 Kyle T. Amber
Kyle T. Amber Manuel Valdebran
Manuel Valdebran Khalaf Kridin
Khalaf Kridin Sergei A. Grando
Sergei A. Grando- 1Department of Dermatology, University of California, Irvine, Irvine, CA, United States
- 2Department of Dermatology, Rambam Healthcare Campus, Haifa, Israel
- 3Departments of Dermatology and Biological Chemistry, Institute for Immunology, University of California, Irvine, Irvine, CA, United States
Bullous pemphigoid (BP) is an autoimmune blistering disease which carries a significant mortality and morbidity. While historically BP has been characterized as an IgG driven disease mediated by anti-BP180 and BP230 IgG autoantibodies, developments in recent years have further elucidated the role of eosinophils and IgE autoantibodies. In fact, eosinophil infiltration and eosinophilic spongiosis are prominent features in BP. Several observations support a pathogenic role of eosinophils in BP: IL-5, eotaxin, and eosinophil-colony stimulating factor are present in blister fluid; eosinophils line the dermo-epidermal junction (DEJ) in the presence of BP serum, metalloprotease-9 is released by eosinophils at the site of blisters; eosinophil degranulation proteins are found on the affected basement membrane zone as well as in serum corresponding with clinical disease; eosinophil extracellular DNA traps directed against the basement membrane zone are present, IL-5 activated eosinophils cause separation of the DEJ in the presence of BP serum; and eosinophils are the necessary cell required to drive anti-BP180 IgE mediated skin blistering. Still, it is likely that eosinophils contribute to the pathogenesis of BP in numerous other ways that have yet to be explored based on the known biology of eosinophils. We herein will review the role of eosinophils in BP and provide a framework for understanding eosinophil pathogenic mechanisms in mucocutaneous disease.
Introduction to Bullous Pemphigoid
Clinical Presentation of Bullous Pemphigoid
Bullous pemphigoid (BP) is the most common autoimmune blistering disease with an estimated annual incidence between 2 and 22 new cases per million people (1–8). BP mainly affects the elderly with an age of onset in the late 70s (8–10). Association with neurological disorders such as dementia, Parkinson's disease and cerebrovascular disease is seen in between 28–56% of BP patients (8, 11).
Most commonly, BP presents with chronic and recurrent blisters, usually arising on urticarial or eczematized skin, favoring the abdomen, and flexural aspects of the extremities. Blisters turn into erosions by mechanical friction with subsequent crust formation and healing. Prior to the development of the blisters, a prodrome of pruritus with or without urticarial lesions commonly occurs (12). Oral involvement is seen in 10–20% of BP patients (13, 14). Pruritus alone may be the only symptom of BP in some of the cases, though it is controversial whether these patients represent falsely seropositive elderly patients with other causes of pruritus, or are in fact pre-clinical cases of BP (15, 16).
Numerous clinical variants exist, with atypical clinical variants accounting for approximately 20% of cases (8, 10, 13, 16). Likewise, medications can also induce bullous pemphigoid, (8, 17–20) with a more atypical clinical and immunologic phenotype seen particularly in patients with dipeptidyl-4 inhibitor induced BP, who demonstrated a decrease in peripheral eosinophil infiltration (21–24).
Diagnosis of Bullous Pemphigoid
Histological sections of BP typically show a subepidermal blister with variable degree of inflammatory infiltrate composed of lymphocytes, neutrophils, and characteristically eosinophils. Histological presentation may vary depending on the clinical presentation. Urticarial lesions may present with spongiosis and eosinophils infiltrating the epidermis, also termed eosinophilic spongiosis, with an absence of subepidermal clefting (Figure 1) (25, 26). Peripheral eosinophilia is present in around 50% of treated patients (27–29).
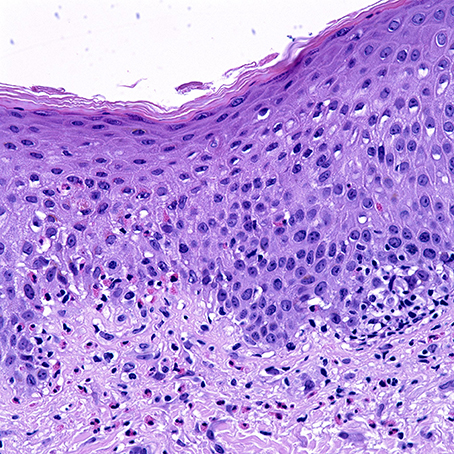
Figure 1. Uriticarial bullous pemphigoid. Histological section shows dermal inflammatory cells, predominantly composed of eosinophils, which line up at the dermoepidermal junction. Notice the spongiosis and exocytosis of eosinophils through the basement membrane into the spinous layer. H&E × 4000. (Courtesy of Dr. Philip LeBoit and the Dermatopathology Service at UCSF).
The diagnosis of BP requires further immunology workup in the form, of immunofluorescence and serologic studies. Direct immunofluorescence reveals linear deposition of IgG and complement component 3 (C3) at the dermal-epidermal junction (DEJ); linear IgA or IgE positivity is sometimes appreciated (8, 30, 31). Indirect immunofluorescence (IIF) shows linear deposition of IgG at the basement membrane on monkey esophagus or the epidermal side of salt-split human skin (32). Circulating antibodies against the proteins BP180 and BP230 can be detected in serum samples by ELISA, with sensitivities ranging from 66 to 100% (33–36). Sensitivity of the BP180 non-collagenous 16A (NC16A) domain ELISA is comparable with that of IIF with salt-split skin (37).
Pathogenesis of Bullous Pemphigoid
Evidence points to formation of autoantibodies against the hemidesmosomal proteins BP180 and BP230 as the leading events in blister formation in BP both clinically and experimentally (38, 39). BP180 (type XVII collagen) is a transmembrane glycoprotein with an extracellular C-terminus that mediates adhesion between the epidermis and the basement membrane (28). Association of disease activity has been demonstrated to clinically correlate with serum concentration of IgG antibodies against NC16A which is considered to contain the main pathogenic epitope of BP (40). Experiments with cultured human keratinocytes have shown cell detachment with reduced BP180 expression after tissue was incubated with antibodies against BP180 protein (41); moreover, anti-BP180 IgG and IgE induce signal transduction events with upregulation of interleukin-6 and interleukin-8 confirmed at the protein and mRNA levels (42–44). These results have been reproduced by transgenic mice expressing human BP180 (45).
Utilizing this humanized BP180 model, investigators demonstrated the role of T-cells as helpers of B-cells to differentiate into plasma cells and produce autoreactive IgG. Particularly they showed that NC16A-reactive CD4+ T cells in these mouse model could activate B-cells to produce anti-NC16A IgG via CD40-CD40L interaction (46). In humans, there is an association between BP and the HLA-DQB1*03:01 allele, as stimulation with BP180 to both healthy and BP patients with this allele can exert a T-cell response. Patients with BP, however, demonstrate a Th2 response while healthy HLA matched controls demonstrate a Th1 response (47–49).
BP230 is an intracellular plakin-like protein of the hemidesmosomal plaque (28). The pathogenic role of anti-BP230 antibody has not been found to be as conclusive as anti-BP180 antibody because blisters have not been observed consistently in animal models in which antibodies to BP230 are present. Furthermore, relationship between serum anti-BP230 autoantibody and disease activity has not been clearly established (12). In clinical practice, the use of ELISA for antibody to BP230 protein increases the sensitivity only 5–10% when combined with that for BP180 protein (50–52). Still, anti-BP230-type BP has been described a distinct entity, thus suggesting a pathogenic role of these autoantibodies as well as a potential for synergy with other pathogenic anti-basement membrane zone (BMZ) autoantibodies (53).
Despite the clear pathogenic role of anti-BP180 IgG autoantibody, many clinical manifestations and pathways are not easily explained by this alone. Effector cells of cell-mediated autoimmunity must be considered as significant contributors to the pathogenesis of BP. Eosinophils, have several known pathogenic roles in BP. Likewise, several known functions of eosinophils that have yet to be described in BP may play an additional role in the pathogenesis and symptoms of BP. We will review both known contributions of eosinophils to the pathogenesis of BP, as well as known mechanisms of pathogenic action of eosinophils that have yet to be evaluated specifically in BP.
Introduction to Eosinophils
Eosinophils are effector cells found in various organs including the skin. Their impact on biological processes is likely mediated primarily by their cytoplasmic granules. These granules are classified as primary, secondary, small granules, and lipid bodies (54). Secondary granules contain four toxic basic proteins: the major basic protein (MBP), eosinophil peroxidase (EPX), eosinophil derived neurotoxin (EDN) and eosinophil cationic protein (ECP). The crystalloid core of secondary granules is constituted by highly cationic MBP and is covered by the 2 ribonucleases: ECP and EDN (55). ECP is used extensively as a marker to assess activity in various inflammatory diseases (56). Granules are secreted when eosinophils become activated. These granules are highly toxic to microbes, parasites, and tumor cells (57). For instance, ECP is a cytotoxic ribonuclease with the ability to exterminate parasites, bacteria and virus in vitro (56). Moreover, ECP forms pores or transmembrane channels, which ultimately results in cellular damage and death (58). ECP can also lead to epithelial and neuronal apoptosis (59–61). MBP toxicity is mediated by affecting the charge of cellular surface membranes resulting in disruption and altered permeability leading to cellular injury (54, 62). Eosinophils are also implicated in the production of various cytokines, chemokines, lipid mediators, and superoxide. Complex immunomodulatory functions have been attributed to eosinophils which can also act as antigen-presenting cells (APCs) (57).
Peripheral eosinophilia is the result of the secretion of several factors such as IL-5, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-3 (63). Migration of eosinophils from circulation into the skin is mediated at least in part by very late activation antigen-4 (VLA4) which is expressed in eosinophils and binds to vascular cell adhesion molecule 1 (VCAM-1) on vascular endothelium. Other chemoattractants include eotaxin-1 (CCL11), eotaxin-2 (CCL24), eotaxin-3 (CCL26), RANTES (CCL5), and monocyte chemoattractant proteins which can bind to eosinophils and lead them to lesional sites (57, 64, 65).
Known Mechanisms by Which Eosinophils Can Contribute to the Pathogenesis of Bullous Pemphigoid
There are several lines of evidence suggesting the role of eosinophils in the pathogenesis of BP. Peripheral blood eosinophilia is present in ~50% of affected patients (27–29). Furthermore, elevated serum concentrations of secretory granules, such as ECP, are significantly elevated in patients with BP, with levels paralleling disease severity (55, 66–71). A similar relationship has been documented to occur with IL-5 levels which runs parallel not only to disease severity, but also to ECP levels (68, 72–80). This is consistent with an increase in eosinophil activation, as confirmed by expression CD69, in peripheral blood and lesional skin of BP patients (81). While an increase in blood and tissue eosinophils has long been known (82), the actual role of eosinophils in the pathogenesis of BP is becoming more readily understood.
Production of Metalloproteases
Proteases including gelatinase B (92-kD gelatinase, matrix metalloproteinase [MMP]-9) and neutrophil elastase (NE) play a significant role in degrading BP180 and cleaving the DEJ (83). MMP-9 is released as a zymogen and is subsequently activated by a series of proteases including MMP-2, 3, 7, 10, and 13, as well as cathepsin G, plasmin, and trypsin. MMP-9 intervenes in tissue remodeling and facilitates cellular migration, extracellular matrix degradation and tissue destruction (84). Studies in isolated human eosinophils have documented that tumor necrosis factor-α (TNF-α) is a potent stimulator for a rapid release of pro-MMP-9 (85). Experiments in peripheral blood of allergic volunteers demonstrated that IL-3 in combination with TNF-α induces significant MMP-9 synthesis by eosinophils (84).
MMP-9 can cleave the extracellular collagenous domain of recombinant 180-kD BP antigen (86). Eosinophils appear to be the principal culprit in MMP-9 secretion. Strong signal for gelatinase mRNA has been detected in eosinophils but not in neutrophils at site of blister formation. In vitro studies conducted in Matrigel have likewise demonstrated eosinophils' ability to degrade the BMZ, identifying MMP-9 as the key protease. Notably, release of MMP-9 was increased only in the presence of both IL-5 and platelet activating factor (PAF) (87).
The direct role of MMP-9 in cleaving BP180 has been challenged, based on its ability to regulate neutrophil elastase (NE). In mouse models, MMP-9 regulates NE activity by inactivating α1-proteinase inhibitor, thus contributing to further degradation of BP180 and DEJ separation (88). Studies by Verraes et al showed that despite the presence of the proform of MMP-9 in human lesional skin, BP180 degradation could be inhibited by a specific elastase inhibitor, but not by a wide spectrum of matrix metalloproteinase inhibitor, suggesting the importance of the regulatory role of MMP-9 on NE in blister formation (83).
Production of Eosinophil Degranulation Proteins
Eosinophils and neutrophil granule proteins can be detected in the blister fluid and serum of BP patients (55, 66, 67, 89). Moreover, studies have found peroxidase positive eosinophil granules along lamina lucida of the BMZ in BP patients (90). Scanning electron microscopy studies have revealed granule release into basal cells (91). Eosinophil granule protein deposition has been demonstrated not only in fully developed blisters, but also at the earliest stages of blister development and urticarial lesions of BP (73, 92). Eosinophil degranulation has also been observed in pemphigoid gestationis, a gestational variant of BP, whereby MBP is deposited extracellularly in the dermis (93).
More recent in vivo observations have demonstrated the presence of ECP, EDN, EPX, and MBP in skin. In addition, in vivo experiments in guinea pig skin have demonstrated the presence of these granules for weeks after intradermal injection at concentrations seen in human disease. During these experiments, Davis et al demonstrated increased cutaneous vascular permeability as an effect of the degranulation; however, basement membrane and epidermal alterations such as spongiosis were not examined (94).
In vitro experiments have shown that eosinophils may be activated through augmenting cell-surface receptors and receptor-linked oxidative metabolism (95). Upon activation, eosinophils degranulate (55, 66, 67, 89). Tsuda et al. demonstrated that degranulated eosinophils adhered to basal keratinocytes suggesting that eosinophil granules may directly damage basal keratinocytes leading to DEJ separation (89). We have demonstrated that MBP has a concentration dependent cytotoxic effect on cultured keratinocytes (96).
As eosinophils also release tissue factor (TF), an initiator of blood coagulation, Marzano et al, hypothesized a role for local activation of the coagulation cascade in BP. This was confirmed to be the case (97), with subsequent correlation between ECP levels and prothrombotic markers (70). Cases with higher coagulation, like ECP, are associated with more severe disease (97). Whether this contributes to blister development is, however, unclear.
Production of Eosinophil Extracellular Traps
Relevance of extracellular traps produced by eosinophils (EET) have increased, at least in part due to the interesting findings obtained from studies on neutrophils. Extracellular traps consist of network-like structures containing DNA, granule proteins and nuclear proteins. These traps can expand up to 15 times the size of the cell, thereby increasing the effective targeting area (98). Experiments performed on skin biopsies from human participants have shown that EET are present in infectious skin diseases, allergic diseases and autoimmune diseases including BP. The number of eosinophils releasing DNA appear to be around 10%, though this phenomenon was most commonly observed in Well's syndrome, whereby trap formation was seen in up 30% (99). Ex vivo experiments utilizing human skin and isolated human eosinophils have shown that EET may contribute to DEJ splitting, after the observation that DNase significantly reduced DEJ separation (99). Still, the mechanism by which EET contribute to DEJ separation is unclear.
Link Between Anti-BP180 IgE Autoantibodies and Dermo-Epidermal Junction Separation
Evidence supporting the pathogenic role of IgE autoantibodies in BP as well as its relationship with eosinophils has increased in recent years. Passive transfer of anti-BMZ IgE autoantibodies results in erythema, pruritis, eosinophil infiltration, and histologic blistering (100). This study did, however, use the LABD97 portion of the BP180 protein rather than the NC16A portion. Lin et al. created a transgenic mouse model expressing human hNC16A and human high-affinity IgE receptor (FcεRI), showing that anti-NC16A IgE from BP patients induced subepidermal split as well as eosinophil infiltration and IgE deposition at the DEJ. Particularly, they found that eosinophils are essential in order to induce BMZ separation in the presence of anti-NC16A IgE in vivo. This step appears to be key in DEJ separation, thus supporting the pathogenic role of IgE autoantibodies against the NC16A region of the BP180. Likewise, eosinophils in this animal model expressed FcεRI, thus providing a further link between IgE autoantibodies, eosinophils and blister formation, occurring independently of neutrophils (101).
The expression of FcεRI in monocytes, mast cells, basophils, eosinophils, dendritic cells, and platelets has been highlighted recently as a link between the biology of these cells in the presence of IgE autoantibodies (102). FcεRI, the high affinity IgE receptor is typically minimally expressed on eosinophils, but it is highly expressed on eosinophils in BP and other diseases characterized by high IgE and eosinophilia (103). FcεRI consist of an α-chain, which controls IgE binding, and β- and γ-chains, which intervene in signal transduction. These chains have been found in either tetrameric (αβγ2) or trimeric (αγ2) form in humans (103). Interestingly, investigators have found mRNA for α-,β-, and γ- chains in eosinophils of BP patients. These FceRI eosinophils appear to predominate in the dermis rather than epidermis (104). The capability of eosinophils to bind IgE may thus influence their subsequent degranulation (103).
Elevated serum IgE in patients with BP was first described in 1974 (105). Since then, several studies have demonstrated elevated IgE levels in up to 40–50% of patients, with the notable discovery of anti-basement-membrane IgE autoantibodies (29, 106–108). Advancements in technology led scientists in the 1990s to identify IgE autoantibodies to different BP molecules such as BP230 (109, 110). Later in the 2000s, IgE autoantibodies targeting NC16A, the principle epitope of BP180 to which IgG4 antibodies preferentially react, were described (111). IgE reactivity with other epitopes, particularly the intracellular domain of BP180 was subsequently described (112). This intracellular domain is critical for incorporating proteins into hemidesmosomes, raising the possibility that autoantibodies against it could impair the interaction of BP180 with other constituents of the hemidesmosome (112). Experiments conducted by Freire et al provided evidence that IgE and BP180 form immune complexes in BP skin. Furthermore, in vitro experiments found that IgE-BP180 complexes have the potential to cross-link FcεRI in basophils leading to activation and degranulation (113). Approximately half of patients demonstrate IgE autoantibodies against BP180 and BP230 (114). Ultimately, the use of an eosinophil deficient mouse demonstrated that eosinophils are the essential link between anti-BP180 IgE antibodies and BP (101).
Despite the prevalence and clear causal role of anti-BP180 IgE in inducing BP, the clinical significance of these autoantibodies remains unclear (114). A systematic review of studies associating disease phenotype with the presence of anti-BMZ IgE found no association with phenotype, though the presence of anti-BMZ IgE was associated with greater disease severity (115). Treatment results with omalizumab, a monoclonal antibody blocking soluble IgE from binding to its receptors, have been mixed with moderate efficacy, showing only a limited role as a monotherapy (116–118). As anti-BP180 IgE ELISA is not routinely available, selecting BP patients for this treatment regimen remains a challenge in the routine clinical practice.
Production of Key Cytokines and Chemokines
Evidence of both inflammatory and anti-inflammatory cytokines have been described to be present in BP. Elevated levels of proinflammatory (IL-6, TNF-alpha, IL-8) and anti-inflammatory cytokines (IL-4, IL-10) have been reported (119). Moreover, Giomi et al has suggested that cytokine milieu varies according to the chronicity of BP. An initial Th0/Th2-like response would be seen in early stage of BP with IL-4, IL-5, and low levels of IFN- γ. Whereas in chronic phases, a Th1 response would follow with significant expression of IFN- γ (120).
A complex network of chemokines also contributes to the development of BP. Both Th1 and Th2 chemokine profiles are exhibited in BP as follows: macrophage inflammatory protein-1 β (MIP-1β) and IFN- γ- inducible protein 10 (IP-10) for Th1; and eotaxin, monocyte chemoattractant protein (MCP)-4 for Th2. Overall, there is predominance of Th2 chemotactic activity compared to Th1 cells (79). Other studies have shown significantly levels of MCP-1, and IP-10, monokine induced by IFN- γ (MIG) for Th1 (121), and, eotaxin, CCR3, and MCP-4 for Th2 (77, 79, 122). Experiments conducted by Gounni-Abdelilah et al found that eotaxin and MCP-4 were present in eosinophil granules in the bullae of patients with BP, as well as in the epidermis and infiltrating eosinophils in skin of BP patients and these were secreted by eosinophils when stimulated by IgG, IgA, or IgE immunocomplexes (79). This autocrine pathway may thus perpetuate the immune response in BP, leading to chronicity of lesions (79). Cytokines and chemokines known to be produced by eosinophils are summarized in Table 1.
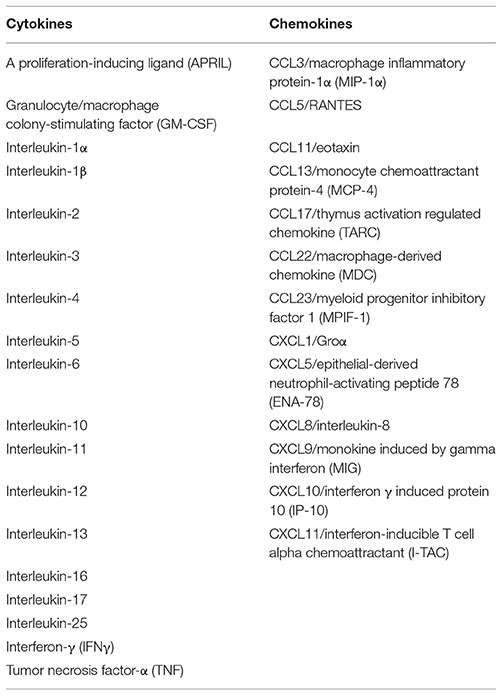
Table 1. Cytokines and chemokines capable of being secreted by eosinophils (65).
Known Mechanism of Eosinophils With Potential Roles in BP (Hypothesis)
Learning From Other Eosinophil-Mediated Conditions
Numerous conditions involve a predominance of eosinophils such as allergic reactions, parasitic infections, and certain malignancies. From these, a large amount of information regarding the function of eosinophils has been discovered. However, it is important to note that not all eosinophils are the same, even within the same disease and organ system. Eosinophils differ in their molecular pattern as observed by Lingblom et al. when investigating differences in children and adults with eosinophilic esophagitis (123). Moreover, investigations in healthy individuals revealed age-dependent differences in levels of eosinophil markers. For instance, levels of CD44 increased with age, while levels of CD54, prostaglandin DP2 receptor (CRTH2), and galectin-10 decreased with age. In addition, they demonstrated that young healthy children express highest levels of galectin-10, CRTH2, and CD54 and that these diminish with age (123). Similarly, the gastrointestinal system hosts substantial number of eosinophils exhibiting differences to eosinophils in the lungs or blood. For example, intestinal eosinophils rarely degranulate, and their lifespan is far longer than of those found in inflammatory sites (124, 125).
Despite these limitations, many functions of eosinophils appear to be retained across allergic diseases. To what degree they contribute specifically to BP, however, remains to be determined. We thus review these known mechanisms, which have a scientific rationale for contributing to the pathogenesis and symptomatology of BP.
The Role of Eosinophils in BP Related Pruritis
Pruritis is a hallmark of BP. In certain cases, it can be the presenting symptom, even when a rash is not present (16). While the depletion of BP180 in itself can generate itch as seen in BP180 knockout mice (126), several other pathways potentially contribute to this cardinal symptom of BP.
Interleukin-31
IL-31 belongs to the IL-6 family of cytokines produced in part by activated Th2 cells (127). It has a significant role in itch, by activating endothelin-1 responsive neurons and by increasing the release of brain natriuretic peptide (BNP), a central mediator of itch (128, 129). IL-31 additionally induces cutaneous nerve growth and branching (130).
In BP, elevated levels of IL-31 have been demonstrated in serum and in lesional skin of patients (131) and has been significantly associated with both eosinophilia and elevated anti BP-180-IgE (132). Eosinophils are capable of producing IL-31 (132, 133). In fact, eosinophils were recently shown to be the primary source of IL-31 in BP (134).
Substance P
Substance P is a major pruritogen and vasodilator released from peripheral nerves. The presence of substance P in BP has, however, varied between studies (135, 136). Substance P can have significant interactions with eosinophils. In mouse models of atopic dermatitis, degranulated eosinophils have been found surrounding an increased number of substance P-positive nerve fibers in lesional skin (137). Also, nasal provocation with substance P in patients with allergic rhinitis leads to an increased number of eosinophils (138, 139). Substance P acts on cells via binding to neurokinin-1 receptor (NK1R) and neurokinin-2 receptor (NK2R). Interestingly, effects of substance P on eosinophils include inhibition of apoptosis in a comparable manner to IL-3, a known apoptosis inhibitor, which contribute to extend eosinophil survival and may perpetuate its biological effects in disease (139).
Substance P can also induce the release of nerve growth factor (NGF) and IL-31 from eosinophils, in addition to mast cells. NGF may play a significant role mediating pruritus due to its ability to sensitize primary itching sensing neurons (140). NGF released from eosinophils may then stimulate neighbor nerves to further release substance P. Other roles of substance P on eosinophils include chemotaxis, activation and survival, thus potentially perpetuating the itch cycle. Mast cell-eosinophil crosstalk can also develop a neuro-immune communication axis and subsequently induce distinctive substance P itch (140).
Direct Interaction With Peripheral Nerves
Eosinophils interact with nerve cells leading to enhanced growth and branching resulting in enhanced innervation of the skin, as documented in cultured dorsal root ganglion neurons (141, 142). Eosinophils also coordinate changes in neurotransmitter release, and protection from cytokine-induced apoptosis. In part, these interactions occur as a result of activation of neural NFκB, activated by adhesion of eosinophils to neural intercellular adhesion molecule-1 (ICAM-1) (143).
The close relationship of eosinophils and nerves has been demonstrated in human skin samples of atopic dermatitis patients where investigators observed increased nerve density near eosinophil granule proteins. These findings were reproduced in mice whereby histological samples of murine skin showed that IL-5-stimulated eosinophils were present in the same epidermal foci of increased nerves. In vitro experiments with cultures of eosinophils have shown a dramatic increase in branching of sensory neurons. Collectively, these findings are in favor of an important role for eosinophils in cutaneous nerve growth (141).
Eosinophil granules may mediate a crosstalk between nerves and eosinophils. In histological samples of prurigo nodularis patients, for example, ECP- and EDN/EPX-containing eosinophils were primarily distributed in the upper dermis where nerves were also increased in number. Some of these nerves were even in direct contact with eosinophils (144).
Interaction With the Autonomic Nervous System
Eosinophils and the autonomic nerve system demonstrate a two-way cross talk. In vitro experiments demonstrate that the adherence of eosinophils to cholinergic nerves triggers a series of molecular events including activation of NFκB and activator protein (AP)-1 in the nerve cells, ultimately promoting nerve growth (145). In guinea pigs, adhesion of eosinophils to parasympathetic nerves results in release of reactive oxygen species (ROS) via neuronal NADPH oxidase, as well as activation of p38 MAP kinase (146). Eosinophils have also been implicated in the remodeling of neurites of the cholinergic nerve cell line (146).
Individual eosinophil derived granule proteins have been shown to affect cholinergic nerves. At non-cytotoxic concentrations, eosinophil cationic proteins have been shown to induce nerve cell signaling pathways by phosphorylation of the MAP kinases ERK 1/2, p38, and AKT and subsequent activation of the nuclear transcription factor NFκB (147). EPX has been shown to upregulate choline acetyltransferase (ChAT) and vesicular acetylcholine transporter (VAChT) gene expression while MBP upregulated VAChT alone. These enzymes coordinate the production of acetylcholine whereby ChAT catalyzes the production of acetylcholine from choline and acetyl-CoA, and VAChT regulates packaging into vesicles for synaptic release (148).
MBP and NGF have also been implicated in upregulating muscarinic M2 receptor expression in vitro; observed changes were associated with a reduction of intracellular neural acetylcholine and an increase in choline content (149, 150). MBP also protects nerve cells from apoptosis by upregulation of adhesion-dependent activation of ERK1/2, inducing expression of the antiapoptotic gene bfl-1 and bfl-2 (147, 149, 151). Thus, MBP released from eosinophils at inflammatory sites may regulate peripheral nerve plasticity by inhibiting apoptosis (147). In animal models, eosinophil MBP is associated with the hyperreactivity of cholinergic nerves (148, 152–155).
Cholingeric nerves additionally can influence eosinophils. Eosinophil degranulation has been demonstrated in tissue taken from patients with inflammatory bowel disease and asthma in which eosinophils adhered to cholinergic nerves (156, 157). Interestingly, nicotinic agonists decrease eosinophil infiltration in lungs and airways of mice (158).
Eosinophils Act as Antigen Presenting Cells
While eosinophils had traditionally been considered an effector cell, recent advances have elucidated the multifaceted nature of eosinophils which can affect tissue homeostasis, metabolism, and immune regulation in both disease and the steady state (159). Eosinophils can effectively process antigen, express co-stimulatory molecules, traffic to the lymph node and induce a T-cell response (160–163). This can be stimulated by GM-CSF. Studies in wild-type mice have demonstrated that eosinophils in the lamina propria of the intestine express surface markers such as MHC II and CD80 suggesting that eosinophils in this location are capable of functioning as APCs. Moreover, investigators identified intraepithelial eosinophils exhibiting dendrites with extensive reaches. Further experiments with antigen sensitized mice revealed that despite the presence of two distinct populations, both populations of eosinophils acquired intestinal antigen in vivo (159). Double stain-immunohistochemistry has likewise been used to demonstrate T-cell activation and tissue eosinophils expressing MHC-II in specimens of eosinophilic esophagitis (164). Still, the efficiency of eosinophils as APCs, their function in the skin, and their ability to process key antigens in BP remains unknown.
Direct Pathogenic Actions of Degranulation Proteins and Reactive Oxygen Species
Eosinophil degranulation proteins are capable of inducing cytotoxicity through several mechanisms. ECP induces pore formation in the cell membranes contributing to inflict cellular damage (58), while MBP increases smooth muscle reactivity due to selective allosteric antagonism of vagal muscarinic M2 receptors (165) and triggers degranulation of mast cells and basophils (166, 167). While we demonstrated a cytotoxic effect of degranulation proteins on keratinocytes at physiologic doses seen in BP (96), it is unclear the extent of damage in vivo as necrosis is not a histologic feature of BP.
Eosinophil peroxidase generates hydrogen peroxidase as well as superoxide, causing additional damage (168). Interestingly, blockade of ROS was capable of inhibiting blister formation in an ex vivo model of BP (169). Limitations to the cryosection model however may overstate the role of ROS in DEJ separation. A similar study in neutrophil mediated BP evaluated luteolin, a plant-derived flavonoid with potent anti-oxidative and anti-inflammatory properties effects in ex vivo cryosection model of BP, resulting in a significant reduction of autoantibody-induced DEJ separation. However in vivo mouse experiments did not yield comparable results (170). Thus, further in vivo studies are needed to determine the role of antioxidants in inhibiting eosinophil induced ROS in BP.
Can Eosinophils Sustain Local Immune Response?
Eosinophils express a series of cytokines involved in plasma cell survival such as activation and proliferation-induced ligand (APRIL), IL-6, IL-4, IL-5, IL-13, IL-10, and TNF (171, 172). Therefore, they could in theory provide a local stimulus sustaining Ig producing plasma cells in the dermis. Eosinophil IL-16 a key cytokine in T-cell recruitment, as well as IL-4, IL-5, and IL-13 can stimulate Th2 immunotype. Thus, eosinophils could also in theory perpetuate T-cell stimulation and a Th2 milieu.
Effect of Eosinophils on B-Cells
B-cell responses are regulated by a series of signals including IL-2, IL-4, IL-7, IL-15, and members of the TNF family such as CD40 ligand (173). In the past two decades, two TNF family molecules: B cell-activating factor of the TNF family (BAFF) and APRIL have been recognized as key regulators of normal B cell functions and autoimmune B cell induction, both of which are expressed by eosinophils (174).
APRIL binds to receptors such as transmembrane activator and calcium modulator ligand interactor (TACI) and B cell maturation antigen (BCMA) (175, 176). Normal functions of APRIL include: increasing B-cell antigen presentation, stimulation of antigen-activated B-cells, enabling isotype switching in B cells, and augmenting plasma cell survival (174, 177, 178). BAFF acts as a potent B-cell growth factor as well as stimulus for immunoglobulin production (173, 179).
BAFF levels are significantly elevated in patients with BP. Interestingly, BAFF levels increased before the anti-BP180 antibody level and quickly decreased in response to treatment, making it a useful marker for early disease (180). Levels of APRIL are likewise elevated in BP, and closely correlate with BAFF. Levels of APRIL similarly occur extremely early on in the development of disease, and thus appear to be a key mediator prior to the development of detectable levels of autoantibodies (181). Whether the APRIL and BAFF in BP primarily comes from eosinophils or other immune cells remains unknown.
Effect of Eosinophils on T-Cell Recruitment
CCL5 (RANTES) is a chemoattractant for CD4+ memory T cells, monocytes, and eosinophils (182, 183). In vitro studies have shown that RANTES activates T lymphocytes in an antigen-independent manner (184). RANTES may also activate eosinophils, upregulating their expression of adhesion molecules and enhance transendothelial migration (182, 183, 185, 186). IL-16 is likewise a strong attractant of CD4+ T-cells. Eosinophils release both IL-16 and RANTES. Even at very low concentrations, RANTES and IL-16 induce migration of T-lymphocytes. Thus, eosinophils can potentially amplify the immune response by recruitment of CD4+ lymphocytes as well as additional eosinophils (187). These interactions have yet to be elucidated in BP.
Effect of Eosinophils on Th2 Polarization
Th2 polarization may be driven by eosinophil production of IL-4, IL-5, and IL-13 (188). In addition, eosinophil expression of indoleamine 2,3-dioxygenase (IDO) which catalyzes the conversion of tryptophan to kynurenines, can regulate T cell subset selection toward Th2 (189). Thus, eosinophil products can serve as Th2 adjuvants via dendritic cell regulation (190, 191).
Eotaxin and MCP-4 are two chemokines that play a significant role in the selective recruitment of not only Th2 effector cells, but also eosinophils to the inflammatory site of BP, both of which are present at elevated levels in tissue and blister fluid (79, 122, 192). In a series of experiments investigators have found that eotaxin and MCP-4 mRNA were expressed in all biopsies of BP patients, present in the epidermis, and were also expressed in eosinophils. Immunohistochemical studies confirmed that these chemokines were localized to the granules of eosinophils (79). Overall, the levels of Th2 associated chemokines (eotaxin and MCP-4) in blister fluid are significantly greater than Th1 associated chemokines (MIP-1B and IP-10). Whether eosinophils or keratinocytes are the primary source of these chemokines is unclear (193).
Keratinocytes Exposed to Anti-BP180 Antibodies Express Key Cytokines and Chemokines Involved in Eosinophil Chemotaxis
In BP, eosinophils are classically aligned along the basement membrane, with several eosinophils often traveling into the epidermis via exocytosis. While eosinophil binding to the BMZ is known to require IgG and complement (not IgE), the ability of eosinophils to exocytose into the epidermis likewise cannot sufficiently be explained by this. Thus, keratinocyte signaling is likely to have a pivotal role, as keratinocytes can express key chemokines such as IL-8 and eotaxins.
Interleukin-8
IL-8 is produced by keratinocytes when exposed to BP autoantibodies (42, 44, 194–197). IL-8 is a known chemoattractant for neutrophils (198, 199). Studies investigating relationship of neutrophils, eosinophils and IL-8 have shown that IL-8 stimulates neutrophils to induce trans-basement membrane migration of eosinophils in the airways of asthmatic patients (200). Upon lipopolysaccharide (LPS) and IL-8 stimulation, neutrophils produce several chemoattractants for eosinophils such as leukotriene B4 and PAF that can recruit eosinophils and induce trans-BMZ migration (201, 202). Interestingly, IL-8 does not stimulate eosinophils alone to migrate through artificial BMZ (Matrigel) (203). Therefore IL-8 seems to play a key role to stimulate neutrophils resulting in subsequent trans-basement membrane migration of eosinophils (197).
Autoantibodies to BP180 mediate the release of IL-8 from human keratinocytes in a dose and time dependent manner (42, 195). In fact, this cytokine is known to be elevated in sera and blister of BP patients with significantly higher levels of IL-8 in blister fluid as compared to serum (42, 194, 196).
Eotaxins
Aside from stimulating Th2 polarization, eotaxins have a key role in the attraction of eosinophils into their target tissues. Eotaxins consists of three chemokines CCL11, CCL24, and CCL26 (204). The main eotaxin receptor is CCR3 which is expressed on all eosinophils in peripheral circulation (205).
The presence of elevated levels of eotaxin, IL-5 and CCR3 has been demonstrated in blister fluid, lesional and perilesional skin in BP (77, 122). Significant correlation with these markers and the number of dermal infiltrating eosinophils has also been demonstrated (77). In addition, studies investigating specific ligands have shown that CCL11 and CCL26 are significantly associated with activated eosinophils (204). Epidermal expression of eotaxin appears to be a consistent feature among all eosinophilic dermatoses (193).
Conclusion
Eosinophils are complex cells with numerous functions. They generally make up the predominant inflammatory cell-type seen in BP. In recent years, the overall understanding of eosinophils has significantly improved, leading to new avenues to pursue in the pathogenesis of BP. Several eosinophil pathways have well-defined roles in the pathogenesis of BP that demonstrate not only a correlative role in disease and severity, but rather a causative.
• MMP9 is secreted from eosinophils and is capable of cleaving BP180 and activating NE
• Eosinophil degranulation proteins are deposited on basal keratinocytes
• Eosinophil extracellular traps can contribute to DEJ separation. This can be abrogated with DNAse treatment
• BP180 IgE autoantibodies need eosinophils in order to mediate DEJ separation in vivo. This requires FcεRI which while not typically expressed on eosinophils, is significantly overexpressed in BP.
• Eotaxin and MCP-4 are seen in eosinophil granules in BP patients, thus perpetuating tissue eosinophilia.
Aside from these known direct roles in the pathogenesis of BP, several known functions of eosinophils have a scientific rationale to contribute to symptomatology and the pathogenesis of BP. Limitations to drawing further conclusions are summarized below:
• Eosinophils are the key producer of IL-31, a known pruritogen, in BP. Whether this is the primary pruritogen in BP is not known.
• Eosinophils can directly attach or degranulate onto peripheral and autonomic nerves, inducing branching and nerve growth which can lead to pruritis. While known to occur in other skin disease, this has not been studied in BP.
• Eosinophils can act as functional APCs. It is not known whether eosinophils in the skin can function as APCs, and whether they can effectively process the BP180 antigen
• Eosinophil degranulation proteins are known to be cytotoxic and have been shown to be cytotoxic to keratinocytes. Whether this cytotoxicity has in vivo contributions to the pathogenesis of BP has not been studied.
• Eosinophils cause generation of ROS which when blocked in ex vivo models, can prevent DEJ separation. Whether eosinophil induced ROS is sufficient to lead to disease in vivo has not been studied. In neutrophils, this is not sufficient to prevent blister formation.
• Eosinophils are known to produce BAFF and APRIL, two key regulators of autoimmune B-cells. Whether they produce BAFF and APRIL in BP, and whether this is indispensable in promoting B-cell responses in BP is not known.
• Eosinophils are known to secrete IL-16 and RANTES, two key T-cell recruiting molecules. Whether this occurs in BP, and whether this is indispensable for T-cell involvement in BP is not known.
• Eosinophils are known to secrete IL-4, IL-5, and IL-13 which can promote Th2 polarization. Whether they are the primary source of IL-4, IL-5, and IL-13 in BP, as well as whether they are indispensable in promoting Th2 polarization in BP is not known.
• Keratinocytes express Eotaxin and IL-8, both strong attractants for eosinophil migration. Whether this has pathologic significance is not known.
The principle mechanism by which eosinophils can potentially contribute to the pathogenesis of BP are summarized as a schematic in Figure 2. Future studies addressing these uncertainties will provide a more thorough understanding of the roles of eosinophils in BP, as well as eosinophils in the skin.
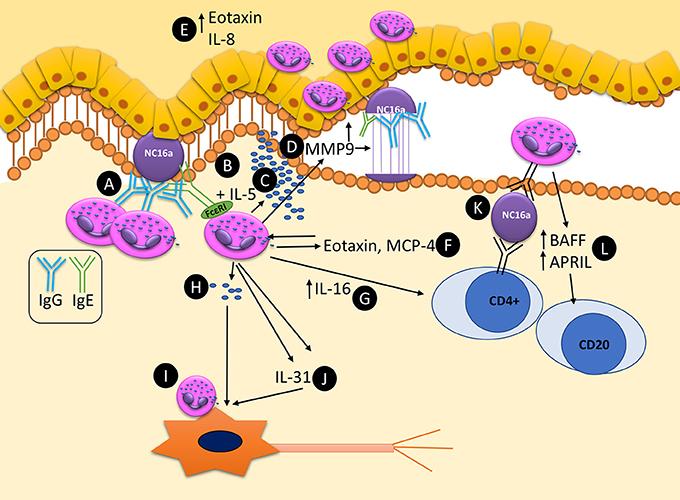
Figure 2. Schematic representation of known and potential pathways by which eosinophils can contribute to the pathogenesis and maintenance of autoimmunity in bullous pemphigoid. (A) Eosinophils bind anti-BP180 IgG, aligning along the BMZ. (B) Dermal eosinophils express FcεRI which can bind to anti-BP180 IgE leading to DEJ separation. (C) Upon activation with IL-5 eosinophils can lead to DEJ separation and degranulation. (D) MMP9 is secreted from eosinophils and is capable of cleaving BP180. (E) Eotaxin and IL-8 are expressed in the epidermis, acting as eosinophil chemotactic chemokines, attracting further tissue eosinophilia. (F) Eotaxin and MCP-4 are released from eosinophil granules, further driving tissue eosinophilia and Th2 polarization. (G) IL-16 is released from eosinophils and is capable of stimulating T-cell response. (H) Eosinophils can directly degranulate on and (I) directly bind to neurons leading to increase branching and potentially pruritis. (J) Eosinophils secrete IL-31, a major pruritogen which can stimulate nerves. (K) Eosinophils are capable of acting as antigen presenting cells, potentially leading to T-cell responses by binding bound antigen via MHC-II to T-cell receptors. (L) Eosinophils express BAFF and APRIL, potentially stimulating local autoimmune B-cells.
Author Contributions
KA: Substantial contributions to the conception or design of the work, or the acquisition, analysis, or interpretation of data for the work; KA, MV, KK, SG: Drafting the work or revising it critically for important intellectual content, Final approval of the version to be published, Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
KA would like to thank the International Pemphigus and Pemphigoid Foundation for support of the project “Identifying novel pharmacologic targets in bullous pemphigoid: Unraveling the mechanisms of eosinophils.”
Abbreviations
AP-1, activator protein-1; APC, antigen presenting cell; APRIL, activation and proliferation-induced ligand; BAFF, B cell-activating factor; BMZ, basement membrane zone; BNP, brain natriuretic peptide; BP, bullous pemphigoid; C3, Complement 3; CCL5, RANTES (regulated on activation, normal T-cell expressed and secreted); CCL11, Eotaxin-1; CCL24, Eotaxin 2; CCL26, Eotaxin 3; ChAT, choline acetyltransferase; CRTH2, prostaglandin DP2 receptor; DEJ, Dermal-epidermal junction; ECP, eosinophil cationic protein; EDN, eosinophil derived neurotoxin; EET, eosinophil extracellular traps; EPX, eosinophil peroxidase; FcεRI, human high-affinity IgE receptor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IIF, indirect immunofluorescence; ICAM-1, intercellular adhesion molecule-1; IP-10, IFN- γ- inducible protein 10; MBP, major basic protein; MCP, monocyte chemoattractant protein; MIG, monokine induced by IFN- γ; MMP-9, Metalloprotease 9, Gelatinase B; NC16a, non-collagenous 16A domain of BP180; NE, neutrophil elastase; NGF, nerve growth factor; NK1R, neurokinin-1 receptor; NK2R, neurokinin-2 receptor; PAF, platelet activating factor; ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α; VAChT, vesicular acetylcholine transporter; VCAM-1, vascular cell adhesion molecule-1.
References
1. Gudi VS, White MI, Cruickshank N, Herriot R, Edwards SL, Nimmo F, et al. Annual incidence and mortality of bullous pemphigoid in the Grampian Region of North-east Scotland. Br J Dermatol. (2005) 153:424–7. doi: 10.1111/j.1365-2133.2005.06662.x
2. Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJ, West J. Bullous pemphigoid and pemphigus vulgaris–incidence and mortality in the UK: population based cohort study. BMJ (2008) 337:a180. doi: 10.1136/bmj.a180
3. Bertram F, Brocker EB, Zillikens D, Schmidt E. Prospective analysis of the incidence of autoimmune bullous disorders in Lower Franconia, Germany. J Dtsch Dermatol Ges. (2009) 7:434–40. doi: 10.1111/j.1610-0387.2008.06976.x
4. Marazza G, Pham HC, Scharer L, Pedrazzetti PP, Hunziker T, Trueb RM, et al. Incidence of bullous pemphigoid and pemphigus in Switzerland: a 2-year prospective study. Br J Dermatol. (2009) 161:861–8. doi: 10.1111/j.1365-2133.2009.09300.x
5. Brick KE, Weaver CH, Lohse CM, Pittelkow MR, Lehman JS, Camilleri MJ, et al. Incidence of bullous pemphigoid and mortality of patients with bullous pemphigoid in Olmsted County, Minnesota, 1960 through 2009. J Am Acad Dermatol. (2014) 71:92–9. doi: 10.1016/j.jaad.2014.02.030
6. Forsti AK, Jokelainen J, Timonen M, Tasanen K. Increasing incidence of bullous pemphigoid in Northern Finland: a retrospective database study in Oulu University Hospital. Br J Dermatol. (2014) 171:1223–6. doi: 10.1111/bjd.13189
7. Serwin AB, Musialkowska E, Piascik M. Incidence and mortality of bullous pemphigoid in north-east Poland (Podlaskie Province), 1999-2012: a retrospective bicentric cohort study. Int J Dermatol. (2014) 53:e432–7. doi: 10.1111/ijd.12492
8. Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol. (2017) 54:26–51. doi: 10.1007/s12016-017-8633-4
9. Jung M, Kippes W, Messer G, Zillikens D, Rzany B. Increased risk of bullous pemphigoid in male and very old patients: a population-based study on incidence. J Am Acad Dermatol. (1999) 41:266–8. doi: 10.1016/S0190-9622(99)70061-7
10. Joly P, Baricault S, Sparsa A, Bernard P, Bedane C, Duvert-Lehembre S, et al. Incidence and mortality of bullous pemphigoid in France. J Invest Dermatol. (2012) 132:1998–2004. doi: 10.1038/jid.2012.35
11. Kridin K. Subepidermal autoimmune bullous diseases: overview, epidemiology, and associations. Immunol Res. (2018) 66:6–17. doi: 10.1007/s12026-017-8975-2
12. Schmidt E, Zillikens D. Pemphigoid diseases. Lancet (2013) 381:320–32. doi: 10.1016/s0140-6736(12)61140-4
13. della Torre R, Combescure C, Cortes B, Marazza G, Beltraminelli H, Naldi L, et al. Clinical presentation and diagnostic delay in bullous pemphigoid: a prospective nationwide cohort. Br J Dermatol. (2012) 167:1111–7. doi: 10.1111/j.1365-2133.2012.11108.x
14. Di Zenzo G, Della Torre R, Zambruno G, Borradori L. Bullous pemphigoid: from the clinic to the bench. Clin Dermatol (2012) 30:3–16. doi: 10.1016/j.clindermatol.2011.03.005
15. Bakker CV, Terra JB, Pas HH, Jonkman MF. Bullous pemphigoid as pruritus in the elderly: a common presentation. JAMA Dermatol. (2013) 149:950–3. doi: 10.1001/jamadermatol.2013.756
16. Schmidt T, Sitaru C, Amber K, Hertl M. BP180- and BP230-specific IgG autoantibodies in pruritic disorders of the elderly: a preclinical stage of bullous pemphigoid? Br J Dermatol. (2014) 171:212–9. doi: 10.1111/bjd.12936
17. Stavropoulos PG, Soura E, Antoniou C. Drug-induced pemphigoid: a review of the literature. J Eur Acad Dermatol Venereol. (2014) 28:1133–40. doi: 10.1111/jdv.12366
18. Amber KT, Korta DZ, de Feraudy S, Grando SA. Vesiculobullous eruption in a patient receiving psoralen ultraviolet A (PUVA) treatment for prurigo nodules: a case of PUVA-aggravated pemphigoid nodularis. Clin Exp Dermatol. (2017) 42:833–5. doi: 10.1111/ced.13172
19. Amber KT, Valdebran M, Lu Y, De Feraudy S, Linden KG. Localized pretibial bullous pemphigoid arising in a patient on pembrolizumab for metastatic melanoma. J Dtsch Dermatol Ges. (2018) 16:196–8. doi: 10.1111/ddg.13411
20. Lopez AT, Khanna T, Antonov N, Audrey-Bayan C, Geskin L. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. (2018) 57:664–9. doi: 10.1111/ijd.13984
21. Izumi K, Nishie W, Mai Y, Wada M, Natsuga K, Ujiie H, et al. Autoantibody profile differentiates between inflammatory and noninflammatory bullous pemphigoid. J Invest Dermatol. (2016) 136:2201–10. doi: 10.1016/j.jid.2016.06.622
22. Benzaquen M, Borradori L, Berbis P, Cazzaniga S, Valero R, Richard MA, et al. Dipeptidyl peptidase IV inhibitors, a risk factor for bullous pemphigoid: retrospective multicenter case-control study from France and Switzerland. J Am Acad Dermatol. (2017) 78:1090–6. doi: 10.1016/j.jaad.2017.12.038
23. Chijiwa C, Takeoka S, Kamata M. Decrease in eosinophils infiltrating into the skin of patients with dipeptidyl peptidase-4 inhibitor-related bullous pemphigoid. J Dermatol. (2018) 45:596–9. doi: 10.1111/1346-8138.14245
24. Horikawa H, Kurihara Y, Funakoshi T, Umegaki-Arao N, Takahashi H, Kubo A, et al. Unique clinical and serological features of bullous pemphigoid associated with dipeptidyl peptidase-4 inhibitors. Br J Dermatol. (2018) 178:1462–3. doi: 10.1111/bjd.16479
25. Crotty C, Pittelkow M, Muller SA. Eosinophilic spongiosis: a clinicopathologic review of seventy-one cases. J Am Acad Dermatol. (1983) 8:337–43. doi: 10.1016/S0190-9622(83)70036-8
26. Nishioka K, Hashimoto K, Katayama I, Sarashi C, Kubo T, Sano S. Eosinophilic spongiosis in bullous pemphigoid. Arch Dermatol. (1984) 120:1166–8. doi: 10.1001/archderm.1984.01650450048015
27. Bernard P, Venot J, Constant F, Bonnetblanc JM. Blood eosinophilia as a severity marker for bullous pemphigoid. J Am Acad Dermatol. (1987) 16:879–81. doi: 10.1016/S0190-9622(87)80227-X
28. van Beek N, Schulze FS, Zillikens D, Schmidt E. IgE-mediated mechanisms in bullous pemphigoid and other autoimmune bullous diseases. Expert Rev Clin Immunol. (2016) 12:267–77. doi: 10.1586/1744666x.2016.1123092
29. Kridin K. Peripheral eosinophilia in bullous pemphigoid: prevalence and influence on the clinical manifestation. Br J Dermatol (2018) doi: 10.1111/bjd.16679. [Epub ahead of print].
30. Mihai S, Sitaru C. Immunopathology and molecular diagnosis of autoimmune bullous diseases. J Cell Mol Med. (2007) 11:462–81. doi: 10.1111/j.1582-4934.2007.00033.x
31. Moriuchi R, Nishie W, Ujiie H, Natsuga K, Shimizu H. In vivo analysis of IgE autoantibodies in bullous pemphigoid: a study of 100 cases. J Dermatol Sci. (2015) 78:21–5. doi: 10.1016/j.jdermsci.2015.01.013
32. Kershenovich R, Hodak E, Mimouni D. Diagnosis and classification of pemphigus and bullous pemphigoid. Autoimmun Rev. (2014) 13:477–81. doi: 10.1016/j.autrev.2014.01.011
33. Lee EH, Kim YH, Kim S, Kim SE, Kim SC. Usefulness of enzyme-linked immunosorbent assay using recombinant BP180 and BP230 for serodiagnosis and monitoring disease activity of bullous pemphigoid. Ann Dermatol. (2012) 24:45–55. doi: 10.5021/ad.2012.24.1.45
34. Yang B, Wang C, Chen S, Chen X, Lu X, Tian H, et al. Evaluation of the combination of BP180-NC16a enzyme-linked immunosorbent assay and BP230 enzyme-linked immunosorbent assay in the diagnosis of bullous pemphigoid. Indian J Dermatol Venereol Leprol. (2012) 78:722–7. doi: 10.4103/0378-6323.102364
35. Ingen-Housz-Oro S, Plee J, Belmondo T, Maizieres M, Pham BN, Hue S, et al. Positive direct immunofluorescence is of better value than ELISA-BP180 and ELISA-BP230 values for the prediction of relapse after treatment cessation in bullous pemphigoid: a retrospective study of 97 patients. Dermatology (2015) 231:50–5. doi: 10.1159/000381143
36. Keller JJ, Kittridge AL, Debanne SM, Korman NJ. Evaluation of ELISA testing for BP180 and BP230 as a diagnostic modality for bullous pemphigoid: a clinical experience. Arch Dermatol Res. (2016) 308:269–72. doi: 10.1007/s00403-016-1631-1
37. Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. (2017) 18:513–28. doi: 10.1007/s40257-017-0264-2
38. Bagci IS, Horvath ON, Ruzicka T, Sardy M. Bullous pemphigoid. Autoimmun Rev. (2017) 16:445–55. doi: 10.1016/j.autrev.2017.03.010
39. Liu Y, Li L, Xia Y. BP180 is critical in the autoimmunity of bullous pemphigoid. Front Immunol. (2017) 8:1752. doi: 10.3389/fimmu.2017.01752
40. Schmidt E, Obe K, Brocker EB, Zillikens D. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol. (2000) 136:174–8. doi: 10.1001/archderm.136.2.174
41. Iwata H, Kamio N, Aoyama Y, Yamamoto Y, Hirako Y, Owaribe K, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Invest Dermatol. (2009) 129:919–26. doi: 10.1038/jid.2008.305
42. Schmidt E, Reimer S, Kruse N, Jainta S, Brocker EB, Marinkovich MP, et al. Autoantibodies to BP180 associated with bullous pemphigoid release interleukin-6 and interleukin-8 from cultured human keratinocytes. J Invest Dermatol. (2000) 115:842–8. doi: 10.1046/j.1523-1747.2000.00141.x
43. Messingham KN, Srikantha R, DeGueme AM, Fairley JA. FcR-independent effects of IgE and IgG autoantibodies in bullous pemphigoid. J Immunol. (2011) 187:553–60. doi: 10.4049/jimmunol.1001753
44. Van den Bergh F, Eliason SL, Burmeister BT, Giudice GJ. Collagen XVII (BP180) modulates keratinocyte expression of the proinflammatory chemokine, IL-8. Exp Dermatol. (2012) 21:605–11. doi: 10.1111/j.1600-0625.2012.01529.x
45. Sasaoka T, Ujiie H, Nishie W, Iwata H, Ishikawa M, Higashino H, et al. Intravenous IgG reduces pathogenic autoantibodies, serum IL-6 levels, and disease severity in experimental bullous pemphigoid models. J Invest Dermatol. (2018) 138:1260–7. doi: 10.1016/j.jid.2018.01.005
46. Ujiie H, Shibaki A, Nishie W, Shinkuma S, Moriuchi R, Qiao H, et al. Noncollagenous 16A domain of type XVII collagen-reactive CD4+ T cells play a pivotal role in the development of active disease in experimental bullous pemphigoid model. Clin Immunol. (2012) 142:167–75. doi: 10.1016/j.clim.2011.10.002
47. Budinger L, Borradori L, Yee C, Eming R, Ferencik S, Grosse-Wilde H, et al. Identification and characterization of autoreactive T cell responses to bullous pemphigoid antigen 2 in patients and healthy controls. J Clin Invest. (1998) 102:2082–9. doi: 10.1172/jci3335
48. Hertl M, Eming R, Veldman C. T cell control in autoimmune bullous skin disorders. J Clin Invest. (2006) 116:1159–66. doi: 10.1172/jci28547
49. Amber KT, Zikry J, Hertl M. A multi-hit hypothesis of bullous pemphigoid and associated neurological disease: is HLA-DQB1*03:01, a potential link between immune privileged antigen exposure and epitope spreading? Hla (2017) 89:127–34. doi: 10.1111/tan.12960
50. Yoshida M, Hamada T, Amagai M, Hashimoto K, Uehara R, Yamaguchi K, et al. Enzyme-linked immunosorbent assay using bacterial recombinant proteins of human BP230 as a diagnostic tool for bullous pemphigoid. J Dermatol Sci. (2006) 41:21–30. doi: 10.1016/j.jdermsci.2005.11.002
51. Charneux J, Lorin J, Vitry F, Antonicelli F, Reguiai Z, Barbe C, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid: a retrospective study of 138 patients. Arch Dermatol. (2011) 147:286–91. doi: 10.1001/archdermatol.2011.23
52. Blocker IM, Dahnrich C, Probst C, Komorowski L, Saschenbrecker S, Schlumberger W, et al. Epitope mapping of BP230 leading to a novel enzyme-linked immunosorbent assay for autoantibodies in bullous pemphigoid. Br J Dermatol. (2012) 166:964–70. doi: 10.1111/j.1365-2133.2012.10820.x
53. Hayakawa T, Teye K, Hachiya T, Uehara R, Hashiguchi M, Kawakami T, et al. Clinical and immunological profiles of anti-BP230-type bullous pemphigoid: restriction of epitopes to the C-terminal domain of BP230, shown by novel ELISAs of BP230-domain specific recombinant proteins. Eur J Dermatol. (2016) 26:155–63. doi: 10.1684/ejd.2015.2719
54. Simon D, Borradori L, Simon HU. Eosinophils as putative therapeutic targets in bullous pemphigoid. Exp Dermatol. (2017) 26:1187–92. doi: 10.1111/exd.13416
55. Giusti D, Gatouillat G, Le Jan S, Plee J, Bernard P, Antonicelli F, et al. Eosinophil Cationic Protein (ECP), a predictive marker of bullous pemphigoid severity and outcome. Sci Rep. (2017) 7:4833. doi: 10.1038/s41598-017-04687-5
56. Bystrom J, Amin K, Bishop-Bailey D. Analysing the eosinophil cationic protein–a clue to the function of the eosinophil granulocyte. Respir Res. (2011) 12:10. doi: 10.1186/1465-9921-12-10
57. Long H, Zhang G, Wang L, Lu Q. Eosinophilic skin diseases: a comprehensive review. Clin Rev Allergy Immunol. (2016) 50:189–213. doi: 10.1007/s12016-015-8485-8
58. Young JD, Peterson CG, Venge P, Cohn ZA. Mechanism of membrane damage mediated by human eosinophil cationic protein. Nature (1986) 321:613–6. doi: 10.1038/321613a0
59. Trautmann A, Schmid-Grendelmeier P, Kruger K, Crameri R, Akdis M, Akkaya A, et al. T cells and eosinophils cooperate in the induction of bronchial epithelial cell apoptosis in asthma. J Allergy Clin Immunol. (2002) 109:329–37. doi: 10.1067/mai.2002.121460
60. Chang KC, Lo CW, Fan TC, Chang MD, Shu CW, Chang CH, et al. TNF-alpha mediates eosinophil cationic protein-induced apoptosis in BEAS-2B cells. BMC Cell Biol. (2010) 11:6. doi: 10.1186/1471-2121-11-6
61. Navarro S, Boix E, Cuchillo CM, Nogues MV. Eosinophil-induced neurotoxicity: the role of eosinophil cationic protein/RNase 3. J Neuroimmunol. (2010) 227:60–70. doi: 10.1016/j.jneuroim.2010.06.012
62. Kato M, Ishioka T, Kita H, Kozawa K, Hayashi Y, Kimura H. Eosinophil granular proteins damage bronchial epithelial cells infected with respiratory syncytial virus. Int Arch Allergy Immunol. (2012) 158(Suppl. 1):11–18. doi: 10.1159/000337752
63. Akuthota P, Weller PF. Eosinophils and disease pathogenesis. Semin Hematol. (2012) 49:113–9. doi: 10.1053/j.seminhematol.2012.01.005
64. Ying S, Meng Q, Zeibecoglou K, Robinson DS, Macfarlane A, Humbert M, et al. Eosinophil chemotactic chemokines (eotaxin, eotaxin-2, RANTES, monocyte chemoattractant protein-3 (MCP-3), and MCP-4), and C-C chemokine receptor 3 expression in bronchial biopsies from atopic and nonatopic (Intrinsic) asthmatics. J Immunol. (1999) 163:6321–9.
65. Davoine F, Lacy P. Eosinophil cytokines, chemokines, and growth factors: emerging roles in immunity. Front Immunol. (2014) 5:570. doi: 10.3389/fimmu.2014.00570
66. Czech W, Schaller J, Schopf E, Kapp A. Granulocyte activation in bullous diseases: release of granular proteins in bullous pemphigoid and pemphigus vulgaris. J Am Acad Dermatol. (1993) 29:210–5. doi: 10.1016/0190-9622(93)70170-X
67. Caproni M, Palleschi GM, Falcos D, D'Agata A, Cappelli G, Fabbri P. Serum eosinophil cationic protein (ECP) in bullous pemphigoid. Int J Dermatol. (1995) 34:177–80. doi: 10.1111/j.1365-4362.1995.tb01562.x
68. D'Auria L, Pietravalle M, Mastroianni A, Ferraro C, Mussi A, Bonifati C, et al. IL-5 levels in the serum and blister fluid of patients with bullous pemphigoid: correlations with eosinophil cationic protein, RANTES, IgE and disease severity. Arch Dermatol Res. (1998) 290:25–7. doi: 10.1007/s004030050272
69. Frezzolini A, Cianchini G, Ruffelli M, Cadoni S, Puddu P, De Pita O. Interleukin-16 expression and release in bullous pemphigoid. Clin Exp Immunol. (2004) 137:595–600. doi: 10.1111/j.1365-2249.2004.02570.x
70. Tedeschi A, Marzano AV, Lorini M, Balice Y, Cugno M. Eosinophil cationic protein levels parallel coagulation activation in the blister fluid of patients with bullous pemphigoid. J Eur Acad Dermatol Venereol. (2015) 29:813–7. doi: 10.1111/jdv.12464
71. Bieber K, Ernst AL, Tukaj S, Holtsche MM, Schmidt E, Zillikens D, et al. Analysis of serum markers of cellular immune activation in patients with bullous pemphigoid. Exp Dermatol. (2017) 26:1248–52. doi: 10.1111/exd.13382
72. Endo H, Iwamoto I, Fujita M, Okamoto S, Yoshida S. Increased immunoreactive interleukin-5 levels in blister fluids of bullous pemphigoid. Arch Dermatol Res. (1992) 284:312–4. doi: 10.1007/BF00372588
73. Borrego L, Maynard B, Peterson EA, George T, Iglesias L, Peters MS, et al. Deposition of eosinophil granule proteins precedes blister formation in bullous pemphigoid. Comparison with neutrophil and mast cell granule proteins. Am J Pathol. (1996) 148:897–909.
74. Inaoki M, Takehara K. Increased serum levels of interleukin (IL)-5, IL-6 and IL-8 in bullous pemphigoid. J Dermatol Sci. (1998) 16:152–7. doi: 10.1016/S0923-1811(97)00044-3
75. Feliciani C, Toto P, Mohammad Pour S, Coscione G, Amerio P, Amerio P. A Th2-like cytokine response is involved in bullous pemphigoid. the role of IL-4 and IL-5 in the pathogenesis of the disease. Int J Immunopathol Pharmacol. (1999) 12:55–61.
76. Shrikhande M, Hunziker T, Braathen LR, Pichler WJ, Dahinden CA, Yawalkar N. Increased coexpression of eotaxin and interleukin 5 in bullous pemphigoid. Acta Derm Venereol. (2000) 80:277–80. doi: 10.1080/000155500750012162
77. Wakugawa M, Nakamura K, Hino H, Toyama K, Hattori N, Okochi H, et al. Elevated levels of eotaxin and interleukin-5 in blister fluid of bullous pemphigoid: correlation with tissue eosinophilia. Br J Dermatol. (2000) 143:112–6. doi: 10.1046/j.1365-2133.2000.03599.x
78. Engineer L, Bhol K, Kumari S, Razzaque Ahmed A. Bullous pemphigoid: interaction of interleukin 5, anti-basement membrane zone antibodies and eosinophils. A preliminary observation. Cytokine (2001) 13:32–8. doi: 10.1006/cyto.2000.0791
79. Gounni Abdelilah S, Wellemans V, Agouli M, Guenounou M, Hamid Q, Beck LA, et al. Increased expression of Th2-associated chemokines in bullous pemphigoid disease. Role of eosinophils in the production and release of these chemokines. Clin Immunol. (2006) 120:220–31. doi: 10.1016/j.clim.2006.03.014
80. Yamashita C, Nakamizo S, Honda Y, Dainichi T, Kabashima K. Combination therapy of prednisolone and iv immunoglobulin treatment decreases circulating interleukin-5 and eosinophils in a patient with bullous pemphigoid. J Dermatol. (2017) 44:101–2. doi: 10.1111/1346-8138.13435
81. Engmann J, Rudrich U, Behrens G, Papakonstantinou E, Gehring M, Kapp A, et al. Increased activity and apoptosis of eosinophils in blister fluids, skin and peripheral blood of patients with bullous pemphigoid. Acta Derm Venereol. (2017) 97:464–71. doi: 10.2340/00015555-2581
82. Bowszyc-Dmochowska M, Dmochowski M. Immediate hypersensitivity phenomena in bullous pemphigoid: critical concepts. J Med. (2002) 33:189–98.
83. Verraes S, Hornebeck W, Polette M, Borradori L, Bernard P. Respective contribution of neutrophil elastase and matrix metalloproteinase 9 in the degradation of BP180 (type XVII collagen) in human bullous pemphigoid. J Invest Dermatol. (2001) 117:1091–6. doi: 10.1046/j.0022-202x.2001.01521.x
84. Kelly EA, Liu LY, Esnault S, Quinchia Johnson BH, Jarjour NN. Potent synergistic effect of IL-3 and TNF on matrix metalloproteinase 9 generation by human eosinophils. Cytokine (2012) 58:199–206. doi: 10.1016/j.cyto.2012.01.009
85. Wiehler S, Cuvelier SL, Chakrabarti S, Patel KD. p38 MAP kinase regulates rapid matrix metalloproteinase-9 release from eosinophils. Biochem Biophys Res Commun. (2004) 315:463–70. doi: 10.1016/j.bbrc.2004.01.078
86. Stahle-Backdahl M, Inoue M, Guidice GJ, Parks WC. 92-kD gelatinase is produced by eosinophils at the site of blister formation in bullous pemphigoid and cleaves the extracellular domain of recombinant 180-kD bullous pemphigoid autoantigen. J Clin Invest. (1994) 93:2022–30. doi: 10.1172/jci117196
87. Okada S, Kita H, George TJ, Gleich GJ, Leiferman KM. Migration of eosinophils through basement membrane components in vitro: role of matrix metalloproteinase-9. Am J Respir Cell Mol Biol. (1997) 17:519–28. doi: 10.1165/ajrcmb.17.4.2877
88. Liu Z, Shapiro SD, Zhou X, Twining SS, Senior RM, Giudice GJ, et al. A critical role for neutrophil elastase in experimental bullous pemphigoid. J Clin Invest. (2000) 105:113–23. doi: 10.1172/jci3693
89. Tsuda S, Miyasato M, Iryo K, Nakama T, Kato K, Sasai Y. Eosinophil phenotypes in bullous pemphigoid. J Dermatol. (1992) 19:270–9. doi: 10.1111/j.1346-8138.1992.tb03224.x
90. Dubertret L, Bertaux B, Fosse M, Touraine R. Cellular events leading to blister formation in bullous pemphigoid. Br J Dermatol. (1980) 103:615–24. doi: 10.1111/j.1365-2133.1980.tb01683.x
91. Iryo K, Tsuda S, Sasai Y. Ultrastructural aspects of infiltrated eosinophils in bullous pemphigoid. J Dermatol. (1992) 19:393–9. doi: 10.1111/j.1346-8138.1992.tb03247.x
92. Dvorak AM, Mihm MC Jr, Osage JE, Kwan TH, Austen KF, Wintroub BU. Bullous pemphigoid, an ultrastructural study of the inflammatory response: eosinophil, basophil and mast cell granule changes in multiple biopsies from one patient. J Invest Dermatol. (1982) 78:91–101. doi: 10.1111/1523-1747.ep12505711
93. Scheman AJ, Hordinsky MD, Groth DW, Vercellotti GM, Leiferman KM. Evidence for eosinophil degranulation in the pathogenesis of herpes gestationis. Arch Dermatol. (1989) 125:1079–83. doi: 10.1001/archderm.1989.01670200055008
94. Davis MD, Plager DA, George TJ, Weiss EA, Gleich GJ, Leiferman KM. Interactions of eosinophil granule proteins with skin: limits of detection, persistence, and vasopermeabilization. J Allergy Clin Immunol. (2003) 112:988–94. doi: 10.1016/j.jaci.2003.08.028
95. Miyasato M, Tsuda S, Kasada M, Iryo K, Sasai Y. Alteration in the density, morphology, and biological properties of eosinophils produced by bullous pemphigoid blister fluid. Arch Dermatol Res. (1989) 281:304–9. doi: 10.1007/BF00412972
96. Amber K, Agnoletti AF, Chernyavsky A, Grando S. Eosinophil major basic protein has a concentration-dependent cytotoxic effect on cultured keratinocytes. J Invest Dermatol. (2017) 5:A615 doi: 10.1016/j.jid.2017.02.637
97. Marzano AV, Tedeschi A, Fanoni D, Bonanni E, Venegoni L, Berti E, et al. Activation of blood coagulation in bullous pemphigoid: role of eosinophils, and local and systemic implications. Br J Dermatol. (2009) 160:266–72. doi: 10.1111/j.1365-2133.2008.08880.x
98. Cortjens B, van Woensel JB, Bem RA. Neutrophil extracellular traps in respiratory disease: guided anti-microbial traps or toxic webs? Paediatr Respir Rev. (2017) 21:54–61. doi: 10.1016/j.prrv.2016.03.007
99. Simon D, Hoesli S, Roth N, Staedler S, Yousefi S, Simon HU. Eosinophil extracellular DNA traps in skin diseases. J Allergy Clin Immunol. (2011) 127:194–9. doi: 10.1016/j.jaci.2010.11.002
100. Zone JJ, Taylor T, Hull C, Schmidt L, Meyer L. IgE basement membrane zone antibodies induce eosinophil infiltration and histological blisters in engrafted human skin on SCID mice. J Invest Dermatol. (2007) 127:1167–74. doi: 10.1038/sj.jid.5700681
101. Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils mediate tissue injury in autoimmune skin disease bullous pemphigoid. J Invest Dermatol. (2017) 138:1032–43. doi: 10.1016/j.jid.2017.11.031
102. Kinet JP. The high-affinity IgE receptor (Fc epsilon RI): from physiology to pathology. Annu Rev Immunol. (1999) 17:931–72. doi: 10.1146/annurev.immunol.17.1.931
103. Messingham KN, Holahan HM, Frydman AS, Fullenkamp C, Srikantha R, Fairley JA. Human eosinophils express the high affinity IgE receptor, FcepsilonRI, in bullous pemphigoid. PLoS ONE (2014) 9:e107725. doi: 10.1371/journal.pone.0107725
104. Kasahara-Imamura M, Hosokawa H, Maekawa N, Horio T. Activation of Fc epsilon RI-positive eosinophils in bullous pemphigoid. Int J Mol Med. (2001) 7:249–53. doi: 10.3892/ijmm.7.3.249
105. Arbesman CE, Wypych JI, Reisman RE, Beutner EH. IgE levels in sera of patients with pemphigus or bullous pemphigoid. Arch Dermatol. (1974) 110:378–81. doi: 10.1001/archderm.1974.01630090016003
106. Provost TT, Tomasi TB Jr. Immunopathology of bullous pemphigoid. Basement membrane deposition of IgE, alternate pathway components and fibrin. Clin Exp Immunol. (1974) 18:193–200.
107. Bushkell LL, Jordon RE. Bullous pemphigoid: a cause of peripheral blood eosinophilia. J Am Acad Dermatol. (1983) 8:648–51. doi: 10.1016/S0190-9622(83)70073-3
108. Pomponi D, Di Zenzo G, Zennaro D, Calabresi V, Eming R, Zuzzi S, et al. Detection of IgG and IgE reactivity to BP180 using the ISAC(R) microarray system. Br J Dermatol. (2013) 168:1205–14. doi: 10.1111/bjd.12161
109. Delaporte E, Dubost-Brama A, Ghohestani R, Nicolas JF, Neyrinck JL, Bergoend H, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol. (1996) 157:3642–7.
110. Ghohestani RF, Cozzani E, Delaporte E, Nicolas JF, Parodi A, Claudy A. IgE antibodies in sera from patients with bullous pemphigoid are autoantibodies preferentially directed against the 230-kDa epidermal antigen (BP230). J Clin Immunol. (1998)18:202–9. doi: 10.1023/A:1020531005776
111. Dopp R, Schmidt E, Chimanovitch I, Leverkus M, Brocker EB, Zillikens D. IgG4 and IgE are the major immunoglobulins targeting the NC16A domain of BP180 in bullous pemphigoid: serum levels of these immunoglobulins reflect disease activity. J Am Acad Dermatol. (2000) 42:577–83. doi: 10.1067/mjd.2000.103986
112. Dresow SK, Sitaru C, Recke A, Oostingh GJ, Zillikens D, Gibbs BF. IgE autoantibodies against the intracellular domain of BP180. Br J Dermatol. (2009) 160:429–32. doi: 10.1111/j.1365-2133.2008.08858.x
113. Freire PC, Munoz CH, Stingl G. IgE autoreactivity in bullous pemphigoid: eosinophils and mast cells as major targets of pathogenic immune reactants. Br J Dermatol. (2017) 177:1644–53. doi: 10.1111/bjd.15924
114. Hashimoto T, Ohzono A, Teye K, Numata S, Hiroyasu S, Tsuruta D, et al. Detection of IgE autoantibodies to BP180 and BP230 and their relationship to clinical features in bullous pemphigoid. Br J Dermatol. (2016) 177:141–51. doi: 10.1111/bjd.15114
115. Saniklidou AH, Tighe PJ, Fairclough LC, Todd I. IgE autoantibodies and their association with the disease activity and phenotype in bullous pemphigoid: a systematic review. Arch Dermatol Res. (2017) 310:11–28. doi: 10.1007/s00403-017-1789-1
116. Fairley JA, Baum CL, Brandt DS, Messingham KA. Pathogenicity of IgE in autoimmunity: successful treatment of bullous pemphigoid with omalizumab. J Allergy Clin Immunol. (2009) 123:704–5. doi: 10.1016/j.jaci.2008.11.035
117. Dufour C, Souillet AL, Chaneliere C, Jouen F, Bodemer C, Jullien D, et al. Successful management of severe infant bullous pemphigoid with omalizumab. Br J Dermatol. (2012) 166:1140–2. doi: 10.1111/j.1365-2133.2011.10748.x
118. Yu KK, Crew AB, Messingham KA, Fairley JA, Woodley DT. Omalizumab therapy for bullous pemphigoid. J Am Acad Dermatol. (2014) 71:468–74. doi: 10.1016/j.jaad.2014.04.053
119. D'Auria L, Cordiali Fei P, Ameglio F. Cytokines and bullous pemphigoid. Eur Cytokine Netw. (1999) 10:123–34.
120. Giomi B, Caproni M, Calzolari A, Bianchi B, Fabbri P. Th1, Th2 and Th3 cytokines in the pathogenesis of bullous pemphigoid. J Dermatol Sci. (2002) 30:116–28. doi: 10.1016/S0923-1811(02)00067-1
121. Nakashima H, Fujimoto M, Asashima N, Watanabe R, Kuwano Y, Yazawa N, et al. Serum chemokine profile in patients with bullous pemphigoid. Br J Dermatol. (2007) 156:454–9. doi: 10.1111/j.1365-2133.2006.07601.x
122. Frezzolini A, Teofoli P, Cianchini G, Barduagni S, Ruffelli M, Ferranti G, et al. Increased expression of eotaxin and its specific receptor CCR3 in bullous pemphigoid. Eur J Dermatol. (2002) 12:27–31.
123. Lingblom C, Kappi T, Bergquist H, Bove M, Arkel R, Saalman R, et al. Differences in eosinophil molecular profiles between children and adults with eosinophilic esophagitis. (2017) 72:1406–14. doi: 10.1111/all.13140
124. Carlens J, Wahl B, Ballmaier M, Bulfone-Paus S, Forster R, Pabst O. Common gamma-chain-dependent signals confer selective survival of eosinophils in the murine small intestine. J Immunol. (2009) 183:5600–7. doi: 10.4049/jimmunol.0801581
125. Verjan Garcia N, Umemoto E, Saito Y, Yamasaki M, Hata E, Matozaki T, et al. SIRPalpha/CD172a regulates eosinophil homeostasis. J Immunol. (2011) 187:2268–77. doi: 10.4049/jimmunol.1101008
126. Hurskainen T, Kokkonen N, Sormunen R, Jackow J, Loffek S, Soininen R, et al. Deletion of the major bullous pemphigoid epitope region of collagen XVII induces blistering, autoimmunization, and itching in mice. J Invest Dermatol. (2015) 135:1303–10. doi: 10.1038/jid.2014.443
127. Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. (2004) 5:752–60. doi: 10.1038/ni1084
128. Meng J, Moriyama M, Feld M, Buddenkotte J, Buhl T, Szollosi A, et al. New mechanism underlying IL-31-induced atopic dermatitis. J Allergy Clin Immunol. (2018) 141:1677–89.e1678. doi: 10.1016/j.jaci.2017.12.1002
129. Takamori A, Nambu A. IL-31 is crucial for induction of pruritus, but not inflammation, in contact hypersensitivity. Sci Rep. (2018) 8:6639. doi: 10.1038/s41598-018-25094-4
130. Feld M, Garcia R, Buddenkotte J, Katayama S, Lewis K, Muirhead G, et al. The pruritus- and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J Allergy Clin Immunol. (2016) 138:500–8.e524. doi: 10.1016/j.jaci.2016.02.020
131. Bonciani D, Quintarelli L, Del Bianco E, Bianchi B, Caproni M. Serum levels and tissue expression of interleukin-31 in dermatitis herpetiformis and bullous pemphigoid. J Dermatol Sci. (2017) 87:210–2. doi: 10.1016/j.jdermsci.2017.04.008
132. Salz M, Haeberle S, Hoffmann J, Enk AH, Hadaschik EN. Elevated IL-31 serum levels in bullous pemphigoid patients correlate with eosinophil numbers and are associated with BP180-IgE. J Dermatol Sci. (2017) 87:309–11. doi: 10.1016/j.jdermsci.2017.07.019
133. Kunsleben N, Rudrich U, Gehring M, Novak N, Kapp A, Raap U. IL-31 induces chemotaxis, calcium mobilization, release of reactive oxygen species, and CCL26 in eosinophils, which are capable to release IL-31. J Invest Dermatol. (2015) 135:1908–11. doi: 10.1038/jid.2015.106
134. Rudrich U, Gehring M, Papakonstantinou E, Rabenhorst A, Engmann J, Kapp A, et al. Eosinophils are a major source of interleukin-31 in bullous pemphigoid. Acta Derm Venereol. (2018) doi: 10.2340/00015555-2951. [Epub ahead of print].
135. Wallengren J, Ekman R, Moller H. Substance P and vasoactive intestinal peptide in bullous and inflammatory skin disease. Acta Derm Venereol. (1986) 66:23–8.
136. Cynkier A, Zebrowska A, Wagrowska-Danilewicz M, Danilewicz M, Erkiert-Polguj A, Stasikowska-Kanicka O, et al. Expression of selected neuropeptides in pathogenesis of bullous pemphigoid and dermatitis herpetiformis. Pol J Pathol. (2012) 63:31–9.
137. Pavlovic S, Daniltchenko M, Tobin DJ, Hagen E, Hunt SP, Klapp BF, et al. Further exploring the brain-skin connection: stress worsens dermatitis via substance P-dependent neurogenic inflammation in mice. J Invest Dermatol. (2008) 128:434–46. doi: 10.1038/sj.jid.5701079
138. Fajac I, Braunstein G, Ickovic MR, Lacronique J, Frossard N. Selective recruitment of eosinophils by substance P after repeated allergen exposure in allergic rhinitis. Allergy (1995) 50:970–5. doi: 10.1111/j.1398-9995.1995.tb02509.x
139. Raap M, Rudrich U, Stander S, Gehring M, Kapp A, Raap U. Substance P activates human eosinophils. Exp Dermatol. (2015) 24:557–9. doi: 10.1111/exd.12717
140. Friedman S, Levi-Schaffer F. Substance P and eosinophils: an itchy connection. Exp Dermatol. (2015) 24:918–9. doi: 10.1111/exd.12806
141. Foster EL, Simpson EL, Fredrikson LJ, Lee JJ, Lee NA, Fryer AD, et al. Eosinophils increase neuron branching in human and murine skin and >in vitro. PLoS ONE (2011) 6:e22029. doi: 10.1371/journal.pone.0022029
142. Lee JJ, Protheroe CA, Luo H, Ochkur SI, Scott GD, Zellner KR, et al. Eosinophil-dependent skin innervation and itching following contact toxicant exposure in mice. J Allergy Clin Immunol. (2015) 135:477–87. doi: 10.1016/j.jaci.2014.07.003
143. Curran DR, Morgan RK, Kingham PJ, Durcan N, McLean WG, Walsh MT, et al. Mechanism of eosinophil induced signaling in cholinergic IMR-32 cells. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L326–32. doi: 10.1152/ajplung.00254.2004
144. Johansson O, Liang Y, Marcusson JA, Reimert CM. Eosinophil cationic protein- and eosinophil-derived neurotoxin/eosinophil protein X-immunoreactive eosinophils in prurigo nodularis. Arch Dermatol Res. (2000) 292:371–8. doi: 10.1007/s004030000142
145. Walsh MT, Curran DR, Kingham PJ, Morgan RK, Durcan N, Gleich GJ, et al. Effect of eosinophil adhesion on intracellular signaling in cholinergic nerve cells. Am J Respir Cell Mol Biol. (2004) 30:333–41. doi: 10.1165/rcmb.2003-0188OC
146. Kingham PJ, McLean WG, Walsh MT, Fryer AD, Gleich GJ, Costello RW. Effects of eosinophils on nerve cell morphology and development: the role of reactive oxygen species and p38 MAP kinase. Am J Physiol Lung Cell Mol Physiol. (2003) 285:L915–24. doi: 10.1152/ajplung.00094.2003
147. Morgan RK, Costello RW, Durcan N, Kingham PJ, Gleich GJ, McLean WG, et al. Diverse effects of eosinophil cationic granule proteins on IMR-32 nerve cell signaling and survival. Am J Respir Cell Mol Biol. (2005) 33:169–77. doi: 10.1165/rcmb.2005-0056OC
148. Akasheh N, Walsh MT, Costello RW. Eosinophil peroxidase induces expression of cholinergic genes via cell surface neural interactions. Mol Immunol. (2014) 62:37–45. doi: 10.1016/j.molimm.2014.05.014
149. Durcan N, Costello RW, McLean WG, Blusztajn J, Madziar B, Fenech AG, et al. Eosinophil-mediated cholinergic nerve remodeling. Am J Respir Cell Mol Biol. (2006) 34:775–86. doi: 10.1165/rcmb.2005-0196OC
150. Mondino BJ, Manthey R. Dermatological diseases and the peripheral cornea. Int Ophthalmol Clin. (1986) 26:121–36. doi: 10.1097/00004397-198602640-00012
151. Morgan RK, Kingham PJ, Walsh MT, Curran DR, Durcan N, McLean WG, et al. Eosinophil adhesion to cholinergic IMR-32 cells protects against induced neuronal apoptosis. J Immunol. (2004) 173:5963–70. doi: 10.4049/jimmunol.173.10.5963
152. Coyle AJ, Perretti F, Manzini S, Irvin CG. Cationic protein-induced sensory nerve activation: role of substance P in airway hyperresponsiveness and plasma protein extravasation. J Clin Invest. (1994) 94:2301–6. doi: 10.1172/jci117594
153. Lee LY, Gu Q, Gleich GJ. Effects of human eosinophil granule-derived cationic proteins on C-fiber afferents in the rat lung. J Appl Physiol. (2001) 91:1318–26. doi: 10.1152/jappl.2001.91.3.1318
154. Gu Q, Wiggers ME, Gleich GJ, Lee LY. Sensitization of isolated rat vagal pulmonary sensory neurons by eosinophil-derived cationic proteins. Am J Physiol Lung Cell Mol Physiol. (2008) 294:L544–52. doi: 10.1152/ajplung.00271.2007
155. Gu Q, Lim ME, Gleich GJ, Lee LY. Mechanisms of eosinophil major basic protein-induced hyperexcitability of vagal pulmonary chemosensitive neurons. Am J Physiol Lung Cell Mol Physiol. (2009) 296:L453–61. doi: 10.1152/ajplung.90467.2008
156. Dvorak AM, Onderdonk AB, McLeod RS, Monahan-Earley RA, Antonioli DA, Cullen J, et al. Ultrastructural identification of exocytosis of granules from human gut eosinophils in vivo. Int Arch Allergy Immunol. (1993) 102:33–45. doi: 10.1159/000236548
157. Costello RW, Schofield BH, Kephart GM, Gleich GJ, Jacoby DB, Fryer AD. Localization of eosinophils to airway nerves and effect on neuronal M2 muscarinic receptor function. Am J Physiol. (1997) 273(1 Pt 1):L93–103. doi: 10.1152/ajplung.1997.273.1.L93
158. Blanchet MR, Langlois A, Israel-Assayag E, Beaulieu MJ, Ferland C, Laviolette M, et al. Modulation of eosinophil activation in vitro by a nicotinic receptor agonist. J Leukoc Biol. (2007) 81:1245–51. doi: 10.1189/jlb.0906548
159. Xenakis JJ, Howard ED, Smith KM, Olbrich CL, Huang Y, Anketell D, et al. Resident intestinal eosinophils constitutively express antigen presentation markers and include two phenotypically distinct subsets of eosinophils. Immunology (2017) 154:298–308. doi: 10.1111/imm.12885
160. Lucey DR, Nicholson-Weller A, Weller PF. Mature human eosinophils have the capacity to express HLA-DR. Proc Natl Acad Sci USA. (1989) 86:1348–51. doi: 10.1073/pnas.86.4.1348
161. Shi HZ, Humbles A, Gerard C, Jin Z, Weller PF. Lymph node trafficking and antigen presentation by endobronchial eosinophils. J Clin Invest. (2000) 105:945–53. doi: 10.1172/jci8945
162. Wang HB, Ghiran I, Matthaei K, Weller PF. Airway eosinophils: allergic inflammation recruited professional antigen-presenting cells. J Immunol. (2007) 179:7585–92. doi: 10.4049/jimmunol.179.11.7585
163. Lin A, Lore K. Granulocytes: new members of the antigen-presenting cell family. Front Immunol. (2017) 8:1781. doi: 10.3389/fimmu.2017.01781
164. Le-Carlson M, Seki S, Abarbanel D, Quiros A, Cox K, Nadeau KC. Markers of antigen presentation and activation on eosinophils and T cells in the esophageal tissue of patients with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. (2013) 56:257–62. doi: 10.1097/MPG.0b013e3182758d49
165. Jacoby DB, Gleich GJ, Fryer AD. Human eosinophil major basic protein is an endogenous allosteric antagonist at the inhibitory muscarinic M2 receptor. J Clin Invest. (1993) 91:1314–8. doi: 10.1172/jci116331
166. O'Donnell MC, Ackerman SJ, Gleich GJ, Thomas LL. Activation of basophil and mast cell histamine release by eosinophil granule major basic protein. J Exp Med. (1983) 157:1981–91. doi: 10.1084/jem.157.6.1981
167. Thomas LL, Zheutlin LM, Gleich GJ. Pharmacological control of human basophil histamine release stimulated by eosinophil granule major basic protein. Immunology (1989) 66:611–15.
168. Rothenberg ME. Eosinophilia. N Engl J Med (1998) 338:1592–600. doi: 10.1056/nejm199805283382206
169. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon HU, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy (2017) 72:1105–13. doi: 10.1111/all.13131
170. Oswald E, Sesarman A, Franzke CW, Wolfle U, Bruckner-Tuderman L, Jakob T, et al. The flavonoid luteolin inhibits Fcgamma-dependent respiratory burst in granulocytes, but not skin blistering in a new model of pemphigoid in adult mice. PLoS ONE (2012) 7:e31066. doi: 10.1371/journal.pone.0031066
171. Chu VT, Frohlich A, Steinhauser G, Scheel T, Roch T, Fillatreau S, et al. Eosinophils are required for the maintenance of plasma cells in the bone marrow. Nat Immunol. (2011) 12:151–9. doi: 10.1038/ni.1981
172. Chu VT, Berek C. Immunization induces activation of bone marrow eosinophils required for plasma cell survival. Eur J Immunol. (2012) 42:130–7. doi: 10.1002/eji.201141953
173. Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. (2002) 2:465–75. doi: 10.1038/nri844
174. Dillon SR, Gross JA, Ansell SM, Novak AJ. An APRIL to remember: novel TNF ligands as therapeutic targets. Nat Rev Drug Discov. (2006) 5:235–46. doi: 10.1038/nrd1982
175. Gras MP, Laabi Y, Linares-Cruz G, Blondel MO, Rigaut JP, Brouet JC, et al. BCMAp: an integral membrane protein in the Golgi apparatus of human mature B lymphocytes. Int Immunol. (1995) 7:1093–106. doi: 10.1093/intimm/7.7.1093
176. von Bulow GU, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science (1997) 278:138–41.
177. O'Connor BP, Raman VS, Erickson LD, Cook WJ, Weaver LK, Ahonen C, et al. BCMA is essential for the survival of long-lived bone marrow plasma cells. J Exp Med. (2004) 199:91–8. doi: 10.1084/jem.20031330
178. Yang M, Hase H, Legarda-Addison D, Varughese L, Seed B, Ting AT. B cell maturation antigen, the receptor for a proliferation-inducing ligand and B cell-activating factor of the TNF family, induces antigen presentation in B cells. J Immunol. (2005) 175:2814–24. doi: 10.4049/jimmunol.175.5.2814
179. Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science (1999) 285:260–3.
180. Asashima N, Fujimoto M, Watanabe R, Nakashima H, Yazawa N, Okochi H, et al. Serum levels of BAFF are increased in bullous pemphigoid but not in pemphigus vulgaris. Br J Dermatol. (2006) 155:330–6. doi: 10.1111/j.1365-2133.2006.07305.x
181. Watanabe R, Fujimoto M, Yazawa N, Nakashima H, Asashima N, Kuwano Y, et al. Increased serum levels of a proliferation-inducing ligand in patients with bullous pemphigoid. J Dermatol Sci. (2007) 46:53–60. doi: 10.1016/j.jdermsci.2006.12.008
182. Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature (1990) 347:669–71. doi: 10.1038/347669a0
183. Alam R, Stafford S, Forsythe P, Harrison R, Faubion D, Lett-Brown MA, et al. RANTES is a chemotactic and activating factor for human eosinophils. J Immunol. (1993) 150:3442–8.
184. Bacon KB, Premack BA, Gardner P, Schall TJ. Activation of dual T cell signaling pathways by the chemokine RANTES. Science (1995) 269:1727–30.
185. Kameyoshi Y, Dorschner A, Mallet AI, Christophers E, Schroder JM. Cytokine RANTES released by thrombin-stimulated platelets is a potent attractant for human eosinophils. J Exp Med. (1992) 176:587–92. doi: 10.1084/jem.176.2.587
186. Rot A, Krieger M, Brunner T, Bischoff SC, Schall TJ, Dahinden CA. RANTES and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granulocytes. J Exp Med. (1992) 176:1489–95. doi: 10.1084/jem.176.6.1489
187. Lim KG, Wan HC, Bozza PT, Resnick MB, Wong DT, Cruikshank WW, et al. Human eosinophils elaborate the lymphocyte chemoattractants. IL-16 (lymphocyte chemoattractant factor) and RANTES. J Immunol. (1996) 156:2566–70.
188. Spencer LA, Szela CT, Perez SA, Kirchhoffer CL, Neves JS, Radke AL, et al. Human eosinophils constitutively express multiple Th1, Th2, and immunoregulatory cytokines that are secreted rapidly and differentially. J Leukoc Biol. (2009) 85:117–23. doi: 10.1189/jlb.0108058
189. Odemuyiwa SO, Ghahary A, Li Y, Puttagunta L, Lee JE, Musat-Marcu S, et al. Cutting edge: human eosinophils regulate T cell subset selection through indoleamine 2,3-dioxygenase. J Immunol. (2004) 173:5909–13. doi: 10.4049/jimmunol.173.10.5909
190. O'Connell AE, Hess JA, Santiago GA, Nolan TJ, Lok JB, Lee JJ, et al. Major basic protein from eosinophils and myeloperoxidase from neutrophils are required for protective immunity to Strongyloides stercoralis in mice. Infect Immun. (2011) 79:2770–8. doi: 10.1128/iai.00931-10
191. Wen T, Rothenberg ME. The regulatory function of eosinophils. Microbiol Spectr (2016) 4:5. doi: 10.1128/microbiolspec.MCHD-0020-2015
192. Gunther C, Wozel G, Dressler J, Meurer M, Pfeiffer C. Tissue eosinophilia in pemphigoid gestationis: association with eotaxin and upregulated activation markers on transmigrated eosinophils. Am J Reprod Immunol. (2004) 51:32–9. doi: 10.1046/j.8755-8920.2003.00118.x
193. Jean-Baptiste S, O'Toole EA, Chen M, Guitart J, Paller A, Chan LS. Expression of eotaxin, an eosinophil-selective chemokine, parallels eosinophil accumulation in the vesiculobullous stage of incontinentia pigmenti. Clin Exp Immunol. (2002) 127:470–8. doi: 10.1046/j.1365-2249.2002.01755.x
194. Sun CC, Wu J, Wong TT, Wang LF, Chuan MT. High levels of interleukin-8, soluble CD4 and soluble CD8 in bullous pemphigoid blister fluid. The relationship between local cytokine production and lesional T-cell activities. Br J Dermatol. (2000) 143:1235–40. doi: 10.1046/j.1365-2133.2000.03894.x
195. Schmidt E, Reimer S, Kruse N, Brocker EB, Zillikens D. The IL-8 release from cultured human keratinocytes, mediated by antibodies to bullous pemphigoid autoantigen 180, is inhibited by dapsone. Clin Exp Immunol. (2001) 124:157–62. doi: 10.1046/j.1365-2249.2001.01503.x
196. Tukaj S, Gruner D, Zillikens D, Kasperkiewicz M. Hsp90 blockade modulates bullous pemphigoid IgG-induced IL-8 production by keratinocytes. Cell Stress Chaperones (2014) 19:887–94. doi: 10.1007/s12192-014-0513-8
197. Nishihara F, Nakagome K, Kobayashi T, Noguchi T, Araki R, Uchida Y, et al. Trans-basement membrane migration of eosinophils induced by LPS-stimulated neutrophils from human peripheral blood in vitro. ERJ Open Res. (2015) 1:2. doi: 10.1183/23120541.00003-2015
198. Gearing AJ, Fincham NJ, Bird CR, Wadhwa M, Meager A, Cartwright JE, et al. Cytokines in skin lesions of psoriasis. Cytokine (1990) 2:68–75. doi: 10.1016/1043-4666(90)90045-U
199. Schroder JM, Gregory H, Young J, Christophers E. Neutrophil-activating proteins in psoriasis. J Invest Dermatol. (1992) 98:241–7. doi: 10.1111/1523-1747.ep12556058
200. Kikuchi I, Kikuchi S, Kobayashi T, Hagiwara K, Sakamoto Y, Kanazawa M, et al. Eosinophil trans-basement membrane migration induced by interleukin-8 and neutrophils. Am J Respir Cell Mol Biol. (2006) 34:760–5. doi: 10.1165/rcmb.2005-0303OC
201. Bruijnzeel PL, Warringa RA, Kok PT, Hamelink ML, Kreukniet H, Koenderman L. Effects of nedocromil sodium on in vitro induced migration, activation, and mediator release from human granulocytes. J Allergy Clin Immunol. (1993) 92:159–64. doi: 10.1016/0091-6749(93)90099-2
202. Liu L, Zuurbier AE, Mul FP, Verhoeven AJ, Lutter R, Knol EF, et al. Triple role of platelet-activating factor in eosinophil migration across monolayers of lung epithelial cells: eosinophil chemoattractant and priming agent and epithelial cell activator. J Immunol. (1998) 161:3064–70.
203. Okada S, Kita H, George TJ, Gleich GJ, Leiferman KM. Transmigration of eosinophils through basement membrane components in vitro: synergistic effects of platelet-activating factor and eosinophil-active cytokines. Am J Respir Cell Mol Biol. (1997) 16:455–63. doi: 10.1165/ajrcmb.16.4.9115757
204. Gunther C, Wozel G, Meurer M, Pfeiffer C. Up-regulation of CCL11 and CCL26 is associated with activated eosinophils in bullous pemphigoid. Clin Exp Immunol. (2011) 166:145–53. doi: 10.1111/j.1365-2249.2011.04464.x
Keywords: bullous pemphigoid, eosinophils, eosinophilia, major basic protein, eosinophil cationic protein, pruritus, cytokines
Citation: Amber KT, Valdebran M, Kridin K and Grando SA (2018) The Role of Eosinophils in Bullous Pemphigoid: A Developing Model of Eosinophil Pathogenicity in Mucocutaneous Disease. Front. Med. 5:201. doi: 10.3389/fmed.2018.00201
Received: 25 May 2018; Accepted: 25 June 2018;
Published: 10 July 2018.
Edited by:
Mette Søndergaard Deleuran, Department of Dermatology, Aarhus University Hospital, DenmarkReviewed by:
Takashi Hashimoto, Graduate School of Medicine, Faculty of Medicine, Osaka University, JapanHiroaki Iwata, Hokkaido University, Japan
Copyright © 2018 Amber, Valdebran, Kridin and Grando. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kyle T. Amber, S3lsZUFtYmVyTURAZ21haWwuY29t