 Jennifer A. Bridge
Jennifer A. Bridge James C. Lee
James C. Lee Adil Daud2
Adil Daud2 James W. Wells
James W. Wells Jeffrey A. Bluestone
Jeffrey A. Bluestone- 1Diabetes Center, University of California, San Francisco, San Francisco, CA, United States
- 2Helen Diller Family Comprehensive Cancer Center, UCSF, San Francisco, CA, United States
- 3The Faculty of Medicine, The University of Queensland Diamantina Institute, The University of Queensland, Translational Research Institute, Brisbane, QLD, Australia
- 4Sean N. Parker Autoimmune Research Laboratory, Diabetes Center, University of California, San Francisco, San Francisco, CA, United States
Immunotherapy for skin malignancies has ushered in a new era for cancer treatments by demonstrating unprecedented durable responses in the setting of metastatic Melanoma. Consequently, checkpoint inhibitors are now the first-line treatment of metastatic melanoma and widely used as adjuvant therapy for stage III disease. With the observation that higher tumor mutational burden correlates with a better response, checkpoint inhibitors are tested in other skin cancer types of known high tumor mutational burden with promising results and recently became the first-ever FDA-approved treatment for metastatic Merkel cell carcinoma. The emerging new standards-of-care will necessitate more precise biomarkers and predictors for treatment response and immune-related adverse events. Measurable immune-related mediators are currently under investigation as factors that promote or block the response to cancer immunotherapy and may provide insights into the underlying immune response to the tumor. Cytokines and chemokines are such mediators and are crucial for facilitating the recruitment and activation of specific subsets of leukocytes within the microenvironment of skin cancers. The exact mechanisms of how these meditators, both immunological and non-immunological, operate in the tumor microenvironment is an area of active research, so to reliable biomarkers of responses to cancer immunotherapy. Here, we will review and summarize the expanding body of literature for immune-related biomarkers pertaining to Melanoma, Basal cell carcinoma, Squamous cell carcinoma, and Merkel cell carcinoma, highlighting clinically relevant checkpoint inhibitor therapy biomarker advancements.
Introduction
In recent years, the field of cutaneous oncology has been invigorated by novel therapies that modulate the immune system, and shifted away from conventional chemotherapy, radiation, and targeted therapy. Immune checkpoint inhibitors (CPIs) are at the center of this new treatment paradigm, providing unprecedented rates of response in Malignant Melanoma (MM), Basal cell carcinoma (BCC), Squamous cell carcinoma (SCC) and metastatic Markel cell carcinoma (mMCC). The most impressive clinical difference of immune-based therapies in comparison with targeted therapy is the durability, while the majority of the complete responder treated with CPIs still cancer free (>60%) (1) vs. <30% at 4 years treated with dabrafenib plus trametinib, a regimen for BRAF-mutated patients (2). However, despite the promising results of CPI treatment in several cancers, only a subset of patients show long-term response, and CPI-based therapies can result in severe and potentially life-threating side effects termed immune-related adverse events (irAEs). The overall incidence rate of irAEs has been reported occur in up to 90% of patients treated with anti-CTLA-4 (3) and 70% of patients treated with PD-1 or PD-L1 antibodies (4, 5), with serious toxicities (> grade 3) needing treatment discontinuation for anti-PD-1 antibody therapy, anti-CTLA-4 antibody therapy, and combination therapy at 21, 28, and 59%, respectively, in large phase III trial analyses(6, 7). It is still unclear of the mechanisms of irAEs and why some patients develop them, nor why certain CPIs cause certain irAEs or their effect on patient outcomes. Hence, predictive biomarkers that can aid in the more precise delivery of immunotherapy to patients are urgently needed. Thus, far, few biomarkers have been established that can predict treatment success and positive clinical outcomes for patients. Mutations in tumor cells are thought to be a mechanistically relevant and an important initial event for the generation of neoantigens that contribute to the initial anti-tumor immune response (8–12). Tumor mutational burden (TMB) became one of the first analytic tools to have a significant correlation with response rate and prognostication (10, 11, 13). Programmed death-ligand 1(PD-L1) is a transmembrane protein, capable of being expressed by most tissues, that plays a major role in suppressing the immune system via engagement of programmed death protein 1 (PD-1) expressed on activated T cells. The inflammatory cytokines produced following T cell activation results in the expression of PD-L1 in the surrounding tissue and promote selective immune tolerance. In the tumor microenvironment, PD-L1 can be overexpressed by the tumor cells and antigen-presenting cells (APCs), in turn blocking the appropriate T cell immune responses required for tumor rejection. PD-L1 expression is currently the only U.S. FDA approved, commercially available predictive tool that helps identify patients who are likely to benefit from therapy that blocks the programmed death protein 1 (PD-1)/PD-L1 axis pathway (14). Both biomarkers serve to estimate the statistical likelihood of treatment success, but seldom are either used in practice as the major clinical decision factor for which to deploy CPIs for a patient, perhaps due to their relatively low positive predictive value and undefined or sometimes controversial correlation with overall survival (15, 16). As more sophisticated means of patient immune monitoring, both on and off therapy, are being developed, other potential biomarkers are being investigated. In addition, it is worth noting that although a number of reviews have addressed the importance of chemokines and cytokines in skin cancer progression and metastasis (17–23), fewer, if any, have addressed their potential as biomarkers for treatment outcomes in skin malignances. Here, we offer a brief overview of the current and emerging standard-of-care drugs used in cancer immunotherapy for metastatic skin cancers, and provide updates on immune-related biomarker advancements pertaining to MM, BCC, SCC, and mMCC, including TMB, PD-L1, novel immune activation/exhaustion markers, as well as relevant cytokines/chemokines, highlighting research that may be invaluable for the diagnostic, prognostic and predictive information helpful in establishing better clinical outcome.
Mechanism of Tumor Immune Escape and Clinical Impact of Immune Checkpoints in Metastatic Skin Cancer
CTLA-4 and PD-1 are both elements of a naturally evolved network of peripheral tolerance responsible for maintaining immune homeostasis and preventing overt autoimmunity. Early studies in animal models showed that cancers exploit these regulatory mechanisms, as checkpoint proteins are frequently over expressed in the tumor microenvironment. CTLA-4 is upregulated during T cell activation and eventually outcompetes costimulatory molecule CD28 for CD86 and CD80 expressed on antigen-presenting cells, halting further activation (24–26). CD86 and CD80 are known to be up-regulated at sites of inflammation and are capable of being removed from the cell surface through process known as trans-endocytosis by CTLA-4-expressing cells as a method also of block CD28 co-stimulation (27). In addition, CTLA-4 engagement enhances Treg suppressive function (28). Anti-CTLA-4 antibodies are capable of blocking and preventing this interaction, effectively shifting the balance back toward T cell activation (3, 4, 29). Pre-clinical and clinical data show CTLA-4 blockade results in the activation of both CD4 and CD8 effector cells in favor of anti-tumor immunity. The effect on the suppressive capacity of regulatory T cells in the tumor microenvironment remains controversial and is an area of active investigation (30–32). While the CTLA-4 pathway appears to regulate anti-tumor immunity in the draining lymph nodes, the PD-1/PD-L1 axis appear to take place in the local tumor microenvironment (33–35), PD-1 is expressed highly on T cells following repeated activation and chronic stimulation (36). As opposed to the draining lymph nodes, the PD-1 ligands PD-L1 and PD-L2 are more widely expressed and upregulated at effector sites of immune responses such as at inflamed tissues or the tumor microenvironment itself where they function to suppress T cell responses (36). The importance of interferon signaling in regulating the expression of PD-L1 and PD-L2, suggests that their expression patterns in advanced cancer and following CPI treatment may reflect a mechanism of primary or acquired resistance (34, 37). PD-1 and PD-L1 interaction inhibits T cell proliferation, survival, and effector function including cytokine release and tumor-targeted killing and can promote regulatory T cells differentiation (36, 38–41). Blocking this interaction via anti-PD-1 or anti-PD-L1 monoclonal antibodies restores the T cell from its “exhausted” phenotype, improves local T cells proliferation and effector function, and ultimately results in anti-tumor immunity (34).
The tumor microenvironment is a key location where tumor cells and the host immune system interact. When a cancer-associated antigen triggers an initial immune response, a series of tumor microenvironment modifications occur and impact the fate of antitumor immunity (42). The mutation-derived neoantigens are presented by the patients' major histocompatibility complex (MHC) as “foreign” antigens, recognized as such by the patients' immune system. The priming and activation of these cancer-reactive T cells is thought to occur in draining lymph nodes, generating helper, and cytotoxic T cells which traffic into the tumor, exert effector functions, and carry with them the potential to reject the tumor (43–45). At this phase, however, the pro-inflammatory environment generated by the inciting immune response favors the upregulation of reactive “shut-down” mechanisms that function to restore immune homeostasis, likely evolved to prevent aberrant immune reactions and the destruction of healthy host cells Figure 1.
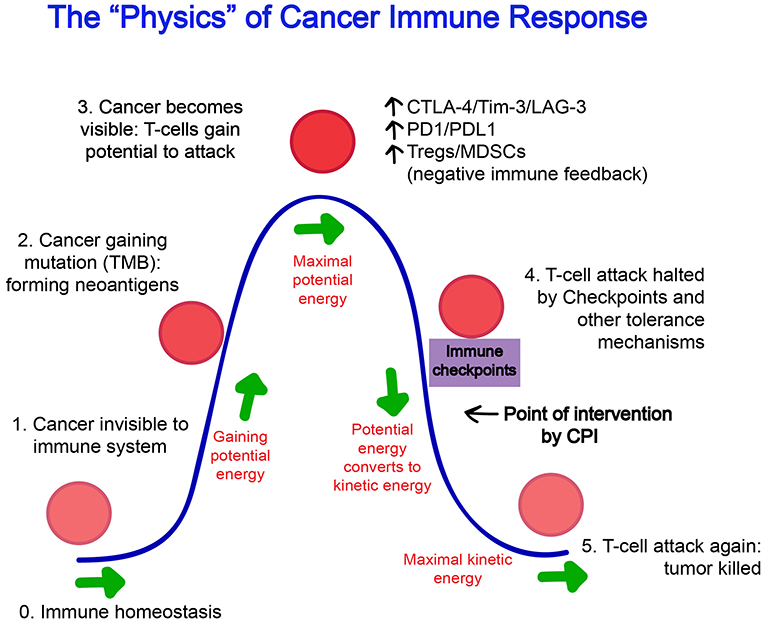
Figure 1. The “Physics” of Cancer Immune Response. (0) The steady state of the immune system at homeostasis is without effector cell activation and inflammation. (1) At its inception, a cancer cell may be invisible to the immune system and not trigger any response. (2) As cancer cells gain mutations over time, protein products foreign to the host are formed and these neoantigens increasingly gain recognition by the immune system. (3) “Potential energy” increases for the immune system to act, and reaches a threshold maximum where an immune attack on the cancer cells begins. (4) Naturally evolved feedback mechanisms such as Tregs, CTLA-4, and PD-1 attempt to restore the immune system back to homeostasis and halts ongoing immune response, however, unlike infectious elements that have been cleared, the immune suppression is often premature as antigens generated from cancer cells persist with the growing tumor. This is the point of intervention where agents such as checkpoint inhibitors are thought to exert its effect. (5) After successful blockade of immune checkpoints, the “kinetic energy” of the immune system resumes and can reach its maximum and tumor can be fully eradicated.
These checkpoint and tolerance mechanisms often prevent successful tumor eradication, but their presence and detection in the tumor microenvironment also provide evidence that an initial immune recognition has occurred and a battle between immune cells and tumor has begun. This is the scenario in which immunotherapy can help offset the balance toward tumor killing. This principle is behind the development of PD-L1 testing as a companion predictive biomarker for PD-1/PD-L1 based CPI treatment and is currently the only commercially available FDA approved predictive biomarker for cancer immunotherapy (section Tumor mutational burden, PD-L1, and other tumor microenvironment-associated biomarkers for checkpoint inhibitor treatment; Table 1). PD-L1 expression in pre-treatment tumor or immune cells is upregulated as a consequence of proinflammatory cytokine interferon-gamma (IFN-γ) released from T cells activation, presumably by tumor-associated antigens within the microenvironment (44). The presence of this immune response is correlated with a significantly higher chance that PD-1 based treatment will work in metastatic melanoma and a variety of other tumor types (46, 47).
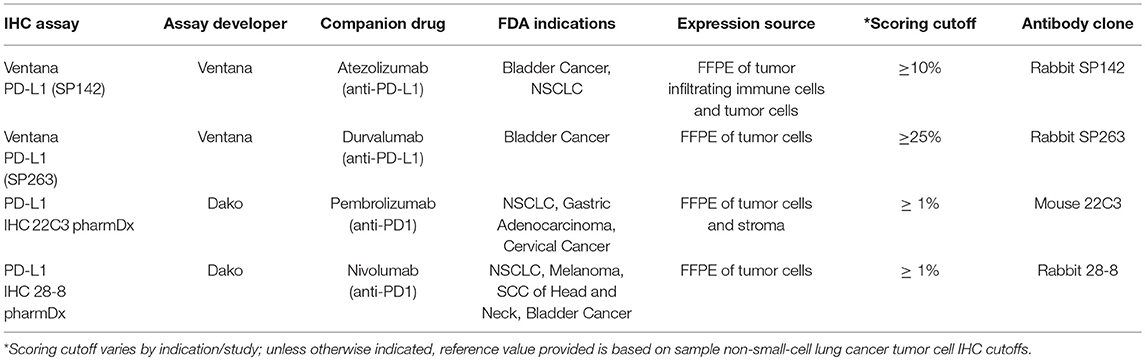
Table 1. Commercially available PD-L1 diagnostic tests.
For metastatic skin malignancies, the durable response rates from checkpoint blockade are unprecedented, with PD-1 inhibitor monotherapy approaching 40% and the hallmark combination approach of dual CTLA-4 and PD-1 blockade providing objective responses approaching 60% in patients with metastatic melanoma, a cohort with a historically dismal treatment response rate and prognosis (48). Furthermore, of the patients that achieve a complete response, >90% remain disease free even with monotherapy, with patients from the largest KEYNOTE-01 (NCT01295827) study still not reaching median treatment response duration at 5 years (49). Encouragingly, the response rates in non-melanoma skin cancers follow a similar trend, with avelamub (anti-PD-L1 antibody) now approved as first-line therapy for metastatic MCC, and cemiplimab (anti-PD-1 antibody) granted FDA priority review in 2018 for SCC, with clinical trials are underway for BCC (NCT03132636, NCT02690948). Despite these advancements, the observation that durable objective response can occur in these difficult to treat metastatic solid tumors with the blockade of checkpoint pathways comes with the frustration that treatment responses and their associated irAEs are highly variable and unpredictable. Nevertheless, there are several irAES with reported correlations to clinical response, notable examples include autoimmune skin depigmentation (vitiligo) and Type 1 Diabetes (8, 50–59). The precise mechanism of irAEs, whether they are driven solely by the reduction of T cell exhaustion and aberrant activation to autoantigens via CPI treatments or by changes in the epigenetic transcriptional control in the effector T cells, is currently unknown (52). As detailed below, research efforts are rapidly underway to help uncover the determinants of anti-tumor response for immunotherapy and their associated predictive biomarkers.
Overview of Current and Emerging Standard-of-care Treatments in Metastatic Skin Cancers
Metastatic melanoma is associated with the greatest mortality of all the skin malignancies and is often considered one of the deadliest metastatic cancers. Prior to the US FDA approval of the CPI ipilimumab in 2011, even with multi-modality systemic therapy, including surgery, radiation, and chemotherapy, the prognosis was grim, with an estimated 5-year survival of <20% and median survival of <1 year (60, 61). Fortunately, the standard-of-care has been shifting over the last decade, as a direct consequence of the success of CPIs. Drugs that block the PD-1 pathway (nivolumab, pembrolizumab, etc.) have been shown to provide higher response rates with a comparable durability of response and less toxicity than the cytotoxic T-lymphocyte associated protein 4 (CTLA-4) blocker ipilimumab (49, 62–64). Consequently, PD-1 blocking CPIs, with or without CTLA-4 blockade (ipilimumab) are now the preferred US FDA approved first-line therapy (Table 2). Chemotherapy, surgery, radiation, MEK inhibitors, and BRAF-mutation targeted therapy rarely produce durable responses, as mentioned above (2). They are options reserved for cases where rapid debulking and early response is necessary, often due to cancer impingement on vital structures (i.e., large brain metastasis), as they are able to achieve a faster, albeit often temporary, regression of large masses (NCCN guidelines version 3.2018). Older generation immunotherapy drugs such as systemic high-dose interleukin-2 (IL-2) or interferon alpha (IFNα) and bio-chemotherapy combinations (decarbonize, cisplatin, vinblastine, IL-2, and IFNα) are rarely recommended due to low efficacy and high toxicity, even in the adjuvant or second-line settings. While we await updated metastatic melanoma survival data in the age of modern immunotherapy, CPIs are expected to significantly improve and thus transform the standard-of-care and outlook for patients with this devastating cancer.
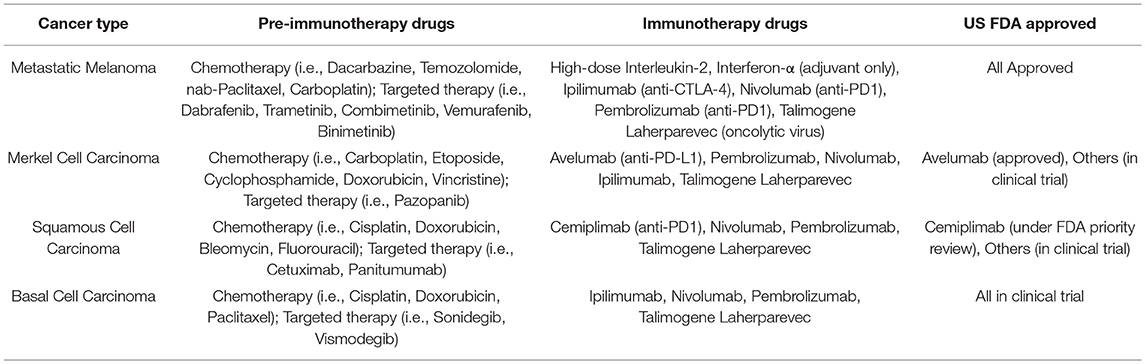
Table 2. Current status of immunotherapy drugs in metastatic skin cancer.
BCC, SCC, and mMCC comprise the vast majority of non-melanoma skin cancers. Like melanoma, at the early stage, local and surgical therapies are often curative and systemic therapies are only considered in settings where the tumor is unresectable or has metastasized. When disseminated, these cancers are comparable in mortality to metastatic melanoma and are notoriously difficult to treat. Similarly, the paradigm for their systemic treatment has shifted away from conventional cytotoxic modalities, which are often ineffective, to CPIs. In 2017, the FDA accelerated approval avelumab, a PD-L1 blocker, as the first-ever and only FDA approved drug for the aggressive metastatic MCC highlighting this trend (Table 2). For metastatic BCC and SCC, currently approved targeted therapies such as hedgehog pathway inhibitors vismodegib and sonidegib for BCC and epidermal growth factor receptor pathway inhibitors (cetuxmimab, pantimumumab, gefitinib, erlotinib) for SCC, although with proven response rates, face the challenge primary or acquired resistance in their respective pathways and have response durations that are often short-lived (65–69). Due to their shared feature of high tumor mutational burden (70–72), PD-1 blockade has demonstrated significant numbers of durable responses for SCC and BCC (73–76) and may likely be considered as a new standard-of-care for these indications.
As the standard-of-care for these different types of metastatic skin cancers converge on CPIs, the treatment decision process of BCC, SCC, and MCCs will likely share the clinical experience gained from MM. Some major challenges remain in the clinical deployment of CPIs for metastatic skin cancers: (1) The response rate for PD-1 inhibitor monotherapy tops out at around 40%, and while in a recent phase 3 trial combination therapy with PD-1 and CTLA-4 blockade found the response rate closer to 60%, it carries a significantly more serious toxicity profile leading to the frequent need for early treatment discontinuation (6, 48, 63, 77). (2) The general unpredictability of responses/toxicities of CPIs makes monitoring challenging and the determinants on how to shift the balance toward more response and less toxicity are largely unknown. (3) What happens if there is a relapse? Are you able to re-start CPI treatment again and get a response? Does resistance to one CPI treatment pathway infer resistance to all? (4) When to stop treatment? The high cost associated with CPIs makes it challenging to continue treatment indefinitely (78), and despite some reports of prolonged efficacy after discontinuation, the optimal time to stop treatment to ensure sustained complete response is currently unknown (49). In practice, in responding patients, many clinicians continue treatment until complete response plus at minimum 1 year after, but there have been no randomized clinical trials designed to address this important question. These and other considerations necessitate the interest and resources dedicated to better the mechanistic understanding of how CPIs work as well as develop biomarkers for treatment so that we can more rationally and efficiently deploy them. Ideally, we can improve upon current overall response to CPIs while lessening toxicity, but until then, predictive biomarkers could help target PD-1 monotherapy to those who have a higher probability of response and help identify those for whom a more aggressive combination of immunotherapy is needed.
Tumor Mutational Burden, PD-L1, and Other Tumor Microenvironment-associated Biomarkers for Checkpoint Inhibitor Treatment
TMB is a mechanistically rational biomarker for CPI treatment efficacy. As the skin is the first line of host defense against environmental assaults, providing the physical barrier against the mutagenic effects of UV radiation, invasion of viral pathogens, and colonization of commensal bacteria, it stands to reason that this organ system and cancers that arise from it should accumulate higher antigenic mutational burden for T cell detection (79, 80). Indeed, all metastatic skin cancers have demonstrated significant responses to CPI treatment (9), and TMB analysis has shown that melanoma, SCC, BCC, and MCC, typically harbor mutational and antigenic loads at the higher end of the mutational spectrum (8, 9, 70–72). Over time, the environmental assaults on the skin accumulate mutations and result in a “tug-of-war” between oncogenic driver mutations that create cancerous lesions and immunogenic passenger mutations that may then cause its regression from immune recognition (81–83). In theory, high tumor mutational burden provides a source of tumor-associated peptides that become neoantigens which trigger the initial differential T cell recognition of cancer cells from normal cells and provide the requisite anti-tumor immune response that allows patients to respond to immune- based treatments (79, 84). Clinical evidence is supportive for TMB as a biomarker of checkpoint response, as there is a correlation with the number of mutations and the chance such a mutation will provide a productive immunogenic antigen. In addition, clinical observations correlating cancers with high tumor mutational load with a better checkpoint blockade response raised the initial possibility of an association (10, 11). This hypothesis was further validated after reports that DNA mismatch-repair deficiency associated cancers (microsatellite instability-high) harbor some of the highest known tumor mutational burdens. Patients with this class of tumors have one of the highest response rates to PD-1 inhibitors (85, 86). These findings eventually led to accelerated FDA approval in 2017 for all cancers that are microsatellite instability-high.
PD-L1 is currently a U.S. FDA approved biomarker for PD-1 based CPI therapies. In general, patients with higher PD-L1 levels by immunohistochemistry have higher chances of responding to PD-1 based CPI therapy (87, 88). However, the predictive value for response is more accurate for patients with lung cancer than for skin cancers such as melanoma (89, 90), as durable responses to PD-1 CPI treatment has been observed in some melanoma patients with low expression of PD-L1. In terms of correlation to overall survival, interestingly, the majority of prior reports (before the wide usage of immunotherapy) suggest that high expression of PD-L1 correlates with shorter survival, even in settings of a high tumor mutational burden (16, 91–94). The paradoxical relationship between lower historical overall survival and higher PD-1 therapy response rate can be reconciled with the explanation that tumors with high tumor mutational burden generally leads to higher local expression of PD-L1, because more T cells sensitive to the tumor are likely to be present to be activated. On the other hand, highly mutated tumors would also have a higher potential to evolve driver mutations or pathways for adaptive resistance (81, 95–97) (Figure 1), therefore bypassing mechanisms of tumor rejection. Thus, while the value of PD-L1 as a biomarker for predicting better PD-1 therapy response is established, its use as a predictor for overall survival remains controversial and is to be determined in the era of immunotherapy. Neither the use of TMB nor PD-L1 is perfect, and the use of the combination of TMB and PD-L1 to identify likely responders to CPIs may be costly. Until more effective biomarkers are available, one clinical strategy for their use in skin cancers that has demonstrated some success is to utilize TMB and PD-L1 together as negative instead of positive predictors of response- when a low TMB and PD-L1 immune signature is present, this indicates that stronger immunotherapy such as PD-1 and CTLA-4 combination therapy should be utilized (98).
As more sophisticated means of patient immune monitoring both on and off therapy are being developed, other potential biomarkers are being investigated. For example, various other T cell activation and exhaustion markers including inducible T cell costimulatory (ICOS), 4-1BB (CD137), PD-1, CTLA-4, lymphocyte activation gene 3 (LAG-3), T cell immunoglobulin mucin-3 (Tim-3) and CD39, whether in circulation or within the tumor microenvironment, are now being correlated with response rates (46, 99–104) These, and other immune activation related biomarkers are biological surrogate markers that collectively signify an active immune response within the tumor microenvironment for which drugs such as CPIs can potentially take advantage, and may add to the predictive value and reliability of tumor mutational burden and PD-L1 expression (Figure 1). For biomarkers that are not directly associated with immune activation, serum lactate dehydrogenase level assessments have traditionally been made in the clinic as a surrogate indicator of disease progression and poor prognosis in melanoma, based on the rationale that lactate dehydrogenase is released upon cell damage and death associated with elevated tumor burden. Low baseline levels further demonstrate a favorable response correlation with CPI usage (105). Additionally, several novel non-immune activation related biomarkers of CPI response have been proposed, and include total tumor volume (101, 106), the presence of liver metastasis (98, 107), MHC protein expression (108), and the composition of the gut microbiome (109–112). Total tumor volume and liver metastasis assessment offer the benefit of cost-effectiveness and easier accessibility in clinical use, as they can be readily identified with standard-of-care imaging. Both high tumor volume and liver metastasis have independently been reported to result in unfavorable alterations in systemic T cell profiles (98, 101), suggesting yet-to-be discovered mechanisms of tumor immune regulation. Gut microbiota composition of certain bacterial strains and overall microbiome diversity appear to correlate with response rates (110–112), but larger and more geographical and demographically diverse studies are likely needed before conclusions can be finalized to influence clinical practice. Finally, the accumulation of the variety of rationally developed biomarkers may eventually contribute to patient-individualized “immunoscoringm,” a promising precision-medicine strategy that may be immunotherapy-focused and disease-agnostic, and effectively allow for constant updates and dynamic improvements in the fast-paced predictive biomarker field (113).
Cytokines and Chemokines as Prognostic Indicators for Skin Cancer
Cytokines and chemokines influence a complex network of regulation regarding immune cell function and trafficking. They can be divided readily into pro-inflammatory, anti-inflammatory, mitogenic, and chemotactic subgroups and are capable of contributing to particular functions dependent on their microenvironment. In addition to these properties, cytokines and chemokines and their receptors play a part in biological functions relevant to oncogenesis, including tumor cell proliferation, protease induction, and angiogenesis. Thus, cytokines and chemokines can facilitate immune-mediated tumor rejection or promote tumor progression as well as metastasis. These dichotomous associations and their underlying mechanism have become an area of active research not only for biomarker development but also for novel therapeutic targeting. Assessment of cytokine and chemokine profiles within the tumor microenvironment or in circulation, may greatly increase the resolution of our understanding of the status of the immune response to the tumor. The ability to tease apart the delicate balance of effector vs. regulatory elements in the tumor microenvironment will be essential to patient therapeutic design targeting a variety of critical aspects of the immune axis that could be dysregulated in cancer disease progression.
A number of chemokines and cytokines have been investigated for their roles in skin malignancies. Pro-inflammatory cytokines including TNF-α, Interleukin-16 (IL-16), IL-17, IL-21, IL-22, and IL-23 have all been associated with tumorigenesis and an inflammatory environment. Cytokines like IFN-γ and IL-12 are cytotoxic T cell related cytokines associated with mounting a cytolytic immune response. Their anti-tumor response is well-characterized in limiting skin cancer development (114–117). Immune-modulating cytokines including IL-10 and TGF-β are reported to oppose anti-tumor response since they are made predominantly by T regulatory cells to suppress cytotoxic responses against the skin malignancies (114, 118–120). However, like all cytokines, their roles are context-specific. For example despite circulating IL-10 levels have been associated with poor patient prognosis in a number of cancer settings, yet IL-10 expressing CD8 T cells confer a positive prognosis in patients with lung cancer (121–123). Chemokines, similarly, have long been associated with the recruitment of cells into the skin and can be sub-grouped as homeostatic or pro-inflammatory chemokines. Homeostatic chemokines are expressed constitutively and play key roles in normal leukocyte development and trafficking, pro-inflammatory chemokines are inducible and are responsible for trafficking to inflammation associated events (124). The chemokine receptor CXCR3 is expressed on inflammatory infiltrates and mediates attraction to the IFN-γ-inducible chemokines CXCL9 (or MIG), CXCL10 (or IP-10), and CXCL11(I-TAC) into the skin (125–129). These pathways of recruitment are also capable of regulating immune cell differentiation and activation, favoring Th1 and IFN-γ-production, respectively. The IFN-γ/CXCL9, CXCL-10, CXCL-11/CXCR3 axis can be a double-edged sword in cancer, promoting an anti-tumor effect but also increasing the capability of cancer cell proliferation, angiogenesis and metastasis (125). CXCL1 (melanoma growth-stimulatory activity/growth-regulatory protein α) and CXCL8 (IL-8) have important roles in inflammation, angiogenesis, tumorigenesis, and wound healing (130). However, these pathways can also be hijacked in cancer, promoting inflammation, inducing angiogenesis, tumor growth, and metastasis (131). For melanoma the expression of chemokine receptors CCR10, CXCR4, and CCR7 have been attributed to tumor escape and preference for metastasis (132). A number of reviews have addressed the importance of chemokines in skin cancer growth and metastasis (17–22, 133). Fewer, if any, have addressed their potential as biomarkers for prognosis or treatment outcomes.
Cytokines and Chemokines as Biomarkers for Checkpoint Inhibitor Treatment in Malignant Skin Cancers
Although limited, a number of groups that have studied cytokines and chemokines as biomarkers for predicting clinical outcomes to CPI therapies for skin malignancies. The prototypical T cell growth factor, IL-2, and the major anti-inflammatory cytokine, IL-10, have yielded surprisingly little correlation with actual clinical response rates, possibly due to the transient and locally expressed nature of these cytokines (88, 134). Available research, with many focused on patients treated with CTLA-4 inhibitors that were approved much earlier, with their main findings and predicted clinical outcomes are summarized in Table 3 and discussed below.
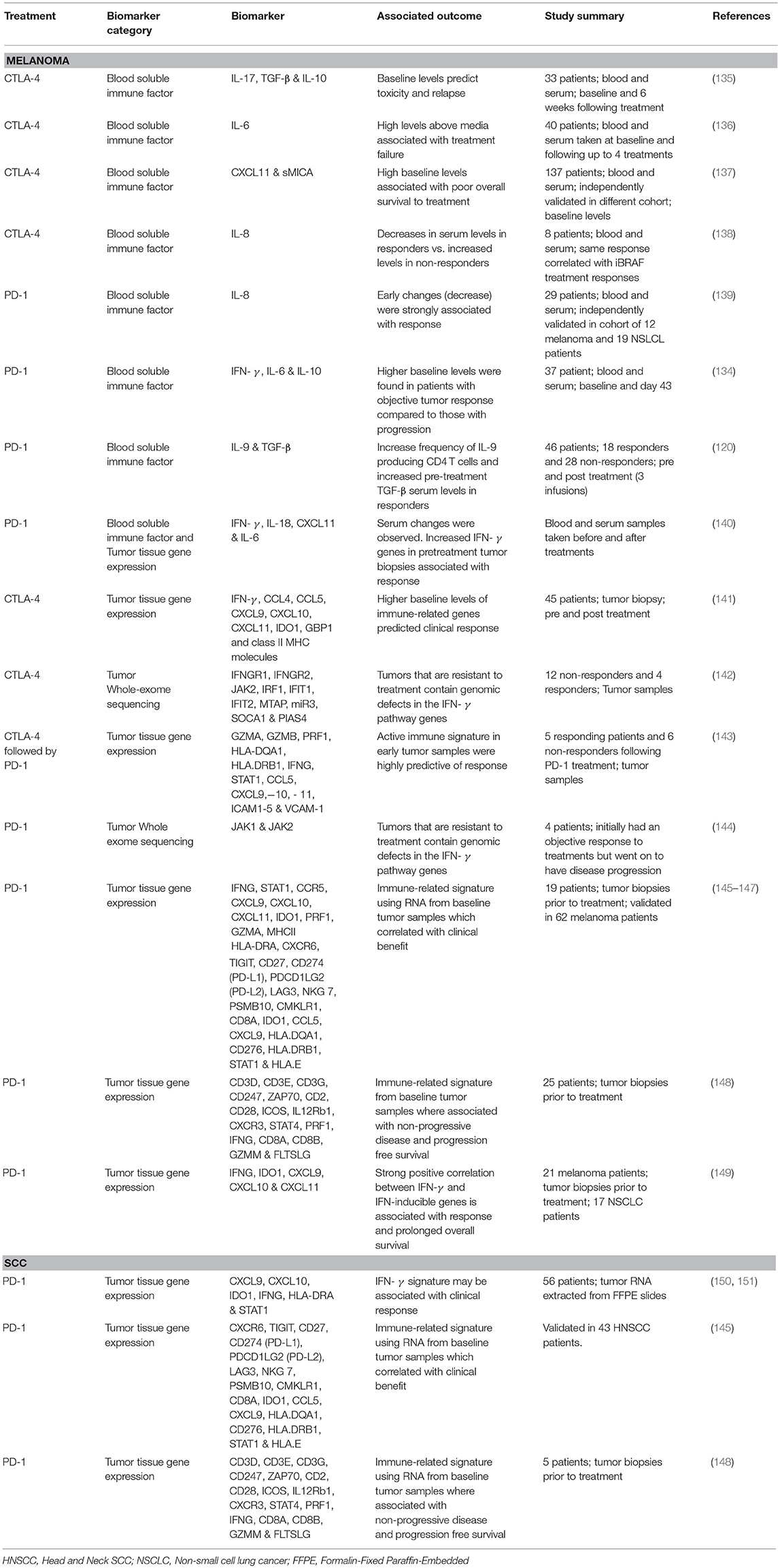
Table 3. Chemokine & Cytokine biomarkers investigated for CPI treatment outcomes.
TGF-β and IFN-γ
Tarhini et al. (135) evaluated circulating serum levels of cytokines, chemokines and a number of growth factors in response to CTLA-4 therapy (ipilimumab) finding a strong correlation between baseline levels of IL-17 and irAEs, and the combination of TGF-β and IL-10 baseline levels were predictive of relapse against therapy (135). Interesting, TGF-β has also been proposed as a serum marker of response to PD-1 (nivolumab) therapy in a cohort of MM patients by Nonomura et al. (120), with significantly increased pre-treatment levels, but not post-treatment levels, reported between responders and non-responders (120). The group found increased levels of newly characterized CD4 population, Th9 cells, that are generated in the presence of TGF-β and IL-4, suggesting that the blockade of PD-1/PD-L1 axis promoted Th9 cell differentiation, which in turn suppressed melanoma progression and increases cytotoxic activities of CD8 T cells (120). Although conflicting, the presence of TGF-β is reflective of a reduced immune response and therefore more likely to represent resistance to CPI treatments (152–154). In melanoma TGF-β production is positively correlated relative to disease progression, acting as a tumor promoter rather than a suppressor, and negatively regulating the activity of T cells by blocking IL-2 production (119).
The importance of an immune-active tumor microenvironment in clinical responses for CPI treatment has been shown in a number of melanoma patient cohorts. Early work performed Herbst et al. (140) to understanding predictive correlates of response to PD-1 therapy (MPD3280A) examined blood-based biomarkers. However, despite increases in IL-18, CXCL11 (ITAC), and IFN-γ found during the initial stages of treatment, there was no correlation to patients' long-term response and outcomes (140). Examining tumor tissue gene expression, however, they did find a number of genes associated with enhanced T-effector cell activity in pre-treatment responsive tumors compared to non-responsive tumors, including IFN-γ, IDO-1, and CXCL9 (140). In 2012, Ji et al. reported a phase II clinical trial in which 45 melanoma patients were treated with CTLA-4 (ipilimumab), and treatment was found to induce IFN-γ-inducible genes IDO1, GBP1, and class II MHC molecules and a number of Th1-associated marker genes IFN-γ, CCL4, CCL5, CXCL9, CXCL10, and CXCL11 in patients with a clinical response (141). The importance of IFN-γ gene signatures for response has been published similarly by Ribas et al. (146) in a test cohort of 19 melanoma patients treated with PD-1. Comparing the gene signature of responders vs. non-responders revealed a number of top-ranking genes that were strongly associated with IFN-γ signaling and correlation with IFN-γ expression. Their preliminary IFN-γ gene signature included IFNG, STAT1, CCR5, CXCL9, CXCL10, CXCL11, IDO1, PRF1, GZMA, and MHCII HLA-DRA and an expanded immune signature of genes related to cytolytic activity (e.g., granzyme A/B/K and PRF1), cytokines and chemokines (e.g., CXCR6, CXCL9, CCL5, and CCR5), T cell markers (e.g., CD3D, CD3E, CD2, IL2RG), NK cell activity (e.g., NKG &, HLA-E), antigen presentation (e.g., CIITA, HLA-DRA) and other immunomodulatory factors (e.g., LAG3, IDO1, and SLAMF6) were able to differentiate between objective responses and non-responders. Additionally, the group was able to further refine their IFN-γ and expanded immune gene signatures within another cohort of 62 melanoma patients (145–147), in which the baseline immune-related tumor samples correlated with clinical benefit from treatment. The importance of activated Th1 and cytotoxic T cells in mediating CPI mediated tumor responses irrespective of the tissue environment was also used to test and further refine the gene signature, with the group finding these signatures capable of associating with treatment response to pembrolizumab in HNSCC and gastric cancer. Furthermore, the final T cell-inflamed gene expression profile consisting of 18 genes, (CXCR6, TIGIT, CD27, CD274 (PD-L1), PDCD1LG2 (PD-L2), LAG3, NKG 7, PSMB10, CMKLR1, CD8A, IDO1, CCL5, CXCL9, HLA.DQA1, CD276, HLA.DRB1, STAT1, and HLA.E), was tested against 9 tumor types (KEYNOTE-012; bladder, gastric, HNSCC, and triple-negative breast cancer, KEYNOTE-028; anal canal, biliary, colorectal, esophageal, and ovarian cancer) with most of the gene signature positive association with response. The data present by Ayers et al. (145) highlights the importance of the T cell-inflamed microenvironment common to patients who respond to anti-PD-1 CPI treatment regardless of the tumor type or tissue type and also proposes a difference between resistance mechanisms between non-responding patients that lack T cell inflammation and those who have infiltration but lack clinical response (145).
Chen et al. (143) followed a unique set of melanoma patients that were initially treated with CTLA-4 followed by PD-1 at progression, identifying a unique immune signature in responders not evident in non-responding patients significant after 2 to 3 cycles of treatment (143). Within the 411 significantly differentially expressed genes of responders where a number of cytolytic makers (i.e., GZMA, GZMB, and PRF1), HLA molecules (e.g., HLA-DQA1 HLA.DRB1) IFN-γ pathway genes (e.g., IFNG, STAT1), chemokines (e.g., CCL5, CXCL9,−10, and 11) and select adhesion molecules (i.e., ICAM1-5 and VCAM-1). The dataset also allow for the comparison of pretreatment samples and after treatment fold changes clearly differentiating responders from non-responders, regardless of the pretreatment with CTLA-4. These finding clearly support the assessment of early immune responses can be highly predictive of overall response to therapy. However, it should also be noted that these on treatment changes maybe associated solely with treatment and not associated with a mechanism of response. In additional Chen et al. also highlight an angiogenic phenotype in non-responding patients (decreased VEGFA), downregulation of antigen processing and presentation (HLA genes) and defects in the interferon signaling pathways, all of which are consistent with a number of previously published works (142, 143, 155–159).
Prat et al. (148) evaluated pre- and post-PD-1 (pembrolizumab or nivolumab) treated tumor samples from a range of cancers, including melanoma patients. Twenty three immune related genes or signatures where identified to be associated with non-progressive disease and progression free survival, these included genes associated with the formation of the TCR complex (e.g., CD3D, CD3E, CD3G, CD247, and ZAP70), co-stimulatory factors (e.g., CD2, CD28, and ICOS) and genes involved in differentiation (e.g., IL12Rb1, CXCR3, and STAT4). In addition to apoptosis pathway genes involving granzyme A, B and perforin 1, T cell receptor signaling (e.g., CD8A, CD8B, and IFN-γ), cell adhesion molecules (e.g., CD4, CD86, and integrinβ2), a number of toll-like receptors (TLR1,4, 7, and 8) and checkpoint molecules PD-1, PDL-2, and LAG3. These signatures were found across all the different cancer types assessed and 12 signatures tracking immune cells including CD8 T cell associated genes PRF1, CD8A, CD8B, GZMM, FLTSLG, CD4 T cell activated genes IL26 and IL17A as well as NK cell and B cell genes and PDL-1, PD1, and CTLA4 and IFN-signaling pathway activation (148). These findings are in support of the Ribbas et al. (146), Ayer et al. (147), and Ayer et al. (145) T cell and IFN activation predicting response to the PD-1/PD-L1 treatment across multiple tumor types and tumors with strong pre-treatment immunity.
Gao et al. (142) used whole-exome sequencing of tumor tissues to show that tumors that are non-responsive to CTLA-4 (ipilimumab) therapy have defects in IFN-γ pathway via significantly higher somatic mutations including copy-number variations and single-nucleotide variants in IFN-γ signaling genes including IFNGR1, IFNGR2, JAK2, IRF1, IFIT1, IFIT2, MTAP, and miR31 and amplification of IFN-γ suppressor genes (SOCA1 and PIAS4) compared to responders (142). Similarly, Zaretsky et al. (144) published a similar finding in melanoma patients treated with anti-PD-1 antibody (Pembrolizumab) finding that loss of function mutations within the IFN receptor associated Janus kinase 1 (JAK1) or Janus kinase 2 (JAK2) lead to lack of response to IFN-γ leading to acquired resistance to PD-1 blockade (144). However, both cohorts of patients were small, 16 and 4, respectively, and require validation in larger cohorts of patients and treatments. Similar observations have been previously published by Tumeh et al. (45) in a MM patient cohort treated with anti-PD-1 (pembrolizumab), with baseline and post-dosing biopsies for phospho-STAT1 (pSTAT1), an immediate downstream effector upon IFN-γ binding (45). Proposing that the PD-L1 tumor expression can be linked to interferon production within the tumor microenvironment via T-cell recognition, patients that were found to have a response to therapy had significantly higher expression of pSTAT1 at baseline and during treatment compared to progression group (45). Highlighting again the importance of a pre-existing tumor immune response in patient outcomes.
Recently, a retrospective study conducted in melanoma samples found that significantly longer progression-free survival and overall survival were observed in patients with pre-treatment high IFN-γ expression (149). High pre-treatment expression of IFN-γ-inducible genes (e.g., IDO1, CXCL9, CXCL10, and CXCL11 among others) was associated with response and prolonged overall survival for anti-PD-1 (pembrolizumab) treatment of melanoma (149). In a cohort melanoma patients treated with anti-CTLA-4 (ipilimumab; NCT00094653) Koguchi et al. (137) found elevated pre-treatment serum levels of CXCL11 and a soluble MHC class I polypeptide-related chain (sMICA) to be strong predictors of poor survival benefit, this finding was confirmed in an independent cohort of melanoma patients treated with ipilimumab (137). This finding is in contrast with tumor tissue samples that favor CXCL11 as a biomarker of T cell infiltration and therefore a favorable prognostic marker, as mentioned above. CXCL11 also has distinct immunoregulatory functions through the higher affinity to its receptor CXCR3 (125). CXCL11 is also capable of binding to CXCR7, more commonly associated with tumor growth, making its functional role controversial (125, 137, 149). In addition, the contrast in findings between protein in the serum to mRNA levels in the tissue microenvironment suggests that parallel analysis of both serum and tissue, and protein and mRNA, maybe required further clarify these differences (137).
Despite strong preclinical support of checkpoint inhibitor for the treatment of non-melanoma skin cancers, little is still known about blocking the PD-1/PD-L1 and CTLA-4 pathways in patients. A number of single case studies have been published on the use CPI in the treatment of these cancers, however due to patient numbers investigation into biomarkers for predicting treatment response has been limited. Chow et al. (150) were the first to clinically assess the use of PD-1 and PD-L1 CPIs in head and neck SCC. Keynote-012 stratified patients to assess the response of PD-1 (pembrolizumab) on PD-L1-positive head and neck SCC patients (150, 151). Using the identified gene signature (CXCL9, CXCL10, IDO1, IFNG, HLA-DRA, and STAT1) that was identified in the melanoma cohort and validated in a head and neck SCC cohort (146, 147), all six of the IFN-γ-related genes were statistically higher in responding patients compared to non-responders. However, it should be noted that the study only included PD-L1 positive patients. Further work is needed to elucidate the interaction with PD-L1 and an IFN-γ gene signature (151). Despite these initial findings, little has been published regarding biomarker discovery, outside of HPV status and PD-L1 expression (150, 160–163). Tumor tissue gene expression of similar IFN-γ gene signatures identified in independent melanoma cohorts has also been correlated with clinical benefit from anti-PD-1 treatment in a small number of head and neck SCC patient cohorts (145, 148).
The use of CPI therapies for the treatment of BCC are currently under clinical trial. A few promising case studies have been published recently (73, 76, 164) with treated efforts based predominately on the success of CPIs in similar tumors with high mutational burdens, and from the finding that ~90% of BCCs stain positively for PD-L1 expression (165). Recently, Lipson et al. (75) correlated the expression of PD-L1 in the tumor microenvironment with predictability for objective responses in PD-1-pathway-directed therapies (75). It has also been established that BCCs undergoing spontaneous regression (presumably immune-mediated) contain elevated levels of Th1 cytokines (e.g., IFN-γ) and infiltrating activated T cells (166, 167). Additionally, treatments that have focused on interferon therapy have shown efficacy in BCC (168). MCC is a rare but aggressive skin cancer, with several published studies finding expressed PD-L1 in the tumor and also PD-1 expression on tumor-infiltrating lymphocytes (159, 169–172). These observations provide a strong rationale for assessing CPIs on patients with advanced disease that has few therapies to extend survival. Current clinical trial by Nghiem and colleagues treated patients with PD-1 (pembrolizumab), response to treatment did not correlate with PD-L1 expression, so too in both virus negative and positive cancers (172).
Although strongly biased by melanoma patient data, there is strong evidence supporting the importance of an active immune microenvironment for successful CPI therapy outcomes in metastatic skin cancer. IFN-γ production by activated T cells is capable of multitude of downstream affects, including activation of dendritic cells and macrophages (like, STAT1 and CMKLR1), which in turn produce their own chemokines and cytokines (like, CCL5, CXCL9,-10,-11) to further recruit CD8 T cells. Upregulation of co-stimulatory molecules (CD27) and effector molecules (IFN-γ, perforin, and granzymes) further contribute to the immune response. IFN-γ induces upregulation of HLA molecules and other pathway associated with antigen processing and presenting. IFN-γ also drives upregulation of PD-L1 and PD-L2 on the surface of macrophages, DCs and tumor cells. Other checkpoint molecules like IDO1, TIGIT and LAG3 are similarly associated with T cell activation and IFN-γ signaling to help restrain antitumor responses (145). The use of gene expression signature highlights the complexity within the immune response associated with CPI treatment and response and suggests the importance of multiple cell types within the tumor microenvironment. These gene expression profiles provide an interesting insight into the immune responses to CPI therapy and highlight a number of possible targets that can be manipulated for better therapeutic outcomes.
IL-6 and IL-8
IL-6 is a pleiotropic cytokine that is associated similarly with disease pathogenesis and metastasis and has already been published as a serum marker for melanoma patients. IL-6 has also been linked to IL-10 production within the melanoma tumor microenvironment. For head and neck SCC, BCC, and mMCC high IL-6 levels are also associated with a poor prognosis and treatment resistance (173). The expression of PD-L1 has also been linked to IL-6 and IL-10 cytokines, which may explain their role in tumor progression and as a marker of non-response. Bjoern et al. (136) found MM patient cohort following CTLA-4 treatment (ipilimumab) who had lower serum baseline levels and lower levels at the 4th dose of pro-inflammatory cytokine IL-6 responded better to therapy (136). This observation of serum IL-6 as a poor prognostic biomarker for MM immunotherapy responses has been previously suggested in IL-2-based and bio-chemotherapy therapies (174, 175). Pre-treatment serum levels of IL-6, IFN-γ, and IL-10 were found to be significantly higher in patients with objective tumor responses in a cohort of phase 2 advanced melanoma patients treated with PD-1 (nivolumab) in comparison to non-responders with progressive disease (134). The increased cytokine levels were also positively correlated with each other, suggesting both a spontaneous activation and suppressive response at the same time (134). In addition, the group found increases in CXCL9 and CXCL10 between pre-treatment and post-treatment (Day 43) serum samples in response to treatment, postulated to be associated with IFN-γ production by activated T cells in the blood (134).
IL-8 serum levels have been proposed to reflect tumor burden and decreased levels in a small cohort of MM patients during treatment with CTLA-4 (ipilimumab) correlated with patient benefit from treatment (138). Sanmamed et al. found that serum IL-8 levels are a consequence of tumor burden in a number of cancer types including melanoma and could be monitored to predict response to BRAF inhibitors and correlated to overall survival (138). In addition, the authors recently published that changes in serum IL-8 levels reflect tumor response to PD-1 treatment (nivolumab or pembrolizumab) in MM patients in both identification cohort and independent validation cohort (139). Decreases in IL-8 serum levels were found at patient's best response, additionally, they were able to distinguish pseudo-progression (decreased) and non-responders (increased) and also monitor responses via IL-8 serum fluxes (139). IL-8 has been similarly implicated as a biomarker for head and neck SCC patients receiving chemoradiation with or without novel hypoxic cytotoxins (176). A recently published case report describing a recurrent head and neck SCC patient on PD-1 (nivolumab) treatment found increased levels of IL-8 and IL-6 following treatment (from pre-treatment levels) and decreased levels of IL-10 and CXC3C1, the patient was found to have progressive disease (177), this cytokine and chemokine profile is constant previously published work from a cohort of melanoma and non-small-cell lung cancer patients (139). The role of IL-8 and its receptor CXCR2 in tumor development and progression has been well-documented in a wide range cancer cells including melanoma, SCC, BCC, and MCC (18, 146, 178–182). IL-8, and its receptor CXCR2, are poised to be examined as potential biomarkers in both BCC and mMCC treated with CPIs. The induction of pro-inflammatory cytokines like IL-6 and IL-8, appear strong indicators of unsuccessful CPI therapy and highlight the need for biomarkers for patients who will not respond to treatment as well as for those who will.
Predictive Biomarkers for Immune-Related Adverse Events
As stated above, checkpoint inhibitor therapy can be associated with severe or even life-threatening irAEs. Generally, irAEs occur within weeks to 3 months after initiation of treatment but have been documented to occur months after discontinuation of treatment. Fatigue is the most common irAE reported following treatment with either anti-CTLA-4 and anti-PD1/anti-PDL-1 antibodies and can range up to 50% (183). For PD-1 based therapy, the next most common (10–20%) are grade 1-2 skin rash, transaminitis, arthralgia, colitis, low-grade fevers, thyroiditis, and other endocrinopathies. For CTLA-4 based therapies, the next most common (20–30%) are grade 1–2 colitis, anemia, transaminitis, skin rash, arthralgia, and low-grade fevers(184). Grade 3–4 irAEs are rare for PD-1 based therapies but are more common in CTLA-4 based or combination CTLA-4 plus PD-1 therapies, and are mostly commonly colitis, transaminitis, and endocrinopathies. In addition, there are also a number of rare (<1%) immune-related adverse events that have also been reported following CPI treatment including Type I Diabetes and systemic lupus erythematosus (50, 185, 186). The causes and mechanisms of these various irAEs are an area of active investigation. We need to have predictive biomarkers for therapy response to maximize benefit as well as predictive biomarkers for irAEs to minimize risk, however, biomarkers for irAEs have been less vigorously investigated than biomarkers that predict therapy response. Nevertheless, as more and more immunotherapy are being deployed, some predictive immune signals are beginning to surface. For example, increased overall white blood cell count and eosinophil count with decreased relative lymphocyte count have been associated with higher grade irAEs (187, 188). Increased T-cell repertoire diversity is associated with more irAEs in patients treated with ipilimumab (189, 190). A post combination anti-CTLA-4 and anti-PD-1 therapy reduction in total peripheral B cells with an enrichment of CD21lo PD-1+ memory B cells correlated with irAE development (191). Increases in circulating autoantibodies against self-antigens and mRNA gene expression signatures of immune activation have been correlated with impending irAEs (192–195). Finally, as mentioned, IL-17 levels may be associated with gastrointestinal toxicities (135). As most studies are small and not repeated in large-scale clinical trials, more work is needed before effective predictors of irAEs are clinically available.
Novel Technologies and Assays Under Investigation
Biomarker assays that require the smallest amounts of accessible samples, for instance, blood and other bodily fluids from patient clinical samples, will inherently be less invasive and more likely to be implemented in a clinical setting. Blood biomarkers such as circulating tumor DNA may allow for more meaningfully analysis of patients tumor response with smaller blood volumes in comparison to approaches capturing circulating tumor cells and have been recently reviewed (196, 197). These approaches have the potential to overcome the tumor heterogeneity and sample quantity procurement limitations of tumor biopsies. A number of biotech companies including Grail, Freenome, and Guardant Health are utilizing this approach to create assay platforms that improve early cancer detection and immunotherapy responses for better outcomes. In addition, recent advancements in imaging are allowing for non-invasive evaluation of the tumor immune-microenvironment and have been recently well-reviewed (198). Using PET scan in conjunction with antibodies or antibody fragments labeled with PET-based radionuclides, scanners have the potential to detect T-cell subsets and effector molecules within tumors or lymphoid tissues and non-invasively monitor changes within the tumor microenvironment throughout the immunotherapy treatment process. Technologies such as this may help clinicians to better distinguish patients with true disease progression, which requires the timely transition to alternative treatments, from pseudoprogression, a phenomenon characterized by the transient increase in tumor size followed by a decrease, where immunotherapy should be continued.
Discussion
Immunotherapies such as CPIs are demonstrating unprecedented response rates across all major types of metastatic skin cancers. Unlike traditional cancer treatment modalities, the durability of response seen in CPIs enabled clinician and patients alike to consider the possibility of cures in a historically highly resistant and fatal group of cancers. Building upon that success, efforts are underway to further improve response rates and more precisely deliver treatment to patients who are most likely to respond while monitoring treatment outcomes in a timely fashion. These efforts will likely serve to ameliorate the problem of high cost and unpredictability of cancer immunotherapy. This review attempts to summarize the tools currently available for clinical practice as well as technologies in emerging areas of discovery. As combination immunotherapy and associated biomarkers gain sophistication, efforts to prioritize the tools available and to standardize practice methods will require dedicated large studies. It is established that immune impairment is common among cancer patients, with the entity of metastatic skin cancer every so close to finally achieving improved clinical outcomes for its patients using immunotherapy, the mechanistic understanding of the tumor microenvironment and lessons learned from improving this field will hopefully benefit other disease entities as well.
Author Contributions
JBr and JL contributed to the writing, figure design, and literature review. AD, JW, and JBl provided expert opinion and critical revision of the review. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
The research was also supported by the Parker Institute for Cancer Immunotherapy and the Sean N. Parker Autoimmune Research Laboratory. JBr funded by Jeffrey G. Klein Family Fellowship in diabetes award. JL funded by A.P. Giannini Foundation Fellowship Award and Parker Institute for Cancer Immunotherapy (A130504). AD research grant from Genentech, Novartis, Pfizer, Merck, BMS, Incyte; advisory board for Roche, Novartis, OncoSec. This work was supported by Cancer Council Queensland project grants APP1069748 and APP1128447, National Health and Medical Research Council project grant APP1099999, and a Perpetual Trustees Fellowship to JW. JBl is supported by grants awarded by the Parker Institute for Cancer Immunotherapy (A130504 & A132273).
Conflict of Interest Statement
JB is CEO and President of Parker Institute for Cancer Immunotherapy; reports receiving commercial research funding from Juno/Celgene; has ownership interest in Celsius Therapeutics, Rheos Medicines, Arcus Biosciences, Solid Biosciences, Vir Biotechnology, and Quentis Therapeutics; and is a consultant/advisory board member for Quentis Therapeutics, Vir Biotechnology, Solid Biosciences, Arcus Biosciences, Rheos Medicines, Celsius Therapeutics, Pfizer, and Merck.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Long GV, Schachter J, Ribas A, Arance AM, Grob J-J, Mortier L, et al. 4-year survival and outcomes after cessation of pembrolizumab (pembro) after 2-years in patients (pts) with ipilimumab (ipi)-naive advanced melanoma in KEYNOTE-006. J Clin Oncol. (2018) 36(15 Suppl.):9503. doi: 10.1200/JCO.2018.36.15_suppl.9503
2. Long GV, Eroglu Z, Infante J, Patel S, Daud A, Johnson DB, et al. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma who received dabrafenib combined with trametinib. J Clin Oncol. 36:667–73 (2018) doi: 10.1200/JCO.2017.74.1025
3. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
4. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
5. Brahmer JR, Tykodi SS, Chow LQM, Hwu W-J, Topalian SL, Hwu P, et al. Safety and activity of anti–PD-L1 Antibody in patients with advanced cancer. N Engl J Med. (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
6. Schadendorf D, Wolchok JD, Stephen Hodi F, Chiarion-Sileni V, Gonzalez R, Rutkowski P, et al. Efficacy and safety outcomes in patients with advanced melanoma who discontinued treatment with nivolumab and ipilimumab because of adverse events: A pooled analysis of randomized phase II and III trials. J Clin Oncol. (2017) 35:3807–14. doi: 10.1200/JCO.2017.73.2289
7. Shoushtari AN, Friedman CF, Navid-Azarbaijani P, Postow MA, Callahan MK, Momtaz P, et al. Measuring toxic effects and time to treatment failure for nivolumab plus ipilimumab in melanoma. JAMA Oncol. (2018) 4:98–101. doi: 10.1001/jamaoncol.2017.2391
8. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. (2017) 16:2598–609. doi: 10.1158/1535-7163.MCT-17-0386
9. Jaffee E, Hopkins A, Yarchoan M. Tumor mutational burden and response rate to PD-1 Inhibition. N Engl J Med. (2017) 377:2500–1. doi: 10.1056/NEJMc1713444
10. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med. (2014) 371:2189–99. doi: 10.1056/NEJMoa1406498
11. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
12. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell (2015) 160:48–61. doi: 10.1016/j.cell.2014.12.033
13. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (2015) 350:207–11. doi: 10.1126/science.aad0095
14. Hersom M, Jørgensen JT. Companion and complementary diagnostics–focus on PD-L1 expression assays for PD-1/PD-L1 checkpoint inhibitors in NSCLC. Ther Drug Monit. (2017) 40:1. doi: 10.1097/FTD.0000000000000460
15. Mathew M, Safyan RA, Shu CA. PD-L1 as a biomarker in NSCLC: challenges and future directions. Ann Transl Med. (2017) 5:375. doi: 10.21037/atm.2017.08.04
16. Wang Q, Liu F, Liu L. Prognostic significance of PD-L1 in solid tumor: an updated meta-analysis. Medicine (2017) 96:e6369. doi: 10.1097/MD.0000000000006369
17. Sarvaiya PJ, Guo D, Ulasov I, Gabikian P, Lesniak MS. Chemokines in tumor progression and metastasis. Oncotarget (2013) 4:2171–85. doi: 10.18632/oncotarget.1426
18. Singh R, Lilladr JW, Singh S. Chemokines: key players in cancer progression and metastasis. Front Biosci. (2011) 3:1569–82.
19. Chow MT, Luster AD. Chemokines in cancer. Cancer Immunol Res. (2014) 2:1125–31. doi: 10.1158/2326-6066.CIR-14-0160
20. Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol. (2002) 118:915–22. doi: 10.1046/j.1523-1747.2002.01725.x
21. Richmond A, Yang J, Su Y. The good and the bad of chemokines/chemokine receptors in melanoma. Pigment Cell Melanoma Res. (2009) 22:175–86. doi: 10.1111/j.1755-148X.2009.00554.x
23. Triozzi PL, Fernandez AP. The role of the immune response in merkel cell carcinoma. Cancers (2013) 5:234–54. doi: 10.3390/cancers5010234
24. Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. (1996) 183:2541–50.
25. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 Blockade. Science (1996) 271:1734–36.
26. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity (1994) 1:405–13. doi: 10.1016/1074-7613(94)90071-X
27. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science (80- ). (2011)
28. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 System of T cell costimulation. Annu Rev Immunol. (1996) 14:233–58. doi: 10.1146/annurev.immunol.14.1.233
29. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252. doi: 10.1038/nrc3239
30. Maker AV, Attia P, Rosenberg SA. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J Immunol. (2005) 175:7746–54. doi: 10.4049/jimmunol.175.11.7746
31. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 Antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. (2013) 1:32–42. doi: 10.1158/2326-6066.CIR-13-0013
32. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell. (2018) 33:649–63.e4. doi: 10.1016/j.ccell.2018.02.010
33. Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar S, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. (2017) 18:e143–52. doi: 10.1016/S1470-2045(17)30074-8
34. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060.
35. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/COC.0000000000000239.
36. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the Pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
37. Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. (2017) doi: 10.1016/j.celrep.2017.04.031
38. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM Motif-carrying immunoreceptor. Immunity (1999) 11:141–51.
39. Okazaki T, Honjo T. PD-1 and PD-1 ligands: from discovery to clinical application. Int Immunol. (2007) 19:813–24. doi: 10.1093/intimm/dxm057
40. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
41. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. (2006) 203:883–95. doi: 10.1084/jem.20051776
42. Chen DS. Mellman oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
43. Spitzer MH, Gherardini PF, Fragiadakis GK, Bhattacharya N, Yuan RT, Hotson AN, et al. IMMUNOLOGY. An interactive reference framework for modeling a dynamic immune system. Science (2015) 349:1259425. doi: 10.1126/science.1259425
44. Blank CU, Haanen JB, Ribas A, Schumacher TN. The "cancer immunogram”. Science (2016) 352:658–60. doi: 10.1126/science.aaf2834
45. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568. doi: 10.1038/nature13954
46. Daud AI, Wolchok JD, Robert C, Hwu WJ, Weber JS, Ribas A, et al. Programmed death-ligand 1 expression and response to the anti-programmed death 1 antibody pembrolizumab in melanoma. J Clin Oncol. (2016) 34:4102–9. doi: 10.1200/JCO.2016.67.2477
47. Khunger M, Hernandez AV., Pasupuleti V, Rakshit S, Pennell NA, Stevenson J, et al. Programmed Cell Death 1 (PD-1) Ligand (PD-L1) Expression in Solid Tumors As a Predictive Biomarker of Benefit From PD-1/PD-L1 Axis Inhibitors: a systematic review and meta-analysis. JCO Precis Oncol. (2017) 1:1–15. doi: 10.1200/PO.16.00030
48. Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. (2017) 377:1345–56. doi: 10.1056/NEJMoa1709684
49. Robert C, Ribas A, Hamid O, Daud A, Wolchok JD, Joshua AM, et al. Durable complete response after discontinuation of pembrolizumab in patients with metastatic melanoma. J Clin Oncol. (2018) 36:1668-1674. doi: 10.1200/JCO.2017.75.6270
50. Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, et al. Collateral damage: insulin-dependent diabetes induced with checkpoint inhibitors. Diabetes (2018) 67:1471–80. doi: 10.2337/dbi18-0002
51. Byrne EH, Fisher DE. Immune and molecular correlates in melanoma treated with immune checkpoint blockade. Cancer (2017) 123:2143–53. doi: 10.1002/cncr.30444
52. June CH, Warshauer JT, Bluestone JA. Is autoimmunity the Achilles' heel of cancer immunotherapy? Nature Med. (2017) 23:540–7. doi: 10.1038/nm.4321
53. Clotman K, Janssens K, Specenier P, Weets I, De Block CEM. Programmed cell death-1 (PD-1) inhibitor induced type 1 diabetes mellitus: mini-review. J Clin Endocrinol Metab. (2018) 103:3144–54. doi: 10.1210/jc.2018-00728
54. Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, et al. Ipilimumab (Anti-CTLA4 Antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother (2007) 30:825–30. doi: 10.1097/CJI.0b013e318156e47e
55. Hua C, Boussemart L, Mateus C, Routier E, Boutros C, Cazenave H, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. (2016) 152:45–51. doi: 10.1001/jamadermatol.2015.2707
56. Boasberg PD, Hoon DSB, Piro LD, Martin MA, Fujimoto A, Kristedja TS, et al. Enhanced survival associated with vitiligo expression during maintenance biotherapy for metastatic melanoma. J Invest Dermatol. (2006) 126:2658–63. doi: 10.1038/sj.jid.5700545
57. Gogas H, Ioannovich J, Dafni U, Stavropoulou-Giokas C, Frangia K, Tsoutsos D, et al. Prognostic Significance of Autoimmunity during Treatment of Melanoma with interferon. N Engl J Med. (2006) 354:709–18. doi: 10.1007/s00281-011-0247-y
58. Rosenberg SA, White DE. Vitiligo in patients with melanoma: normal tissue antigens can be targets for cancer immunotherapy. J Immunother. (1996) 19:81–4. doi: 10.1097/00002371-199601000-00009
59. Sibaud V, David I, Lamant L, Resseguier S, Radut R, Attal J, et al. Acute skin reaction suggestive of pembrolizumab-induced radiosensitization. Melanoma Res. (2015) 25:555–8. doi: 10.1097/CMR.0000000000000191
60. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. (2009) 27:6199–206. doi: 10.1200/JCO.2009.23.4799
61. Garbe C, Eigentler TK, Keilholz U, Hauschild A, Kirkwood JM. Systematic review of medical treatment in melanoma: current status and future prospects. Oncologis (2011) 16:5–24. doi: 10.1634/theoncologist.2010-0190
62. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol. (2015) 33:4015–22. doi: 10.1200/JCO.2015.62.3397
63. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. (2015) 373:23–34. doi: 10.1056/NEJMoa1504030
64. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. (2015) 33:1974–82. doi: 10.1200/JCO.2014.59.4358
65. Chang ALS, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. (2012) 148:1324–5. doi: 10.1001/archdermatol.2012.2354
66. Atwood SX, Sarin KY, Whitson RJ, Li JR, Kim G, Rezaee M, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. (2015) 27:342–53. doi: 10.1016/j.ccell.2015.02.002
67. Maubec E, Petrow P, Scheer-Senyarich I, Duvillard P, Lacroix L, Gelly J, et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J Clin Oncol. (2011) 29:3419–26. doi: 10.1200/JCO.2010.34.1735
68. William WN, Feng L, Ferrarotto R, Ginsberg L, Kies M, Lippman S, et al. Gefitinib for patients with incurable cutaneous squamous cell carcinoma: a single-arm phase II clinical trial. J Am Acad Dermatol. (2017) 77:1110–3. doi: 10.1016/j.jaad.2017.07.048
69. Gold KA, Kies MS, William WN, Johnson FM, Lee JJ, Glisson BS. Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: a single-arm phase 2 clinical trial. Cancer (2018) 124:2169–73. doi: 10.1002/cncr.31346
70. Pickering CR, Zhou JH, Lee JJ, Drummond JA, Peng SA, Saade RE, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. (2014) 20:6582–93. doi: 10.1158/1078-0432.CCR-14-1768
71. Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS. Mutational landscape of basal cell carcinomas by whole-exome sequencing. J Invest Dermatol. (2014) 134:213–20. doi: 10.1038/jid.2013.276
72. Goodman AM, Kato S, Cohen PR, Stephens PJ, Daniels GA, Kurzrock R. Genomic landscape of advanced basal cell carcinoma : Implications for precision treatment with targeted and immune therapies. (2018) 7:e1404217. doi: 10.1080/2162402X.2017.1404217
73. Falchook GS, Leidner R, Stankevich E, Piening B, Bifulco C, Lowy I, et al. Responses of metastatic basal cell and cutaneous squamous cell carcinomas to anti-PD1 monoclonal antibody REGN2810. J Immunother Cancer (2016) 4:70. doi: 10.1186/s40425-016-0176-3
74. Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD, et al. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med. (2018) 379:341–51. doi: 10.1056/NEJMoa1805131
75. Lipson EJ, Lilo MT, Ogurtsova A, Esandrio J, Xu H, Brothers P, et al. Basal cell carcinoma: PD-L1/PD-1 checkpoint expression and tumor regression after PD-1 blockade. J Immunother Cancer (2017) 5:23. doi: 10.1186/s40425-017-0228-3
76. Ikeda S, Goodman AM, Cohen PR, Jensen TJ, Ellison CK, Frampton G, et al. Metastatic basal cell carcinoma with amplification of PD-L1: exceptional response to anti-PD1 therapy. NPJ Genomic Med. (2016) 1:16037. doi: 10.1038/npjgenmed.2016.37
77. Callahan MK, Kluger H, Postow MA, Segal NH, Lesokhin A, Atkins MB, et al. Nivolumab plus ipilimumab in patients with advanced melanoma: Updated survival, response, and safety data in a phase i dose-escalation study. J Clin Oncol. (2018). 391–8. doi: 10.1200/JCO.2017.72.2850
78. Andrews A. Treating with checkpoint inhibitors-Figure $1 million per patient. Am Heal drug Benefits (2015) 8:9.
79. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature (2013) 500:415–21. doi: 10.1038/nature12477
80. Pfeifer GP, You Y-H, Besaratinia A. Mutations induced by ultraviolet light. Mutat Res. (2005) 571:19–31. doi: 10.1016/j.mrfmmm.2004.06.057
81. McFarland CD, Mirny LA, Korolev KS. Tug-of-war between driver and passenger mutations in cancer and other adaptive processes. Proc Natl Acad Sci USA. (2014) 111:15138–43. doi: 10.1073/pnas.1404341111
82. McFarland CD, Korolev KS, Kryukov GV, Sunyaev SR, Mirny LA. Impact of deleterious passenger mutations on cancer progression. Proc Natl Acad Sci USA. (2013) 110:2910–5. doi: 10.1073/pnas.1213968110
83. Vesely MD, Schreiber RD. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci. (2013) 1284:1–5. doi: 10.1111/nyas.12105
84. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Front Immunol. 8:1848. doi: 10.3389/fimmu.2017.01848
85. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. (2018) 36:773–9. doi: 10.1200/JCO.2017.76.9901
86. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
87. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer (2018) 17:129. doi: 10.1186/s12943-018-0864-3
88. Buder-Bakhaya K, Hassel JC. Biomarkers for clinical benefit of immune checkpoint inhibitor treatment-A review from the melanoma perspective and beyond. Front Immunol. (2018) 9:1474. doi: 10.3389/fimmu.2018.01474
89. Carbognin L, Pilotto S, Milella M, Vaccaro V, Brunelli M, Caliò A, et al. Differential activity of nivolumab, pembrolizumab and MPDL3280A according to the tumor expression of programmed death-ligand-1 (PD-L1): sensitivity analysis of trials in melanoma, lung and genitourinary cancers. PLoS ONE (2015) 10:1–16. doi: 10.1371/journal.pone.0130142
90. Hirsch FR, McElhinny A, Stanforth D, Ranger-Moore J, Jansson M, Kulangara K, et al. PD-L1 immunohistochemistry assays for lung cancer: results from phase 1 of the blueprint PD-L1 IHC assay comparison project. J Thorac Oncol. (2017) 12:208–22. doi: 10.1016/j.jtho.2016.11.2228
91. Xue S, Song G, Yu J. The prognostic significance of PD-L1 expression in patients with glioma: a meta-analysis. Sci Rep. (2017) 7:1–8. doi: 10.1038/s41598-017-04023-x
92. Gao Q, Wang XY, Qiu SJ, Yamato I, Sho M, Nakajima Y, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. (2009) 15:971–9. doi: 10.1158/1078-0432.CCR-08-1608
93. Mu CY, Huang JA, Chen Y, Chen C, Zhang XG. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med Oncol. (2011) 28:682–8. doi: 10.1007/s12032-010-9515-2
94. Osmani L, Askin F, Gabrielson E, Li QK. Current WHO guidelines and the critical role of immunohistochemical markers in the subclassification of non-small cell lung carcinoma (NSCLC): Moving from targeted therapy to immunotherapy. Semin Cancer Biol. (2017) 52(Pt 1):103–9. doi: 10.1016/j.semcancer.2017.11.019
95. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
96. Ribas A. Adaptive immune resistance: how cancer protects from immune attack. Cancer Discov. (2015) 5:915–9. doi: 10.1158/2159-8290.CD-15-0563
97. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to PD-1 blockade mediated by JAK mutations. Cancer Discov. (2016) 7:188–201. doi: 10.1158/2159-8290.CD-16-1223
98. Loo K, Tsai KK, Mahuron K, Liu J, Pauli ML, Sandoval PM, et al. Partially exhausted tumor-infiltrating lymphocytes predict response to combination immunotherapy. JCI Insight (2017) 2:93433. doi: 10.1172/jci.insight.93433
99. Daud AI, Loo K, Pauli ML, Sanchez-rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti – PD-1 therapy in human melanoma. J Clin Invest. (2016) 126:1–6. doi: 10.1172/JCI87324
100. Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. (2014) 211:715–25. doi: 10.1084/jem.20130590
101. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature (2017) 545:60–5. doi: 10.1038/nature22079
102. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
103. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discovery. (2014) 4:879–88. doi: 10.1158/2159-8290.CD-14-0341
104. Jacquelot N, Roberti MP, Enot DP, Rusakiewicz S, Ternès N, Jegou S, et al. Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat Commun. (2017) 8:592. doi: 10.1038/s41467-017-00608-2
105. Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res. (2016) 22:5487–96. doi: 10.1158/1078-0432.CCR-16-0127
106. Joseph RW, Elassaiss-Schaap J, Kefford RF, Hwu W-J, Wolchok JD, Joshua AM, et al. Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin Cancer Res. (2018) 24:4960–7.doi: 10.1158/1078-0432.CCR-17-2386
107. Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, et al. Liver metastasis and treatment outcome with Anti-PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res. (2017) 5:417–24. doi: 10.1158/2326-6066.CIR-16-0325
108. Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med. (2018) 10:eaar3342. doi: 10.1126/scitranslmed.aar3342
109. Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science (2018) 359:1366–70. doi: 10.1126/science.aar6918
110. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, et al. The commensal microbiome is associated with anti PD-1 efficacy in metastatic melanoma patients. Science (2018) 359:104–8. doi: 10.1126/science.aao3290
111. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science (2017) 359:97–103. doi: 10.1126/science.aan4236
112. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science (2017) 359:91–7. doi: 10.1126/science.aan3706.E
113. Pagès F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet (2018) 391:2128–39. doi: 10.1016/S0140-6736(18)30789-X
114. Tang L, Wang K. Chronic inflammation in skin malignancies. J Mol Signal. (2016) 11:2. doi: 10.5334/1750-2187-11-2
115. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
116. Nicholas C, Lesinski GB. Immunomodulatory cytokines as therapeutic agents for melanoma. Immunotherapy (2011) 3:673–90. doi: 10.2217/imt.11.45
117. Weiss JM, Subleski JJ, Wigginton JM, Wiltrout RH. Immunotherapy of cancer by IL-12-based cytokine combinations. Expert Opin Biol Ther. (2007) 7:1705–21. doi: 10.1517/14712598.7.11.1705
118. Teng MWL, Darcy PK, Smyth MJ. Stable IL-10: A new therapeutic that promotes tumor immunity. Cancer Cell (2011) 20:691–3. doi: 10.1016/j.ccr.2011.11.020
119. Perrot CY, Javelaud D, Mauviel A. Insights into the transforming growth factor-β signaling pathway in cutaneous melanoma. Ann Dermatol. (2013) 25:135–44. doi: 10.5021/ad.2013.25.2.135
120. Nonomura Y, Otsuka A, Nakashima C, Seidel JA, Kitoh A, Dainichi T, et al. Peripheral blood Th9 cells are a possible pharmacodynamic biomarker of nivolumab treatment efficacy in metastatic melanoma patients. Oncoimmunology (2016) 5:e1248327. doi: 10.1080/2162402X.2016.1248327
121. Miotto D, Cascio N Lo, Stendardo M, Querzoli P, Pedriali M, De Rosa E, et al. CD8+ T cells expressing IL-10 are associated with a favourable prognosis in lung cancer. Lung Cancer (2010) 69:355–60. doi: 10.1016/j.lungcan.2009.12.012
122. Mannino MH, Zhu Z, Xiao H, Bai Q, Wakefield MR, Fang Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. (2015) 367:103–7. doi: 10.1016/j.canlet.2015.07.009
123. Zhang H, Wang Y, Hwang ES, He YW. Interleukin-10: an immune-activating cytokine in cancer immunotherapy. J Clin Oncol. (2016) 34:3576–78. doi: 10.1200/JCO.2016.69.6435
124. Martins-Green M, Petreaca M, Wang L. Chemokines and their receptors are key players in the orchestra that regulates wound healing. Adv Wound Care (2013) 2:327–47. doi: 10.1089/wound.2012.0380
125. Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation A target for novel cancer therapy. Cancer Treat Rev. (2018) 63:40–7. doi: 10.1016/j.ctrv.2017.11.007
126. Yan Y, Cao S, Liu X, Harrington SM, Bindeman WE, Adjei AA, et al. CX3CR1 identifies PD-1 therapy–responsive CD8+ T cells that withstand chemotherapy during cancer chemoimmunotherapy. JCI Insight (2018) 3:97828. doi: 10.1172/jci.insight.97828
127. Jenkins MH, Brinckerhoff CE, Mullins DW. CXCR3 Signaling in BRAF(WT) Melanoma Increases IL-8 Expression and Tumorigenicity. Chen S, editor. PLoS ONE (2015) 10:e0121140. doi: 10.1371/journal.pone.0121140
128. Kawada K, Sonoshita M, Sakashita H, Takabayashi A, Yamaoka Y, Manabe T, et al. Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res. (2004) 64:4010–7. doi: 10.1158/0008-5472.CAN-03-1757
129. Monteagudo C, Martin JM, Jorda E, Llombart-Bosch A. CXCR3 chemokine receptor immunoreactivity in primary cutaneous malignant melanoma: Correlation with clinicopathological prognostic factors. J Clin Pathol. (2007) 60:596–9. doi: 10.1136/jcp.2005.032144
130. Dhawan P, Richmond A. Role of CXCL1 in tumorigenesis of melanoma. J Leukoc Biol. (2002) 72:9–18.
131. Liu Q, Li A, Tian Y, Wu JD, Liu Y, Li T, et al. The CXCL8-CXCR1/2 pathways in cancer. Cytokine Growth Factor Rev. (2016) 31:61–71. doi: 10.1016/j.cytogfr.2016.08.002
132. Müller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature (2001) 410:50. doi: 10.1038/35065016
133. Byrne SN, Halliday GM. UV-Induced Chemokines as Emerging Targets for Skin Cancer Photochemoprevention. In: Wondrak GT, editor Skin Stress Response Pathways: Environmental Factors and Molecular Opportunities, Cham: Springer International Publishing (2016), p. 211–34.
134. Yamazaki N, Kiyohara Y, Uhara H, Iizuka H, Uehara J, Otsuka F, et al. Cytokine biomarkers to predict antitumor responses to nivolumab suggested in a phase 2 study for advanced melanoma. Cancer Sci. (2017) 108:1022–31. doi: 10.1111/cas.13226
135. Tarhini AA, Zahoor H, Lin Y, Malhotra U, Sander C, Butterfield LH, et al. Baseline circulating IL-17 predicts toxicity while TGF-β1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J Immunother Cancer (2015) 3:39. doi: 10.1186/s40425-015-0081-1
136. Bjoern J, Juul Nitschke N, Zeeberg Iversen T, Schmidt H, Fode K, Svane IM. Immunological correlates of treatment and response in stage IV malignant melanoma patients treated with Ipilimumab. Oncoimmunology (2016) 5:e1100788. doi: 10.1080/2162402X.2015.1100788
137. Koguchi Y, Hoen HM, Bambina SA, Rynning MD, Fuerstenberg RK, Curti BD, et al. Serum immunoregulatory proteins as predictors of overall survival of metastatic melanoma patients treated with ipilimumab. Cancer Res. (2015) 75:5084–92. doi: 10.1158/0008-5472.CAN-15-2303
138. Sanmamed MF, Carranza-Rua O, Alfaro C, O-ate C, Martín-Algarra S, Perez G, et al. Serum interleukin-8 reflects tumor burden and treatment response across malignancies of multiple tissue origins. Clin Cancer Res. (2014) 20:5697–707. doi: 10.1158/1078-0432.CCR-13-3203
139. Sanmamed MF, Perez-Gracia JL, Schalper KA, Fusco JP, Gonzalez A, Rodriguez-Ruiz ME, et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann Oncol. (2017) 28:1988–95. doi: 10.1093/annonc/mdx190
140. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–7. doi: 10.1038/nature14011
141. Ji R-R, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. (2012) 61:1019–31. doi: 10.1007/s00262-011-1172-6
142. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell (2016) 167:397–404.e9. doi: 10.1016/j.cell.2016.08.069
143. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. (2016) 6:827–37. doi: 10.1158/2159-8290.CD-15-1545
144. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
145. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. (2017) 127:2930–40. doi: 10.1172/JCI91190
146. Ribas A, Robert C, Hodi FS, Wolchok JD, Joshua AM, Hwu WJ, et al. Association of response to programmed death receptor 1 (PD-1) blockade with pembrolizumab (MK-3475) with an interferon-inflammatory immune gene signature. J Clin Oncol. (2015) 33(Suppl. 15):3001. doi: 10.1200/jco.2015.33.15_suppl.3001
147. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Albright A, et al. Relationship between immune gene signatures and clinical response to PD-1 blockade with pembrolizumab (MK-3475) in patients with advanced solid tumors. J Immunother Cancer (2015) 3(Suppl. 2):P80. doi: 10.1186/2051-1426-3-S2-P80
148. Prat A, Navarro A, Paré L, Reguart N, Galván P, Pascual T, et al. Immune-related gene expression profiling after PD-1 blockade in non–small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res. (2017) 77:3540–50. doi: 10.1158/0008-5472.CAN-16-3556
149. Karachaliou N, Gonzalez-Cao M, Crespo G, Drozdowskyj A, Aldeguer E, Gimenez-Capitan A, et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther Adv Med Oncol. (2018) 10. doi: 10.1177/1758834017749748
150. Chow LQM, Mehra R, Haddad RI, Mahipal A, Weiss J, Berger R, et al. Biomarkers and response to pembrolizumab (pembro) in recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC). J Clin Oncol. (2016) 34(Suppl. 15):6010. doi: 10.1200/JCO.2016.34.15_suppl.6010
151. Seiwert TY, Burtness B, Mehra R, Weiss J, Berger R, Eder JP, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. (2016) 17:956–65. doi: 10.1016/S1470-2045(16)30066-3
152. O'Donnell JS, Long GV, Scolyer RA, Teng MWL, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev. (2017) 52:71–81. doi: 10.1016/j.ctrv.2016.11.007
153. Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP. Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res. (1999) 5:2963–70.
154. Hammer GE, Ma A. Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu Rev Immunol. (2013) 31:743–91. doi: 10.1146/annurev-immunol-020711-074929
155. Ferrara N, Gerber HP, LeCouter J, Kerbel RS. Angiogenesis as a therapeutic target. Nature (2003) 438:967–74. doi: 10.1038/nature04483
156. Yuan J, Zhou J, Dong Z, Tandon S, Kuk D, Panageas KS, et al. Pretreatment serum VEGF is associated with clinical response and overall survival in advanced melanoma patients treated with ipilimumab. Cancer Immunol Res. (2014) 2:127–32. doi: 10.1158/2326-6066.CIR-13-0163
157. Kageshita T, Hirai S, Ono T, Hicklin DJ, Ferrone S. Down-regulation of HLA Class I antigen-processing molecules in malignant melanoma: association with disease progression. Am J Pathol. (1999) 154:745–54 doi: 10.1016/S0002-9440(10)65321-7
158. Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class i HLA genes. Nat Biotechnol. (2015) 33, 1152–8. doi: 10.1038/nbt.3344
159. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape Sci Transl Med. (2012) 4:127ra37. doi: 10.1126/scitranslmed.3003689
160. Bauml J, Seiwert TY, Pfister DG, Worden F, Liu SV, Gilbert J, et al. Pembrolizumab for platinum- and cetuximab-refractory head and neck cancer: results from a single-Arm, phase II study. J Clin Oncol. (2017) 35:1542–9. doi: 10.1200/JCO.2016.70.1524
161. Segal NH, Ou S-HI, Balmanoukian AS, Fury MG, Massarelli E, Brahmer JR, et al. Safety and efficacy of MEDI4736, an anti-PD-L1 antibody, in patients from a squamous cell carcinoma of the head and neck (SCCHN) expansion cohort. J Clin Oncol. (2015) 33(Suppl. 15):3011. doi: 10.1200/jco.2015.33.15_suppl.3011
162. Ferris RL, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. (2016) 375:1856–67. doi: 10.1056/NEJMoa1602252
163. Saleh K, Eid R, Haddad FGH, Khalife-Saleh N, Kourie HR. New developments in the management of head and neck cancer – impact of pembrolizumab. Ther Clin Risk Manage. (2018) 14:295–303. doi: 10.2147/TCRM.S125059
164. Winkler JK, Schneiderbauer R, Bender C, Sedlaczek O, Fröhling S, Penzel R, et al. Anti-programmed cell death-1 therapy in nonmelanoma skin cancer. Br J Dermatol. (2016) 176:498–502. doi: 10.1111/bjd.14664
165. Ghafouri-Fard S, Ghafouri-Fard S. Immunotherapy in nonmelanoma skin cancer. Immunotherapy (2012) 4:499–510. doi: 10.2217/imt.12.29
166. Hunt MJ, Halliday GM, Weedon D, Cooke BE, Barnerson RS. Regression in basal cell carcinoma: an immunohistochemical analysis. Br J Dermatol. (2018) 130:1–8. doi: 10.1111/j.1365-2133.1994.tb06873.x
167. Wong DA, Bishop GA, Lowes MA, Cooke B, Barnetson RSC, Halliday GM. Cytokine profiles in spontaneously regressing basal cell carcinomas. Br J Dermatol. (2001) 143:91–8. doi: 10.1046/j.1365-2133.2000.03596.x
168. Fenton SE, Sosman JA, Chandra S. Current therapy for basal cell carcinoma and the potential role for immunotherapy with checkpoint inhibitors. Clin Ski Cancer (2017) 2:59–65. doi: 10.1016/j.clsc.2017.11.001
169. Afanasiev OK, Yelistratova L, Miller N, Nagase K, Paulson K, Iyer JG, et al. Merkel polyomavirus-specific T cells fluctuate with merkel cell carcinoma burden and express therapeutically targetable PD-1 and Tim-3 exhaustion markers. Clin Cancer Res. (2013) 19:5351–60. doi: 10.1158/1078-0432.CCR-13-0035
170. Lipson EJ, Vincent JG, Loyo M, Kagohara LT, Luber BS, Wang H, et al. PD-L1 expression in the merkel cell carcinoma microenvironment: association with inflammation, merkel cell polyomavirus, and overall survival. Cancer Immunol Res. (2013) 1:54–63. doi: 10.1158/2326-6066.CIR-13-0034
171. Dowlatshahi M, Huang V, Gehad AE, Jiang Y, Calarese A, Teague JE, et al. Tumor-specific T cells in human merkel cell carcinomas: a possible role for tregs and T-cell exhaustion in reducing T-cell responses. J Invest Dermatol. (2013) 133:1879–89. doi: 10.1038/jid.2013.75
172. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med. (2016) 374:2542–52. doi: 10.1056/NEJMoa1603702
173. Gao J, Zhao SEN, Halstensen TS. Increased interleukin-6 expression is associated with poor prognosis and acquired cisplatin resistance in head and neck squamous cell carcinoma. Oncol Rep. (2016) 35:3265–74. doi: 10.3892/or.2016.4765
174. Hoejberg L, Bastholt L, Johansen J, Jarle Christensen I, Gehl J, Schmidt H. Serum interleukin-6 as a prognostic biomarker in patients with metastatic melanoma. Melanoma Res. (2012) 22:287–93. doi: 10.1097/CMR.0b013e3283550aa5
175. Soubrane C, Rixe O, Meric JB, Khayat D, Mouawad R. Pretreatment serum interleukin-6 concentration as a prognostic factor of overall survival in metastatic malignant melanoma patients treated with biochemotherapy: a retrospective study. Melanoma Res. (2005) 15:199–204. doi: 10.1097/00008390-200506000-00009
176. Le QT, Fisher R, Oliner KS, Young RJ, Cao H, Kong C, et al. Prognostic and predictive significance of plasma HGF and IL-8 in a phase III trial of chemoradiation with or without tirapazamine in locoregionally advanced head and neck cancer. Clin Cancer Res. (2012) 18:1798–1807. doi: 10.1158/1078-0432.CCR-11-2094
177. Merhi M, Raza A, Inchakalody VP, Nashwan AJJ, Allahverdi N, Krishnankutty R, et al. Squamous cell carcinomas of the head and neck cancer response to programmed cell death protein-1 targeting and differential expression of immunological markers: a case report. Front Immunol. (2018) 9:1769. doi: 10.3389/fimmu.2018.01769
178. Singh S, Varney M, Singh RK. Host CXCR2-Dependent regulation of melanoma growth, angiogenesis, and experimental lung metastasis. Cancer Res. (2009) 69:411–5. doi: 10.1158/0008-5472
179. Yen HT, Chiang LC, Wen KH, Tsai CC, Yu CL, Yu HS. The expression of cytokines by an established basal cell carcinoma cell line (BCC-1/KMC) compared with cultured normal keratinocytes. Arch Dermatol Res. (1996) 288:157–61. doi: 10.1007/BF02505826
180. Aoki M, Pawankar R, Niimi Y, Kawana S. Mast cells in basal cell carcinoma express VEGF, IL-8 and RANTES. Int Arch Allergy Immunol. (2003) 130:216–23. doi: 10.1159/000069515
181. Cohen RF, Contrino J, Spiro JD, Mann EA, Chen LL, Kreutzer DL. Interleukin-8 expression by head and neck squamous cell carcinoma. Arch Otolaryngol Neck Surg. (1995) 121:202–9. doi: 10.1001/archotol.1995.01890020064013
182. Richards KF, Guastafierro A, Shuda M, Toptan T, Moore PS, Chang Y. Merkel cell polyomavirus T antigens promote cell proliferation and inflammatory cytokine gene expression. J Gen Virol. (2015) 96(Pt 12):3532–44. doi: 10.1099/jgv.0.000287
183. Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB. Current diagnosis and management of Immune Related Adverse Events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol. (2017) 8:49. doi: 10.3389/fphar.2017.00049
184. Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. (2018) 4: 36(17):1714-1768. doi: 10.1200/JCO.2017.77.6385
185. Byun DJ, Wolchok JD, Rosenberg LM, Girotra M. Cancer immunotherapy — immune checkpoint blockade and associated endocrinopathies. Nat Rev Endocrinol. (2017) 13:195–207. doi: 10.1038/nrendo.2016.205
186. Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer (2016) 54:139–48. doi: 10.1016/j.ejca.2015.11.016
187. Fujisawa Y, Yoshino K, Otsuka A, Funakoshi T, Fujimura T, Yamamoto Y, et al. Fluctuations in routine blood count might signal severe immune-related adverse events in melanoma patients treated with nivolumab. J Dermatol Sci. (2017) 88:225–31. doi: 10.1016/j.jdermsci.2017.07.007
188. Hopkins AM, Rowland A, Kichenadasse G, Wiese MD, Gurney H, McKinnon RA, et al. Predicting response and toxicity to immune checkpoint inhibitors using routinely available blood and clinical markers. Br J Cancer (2017) 117:913–20. doi: 10.1038/bjc.2017.274
189. Oh DY, Cham J, Zhang L, Fong G, Kwek SS, Klinger M, et al. Immune toxicities elicted by CTLA-4 blockade in cancer patients are associated with early diversification of the T-cell repertoire. Cancer Res. (2017) 77:1322–30. doi: 10.1158/0008-5472.CAN-16-2324
190. Fong L, Oh DY, Cham J, Zhang L, Fong G, Kwek SS, et al. T cell repertoire diversification is associated with immune related toxicities following CTLA-4 blockade in cancer patients. Cancer Res. (2016) 77:1322–30. doi: 10.1158/0008-5472.CAN-16-2324
191. Das R, Bar N, Ferreira M, Newman AM, Zhang L, Bailur JK, et al. Early B cell changes predict autoimmunity following combination immune checkpoint blockad. J Clin Invest. (2018) 128:715–20. doi: 10.1172/JCI96798
192. Shahabi V, Berman D, Chasalow SD, Wang L, Tsuchihashi Z, Hu B, et al. Gene expression profiling of whole blood in ipilimumab-treated patients for identification of potential biomarkers of immune-related gastrointestinal adverse events. J Transl Med. (2013) 11:75. doi: 10.1186/1479-5876-11-75
193. Friedlander P, Wassmann K, Christenfeld AM, Fisher D, Kyi C, Kirkwood JM, et al. Whole-blood RNA transcript-based models can predict clinical response in two large independent clinical studies of patients with advanced melanoma treated with the checkpoint inhibitor, tremelimumab. J Immunother Cancer (2017) 5:67. doi: 10.1186/s40425-017-0272-z
194. Duarte JDG, Parakh S, Andrews MC, Woods K, Pasam A, Tutuka C, et al. Autoantibodies may predict immune-related toxicity: Results from a phase I study of intralesional Bacillus Calmette-Guérin followed by ipilimumab in patients with advanced metastatic melanoma. Front Immunol. (2018) 9:411. doi: 10.3389/fimmu.2018.00411
195. Friedlander P, Wood K, Wassmann K, Christenfeld AM, Bhardwaj N, Oh WK, et al. A whole-blood RNA transcript-based gene signature is associated with the development of CTLA-4 blockade-related diarrhea in patients with advanced melanoma treated with the checkpoint inhibitor tremelimumab. J Immunother Cancer (2018) 6:90. doi: 10.1186/s40425-018-0408-9
196. Cabel L, Proudhon C, Romano E, Girard N, Lantz O, Stern MH, et al. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat Rev Clin Oncol. (2018) 15:639–50. doi: 10.1038/s41571-018-0074-3
197. Pantel K. Blood-based analysis of circulating cell-free DNA and tumor cells for early cancer detection. PLoS Med. (2016) 13:e1002205. doi: 10.1371/journal.pmed.1002205
Keywords: biomarkers, checkpoint inhibitors, cytokines, chemokines, melanoma, Squamous cell carcinoma, Basal cell carcinoma, Merkel cell carcinoma
Citation: Bridge JA, Lee JC, Daud A, Wells JW and Bluestone JA (2018) Cytokines, Chemokines, and Other Biomarkers of Response for Checkpoint Inhibitor Therapy in Skin Cancer. Front. Med. 5:351. doi: 10.3389/fmed.2018.00351
Received: 27 August 2018; Accepted: 29 November 2018;
Published: 12 December 2018.
Edited by:
Günther F. L. Hofbauer, University of Zurich, SwitzerlandReviewed by:
Annalisa Saltari, UniversitätsSpital Zürich, SwitzerlandOleg E. Akilov, University of Pittsburgh, United States
Elisabetta Palazzo, Università degli Studi di Modena e Reggio Emilia, Italy
Copyright © 2018 Bridge, Lee, Daud, Wells and Bluestone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer A. Bridge, amVubmlmZXIuYnJpZGdlQHVjc2YuZWR1
James C. Lee, amFtZXMubGVlNEB1Y3NmLmVkdQ==
†These authors have contributed equally to this work