 César Magro-Checa
César Magro-Checa Gerda M. Steup-Beekman1
Gerda M. Steup-Beekman1 Mark A. van Buchem
Mark A. van Buchem Itamar Ronen
Itamar Ronen- 1Department of Rheumatology, Leiden University Medical Center, Leiden, Netherlands
- 2Department of Rheumatology, Zuyderland Medical Center, Heerlen, Netherlands
- 3Department of Radiology, Leiden University Medical Center, Leiden, Netherlands
- 4Department of Radiology, C.J. Gorter Center for High Field MRI, Leiden University Medical Center, Leiden, Netherlands
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by multi-systemic involvement. Nervous system involvement in SLE leads to a series of uncommon and heterogeneous neuropsychiatric (NP) manifestations. Current knowledge on the underlying pathogenic processes and their subsequent pathophysiological changes leading to NP-SLE manifestations is incomplete. Several putative laboratory biomarkers have been proposed as contributors to the genesis of SLE-related nervous system damage. Alongside the laboratory biomarkers, several neuroimaging tools have shown to reflect the nature of tissue microstructural damage associated with SLE, and thus were suggested to contribute to the understanding of the pathophysiological changes and subsequently help in clinical decision making. However, the number of useful biomarkers in NP-SLE in clinical practice is disconcertingly modest. In some cases it is not clear whether the biomarker is truly involved in pathogenesis, or the result of non-specific pathophysiological changes in the nervous system (e.g., neuroinflammation) or whether it is the consequence of a concomitant underlying abnormality related to SLE activity. In order to improve the diagnosis of NP-SLE and provide a better targeted care to these patients, there is still a need to develop and validate a range of biomarkers that reliably capture the different aspects of disease heterogeneity. This article critically reviews the current state of knowledge on laboratory and neuroimaging biomarkers in NP-SLE, discusses the factors that need to be addressed to make these biomarkers suitable for clinical application, and suggests potential future research paths to address important unmet needs in the NP-SLE field.
Introduction
For the last several years, clinicians and researchers in the field of neuropsychiatric systemic lupus erythematosus (NP-SLE) have been emphasizing the need for standardized and validated biomarkers to be used in clinical practice. Dozens of putative molecular mechanisms, such as serum and cerebrospinal fluid (CSF) antibodies to neuronal cell and cellular components, cytokines, complement and other immunochemical phenomena have been proposed to play a role in the genesis of nervous system involvement in SLE (1). Moreover, several neuroimaging techniques such as magnetic resonance imaging (MRI) and nuclear medicine techniques have allowed the characterization of structural and functional abnormalities in SLE patients therefore helping to better understand the underlying pathogenesis and subsequent pathophysiological changes (2, 3). Ideally, these laboratory and neuroimaging biomarkers, or a combination of them, would be used in clinical practice for the attribution of NP symptoms to SLE, as well as prognostic factors, predictors of response to therapies or even to develop new targeted therapies (Figure 1) (2). Despite these efforts, so far none of the postulated biomarkers has been demonstrated reproducible or ubiquitous enough to become a specific biomarker for NP-SLE. In clinical practice, the presentation of NP symptoms in SLE patients still poses an important diagnostic and therapeutic challenge to the physician. So far, the best strategy for diagnosing NP-SLE remains multidisciplinary expert consensus after standardized assessment (4, 5). An important goal of ongoing research is the development and validation of a range of laboratory and imaging biomarkers that reliably capture the different aspects of nervous system involvement in SLE and enable clinicians to attribute these symptoms either to SLE or to other etiologies.
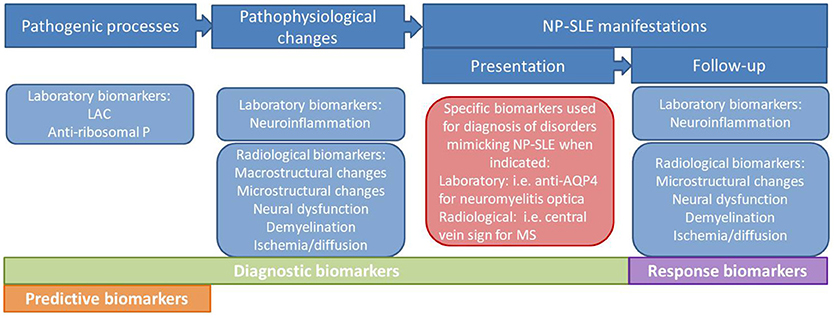
Figure 1. Types of biomarkers in NP-SLE.
This review compiles current knowledge on candidate biomarkers on NP-SLE. We discuss the strength of evidence for proposed laboratory and neuroimaging biomarkers; hypothesize about the desired properties of a biomarker in NP-SLE; and comment about the obstacles for biomarker development and its application in clinical practice. We conclude by highlighting some promising avenues for such biomarkers and suggest some strategies to increase their specificity and therefore increase their diagnostic role in clinical practice.
What We Have Learned From Animal Studies
Most if not all current knowledge about NP-SLE pathogenesis comes from mice models. Passive-transfer experiments in mice, consisting of injection of a certain substance or autoantibody, are the most common preclinical disease model in NP-SLE. Three different families of SLE mice models are currently used: induced models, spontaneous models and genetically engineered knockout and transgenic models. In the case of spontaneous models, these mice spontaneously manifest over varying periods of time with a series of clinical and serological features comparable to human SLE. NP manifestations are only described to appear in these models. Several lupus-prone strains have been so far the most commonly used for modeling NP-SLE manifestations: MRL/lpr, NZB/NZW F1 and 564Igi mice models (Panel 1 in Supplementary Material) (6, 7).
Studies using these mice models have provided some insight into the underlying pathogenic mechanisms contributing to NP-SLE in humans. Autoantibodies against neuronal auto-antigens, as well as other molecules are among the proposed mechanisms:
- Anti-ribosomal P (RP) antibody: Several studies have tried to explain the neuro-pathogenic effect of this antibody. After injection into the ventricles or hippocampus in mice, anti-RP antibodies induced depression-like behavior and memory impairment in mice, respectively, and were found to target the limbic system, especially the neurons in the hippocampus, cingulate cortex, and the primary olfactory piriform cortex (7, 8). Segovia-Miranda et al. have proposed that anti-RP targets the neuronal cell surface P antigen, or NSPA, in specific brain regions. This interaction may alter glutamatergic synaptic transmission in the hippocampus by involving both α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and N-methyl-D-aspartate receptor (NMDAR) activation and compromise synaptic plasticity involved in memory, hypothetically leading to cognitive dysfunction and other diffuse NP manifestations (9).
- Antibodies against the NMDAR subtypes 2a and 2b (anti-NR2 antibodies): Previous studies by Diamond et al have demonstrated how a murine monoclonal anti-dsDNA antibody cross-reacts with an amino-acid present in the subunits NR2a and NR2b of NMDAR, and how injecting these antibodies into mice leads to hippocampal neuronal death by apoptosis and cognitive impairment (10). Anti-NR2 antibodies have been also related to neuronal dysfunction and death in the hippocampus and amygdala in MRL/lpr mice (11). However, the mere presence of anti-NR2 antibodies in the blood of mice does not lead to neural death or subsequent NP symptoms; it is proposed in this work that to exert an effect upon neurons, anti-NR2 antibodies need to gain access to the brain through a disrupted blood brain barrier (BBB) (12, 13). Once these antibodies reach the brain they may produce several interactions. The acute exposure to the NMDAR may depend on the dose: at low concentrations they alter synaptic function; higher concentrations lead to excessive NMDAR activation causing neuronal cell death by apoptosis. Furthermore, chronic irreversible functional and structural damage of surviving neurons has been described to persist even when antibodies are no longer present. Impaired memory and hippocampal atrophy, as well as emotional disturbances and atrophy of the amygdala have been described to follow in mice (14, 15).
- An anti-dsDNA idiotype (Id) antibody in SLE, 16/6-Id antibody, hampers visual-recognition and results in cognitive impairments via cross-reaction with cytoskeletal proteins, glycoproteins and brain glycolipids. Kivity et al. showed an increased number of activated astrocytes and microglial cells as markers of brain inflammation after ventricular injection of this antibody in mice (16).
- Neurofilament alpha-internexin (INA): A murine model developed by INA immunization demonstrated cortical and hippocampal neuron apoptosis that resulted in pronounced cognitive dysfunction and memory loss (17).
Other molecules have been more recently also involved in NP-SLE pathogenesis in mice:
- Interferon (IFN) alpha: This inflammatory mediator has been postulated as one of the most promising targets in NP-SLE. Recently, Bialas et al have demonstrated the way in which peripheral type I INF enters the brain of 5641gi and NZB/NZW F1 strains and stimulates microglial engulfment of synaptic material. It was shown how reactive microglia lead to synapse loss in the frontal cortex of these mice, and these findings correlated with the appearance of behavioral deficits. Furthermore, it was shown how targeting the INF with anti-IFNAR antibodies prevented these symptoms and also mitigated synapse loss and microglial dysfunction (18).
- Complement cascade: Complement has been proposed as a candidate mechanism participating in microglial engulfing of synapses in the lupus brain. Recent discoveries on mouse models of Alzheimer's disease confirmed a critical role of the classical complement cascade in early synapse loss (19, 20). Other alternative role of complement is the regulation of brain inflammation. Alexander et al. showed that the deletion of a key alternative pathway protein known as factor B, and the inhibition of the classical and alternative complement cascades with the soluble complement inhibitor Crry-Ig alleviated NP symptoms of MRL-lpr mice (21, 22). Ulterior studies of the same group using the same mice model showed that selective inhibition of complement receptors C3aR and C5aR resulted, respectively in a reduction in neuronal degeneration and a drop in NP symptoms (23, 24). The same group also showed that C5 plays an important role in the maintenance of BBB in MRL-lpr mice (25).
- TWEAK: Apart from the alternative complement cascade, other molecules have shown to be important modulators of the integrity of the BBB and subsequently the transport into and out of the brain parenchyma (26). A pro-inflammatory cytokine member of the TNF superfamily called TWEAK (TNF-like weak inducer of apoptosis) induced cellular proliferation, angiogenesis, apoptosis, and production of inflammatory cytokines and chemokines through its receptor Fn14 (27). MRL/lpr mice lacking Fn14 improved cognitive function and exhibited less depression symptoms such as anhedonia (28).
Laboratory Biomarkers in Human NP-SLE: Looking for a Crystal Ball?
In 1998, The National Institutes of Health Biomarkers Definitions Working Group established by consensus the definition of a biomarker as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes or pharmacological responses to a therapeutic intervention”(29). This definition was later expanded by the World Health Organization (WHO) through the International Program of Chemical Safety to include “any substance, structure, or process that can be measured in the body or its products and influence or predict the incidence of outcome or disease” (30). This definitions are mainly applicable for laboratory-measured biomarkers. The first step in the path of biomarker development is invariably the identification and proposition as candidate of a marker of disease or any of its manifestations (exploratory biomarker). Subsequently, the reproducibility of findings in other studies across different populations will test the actual effectiveness of this biomarker (validated biomarker). Ultimately, these biomarkers may become clinically relevant (clinically useful biomarker) (31). In multiple sclerosis (MS), a neurological disease sharing some similarities with NP-SLE, biomarker research has significantly progressed in the last years; biomarkers for MS have been categorized into predictive, diagnostic, disease activity and treatment-response biomarkers (32). In current medical literature we find many studies directly identifying new potential biomarkers associated with NP-SLE or a specific neuropsychiatric symptom (e.g., cognitive dysfunction) due to SLE. Production of autoantibodies is a hallmark of SLE, with around 200 different antibodies more often present in SLE than in controls described so far (33). Based on data from the mouse models mentioned above, it is reasonable to assume that the target of certain antigens in the central nervous system by one or more antibodies may play a causal role in the genesis of NP-SLE. In humans, the concept that serum antibodies can lead to neurological symptoms is not new and auto-immune limbic encephalitis (LE) is a good example for that. LEs are associated with three neuronal cell-surface antibodies targeting leucine-rich glioma-inactivated 1 (anti-LGI1), γ-amino-butyric acid B-receptor (anti-GABAbR), and anti-AMPAR, usually leading to irreversible neurological deficits by altering the structure or function of the target antigen (34). In NP-SLE, several auto-antibodies targeting antigens located in brain tissue, as well as auto-antibodies directed against ubiquitous cellular components have been proposed as exploratory biomarkers (Table 1). Most studies report a higher proportion of positivity for a certain antibody in the serum or CSF in NP-SLE patients when compared with SLE and suggest a role for that antibody as diagnostic biomarker. An important number of these antibodies have only been found in discovery studies while reproducibility of findings across different patient populations is lacking. Furthermore, contradictory results among studies are common. In other cases the association has not been found in reproducibility studies (Supplementary Table 1); therefore so far only few antibodies have been validated as biomarkers. In a prospective study including 1,047 SLE patients, Hanly et al. found a role for lupus anticoagulant (LAC) and for anti-ribosomal P as predictive biomarkers for intracranial thrombosis and lupus psychosis, respectively (35). Three meta-analyses evaluating the role of antibodies as diagnostic biomarkers in NP-SLE have been published so far (36–38). Compared with SLE, NP-SLE patients had higher proportion of elevated serum levels of anticardiolipin, LAC, anti-ribosomal P and anti-neuronal antibodies, and an increased prevalence of positive titers for CSF anti-neuronal antibodies (37). These results are conflicting with a previous meta-analysis that combined data from 1,537 SLE patients from 14 centers and suggested a limited value for anti-P ribosomal antibodies as diagnostic biomarker in NP-SLE or any of its manifestations (38). Also the role of serum anti-NMDAR antibodies has been evaluated in a meta-analysis that included data from a total of 2,212 SLE patients. A higher positivity for serum anti-NMDAR was found in NP-SLE compared with SLE; this study concluded that serum anti-NMDAR antibodies may have a diagnostic value collectively but cannot distinguish among the different NP-SLE manifestations (36). From our point of view, only LAC may have a place as a clinically useful biomarker, since its positivity may help in the classification into disease phenotypes and might affect clinical decision making (Table 1). Other molecular biomarkers measured in the serum and in CSF, such as interleukins, chemokines, hormones and complement components, have also been proposed to have a role as diagnostic biomarkers in NP-SLE. Among them we find higher degree of agreement (at least in three different studies) in findings of higher levels of intrathecal IL-6 in NP-SLE patients, especially those with diffuse NP-SLE and acute confusional state (39–45). Although not specific, IL-6 is used in some clinics to assist the diagnosis of such cases and even a role for IL-6 in monitoring of disease activity and response to treatment has been proposed (43, 46). Besides the promising data from mouse studies, studies in humans on the complement cascade have been disappointing so far. The value of INF-α as diagnostic biomarkers in human NP-SLE merits further research (47).

Table 1. Strength of evidence for laboratory biomarkers in neuropsychiatric systemic lupus erythematosus in humans.
The Challenges of Establishing a Neuroimaging Biomarker in NP-SLE
Neuroimaging tools provide the ultimate means of obtaining information about the brain. Most neuroimaging tools are low to non-invasive, and are in general subdivided in four categories, employing four different physical principles: reflection and scattering of high frequency sound waves (Ultrasound), mapping the distribution of exogenous molecules in which radioactive atom or atoms are incorporated [Positron Emission Tomography (PET) and Single Photon Emission Computed Tomography (SPECT)], mapping of the attenuation of x-rays through the body (Computer Tomography) and mapping of the radiofrequency signal generated by the nuclei of hydrogen atoms, mostly those incorporated in water (Magnetic Resonance Imaging or MRI). Of these four, two have become the main staple of neuroimaging of disease—MRI and PET. These methods are in many ways complementary to each other, and each provides several means to monitor disease—from gross changes in brain shape and volume, to subtle changes in brain physiology, neurochemistry, and tissue microstructure. These two neuroimaging methods yield clear and unequivocal diagnostic evidence when the source of the damage to the brain is well-delineated and well-defined, as is the case in brain tumors and stroke. Yet, the diagnostic utility of both MRI and PET term “neuroimaging biomarker” in conjunction with brain diseases that involve multiple pathomechanisms that affect the brain in a more subtle way is less obvious. For example, neuroimaging contributes significantly to diagnosis of diseases such as MS (48, 49), Parkinson's disease (50), and Alzheimer's disease (51) but rarely can the diagnosis rely on the findings of neuroimaging alone, and often the neuroimaging results are used in a confirmatory or exclusionary way. Both PET and MRI have been intensely applied to study the brain of SLE and NP-SLE patients, and their role in understanding the disease process and contributing to diagnosis is undisputed. However, the term “neuroimaging biomarker” elicits a degree of sensitivity and most of all specificity that current neuroimaging methods do not provide in NP-SLE.
Magnetic Resonance Imaging (MRI)
Since its inception in the 1970s, MRI has become the most commonly used neuroimaging tool in diagnosis and research, providing data of high diagnostic value as well as insights into pathological mechanisms underlying these diseases. The astounding versatility of MRI, combined with its non-invasive nature, led to its primary status as the first-stop radiological diagnostic tool, including in SLE patients presenting with neuropsychiatric manifestations. Abnormal brain MRI in SLE and NP-SLE has been reported since the 1980s (52, 53). The most visible aspects of brain pathology available to the radiologist are brain lesions, reflected via hyper/hypointense areas on the images, and gross morphological changes such as global atrophy. Visible changes on MRI are the most immediate tool for a radiological evaluation of brain involvement, and in SLE and NP-SLE these have been regularly used as a standard clinical evaluation tool and as part of NP-SLE diagnosis (54).
Conventional MRI and the Clinico-Radiological Paradox in NP-SLE
Despite providing vital evidence about CNS involvement, both morphological changes and brain lesions have not provided a robust link neither to symptoms and disease outcome, nor to the pathological mechanisms underlying NP-SLE. A significant portion (around 40%) of those diagnosed with NP-SLE show no abnormalities on conventional MRI (cMRI), and global measures such as lesion load or brain atrophy do not scale with symptom severity (54–56). The reasons for this apparent failure are not completely understood. Brain tissue damage in NP-SLE is caused by a multitude of pathological processes, the endpoint of which are the visible changes on the MRI. In the most comprehensive prospective study to date that correlated pre-mortem cMRI in NP-SLE with post-mortem histopathology, visible MRI findings reflected global ischemic changes, parenchymal edema, microhemorrhages, gliosis, diffuse neuronal/axonal loss, cerebral infarction, microthromboemboli, and other findings, many of which were attributed to vascular origins (57). This apparent clinico-radiological paradox is the most powerful driving force for establishing neuroimaging biomarkers that not only correlate better with disease outcome, but also provide better insight into the underlying pathology of NP-SLE.
Quantitative MRI – Attaching Numbers to Subtle Disease-Modulated Image Changes
In addition to visible changes in brain tissue integrity, structure and morphology, MRI can report on diffuse changes in brain tissue microstructure, neurochemical composition and physiology. These changes cannot be reported immediately from radiological observations and require additional analysis of the images, typically resulting in a quantitative measure. In quantitative MRI (qMRI), the numeric value assigned to each image unit, or voxel, is not the one resulting directly from the image measurement but rather a calculated value, typically derived from two or more images. These values can represent intrinsic properties of the MRI signal, or a proxy to a local physiological, neurochemical or a microstructural property of brain tissue. Analyses can be performed based on regions of interest (ROI), or on voxel-by-voxel comparison following registration of individual quantitative images (or maps) to a common brain template. qMRI measures are particularly useful in assessing subtle inter-subject differences in brain regions where there is no apparent damage seen on conventional MRI. In many diseases, these subtle, more diffuse alterations are believed to reflect changes at early stages of the disease (58). Although qMRI can be useful on a single-subject level, most qMRI analyses focus on group differences. Such qMRI methods include relaxometric analyses and magnetization transfer imaging, diffusion weighted and diffusion tensor imaging, perfusion based imaging, and magnetic resonance spectroscopy. See Supplementary Table 2 for a brief description of quantitative MRI methods.
qMRI has been used in the study of NP-SLE and in earnest attempt to provide quantitative measures for the effects of NP-SLE on the brain:
- MR Relaxometry: Early studies looked at the usefulness of an automated evaluation of mean cortical T2 values as a possible marker for cortical changes in NP-SLE (59). The study acknowledged the potential confound of increased CSF fraction due to atrophy, which was more prominent in the NP-SLE population. T2 relaxometry was later incorporated in a multisequence study of NP-SLE in a small group of patients, in which MTR, MRS and DWI were also recorded (60). This is the first study in which several qMRI methods were applied, and whole brain values of the various modalities were correlated with each other. Several significant correlations were detected among modalities, but the study did not report on correlations with clinical parameters, thus the relationship of the quantitative MRI measures with any pathomechanism in NP-SLE is not explored.
- Magnetization transfer imaging (MTI) and magnetization transfer ratio (MTR): Several studies have highlighted the usefulness of MTR, an MTI-derived parameter, as a potential marker for brain microstructural changes in NP-SLE. Most studies focused on analysis of whole brain or tissue-specific (gray matter, white matter) histograms of MTR values (Figure 2) (61). Histograms of qMRI values in neurological disorders, development and aging are commonly applied (62–68). They provide a cumulative estimation of a quantitative measure, and thus lack any spatial information. They are, however, sensitive to diffuse, global effects, and successive studies have shown the sensitivity of MTR histograms to a variety of disease related clinical factors (69–73), including to the presence of specific antibodies. It was recently shown that white matter MTR histogram peak heights (HPH) were significantly lower in inflammatory NP-SLE patients than in NP-SLE patients diagnosed as ischemic, as well as than SLE patients with no NP complaints and healthy controls (74). The mechanism by which the MTR is decreased in NP-SLE is not clear, but its reversibility upon successful treatment suggests intracellular edema and gliosis as the associated pathomechanisms (75).
- Diffusion weighted imaging (DWI) and diffusion tensor imaging (DTI):Abnormal water diffusivity in brain white matter in NP-SLE has been reported in several studies (2, 76–80) and a review that summarizes many of these findings has been recently published (81). Global and local microstructural abnormalities, reflected in increase in mean diffusivity (MD) and decrease in fractional anisotropy (FA), were reported in most of the studies selected in this review. As in most MTI studies, most DWI and DTI studies did not explicitly exclude “visible” white matter hyperintensities from the analysis, albeit some of the studies made the point that the data suggests microstructural differences in NP-SLE in normal appearing white matter (82). Of importance is to note that several studies reported DTI abnormalities in SLE patients with no NP symptoms (non-NP-SLE). The review mentioned above dedicates a paragraph to DTI findings in non-NP-SLE compared to healthy controls, citing several studies that report positive findings, such as reduced FA in frontal white matter (78) and increased MD and reduced FA in the corpus callosum (83).
- Proton Magnetic resonance spectroscopy (1H-MRS): This technique is a non-invasive test that permits chemically specific, non-invasive measurements of the concentration of neuronal metabolites. In the brain there are about 20 such metabolites on which MRS can reliably report. Some have known functions such as neurotransmitters (glutamate, GABA), some are involved in energy metabolism [Lactate, creatine (tCr)] and some are uniquely (or preferentially) located in specific cell types (N-acetylasparate (NAA) in neurons, myo-inositol (MI) in astrocytes) (84, 85). Concentrations of metabolites are sometime modulated by disease, as well as their ability to freely move (or diffuse) in the cytoplasm. MRS measurements are either performed on a single volume of interest (VOI) positioned on an area of interest, or in multivoxel mode (spectroscopic imaging). This technique has been used in SLE studies where differences in the concentrations of several metabolites (relative to tCr) have been reported (86). Lower NAA and higher Cho and MI levels have been reported in SLE and NP-SLE patients when compared to healthy controls, suggesting decreased neuronal function and glial activation, respectively (73, 80, 87–90). More recently, lower NAA changes in NP-SLE patients when compared with SLE and in SLE with high disease activity when compared with low activity were found (91, 92). Diffusion weighted MRS (DWS) probes the mobility of intracellular metabolites in the cytoplasm, and is thus able to detect cytomorphological changes in disease (93). One such study reported increased diffusivity of glial metabolites in NP-SLE compared to healthy controls, with no concomitant change in neuronal metabolites, suggesting glial involvement (94).
- Perfusion weighted MRI: Despite the strong vascular aspect of NP-SLE, perfusion-based neuroimaging methods do not provide unequivocal information regarding NP-SLE-related modulation of cerebral blood flow and cerebral blood volume. Some studies report global and regional increase in cerebral blood flow (CBF) in NP-SLE as measured by MRI (95, 96). A recent study reported decreases in CBF in a well-defined white matter region, the centrum semiovale, with good sensitivity and specificity. The systematic attribution of the NP symptoms to SLE in this study was particularly tight and the high statistical significance of the results show big promise (97).
- Functional MRI (fMRI): Several studies investigated functional deficits in SLE and NP-SLE, and comprehensive review that summarizes the findings from these studies was published in 2016 (98). Task-related studies spanned a variety of tasks aimed at investigating working memory (99), sensory integration (100), and emotional responses (101). Resting state fMRI studies focused on functional networks that can be linked to cognitive and behavioral deficits (102, 103). Findings of hypo- and hyperactivation linked to symptoms and disease state were reported, and overall shed light on potential links between NP symptoms and brain functional deficits on local and network levels. Despite the usefulness of these studies, the possibility of generating a neuroimaging biomarker specific to NP-SLE based on brain function is not particularly promising, given the heterogeneity of the underlying pathomechanisms of NP-SLE and the wide range of NP symptoms.
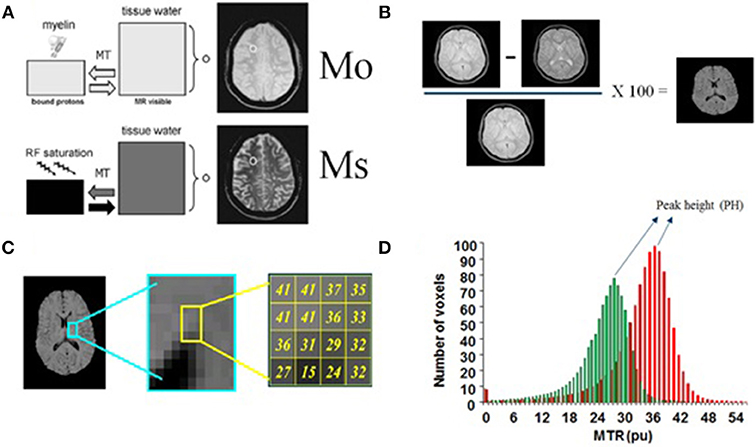
Figure 2. Basis of Magnetization Transfer Imaging. (A) This technique is based on the application of off-resonance radiofrequency pulses. M0: proton density image or intensity of voxels without saturation, Ms: Bound protons or intensity of voxels saturated. (B) Measurement of signal intensity with and without the application of these pulses allows the calculation of an index called the magnetization transfer ratio (MTR) which is defined as (M0-Ms/M0) × 100%. (C) MTR histogram: this technique takes a ratio of the two images on a voxel-by-voxel basis (brain pixels). (D) The histogram peak height (HPH), a MTR histogram-derived measure, accounts for the proportion of brain pixels at the most common MTR value. (A) partially adapted from Grossman et al. (61).
Positron Emission Tomography (PET) and Single Photon Emission Computed Tomography (SPECT)
Positron Emission Tomography is widely regarded as a highly useful neuroimaging tool in the clinic, in particular in oncology, where radioligands with high specificity to tumors have been developed and can provide early vital information on the presence of tumors in early stages of the disease (104). In addition, PET provides also useful information on physiological as well as on tissue microstructural and composition changes in neurological disorders, either through ligands that bind to abnormal formations of proteins involved in diseases such as Alzheimer's disease (105) or via reporting on abnormal metabolism, sometime associated with early signs of neurodegeneration and inflammation (106). Positron Emission Tomography studies in SLE and NP-SLE showed potential usefulness, in particular studies with 18F-FDG (fluorodeoxyglucose) showed increased metabolism in response to neuroinflammation and correlated with inflammatory status also under follow up (107). Positron Emission Tomography radioligands that bind to translocator protein (TSPO) are currently being investigated as potential markers for microglial activation in response to inflammation (108) and may provide an additional probe for neuroinflammation in NP-SLE (109).
Single photon emission computed tomography (SPECT) has also been used in SLE patients. Some SPECT studies report decreases in blood flow in NP-SLE in watershed regions (110). Glucose uptake, as detected by PET using 18-FDG, is intimately linked to blood flow, as they are both modulated by cellular metabolism (111). Several 18-FDG PET studies reported regional hypometabolism in NP-SLE (107, 112), but others provide evidence also for increase in glucose metabolism in white matter regions, consistent with inflammatory response (96, 113). Taken together, it appears that hypo- and hypermetabolism may relate to two different yet coexisting aspects of brain involvement in SLE.
Biomarkers in NP-SLE: Future Perspectives
As previously shown, yet despite years of efforts of the NP-SLE scientific community, the number of clinically useful biomarkers and even of validated biomarkers is embarrassingly modest. From our point of view a series of scientific challenges in the field have yet to be overcome:
- The NP-SLE definition is a challenge by itself. Since 1999, research in this field has been guided by the ACR case definitions for NP-SLE syndromes including a group of 19 complex and uncommon neuropsychiatric manifestations involving both the central (12 syndromes) and peripheral (7 syndromes) nervous system (114). Researchers have mainly focused on analyzing biomarkers in NP-SLE defined as a group based on these definitions without taking into account the underlying pathophysiological mechanism. Using such heterogeneous manifestations as a group may be problematic since it may include manifestations with obviously different underlying pathophysiological mechanisms, e.g., stroke and acute confusional state. Clinicians and researchers in the field would benefit from resolving the problem of heterogeneity by using biomarkers capturing the different aspects of nervous involvement in SLE. Borowoy et al. demonstrated how autoantibody associations depend on the NP-SLE definition used (115). In clinical practice, the gold standard is a diagnosis conducted by a multidisciplinary expert clinical team. Furthermore, the diagnosis in NP-SLE is made phenotypically according to the suspected underlying pathophysiological mechanism (inflammatory, thrombotic or infrequently coexistence of both) which is critical for guiding treatment. Phenotypic characterization is important in clinical practice but may be also in research. A given phenotype may arise from a diverse set of biochemical processes and its changes in the brain may be captured by a diverse set of neuroimaging techniques. The identification of a biochemical and neuroimaging subset of factors that underlie a specific phenotype or certain NP-SLE manifestation should be preferable in future research and more applicable in clinical practice.
- Apart from the heterogeneity of the groups, the small sample size due to the low prevalence is one of the common denominators of studies describing new potential biomarkers in NP-SLE. Given the rarity and complexity of NP-SLE, collaborative efforts, using pooled clinical, laboratory, and neuroimaging data sets are needed. Much larger studies will allow for more specific hypothesis about for example a specific phenotype or NP-SLE manifestation, permit the use of biomarker combinations and analyze the relations among them. Furthermore, collaboration will facilitate performing the first serious trial with well-known drugs or even additional therapeutic choices, giving the opportunity to assess the role of these biomarkers in monitoring of disease activity and response to treatment.
- Study design: A reason for the minimal clinical impact of reported biomarkers may be that most of these studies report differences between NP-SLE patients and SLE at a group level while physicians have to make clinical decisions individually. Furthermore, in clinical practice, when a SLE patient presents with NP complaints it is obligatory first to exclude other potential causes before these symptoms are attributed to SLE. Most of the studies compare the higher presence of a certain biomarker in SLE patients with and without NP-SLE manifestations, remaining uncertain if this biomarker profiles are unique to NP-SLE or may be present in other mimicking neuropsychiatric disorders; only a few studies have used a group of patients with other neuropsychiatric disease (e.g., MS or septic meningitis) as control groups (116). For example, B-cell activating factor of TNF family or matrix metalloprotease-9 have been proposed as exploratory biomarker in NP-SLE because its higher positivity when compared with SLE; however both biomarkers have also been proposed as biomarkers in patients with MS (117).
In the particular case of laboratory biomarkers, the next promising aspects in NP-SLE should be addressed in the near future.
- Identification of new neuronal surface antigens which are responsible for NP-SLE. The antigen identification paradigm which has been successfully used with limbic encephalitis may be applied on NP-SLE to recognize unknown neuronal cell-surface protein(s) (34). Determination of neuronal immunoreactivity in different areas of brain and cerebellum of homogeneous clinical and radiological NP-SLE groups may be analyzed and afterwards correlated with clinical symptoms and MRI characteristics. To identify the target antigen cultured neurons and mass spectrometry could be used. Lastly, brains of knock out animal models or cells deprived of the suspect antigen by siRNA knock down may confirm the specificity of these candidate autoantibodies (118).
- Omics: In the last years, laboratory biomarker discovery has benefit from the development of omics technologies such as genomics or immune-proteomics, which has successfully increased the list of exploratory biomarkers in many diseases (119). These techniques give the opportunity to explore a wide spectrum of biomarkers in a more comprehensive and unbiased way. Autoantigen microarrays have already been used in NP-SLE (120, 121). For example, van der Meulen et al have shown how a profile of IgG and IgM autoantibodies against 15 antigens may help to differentiate NP-SLE from non-NP-SLE (120). The potential for false positive discoveries using these techniques is high; reproduction of this data and selection of best candidates may be a next step before validation in large-scale independent cohorts (122).
- Complement cascade and IFN-alpha: The exciting area of research in NP-SLE mice models on complement cascade and IFN-alpha needs to be translated to human NP-SLE. The study of these two biomarkers may lead to a better understanding of pathogenic underlying mechanisms of synapse loss and will probably open the door to the use of new therapeutic strategies in NP-SLE (e.g., Eculizumab, a humanized monoclonal antibody blocking the generation of terminal complement components C5a and C5b-9, and Sifalimumab, a human anti-IFN-a monoclonal antibody).
- Understanding of the BBB in NP-SLE. Brain tissue-reactive antibodies in NP-SLE are thought to be synthesized in the CNS, but also in peripheral organs (lymph nodes and bone marrow). In the last case it was proposed that these autoantibodies must pass through pathologically permeable BBB to exert an effect upon neurons. Although an important role of BBB has been supposed, we need better understand the role of BBB in human NP-SLE. A recent study in mice questions the widely accepted hypothesis of a disrupted BBB and suggests a dysfunction of the blood-cerebrospinal fluid barrier in the choroid plexus underlying brain exposure to these neuropathic antibodies (123). More studies in humans comparing serum and CSF and using quotients are warranted.
Future research on neuroimaging biomarkers for NP-SLE will need to address the next factors
- Another look at cMRI: the case for more sophisticated characterization of lesions: In recent years, characterization of lesions in neurological disorders has advanced far beyond the basic lesion count or lesion load. Most notable is the work related to lesions and their pathological classification in MS. It has long been postulated that lesion location and not lesion load as a robust surrogate marker of neuropsychological impairment in patients with MS (124). Link between lesion location and cognitive function was shown to be significant in some studies (125, 126). The latest, and most significant step in characterization of white matter lesions in MS came after examining the spatial relationship between lesions and large veins in a visualization method that superimposes FLAIR images containing lesion spatial information, and -weighted images, showing vein distribution in great detail (Figure 3) (127). It was found that concentric co-localization of a white matter lesion with a vein that passes through it, termed central vein sign (CVS), is highly specific to the early stages of MS and has been swiftly adopted as a biomarker mandated for MS diagnosis by the North American Imaging in Multiple Sclerosis Cooperative (NAIMS). The presence and development of CVS are well-explained by a neuroinflammatory mechanism with a vascular origin, and CVS has been shown to differentiate well-between MS and other central nervous system inflammatory vasculopathies including SLE (128), although not without cautionary remarks (129).
- Same picture – multiple views: the role of multimodal neuroimaging in NP-SLE: The multifactorial nature of NP-SLE, combined with the lack of specificity of most imaging modalities to any particular pathomechanism makes the quest for a “silver bullet” diagnostic tool unrealistic. Even PET ligands for TSPO, initially assumed to have high specificity to microglial activation and thus to inflammation, have been shown to be less specific than the initial expectations A natural approach is to combine several neuroimaging markers, each highlighting a different aspect of the disease in an approach that uses a multivariate analysis in one way or the other. This approach has been suggested for other neuropsychiatric and neurological disorders, especially for those with little overt brain damage and complex underlying mechanisms such as major psychiatric disorders. In NP-SLE, most of the clinical MRI protocols are intrinsically multimodal, and in many sites additional imaging data that is not directly clinical are acquired. Several approaches for multimodal data analysis of neuroimaging data have been proposed, aimed primarily at developmental psychiatric and neurodegenerative disorders but also to stroke and tumors (130–136).
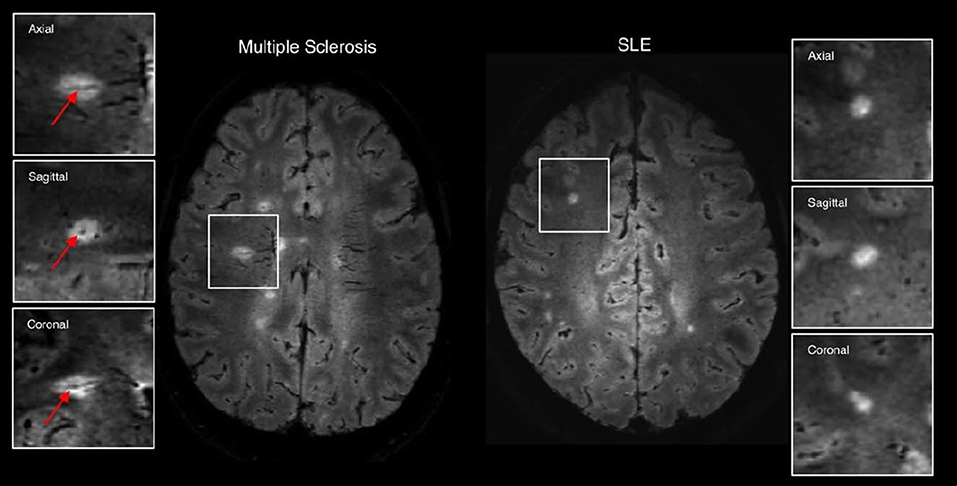
Figure 3. Examples of 3T FLAIR* imaging demonstrating the central vessel sign (arrows) in multiple sclerosis (left) but not in systemic lupus erythematosus (right). Figure adapted with permission from Maggi et al. (119).
The application of the previous laboratory and radiological techniques in NP-SLE will produce hundreds of exploratory biomarkers where complicated statistical methodology is required. Analytical methods such as supervised machine learning (ML) promise help solving this problem and advance the development of biomarkers in the near future. This technique uses algorithms to automatically extract information from data that can be applied at the individual level to make predictions therefore with a higher level of clinical translation. Furthermore, ML can be applied to laboratory biomarkers but also to neuroimaging data, since these methods are sensitive to spatially distributed and subtle effects (137). For example, neuroimaging data are intrinsically large, if it is considered that the number of pixels in MRI data sets is in the order of 1 × 107 (ten million) pixels, and several such sets are combined together to yield the multimodal data set.
Conclusion
Besides the enormous progress made in the area, specific laboratory and neuroimaging biomarkers in NP-SLE are scarce and their validation as useful biomarkers in routine clinical practice is far from becoming reality. In the present paper, we have proposed several research paths that may help to overcome some of the obstacles that hamper the validation of laboratory and neuroimaging features useful as diagnostic, prognostic or response to treatment biomarkers. We are convinced that many of these obstacles will only be overcome thanks to large collaborative efforts. Importantly, the difficulties in establishing a single imaging or laboratory biomarker for NP-SLE point toward a concerted interdisciplinary effort to establish an optimal set of imaging and laboratory markers that will correlate best with disease phenotype, progression and severity.
Author Contributions
CM-C, GS-B, TH, MB, and IR have made significant contributions to the design, the literature research, and the writing of the manuscript. All authors reviewed and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2018.00340/full#supplementary-material
References
1. Jeltsch-David H, Muller S. Neuropsychiatric systemic lupus erythematosus: pathogenesis and biomarkers. Nat Rev Neurol. (2014) 10:579–96. doi: 10.1038/nrneurol.2014.148
2. Ercan E, Ingo C, Tritanon O, Magro-Checa C, Smith A, Smith S, et al. A multimodal MRI approach to identify and characterize microstructural brain changes in neuropsychiatric systemic lupus erythematosus. Neuroimage Clin. (2015) 8:337–44. doi: 10.1016/j.nicl.2015.05.002
3. Curiel R, Akin EA, Beaulieu G, DePalma L, Hashefi M. PET/CT imaging in systemic lupus erythematosus. Ann NY Acad Sci. (2011) 1228:71–80. doi: 10.1111/j.1749-6632.2011.06076.x
4. Magro-Checa C, Zirkzee EJ, Beaart-van de Voorde LJJ, Middelkoop HA, van der Wee NJ, Huisman MV, et al. Value of multidisciplinary reassessment in attribution of neuropsychiatric events to systemic lupus erythematosus: prospective data from the Leiden NPSLE cohort. Rheumatology (2017) 56:1676–83. doi: 10.1093/rheumatology/kex019
5. Magro-Checa C, Zirkzee EJ, Huizinga TW, Steup-Beekman GM. Management of neuropsychiatric systemic lupus erythematosus: current approaches and future perspectives. Drugs (2016) 76:459–83. doi: 10.1007/s40265-015-0534-3
6. Jeltsch-David H, Muller S. Neuropsychiatric systemic lupus erythematosus and cognitive dysfunction: the MRL-lpr mouse strain as a model. Autoimmun Rev. (2014) 13:963–73. doi: 10.1016/j.autrev.2014.08.015
7. Katzav A, Solodeev I, Brodsky O, Chapman J, Pick CG, Blank M, et al. Induction of autoimmune depression in mice by anti-ribosomal P antibodies via the limbic system. Arthritis Rheum. (2007) 56:938–48. doi: 10.1002/art.22419
8. Bravo-Zehnder M, Toledo EM, Segovia-Miranda F, Serrano FG, Benito MJ, Metz C, et al. Anti-ribosomal P protein autoantibodies from patients with neuropsychiatric lupus impair memory in mice. Arthritis Rheumatol. (2015) 67:204–14. doi: 10.1002/art.38900
9. Segovia-Miranda F, Serrano F, Dyrda A, Ampuero E, Retamal C, Bravo-Zehnder M, et al. Pathogenicity of lupus anti-ribosomal P antibodies: role of cross-reacting neuronal surface P antigen in glutamatergic transmission and plasticity in a mouse model. Arthritis Rheumatol. (2015) 67:1598–610. doi: 10.1002/art.39081
10. DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. (2001) 7:1189–93. doi: 10.1038/nm1101-1189
11. Lauvsnes MB, Omdal R. Systemic lupus erythematosus, the brain, and anti-NR2 antibodies. J Neurol. (2012) 259:622–9. doi: 10.1007/s00415-011-6232-5
12. Kowal C, DeGiorgio LA, Nakaoka T, Hetherington H, Huerta PT, Diamond B, et al. Cognition and immunity; antibody impairs memory. Immunity (2004) 21:179–88. doi: 10.1016/j.immuni.2004.07.011
13. Kowal C, Degiorgio LA, Lee JY, Edgar MA, Huerta PT, Volpe BT, et al. Human lupus autoantibodies against NMDA receptors mediate cognitive impairment. Proc Natl Acad Sci USA. (2006) 103:19854–9. doi: 10.1073/pnas.0608397104
14. Chang EH, Volpe BT, Mackay M, Aranow C, Watson P, Kowal C, et al. Selective impairment of spatial cognition caused by autoantibodies to the N-methyl-D-aspartate receptor. EBioMedicine (2015) 2:755–64. doi: 10.1016/j.ebiom.2015.05.027
15. Faust TW, Chang EH, Kowal C, Berlin R, Gazaryan IG, Bertini E, et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proc Natl Acad Sci USA. (2010) 107:18569–74. doi: 10.1073/pnas.1006980107
16. Kivity S, Katzav A, Arango MT, Landau-Rabi M, Zafrir Y, Agmon-Levin N, et al. 16/6-idiotype expressing antibodies induce brain inflammation and cognitive impairment in mice: the mosaic of central nervous system involvement in lupus. BMC Med. (2013) 11:90. doi: 10.1186/1741-7015-11-90
17. Lu XY, Chen XX, Huang LD, Zhu CQ, Gu YY, Ye S. Anti-alpha-internexin autoantibody from neuropsychiatric lupus induce cognitive damage via inhibiting axonal elongation and promote neuron apoptosis. PLoS ONE (2010) 5:e11124. doi: 10.1371/journal.pone.0011124
18. Bialas AR, Presumey J, Das A, van der Poel CE, Lapchak PH, Mesin L, et al. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature (2017) 546:539–43. doi: 10.1038/nature22821
19. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (2016) 352:712–6. doi: 10.1126/science.aad8373
20. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
21. Alexander JJ, Bao L, Jacob A, Kraus DM, Holers VM, Quigg RJ. Administration of the soluble complement inhibitor, Crry-Ig, reduces inflammation and aquaporin 4 expression in lupus cerebritis. Biochim Biophys Acta (2003) 1639:169–76. doi: 10.1016/j.bbadis.2003.09.005
22. Alexander JJ, Jacob A, Vezina P, Sekine H, Gilkeson GS, Quigg RJ. Absence of functional alternative complement pathway alleviates lupus cerebritis. Eur J Immunol. (2007) 37:1691–701. doi: 10.1002/eji.200636638
23. Jacob A, Bao L, Brorson J, Quigg RJ, Alexander JJ. C3aR inhibition reduces neurodegeneration in experimental lupus. Lupus (2010) 19:73–82. doi: 10.1177/0961203309348978
24. Jacob A, Hack B, Bai T, Brorson JR, Quigg RJ, Alexander JJ. Inhibition of C5a receptor alleviates experimental CNS lupus. J Neuroimmunol. (2010) 221:46–52. doi: 10.1016/j.jneuroim.2010.02.011
25. Jacob A, Hack B, Chiang E, Garcia JG, Quigg RJ, Alexander JJ. C5a alters blood-brain barrier integrity in experimental lupus. FASEB J. (2010) 24:1682–8. doi: 10.1096/fj.09-138834
26. Engelhardt B. Neuroscience. Blood-brain barrier differentiation. Science (2011) 334:1652–3. doi: 10.1126/science.1216853
27. Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, et al. TWEAK induces liver progenitor cell proliferation. J Clin Invest. (2005) 115:2330–40. doi: 10.1172/JCI23486
28. Wen J, Xia Y, Stock A, Michaelson JS, Burkly LC, Gulinello M, et al. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J Autoimmun. (2013) 43:44–54. doi: 10.1016/j.jaut.2013.03.002
29. Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. (2001) 69:89–95. doi: 10.1067/mcp.2001.113989
30. WHO. Biomarkers in Risk Assessment: Validity and Validation (2001). Available online at: http://apps.who.int/iris/handle/10665/42363
31. Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. (2006) 24:971–83. doi: 10.1038/nbt1235
32. Comabella M, Montalban X. Body fluid biomarkers in multiple sclerosis. Lancet Neurol. (2014) 13:113–26. doi: 10.1016/S1474-4422(13)70233-3
33. Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum. (2004) 34:501–37. doi: 10.1016/j.semarthrit.2004.07.002
34. Leypoldt F, Armangue T, Dalmau J. Autoimmune encephalopathies. Ann NY Acad Sci. (2015) 1338:94–114. doi: 10.1111/nyas.12553
35. Hanly JG, Urowitz MB, Su L, Bae SC, Gordon C, Clarke A, et al. Autoantibodies as biomarkers for the prediction of neuropsychiatric events in systemic lupus erythematosus. Ann Rheum Dis. (2011) 70:1726–32. doi: 10.1136/ard.2010.148502
36. Tay SH, Fairhurst AM, Mak A. Clinical utility of circulating anti-N-methyl-d-aspartate receptor subunits NR2A/B antibody for the diagnosis of neuropsychiatric syndromes in systemic lupus erythematosus and Sjogren's syndrome: An updated meta-analysis. Autoimmun Rev. (2017) 16:114–22. doi: 10.1016/j.autrev.2016.12.002
37. Ho RC, Thiaghu C, Ong H, Lu Y, Ho CS, Tam WW, et al. A meta-analysis of serum and cerebrospinal fluid autoantibodies in neuropsychiatric systemic lupus erythematosus. Autoimmun Rev. (2016) 15:124–38. doi: 10.1016/j.autrev.2015.10.003
38. Karassa FB, Afeltra A, Ambrozic A, Chang DM, De Keyser F, Doria A, et al. Accuracy of anti-ribosomal P protein antibody testing for the diagnosis of neuropsychiatric systemic lupus erythematosus: an international meta-analysis. Arthritis Rheum. (2006) 54:312–24. doi: 10.1002/art.21539
39. Wang JB, Li H, Wang LL, Liang HD, Zhao L, Dong J. Role of IL-1beta, IL-6, IL-8 and IFN-gamma in pathogenesis of central nervous system neuropsychiatric systemic lupus erythematous. Int J Clin Exp Med. (2015) 8:16658–63.
40. Fragoso-Loyo H, Richaud-Patin Y, Orozco-Narvaez A, Davila-Maldonado L, Atisha-Fregoso Y, Llorente L, et al. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. (2007) 56:1242–50. doi: 10.1002/art.22451
41. Yoshio T, Okamoto H, Kurasawa K, Dei Y, Hirohata S, Minota S. IL-6, IL-8, IP-10, MCP-1 and G-CSF are significantly increased in cerebrospinal fluid but not in sera of patients with central neuropsychiatric lupus erythematosus. Lupus (2016) 25:997–1003. doi: 10.1177/0961203316629556
42. Jara LJ, Irigoyen L, Ortiz MJ, Zazueta B, Bravo G, Espinoza LR. Prolactin and interleukin-6 in neuropsychiatric lupus erythematosus. Clin Rheumatol. (1998) 17:110–4. doi: 10.1007/BF01452255
43. Trysberg E, Carlsten H, Tarkowski A. Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus (2000) 9:498–503. doi: 10.1177/096120330000900704
44. Asano T, Ito H, Kariya Y, Hoshi K, Yoshihara A, Ugawa Y, et al. Evaluation of blood-brain barrier function by quotient alpha2 macroglobulin and its relationship with interleukin-6 and complement component 3 levels in neuropsychiatric systemic lupus erythematosus. PLoS ONE (2017) 12:e0186414. doi: 10.1371/journal.pone.0186414
45. Hirohata S, Kanai Y, Mitsuo A, Tokano Y, Hashimoto H. Accuracy of cerebrospinal fluid IL-6 testing for diagnosis of lupus psychosis. A multicenter retrospective study. Clin Rheumatol. (2009) 28:1319–23. doi: 10.1007/s10067-009-1226-8
46. Lu XY, Zhu CQ, Qian J, Chen XX, Ye S, Gu YY. Intrathecal cytokine and chemokine profiling in neuropsychiatric lupus or lupus complicated with central nervous system infection. Lupus (2010) 19:689–95. doi: 10.1177/0961203309357061
47. Fragoso-Loyo H, Atisha-Fregoso Y, Nunez-Alvarez CA, Llorente L, Sanchez-Guerrero J. Utility of interferon-alpha as a biomarker in central neuropsychiatric involvement in systemic lupus erythematosus. J Rheumatol. (2012) 39:504–9. doi: 10.3899/jrheum.110983
48. Filippi M, Rocca MA, Ciccarelli O, De Stefano N, Evangelou N, Kappos L, et al. MRI criteria for the diagnosis of multiple sclerosis: MAGNIMS consensus guidelines. Lancet Neurol. (2016) 15:292–303. doi: 10.1016/S1474-4422(15)00393-2
49. Louapre C, Bodini B, Lubetzki C, Freeman L, Stankoff B. Imaging markers of multiple sclerosis prognosis. Curr Opin Neurol. (2017) 30:231–6. doi: 10.1097/WCO.0000000000000456
50. Meijer FJA, Goraj B, Bloem BR, Esselink RAJ. Clinical application of brain MRI in the diagnostic work-up of parkinsonism. J Parkinsons Dis. (2017) 7:211–7. doi: 10.3233/JPD-150733
51. Wurtman R. Biomarkers in the diagnosis and management of Alzheimer's disease. Metabolism (2015) 64:S47–50. doi: 10.1016/j.metabol.2014.10.034
52. Aisen AM, Gabrielsen TO, McCune WJ. MR imaging of systemic lupus erythematosus involving the brain. AJR Am J Roentgenol. (1985) 144:1027–31. doi: 10.2214/ajr.144.5.1027
53. McCune WJ, MacGuire A, Aisen A, Gebarski S. Identification of brain lesions in neuropsychiatric systemic lupus erythematosus by magnetic resonance scanning. Arthritis Rheum. (1988) 31:159–66. doi: 10.1002/art.1780310202
54. Luyendijk J, Steens SC, Ouwendijk WJ, Steup-Beekman GM, Bollen EL, van der Grond J, et al. Neuropsychiatric systemic lupus erythematosus: lessons learned from magnetic resonance imaging. Arthritis Rheum. (2011) 63:722–32. doi: 10.1002/art.30157
55. Toledano P, Sarbu N, Espinosa G, Bargallo N, Cervera R. Neuropsychiatric systemic lupus erythematosus: magnetic resonance imaging findings and correlation with clinical and immunological features. Autoimmun Rev. (2013) 12:1166–70. doi: 10.1016/j.autrev.2013.07.004
56. Jennings JE, Sundgren PC, Attwood J, McCune J, Maly P. Value of MRI of the brain in patients with systemic lupus erythematosus and neurologic disturbance. Neuroradiology (2004) 46:15–21. doi: 10.1007/s00234-003-1049-2
57. Sibbitt WL Jr, Brooks WM, Kornfeld M, Hart BL, Bankhurst AD, Roldan CA. Magnetic resonance imaging and brain histopathology in neuropsychiatric systemic lupus erythematosus. Semin Arthritis Rheum. (2010) 40:32–52. doi: 10.1016/j.semarthrit.2009.08.005
58. Crespy L, Zaaraoui W, Lemaire M, Rico A, Faivre A, Reuter F, et al. Prevalence of grey matter pathology in early multiple sclerosis assessed by magnetization transfer ratio imaging. PloS ONE (2011) 6:e24969. doi: 10.1371/journal.pone.0024969
59. Petropoulos H, Sibbitt WL Jr, Brooks WM. Automated T2 quantitation in neuropsychiatric lupus erythematosus: a marker of active disease. J Magn Reson Imaging. (1999) 9:39–43. doi: 10.1002/(SICI)1522-2586(199901)9:1<39::AID-JMRI5>3.0.CO;2-8
60. Bosma GP, Steens SC, Petropoulos H, Admiraal-Behloul F, van den Haak A, Doornbos J, et al. Multisequence magnetic resonance imaging study of neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. (2004) 50:3195–202. doi: 10.1002/art.20512
61. Grossman RI, Gomori JM, Ramer KN, Lexa FJ, Schnall MD. Magnetization transfer: theory and clinical applications in neuroradiology. Radiographics (1994) 14:279–90.
62. Iannucci G, Rovaris M, Giacomotti L, Comi G, Filippi M. Correlation of multiple sclerosis measures derived from T2-weighted, T1-weighted, magnetization transfer, and diffusion tensor MR imaging. AJNR Am J Neuroradiol. (2001) 22:1462–7.
63. van Buchem MA, Steens SC, Vrooman HA, Zwinderman AH, McGowan JC, Rassek M, et al. Global estimation of myelination in the developing brain on the basis of magnetization transfer imaging: a preliminary study. AJNR Am J Neuroradiol. (2001) 22:762–6.
64. Ge Y, Grossman RI, Babb JS, Rabin ML, Mannon LJ, Kolson DL. Age-related total gray matter and white matter changes in normal adult brain. Part II: quantitative magnetization transfer ratio histogram analysis. AJNR Am J Neuroradiol. (2002) 23:1334–41.
65. Inglese M, Salvi F, Iannucci G, Mancardi GL, Mascalchi M, Filippi M. Magnetization transfer and diffusion tensor MR imaging of acute disseminated encephalomyelitis. AJNR Am J Neuroradiol. (2002) 23:267–72.
66. Lee KY, Kim TK, Park M, Ko S, Song IC, Cho IH. Age-related changes in conventional and magnetization transfer MR imaging in elderly people: comparison with neurocognitive performance. Korean J Radiol. (2004) 5:96–101. doi: 10.3348/kjr.2004.5.2.96
67. van den Bogaard SJ, Dumas EM, Milles J, Reilmann R, Stout JC, Craufurd D, et al. Magnetization transfer imaging in premanifest and manifest Huntington disease. AJNR Am J Neuroradiol. (2012) 33:884–9. doi: 10.3174/ajnr.A2868
68. Sala M, de Roos A, van den Berg A, Altmann-Schneider I, Slagboom PE, Westendorp RG, et al. Microstructural brain tissue damage in metabolic syndrome. Diabetes Care (2014) 37:493–500. doi: 10.2337/dc13-1160
69. Bosma GP, Rood MJ, Huizinga TW, de Jong BA, Bollen EL, van Buchem MA. Detection of cerebral involvement in patients with active neuropsychiatric systemic lupus erythematosus by the use of volumetric magnetization transfer imaging. Arthritis Rheum. (2000) 43:2428–36. doi: 10.1002/1529-0131(200011)43:11<2428::AID-ANR9>3.0.CO;2-H
70. Dehmeshki J, Van Buchem MA, Bosma GP, Huizinga TW, Tofts PS. Systemic lupus erythematosus: diagnostic application of magnetization transfer ratio histograms in patients with neuropsychiatric symptoms–initial results. Radiology (2002) 222:722–8. doi: 10.1148/radiol.2223010413
71. Steens SC, Admiraal-Behloul F, Bosma GP, Steup-Beekman GM, Olofsen H, Le Cessie S, et al. Selective gray matter damage in neuropsychiatric lupus. Arthritis Rheum. (2004) 50:2877–81. doi: 10.1002/art.20654
72. Steens SC, Bosma GP, Steup-Beekman GM, le Cessie S, Huizinga TW, van Buchem MA. Association between microscopic brain damage as indicated by magnetization transfer imaging and anticardiolipin antibodies in neuropsychiatric lupus. Arthritis Res Ther. (2006) 8:R38. doi: 10.1186/ar1892
73. Emmer BJ, Steup-Beekman GM, Steens SC, Huizinga TW, van Buchem MA, van der Grond J. Correlation of magnetization transfer ratio histogram parameters with neuropsychiatric systemic lupus erythematosus criteria and proton magnetic resonance spectroscopy: association of magnetization transfer ratio peak height with neuronal and cognitive dysfunction. Arthritis Rheum. (2008) 58:1451–7. doi: 10.1002/art.23452
74. Magro-Checa C, Ercan E, Wolterbeek R, Emmer B, van der Wee NJ, Middelkoop HA, et al. Changes in white matter microstructure suggest an inflammatory origin of neuropsychiatric systemic lupus erythematosus. Arthritis Rheumatol. (2016) 68:1945–54. doi: 10.1002/art.39653
75. Emmer BJ, Steens SC, Steup-Beekman GM, van der Grond J, Admiraal-Behloul F, Olofsen H, et al. Detection of change in CNS involvement in neuropsychiatric SLE: a magnetization transfer study. J Magn Reson Imaging. (2006) 24:812–6. doi: 10.1002/jmri.20706
76. Bosma GP, Huizinga TW, Mooijaart SP, Van Buchem MA. Abnormal brain diffusivity in patients with neuropsychiatric systemic lupus erythematosus. AJNR Am J Neuroradiol. (2003) 24:850–4.
77. Emmer BJ, Veer IM, Steup-Beekman GM, Huizinga TW, van der Grond J, van Buchem MA. Tract-based spatial statistics on diffusion tensor imaging in systemic lupus erythematosus reveals localized involvement of white matter tracts. Arthritis Rheum. (2010) 62:3716–21. doi: 10.1002/art.27717
78. Schmidt-Wilcke T, Cagnoli P, Wang P, Schultz T, Lotz A, McCune WJ, et al. Diminished white matter integrity in patients with systemic lupus erythematosus. Neuroimage Clin. (2014) 5:291–7. doi: 10.1016/j.nicl.2014.07.001
79. Cesar B, Dwyer MG, Shucard JL, Polak P, Bergsland N, Benedict RH, et al. Cognitive and white matter tract differences in ms and diffuse neuropsychiatric systemic lupus erythematosus. AJNR Am J Neuroradiol. (2015) 36:1874–83. doi: 10.3174/ajnr.A4354
80. Zimny A, Szmyrka-Kaczmarek M, Szewczyk P, Bladowska J, Pokryszko-Dragan A, Gruszka E, et al. In vivo evaluation of brain damage in the course of systemic lupus erythematosus using magnetic resonance spectroscopy, perfusion-weighted and diffusion-tensor imaging. Lupus (2014) 23:10–9. doi: 10.1177/0961203313511556
81. Costallat BL, Ferreira DM, Lapa AT, Rittner L, Costallat LTL, Appenzeller S. Brain diffusion tensor MRI in systematic lupus erythematosus: a systematic review. Autoimmun Rev. (2018) 17:36–43. doi: 10.1016/j.autrev.2017.11.008
82. Shastri R, Shah G, Wang P, Cagnoli P, Schmidt-Wilcke T, McCune J, et al. MR diffusion tractography to identify and characterize microstructural white matter tract changes in systemic lupus erythematosus patients. Acad Radiol. (2016) 23:1431–40. doi: 10.1016/j.acra.2016.03.019
83. Shapira-Lichter I, Weinstein M, Lustgarten N, Ash E, Litinsky I, Aloush V, et al. Impaired diffusion tensor imaging findings in the corpus callosum and cingulum may underlie impaired learning and memory abilities in systemic lupus erythematosus. Lupus (2016) 25:1200–8. doi: 10.1177/0961203316636471
84. Urenjak J, Williams SR, Gadian DG, Noble M. Proton nuclear magnetic resonance spectroscopy unambiguously identifies different neural cell types. J Neurosci. (1993) 13:981–9. doi: 10.1523/JNEUROSCI.13-03-00981.1993
85. Choi JK, Dedeoglu A, Jenkins BG. Application of MRS to mouse models of neurodegenerative illness. NMR Biomed. (2007) 20:216–37. doi: 10.1002/nbm.1145
86. Appenzeller S, Li LM, Costallat LT, Cendes F. Evidence of reversible axonal dysfunction in systemic lupus erythematosus: a proton MRS study. Brain (2005) 128:2933–40. doi: 10.1093/brain/awh646
87. Appenzeller S, Li LM, Costallat LT, Cendes F. Neurometabolic changes in normal white matter may predict appearance of hyperintense lesions in systemic lupus erythematosus. Lupus (2007) 16:963–71. doi: 10.1177/0961203307084723
88. Brooks WM, Sibbitt WL Jr, Kornfeld M, Jung RE, Bankhurst AD, Roldan CA. The histopathologic associates of neurometabolite abnormalities in fatal neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. (2010) 62:2055–63. doi: 10.1002/art.27458
89. Sibbitt WL Jr, Sibbitt RR. Magnetic resonance spectroscopy and positron emission tomography scanning in neuropsychiatric systemic lupus erythematosus. Rheum Dis Clin North Am. (1993) 19:851–68.
90. Cagnoli P, Harris RE, Frechtling D, Berkis G, Gracley RH, Graft CC, et al. Reduced Insular Glutamine and N-acetylaspartate in systemic lupus erythematosus: a single-voxel (1)H-MR spectroscopy study. Acad Radiol. (2013) 20:1286–96. doi: 10.1016/j.acra.2013.07.011
91. Sibbitt WL Jr, Haseler LJ, Griffey RR, Friedman SD, Brooks WM. Neurometabolism of active neuropsychiatric lupus determined with proton MR spectroscopy. AJNR Am J Neuroradiol. (1997) 18:1271–7.
92. Zhang Z, Wang Y, Shen Z, Yang Z, Li L, Chen D, et al. The Neurochemical and microstructural changes in the brain of systemic lupus erythematosus patients: a multimodal MRI study. Sci Rep. (2016) 6:19026. doi: 10.1038/srep19026
93. Nicolay K, Braun KP, Graaf RA, Dijkhuizen RM, Kruiskamp MJ. Diffusion NMR spectroscopy. NMR Biomed. (2001) 14:94–111. doi: 10.1002/nbm.686
94. Ercan E, Magro-Checa C, Valabregue R, Branzoli F, Wood ET, Steup-Beekman GM, et al. Glial and axonal changes in systemic lupus erythematosus measured with diffusion of intracellular metabolites. Brain (2016) 139:1447–57. doi: 10.1093/brain/aww031
95. Wang PI, Cagnoli PC, McCune WJ, Schmidt-Wilcke T, Lowe SE, Graft CC, et al. Perfusion-weighted MR imaging in cerebral lupus erythematosus. Acad Radiol. (2012) 19:965–70. doi: 10.1016/j.acra.2012.03.023
96. Gasparovic CM, Roldan CA, Sibbitt WL Jr, Qualls CR, Mullins PG, Sharrar JM, et al. Elevated cerebral blood flow and volume in systemic lupus measured by dynamic susceptibility contrast magnetic resonance imaging. J Rheumatol. (2010) 37:1834–43. doi: 10.3899/jrheum.091276
97. Papadaki E, Fanouriakis A, Kavroulakis E, Karageorgou D, Sidiropoulos P, Bertsias G, et al. Neuropsychiatric lupus or not? Cerebral hypoperfusion by perfusion-weighted MRI in normal-appearing white matter in primary neuropsychiatric lupus erythematosus. Ann Rheum Dis. (2018) 77:441–8. doi: 10.1136/annrheumdis-2017-212285
98. Mikdashi JA. Altered functional neuronal activity in neuropsychiatric lupus: a systematic review of the fMRI investigations. Semin Arthritis Rheum. (2016) 45:455–62. doi: 10.1016/j.semarthrit.2015.08.002
99. DiFrancesco MW, Holland SK, Ris MD, Adler CM, Nelson S, DelBello MP, et al. Functional magnetic resonance imaging assessment of cognitive function in childhood-onset systemic lupus erythematosus: a pilot study. Arthritis Rheum. (2007) 56:4151–63. doi: 10.1002/art.23132
100. Mak A, Ren T, Fu EH, Cheak AA, Ho RC. A prospective functional MRI study for executive function in patients with systemic lupus erythematosus without neuropsychiatric symptoms. Semin Arthritis Rheum. (2012) 41:849–58. doi: 10.1016/j.semarthrit.2011.11.010
101. Mackay M, Bussa MP, Aranow C, Ulug AM, Volpe BT, Huerta PT, et al. Differences in regional brain activation patterns assessed by functional magnetic resonance imaging in patients with systemic lupus erythematosus stratified by disease duration. Mol Med. (2011) 17:1349–56. doi: 10.2119/molmed.2011.00185
102. Hou J, Lin Y, Zhang W, Song L, Wu W, Wang J, et al. Abnormalities of frontal-parietal resting-state functional connectivity are related to disease activity in patients with systemic lupus erythematosus. PLoS ONE (2013) 8:e74530. doi: 10.1371/journal.pone.0074530
103. Lin Y, Zou QH, Wang J, Wang Y, Zhou DQ, Zhang RH, et al. Localization of cerebral functional deficits in patients with non-neuropsychiatric systemic lupus erythematosus. Hum Brain Mapp. (2011) 32:1847–55. doi: 10.1002/hbm.21158
104. Heiss WD. Positron emission tomography imaging in gliomas: applications in clinical diagnosis, for assessment of prognosis and of treatment effects, and for detection of recurrences. Eur J Neurol. (2017) 24:1255-e70. doi: 10.1111/ene.13385
105. Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, et al. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer's disease. Brain (2011) 134:1089–100. doi: 10.1093/brain/awr038
106. Kantarci K, Senjem ML, Lowe VJ, Wiste HJ, Weigand SD, Kemp BJ, et al. Effects of age on the glucose metabolic changes in mild cognitive impairment. AJNR Am J Neuroradiol. (2010) 31:1247–53. doi: 10.3174/ajnr.A2070
107. Lee SW, Park MC, Lee SK, Park YB. The efficacy of brain (18)F-fluorodeoxyglucose positron emission tomography in neuropsychiatric lupus patients with normal brain magnetic resonance imaging findings. Lupus (2012) 21:1531–7. doi: 10.1177/0961203312459104
108. Airas L, Rissanen E, Rinne JO. Imaging neuroinflammation in multiple sclerosis using TSPO-PET. Clin Transl Imaging. (2015) 3:461–73. doi: 10.1007/s40336-015-0147-6
109. Wang Y, Coughlin JM, Ma S, Endres CJ, Kassiou M, Sawa A, et al. Neuroimaging of translocator protein in patients with systemic lupus erythematosus: a pilot study using [(11)C]DPA-713 positron emission tomography. Lupus (2017) 26:170–8. doi: 10.1177/0961203316657432
110. Driver CB, Wallace DJ, Lee JC, Forbess CJ, Pourrabbani S, Minoshima S, et al. Clinical validation of the watershed sign as a marker for neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. (2008) 59:332–7. doi: 10.1002/art.23308
111. Payoux P, Salabert AS. New PET markers for the diagnosis of dementia. Curr Opin Neurol. (2017) 30:608–16. doi: 10.1097/WCO.0000000000000489
112. Otte A, Weiner SM, Peter HH, Mueller-Brand J, Goetze M, Moser E, et al. Brain glucose utilization in systemic lupus erythematosus with neuropsychiatric symptoms: a controlled positron emission tomography study. Eur J Nucl Med. (1997) 24:787–91. doi: 10.1007/BF00879668
113. Ramage AE, Fox PT, Brey RL, Narayana S, Cykowski MD, Naqibuddin M, et al. Neuroimaging evidence of white matter inflammation in newly diagnosed systemic lupus erythematosus. Arthritis Rheum. (2011) 63:3048–57. doi: 10.1002/art.30458
114. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. (1999) 42:599–608. doi: 10.1002/1529-0131(199904)42:4<599::AID-ANR2>3.0.CO;2-F
115. Borowoy AM, Pope JE, Silverman E, Fortin PR, Pineau C, Smith CD, et al. Neuropsychiatric lupus: the prevalence and autoantibody associations depend on the definition: results from the 1000 faces of lupus cohort. Semin Arthritis Rheum. (2012) 42:179–85. doi: 10.1016/j.semarthrit.2012.03.011
116. Chen J, Feng X, Wang H, Hua B, Ding C, Liu B, et al. Discriminating infectious meningitis versus neuropsychiatric involvement in patients with systemic lupus erythematosus: a single-center experience. Clin Rheumatol. (2015) 34:365–9. doi: 10.1007/s10067-014-2726-8
117. Magro Checa C, Cohen D, Bollen EL, van Buchem MA, Huizinga TW, Steup-Beekman GM. Demyelinating disease in SLE: is it multiple sclerosis or lupus? Best Pract Res Clin Rheumatol. (2013) 27:405–24. doi: 10.1016/j.berh.2013.07.010
118. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. (2010) 9:776–85. doi: 10.1016/S1474-4422(10)70137-X
119. Brooks JD. Translational genomics: the challenge of developing cancer biomarkers. Genome Res. (2012) 22:183–7. doi: 10.1101/gr.124347.111
120. van der Meulen PM, Barendregt AM, Cuadrado E, Magro-Checa C, Steup-Beekman GM, Schonenberg-Meinema D, et al. Protein array autoantibody profiles to determine diagnostic markers for neuropsychiatric systemic lupus erythematosus. Rheumatology (2017) 56:1407–16. doi: 10.1093/rheumatology/kex073
121. Lefranc D, Launay D, Dubucquoi S, de Seze J, Dussart P, Vermersch M, et al. Characterization of discriminant human brain antigenic targets in neuropsychiatric systemic lupus erythematosus using an immunoproteomic approach. Arthritis Rheum. (2007) 56:3420–32. doi: 10.1002/art.22863
122. Comabella M, Martin R. Genomics in multiple sclerosis–current state and future directions. J Neuroimmunol. (2007) 187:1–8. doi: 10.1016/j.jneuroim.2007.02.009
123. Gelb S, Stock AD, Anzi S, Putterman C, Ben-Zvi A. Mechanisms of neuropsychiatric lupus: The relative roles of the blood-cerebrospinal fluid barrier versus blood-brain barrier. J Autoimmun. (2018) 91:34–44. doi: 10.1016/j.jaut.2018.03.001
124. Fulton JC, Grossman RI, Udupa J, Mannon LJ, Grossman M, Wei L, et al. MR lesion load and cognitive function in patients with relapsing-remitting multiple sclerosis. AJNR Am J Neuroradiol. (1999) 20:1951–5.
125. Louapre C, Govindarajan ST, Gianni C, Madigan N, Nielsen AS, Sloane JA, et al. The association between intra- and juxta-cortical pathology and cognitive impairment in multiple sclerosis by quantitative T2* mapping at 7 T MRI. Neuroimage Clin. (2016) 12:879–86. doi: 10.1016/j.nicl.2016.11.001
126. Tobyne SM, Ochoa WB, Bireley JD, Smith VM, Geurts JJ, Schmahmann JD, et al. Cognitive impairment and the regional distribution of cerebellar lesions in multiple sclerosis. Mult Scler. (2017) 24:1687–95. doi: 10.1177/1352458517730132
127. Sati P, George IC, Shea CD, Gaitan MI, Reich DS. FLAIR*: a combined MR contrast technique for visualizing white matter lesions and parenchymal veins. Radiology (2012) 265:926–32. doi: 10.1148/radiol.12120208
128. Maggi P, Absinta M, Grammatico M, Vuolo L, Emmi G, Carlucci G, et al. Central vein sign differentiates Multiple Sclerosis from central nervous system inflammatory vasculopathies. Ann Neurol. (2018) 83:283–94. doi: 10.1002/ana.25146
129. Miller AE, Calabresi PA. Central vein sign in multiple sclerosis: ready for front and center? Neurology (2018) 90:631–2. doi: 10.1212/WNL.0000000000005241
130. Fusar-Poli P, McGuire P, Borgwardt S. Mapping prodromal psychosis: a critical review of neuroimaging studies. Eur Psychiatry (2012) 27:181–91. doi: 10.1016/j.eurpsy.2011.06.006
131. Uludag K, Roebroeck A. General overview on the merits of multimodal neuroimaging data fusion. Neuroimage (2014) 102:3–10. doi: 10.1016/j.neuroimage.2014.05.018
132. Libero LE, DeRamus TP, Lahti AC, Deshpande G, Kana RK. Multimodal neuroimaging based classification of autism spectrum disorder using anatomical, neurochemical, and white matter correlates. Cortex (2015) 66:46–59. doi: 10.1016/j.cortex.2015.02.008
133. Chang K, Zhang B, Guo X, Zong M, Rahman R, Sanchez D, et al. Multimodal imaging patterns predict survival in recurrent glioblastoma patients treated with bevacizumab. Neuro Oncol. (2016) 18:1680–7. doi: 10.1093/neuonc/now086
134. O'Halloran R, Kopell BH, Sprooten E, Goodman WK, Frangou S. Multimodal neuroimaging-informed clinical applications in neuropsychiatric disorders. Front Psychiatry (2016) 7:63. doi: 10.3389/fpsyt.2016.00063
135. Liem F, Varoquaux G, Kynast J, Beyer F, Kharabian Masouleh S, Huntenburg JM, et al. Predicting brain-age from multimodal imaging data captures cognitive impairment. Neuroimage (2017) 148:179–88. doi: 10.1016/j.neuroimage.2016.11.005
136. Zeestraten EA, Lawrence AJ, Lambert C, Benjamin P, Brookes RL, Mackinnon AD, et al. Change in multimodal MRI markers predicts dementia risk in cerebral small vessel disease. Neurology (2017) 89:1869–76. doi: 10.1212/WNL.0000000000004594
Keywords: systemic lupus erythematosus, neuropsychiatric systemic lupus erythematosus, NP-SLE, neuroimaging, magnetic resonance imaging, biomarkers
Citation: Magro-Checa C, Steup-Beekman GM, Huizinga TW, van Buchem MA and Ronen I (2018) Laboratory and Neuroimaging Biomarkers in Neuropsychiatric Systemic Lupus Erythematosus: Where Do We Stand, Where To Go? Front. Med. 5:340. doi: 10.3389/fmed.2018.00340
Received: 05 October 2018; Accepted: 19 November 2018;
Published: 04 December 2018.
Edited by:
Antonis Fanouriakis, University General Hospital Attikon, GreeceReviewed by:
Alessandra Bortoluzzi, University of Ferrara, ItalyCheng-De Yang, Ruijin Hospital, China
Copyright © 2018 Magro-Checa, Steup-Beekman, Huizinga, van Buchem and Ronen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: César Magro-Checa, Qy5NYWdyb19DaGVjYUBsdW1jLm5s