
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Med., 28 September 2018
Sec. Translational Medicine
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00276
This article is part of the Research TopicRecent Advances in Endothelial Progenitor Cells Toward Their use in Clinical TranslationView all 10 articles
 Erica Carolina1†
Erica Carolina1† Toshiki Kato1,2†
Toshiki Kato1,2† Vuong Cat Khanh1
Vuong Cat Khanh1 Kana Moriguchi1
Kana Moriguchi1 Toshiharu Yamashita1
Toshiharu Yamashita1 Kosuke Takeuchi1
Kosuke Takeuchi1 Hiromi Hamada3
Hiromi Hamada3 Osamu Ohneda1*
Osamu Ohneda1*Background: Endothelial progenitor cells (EPCs) can be used to treat ischemic disease in cell-based therapy owing to their neovascularization potential. Glucocorticoids (GCs) have been widely used as strong anti-inflammatory reagents. However, despite their beneficial effects, side effects, such as impairing wound healing are commonly reported with GC-based therapy, and the effects of GC therapy on the wound healing function of EPCs are unclear.
Methods: In this study, we investigated how GC treatment affects the characteristics and wound healing function of EPCs.
Results: We found that GC treatment reduced the proliferative ability of EPCs. In addition, the expression of CXCR4 was dramatically impaired, which suppressed the migration of EPCs. A transplantation study in a flap mouse model revealed that GC-treated EPCs showed a poor homing ability to injured sites and a low activity for recruiting inflammatory cells, which led to wound healing dysfunction. Impairment of prostaglandin E2 (PGE2) synthases, cyclooxygenase (COX2) and microsomal PGE2 synthase 1 (mPEGS1) were identified as being involved in the GC-induced impairment of the CXCR4 expression in EPCs. Treatment with PGE2 rescued the expression of CXCR4 and restored the migration ability of GC-treated EPCs. In addition, the PGE2 signal that activated the PI3K/AKT pathway was identified to be involved in the regulation of CXCR4 in EPCs under the effects of GCs. In addition, similar negative effects of GCs were observed in EPCs under hypoxic conditions. Under hypoxic conditions, GCs independently impaired the PGE2 and HIF2α pathways, which downregulated the expression of CXCR4 in EPCs. Our findings highlighted the influences of GCs on the characteristics and functions of EPCs, suggesting that the use of EPCs for autologous cell transplantation in patients who have used GCs for a long time should be considered carefully.
Glucocorticoids (GCs) have been widely used as extremely effective agents for suppressing inflammation associated with various kinds of diseases (1). However, despite its beneficial effects, GC treatment is associated with several deleterious effects, including impaired wound healing (2). This side effect happens either via the transrepression of pro-inflammatory cytokines, growth factors, matrix proteins and matrix protease or via direct inhibitory influence on genes that are important for skin regeneration (3). GCs act as the synthetic analogs of human natural endogenous GCs, which exert their effects by binding to GC receptors (GRs), thereby regulating the expression of target genes that play important roles in the wound healing process (4). In addition, GCs also cause the dysfunction of cells involved in wound healing (5, 6). We recently demonstrated that GC treatment impairs the wound healing function of adipose tissue-derived mesenchymal stem cells (MSCs) via the downregulation of stromal cell-derived factor 1 (SDF-1) (5).
Wound healing is a complex process that requires the contribution of many cell types (7). Central to this, MSCs and endothelial progenitor cells (EPCs) play crucial roles in promoting wound healing (7). While MSCs contributes to the wound healing process mostly via paracrine effects, secreting a mixture of growth factors and vesicles to recruit numerous of cells to the wound sites and support these cells' functions, EPCs are the key effectors of neovascularization and are involved in the formation of blood vessels and maintenance of the function of the vascular endothelium (8). EPCs possess the self-renewal potential, differentiation ability to endothelial cells, and neovascularization capability toward new blood vessel formation (9, 10). A previous study showed that EPCs accelerate wound healing not only as the endothelial substrate of blood vessels but also by migrating to the injured tissue, exerting their effects by excreting proangiogenic factors (10).
The definition of an EPC has been controversial. Originally, EPCs were identified as the cell population isolated from human peripheral blood that expresses the CD34 and Flk1 markers (11). However, recently studies have highlighted that positive with CD34 and Flk1 are insufficient to define EPCs in vitro (8). Several studies have reported the two distinct types of EPCs, early and late EPCs, which early EPCs are characterized by the expression of CD45 and CD14, together with some endothelial cell (EC) markers, and have a short lifespan of 3–4 weeks. Additionally, late EPCs are characterized by EC markers, such as CD31, CD34, VEGFR2, and VE-cadherin, but are negative for myeloid markers (12). We previously reported a novel method for isolating EPCs according to the aldehyde dehydrogenase (ALDH) activity which showed that the expression of CD34 declines during the culturing of DiI-Ac-LDL–positive/CD45−/CD31+ cells; whereas, ALDH activity was retained stably in EPCs in long term culture (13). Of note, EPCs with a low ALDH activity (Alde-low) possess a higher migratory ability toward damaged tissue and show a better recover ability in ischemic wound healing than Alde-high EPCs (13). Under hypoxic conditions, Alde-low EPCs are highly responsive and show the upregulation of hypoxic condition inducible factor 2α (HIF2α), which regulates the expression of CXCR4, a major chemokine receptor for cell migration in response to SDF-1 (13). Therefore, Alde-low EPCs can be considered promising candidates for wound treatment (13).
Autologous transplantation of EPCs is considered a prospective approach for therapeutic revascularization and chronic wound (14). It is reported that autoimmune diseases and its drug-based therapy, such as GC, account for 20% of chronic wound cases (15). Therefore, the patients who have been receiving GC treatment are one of the major targets for EPC therapy. However, similar to other cell sources, the introduction of autologous EPCs transplantation into clinical treatment is still being met with difficulty due to the negative influence of patients' medical backgrounds on the outcome of the treatment (16, 17). Ensuring the EPCs' potential is crucial for achieving the best outcome of EPC-based therapy. A previous study suggested that chronic GC treatment reduces the number of circulating EPCs in the patients (16). However, no report has yet clarified the influences of GCs on the wound healing ability of EPCs. Thus, whether or not the outcomes of EPC-based therapy are worsened in patients who have been chronically treated by GCs remains unclear.
In the present study we examined whether or not GCs interfere with the wound healing ability of Alde-low EPCs. We found that GCs downregulated the expression of CXCR4 in a transplantation flap mouse model, which impaired the migration and wound healing ability of EPCs. Treatment with PGE2 upregulated the EP4 receptor and activated the PI3K/AKT signaling which were involved in rescuing the detrimental effects of GC on the CXCR4 expression of EPCs. In addition, similar detrimental effects of GCs on the PGE2/CXCR4 pathway were noted in EPCs under hypoxic conditions. Of note, under hypoxic conditions, independent with the PEG2 pathway, the HIF2α pathway was also involved in the GC-impaired CXCR4 expression in EPCs. Taken together, these findings highlight the negative effects of GCs on the EPC functions, suggesting that autologous EPC therapy for patients receiving GC treatment be considered carefully.
All experiments involving human subjects were performed in accordance with the Guidelines for Medical and Health Research Involving Human Subjects, Ministry of Education, Culture, Sports, Science and Technology, Japan and the permission of the Institutional Ethics Review Committee of the University of Tsukuba. Human EPCs were isolated from umbilical cord blood (UCB), as previously described (13). Isolated EPCs were sorted based on the ALDH activity using ALDEFLUOR® system reagents (StemCell Technologies, Vancouver, Canada). The EPCs with the low ALDH activity (Alde-low EPCs) were used for further experiments in the present study. Alde-low EPCs were cultured with maintenance medium (Iscove's modified Dulbecco medium-IMDM, Invitrogen, Carlsbad, CA, USA)/10% FBS/5 ng/mL bFGF (PeproTech, NJ, USA), and 0.1% (v/v) penicillin-streptomycin (100 U/mL penicillin, 0.1 mg/mL streptomycin; Invitrogen) and incubated at 37°C in 5% CO2.
EPCs were harvested with trypsin, counted the cell number and a number of 2 × 105 cells were reconstituted in 100 μl 2% FBS-containing PBS. For staining purpose, cells were incubated with the desired antibodies for 30 min at 4°C.
The following antibodies were utilized with the volume of 5μl as the recommendation of the manufacturer: PE-labeled anti-CD105 (323206, BioLegend), PE-labeled anti-CD73 (550257, BD Pharmingen), PE-labeled anti-CD31 (303106, BioLegend), allophycocyanine (APC)–labeled anti-CD45 (555485, BD Biosciences, San Jose, CA), and FITC-labeled anti-CD34 (555821, BD Biosciences). Purified-anti-VEGFR-2 were used as previously reported (13). APC-labeled anti-IgG1 (555751, BD Biosciences), PE-labeled anti-IgG1 (555749, BD Biosciences), FITC-labeled anti-IgG1 (555748, BD Biosciences) were used as the isotype controls. After staining, cells were washed by 2% FBS-containing PBS, centrifuged at 1,800 RPM for 3 min at 4°C, and reconstituted in 300 μl 2% FBS-containing PBS. A flow cytometer (MoFlo XDP; Beckman Coulter, Pasadena, CA, USA) collected 10,000 events for each group, and the isotype control IGg was used as the negative control.
In order to quantify CXCR4 expression in EPCs, the cells were stained with 5μl of PE-labeled anti-CXCR4 (306506, BioLegend) and performed the FACS analysis with the similar above protocol. PE-labeled anti-IgG1 (555749, BD Biosciences) was used as the isotype controls. The threshold for CXCR4-positivity was quantified by means of fluorescent intensity value subtracted with isotype control value IgG of each group.
The proliferation of EPCs was evaluated by the growth curve and proliferation assay staining with Cell Counting Kit-8 (CCK-8, Dojindo, Tokyo, Japan).
For growth curve, EPCs were seeded at a number of 4 × 103 cells/well in the 24 well-plate dishes and cultured at 37°C in 5% CO2 for 9 days. The culture medium was changed every 3 days. The cells were washed with sterile PBS and treated with 0.05% trypsin/EDTA (Invitrogen) at 24-h intervals for 9 days to separate single cells. Dead cells were excluded using trypan-blue staining solution (35525-02; Nacalai Tesque, Kyoto, Japan), and the numbers of live cells in triplicate dishes were counted using a hemocytometer.
For proliferation assay staining with CCK-8, EPCs were seeded at a number of 2.5 × 104, 1.25 × 104, and 6.25 × 103 cells/well in 96-well microplate. Cells were incubated for 48 h and 10 μl of Cell Counting Kit-8 reagent was added to each well, then incubate for 2 h. The number of live cells were measured by colorimetric reading using a microplate reader (Varioskan, ThermoFisher Scientific, Massachusetts, USA) for 450 nm.
EPCs were trypsinized and centrifuged at 1,000 RPM for 3 min at 4°C. Cells were then resuspended in 100 μl of 2% FBS and stained with 5 μl of PE annexin V and 7-AAD (BD BioSciences (CA, USA), and then incubated at 4°C in the dark for 30 min. FBS (2%, 300 μl) was added, and the apoptotic cells were analyzed by flow cytometry.
Scratch assay: An in vitro migration assay was performed as previously reported with minor modifications (18). EPCs with a number of 1.5 × 105 cells were seeded onto 4-well plates with maintenance medium until they reached confluency after 24 h. Scratch wounds ~1 mm wide were created. After gentle washing of the detached cells with PBS, the growth medium was changed to 2% FBS-containing IMDM. The pictures of wound closure were taken every 6 h during 24-h post-scratching at 100 × magnification (10 × objective and 10 × eyepiece) under a microscope (Olympus, Tokyo, Japan). The cell migration was calculated using the ImageJ software program (NIH, MD, USA). The wound closure distance was measured at the beginning (T0) and end of the experiment (Tx). The following formula was used to convert the migrated area to percentage: Percentage (%) of wound closure = T0 – TX = [1 – X/T0 × 100].
A volume of 600 μl SDF-1 (100 ng/ml) in IMDM medium was placed in the lower chamber of each well of 24 well-plates. Afterwards, a volume of 200 μl IMDM medium containing 3 × 104 EPCs added in the upper chamber of 8 μm pore sized of filter membrane. After 24 h of incubation, the lower side of filter was washed with PBS and fixed with 2% PFA. The number of migrated EPCs which migrate toward SDF-1 was determined by Hematoxylin & Eosin staining and counted under the observation using a microscope (Olympus) at 100 × magnification.
Total RNA was isolated from cultured EPCs using extraction reagent (Sepasol-RNA I Super G; Nacalai Tesque), and reverse transcription was performed with 1 μg of the total RNA using a reverse transcription polymerase chain reaction (RT-PCR) kit (Toyobo, Osaka, Japan). The expression of the target genes was analyzed using a GeneAmp PCR System (Life Technologies, CA, USA) with Thunderbird SYBR qPCR Mix (Toyobo). Experiments were carried out in triplicate, and the expression of the target genes was calculated using the 2−ΔΔCT method. β-Actin was used as an internal control. The primer sequences used for quantitative RT-PCR are shown in Table 1.

Table 1. The primer sets for the quantitative polymerase chain reactions.
C57BL/6 mice were purchased from Charles River Japan, Inc. (Kanagawa, Japan). All experiments were performed in compliance with the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology, Japan. Each experiment was repeated triplicate. In order to compare the wound healing ability of untreated EPCs and GC-treated EPCs, total 15 male mice at 12th week of age were used in each experiment and divided into three groups (5 mice/group): PBS, EPCs, GC-treated EPC. In order to examine the role of CXCR4 in the wound healing ability of EPCs, total nine male mice at 12th week of age were used in each experiment and divided into three groups (3 mice/group): untreated EPCs, GC-treated EPCs, and CXCR4 antibody-treated EPCs (AMD3100, Sigma-Aldrich). 100 nM of AMD3100 at 24-h treatment was used to block the CXCR4. The mice were anesthetized using avertin, and a skin incision (3 × 2 cm) was made to create an ischemia gradient as previously described (19). EPCs were treated with 100 nM GC for 24 h prior to surgery then labeled with PKH26 (Sigma-Aldrich, Missouri, USA) as the instruction of the manufacturer before transplantation. PBS and PKH26-labeled untreated EPCs were used as the controls. The EPCs were injected at a number of 5 × 105/mouse through the tail vein. Skin tissue samples were fixed overnight with 4% paraformaldehyde; then, washed with PBS, soaked in sucrose 10% for 2 h, and finally in sucrose 20% overnight. Then, the frozen blocks of samples were made by embedded the samples in the O.C.T compound (Sakura Finetek, Tokyo, Japan) and freeze in liquid nitrogen. After that the tissues were sectioned before immunostaining. For observation, the flap tissues were collected at two different time points: on the third day of transplantation to analyze the inflammatory cell recruitment and on the seventh day of transplantation to analyze the neovascularization. Images of the ischemic flaps were captured on the seventh day of transplantation, and the necrotic areas were quantified using the Image J software program (NIH).
The frozen flap tissue sections were mounted, stained with hematoxylin and eosin (Wako, Osaka, Japan) and observed under a microscope (Olympus, Tokyo Japan). The inflammatory cells recruited in the ischemic area were visualized by immunohistochemical staining with rat anti-mouse CD45 (553078, BD Pharmigen), rat anti-mouse Mac1 (rat anti-mouse CD11b 550282, BD Pharmigen). The neovascularization was analyzed by immunohistochemical staining with rat anti-mouse CD31 (553370, BD Pharmigen) as the instruction of the manufacturer. Briefly, sections were washed in PBS and then rinsed in 10% H2O2 in PBS before blocking with blocking buffer containing Phosphate-Buffered Saline with 0.5% Tween 20 (PBST) and normal rabbit serum at a dilution of 4:1 (v/v) for 1 h at room temperature. After that, the sections were incubated with appropriate antibody overnight. On the following day, the sections were washed with PBST before incubating in the secondary antibody (POD Conjugate Anti-rat, MK-201, Takara, Shiga, Japan) for 30 min at room temperature. The sections were then washed with PBS and covered with 0.05% 3,3′-Diaminobenzidine (DAB) solution (Sigma-Aldrich) in PBS with H2O2 for 3 min then stopped detection by water. Quantification of the number of positive cells was performed at 200 × magnification (20 × objective and 10 × eyepiece) under a microscope (Olympus) by counting the brown dots in 10 fields.
Cultured EPCs were harvested, counted and a number of 106 cells were suspended in low-salt buffer (10 mM HEPES, 10 mM KCL, 1 mM dithiothreitol, 1 mM EDTA, protease inhibitor cocktail (PIC), and 1% Nonidet P-40, Roche Diagnostics, Basel, Switzerland), and nuclear pellets were collected by centrifugation. The nuclear pellets were then suspended in high-salt buffer (20 mM HEPES, 400 mM NaCl, 1 mM dithiothreitol, and 1 mM EDTA, PIC), and the nuclear extract was obtained. The total protein concentration was measured by Bradford assay (Bio-rad, CA, USA) as the instruction of the manufacturer. 30μg protein of each nuclear extract samples were separated on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a PVDF membrane (EMD Millipore, Darmstadt, Germany).
An immunoblotting analysis was performed. Briefly, the membranes were blocked with 5% Bovine Serum Albumin (Sigma-Aldrich, Missouri, USA) in Tris-Buffered Saline with 0.5% Tween 20 (TBST) for an hour at room temperature, then incubated incubation with the appropriate primary antibody overnight. Primary antibodies were diluted at the dilution of 1:1,000 (v/v) as the instruction of the manufacturer, including: Rabbit anti-human AKT (9272S, Cell Signaling Technology, Massachusetts, USA, phosphorylated AKT antibody (9271S, Cell Signaling Technology), rabbit anti-human HIF-2α antibody (NB100-122, Novus Biologicals, CO, USA), and goat anti-Lamin B antibody (M-20, Santa Cruz Biotechnology, Inc., CA, USA. The membranes were washed with TBST buffer then incubated with secondary antibodies, including: Horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (32460, ThermoFisher Scientific) and rabbit anti-goat IgG (31402, ThermoFisher Scientific) with the dilution of 1:10,000 (v/v) for an hour at room temperature. Then, the membranes were washed and the enhanced chemiluminescence (GE Healthcare, Illinois, USA) was used for detection by a luminescent image analyzer (ImageQuant LAS4000, GE Healthcare). The band densities of target proteins were analyzed by ImageJ software (NIH). The relative expressions of target proteins were normalized with the total-LaminB expression. For the phosphorylation analysis, after incubation with phosphorylated antibody and detected the phosphorylated protein, the membranes were striped with Restore Stripping Buffer (ThermoFisher Scientific) for 15 min at room temperature, then, continued to incubate with rabbit Akt antibody. The Akt signals were detected and the membranes were then stripped and re-probed with LaminB antibody.
Data were statistically analyzed using Mann Whitney U test using the GraphPad Prism 5 software program (GraphPad Software, CA, USA) as appropriate. Data are presented as the mean ± standard deviation. P < 0.05 was considered as significant.
A previous report stated that GCs induced apoptosis in lymphocytes, demonstrating the negative effects of GCs on cells (20). However, thus far, no report has described the effects of GCs on EPCs; therefore, in the present study, we examined the characteristics of EPCs, including the cell morphology, viability, surface markers, and proliferation, under treatment with 100 nM dexamethasone as a GC. We found that GC treatment exerted no effects on the adherence or visual appearance of EPCs (Figure 1A). In addition, Annexin-V/7AAD staining assay data showed that 24-h treatment of GC showed no apoptosis induction or any cytotoxicity in EPCs (Figure 1B).
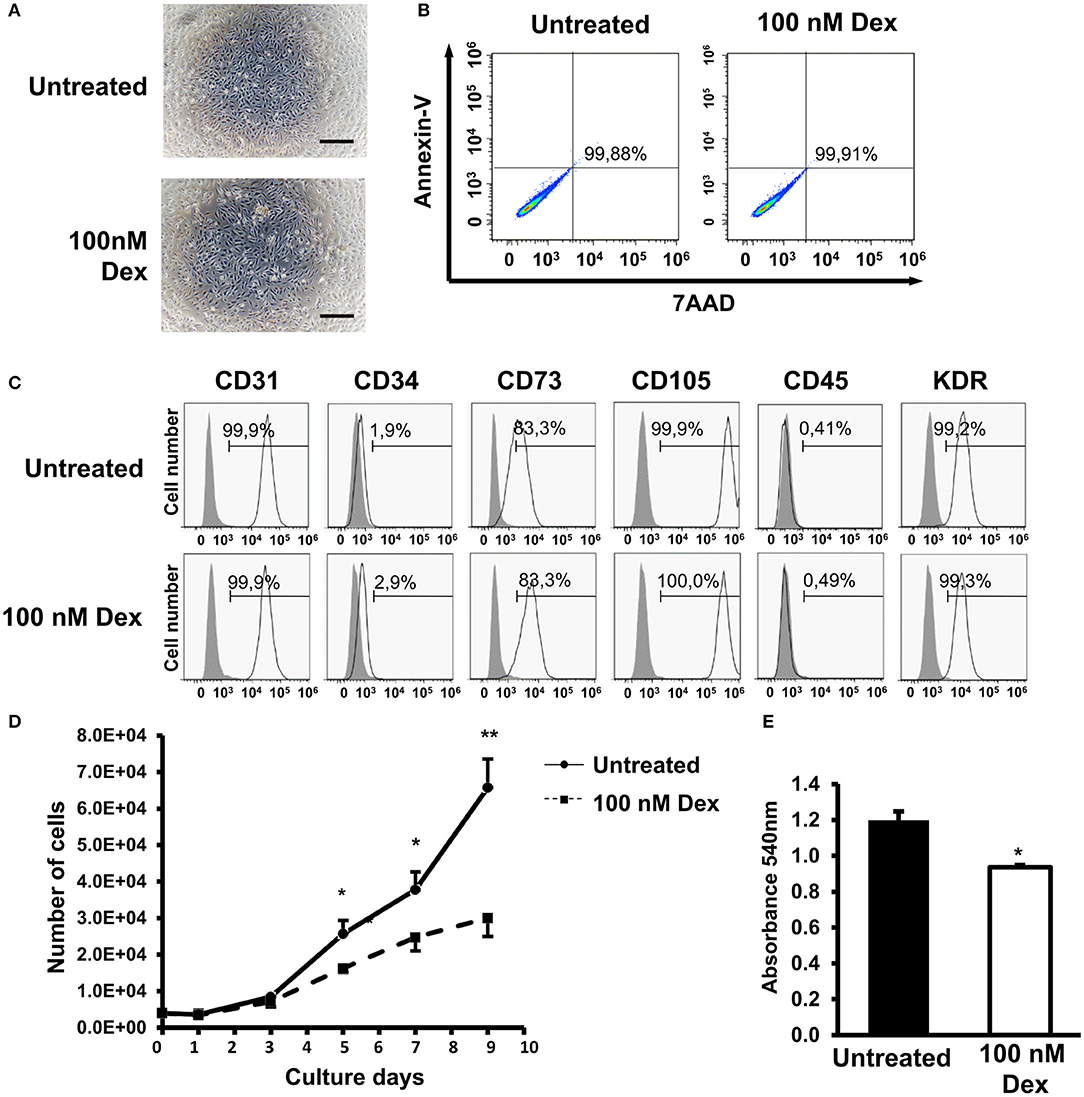
Figure 1. Glucocorticoid (GC) treatment impaired the proliferation ability of EPCs. (A) Cell morphology of untreated and GC-treated EPCs from one representative experiment. (B) Apoptotic cells of untreated and GC-treated EPCs. Cells were stained by Annexin V staining, then analyzed by the FACS analysis. The histograms were from one representative experiment. (C) Cell surface markers of untreated and GC-treated EPCs analyzed by FACS analysis. The histograms were from one representative experiment. (D) The growth curve of untreated and GC-treated EPCs from one representative experiment. (E) The proliferation assay of untreated and GC-treated EPCs examined by Cell counting kit-8. The chart shows the mean data from all experiments. Dexamethasone (100 nM, 24-h treatment) was used as a GC. Untreated: untreated EPCs, 100 nM Dex: GC-treated EPCs. The data represent the mean ± SD. n = 3, **P < 0.01, *P < 0.05. The scale bar indicates 100 μm. The experiment was repeated triplicate.
To examine the effects of GC on the EPC surface markers, a FACS analysis was performed. The data showed that there were no significant changes in the self-surface markers of EPCs under GC treatment, with both control and treated groups expressing high levels of CD31, CD34, CD105, CD73, and VEGFR2 and not expressing CD45 hematopoietic marker at all (Figure 1C). Next, we investigated the effects of GC treatment on the proliferation ability of EPCs. The data showed that the proliferation ability of EPCs treated with GC was significantly impaired compared to the untreated EPCs (Figures 1D,E). Taken together, these data show that GC impaired the proliferation ability of EPCs.
Previous studies have described the negative effects of GCs on the wound healing process (3, 20). Recently, we also reported that GCs impair the wound healing function of mesenchymal stem cell by reducing the expression of SDF-1 (5). Therefore, in the present study, we thought that GCs might also affect the wound healing function of EPCs. To test this hypothesis, we evaluated the wound healing function of EPCs under GC treatment. First, because the EPC migration ability is critical for their biological properties, the effect of GC on the migration ability of EPCs was evaluated by a scratch assay as an in vitro wound healing model (21). As shown in Figure 2A, GC-treated EPCs showed a lower capability of wound closure than the control, indicating a reduced migration ability of EPCs on exposure to GC (Figures 2A,B).
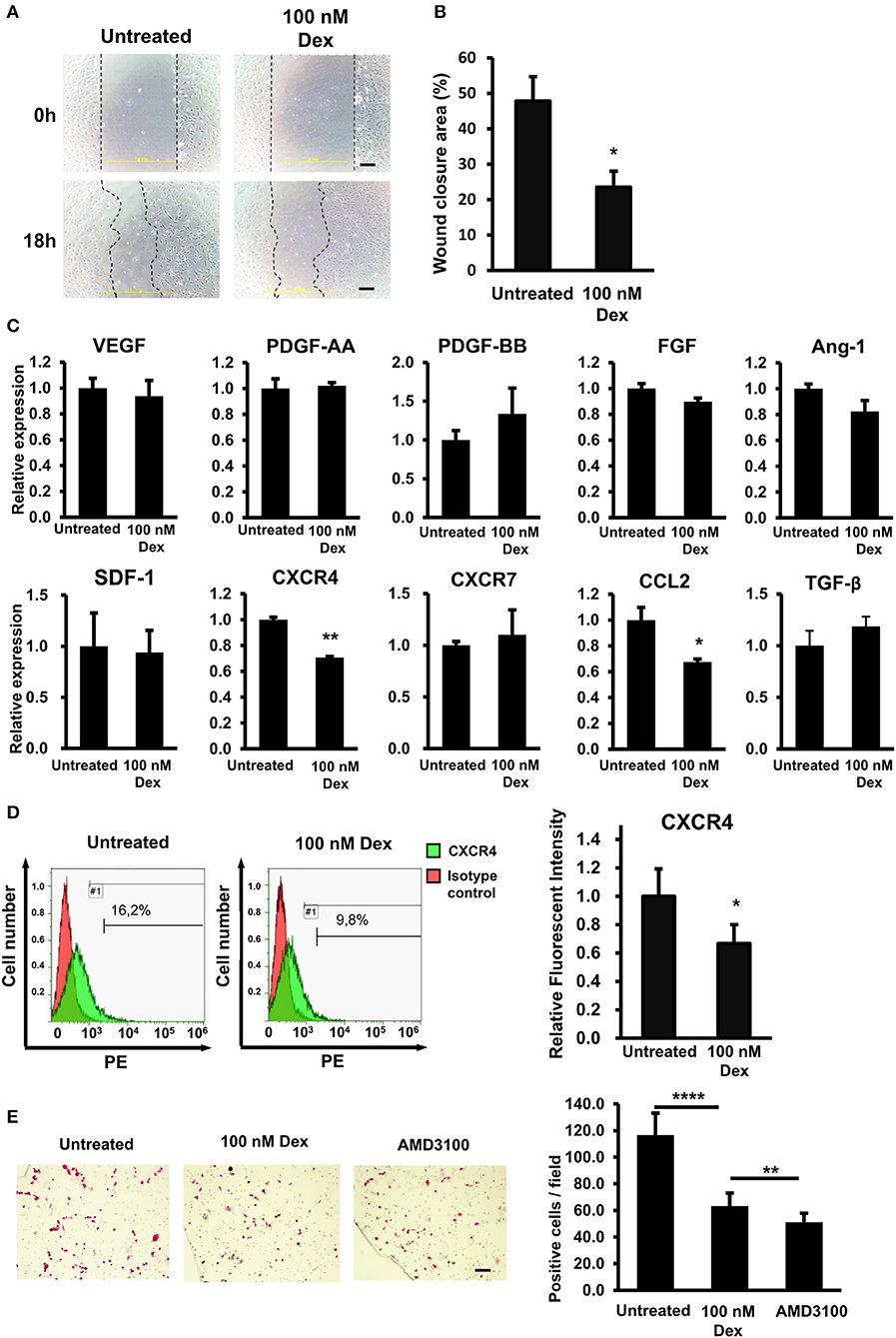
Figure 2. GCs impaired the in vitro migration ability of EPCs via the downregulation of CXCR4. (A) The in vitro wound healing ability of untreated and GC-treated EPCs by a scratch assay. The images showed the wound closures from one representative experiment taken under a microscope at 100× magnification. (B) Quantification of the wound closure area in a scratch assay of untreated and GC-treated EPCs. The data were displayed as the means from all experiments. (C) The relative mRNA expression of wound healing genes in untreated and GC-treated EPCs. The chart shows the mean data from all experiments. (D) The relative protein expression of CXCR4 in untreated and GC-treated EPCs identified by a FACS analysis. The FACS histograms were from one representative experiment. The chart shows the quantification of relative expression of CXCR4 protein. The data were displayed as the means from all experiments. (E) The transwell assay of untreated, GC-treated EPCs, and CXCR4 antibody-treated EPCs toward SDF-1. AMD3100 (100 nM, 24-h treatment) was used as the CXCR4 antibody. The images were the migrated EPCs from one representative experiment taken under a microscope at ×100 magnification. The chart shows the quantification of number of migrated EPCs. The data were displayed as the means calculated from all experiments. Dexamethasone (100 nM, 24-h treatment) was used as a GC. Untreated: untreated EPCs, 100 nM: GC-treated EPCs. The data represent the mean ± SD. n = 3, ****P < 0.0001, **P < 0.01, *P < 0.05. The scale bar indicates 100 μm. The experiment was repeated triplicate.
Next, we assessed the expression profiles of the wound healing-related genes in EPCs. Numerous reports have shown that EPCs express multiple chemokines and their receptors related to mobilization (SDF-1, CXCR4, CXCR7), angiogenesis (Ang-1, TGFβ), immunomodulatory (CCL2), and growth factors (PDGF, VEGF, FGF) that play a critical role in wound healing (22, 23). To clarify how GC interferes with the wound healing capacity of EPCs, we examined the mRNA expression of those genes in GC-treated EPCs.
We observed a significantly impaired expression of CXCR4, which related to the mobility (24) and CCL2, which related to the inflammatory cell recruitment (25, 26) in GC-treated EPCs compared to that in control cells (Figure 2C). In addition, several genes that contribute to EPC migration, such as the extracellular matrix (ECM) protease MMP9 and homing and adherence factor VCAM 1, were also downregulated due to GC treatment (data not shown). Consistent with the mRNA expression data, a further FACS analysis showed a reduction in the CXCR4 surface protein level (Figure 2D).
It is reported that the SDF-1/CXCR4 cascade is responsible for the mobilization of EPCs (10). Therefore, we next performed the transwell assay to examine the effects of GC treatment on the migration ability of EPCs toward the signal of SDF-1. As expected, GC-treated EPCs showed the low migration ability toward SDF-1 compared to the untreated cells (Figure 2E). In addition, treatment EPCs with a CXCR4 antibody, AMD3100, also showed the similar impaired migration ability, suggesting the direct role of CXCR4 that regulates the mobility of EPCs (Figure 2E). Taken together, these results showed that GC impaired wound healing ability in vitro and the migration ability toward SDF-1 of EPCs by the downregulation of CXCR4 and CCL2.
We next examined the effects of GC on the wound healing ability of EPCs by a transplantation study using an in vivo mouse flap model. The wound healing functions of GC-treated EPCs were analyzed and compared to those in untreated cells. We found that the injection of untreated EPCs healed the wounds of mice with a reduction in the necrotic areas at 7 days after transplantation. In contrast, the wound healing ability of the GC-treated EPCs was significantly impaired, with some necrotic areas remaining, similar to control mice without any cell transplantation (Figures 3A,B).
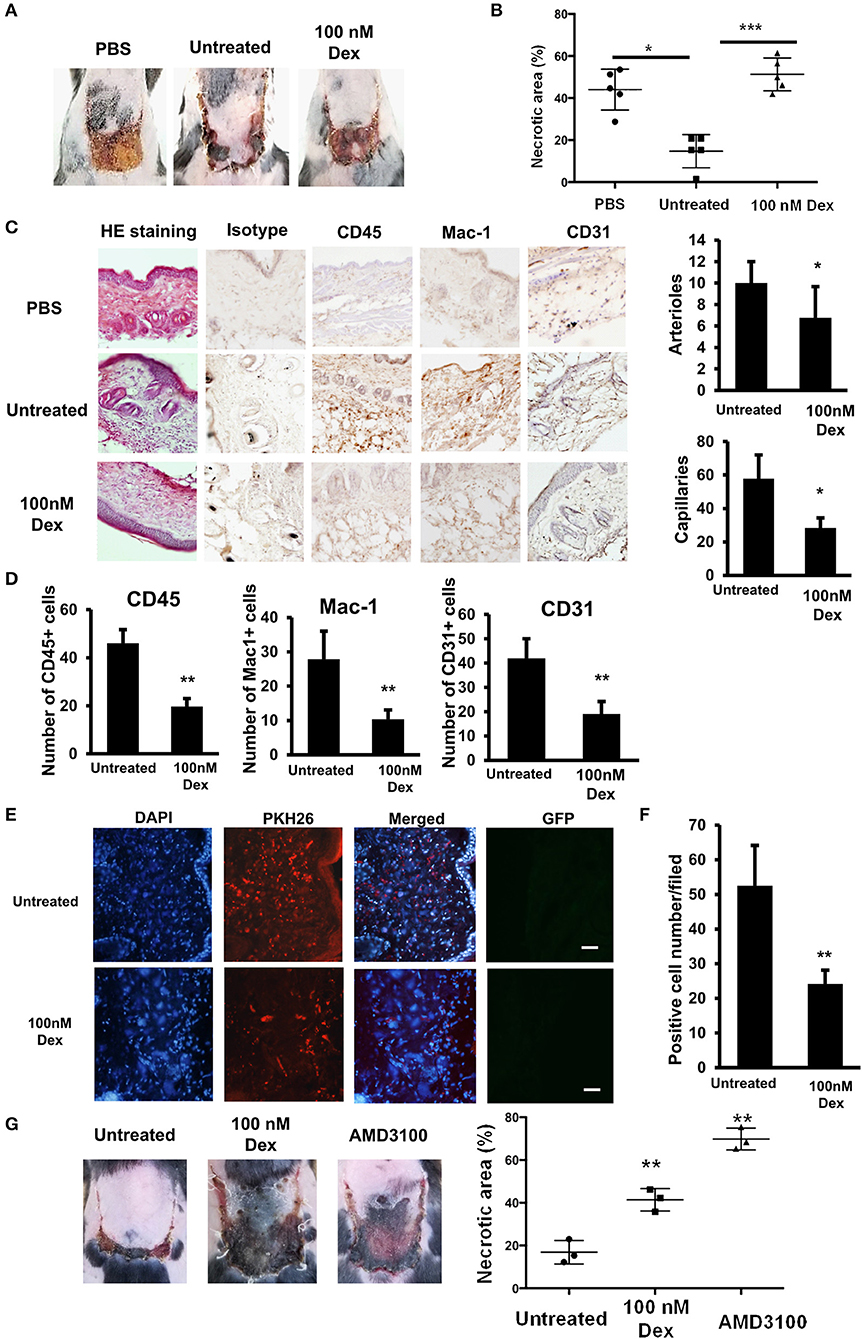
Figure 3. GCs impaired the wound healing ability of EPCs. (A) Wound healing ability of untreated and GC-treated EPCs in flap mouse model. (B) The necrotic area of the flap tissues injected with untreated and GC-treated EPCs. (C) Immunohistochemistry of CD45-, MAC-1- (at day 3 post-transplantation), and CD31-positive cells (at day 7 post-transplantation) in flap tissues injected with untreated and GC-treated EPCs observing under a microscope at 200× magnification. (D) Quantification of CD45-, MAC-1- (at day 3 post-transplantation), and CD31-positive cell numbers (at day 7 post-transplantation) in flap tissues injected with untreated and GC-treated EPCs observing under a microscope at 200× magnification. (E) PKH-positive EPCs in flap tissues injected with untreated and GC-treated EPCs observing under a microscope at 200× magnification. (F) Quantification of PKH-positive EPCs in flap tissues injected with untreated and GC-treated EPCs observing under a microscope at 200× magnification. From A–F, the data were calculated from five individual mice from one representative experiment. The data represent the mean ± SD. n = 5, ***P < 0.001, **P < 0.01, *P < 0.05. (G) Wound healing ability of untreated and CXCR4 antibody-treated EPCs in flap mouse model. AMD3100 (100 nM, 24-h treatment) was used as a CXCR4 antibody. The data were calculated from three individual mice from one representative experiment. The data represent the mean ± SD. n = 3, **P < 0.01. Dexamethasone (100 nM, 24-h treatment) was used as a GC. Untreated: untreated EPCs, 100 nM: GC-treated EPCs. The scale bar indicates 100 μm. The experiment was repeated triplicate.
Because the GC-treated EPCs showed the impaired expression of CCL2 which related to the inflammatory cell recruitment ability (25, 26), we performed a histological analysis of the wound healing process on the third day after transplantation and the data revealed that mice transplanted with untreated EPCs showed an increased recruitment of CD45- and Mac-1 positive cells to the wound sites compared to mice transplanted with GC-treated EPCs (Figures 3C,D). In addition, on the seventh day after transplantation, mice injected with untreated EPCs showed a greater number of CD31-positive vascular endothelial cells than mice injected with GC-treated EPCs (Figures 3C,D). In addition, to examine the effects of GC treatment on the homing and migration ability of EPCs to the wound sites, we quantified the PKH-labeled EPCs in the wound regions on the first day of transplantation. We found that there were fewer PKH-positive cells at the wound sites of mice transplanted with GC-treated EPCs than in those injected with untreated EPCs (Figures 3E,F).
We previously reported the impaired wound healing ability of EPCs with high activity of ALDH (Alde-high EPCs) due to the low expression of CXCR4 compared to those with low activity of ALDH (Alde-low EPCs) (19). In order to clarify the direct role of CXCR4 in the wound healing ability of EPCs, we blocked the activity of CXCR4 by its antibody, AMD3100, and compared the necrotic area in the transplantation study. As expected, similar to the effects of GC, blocking CXCR4 in EPCs showed the impaired wound healing function in the flap mice (Figure 3G), suggesting CXCR4 is responsible for the wound healing ability of EPCs.
Taken together, these data demonstrated that GCs impaired the wound healing functions of EPCs.
Previous studies have shown that GCs are a strong inhibitor of the arachidonic acid cascade, resulting in less production of prostaglandin E2 (PGE2) in macrophages, vascular smooth muscle cells, and MSCs (27–30). In addition, other studies have suggested that the expression of CXCR4 is elevated in the presence of PGE2 (31). Therefore, we next investigated whether or not GC impaired the production of PGE2 to cause the downregulation of CXCR4 expression in EPCs. Cyclooxygenase (COX) and prostaglandin dehydrogenase (mPGES) are reported to be key enzymes in the catabolism of PGE2 (32–34). We found that GC treatment impaired the expression of COX-2 and mPGES-1 in EPCs, implying the negative effects of GCs on the PGE2 production of these cells (Figure 4A).
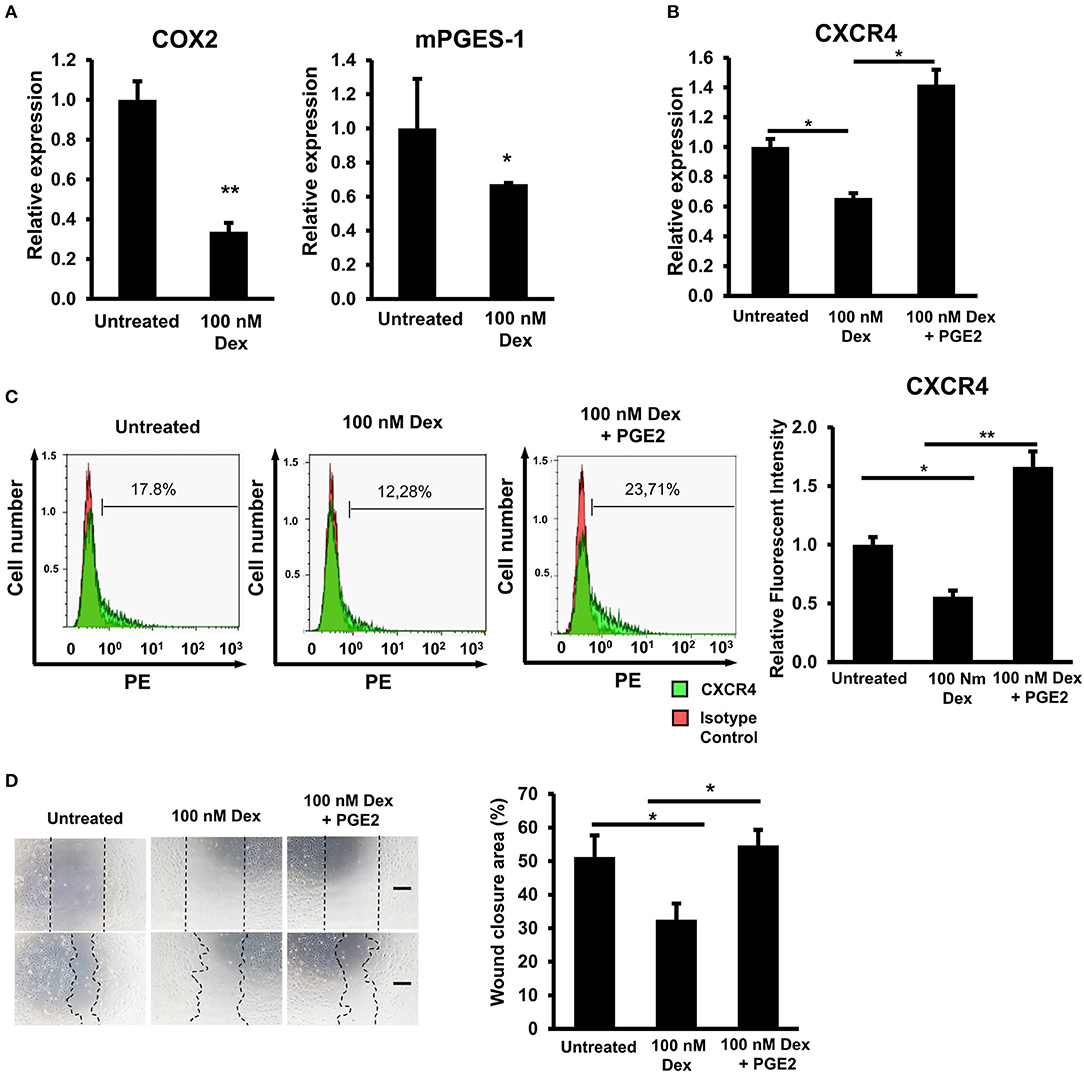
Figure 4. GC-impaired prostaglandin E2 production was involved in the downregulation of CXCR4 in EPCs. (A) The relative mRNA expression of COX2 and mPEGS1 in untreated and GC-treated EPCs. (B) The relative mRNA expression of CXCR4 in untreated, GC-treated EPCs, and GC-treated EPCs in the presence of PGE2. (C) The relative protein expression of CXCR4 in untreated, GC-treated EPCs, and GC-treated EPCs in the presence of PGE2 by a FACS analysis. (D) An in vitro scratch assay of untreated, GC-treated EPCs, and GC-treated EPCs in the presence of PGE2 observing under a microscope at 100× magnification. Dexamethasone (100 nM, 24-h treatment) was used as a GC. Untreated: untreated EPCs, 100 nM: GC-treated EPCs, 100 nM + PGE2: GC-treated EPCs in the presence of PGE2. The data represent the mean ± SD. n = 3, **P < 0.01, *P < 0.05. The scale bar indicates 100 μm. The experiment was repeated triplicate. The charts showed the overall mean data from all experiments.
Next, in order to clarify the involvement of PGE2 in the downregulation of the CXCR4 expression in GC-treated EPCs, we examined whether or not treatment with PGE2 could reverse the expression of CXCR4 of these cells. The data showed that treatment of PGE2 rescued the GC-impaired mRNA and protein expression of CXCR4 in EPCs (Figures 4B,C). Furthermore, consistent with the upregulation of CXCR4 in the presence of PGE2, the migratory capacity of GC-treated EPCs was also recovered by PGE2 treatment (Figure 4D). Taken together, these results suggested that GC downregulated the expression of CXCR4 in EPCs by the impairment of PGE2 synthesis via COX-2 and mPGES-1.
A previous reports showed that PGE2 exerted its activity by interaction with a G-protein-coupled receptor family (GPCRs) consisting of EP1, EP2, EP3, and EP4 subtypes, resulting in the different signal transduction and regulation of the expression of numerous target genes (35). To identify which PGE2 receptors are affected by GC, we analyzed the gene expression of these subtypes in EPCs in the presence of GC. We found that, among these four receptors, GC treatment only impaired the expression of EP4 in EPCs. In addition, treatment with PGE2 reversed the negative effects of GC on the EP4 expression, suggesting that EP4 is the PGE2 receptor related to the effects of GC on EPCs (Figure 5A).
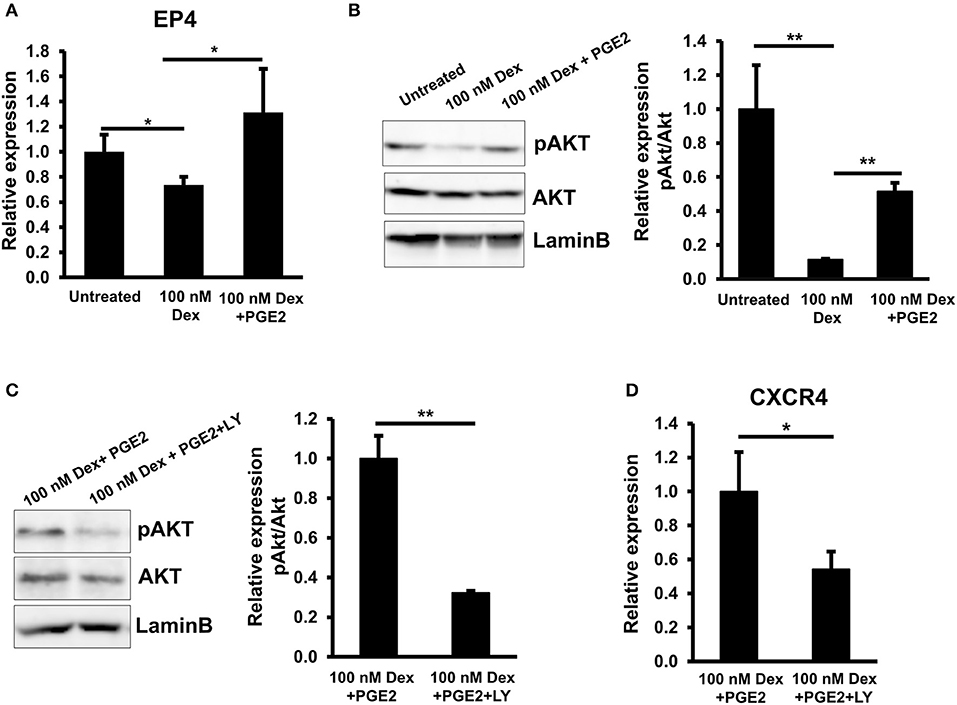
Figure 5. GCs downregulated CXCR4 via the impaired PGE2/EP4/AKT signaling in EPCs. (A) The relative mRNA expression of EP4 in untreated, GC-treated EPCs, and GC-treated EPCs in the presence of PGE2. (B) The relative protein expression of pAKT/AKT in untreated, GC-treated EPCs, and GC-treated EPCs in the presence of PGE2. (C) The inhibition of PI3K/AKT pathway in GC-treated EPCs in the presence of PGE2. LY294002 was used as a PI3K inhibitor. (D) The relative mRNA expression of CXCR4 in GC-treated EPCs in the presence of PGE2 and PI3K inhibitor. Dexamethasone (100 nM, 24-h treatment) was used as a GC. Untreated: untreated EPCs, 100 nM: GC-treated EPCs, 100 nM + PGE2: GC-treated EPCs in the presence of PGE2, 100 nM + PGE2 + LY: GC-treated EPCs in the presence of PGE2 and PI3K inhibitor. LY: LY294002. The data represent the mean ± SD. n = 3, **P < 0.01, *P < 0.05. The experiment was repeated triplicate. The charts showed the overall mean data from all experiments.
The EP2 and EP4 receptors which mediate the increased cAMP concentrations had been thought to have similar effects in some biological process; however, recently, their distinct roles were reported (36, 37). It might be because of the selective expression of either of them in cells and the selective actions on the different signaling pathways (37). It is reported that EP4 but not EP2 couples to PI3K which regulates the migration of dendritic cells in the mouse suggesting the role of EP4/PI3K/AKT in the regulation of migration ability of the cells (37). Therefore, we next examined the role of PI3K/AKT pathways in the GC-impaired CXCR4 expression in EPCs. As expected, we found the impaired phosphorylation of AKT in EPCs in the presence of GC; this effect was rescued by the adding of PGE2 which showed by the upregulation of pAKT (Figure 5B). In order to clarify the role of PGE2/PI3K/AKT pathway in the CXCR4 regulation of GC-treated EPCs, we analyzed the expression of CXCR4 in the present of a PI3K inhibitor. The effect of PI3K inhibitor on the phosphorylation of AKT was confirmed (Figure 5C). The data showed that treatment with PI3K inhibitor abolished the rescued effects of PGE2 on the GC-treated EPCs in which CXCR4 expression was impaired (Figure 5D) indicating that PGE2 reversed the GC-impaired CXCR4 expression in EPCs via the activation of PI3K/AKT pathway.
Taken together, these results demonstrated that GC impaired the EP4/AKT signaling which was involved in the downregulation of CXCR4 expression of EPCs.
We previously reported the HIF2α-dependent upregulation of CXCR4 in Alde-low EPCs under hypoxic conditions, which promotes the migration and wound healing function of the cells (13, 19). To examine whether or not GC causes similar negative effects that impair the CXCR4 expression in EPCs under hypoxic conditions, we next analyzed untreated and GC-treated EPCs under hypoxic conditions. As expected, in the control EPCs without treatment of GC, we observed the upregulation of CXCR4 at both the mRNA and protein levels under hypoxic conditions (Figures 6A,B). However, in the GC-treated EPCs, the upregulation of CXCR4 under hypoxic conditions was abrogated (Figures 6A,B). Interestingly, this detrimental effect of GC on the CXCR4 expression was rescued by treatment with PGE2 (Figures 6C,D), indicating the similar effects of GC on EPCs under both normoxic and hypoxic conditions of downregulating the expression of CXCR4 via the impairment of the PGE2 pathway.
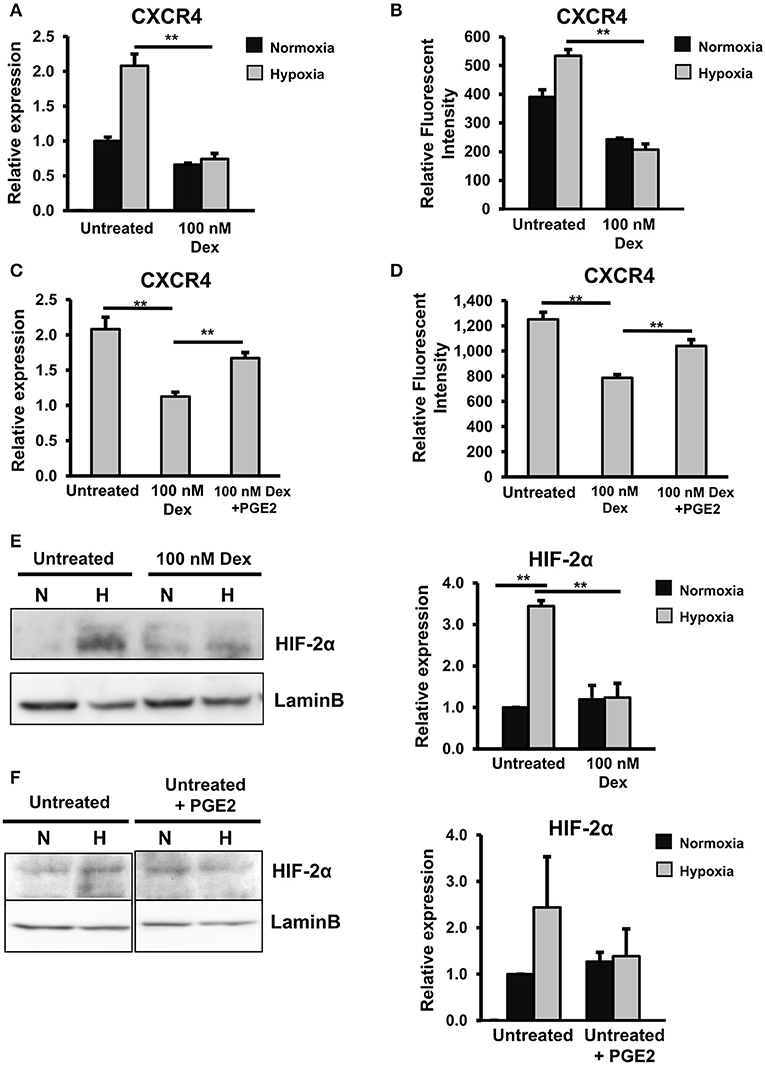
Figure 6. GCs downregulated the expression of CXCR4 via the independent impairment of the HIF2α and PGE2 pathways. (A) The relative mRNA expression of CXCR4 in untreated and GC-treated EPCs under normoxic and hypoxic conditions. (B) The relative protein expression of CXCR4 in untreated and GC-treated EPCs under normoxic and hypoxic conditions. (C) The relative mRNA expression of CXCR4 in untreated EPCs, GC-treated EPCs, and GC-treated EPCs in the presence of PGE2 under hypoxic conditions. (D) The relative protein expression of CXCR4 in untreated EPCs, GC-treated EPCs, and GC-treated EPCs in the presence of PGE2 under hypoxic conditions. (E) The expression of HIF2α protein in untreated and GC-treated EPCs under normoxic and hypoxic conditions. (F) The expression of HIF2α protein in untreated EPCs in the presence of PGE2 under normoxic and hypoxic conditions. Dexamethasone (100 nM, 24-h treatment) was used as a GC. Untreated: untreated EPCs, 100 nM: GC-treated EPCs, 100 nM + PGE2: GC-treated EPCs in the presence of PGE2, Untreated + PGE2: untreated EPCs in the presence of PGE2. The data represent the mean ± SD. n = 3, **P < 0.01. The experiment was repeated triplicate. The charts showed the overall mean data from all experiments.
Our previous report described the functions of the HIF2α pathway, which plays a key role as an upregulator of the CXCR4 expression in EPCs under hypoxic conditions (19). In order to examine whether or not the HIF2α pathway is involved in the GC-impaired reduction of the CXCR4 expression under hypoxic conditions, we next examined the effects of GC on the expression of HIF2α. Interestingly, we found that GC also impaired the expression of HIF2α under hypoxic conditions (Figure 6E).
COX2/PGE2 was reported to upregulate the expression of HIF2α under hypoxic conditions in hepatocellular carcinoma cells (38). Therefore, we thought that PGE2 might also upregulate the expression of HIF2α in EPCs under hypoxic conditions. In order to test this hypothesis, untreated EPCs were cultured in the presence of PGE2 under hypoxic conditions, and the expression of HIF2α was examined. Unexpectedly, we found that PGE2 showed no effect on the expression of HIF2α (Figure 6F). This suggested that the HIF2α and PGE2 pathways independently regulated the expression of CXCR4 in EPCs under hypoxic conditions.
Taken together, these results indicated that GC exerted similar effects on the CXCR4 expression in EPCs under both normoxic and hypoxic conditions. However, under hypoxic conditions, the GC-impaired CXCR4 expression in EPCs was caused independently by the downregulation of the HIF2α or PGE2 pathway.
In the present study, we showed that GC downregulated the expression of CXCR4, thus abrogating the wound healing ability of EPCs. GC-treated EPCs showed a poor migration ability and dysfunction in the recruitment of inflammatory cells and neovascularization. Of note, the production of PGE2 was associated with the detrimental effects of GCs on EPCs. GCs impaired the expression of COX2 and mPGES1, which are the main enzymes of PGE2 synthesis. Treatment with PGE2 upregulated the expression of EP4 receptors and activated the PI3K/AKT signaling pathways, thereby reversing the impaired effects of GCs on the expression of CXCR4 in EPCs. Importantly, our data demonstrated that, GCs similarly impaired the expression of CXCR4 under hypoxic conditions as under normoxic conditions, as proven by the impairment of PGE2 and another pathway related to HIF2α. Of note, PGE2 and HIF2α acted independently in the regulation of CXCR4 in EPCs under hypoxic conditions (Figure 7, proposed model).
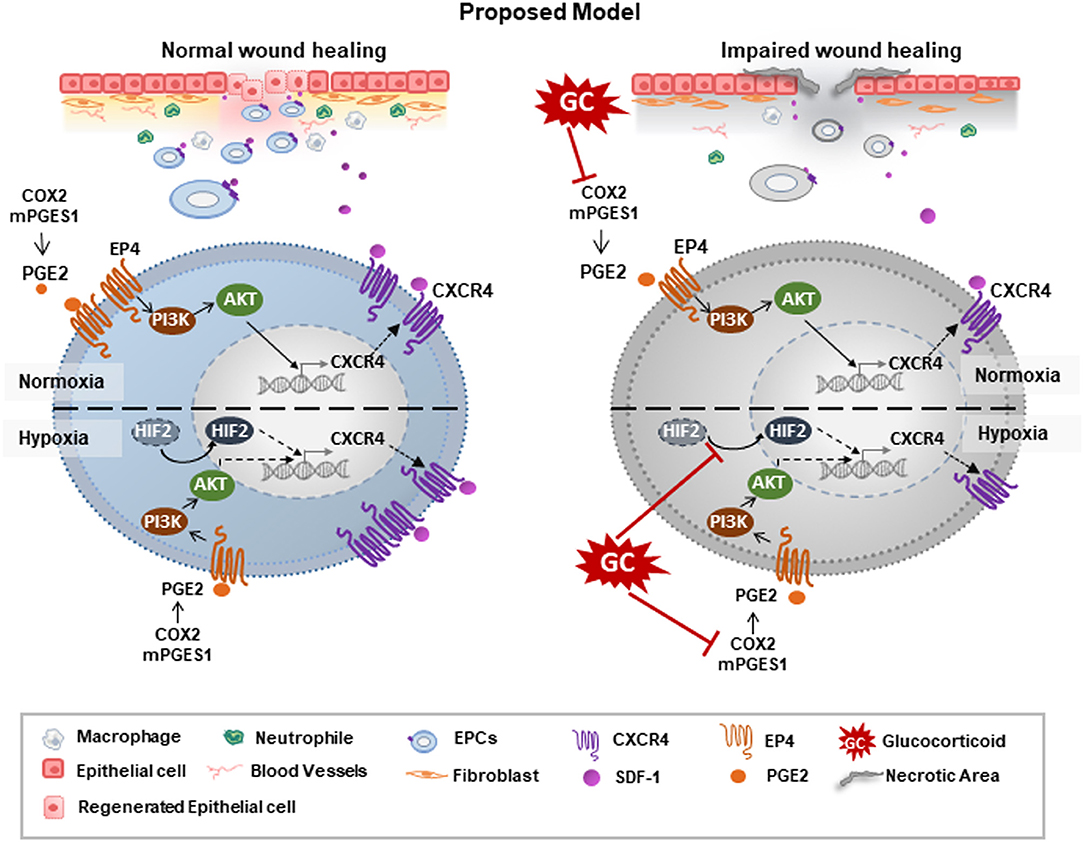
Figure 7. Proposed model: GCs impaired the wound healing functions of EPCs via the impairment of CXCR4 regulated pathways. GCs downregulated the expression of CXCR4 under both normoxic and hypoxic conditions, thereby reducing the migration and wound healing ability of EPCs. Under normoxic condition, GCs impaired the expression of COX2/mPEGS1, thereby suppressing the PGE2/EP4/AKT pathway, which downregulated the CXCR4 expression. Under normoxic conditions, in addition to the PEG2 pathway, GCs independently impaired the HIF2α pathway, consequently decreasing the expression of CXCR4.
Although no significant influence on the EPCs was observed under microscopic observation, GC treatment decreased the proliferation ability of EPCs. GCs are known to exert anti-proliferative activities against various cells, including neural cells, osteoblasts, osteosarcoma cells, hepatoma cells, and lung, ovarian, and prostate cancer cells (39–41). It has been reported that GC inhibits cell proliferation by arresting the cell cycle via several molecular mechanisms, including the upregulation of cyclin-dependent kinase inhibitors (CKIs), such as p21, that inactivate the cyclinD/Ckd4 complex, leading to G1/G0 arrest (42, 43), or affects other regulators, such as cyclinD1 and c-myc, which induce arrest in the G1 phase (44, 45). In addition, although in our present study, GC treatment showed no apoptosis induction in EPCs after 24-h treatment, a further time-course study is necessary to examine the effects of GC to induce EPCs apoptosis at later time points.
Additionally to the impaired-proliferation, GC treatment downregulated the expression of wound healing genes in EPCs, including CCL2 and CXCR4. CCL2 is the proinflammatory chemokine which is responsible for the inflammatory cell recruitment (25). It is reported that CCL2 promotes wound healing in diabetic mice by inducing macrophage (26). The downregulation of CCL2 in GC-treated EPCs might be involved in the low recruitment of CD45 and Mac1-possitive cells to the wound sites on the third day after transplantation of GC-treated EPCs in the mouse model. The SDF-1/CXCR4 cascade is a major cascade underlying the mobilization of EPCs (46). We previously reported on the GC-impaired wound healing function of MSCs by the suppression of SDF-1 (5). In the present study, although no significant effects on the expression of SDF-1 were noted, GC still adversely affected the wound healing function of EPCs via interference with the regulation of CXCR4. CXCR4 is a G-protein coupled receptor that is highly expressed on EPCs (47). In injured tissues, the surrounding cells secrete SDF-1, which recruits the CXCR4-expressing cells to the wound sites and play their functions in wound healing (48). A study in coronary artery disease patients showed that the dysregulation of CXCR4 signaling reduced the migratory capacity of EPCs in these patients compared to healthy subjects (49). In our study, we found that, under GC treatment, the downregulation of CXCR4, together with the contribution of impaired proliferation, led to a reduced in vitro wound healing ability of EPCs in the migration scratch assay. These impaired effects might be involved in the low homing ability of EPCs to the injured tissues in the mouse flap model. These findings suggested the need for further studies on the CXCR4 regulatory effects and functions of EPCs derived from patients receiving GC therapy.
The present findings raised questions about how GCs reduce the CXCR4 expression in EPCs. PGE2 has been proven to induce the expression of CXCR4 in several cells, such as myeloid-derived suppressor cells and microvascular endothelial cells (31, 50). In addition, the inhibitory effects of GCs on prostaglandin synthesis have been well-demonstrated in various cell types and tissues by the mediation of multiple pathways (51, 52). Therefore, we hypothesized that GCs impaired the expression of CXCR4 in EPCs through activity against prostaglandin synthesis. Indeed, our data demonstrated that GCs interfered with the production of PGE2 by the impairment of two enzymes: COX2 and mPGES1. Of note, treatment with PGE2 rescued the expression of CXCR4 in the GC-treated EPCs, which confirmed our hypothesis.
PGE2 is a potent upstream mediator of several genes through interaction with its receptors (EPs), including EP1, EP2, EP3, and EP4, thereby recruiting the transcription factors and modulating the target gene expression (37). Several reports have described the role of PGE2-EPs signaling in the SDF-1-CXCR4 chemokine system (53, 54). EP3 or EP4 knockout suppressed SDF-1 and CXCR4-positive stromal cells in mice (53). PGE2 promotes the homing ability of CD34-positive cells through EP2 and EP4 (54). However, thus far, how GC affects the PGE2 receptors and its effects on the regulation of CXCR4 in EPCs have been unclear. We found that GC impaired the expression of EP4 but not EP1, EP2, or EP3 in EPCs which suggested the involvement of EP4 receptor in this pathway. In addition, previous reports showed that only EP4 regulates the migration of numerous cells (55–57). For instance, EP4 regulates the migration of dendritic cells in mice via selective action on PI3K (55). In addition EP4 is also involved in breast cancer cell migration during tumor invasion (56) and enhances the migration of rat smooth muscle cells (57).
Hypoxia plays a crucial role in modulating the functions of many types of cells via the activation of HIF (58). Hypoxic preconditioning promoted the survival, differentiation, and function of EPCs for the preservation of the left ventricle in acute myocardial ischemia mice (59). In addition, hypoxic treatment upregulated the expression of CXCR4 in EPCs, thereby enhancing the migration of the cells (60). We therefore expected that hypoxic treatment might reverse the negative effects of GCs on the impaired expression of CXCR4. However, our data showed that GC reduced the expression of CXCR4 in EPCs under not only normoxic but also hypoxic conditions, although this impairment was able to be rescued by PGE2. We previously showed that the CXCR4 expression is directly regulated by HIF2α under hypoxic conditions in Alde-low EPCs (19). In our current study, we found that the expression of HIF2α was also downregulated by GC treatment under hypoxic conditions, highlighting another CXCR4-regulated pathway impaired by GC. Previous report described the role of the COX2/PGE2 pathway in the upregulation of HIF2α in carcinoma cells (38); however, in our present study, PGE2 exerted no significant effects on the expression of HIF2α in EPCs, suggesting the cell-specific regulation of HIF2α would exist. In addition, our data indicated the different mechanisms underlying the regulation of CXCR4 under normoxic and hypoxic conditions. Under hypoxic conditions, PGE2 and HIF2α operate as independent pathways that are impaired by GC, consequently suppressing the expression of CXCR4 in EPCs.
Under trauma conditions, peripheral tissues—through myofibroblasts, epithelial cells, and keratinocytes—show an increased production of EPC-mobilizing factors, like VEGF, G-CSF, bFGF, PDGF, and most importantly SDF1, as a potent chemoattractant of EPCs (61). MSCs are considered to contribute to the vascular niche development by providing growth factors (62). SDF-1 production by resident MSCs might therefore encourage the homing ability of EPCs to ischemic sites. A previous report found that the level of SDF-1 protein in the serum of GC-treated patients was significantly decreased compared with untreated patients (63). Because the biologic effects of chemokines are mediated by their corresponding receptors, the interplay between SDF-1 and CXCR4 may provide another way to regulate the distribution of circulating EPCs (64). Similar to our previous finding that GCs reduced SDF-1 production in MSCs (5), the present study showed that GCs also reduced the CXCR4 expression in EPCs. These two discoveries thoroughly show why GC-treated patients have a reduced number of circulating EPCs and a consequently impaired angiogenic ability that leads to dreadful outcomes, like avascular necrotic femoral head (ANFH).
Synthetic GCs have been developed to help treat many different conditions, such as autoimmune disorders, allergies and asthma, cancer, and surgery (24). Despite the multi-functions, GCs causes various side effects including chronic wound (65). It is reported that patients who receive GC treatment for 30 days prior to wounding or operation have a 2-fold increase in wound infection, three times increase in wound dehiscence, and four times increased mortality compared to those who not get the GC treatment (66). In addition, rheumatoid arthritis patients who receive long term GC treatment and surgery have the high risk of delayed wound healing (67). Therefore, it can be implied that the patients with GC-induced chronic wound have become the target of EPC therapy which helps to accelerate wound healing. Our study indicated the negative influences of GC on the wound healing ability of EPCs by the impairment of PGE2/CXCR4, which suggested a strategy to improve the function of EPCs. As autologous cell sources are preferred, the intervention of PGE2 or CXCR4 to improve wound healing ability of GC-treated patients-derived EPCs might be useful before the application to clinical settings.
Previous studies showed that glucocorticoid treatment impairs the corneal neovascularization in mice and rabbit which implied the possibility of abnormal vascularization process, including the dysfunction of EPCs and ECs (68, 69). In addition, EPCs from mice with aldosterone treatment showed the impaired differentiation ability to ECs and migration ability. These studies suggested the in vivo negative effects of GCs on EPCs functions (70). However, up to now, the influences of GCs on wound healing ability of EPCs have not yet reported. Therefore, our present study focused on the effects of in vitro treated GC on EPCs to provide an idea of how GCs influence EPCs wound healing functions. Further studies related to in vivo effects of GCs on EPCs using GC-induced mouse model and GC-treated patients derived EPCs are required to clarify the regulatory ability of GCs on EPCs.
In summary, our study demonstrated that GCs suppress the migration ability and wound healing function of EPCs by the downregulation of CXCR4 under both normoxic and hypoxic conditions. Under normoxic conditions, the impairment of prostaglandin synthases COX2 and mPEGS1 and the prostaglandin receptor EP4 are involved in the detrimental effects on GCs on EPCs. Treatment with PGE2 upregulated the expression of EP4 and consequently activated the PI3K/AKT pathway, which might be involved in rescuing the GC-impaired CXCR4 expression in EPCs. Under hypoxic conditions, in addition to impairment of the PGE2 pathway, GCs exerted similarly detrimental effects on the HIF2α pathway that independently downregulated the expression of CXCR4 in EPCs. Further studies should be performed to carefully assess wound healing functions of EPCs derived from patients who have been receiving long-term treatment with GCs.
EC, TK, and VK contributed to the study concept, design and access to all the data, data analysis and interpretation, and writing of the manuscript. EC, VK, and OO contributed to the writing and editing of the manuscript. KM, KT, and HH provided samples, technical and laboratory support for the research. TY contributed to the study concept, design, and technical support. OO contributed to the study concept and design, editing of the manuscript and final approval.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Japanese Ministry of Education, Culture, Sports, Science & Technology (MEXT) for their support.
1. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. (2005) 353:1711–23. doi: 10.1056/NEJMra050541
2. Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. (2002) 96:23–43. doi: 10.1016/S0163-7258(02)00297-8
3. Slominski AT, Zmijewski MA. Glucocorticoids inhibit wound healing: novel mechanism of action. J Invest Dermatol. (2017) 137:1012–4. doi: 10.1016/j.jid.2017.01.024
4. Xavier AM, Anunciato AK, Rosenstock TR, Glezer I. Gene expression control by glucocorticoid receptors during innate immune responses. Front Endocrinol. (2016) 7:31. doi: 10.3389/fendo.2016.00031
5. Kato T, Khanh VC, Sato K, Takeuchi K, Carolina E, Yamashita T, et al. SDF-1 improves wound healing ability of glucocorticoid-treated adipose tissue-derived mesenchymal stem cells. Biochem Biophys Res Commun. (2017) 493:1010–7. doi: 10.1016/j.bbrc.2017.09.100
6. Logie JJ, Ali S, Marshall KM, Heck MM, Walker BR, Hadoke PW. Glucocorticoid-mediated inhibition of angiogenic changes in human endothelial cells is not caused by reductions in cell proliferation or migration. PLoS ONE (2010) 5:e14476. doi: 10.1371/journal.pone.0014476
7. Cha J, Falanga V. Stem cells in cutaneous wound healing. Clin Dermatol. (2007) 25:73–8. doi: 10.1016/j.clindermatol.2006.10.002
8. Medina RJ, Barber CL, Sabatier F, Dignat-George F, Melero-Martin JM, Khosrotehrani K, et al. Endothelial progenitors: a consensus statement on nomenclature. Stem Cells Transl Med. (2017) 6:1316–20. doi: 10.1002/sctm.16-0360
9. Janic B, Arbab AS. The role and therapeutic potential of endothelial progenitor cells in tumor neovascularization. ScientificWorldJournal (2010) 10:1088–99. doi: 10.1100/tsw.2010.100
10. Liu ZJ, Velazquez OC. Hyperoxia, endothelial progenitor cell mobilization, and diabetic wound healing. Antioxid Redox Signal. (2008) 10:1869–82. doi: 10.1089/ars.2008.2121
11. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science (1997) 275:964–7. doi: 10.1126/science.275.5302.964
12. Hur J, Yoon CH, Kim HS, Choi JH, Kang HJ, Hwang KK, et al. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. (2004) 24:288–93. doi: 10.1161/01.ATV.0000114236.77009.06
13. Nagano M, Yamashita T, Hamada H, Ohneda K, Kimura K, Nakagawa T, et al. Identification of functional endothelial progenitor cells suitable for the treatment of ischemic tissue using human umbilical cord blood. Blood (2007) 110:151–60. doi: 10.1182/blood-2006-10-047092
14. Sukmawati D, Tanaka R. Introduction to next generation of endothelial progenitor cell therapy: a promise in vascular medicine. Am J Transl Res. (2015) 7:411–21.
15. Shanmugam VK, Angra D, Rahimi H, McNish S. Vasculitic and autoimmune wounds. J Vasc Surg Venous Lymphat Disord. (2017) 5:280–92. doi: 10.1016/j.jvsv.2016.09.006
16. Chen C, Yang S, Feng Y, Wu X, Chen D, Yu Q, et al. Impairment of two types of circulating endothelial progenitor cells in patients with glucocorticoid-induced avascular osteonecrosis of the femoral head. Joint Bone Spine (2013) 80:70–6. doi: 10.1016/j.jbspin.2012.02.015
17. Loggers ET, LeBlanc TW, El-Jawahri A, Fihn J, Bumpus M, David J, et al. Pretransplantation supportive and palliative care consultation for high-risk hematopoietic cell transplantation patients. Biol Blood Marrow Transplant. (2016) 22:1299–305. doi: 10.1016/j.bbmt.2016.03.006
18. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. (2007) 2:329–33. doi: 10.1038/nprot.2007.30
19. Tu TC, Nagano M, Yamashita T, Hamada H, Ohneda K, Kimura K, et al. A chemokine receptor, CXCR4, which is regulated by hypoxia-inducible factor 2alpha, is crucial for functional endothelial progenitor cells migration to ischemic tissue and wound repair. Stem Cells Dev. (2016) 25:266–76. doi: 10.1089/scd.2015.0290
20. Planey SL, Derfoul A, Steplewski A, Robertson NM, Litwack G. Inhibition of glucocorticoid-induced apoptosis in 697 pre-B lymphocytes by the mineralocorticoid receptor N-terminal domain. J Biol Chem. (2002) 277:42188–96. doi: 10.1074/jbc.M205085200
21. Ueck C, Volksdorf T, Houdek P, Vidal-y-Sy S, Sehner S, Ellinger B, et al. Comparison of in-vitro and ex-vivo wound healing assays for the investigation of diabetic wound healing and demonstration of a beneficial effect of a triterpene extract. PLoS ONE (2017) 12:e0169028. doi: 10.1371/journal.pone.0169028
22. Balaji S, Watson CL, Ranjan R, King A, Bollyky PL, Keswani SG. Chemokine involvement in fetal and adult wound healing. Adv Wound Care (2015) 4:660–72. doi: 10.1089/wound.2014.0564
23. Wong VW, Crawford JD. Vasculogenic cytokines in wound healing. BioMed Res Int. (2013) 2013:190486. doi: 10.1155/2013/190486
24. Liu D, Ahmet A, Ward L, Krishnamoorthy P, Mandelcorn ED, Leigh R, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol. (2013) 9:30. doi: 10.1186/1710-1492-9-30
25. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762–74. doi: 10.1038/nri3070
26. Wood S, Jayaraman V, Huelsmann EJ, Bonish B, Burgad D, Sivaramakrishnan G, et al. Pro-inflammatory chemokine CCL2 (MCP-1) promotes healing in diabetic wounds by restoring the macrophage response. PLoS ONE (2014) 9:e91574. doi: 10.1371/journal.pone.0091574
27. Goppelt-Struebe M, Wolter D, Resch K. Glucocorticoids inhibit prostaglandin synthesis not only at the level of phospholipase A2 but also at the level of cyclo-oxygenase/PGE isomerase. Br J Pharmacol. (1989) 98:1287–95. doi: 10.1111/j.1476-5381.1989.tb12676.x
28. Lee HY, Acosta TJ, Tanikawa M, Sakumoto R, Komiyama J, Tasaki Y, et al. The role of glucocorticoid in the regulation of prostaglandin biosynthesis in non-pregnant bovine endometrium. J Endocrinol. (2007) 193:127–35. doi: 10.1677/joe.1.06975
29. Lewis GD, Campbell WB, Johnson AR. Inhibition of prostaglandin synthesis by glucocorticoids in human endothelial cells. Endocrinology (1986) 119:62–9. doi: 10.1210/endo-119-1-62
30. Zicari A, Ticconi C, Pontieri G, Loyola G, Piccione E. Effects of glucocorticoids and progesterone on prostaglandin E2 and leukotriene B4 release by human fetal membranes at term gestation. Prostaglandins (1997) 54:539–47. doi: 10.1016/S0090-6980(97)00124-X
31. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. (2011) 71:7463–70. doi: 10.1158/0008-5472.CAN-11-2449
32. Chandrasekharan S, Foley NA, Jania L, Clark P, Audoly LP, Koller BH. Coupling of COX-1 to mPGES1 for prostaglandin E2 biosynthesis in the murine mammary gland. J Lipid Res. (2005) 46:2636–48. doi: 10.1194/jlr.M500213-JLR200
33. Gosset M, Berenbaum F, Levy A, Pigenet A, Thirion S, Saffar JL, et al. Prostaglandin E2 synthesis in cartilage explants under compression: mPGES-1 is a mechanosensitive gene. Arthritis Res Ther. (2006) 8:R135. doi: 10.1186/ar2024
34. Zhang J, Fujii S, Wu Z, Hashioka S, Tanaka Y, Shiratsuchi A, et al. Involvement of COX-1 and up-regulated prostaglandin E synthases in phosphatidylserine liposome-induced prostaglandin E2 production by microglia. J Neuroimmunol. (2006) 172:112–20. doi: 10.1016/j.jneuroim.2005.11.008
35. Markovič T, Jakopin Ž, Dolenc MS, Mlinarič-Raščan I. Structural features of subtype-selective EP receptor modulators. Drug Discov Today (2017) 22:57–71. doi: 10.1016/j.drudis.2016.08.003
36. Miyamoto M, Ito H, Mukai S, Kobayashi T, Yamamoto H, Kobayashi M, et al. Simultaneous stimulation of EP2 and EP4 is essential to the effect of prostaglandin E2in chondrocyte differentiation. Osteoarthritis Cartilage (2003) 11:644–52. doi: 10.1016/S1063-4584(03)00118-3
37. Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. (2007) 282:11613–7. doi: 10.1074/jbc.R600038200
38. Dong XF, Liu TQ, Zhi XT, Zou J, Zhong JT, Li T, et al. COX-2/PGE2 axis regulates HIF2alpha activity to promote hepatocellular carcinoma hypoxic response and reduce the sensitivity of sorafenib treatment. Clin Cancer Res. (2018) 24:3204–16. doi: 10.1158/1078-0432.CCR-17-2725
39. Almawi WY, Melemedjian OK, Rieder MJ. An alternate mechanism of glucocorticoid anti-proliferative effect: promotion of a Th2 cytokine-secreting profile. Clin Transpl. (1999) 13:365–74. doi: 10.1034/j.1399-0012.1999.130501.x
40. Guichard A, Humbert P, Tissot M, Muret P, Courderot-Masuyer C, Viennet C. Effects of topical corticosteroids on cell proliferation, cell cycle progression and apoptosis: in vitro comparison on HaCaT. Int J Pharm. (2015) 479:422–9. doi: 10.1016/j.ijpharm.2014.12.066
41. Mattern J, Buchler MW, Herr I. Cell cycle arrest by glucocorticoids may protect normal tissue and solid tumors from cancer therapy. Cancer Biol Ther. (2007) 6:1345–54. doi: 10.4161/cbt.6.9.4765
42. Fernandes D, Guida E, Koutsoubos V, Harris T, Vadiveloo P, Wilson JW, et al. Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular-regulated kinases in human cultured airway smooth muscle. Am J Respir Cell Mol Biol. (1999) 21:77–88. doi: 10.1165/ajrcmb.21.1.3396
43. Lu J. The anti-proliferation mechanism of glucocorticoid mediated by glucocorticoid receptor-regulating gene expression. Pathophysiology (2009) 16:267–72. doi: 10.1016/j.pathophys.2009.02.009
44. Goya L, Maiyar AC, Ge Y, Firestone GL. Glucocorticoids induce a G1/G0 cell cycle arrest of Con8 rat mammary tumor cells that is synchronously reversed by steroid withdrawal or addition of transforming growth factor-alpha. Mol Endocrinol. (1993) 7:1121–32.
45. Takayama S, Rogatsky I, Schwarcz LE, Darimont BD. The glucocorticoid receptor represses cyclin D1 by targeting the Tcf-beta-catenin complex. J Biol Chem. (2006) 281:17856–63. doi: 10.1074/jbc.M602290200
46. Petit I, Jin D, Rafii S. The SDF-1–CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol. (2007) 28:299–307. doi: 10.1016/j.it.2007.05.007
47. Yan X, Dai X, He L, Ling X, Shao M, Zhang C, et al. A novel CXCR4 antagonist enhances angiogenesis via modifying the ischaemic tissue environment. J Cell Mol Med. (2017) 21:2298–307. doi: 10.1111/jcmm.13150
48. Ding J, Tredget EE. The role of chemokines in fibrotic wound healing. Adv Wound Care (2015) 4:673–86. doi: 10.1089/wound.2014.0550
49. Walter DH, Haendeler J, Reinhold J, Rochwalsky U, Seeger F, Honold J, et al. Impaired CXCR4 signaling contributes to the reduced neovascularization capacity of endothelial progenitor cells from patients with coronary artery disease. Circ Res. (2005) 97:1142–51. doi: 10.1161/01.RES.0000193596.94936.2c
50. Salcedo R, Zhang X, Young HA, Michael N, Wasserman K, Ma WH, et al. Angiogenic effects of prostaglandin E2 are mediated by up-regulation of CXCR4 on human microvascular endothelial cells. Blood (2003) 102:1966–77. doi: 10.1182/blood-2002-11-3400
51. Malcher-Lopes R, Franco A, Tasker JG. Glucocorticoids shift arachidonic acid metabolism toward endocannabinoid synthesis: a non-genomic anti-inflammatory switch. Eur J Pharmacol. (2008) 583:322–39. doi: 10.1016/j.ejphar.2007.12.033
52. Weidenfeld J, Lysy J, Shohami E. Effect of dexamethasone on prostaglandin synthesis in various areas of the rat brain. J Neurochem. (1987) 48:1351–4. doi: 10.1111/j.1471-4159.1987.tb05670.x
53. Katoh H, Hosono K, Ito Y, Suzuki T, Ogawa Y, Kubo H, et al. COX-2 and prostaglandin EP3/EP4 signaling regulate the tumor stromal proangiogenic microenvironment via CXCL12-CXCR4 chemokine systems. Am J Pathol. (2010) 176:1469–83. doi: 10.2353/ajpath.2010.090607
54. Wang Y, Lai S, Tang J, Feng C, Liu F, Su C, et al. Prostaglandin E2 promotes human CD34(+) cells homing through EP2 and EP4 in vitro. Mol Med Rep. (2017) 16:639–46. doi: 10.3892/mmr.2017.6649
55. Kabashima K, Sakata D, Nagamachi M, Miyachi Y, Inaba K, Narumiya S. Prostaglandin E2-EP4 signaling initiates skin immune responses by promoting migration and maturation of Langerhans cells. Nat Med. (2003) 9:744–9. doi: 10.1038/nm872
56. Timoshenko AV, Xu G, Chakrabarti S, Lala PK, Chakraborty C. Role of prostaglandin E2 receptors in migration of murine and human breast cancer cells. Exp Cell Res. (2003) 289:265–74. doi: 10.1016/S0014-4827(03)00269-6
57. Yokoyama U, Minamisawa S, Quan H, Ghatak S, Akaike T, Segi-Nishida E, et al. Chronic activation of the prostaglandin receptor EP4 promotes hyaluronan-mediated neointimal formation in the ductus arteriosus. J Clin Invest. (2006) 116:3026–34. doi: 10.1172/JCI28639
58. Hong WX, Hu MS, Esquivel M, Liang GY, Rennert RC, McArdle A, et al. The role of hypoxia-inducible factor in wound healing. Adv Wound Care (2014) 3:390–9. doi: 10.1089/wound.2013.0520
59. Jian KT, Shi Y, Zhang Y, Mao YM, Liu JS, Xue FL. Time course effect of hypoxia on bone marrow-derived endothelial progenitor cells and their effects on left ventricular function after transplanted into acute myocardial ischemia rat. Eur Rev Med Pharmacol Sci. (2015) 19:1043–54.
60. Dai T, Hu Y, Zheng H. Hypoxia increases expression of CXC chemokine receptor 4 via activation of PI3K/Akt leading to enhanced migration of endothelial progenitor cells. Eur Rev Med Pharmacol Sci. (2017) 21:1820–7.
61. Darby IA, Laverdet B, Bonté F, Desmoulière A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Invest Dermatol. (2014) 7:301–11. doi: 10.2147/CCID.S50046
62. Wu Y, Zhao RCH, Tredget EE. Concise review: bone marrow-derived stem/progenitor cells in cutaneous repair and regeneration. Stem Cells (2010) 28:905–15. doi: 10.1002/stem.420
63. Butler JM, Guthrie SM, Koc M, Afzal A, Caballero S, Brooks HL, et al. SDF-1 is both necessary and sufficient to promote proliferative retinopathy. J Clin Invest. (2005) 115:86–93. doi: 10.1172/JCI22869
64. Lin Y, Luo LL, Sun J, Gao W, Tian Y, Park E, et al. Relationship of circulating CXCR4(+) EPC with prognosis of mild traumatic brain injury patients. Aging Dis. (2017) 8:115–27. doi: 10.14336/AD.2016.0610
65. Moghadam-Kia S, Werth VP. Prevention and treatment of systemic glucocorticoid side effects. Int J Dermatol. (2010) 49:239–48. doi: 10.1111/j.1365-4632.2009.04322.x
66. Ismael H, Horst M, Farooq M, Jordon J, Patton JH, Rubinfeld IS. Adverse effects of preoperative steroid use on surgical outcomes. Am J Surg. (2011) 201:305–8; discussion 308–9. doi: 10.1016/j.amjsurg.2010.09.018
67. Krause ML, Matteson EL. Perioperative management of the patient with rheumatoid arthritis. World J Orthop. (2014) 5:283–91. doi: 10.5312/wjo.v5.i3.283
68. Mello GR, Pizzolatti ML, Wasilewski D, Santhiago MR, Budel V, Moreira H. The effect of subconjunctival bevacizumab on corneal neovascularization, inflammation and re-epithelization in a rabbit model. Clinics (Sao Paulo) (2011) 66:1443–50. doi: 10.1590/S1807-59322011000800023
69. Nakao S, Hata Y, Miura M, Noda K, Kimura NY, Kawahara S, et al. Dexamethasone inhibits interleukin-1β-induced corneal neovascularization: role of nuclear factor-κB-activated stromal cells in inflammatory angiogenesis. Am J Pathol. (2007) 171:1058–65. doi: 10.2353/ajpath.2007.070172
Keywords: endothelial progenitor cells, glucocorticoids, CXCR4, wound healing, prostaglandin E2, HIF2α
Citation: Carolina E, Kato T, Khanh VC, Moriguchi K, Yamashita T, Takeuchi K, Hamada H and Ohneda O (2018) Glucocorticoid Impaired the Wound Healing Ability of Endothelial Progenitor Cells by Reducing the Expression of CXCR4 in the PGE2 Pathway. Front. Med. 5:276. doi: 10.3389/fmed.2018.00276
Received: 16 June 2018; Accepted: 10 September 2018;
Published: 28 September 2018.
Edited by:
Reinhold J. Medina, Queen's University Belfast, United KingdomReviewed by:
Gaia Spinetti, MultiMedica (IRCCS), ItalyCopyright © 2018 Carolina, Kato, Khanh, Moriguchi, Yamashita, Takeuchi, Hamada and Ohneda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Osamu Ohneda, b29obmVkYUBtZC50c3VrdWJhLmFjLmpw
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.