 Robert J. Ju
Robert J. Ju Samantha J. Stehbens
Samantha J. Stehbens Nikolas K. Haass
Nikolas K. Haass
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Med. , 06 November 2018
Sec. Dermatology
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00307
This article is part of the Research Topic Insights into Biomarkers, Cytokines, and Chemokines in Skin Cancer View all 8 articles
The importance of studying cancer cell invasion is highlighted by the fact that 90% of all cancer-related mortalities are due to metastatic disease. Melanoma metastasis is driven fundamentally by aberrant cell motility within three-dimensional or confined environments. Within this realm of cell motility, cytokines, growth factors, and their receptors are crucial for engaging signaling pathways, which both mediate crosstalk between cancer, stromal, and immune cells in addition to interactions with the surrounding microenvironment. Recently, the study of the mechanical biology of tumor cells, stromal cells and the mechanics of the microenvironment have emerged as important themes in driving invasion and metastasis. While current anti-melanoma therapies target either the MAPK signaling pathway or immune checkpoints, there are no drugs available that specifically inhibit motility and thus invasion and dissemination of melanoma cells during metastasis. One of the reasons for the lack of so-called “migrastatics” is that, despite decades of research, the precise biology of metastatic disease is still not fully understood. Metastatic disease has been traditionally lumped into a single classification, however what is now emergent is that the biology of melanoma metastasis is highly diverse, heterogeneous and exceedingly dynamic—suggesting that not all cases are created equal. The following mini-review discusses melanoma heterogeneity in the context of the emergent theme of mechanobiology and how it influences the tumor-stroma crosstalk during metastasis. Thus, highlighting future therapeutic options for migrastatics and mechanomedicines in the prevention and treatment of metastatic melanoma.
In recent years, studies elucidating the biology of metastatic melanoma have revealed highly complex, yet dynamic processes, with equal parts unpredictability. The dynamic behavior of metastatic melanoma is highlighted by the multiple migration modalities employed to navigate the topography of a fluctuating microenvironment (Figures 1, 2). The ability of melanoma to both transition through multiple environments and integrate stromal signals with invasive and proliferative behaviors influences the response of metastatic melanoma to therapies. Advanced stage metastatic disease is particularly important as it is currently incurable by conventional targeted therapies and patient survival does not benefit from surgical resection alone (1). Furthermore, paradigm shifting “immuno-therapies” whilst effective in a subset of patients, are not effective in all (2–5). Thus, many patients diagnosed with metastatic melanoma are left with little to no treatment options, leaving survival prognosis bleak (1, 6–8). This underscores the importance of both studying and understanding the biology of melanoma, with the aim of using this knowledge to produce novel targeted therapies, or “migrastatics” (9), that specifically target both the cancer and the microenvironment to prevent metastatic spread.
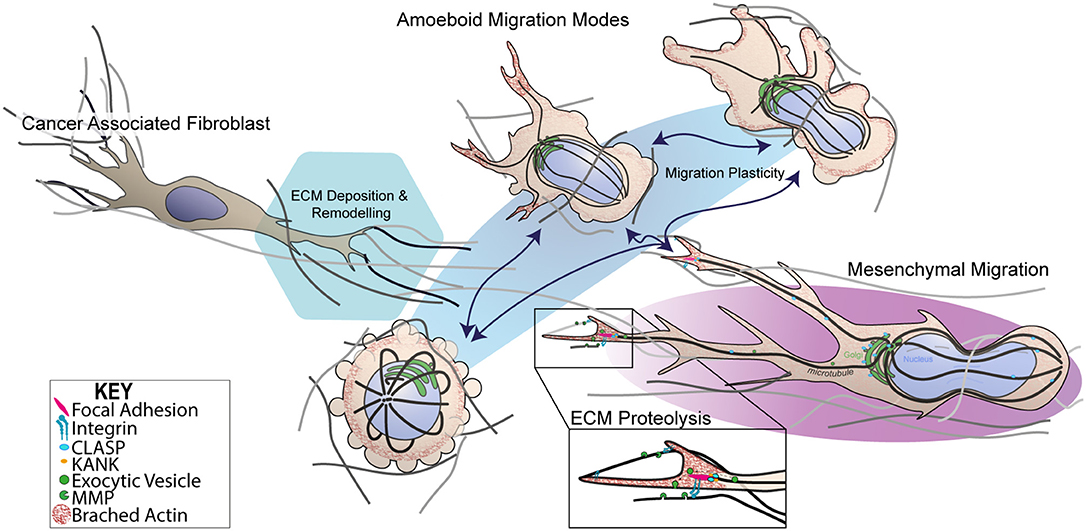
Figure 1. Dynamic switching of 3D migration modes in metastatic melanoma. Schematic representation of 3D mesenchymal and amoeboidal migration mode dynamic switching. Mesenchymal migration relies on RAC signaling to establish front-rear 3D polarity in order to generate a predominant pseudopodia, producing the characteristic spindle like morphology. Mesenchymal migration relies on MMP-dependent ECM degradation and integrin-dependent cell-matrix attachment protein complexes known as focal adhesions (FAs). FAs are thought to be hot spots for exocytic trafficking of MMPs, mediated by cortical microtubule stabilization complexes containing microtubule associated proteins, CLASPs, and FA adaptor proteins, KANKs. In contrast, amoeboid migration subsumes several migration modes from blebbing, chimneying to actin gliding modes. Importantly, amoeboidal migration requires little to no integrin activity and or MMP-mediated matrix degradation. Transformed cancer-associated fibroblasts (CAFs) are known to facilitate ECM changes through deposition of fibronectin that crosslinks collagen I fibers. CAF-dependent contractility further in reorganization of ECM influenceing melanoma migration and survival mechanosensory proteins.
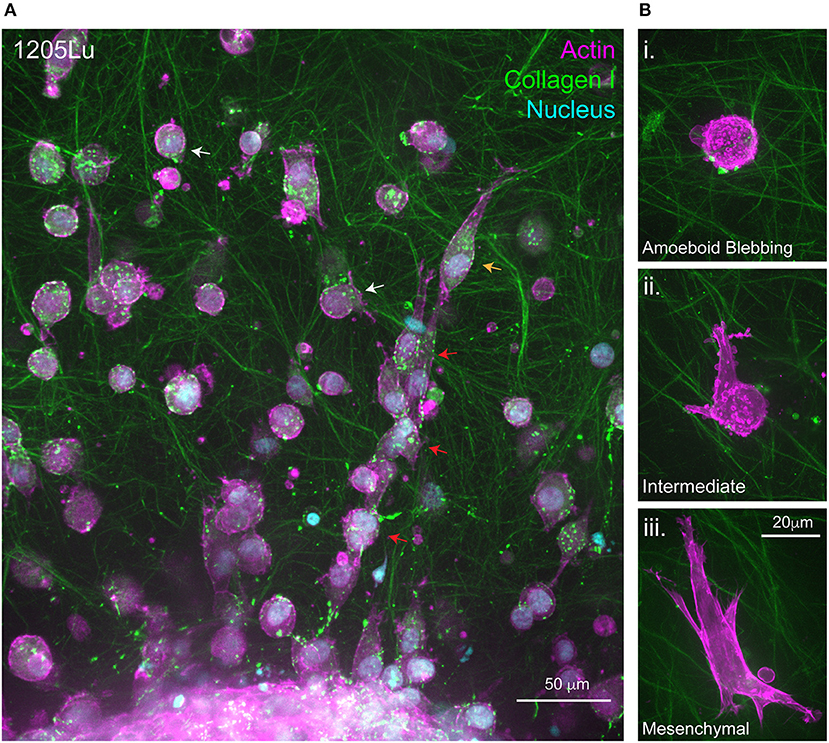
Figure 2. Migration at the 3D Melanoma-collagen interface. (A) Melanoma spheroids (1205Lu cells) embedded in a porous fibrillar collagen I hydrogel show heterogeneous 3D migration phenotypes at the spheroid-matrix interface. Within collagen I hydrogels, melanoma cells exhibit multi-cellular streaming (red arrows), single-cell rounded migration (white arrows) and polarized mesenchymal migration (orange arrow). (B) Representative high resolution spinning disc confocal images of single melanoma cells expressing fluorescently labeled filamentous actin (mScarlet-i-Lifeact) demonstrating several 3D migration phenotypes within the same collagen I hydrogel i. Amoeboid blebbing migration ii. Intermediate blebbing-pseudopodia phenotype and iii. Mesenchymal migration.
Melanoma metastasis is governed by the fundamental process of cell motility, whereby aberrantly transformed cancer cells hi-jack normal cellular processes used in homeostasis and development (10–12). Broadly, tumor cells that have subsumed a pro-invasive phenotype achieve metastatic dissemination through processes of intra- and extravasation to arrest at anatomically distant sites where they regain proliferative programming (13). Although we have progressively elucidated our understanding of metastatic behaviors, particularly those shared across multiple cancer types, the exact molecular mechanisms remain elusive and are of high clinical relevance (14–17). Metastatic melanoma spreads in an unpredictable fashion, often through parallel routes of widespread dissemination to multiple organs (18). Melanoma has been shown to engage in conventional intravasation and subsequent vascular and lymphovascular metastatsis (19) and also unconventional intravasation-independent migration known as “pericytic mimicry” (20, 21), where tumor cells migrate along the exterior of blood vessels. There have even been reports of melanoma cells exhibiting exhibiting “vasculogenic mimicry,” whereby they are proposed to form de novo vascular networks to promote tumor perfusion (22). Interestingly, parallels exist between the highly invasive nature of metastatic melanoma and their neural crest/melanoblast precursors, with the two sharing similar pro-migratory behavior attributes resulting in multiple studies suggesting that melanoma reactivates neural crest migration programs to drive plasticity and invasiveness in melanoma (12, 18, 23, 24).
Despite dissemination to most tissue types, melanoma exhibits metastatic tropism, preferentially metastasizing to the brain, lung, liver, small bowel or skin (25). Although the specific tumor-tissue tropism mechanisms are still unclear; chemokine receptors appear to play a role in tumor-tissue “homing” (26, 27). Recent studies show that cytokines and chemokines are integral to immune detection of melanoma cells by differentially regulating the behavior of monocytes, macrophages and natural killer cells (NK cells) (27, 28). Normally, these immune cells function to detect and kill pre-metastatic tumor cells. This process is mediated by the type 2 tumor suppressor protein, pigment epithelium-derived factor (PEDF), whereby PEDF-positive tumor-derived exosomes circulate the vasculature and mount immune responses. This results in, (1) macrophage differentiation and tumor cell detection through the modulation of the IL-10/12 axis, as well as (2) the recruitment of CX3CR1-expressing patrolling monocytes, which function to clear micro-particles and cellular debris from the microvasculature. Additionally, the recruitment and activation of NK cells has been shown to play an auxiliary role in tumor cell killing. The activation of these three arms results in immune detection of pre-metastatic melanoma cells ensuing in tumor death and clearance (27). However, PEDF expression in tumor cells and circulating exosomes is lost during metastatic melanoma transformation, and thus metastatic cells go undetected, allowing cellular debris and micro-particles to create pre-metastatic niches at distant microenvironments (27, 29–33). This process involves transforming and modulating local inflammatory immune cells, stromal cells and extracellular matrix (ECM) through the secretion of homing factors, inflammatory cytokines, and chemokines (34–36). Reciprocally, melanoma secreted cytokines and progressive increases in chemokine receptor expression during progression act to drive angiogenesis and metastasis to certain organs, respectively (37–39). Specifically, studies have shown that the ectopic expression of the chemokine receptor CCR7 in murine melanoma cells increases tumor-lymph node and -brain tissue homing in vivo (40), whilst CXCR4 promotes melanoma-lung tropism (41). However, melanoma tissue tropism is likely to be more complex as studies using human melanoma xenografts only partially recapitulate this phenomenon (42). Irrespectively, these findings demonstrate that chemokines play a role in the “tissue-homing,” supporting Paget's 1889 “Seed and Soil” hypothesis that postulated tumor metastasis to particular anatomical sites was driven by cellular mechanism, and not at random (43, 44).
The phenotype-switching model of melanoma heterogeneity (45–47) highlights the importance of understanding the influence of the microenvironment on invasive behavior, notably, how do cells move in 3D? 3D cell motility is a complex biophysical process, which occurs through dynamic interplay between cytoskeletal remodeling, plasma membrane deformation, acto-myosin contractility, and cell-matrix adhesion. The functional organization of these molecular components is highly adaptive, mechanically responsive and varies between cell and tissue types (48–50). The theme of mechanoreciprocity encompasses the rapidly growing knowledge that the cell-ECM interaction is in fact a bi-directional relationship resulting in a biophysical reciprocity whereby cancer cells switch between a growing repertoire of migration modalities to adapt to the geometry, topography, elasticity and chemical composition their surrounding microenvironment (Figure 1). The bi-direcitonal nature of mechanoreciprocity arrives from our understanding of the multi-component viscoelastic units (i.e., cells and the ECM), subject to reciprocal mechanical and chemical interactions that catalyze, assist or restrict cell migration, in context dependent manners (51). This relationship acts to facilitate changes in cell shape, position, the decision between single-cell and collective migration, and structural modification to tissue and the microenvironment through the deposition of matrix (52). The question of how melanoma cells interpret particular environmental cues and integrate a response hierarchy to each stimulus remains an exciting area with the capacity to influence therapies (9, 53).
Cell motility is driven by a symphony of cytoskeletal components, which act in fine spatial-temporal orchestration to facilitate movement. The cytoskeleton encompasses actin microfilaments, microtubules, intermediate filaments, and septins (48, 54–58). The spotlight for motility has long been held by the actin cytoskeleton, with many researchers proposing that targeting the actin cytoskeletal network during invasion, through rate limiting mechanisms, may abrogate cancer metastasis and invasion (9, 59–62). A substantial amount of our cell motility knowledge has been uncovered by studying cultured cells on stiff 2D substrates such as plastic and glass (63, 64). However, the greatest limitation that 2D migration biology imposes is that it very poorly recapitulates the biomechanical architecture within tissues and organs in mammalian tissue (63, 65, 66) which varies dramatically in bio-mechanical composition and thus impacts on mechanosensing (67). Similar to other 3D models of cancer, our studies, have demonstrated that 3D models more accurately mirror in vivo biology and tumor drug response when studying melanoma (68–71).
Similarly to 2D, cell shape changes observed in 3D migration are classically attributed to remodeling of the actin cytoskeleton and spatio-temporal regulation of RHO/RAC GTPase signaling that govern cytoskeletal dynamics (72). Traditionally, cancer cells predominantly exploit two modes of 3D or confined migration, categorized as either amoeboidal or mesenchymal migration (Figure 1) (73). More complex models have identified that cancer cells can also behave semi-collectively to exhibit multicellular streaming whereby cells invade in a chain-like fashion following a primary leader cell (Figure 2) (52). It is the ability of cells when subjected to different 3D ECM topographies to dynamically adapt to undergo the most efficient mode of migration (51) (often switching between migration mode sub-types), which highlights the role of mechanoreciprocity in facilitating dynamic plasticity during cancer invasion
One crucial feature that affects the cell determination of migration mode is the degree of ECM confinement (74). Increased confinement imposes a mechanical cell-deformation challenge, which allows cells to traverse through narrow spaces. Several landmark studies have demonstrated that the degree to which cancer cells are able to squeeze to facilitate migration is rate-limited by the degree of nuclear compression or distortion, defined as the “nuclear limit of migration,” which is equal to approximately 10% of the nuclear cross-sectional area (75, 76). If nuclear constriction falls beyond this limit, cells switch to migration modalities that upregulate ECM proteolysis, seen in the process of amoeboid-to-mesenchymal switching (50, 75, 77) or alternatively undergo nuclear envelope rupture which may affect cell viability (78–80).
Blebbing behavior for motility purposes was overlooked until a landmark study identified melanoma cells lacking the actin crosslinking protein, Filamin A, exhibited non-apoptotic blebbing which correlated with motility (81). The classification of amoeboid migration now subsumes a heterogeneous spectrum of migration modes ranging from blebbing, “chimneying” and actin-polymerisation gliding-based modes (Figure 1) (82). Amoeboid-based motility is rapid as it entails little to no integrin activity and does not require proteolytic degradation, opening of junctions, penetration of basement membranes, or breaching endothelial vasculature (50, 83). Instead, surrounding surfaces immobilize cells with the transmission of traction forces alone being sufficient for cells movement. The mechanical drivers of amoeboid migration are attributed to the RHOA-ROCK-myosin II signaling axis which governs acto-myosin contractility to generate membrane blebs which are both reinforced by ROCK-dependent JAK-STAT3 signaling and maintained by IL6-STAT3 (84, 85). Myosin II contractile activity drives membrane blebbing by localized rises in hydrostatic pressure resulting in the rupture of cortical actin networks (86) or localized detachment of plasma membrane from the cortical actin cytoskeleton (87). Both mechanisms that generate hydrostatic pressure gradients are able to produce spherical membrane blebbing as a result of cytosolic content flow that protrudes the plasma membrane. The subsequent membrane bleb retraction occurs once pressure is equilibrated (88). Acto-myosin blebbing activity functions in tandem with RHO-RAC inhibitory crosstalk, which generates tractional force through the inter-digitation of blebs or actin based protrusions into gaps within the matrix environment (89). The cytokine TGFβ has also been shown to promote amoeboid migration and invasion in melanoma via a SMAD/CITED-dependent transcriptional activation of key genes involved in sustained actomyosin contractility (90).
Mesenchymal or proteolytic-dependent modes of migration occur when cells are subjected to highly confined microenvironments, that are not permissible to squeezing-based migration modes, requiring cells to remodel the matrix to facilitate invasion (50). Cells engaged in mesenchymal migration exhibit polarized spindle-like morphologies, which are highly dependent on integrin-based ECM adhesion and proteolysis of ECM (Figure 1) (91). The characteristic polarized phenotype of mesenchymal migration and also the formation of a predominant pseudopodium, analogous to a lamellipodium in 2D, arises from RAC signaling which results in a slow, but highly directional 3D migration (Figure 2iii) (92–94). Interestingly, the RAC1 GTPase activating mutation (P29S) is the third most frequently occurring mutation in sun-exposed melanoma (~9%). RAC1 plays significant roles in lamella actin dynamics, acting upstream of key actin regulatory proteins including Arp2/3 and Cofilin to drive membrane protrusion in 2D. RAC1P29S drives resistance to targeted therapy (84, 85) via mechanosensory-dendritic actin polymerization in low compliance environments which drives proliferation via a MAPK-independent pathway (95). RAC1 also affects 3D cell motility (86) and inhibits RHO-ROCK signaling, further highlighting that the presence and high frequency of RAC1P29S in melanoma cases may have implications on both melanoma metastasis and survival in these patients.
The pseudopodial extensions are created by polarized actin polymerisation which results in the breaking of symmetry and formation of a dominant protrusion. Pseudopodia a facilitates directional 3D migration whilst also allowing cells to mechanosense their microenvironments. Pseudopodial extensions are stabilized by interactions with the matrix through integrin transmembrane receptor interactions, which scaffold into multiprotein complexes known as focal adhesions (Figures 1, 2iii.). These mechanosensory complexes determine changes in substrate composition and stiffness primarily through the force transduction proteins talin and vinculin which scaffold onto the acto-myosin cytoskeleton (Figure 1) (96). Force-dependent conformational changes in talin act to coordinate recruitment of key focal adhesion signaling and scaffolding proteins [i.e., KANKS (97)] by exposing and disrupting binding-sites in a tension-dependent manner (98). Additionally, focal adhesions serve as ECM anchor points, enabling actin-dependent traction force generation via a molecular-clutch to drive cells forward (99). Synchronous acto-myosin contractility occurs at the rear of the cell which facilitates the disassembly of trailing adhesion sites whilst also propelling the cell body and nucleus forward to drive migration (50, 100).
An important facet of mesenchymal migration is the focalization of matrix degradation, which is dependent on targeted matrix metalloprotease (MMP) secretion. A landmark study by Wolf and Friedl, identified MMP focalization to the neck of pseudopodia (101). Although the mechanism of MMP targeting during collagenolysis is still unclear, we have demonstrated that microtubules and the associated +TIP proteins, CLASPs, play a crucial role in the release of ECM-matrix interactions via targeted delivery of MMPs to adhesion sites (Figure 1) (102). We have recently identified that CLASPs are overexpressed in several metastatic melanoma cell lines, supported by gene expression database analysis (103–105), outlining a potential role for this process to be dysregulated in metastatic melanoma. Understanding the mechanisms governing MMP-targeted secretion in the context of 3D models will elucidate how cells co-ordinate polarized proteolysis to drive invasion in response to the microenvironment. Nuclear-deformation is also thought to play a mechanosensitive rate limiting role as nuclear deformation has been shown to arrest migration if proteolysis is inhibited (75, 76). Although microtubules are less well understood in the context of 3D migration, we know that confined migration requires microtubules (106) and microtubules are mechanically coupled to the nuclear envelope (107). This in addition to microtubule roles in trafficking, polarity and spatio-temporal regulation of signaling, highlight that microtubules are likely to play key roles in melanoma invasion (60, 63, 108).
Binary concepts like Epithelial to Mesenchymal Transition (EMT), and the reverse (MET), have been extensively studied in the fields of development and cancer (109). Our current understanding of cell motility in complex environments highlights that the initiation of invasion is not as simple as a transcriptional program switch and is heavily influenced by mechanochemical signaling and the plasticity it evokes. Until recently, the two dichotomous modes of 3D migration, mesenchymal or amoeboid, were thought to be independent. We now know they are likely to be a continuum of a single migration process whereby cells dynamically switch between modes (Figure 1). This adaptive response is highlighted in cases whereby cells are subjected to continuous vs. non-continuous confinement, which catalyze very different migration phenotypes (Figures 2A,B). Continuous confinement is analogous to the cellular movement in a parallel, tube-like, structure or between two planes of tissue, resulting in cellular preference for nuclear piston-driven lobopodial migration (110, 111); whereas, non-continuous confinement is analogous to cells traversing between networks of fibers resulting in squeezing-based amoeboid migration (Figures 2Bi,ii) (50). Importantly, the premise of mechanoreciprocity and migrational plasticity has the potential to encompass many more modes of migration that we do not yet fully understand and have not yet discovered.
Melanoma cell invasion is facilitated by dynamic interactions between melanoma, ECM (112) and stroma (113, 114), to which the mechanics of ECM play key roles (115). We now know the surrounding tumor stroma is considered to be equally important as the tumor itself (116, 117). This understanding encompasses findings that demonstrate that during disease progression, tumor cells do not act in isolation, instead reciprocally interacting with the surrounding stroma to facilitate tumor cell dissemination. This process occurs through tumor recruitment of stromal cells (117–119), causing stromal cells to co-evolve with the primary tumor which triggers the metastatic cascade (116). This tumor-stroma cross talk involves a “melting pot” of extracellular matrices, fibroblasts, adipocytes, pericytes, endothelial, immune, and inflammatatory cells, such as macrophages and neutrophils (117, 120). The process of stromal cell recruitment is facilitated by tumor-secreted growth factors in a continual paracrine fashion (52). This disruption of the finely-tuned balance of stromal homeostasis and elicits transformed stromal cells to alter the tumor microenvironment to facilitate permissive conditions for tumor cell invasion (116). In a recent study, transformed cancer-associated fibroblasts (CAFs), were shown to facilitate tumor invasion through integrin αVβ3-dependent fibronectin secretion, which induced mechanical changes in the ECM through the contraction of collagen fibers (Figure 1) (105). The geometrical and mechanical ECM changes have been shown to affect the initial onset of invasion at the tumor-ECM interface (108, 109). Reciprocally, stromal secretion of growth factors elicits changes in primary tumor cell behavior, resulting in tumor cell secretion of proteolytic enzymes, including MMPs (116, 121) which have both catalytic and non-catalytic functions in melanoma invasion (32). Tumor-stromal crosstalk also plays a role in resistance to therapies where CAF-dependent ECM remodeling provides a therapeutic “safe-haven” for BRAF mutant melanoma from the BRAF inhibitor PLX4720 (122). This protective stromal signaling was mediated by reactivation of ERK and MAPK, via adhesion-dependent β1-integrin-FAK-Src signaling.
In summary, the many targets that drive metastatic melanoma motility are complex, diverse and outline biological heterogeneity. Our review highlights insight into the idea that motility and survival are not mechanistically separate and thus identifying targets which prevent melanoma migration, may also act to interrupt pro-survival pathways, strengthening the idea behind “mechanomedicines” for cancer (53). The search for migrastatics aims to synthesize the many findings that drive aberrant motility in melanoma to identify a crucial mechanism that melanoma cells engage that is not only specifically dysregulated, but the inhibition of such pathway is inescapable for halting melanoma motility.
RJ wrote this mini review under the guidance of SS and NH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
RJ holds a Research Training Program Scholarship for Ph. D. in Biomedical Research, funded by the Australian Government and the University of Queensland. SS is funded by the Rebecca L. Cooper Medical Research Foundation (PG2018168). NH is a Cameron fellow of the Melanoma and Skin Cancer Research Institute, Australia, and currently funded by the National Health and Medical Research Council (APP1084893).
1. Leung AM, Hari DM, Morton DL. Surgery for distant melanoma metastasis. Cancer J. (2012) 18:176–84. doi: 10.1097/PPO.0b013e31824bc981
2. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. (2015) 372:2006–17. doi: 10.1056/NEJMoa1414428
3. Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. (2011) 29:3085–96. doi: 10.1200/JCO.2010.33.2312
4. Redman JM, Gibney GT, Atkins MB. Advances in immunotherapy for melanoma. Mediators Inflamm. (2016) 14:20. doi: 10.1186/s12916-016-0571-0
5. Beaver JA, Hazarika M, Mulkey F, Mushti S, Chen H, He K, et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: a US Food and Drug Administration pooled analysis. Lancet Oncol. (2018) 19:229–39. doi: 10.1016/S1470-2045(17)30846-X
6. Faries MB, Mozzillo N, Kashani-Sabet M, Thompson JF, Kelley MC, DeConti RC, et al. Long-term survival after complete surgical resection and adjuvant immunotherapy for distant melanoma metastases. Ann Surg Oncol. (2017) 24:3991–4000. doi: 10.1245/s10434-017-6072-3
7. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
8. Fellner C. Ipilimumab (Yervoy) prolongs survival in advanced melanoma: serious side effects and a hefty price tag may limit its use. PT. (2012) 37:503–30.
9. Gandalovičová A, Rosel D, Fernandes M, Veselý P, Heneberg P, Cermák V, et al. Migrastatics - anti-metastatic and anti-invasion drugs: promises and challenges. Trends Cancer (2017) 3:391–406. doi: 10.1016/j.trecan.2017.04.008
10. Gupta PB, Kuperwasser C, Brunet JP, Ramaswamy S, Kuo WL, Gray JW, et al. The melanocyte differentiation program predisposes to metastasis after neoplastic transformation. Nat Genet. (2005) 37:1047–54. doi: 10.1038/ng1634
11. Mitra D, Fisher DE. Transcriptional regulation in melanoma. Hematol Oncol Clin North Am. (2009) 23:447–65. doi: 10.1016/j.hoc.2009.03.003
12. Bailey CM, Morrison JA, Kulesa PM. Melanoma revives an embryonic migration program to promote plasticity and invasion. Pigment Cell Melanoma Res. (2012) 25:573–83. doi: 10.1111/j.1755-148X.2012.01025.x
13. Valastyan S, Weinberg Robert A. Tumor metastasis: molecular insights and evolving paradigms. Cell (2011) 147:275–92. doi: 10.1016/j.cell.2011.09.024
14. Zetter BR. The cellular basis of site-specific tumor metastasis. N Engl J Med. (1990) 322:605–12. doi: 10.1056/NEJM199003013220907
15. Zbytek B, Carlson JA, Granese J, Ross J, Mihm MC, Slominski A. Current concepts of metastasis in melanoma. Expert Rev Dermatol. (2008) 3:569–85. doi: 10.1586/17469872.3.5.569
16. Damsky WE, Rosenbaum LE, Bosenberg M. Decoding melanoma metastasis. Cancers (2011) 3:126–63. doi: 10.3390/cancers3010126
18. Arozarena I, Wellbrock C. Targeting invasive properties of melanoma cells. FEBS J. (2017) 284:2148–62. doi: 10.1111/febs.14040
19. Pandya P, Orgaz JL, Sanz-Moreno V. Modes of invasion during tumour dissemination. Mol Oncol. (2017) 11:5–27. doi: 10.1002/1878-0261.12019
20. Landsberg J, Tüting T, Barnhill RL, Lugassy C. The role of neutrophilic inflammation, angiotropism, and pericytic mimicry in melanoma progression and metastasis. J Invest Dermatol. (2016) 136:372–7. doi: 10.1016/j.jid.2015.11.013
21. Lugassy C, Zadran S, Bentolila LA, Wadehra M, Prakash R, Carmichael ST, et al. Angiotropism, pericytic mimicry and extravascular migratory metastasis in melanoma: an alternative to intravascular cancer dissemination. Cancer Microenviron. (2014) 7:139–52. doi: 10.1007/s12307-014-0156-4
22. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LMG, Pe'er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. (1999) 155:739–52. doi: 10.1016/S0002-9440(10)65173-5
23. Kulesa PM, Morrison JA, Bailey CM. The neural crest and cancer: a developmental spin on melanoma. Cells Tissues Organs (2013) 198:12–21. doi: 10.1159/000348418
24. Maguire LH, Thomas AR, Goldstein AM. Tumors of the neural crest: common themes in development and cancer. Dev Dyn. (2015) 244:311–22. doi: 10.1002/dvdy.24226
25. Maxwell R, Garzon-Muvdi T, Lipson EJ, Sharfman WH, Bettegowda C, Redmond KJ, et al. BRAF-V600 mutational status affects recurrence patterns of melanoma brain metastasis. Int J Cancer (2017) 140:2716–27. doi: 10.1002/ijc.30241
26. Richmond A, Yang J, Su Y. The good and the bad of chemokines/chemokine receptors in melanoma. Pigment Cell Melanoma Res. (2009) 22:175–86. doi: 10.1111/j.1755-148X.2009.00554.x
27. Plebanek MP, Angeloni NL, Vinokour E, Li J, Henkin A, Martinez-Marin D, et al. Pre-metastatic cancer exosomes induce immune surveillance by patrolling monocytes at the metastatic niche. Nat Commun. (2017) 8:1319. doi: 10.1038/s41467-017-01433-3
28. Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, et al. Patrolling monocytes control tumor metastasis to the lung. Science (2015) 350:985–90. doi: 10.1126/science.aac9407
29. Dadras SS, Lin RJ, Razavi G, Kawakami A, Du J, Feige E, et al. A novel role for microphthalmia-associated transcription factor-regulated pigment epithelium-derived factor during melanoma progression. Am J Pathol. (2015) 185:252–65. doi: 10.1016/j.ajpath.2014.09.012
30. Fernandez-Barral A, Orgaz JL, Baquero P, Ali Z, Moreno A, Tiana M, et al. Regulatory and functional connection of microphthalmia-associated transcription factor and anti-metastatic pigment epithelium derived factor in melanoma. Neoplasia (2014) 16:529–42. doi: 10.1016/j.neo.2014.06.001
31. Ladhani O, Sanchez-Martinez C, Orgaz JL, Jimenez B, Volpert OV. Pigment epithelium-derived factor blocks tumor extravasation by suppressing amoeboid morphology and mesenchymal proteolysis. Neoplasia (2011) 13:633–42. doi: 10.1593/neo.11446
32. Orgaz JL, Ladhani O, Hoek KS, Fernandez-Barral A, Mihic D, Aguilera O, et al. Loss of pigment epithelium-derived factor enables migration, invasion and metastatic spread of human melanoma. Oncogene (2009) 28:4147–61. doi: 10.1038/onc.2009.284
33. Sarkar D, Leung EY, Baguley BC, Finlay GJ, Askarian-Amiri ME. Epigenetic regulation in human melanoma: past and future. Epigenetics (2015) 10:103–21. doi: 10.1080/15592294.2014.1003746
34. Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell (2009) 15:35–44. doi: 10.1016/j.ccr.2008.11.012
35. Liu Y, Gu Y, Han Y, Zhang Q, Jiang Z, Zhang X, et al. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell (2016) 30:243–56. doi: 10.1016/j.ccell.2016.06.021
36. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol. (2006) 8:1369–75. doi: 10.1038/ncb1507
37. Richmond A. NF-κB, chemokine gene transcription and tumour growth. Nat Rev Immunol. (2002) 2:664–74. doi: 10.1038/nri887
38. Yang J, Amiri KI, Burke JR, Schmid JA, Richmond A. BMS-345541Targets inhibitor of κb kinase and induces apoptosis in melanoma: involvement of nuclear factor κb and mitochondria pathways. Clin Cancer Res. (2006) 12:950–60. doi: 10.1158/1078-0432.CCR-05-1220
39. Yang J, Pan W-H, Clawson GA, Richmond A. Systemic targeting inhibitor of κb kinase inhibits melanoma tumor growth. Cancer Res. (2007) 67:3127–34. doi: 10.1158/0008-5472.CAN-06-3547
40. Wiley HE, Gonzalez EB, Maki W, Wu MT, Hwang ST. Expression of CC chemokine receptor-7 and regional lymph node metastasis of B16 murine melanoma. J Natl Cancer Inst. (2001) 93:1638–43. doi: 10.1093/jnci/93.21.1638
41. Murakami T, Maki W, Cardones AR, Fang H, Tun Kyi A, Nestle FO, et al. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. (2002) 62:7328–34.
42. Izraely S, Klein A, Sagi-Assif O, Meshel T, Tsarfaty G, Hoon DSB, et al. Chemokine–chemokine receptor axes in melanoma brain metastasis. Immunol Lett. (2010) 130:107–14. doi: 10.1016/j.imlet.2009.12.003
43. Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. (1989) 8:98–101.
44. Paget S. The distribution of secondary growths in cancer of the breast. Lancet (1889) 133:571–3. doi: 10.1016/S0140-6736(00)49915-0
45. Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. (2008) 68:650–6. doi: 10.1158/0008-5472.CAN-07-2491
46. Hoek KS, Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. (2010) 23:746–59. doi: 10.1111/j.1755-148X.2010.00757.x
47. Widmer DS, Hoek KS, Cheng PF, Eichhoff OM, Biedermann T, Raaijmakers MIG, et al. Hypoxia contributes to melanoma heterogeneity by triggering HIF1α-dependent phenotype switching. J Invest Dermatol. (2013) 133:2436–43. doi: 10.1038/jid.2013.115
48. Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev. (2014) 94:235–63. doi: 10.1152/physrev.00018.2013
49. Carlier MF, Pernier J, Montaville P, Shekhar S. Kuhn S. Control of polarized assembly of actin filaments in cell motility. Cell Mol Life Sci. (2015) 72:3051–67. doi: 10.1007/s00018-015-1914-2
50. Petrie RJ, Yamada KM. At the leading edge of three-dimensional cell migration. J Cell Sci. (2012) 125:5917–26. doi: 10.1242/jcs.093732
51. van Helvert S, Storm C, Friedl P. Mechanoreciprocity in cell migration. Nat Cell Biol. (2018) 20:8–20. doi: 10.1038/s41556-017-0012-0
52. Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell (2011) 147:992–1009. doi: 10.1016/j.cell.2011.11.016
53. Lampi MC, Reinhart-King CA. Targeting extracellular matrix stiffness to attenuate disease: from molecular mechanisms to clinical trials. Sci Transl Med. (2018) 10:eaao0475. doi: 10.1126/scitranslmed.aao0475
54. Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature (2010) 463:485–92. doi: 10.1038/nature08908
56. Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol. (2012) 13:183–94. doi: 10.1038/nrm3284
57. Weirich CS, Erzberger JP, Barral Y. The septin family of GTPases: architecture and dynamics. Nat Rev Mol Cell Biol. (2008) 9:478–99. doi: 10.1038/nrm2407
58. Bezanilla M, Gladfelter AS, Kovar DR, Lee W-L. Cytoskeletal dynamics: a view from the membrane. J Cell Biol. (2015) 209:329–37. doi: 10.1083/jcb.201502062
59. Davidson AJ, Wood W. Unravelling the actin cytoskeleton: a new competitive edge? Trends Cell Biol. (2016) 26:569–76. doi: 10.1016/j.tcb.2016.04.001
60. Etienne-Manneville S. Actin and microtubules in cell motility: which one is in control? Traffic (2004) 5:470–7. doi: 10.1111/j.1600-0854.2004.00196.x
61. Nürnberg A, Kitzing T, Grosse R. Nucleating actin for invasion. Nat Rev Cancer (2011) 11:177–87. doi: 10.1038/nrc3003
62. Chen L, Yang S, Jakoncic J, Zhang JJ, Huang X-Y. Migrastatin analogues target fascin to block tumour metastasis. Nature (2010) 464:1062–6. doi: 10.1038/nature08978
63. Bouchet BP, Akhmanova A. Microtubules in 3D cell motility. J Cell Sci. (2017) 130:39–50. doi: 10.1242/jcs.189431
64. Doyle AD, Carvajal N, Jin A, Matsumoto K, Yamada KM. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat Commun. (2015) 6:8720. doi: 10.1038/ncomms9720
65. Elsdale T, Bard J. Collagen substrata for studies on cell behaviour. J Cell Biol. (1972) 54:626–37. doi: 10.1083/jcb.54.3.626
66. Zaman MH, Trapani LM, Sieminski AL, MacKellar D, Gong H, Kamm RD, et al. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc Natl Acad Sci USA. (2006) 103:10889–94. doi: 10.1073/pnas.0604460103
67. Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. (1982) 99:31–68. doi: 10.1016/0022-5193(82)90388-5
68. Spoerri L, Beaumont KA, Anfosso A, Haass NK. Real-time cell cycle imaging in a 3D cell culture model of melanoma. Methods Mol Biol. (2017) 1612:401–16. doi: 10.1007/978-1-4939-7021-6_29
69. Haass NK, Beaumont KA, Hill DS, Anfosso A, Mrass P, Munoz MA, et al. Real-time cell cycle imaging during melanoma growth, invasion, and drug response. Pigment Cell Melanoma Res. (2014) 27:764–76. doi: 10.1111/pcmr.12274
70. Smalley KS, Lioni M, Noma K, Haass NK, Herlyn M. In vitro three-dimensional tumor microenvironment models for anticancer drug discovery. Expert Opin Drug Discov. (2008) 3:1–10. doi: 10.1517/17460441.3.1.1
71. Santiago-Walker A, Li L, Haass NK, Herlyn M. Melanocytes: from morphology to application. Skin Pharmacol Physiol. (2009) 22:114–21. doi: 10.1159/000178870
73. Liu Y-J, Le Berre M, Lautenschlaeger F, Maiuri P, Callan-Jones A, Heuzé M, et al. Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell (2015) 160:659–72. doi: 10.1016/j.cell.2015.01.007
74. Paul CD, Mistriotis P, Konstantopoulos K. Cancer cell motility: lessons from migration in confined spaces. Nat Rev Cancer (2017) 17:131–40. doi: 10.1038/nrc.2016.123
75. Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. (2013) 201:1069–84. doi: 10.1083/jcb.201210152
76. McGregor AL, Hsia C-R, Lammerding J. Squish and squeeze – the nucleus as a physical barrier during migration in confining environments. Curr Opin Cell Biol. (2016) 40:32–40. doi: 10.1016/j.ceb.2016.01.011
77. Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, et al. Compensation mechanism in tumor cell migration: mesenchymal–amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. (2003) 160:267–77. doi: 10.1083/jcb.200209006
78. Shah P, Wolf K, Lammerding J. Bursting the bubble - nuclear envelope rupture as a path to genomic instability? Trends Cell Biol. (2017) 27:546–55. doi: 10.1016/j.tcb.2017.02.008
79. Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, et al. Nuclear envelope rupture and repair during cancer cell migration. Science (2016) 352:353–8. doi: 10.1126/science.aad7297
80. Raab M, Gentili M, de Belly H, Thiam H-R, Vargas P, Jimenez AJ, et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science (2016) 352:359–62. doi: 10.1126/science.aad7611
81. Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, et al. Actin-binding protein requirement for cortical stability and efficient locomotion. Science (1992) 255:325–7. doi: 10.1126/science.1549777
82. Lämmermann T, Sixt M. Mechanical modes of 'amoeboid' cell migration. Curr Opin Cell Biol. (2009) 21:636–44. doi: 10.1016/j.ceb.2009.05.003
83. Friedl P, Wolf K. Proteolytic and non-proteolytic migration of tumour cells and leucocytes. Biochem Soc Symp. (2003) 70:277–85.
84. Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell (2011) 20:229–45. doi: 10.1016/j.ccr.2011.06.018
85. Orgaz JL, Pandya P, Dalmeida R, Karagiannis P, Sanchez-Laorden B, Viros A, et al. Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun. (2014) 5:4255. doi: 10.1038/ncomms5255
86. Paluch E, Piel M, Prost J, Bornens M. Sykes, C. Cortical actomyosin breakage triggers shape oscillations in cells and cell fragments. Biophys J. (2005) 89:724–33. doi: 10.1529/biophysj.105.060590
87. Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison TJ. Non-equilibration of hydrostatic pressure in blebbing cells. Nature (2005) 435:365–9. doi: 10.1038/nature03550
88. Charras GT, Hu C-K, Coughlin M, Mitchison TJ. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. (2006) 175:477–90. doi: 10.1083/jcb.200602085
89. Tozluoglu M, Tournier AL, Jenkins RP, Hooper S, Bates PA, Sahai E. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol. (2013) 15:751–62. doi: 10.1038/ncb2775
90. Cantelli G, Orgaz Jose L, Rodriguez-Hernandez I, Karagiannis P, Maiques O, Matias-Guiu X, et al. TGF-β-induced transcription sustains amoeboid melanoma migration and dissemination. Curr Biol. (2015) 25:2899–914. doi: 10.1016/j.cub.2015.09.054
91. Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science (2001) 294:1708–12. doi: 10.1126/science.1064829
92. Friedl P, Zanker KS, Brocker EB. Cell migration strategies in 3-D extracellular matrix: differences in morphology, cell matrix interactions, and integrin function. Microsc Res Tech. (1998) 43:369–78. doi: 10.1002/(SICI)1097-0029(19981201)43:5<369::AID-JEMT3>3.0.CO;2-6
93. Friedl P, Maaser K, Klein CE, Niggemann B, Krohne G, et al. Migration of highly aggressive MV3 melanoma cells in 3-dimensional collagen lattices results in local matrix reorganization and shedding of α2 and β1 integrins and CD44. Cancer Res. (1997) 57:2061–70.
94. Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. (2010) 8:23. doi: 10.1186/1478-811X-8-23
95. Mohan AS, Dean KM, Kasitinon SY, Isogai T, Siruvallur Murali V, Han SJ, et al. Hyperactive Rac1 drives MAPK-independent proliferation in melanoma by assembly of a mechanosensitive dendritic actin network. bioRxiv [preprint] (2018). doi: 10.1101/326710
96. Swaminathan V, Alushin GM, Waterman CM. Mechanosensation: a catch bond that only hooks one way. Curr Biol. (2017) 27:R1158–60. doi: 10.1016/j.cub.2017.09.023
97. Bouchet BP, Gough RE, Ammon Y-C, van de Willige D, Post H, Jacquemet G, et al. Talin-KANK1 interaction controls the recruitment of cortical microtubule stabilizing complexes to focal adhesions. eLife (2016) 5:e18124. doi: 10.7554/eLife.18124
98. Yao M, Goult BT, Klapholz B, Hu X, Toseland CP, Guo Y, et al. The mechanical response of talin. Nat Commun. (2016) 7:11966. doi: 10.1038/ncomms11966
99. Hu K, Ji L, Applegate KT, Danuser G, Waterman-Storer CM. Differential transmission of actin motion within focal adhesions. Science (2007) 315:111–5. doi: 10.1126/science.1135085
100. Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer (2003) 3:362–74. doi: 10.1038/nrc1075
101. Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. (2007) 9:893–904. doi: 10.1038/ncb1616
102. Stehbens SJ, Paszek M, Pemble H, Ettinger A, Gierke S, Wittmann T. CLASPs link focal-adhesion-associated microtubule capture to localized exocytosis and adhesion site turnover. Nat Cell Biol. (2014) 16:561–73. doi: 10.1038/ncb2975
103. Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, et al. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. (2005) 11:7234–42. doi: 10.1158/1078-0432.CCR-05-0683
104. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
105. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. (2013) 6:pl1. doi: 10.1126/scisignal.2004088
106. Balzer EM, Tong Z, Paul CD, Hung W-C, Stroka KM, Boggs AE, et al. Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. (2012) 26:4045–56. doi: 10.1096/fj.12-211441
107. Liu L, Luo Q, Sun J, Song G. Nucleus and nucleus-cytoskeleton connections in 3D cell migration. Exp Cell Res. (2016) 348:56–65. doi: 10.1016/j.yexcr.2016.09.001
108. Bouchet BP, Noordstra I, van Amersfoort M, Katrukha EA, Ammon Y-C, ter Hoeve ND, et al. Mesenchymal cell invasion requires cooperative regulation of persistent microtubule growth by SLAIN2 and CLASP1. Dev Cell (2016) 39:708–23. doi: 10.1016/j.devcel.2016.11.009
109. Sanz-Moreno V. Tumour invasion: a new twist on rac-driven mesenchymal migration. Curr Biol. (2012) 22:R449–51. doi: 10.1016/j.cub.2012.04.024
110. Petrie R, Gavara N, Chadwick R, Yamada K. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol. (2012) 197:439–55. doi: 10.1083/jcb.201201124
111. Petrie RJ, Harlin HM, Korsak LIT, Yamada KM. Activating the nuclear piston mechanism of 3D migration in tumor cells. J Cell Biol. (2016) 216:93–100. doi: 10.1083/jcb.201605097
112. Miskolczi Z, Smith MP, Rowling EJ, Ferguson J, Barriuso J, Wellbrock C. Collagen abundance controls melanoma phenotypes through lineage-specific microenvironment sensing. Oncogene (2018) 37:3166–82. doi: 10.1038/s41388-018-0209-0
113. Roh-Johnson M, Shah AN, Stonick JA, Poudel KR, Kargl J, Yang GH, et al. Macrophage-dependent cytoplasmic transfer during melanoma invasion in vivo. Dev Cell (2017) 43:549–62.e6. doi: 10.1016/j.devcel.2017.11.003
114. Chapman A, Fernandez del Ama L, Ferguson J, Kamarashev J, Wellbrock C, Hurlstone A. Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep. (2014) 8:688–95. doi: 10.1016/j.celrep.2014.06.045
115. Tanner K. Perspective: the role of mechanobiology in the etiology of brain metastasis. APL Bioeng. (2018) 2:031801. doi: 10.1063/1.5024394
116. Mueller MM, Fusenig NE. Friends or foes — bipolar effects of the tumour stroma in cancer. Nat Rev Cancer (2004) 4:839–49. doi: 10.1038/nrc1477
117. Pietras K, Östman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. (2010) 316:1324–31. doi: 10.1016/j.yexcr.2010.02.045
118. Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
119. Attieh Y, Clark AG, Grass C, Richon S, Pocard M, Mariani P, et al. Cancer-associated fibroblasts lead tumor invasion through integrin-β3–dependent fibronectin assembly. J Cell Biol. (2017) 216:3509–20. doi: 10.1083/jcb.201702033
120. Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov (2018) 8:1006–25. doi: 10.1158/2159-8290.CD-17-1371
121. Santi A, Kugeratski Fernanda G, Zanivan S. Cancer associated fibroblasts: the architects of stroma remodeling. Proteomics (2017) 18:1700167. doi: 10.1002/pmic.201700167
Keywords: melanoma, motility, invasion, metastasis, microenviroment, cytokines, chemokines, migrastatics
Citation: Ju RJ, Stehbens SJ and Haass NK (2018) The Role of Melanoma Cell-Stroma Interaction in Cell Motility, Invasion, and Metastasis. Front. Med. 5:307. doi: 10.3389/fmed.2018.00307
Received: 20 August 2018; Accepted: 16 October 2018;
Published: 06 November 2018.
Edited by:
Jason Waithman, Telethon Kids Institute, AustraliaReviewed by:
Deirdre R. Coombe, Curtin University, AustraliaCopyright © 2018 Ju, Stehbens and Haass. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samantha J. Stehbens, cy5zdGVoYmVuc0B1cS5lZHUuYXU=
Nikolas K. Haass, bi5oYWFzczFAdXEuZWR1LmF1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.