Th1/Th17 cytokine profiles in patients with reactive arthritis/undifferentiated spondyloarthropathy. J Rheumatol. (2007) 34:2285–90.
101. Bernatsky SM, Pfau JC, Fritzler MJ. Environmental exposures and biomarkers predictive of rheumatoid arthritis and the pathway to precision medicine. J Lab Precis Med. (2017) 2:1–4. doi: 10.21037/jlpm.2017.02.01
102. Kunwar S, Dahal K, Sharma S. Anti-IL-17 therapy in treatment of rheumatoid arthritis: a systematic literature review and meta-analysis of randomized controlled trials. Rheumatol Int. (2016) 36:1065–75. doi: 10.1007/s00296-016-3480-9
103. van Baarsen LG, Lebre MC, van der Coelen D, Aarrass S, Tang MW, Ramwadhdoebe TH, et al. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res Ther. (2014) 16:426. doi: 10.1186/s13075-014-0426-z
104. Silacci M, Lembke W, Woods R, Attinger-Toller I, Baenziger-Tobler N, Batey S, et al. Discovery and characterization of COVA322, a clinical-stage bispecific TNF/IL-17A inhibitor for the treatment of inflammatory diseases. MAbs. (2016) 8:141–9. doi: 10.1080/19420862.2015.1093266
105. Genovese MC, Weinblatt ME, Aelion JA, Mansikka HT, Peloso PM, Chen K, et al. ABT-122, a bispecific dual variable domain immunoglobulin targeting tumor necrosis factor and Interleukin-17A, in patients with rheumatoid arthritis with an inadequate Response to methotrexate: a randomized, double-blind study. Arthritis Rheumatol. (2018) 70:1710–20. doi: 10.1002/art.40580
Keywords: synovitis, rheumatoid arthritis, interleukin-17, interleukin-17 inhibition, precision medicine
Citation: Robert M and Miossec P (2019) IL-17 in Rheumatoid Arthritis and Precision Medicine: From Synovitis Expression to Circulating Bioactive Levels. Front. Med. 5:364. doi: 10.3389/fmed.2018.00364
Received: 16 November 2018; Accepted: 24 December 2018;
Published: 14 January 2019.
Edited by:
João Eurico Fonseca, Universidade de Lisboa, PortugalReviewed by:
Alexandre Sepriano, Leiden University Medical Center, NetherlandsCopyright © 2019 Robert and Miossec. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pierre Miossec, cGllcnJlLm1pb3NzZWNAdW5pdi1seW9uMS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.