 Roberta Bertelli1
Roberta Bertelli1 G. M. Ghiggeri
G. M. Ghiggeri
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Med. , 11 June 2018
Sec. Nephrology
Volume 5 - 2018 | https://doi.org/10.3389/fmed.2018.00170
This article is part of the Research Topic Molecular Mechanisms of Proteinuria View all 8 articles
Minimal Change Disease (MCD) is a clinical condition characterized by acute nephrotic syndrome, no evident renal lesions at histology and good response to steroids. However, frequent recurrence of the disease requires additional therapies associated with steroids. Such multi-drug dependence and frequent relapses may cause disease evolution to focal and segmental glomerulosclerosis (FSGS) over time. The differences between the two conditions are not well defined, since molecular mechanisms may be shared by the two diseases. In some cases, genetic analysis can make it possible to distinguish MCD from FSGS; however, there are cases of overlap. Several hypotheses on mechanisms underlying MCD and potential molecular triggers have been proposed. Most studies were conducted on animal models of proteinuria that partially mimic MCD and may be useful to study glomerulosclerosis evolution; however, they do not demonstrate a clear-cut separation between MCD and FSGS. Puromycin Aminonucleoside and Adriamycin nephrosis are models of glomerular oxidative damage, characterized by loss of glomerular basement membrane polyanions resembling MCD at the onset and, at more advanced stages, by glomerulosclerosis resembling FSGS. Also Buffalo/Mna rats present initial lesions of MCD, subsequently evolving to FSGS; this mechanism of renal damage is clearer since this rat strain inherits the unique characteristic of overexpressing Th2 cytokines. In Lipopolysaccharide nephropathy, an immunological condition of renal toxicity linked to B7-1(CD80), mice develop transient proteinuria that lasts a few days. Overall, animal models are useful and necessary considering that they reproduce the evolution from MCD to FSGS that is, in part, due to persistence of proteinuria. The role of T/Treg/Bcells on human MCD has been discussed. Many cytokines, immunomodulatory mechanisms, and several molecules have been defined as a specific cause of proteinuria. However, the hypothesis of a single cell subset or molecule as cause of MCD is not supported by research and an interactive process seems more logical. The implication or interactive role of oxidants, Th2 cytokines, Th17, Tregs, B7-1(CD80), CD40/CD40L, c-Mip, TNF, uPA/suPAR, Angiopoietin-like 4 still awaits a definitive confirmation. Whole genome sequencing studies could help to define specific genetic features that justify a definition of MCD as a “clinical-pathology-genetic entity.”
Minimal Change Disease (MCD) is a clinical condition characterized by heavy proteinuria and nephrotic syndrome that classically arises acutely in patients aged under 18 years. Histologically, immunofluorescence staining is typically negative, even though MCD is characterized by fusion of foot process in podocytes that can be detected only by Electron Microscopy. Concerning therapy, most MCD patients respond to steroids that usually reverse the clinical picture in a few days. Steroid dependence is also frequent and, when present, it requires additional therapy. Besides steroids, the natural history of MCD is influenced by other drugs that are essential to obtain a good stability over months/years allowing withdrawal of the initial therapy. Renal histology justifies the definition of “minimal changes” and the good sensitivity to drugs supports the concept that MCD is a benign condition.
As regards MCD, there is not a complete correspondence between sensitivity to steroids and pathology findings, since not all patients respond to steroids, while a minority of patients responding to steroids present sclerotic lesions that involve focal segments of glomeruli and are associated with diffuse fusion of foot process. When these pathological features are well structured, then the condition is defined as primary Focal Segmental Glomerulosclerosis (FSGS), that represents a separate entity with different clinical and prognostic features (Table 1). Moreover, there are conditions in which the clinical-pathology trait is less definite and renal lesions evolve from initial pathological findings of minimal lesions to FSGS. Reduced responsiveness to therapies often characterizes these patients who usually require the association of steroids and calcineurin inhibitors. The existence of clinical and pathology overlaps between MCD and FSGS supports the definition of MCD as not a completely separate entity, which may evolve into FSGS over years. This suggests that MCD and FSGS could represent, in a proportion of patients, two phases of the same disease (1, 2). However, the actual link between MCD and FSGS is still unclear (3, 4). A correct definition of MCD should be based on the observation of the concomitant absence of histological lesions and a good clinical response to steroids in the same patient.
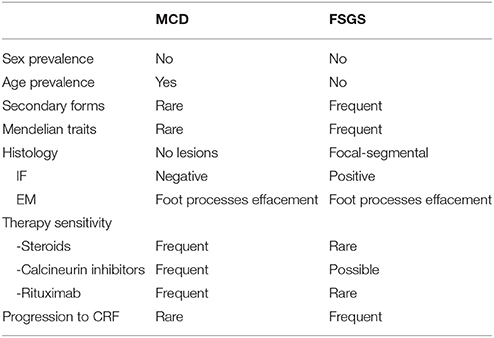
Table 1. Clinical features of Minimal Change disease (MCD) and Focal Segmental Glomerulosclerosis (FSGS).
While a genetic origin is frequent in FSGS and, more in general, in nephrotic syndrome not responding to any therapy, genetic data in MCD are limited and their interpretation should consider the discrimination between MCD and FSGS. Genetic studies could offer more solid criteria for discriminating between the two conditions.
Familial aggregation and cases with siblings or parent-child aggregation were described for MCD (5), which suggests a possible genetic origin of the disease. Structured studies on genetics of MCD are now in progress and some interesting associations have been reported. In the case of FSGS, mutations in several genes causative of the disease were detected in the last decade (Table 2). MCD is considered a disease with complex inheritance where HLA genes are susceptibility loci (6–9) and inflammatory triggers, or modifiers of the genetic milieu, act as second hit. In general, there are ethnic trends according to which MCD (or better saying steroid sensitive nephrotic syndrome) is more frequent in Asia and Asian children have a seven-fold risk of developing the disease compared to non-Asian races (10). Variants of the HLA-DQA1 and of other MHC loci, associated with other gene variants (PLCG2) (11) and/or with environmental factors should contribute as second hits to the pathogenesis (12). It is of interest that a model implying the co-existence of HLA and other genes or molecules has been proposed also for other glomerular diseases such as IgA and membranous nephropathy (13). We expect some evolution from whole genome sequencing and it is probable that, in a near future, MCD will be defined also on the basis of specific genetic features.
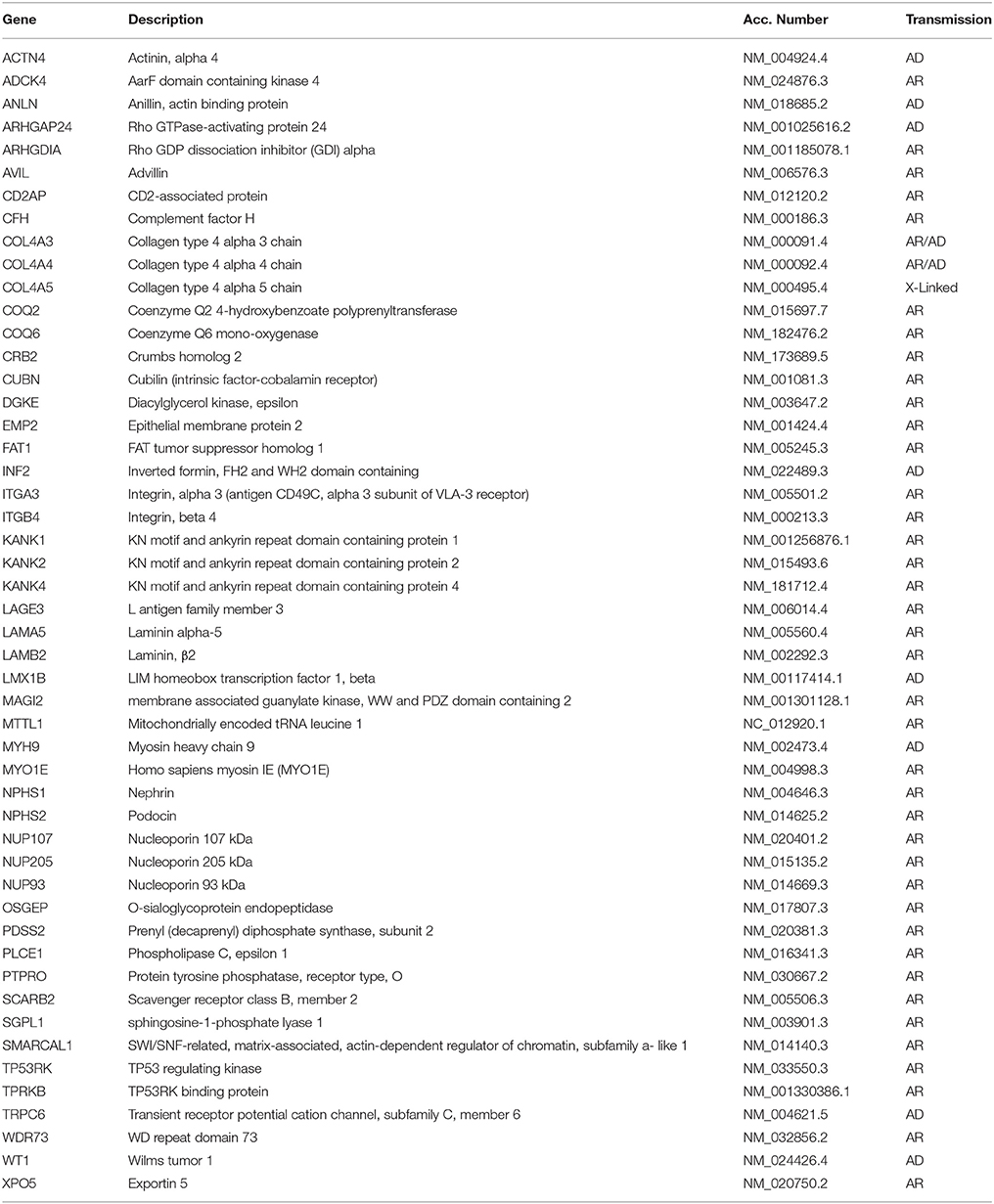
Table 2. Genes associated to MCD and FSGS.
MCD as a part of a syndrome has been uniquely reported in association with EXT1 mutations that cause autosomal dominant exostoses (OMIM 133700) (14). Isolated MCD with autosomal recessive inheritance has been reported in association with mutations of EMP2 gene encoding a regulatory protein of caveolin-1, that is expressed in podocytes and in renal endothelium (15). Rare cases of MCD associated with mutations of KANK1 and KANK2 genes have also been described in two families with a mixed pattern of MCD and FSGS (16). Given the similarities between MCD and FSGS, we cannot exclude that mutations in one of the recognized gene panels reported for FSGS may coexist in MCD. An example is NPHS1 mutation, that is causative of one of the most clinically relevant forms of congenital nephrotic syndrome. Three cases of NPHS1 mutation in children with nephrotic syndrome responsive to steroids and cyclosporine have been described, suggesting that hypomorphic mutations (such as those here reported) may coexist with the more frequent and severe clinical phenotypes (17).
There are cases of secondary MCD for which a direct relationship with a definite trigger or cause has been hypothesized. Cases of an association with drugs such as salazopyrin, D-penicillinamine, mercury, and gold have been reported, in which proteinuria remits following their withdrawal. In this case, the conclusion is that simple molecules may modify podocyte structure inducing a reversible defect that underlies the transitory nature of foot process fusion phenomenon. MCD may occur in parallel with major blood disorders such as Hodgkin (18, 19) and non-Hodgkin lymphoma (20–22) (mainly marginal zone B-cell lymphoma and chronic lymphocytic leukemia) and other lymphoproliferative disorders (such as multiple myeloma) and cancer (18, 23) (i.e., thymoma, bronchogenic cancer, and colon cancer). MCD is the most frequent glomerular disease associated with Hodgkin lymphoma (19, 24). C-Mip overexpression in tumor cells and in podocytes is a molecular signature of the association (25), that suggests a pathogenetic implication (for c-Mip see below). The association with non-Hodgkin lymphoma has been less frequently described; Kofman et al. (22) studied a large series of 13,992 patients and reported on 18 cases with this association, 50% of them presenting marginal B cell lymphoma or chronic lymphocytic leukemia. These are very rare associations in which the nephrotic syndrome generally remits after resolution of the primary problem.
The association of MCD with multiple myeloma is also rare. In myeloma, immunoglobulins or immunoglobulin fragments such as light chains present structural modifications that alter their molecular charge, often in terms of cationization and glycosylation. Cationic and/or hyper-glycosylated immunoglobulins are potential pathological effectors of proteinuria. Studies performed several decades ago clearly demonstrated that the charge of molecules is potentially able to modify the mechanism of repulsion of proteins at the glomerular level that, for molecules with a molecular mass >46 KDa, is mainly based on their charge (26, 27). Experimental models of proteinuria induced by infusion of cationic proteins strongly support this concept, since these molecules are not repulsed by glomerular polyanions and are freely excreted into urines. The possibility that a modification of protein charges could induce MCD has been explored in the past, without consistent results. There is now an increasing interest in the role of IgM glycosylation, that is now under study and could lead to more structured conclusions.
One of the clinically relevant characteristics of MCD is that it has been successfully treated from many years in spite of lack of any knowledge about mechanisms. Cyclosporine had historically a main role in successful treatment of MCD dependent and/or resistant to steroids (28). Cyclosporine is an inhibitor of calcineurin that inhibits, in turn, a nuclear factor in T lymphocytes. It was only after the study by Faul et al. (29) that it became clear that cyclosporine may also have a direct effect on podocytes, which blunts its importance as immunosuppressor. It blocks, in fact, the calcineurin dependent dephosphorylation of synaptopodin in podocytes and this protects the enzymatic degradation of synaptopodin by cathepsin L: the final result is the stabilization of the cytoskeleton. The study above also showed that the cytoskeleton in podocytes is crucial for their stability and that its perturbation leads to proteinuria. Molecular studies on genetic inherited nephrotic syndrome showing mutations in genes encoding cytoskeleton proteins confirmed this key function (30).
The possible implication of T cell effectors in MCD is mainly supported by the finding that supernatants of T cell hybridoma deriving from blood of patients with MCD determine foot process effacement and proteinuria in rats (31). This is a strong observation dating back to 1991; since then, no new evidence on specific proteinuric factors deriving from the T hybridoma was added to the original report, leaving this study field open to speculation and evolution. The existence of an imbalance in T cell subsets, with predominance of CD8+ over CD4+ and an increase in Natural Killer (NK) cells was the main hypothesis in MCD pathogenesis until the early 2000s (32). However, it is also clear that a single cell compartment as cause of MCD is not supported by research. The overview on circulating cells and cytokines potentially implicated in MCD that is reported below will give an idea of the complexity of the pathogenesis. Two specialized T cell lineages potentially involved in MCD are Th2 and Th17 (33–35). Th2 role is supported by the observation of a specific cytokine profile in patients with MCD (i.e., upregulation of IL4, IL5, IL9, IL10, IL13) (34, 36–38). Furthermore, Buffalo/Mna rats were characterized by Th2 polarization and presented a predominant increase in IL4 and IL13 levels, preceding the development of nephrotic syndrome (39, 40). A selective IL13-Th2 profile characterized children with nephrotic syndrome, who also presented upregulation of IL13 (but not IL4) mRNA by CD4+ and CD8+ cells during relapse (34). On the other hand, it is known that IL13 overexpression may directly induce proteinuria and podocyte lesions in rats (41) (see below). Th17 cells derive from naïve CD4+ upon stimulation with IL6 and IL13 (42). Th17 levels rise in several glomerulonephritides characterized by inflammatory glomerular lesions. This is not observed in MCD and raises doubts on a selective role of Th17 in renal disease (42). In general, previous studies focusing on specific cytokines reached fragmentary conclusions and the interpretation of their results must take into account that different technologies were used in the separation of cellular subsets and in cytokine determination. For this reason, IL8 and IL12 were reported both as high and low by different Authors and other cytokines such as IL10, IL2, IL4, and IL6 were described as low in serum despite their synthesis by purified polymorphonucleates and monocytes was high.
T regulatory cells (Tregs) are a dynamic cell population: at equilibrium, their level is low but they are rapidly generated from CD4 T cells, that expand in response to T cell Receptor (TCR) stimulation by class II HLA peptides. CD4 T cells differentiate into Tregs (CD4+ CD25+ Foxp3+) or into T effector cells (CD4+ CD25+ Foxp3−), depending on the cytokine milieu [for a review see Josefowicz et al. (43)]. Since Tregs and Teffs have anti-inflammatory and pro-inflammatory effects, respectively, it is clear that the balance between Tregs and Teffs derived from CD4+ has a crucial role in adaptive response.
Several studies in experimental models support the association between low Tregs and proteinuria [for a review see Bertelli et al. (44)]. Depending on the timing of the observation, animals expressing low Treg titers may develop alternatively MCD or FSGS. The hypothesis of an association between low Tregs and proteinuria has been confirmed in almost all models of proteinuria. In particular, (i) Adriamycin nephrosis can be modulated by infusing Foxp3-transduced T cells and/or by infusion of adenosine and/or by administration of low dose IL2 (45, 46); in all these cases, circulating Tregs were upregulated and proteinuria downmodulated; (ii) direct infusion of Tregs into Buffalo/Mna rats (40) was associated with proteinuria reduction and regression of renal lesions; (iii) Tregs role has also been investigated in LPS nephropathy that represents, as already described, a model of transient proteinuria; in this case, Tregs level was modulated by the administration of IL2/anti-IL2 immunocomplexes (Figure 1), that induces differentiation of CD4+ cells into Tregs (47). Polhill et al. (46) showed that infusion of IL2/anti-IL2 can reduce proteinuria and improves renal function in rats with Adriamycin nephrosis. In mice treated with LPS, Bertelli et al. (47) obtained a reduction of proteinuria utilizing the same therapeutic approach, but results were less clear and were inconclusive regarding the renal outcome since IL2 did not modify renal lesions.
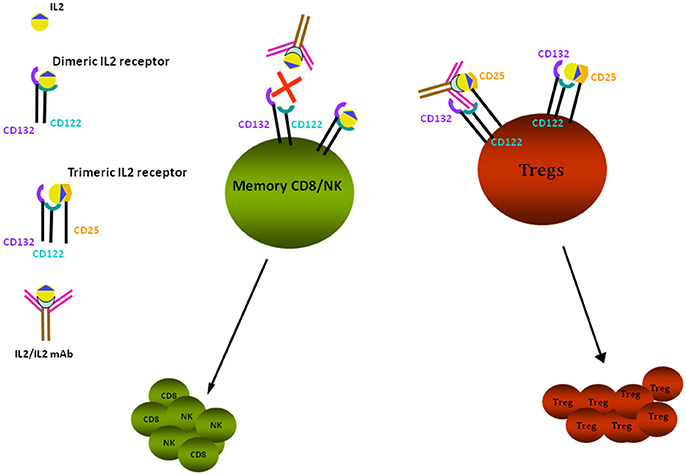
Figure 1. IL2 effects on Tregs. Tregs proliferation is stimulated through binding of IL2/anti-IL2 Ab to the trimeric IL2 receptor composed by CD122-CD132-CD25 subunits, having high affinity for the IL2 and IL2-anti-IL2 complex; the high affinity receptor is expressed by CD4+ cells, including Tregs. Low affinity receptors are expressed by memory CD8+ and NK cells: they are composed of the two subunits CD122 and CD132 that bind free IL2. In presence of anti-IL2Ab, the free quota of IL2 is decreased and proliferation of CD8+/NK is reduced. Tregs are dependent from IL2 for their survival and proliferation. In vivo administration of IL2 and of IL2 coupled to a monoclonal anti-IL2 antibody (JES6-1), specific for the CD132 subunit, prevents the activation of CD8+ and NK cells and allows the selective stimulation and proliferation of Tregs by the interaction with CD25 receptor subunit.
Evidence for a role of Tregs in human MCD is scanty but deserves a careful analysis in view of the possibility that these cells may be modulated in vivo. An interesting starting point is the association of MCD with IPEX (48), a syndrome characterized by immunodeficiency, polyendocrinopathy, and enteropathy, caused by a genetic mutation of FOXP3 that inactivates Tregs. It is of interest that, in a child with IPEX, nephrotic syndrome disappeared after bone marrow transplantation that normalized Treg functions (48). Studies in human pathology demonstrated that circulating levels of Tregs were low in patients with active MCD (49) and that low Treg levels correlated with high oxidant production deriving from ATP in polymorphonucleates. The balance between ATP and ADP is crucial to oxidant generation by circulating cells and apyrase has a key role in it. Apyrase is, in fact, the enzyme that reduces ATP and, as a consequence, it reduces oxidants. Studies by Bertelli et al. (49) produced evidence about the link among Tregs, apyrase, ATP, and oxidant generation in MCD. Modulation of Treg levels has become an interesting new potential treatment of MCD following the observation that low dose recombinant IL2 is safe and can be utilized in humans. In two important studies, low dose recombinant IL2 was utilized in patients with HCV-induced vasculitis and in Graft versus Host disease, showing that modulation of Treg levels was obtained in both cases (50, 51). So far, the unique pilot study in patients with renal diseases utilizing low dose recombinant IL2 was conducted in 4 patients with FSGS and nephrotic syndrome resistant to drugs (52). These patients were treated with six cycles of five IL2 infusions (1 × 106 U/m2 1st month, 1.5 × 106 U/m2 subsequent months) and had a stable 4- to 10-fold increment of Treg levels. Proteinuria, however, remained unchanged over 6 months, with no effects of IL2 at least in this set of patients. Good safety and tolerability of the drug suggested that low dose IL2 could be extended to MCD. However, given the better efficacy and tolerability of other drugs (i.e., anti-CD20) compared to IL2 (see below), there seems to be no reason to design a new study in this class of patients.
This is a field of recent interest. In the last few years, anti-CD20 monoclonal antibodies (Rituximab) were successfully utilized in MCD (53), representing at this time the most effective treatment of MCD patients with steroid dependence (54). Anti-CD20 are not synonymous of B cell inhibitors, since it has been shown that, within in vitro podocytes, these antibodies bind to Sphingomyelin Phosphodiesterase acid-like 3b (SMPDL3B), which represents a potential second receptor for the drug (55). Molecular studies showed that Rituximab binding to SMPDL3B in podocytes modifies the interaction with synaptopodin and prevents cytoskeleton remodeling induced by serum of patients with FSGS (55, 56). There are also chances that other cell lineages, such as Th17, could be modified by Rituximab infusion (57). The interaction between anti-CD20 and circulating Th17 may have a direct pathogenic implication (58).
Besides the clinical interest in Rituximab, a putative role of B cells in MCD was studied in the past. This hypothesis is supported by some indirect associations. A point of interest is the association of MCD with multiple myeloma discussed above (22), for which was considered a possible mechanism linked to the charge of immunoglobulins. On the whole, the key question of the role of B cells in MCD seems today of renewed interest but needs further study and discussion.
It should be emphasized that a reliable animal model of MCD has not been described yet. The main difference is the abrupt onset of massive proteinuria in human MCD that is almost absent in animal models. Another important characteristic of experimental models is that initial minimal lesions usually evolve to histological alterations typical of focal glomerulosclerosis. Therefore, it appears that studies in experimental models offer the opportunity to elucidate mechanisms of progression rather than of acute renal damage. Slow and constant increment of proteinuria would emerge as a progression factor in many of the descriptions below. The discordances between human MCD and models of proteinuria raise the main question of why MCD may maintain a normal glomerular structure in spite of podocyte effacement and heavy proteinuria. Brief persistence of proteinuria due to sensitivity to treatments and/or the possibility that protective factors occur in human MCD emerge as possible explanations but they remain uniquely as potential issues for future studies. LPS nephropathy is different from other models in terms of timing of proteinuria, that is typically transient, while renal lesions develop in spite of normal urinalysis. With this in mind, we consider the description of experimental models of proteinuria (more than of MCD) useful to facilitate comprehension of the overall problem of the pathogenesis of non-immunologic renal lesions.
Puromycin Aminonucleoside (PAN) was the first molecule utilized for inducing nephrotic syndrome in rats. It causes loss of glomerular basement membrane (GBM) polyanions and proteinuria within a few days from injection (59). PAN is partially metabolized into hypoxantine in glomeruli by adenosine deaminase, purine nucleoside phosphorylase and xanthine oxidase, which suggests the implication of a metabolic pathway ending with formation (60). However, free radical scavengers did not modify PAN toxicity in vivo and in podocytes in vitro (60, 61) suggesting the existence of parallel toxic pathways. Recent studies showed that, in glomeruli, PAN upregulates B7-1, a molecule known to induce foot process effacement and disruption of the slit-diaphragm barrier. Adriamycin (ADR) is an anthracycline antibiotic that has pleiotropic effects on podocytes. It may alter DNA, induces lipid peroxidation (62), and/or directly alters cell cytoskeleton (63); it also produces depletion of mitochondrial DNA (64). ADR was introduced in experimental nephrology in 1982 (65), when it was observed that its infusion in rats determined rapid loss of GBM polyanions, proteinuria, and initial minimal glomerular changes. In mice, sensitivity to ADR was linked with maintenance of a functioning mitochondrial DNA system (66). It is of interest that ADR nephropathy occurred only in specific inbred strains in association with mutations of Prkdc gene encoding a protein critical for DNA repair (66). In vitro experiments suggest that free radicals are the main effectors of the toxic effect induced by the drug on podocytes and in vivo inhibition of proteinuria by free radical scavengers supports this concept (67). For the above reasons, ADR was considered as a mixed model of nephrosis induced by oxidants in which susceptibility to mitochondrial DNA damage plays a role (66). MCD is the starting pathological feature whereas FSGS develops over days and weeks of stable proteinuria. Overall, PAN and ADR were described in view of a possible parallel mechanism of renal damage: even if the participation of oxidants in PAN is supported by the biochemistry of metabolites, concerns still exist due to lack of inhibitory effects of anti-oxidants. B7-1 may represent the target of PAN action on podocytes (68) and as it has proved to be induced by LPS in a transient model of proteinuria (see below).
Buffalo/Mna rats were first studied as a genetic model of thymoma with associated myasthenia (69, 70). Only subsequently it was found that Buffalo/Mna rats at 8 weeks of age developed proteinuria, associated with nephrotic syndrome at advanced stages, and renal lesions typical of FSGS (71). Based on segregation analysis, FSGS was initially considered genetically inherited on a recessive basis and two genes at chromosome 13 were identified as determining susceptibility (72). Further experiments clearly showed the existence of extra-renal factors correlated with a Th2 cytokine profile (39, 73). On this basis, Buffalo/Mna nephrosis was utilized to reinforce the view on immunologic factors as cause of proteinuria in conditions of genetically mediated susceptibility (40).
LPS is an inducer of transient proteinuria, typically beginning after 24 h from injection and lasting for 72 h; after this period, proteinuria normalizes. Renal lesions are instead progressive and after a first phase characterized by minimal lesions, FSGS develops and becomes evident after 1–2 weeks (47). LPS nephropathy is of particular interest for the evaluation of the immunomodulatory mechanism of proteinuria, principally linked to B7-1 (68). This aspect will be discussed in a dedicated section.
In the following sections, several animal models overexpressing single molecules or having undergone specific genes silencing are presented and discussed. These transgenic and knockout models represent a fascinating evolution. Their limit is that single factors or cytokines that are studied in these models are probably a part of a multi-factorial system causing proteinuria and therefore single factors per se play a limited role. This is demonstrated by the fact that proteinuria caused by foot process effacement is the unequivocal consequence in almost all cases. Another potential limit of these models is that they do not mirror the in vivo situation in terms of quantitative changes that are usually massive.
Oxidants represent a potential hit for proteinuria. They are generated by polymorphonucleates interacting with bacteria or viruses and could explain why MCD is often determined by or follows an infectious episode. Animal models support oxidant implication. As already pointed out, Adriamycin and Puromycin Aminonucleoside, two molecules that have historically been utilized to induce non-immunologic proteinuria in rats, are, in fact, oxidants (60, 67).
The bulk of evidence suggesting an implication of oxidants in MCD derives from studies on the generation of oxidants by polymorphonuclear cells obtained from patients with MCD (49), or from studies on the status of anti-oxidant systems during proteinuria. Generation of ex vivo oxidants has already been discussed in relation to Tregs and apyrase, (49). The status of the anti-oxidant system in MCD is directly correlated with loss of albumin in urine. It is known that albumin is the most important anti-oxidant buffer in blood since chloramines, that are the product of the interaction between and HCO3, are extremely reactive with the unique free sulfhydryl group of albumin (located at the 34 Cys of the sequence). The interaction of chloramines with albumin induces the formation of a sulfonic acid. Therefore, oxidized albumin contains no free sulfhydril residue and has a modified electric charge owing to the addition of an acidic residue. Musante et al. (74) demonstrated the presence of massive amounts of oxidized albumin in active nephrotic syndrome and, considering its reduced concentration in the serum of patients with MCD, these authors proposed that massive oxidation of albumin implies that oxidants are active in MCD and that the reduced buffering system implicates reduced defense. Taken together, these two mechanisms concur to proteinuria and/or have a negative role in it.
The oxidant connection seems an interesting interpretation of events leading to proteinuria in MCD. However, for their own nature, data from ex vivo experiments and observations on oxidized albumin should be read with caution. We need data more directly correlated with the evolution of this disease in humans, showing in particular a positive effect of anti-oxidant therapies in nephrotic syndrome. We also need more data on the role of potentially anti-oxidant substances such as adenosine that should modify the balance of oxidants deriving from ATP. It would be easy to carry out randomized clinical trials with anti-oxidants in association with canonical approaches: however, these studies require a large number of patients and do not seem a priority at the moment. Moreover, studies on adenosine metabolism are difficult since this molecule is rapidly degraded in vivo.
Several single molecules have been proposed to be involved in the pathogenesis of proteinuria/MCD and should be considered outside the general context of cells and cytokines. An attractive effect of studies demonstrating that specific stimuli acting on podocytes may induce MCD is the potential development of biomarkers and/or of new strategies based on targeted drugs. Animal models facilitate this task and several specific approaches have been developed in experimental MCD. However, we still lack a homogeneous view on the context and, above all, we need the demonstration that the same mechanisms and therapies that are active in experimental models are also effective in humans.
Cathepsin-L has already been mentioned in relation to cyclosporine. It is a potent endoprotease responsible of the breakdown of lysosomal proteins; its secreted form is involved in extracelluar matrix degradation (75). The actin-binding protein synaptopodin is a substrate for Cathepsin-L, having an effect on cytoskeleton organization (29). Renal Cathepsin-A is overexpressed in two animal models of proteinuria, i.e., PAN nephrosis and LPS nephropathy (76), and proteinuria is reduced through administration of the Cathepsin inhibitor E-64 (76). Similarly, Cathepsin-L knockout mice are protected against LPS nephropathy. Finally, in addition to MCD, increased podocyte expression of Cathepsin-L was reported in human conditions characterized by proteinuria including membranous and diabetic nephropathy (76). This finding suggests that this protein could represent a second hit following different stimuli, from autoantibodies to metabolic conditions.
Wistar rats overexpressing IL13 develop ultrastructural glomerular changes that are reminiscent of MCD and support a role of IL13 in the pathogenesis of experimental MCD (41). Even though IL13 mRNA is upregulated and the production of IL13 protein by CD3+ cells is increased in children with MCD (34), there is some concern about the above IL13 role in human beings. Of note, high levels of IL13 characterize patients with asthma and with atopic dermatitis, two very frequent conditions never associated with proteinuria (77, 78). Several anti-IL13 drugs are now available for human use and are ready for being tested in clinical trials on asthma (79, 80) and atopic dermatitis (80) in clinical trials. The inclusion of MCD in a small pilot study could resolve the open problem of IL13 role in MCD pathogenesis.
B7-1, also called CD80, is a trans-membrane molecule present on the surface of both B cells and other antigen presenting cells. It is a costimulatory molecule that is activated by various stimuli via Toll-like Receptors (TLRs) (81) After activation, B7-1 may interact with CD28 in Teffs and/or with CTLA4 in Tregs, potentially determining their activation (CD28) or inhibition (CTLA4) (82, 83). In podocytes, B7-1 activation is followed by effacement of foot process and proteinuria (68). Various stimuli including oxidants, as in the case of PAN, and/or inflammatory stimuli such as LPS, may activate B7-1 in glomeruli (68). Activation of the TLRs/B7-1 axis by LPS is of interest since humans are particularly exposed to potential inflammatory triggers. The induction of B7-1 by LPS and the resulting nephropathy that mimics, in the early phase, MCD, are already been described in the section dedicated to animal models. Studies in patients with MCD are also available. Garin et al. (84, 85) first showed that urinary B7-1 levels are high in patients with active MCD compared to MCD in remission and B7-1 was also detected in glomeruli of the same patients. These results were confirmed in other populations with MCD (86), thus leading to the conclusion that high B7-1 in urine and in glomeruli is a biomarker of MCD and that MCD and FSGS patients can be differentiated on this basis. This is interesting, since in animal models (here including LPS nephropathy), there is not a clear-cut difference between minimal lesions and FSGS. Other studies documented B7-1 glomerular upregulation in patients with other diseases such as diabetic nephropathy (87) and/or in patients with post-transplant FSGS recurrence (88). This observation led to the use of Abatacept, a B7-1 inhibitor, as a specific therapeutic agent in post-transplant FSGS recurrence. However, results from studies on this molecule were discordant and did not confirm the original observation (89). Currently, B7-1 blockade is not considered a proper pharmacological approach to FSGS recurrence. Overall, data on urinary and glomerular B7-1 and, in particular, the conclusion that this marker is specific for MCD require further demonstrations. Targeted therapies with Abatacept also need further studies, especially in patients with post-transplant recurrence of proteinuria.
CD40 is another costimulatory molecule expressed by B cells, professional antigen presenting cells, monocytes/macrophages, platelets, and also non-immune cells such as endothelial and smooth muscle cells (90). Its ligand CD40L (transiently expressed on T cells and other non-immune cells under inflammatory conditions) may also exist in a soluble circulating form. The CD40/CD40L complex mediates, at several sites, pro-inflammatory events (91), including production of cytokines, chemoattractants, oxygen radicals, etc. CD40 is also constitutively expressed in podocytes, where activation by CD40L promotes redistribution of nephrin and increases permeability to albumin in isolated glomeruli (92, 93). Previous studies showed that, in murine models of membranous nephropathy and lupus nephritis, CD40L blockade by anti-CD40L antibodies protects from autoimmune glomerulonephritis (94, 95). In a similar vein, infusion of anti-CD40L antibodies reduced proteinuria and progression of ADR nephrosis (96). Anti-CD40 antibodies were detected in FSGS patients and their levels were found to correlate with post-transplant recurrence of the disease (97); injection of the CD40/anti-CD40 complex induced proteinuria in mice. This interesting observation suggests that in some way CD40 is linked to post-transplant recurrence of FSGS, even if its exact implication is presently unclear.
C-Mip (C-maf inducing protein) is an 86 KDa protein with unclear function (98) that is scarcely detected in normal human kidney. Transgenic mice overexpressing c-Mip develop a disease resembling human MCD that suggests the role of c-Mip as an effector of the disease in human beings (99). Other observations support this possibility. Overexpression of c-Mip also occurs in oncologic patients treated with inhibitors of Vascular Endothelial Growth Factor-receptor tyrosine kinase (VEGF-TKIs), such as sorafenib and sunitinib, that are utilized as inhibitors of angiogenesis (100, 101). These patients frequently develop MCD/FSGS and c-Mip is markedly increased in podocytes of patients who develop the disease. RelA, a factor belonging to NFk-B family that represses c-Mip transcription, is down-regulated by VEGF-TKIs (102) and this could explain why MCD is induced by VEGF (103). The implication of other factors interacting with c-Mip such as Fyn, a Src kinase involved in nephrin phosphorylation (104, 105), is now under discussion since activation of the c-Mip-Fyn axis could modify nephrin phosphorylation and eventually determine alterations of the cytoskeletal architecture and proteinuria. Overall, c-Mip seems a strong candidate as a factor implicated in the pathogenesis of MCD and studies in human beings are now in progress to conclude on this possibility.
The plasminogen activator receptor (uPAR) pathway has been intensely discussed in the last few years. We know from physiology that uPAR assembles αvβ3integrin and activates a signal cascade modifying adhesion to extracellular matrix (106), that is functional in maintaining podocyte shape and sieving properties of glomeruli (107). Mice lacking uPAR (PLAUR−/−) are protected from developing LPS experimental proteinuria, which implies that an excess of circulating soluble uPAR (suPAR), derived from uPAR shedding from the cell surface, may be a determinant of proteinuria associated with LPS (107, 108). However, serum levels of suPAR are not increased in nephrotic patients, while high serum levels correlate with renal function (109). Apolipoproptein (APOL1) genotype has been recently proposed as third actor of the pathway involving suPAR and integrins (110), in which case G1 or G2 variants of APOL1 would have affinity for αvβ3integrin and suPAR and might cause proteinuria on a suPAR dependent basis. This finding follows the demonstration that APOL1 is a haplotype of risk of developing glomerulosclerosis and HIV-nephropathy (111) and could explain the reason why not all subjects carrying this haplotype develop renal failure. Presently, further studies are required before concluding on suPAR pathogenetic role.
ANGPTL4 is a glycoprotein, expressed by several tissues and organs (i.e., adipose tissue, liver, skeletal muscle, heart), that exists in two isoforms with high (pI > 8) and neutral (p = I7) isoelectric point, depending on the different contents of sialic acid. The idea of ANGPTL4 involvement in MCD derives from the observation of transgenic rats which have a generalized (aP2-ANGPTL4) or site-specific overexpression of ANGPTL4 in podocytes (NPHS2-ANGPTL4) (112). It is of interest that the nephrotic syndrome does not develop in all cases but only in mice with podocyte overexpression of ANGPTL4, having normal circulating levels of the protein. Rats overexpressing ANGPTL4 outside the kidney have instead high circulating levels of this factor but do not develop proteinuria. Circulating and glomerular ANGPTL4 are markedly high in PAN nephrosis and are reduced by 70% by glucocorticoids; reduced serum levels are not followed by resolution of proteinuria. Therefore, there are concerns about the meaning and implication of circulating ANGPTL4 in experimental nephrosis. Data in human beings are more homogeneous and follow a logical sequence: ANGPTL4 is minimally expressed in normal glomeruli, but it is upregulated in MCD (113) and serum and urine ANGPTL4 levels are high in these patients (113). Therefore, high circulating ANGPTL4 levels and high renal expression coincide in human MCD and correspond to proteinuria. A possible explanation reconciling data deriving from animal models is that ANGPTL4 may determine proteinuria acting specifically on podocytes and that a site specific effect does not necessarily mean high circulating levels. ANGPTL4 remains a strong candidate for explaining MCD both in human beings and in experimental models.
Plasma hemopexin is a glycoprotein involved in the binding and transport of free heme and iron homeostasis. It is considered an acute phase protein with anti-oxidant function (114, 115). When infused into rats, hemopexin induces reversible proteinuria supported by actin and nephrin reorganization in glomeruli (116) Studies in human MCD showed decreased titer of plasma and urine hemopexin and increased activity exclusively during disease relapse. These changes were ascribed to an altered configuration of hemopexin in relapsing MCD (117). Conclusions on hemopexin are still far from being reached and its implication in MCD seems, at the moment, a secondary effect.
The definition of molecular and cellular mechanisms for proteinuria in MCD has been the topic of many studies in the last few decades (Table 3) and several interesting and plausible conclusions have been reached. Most data on these mechanisms are based on animal models and human studies have also been carried out. A major problem is the definition of MCD, that is based on pathology characteristics, while it is still not clear whether MCD is an autonomous disease or represents a phase of a progressive condition leading to FSGS. In animal models, the continuum from MCD to FSGS has been clearly documented, but data in human beings are of difficult interpretation and we lack a definite conclusion on this aspect. Studies showed an implication of different circulating cells and documented the role of cytokines and of other soluble factors. Based on this complex picture, the unique possible conclusion is that the pathogenesis of MCD is multifactorial and here we propose the concept that MCD is a “clinical-pathological-genetic entity” in which many molecules are involved. Indeed, it is also clear that we lack a single causative factor for human MCD. Good news come from therapeutic developments that are changing the clinical impact of the disease, shifting from a day by day approach to the administration of drugs active in the mid/long-term that are better tolerated by patients (53, 54, 118, 119). These evolutions reinforce our hope for a conclusive solution deriving from pharmacological developments.

Table 3. Principal fields of research on mechanisms for Minimal Change Disease.
Clinical studies cited in this article and conducted by our research group were approved by local Ethics Committee. A written informed consent was in all cases obtained from parents of patients. All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki Experiments in animals were done by our research group according to the principles expressed in the Declaration of Helsinki and were approved by the Institutional Review Board of IRCCS S. Martino (Genoa) and by the local authorities according to the legal requirements.
AB and AC performed clinical observations, and GC genetic testing and correlations of some patients with MCN cited in the study. RB and GG prepared the manuscript, and endorsed the final draft submitted.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Giannina Gaslini Institute had provided logistic and financial support to studies through grants from the ministry of health (Cinque per mille of IRPEF-Finanziamento della ricerca sanitaria). People working at the project on iMN belongs to the Fondazione Malattie Renali del Bambino of which we acknowledge the financial support. Grant n ROL 9849 was received from Compagnia di San Paolo.
1. Hayslett JP, Krassner LS, Bensch KG, Kashgarian M, Epstein FH. Progression of “lipoid nephrosis” to renal insufficiency. N Engl J Med. (1969) 281:181–7.
2. Tejani A, Phadke K, Nicastri A, Adamson O, Chen CK, Trachtman H, et al. Efficacy of cyclophosphamide in steroid-sensitive childhood nephrotic syndrome with different morphological lesions. Nephron (1985) 41:170–3.
3. Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF. Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol. (2016) 12:768–76. doi: 10.1038/nrneph.2016.147
4. Fogo AB. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat Rev Nephrol. (2015) 11:76–87. doi: 10.1038/nrneph.2014.216
5. Motoyama O, Sugawara H, Hatano M, Fujisawa T, Iitaka K. Steroid-sensitive nephrotic syndrome in two families. Clin Exp Nephrol. (2009) 13:170–3. doi: 10.1007/s10157-008-0117-7
6. Konrad M, Mytilineos J, Bonissou F, Scherer S, Gulli MP, Meissner I. HLA class II associations with idiopathic nephrotic syndrome in children. Tissue Antigens (1994) 43:275–80.
7. Clark AG, Vaughan RW, Stephens HA, Chantler C, Williams DG, Welsh KI. Genes encoding the beta-chains of HLA-DR7 and HLA-DQw2 define major susceptibility determinants for idiopathic nephrotic syndrome. Clin Sci. (1990) 78:391–7.
8. Lagueruela CC, Buettner TL, Cole BR, Kissane JM, Robson AM. HLA extended haplotypes in steroid-sensitive nephrotic syndrome of childhood. Kidney Int. (1990) 38:145–50.
9. Abe KK, Michinaga I, Hiratsuka T, Ogahara S, Naito S, Arakawa K, et al. Association of DQB1*0302 alloantigens in Japanese pediatric patients with steroid-sensitive nephrotic syndrome. Nephron (1995) 70:28–34.
10. Feehally J, Kendell NP, Swift PG, Walls J. High incidence of minimal change nephrotic syndrome in Asians. Arch Dis Child (1985) 60:1018–20.
11. Hinkes B, Wiggins RC, Gbadegesins RA, Vlangos CN, Seelow D, Nürberg G. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet. (2006) 38:1397–405. doi: 10.1038/ng1918
12. Gbadegesin RA, Adeyamo A, Webb NJ, Greenbaum LA, Abeyagunawardena A, Thalgahagada S. HLA-DQA1 and PLCG2 are candidate risk loci for childhood-onset steroid-sensitive nephrotic syndrome. J Am Soc Nephrol. (2015) 26:1701–10. doi: 10.1681/ASN.2014030247
13. Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. (2014) 46:1187–96. doi: 10.1038/ng.3118
14. Roberts IS, Gleadle JM. Familial nephropathy and multiple exostoses with exostosin-1 (EXT1) gene mutation. J Am Soc Nephrol. (2008) 19:450–3. doi: 10.1681/ASN.2007080842
15. Gee HY, Ashraf S, Wan X, Vega-Warner V, Esteve-Rudd J, Lovric S. Mutations in EMP2 cause childhood-onset nephrotic syndrome. Am J Hum Genet. (2014) 94:884–90. doi: 10.1016/j.ajhg.2014.04.010
16. Gee HY, Zhang F, Ashraf S, Kohl S, Sadowski CE, Vega-Warner V. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J Clin Invest. (2015) 125:2375–84. doi: 10.1172/JCI79504
17. Lahdenkari AT, Kestila M, Holmberg C, Koskimies O, Jalanko H. Nephrin gene (NPHS1) in patients with minimal change nephrotic syndrome (MCNS). Kidney Int. (2004) 65:1856–63. doi: 10.1111/j.1523-1755.2004.00583.x
18. Cambier JF, Ronco P. Onco-nephrology: glomerular diseases with cancer. Clin J Am Soc Nephrol. (2012) 7:1701–12. doi: 10.2215/CJN.03770412
19. Lien YH, Lai LW. Pathogenesis, diagnosis and management of paraneoplastic glomerulonephritis. Nat Rev Nephrol. (2011) 7:85–95. doi: 10.1038/nrneph.2010.171
20. Cohen LJ, Rennke HG, Laubach JP, Humphreys BD. The spectrum of kidney involvement in lymphoma: a case report and review of the literature. Am J Kidney Dis. (2010) 56:1191–6. doi: 10.1053/j.ajkd.2010.07.009
21. Ronco PM. Paraneoplastic glomerulopathies: new insights into an old entity. Kidney Int. (1999) 56:355–77.
22. Kofman T, Zhang SY, Copie-Bergman C, Moktefi A, Rainbourg Q, Francois H. Minimal change nephrotic syndrome associated with non-Hodgkin lymphoid disorders: a retrospective study of 18 cases. Medicine (2014) 93:350–8. doi: 10.1097/MD.0000000000000206
23. Karras A, de Montpreville V, Fakhouri F, Grünfeld JP, Lesavre P. Group d'Etudes des nephropaties associees aux thymomes renal and thymic pathology in thymoma-associated nephropathy: report of 21 cases and review of the literature. Nephrol Dial Transplant. (2005) 20:1075–82. doi: 10.1093/ndt/gfh615
24. Audard V, Larousserie F, Grimbert P, Abtahi M, Sotto JI, Delmer A. Minimal change nephrotic syndrome and classical Hodgkin's lymphoma: report of 21 cases and review of the literature. Kidney Int. (2006) 69:2251–60. doi: 10.1038/sj.ki.5000341
25. Audard V, Zhang SY, Copie-Bergman C, Rucker-Martin C, Ory V, Candelier M. Occurrence of minimal change nephrotic syndrome in classical Hodgkin lymphoma is closely related to the induction of c-mip in Hodgkin-Reed Sternberg cells and podocytes. Blood (2010) 115:3756–62. doi: 10.1182/blood-2009-11-251132
26. Brenner BM, Hostetter TH, Humes HD. Molecular basis of proteinuria of glomerular origin. N Engl J Med. (1978) 298:826–33.
27. Ghiggeri GM, Ginevri F, Candiano G, Oleggini R, Perfumo F, Queirolo C, et al. Characterization of cationic albumin in minimal change nephropathy. Kidney Int. (1987) 32:547–53.
28. Meyrier A. Treatment of focal segmental glomerulosclerosis. Expert Opin Pharmacother. (2005) 6:1539–49. doi: 10.1517/14656566.6.9.1539
29. Faul C, Donnelly M, Merscher-Gomez S, Chang YH, Franz S, Delfgaauw J. The actin cytoskeleton of kidney podocytes is a direct target of the antiproteinuric effect of cyclosporine A. Nat Med. (2008) 14:931–8. doi: 10.1038/nm.1857
30. Sanna-Cherchi S, Somenzi D, Carnevali ML, Pilato FP, Carraro M, Ghiggeri GM, et al. Recurrent autosomal-dominant focal segmental glomerulosclerosis. Kidney Int. (2006) 70:1664–5. doi: 10.1038/sj.ki.5001796
31. Koyama A, Fujisaki M, Kobayashi M, Igarashi M, Narita M. A glomerular permeability factor produced by human T cell hybridomas. Kidney Int. (1991) 40:453–60.
32. Frank C, Herrmann M, Fernandez M, Dirnecker S, Böswald M, Kolowos W. Dominant T cells in idiopathic nephrotic syndrome of childhood. Kidney Int. (2000) 57:510–7. doi: 10.1046/j.1523-1755.2000.00870.x
33. Fiser RT, Arnold WC, Charlton RK, Steele RW, Childress SH, Shirkey B. T-lymphocyte subsets in nephrotic syndrome. Kidney Int. (1991) 40:913–6.
34. Yap HK, Cheung W, Murugasu B, Sim SK, Seah CC, Jordan SC. Th1 and Th2 cytokine mRNA profiles in childhood nephrotic syndrome: evidence for increased IL-13 mRNA expression in relapse. J Am Soc Nephrol. (1999) 10:529–37.
35. Liu LL, Qin V, Cai JF, Wang HY, Tao JL, Li H. Th17/Treg imbalance in adult patients with minimal change nephrotic syndrome. Clin Immunol. (2011) 139:314–20. doi: 10.1016/j.clim.2011.02.018
36. Lama G, Luongo I, Tirino G, Borriello A, Carangio C, Salsamo ME. T-lymphocyte populations and cytokines in childhood nephrotic syndrome. Am J Kidney Dis. (2002) 39:958–65. doi: 10.1053/ajkd.2002.32769
37. Araya C, Diaz L, Wasserfall C, Atkinson M, Mu W, Johnson R, et al. T regulatory cell function in idiopathic minimal lesion nephrotic syndrome. Pediatr Nephrol. (2009) 24:1691–8. doi: 10.1007/s00467-009-1214-x
38. Cho BS, Yoon SR, Jang JY, Pyun KH, Lee CE. Up-regulation of interleukin-4 and CD23/FcepsilonRII in minimal change nephrotic syndrome. Pediatr Nephrol. (1999) 13:199–204.
39. Le Berre L, Hervè C, Buzelin F, Usai C, Soulillou JP, Dantal J. Renal macrophage activation and Th2 polarization precedes the development of nephrotic syndrome in Buffalo/Mna rats. Kidney Int. (2005) 68:2079–90. doi: 10.1111/j.1523-1755.2005.00664.x
40. Le Berre L, Bruneau S, Naulet J, Renaudin K, Buzelin F, Usai C. Induction of T regulatory cells attenuates idiopathic nephrotic syndrome. J Am Soc Nephrol. (2009) 20:57–67. doi: 10.1681/ASN.2007111244
41. Lai KW, Wei CL, Tan LK, Tan PH, Chiang GS, Lee CG. Overexpression of interleukin-13 induces minimal-change-like nephropathy in rats. J Am Soc Nephrol. (2007) 18:1476–85. doi: 10.1681/ASN.2006070710
42. Couser WG. Basic and translational concepts of immune-mediated glomerular diseases. J Am Soc Nephrol. (2012) 23:381–99. doi: 10.1681/ASN.2011030304
43. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
44. Bertelli R, Bonanni A, Di Donato A, Cioni M, Ravani PG, Ghiggeri M. Regulatory T cells and minimal change nephropathy: in the midst of a complex network. Clin Exp Immunol. (2016) 183:166–74. doi: 10.1111/cei.12675
45. Wang YM, Zhang GY, Hu M, Polhill T, Sawyer A, Zhou JJ. CD8+ regulatory T cells induced by T cell vaccination protect against autoimmune nephritis. J Am Soc Nephrol. (2012) 23:1058–67. doi: 10.1681/ASN.2011090914
46. Polhill T, Zhang GY, Hu M, Sawyer A, Zhou JJ, Saito M. IL-2/IL-2Ab complexes induce regulatory T cell expansion and protect against proteinuric CKD. J Am Soc Nephrol. (2012) 23:1303–8. doi: 10.1681/ASN.2011111130
47. Bertelli R, Di Donato A, Cioni M, Grassi F, Ikehata M, Bonanni A, et al. LPS nephropathy in mice is ameliorated by IL-2 independently of regulatory T cells activity. PLoS ONE (2014) 9:e111285. doi: 10.1371/journal.pone.0111285
48. Bertelli R, Trivelli A, Magnasco A, Cioni M, Bodria M, Carrea A. Failure of regulation results in an amplified oxidation burst by neutrophils in children with primary nephrotic syndrome. Clin Exp Immunol. (2010) 161:151–8. doi: 10.1111/j.1365-2249.2010.04160.x
49. Bertelli R, Bodria M, Nobile M, Alloisio S, Barbieri R, Montobbio G, et al. Regulation of innate immunity by the nucleotide pathway in children with idiopathic nephrotic syndrome. Clin Exp Immunol. (2011) 166:55–63. doi: 10.1111/j.1365-2249.2011.04441.x
50. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med. (2011) 365:2067–77. doi: 10.1056/NEJMoa1105143
51. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
52. Bonanni A, Bertelli R, Rossi R, Bruschi M, Di Donato A, Ravani P, et al. A pilot study of IL2 in drug-resistant idiopathic nephrotic syndrome. PLoS ONE (2015) 10:e0138343. doi: 10.1371/journal.pone.0138343
53. Ravani P, Bonanni A, Rossi R, Caridi G, Ghiggeri GM. Anti-CD20 antibodies for idiopathic nephrotic syndrome in children. Clin J Am Soc Nephrol. (2016) 11:710–20. doi: 10.2215/CJN.08500815
54. Ravani P, Rossi R, Bonanni A, Quinn RR, Sica F, Bodria M. Rituximab in children with steroid-dependent nephrotic syndrome: a multicenter, open-label, noninferiority, randomized controlled trial. J Am Soc Nephrol. (2015) 26:2259–66. doi: 10.1681/ASN.2014080799
55. Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med. (2011) 3:85ra46. doi: 10.1126/scitranslmed.3002231
56. Yoo TH, Pedigo CE, Guzman J, Correa-Medina M, Wei C, Villareal R. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. J Am Soc Nephrol. (2015) 26:133–47. doi: 10.1681/ASN.2013111213
57. Alunno A, Carubbi F, Bistoni O, Caterbi S, Bartoloni E, Di Benedetto P. Interleukin (IL)-17-producing pathogenic T lymphocytes co-express CD20 and are depleted by rituximab in primary Sjogren's syndrome: a pilot study. Clin Exp Immunol. (2016) 184:284–92. doi: 10.1111/cei.12771
58. van de Veerdonk FL, Lauwerys B, Marijnissen RJ, Timmermans K, Di Padova F, Koenders MI. The anti-CD20 antibody rituximab reduces the Th17 cell response. Arthritis Rheum. (2011) 63:1507–16. doi: 10.1002/art.30314
59. Frenk S, Antonowicz I, Craig JM, Metcoff J. Experimental nephrotic syndrome induced in rats by aminonucleoside; renal lesions and body electrolyte composition. Proc Soc Exp Biol Med. (1955) 89:424–7.
60. Ghiggeri GM, Cercignani G, Ginevri F, Bertelli R, Zetta L, Greco F. Puromycin aminonucleoside metabolism by glomeruli and glomerular epithelial cells in vitro. Kidney Int. (1991) 40:35–42.
61. Ginevri F, Gusmano R, Oleggini R, Acerbo S, Bertelli R, Perfumo F. Renal purine efflux and xanthine oxidase activity during experimental nephrosis in rats: difference between puromycin aminonucleoside and adriamycin nephrosis. Clin Sci. (1990) 78:283–93.
62. Fukuda F, Kitada M, Horie T, Awazu S. Evaluation of adriamycin-induced lipid peroxidation. Biochem Pharmacol. (1992) 44:755–60.
63. Coers W, Huitema S, van der Horst ML, Weening JJ. Puromycin aminonucleoside and adriamycin disturb cytoskeletal and extracellular matrix protein organization, but not protein synthesis of cultured glomerular epithelial cells. Exp Nephrol. (1994) 2:40–50.
64. Adachi K, Fujiura Y, Mayumi F, Nozuhara A, Sugiu Y, Sakanashi T. A deletion of mitochondrial DNA in murine doxorubicin-induced cardiotoxicity. Biochem Biophys Res Commun. (1993) 195:945–51.
65. Bertani T, Poggi A, Pozzoni R, Delaini F, Sacchi G, Thoua Y. Adriamycin-induced nephrotic syndrome in rats: sequence of pathologic events. Lab Invest. (1982) 46:16–23.
66. Papeta N, Zheng Z, Schon EA, Brosel S, Altintas MM, Nasr SH. Prkdc participates in mitochondrial genome maintenance and prevents Adriamycin-induced nephropathy in mice. J Clin Invest. (2010) 120:4055–64. doi: 10.1172/JCI43721
67. Morgan WA, Kaler B, Bach PH. The role of reactive oxygen species in adriamycin and menadione-induced glomerular toxicity. Toxicol Lett. (1998) 94:209–15.
68. Reiser J, von Gersdorff G, Loos M, Oh J, Asanuma K, Giardino L. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J Clin Invest. (2004) 113:1390–7. doi: 10.1172/JCI20402
69. Matsuyama M, Yamada C, Hiai H. A single dominant susceptible gene determines spontaneous development of thymoma in BUF/Mna rat. Jpn J Cancer Res. (1986) 77:1066–8.
70. Iwasa K, Komai K, Takamori M. Spontaneous thymoma rat as a model for myasthenic weakness caused by anti-ryanodine receptor antibodies. Muscle Nerve (1998) 21:1655–60.
71. Matsuyama M, Ogiu T, Kontani K, Yamada C, Kawai M, Hiai H. Genetic regulation of the development of glomerular sclerotic lesions in the BUF/Mna rat. Nephron (1990) 54:334–7.
72. Murayama S, Yagyu S, Higo K, Ye C, Mizuno T, Oyabu A. A genetic locus susceptible to the overt proteinuria in BUF/Mna rat. Mamm Genome (1998) 9:886–8.
73. Le Berre L, Godfrin Y, Günther E, Buzelin F, Perretto S, Smit H. Extrarenal effects on the pathogenesis and relapse of idiopathic nephrotic syndrome in Buffalo/Mna rats. J Clin Invest. (2002) 109:491–8. doi: 10.1172/JCI12858
74. Musante L, Candiano G, Petretto A, Bruschi M, Dimasi N, Caridi G. Active focal segmental glomerulosclerosis is associated with massive oxidation of plasma albumin. J Am Soc Nephrol. (2007) 18:799–810. doi: 10.1681/ASN.2006090965
75. Ishidoh K, Kominami E. Procathepsin L degrades extracellular matrix proteins in the presence of glycosaminoglycans in vitro. Biochem Biophys Res Commun. (1995) 217:624–31.
76. Sever S, Altintas MM, Nankoe SR, Möller CC, Ko D, Wei C. Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest. (2007) 117:2095–104. doi: 10.1172/JCI32022
77. Bagnasco D, Ferrando M, Varricchi G, Passalacqua G, Canonica GW. A critical evaluation of anti-IL-13 and anti-IL-4 strategies in severe asthma. Int Arch Allergy Immunol. (2016) 170:122–31. doi: 10.1159/000447692
78. Matsunaga MC, Yamauchi PS. IL-4 and IL-13 inhibition in atopic dermatitis. J Drugs Dermatol. (2016) 15:925–9.
79. Chung KF. Targeting the interleukin pathway in the treatment of asthma. Lancet (2015) 386:1086–96. doi: 10.1016/S0140-6736(15)00157-9
80. Luo J, Liu D, Liu CT. The efficacy and safety of antiinterleukin 13, a monoclonal antibody, in adult patients with asthma: a systematic review and meta-analysis. Medicine (2016) 95:e2556. doi: 10.1097/MD.0000000000002556
81. Shimada M, Ishimoto T, Lee PY, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA. Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-kappaB-dependent pathway. Nephrol Dial Transplant. (2012) 27:81–9. doi: 10.1093/ndt/gfr271
83. Chambers CA, Allison JP. Costimulatory regulation of T cell function. Curr Opin Cell Biol. (1999) 11:203–10.
84. Garin EH, Diaz LN, Mu W, Wasserfall C, Araya C, Segal M, et al. Urinary CD80 excretion increases in idiopathic minimal-change disease. J Am Soc Nephrol. (2009) 20:260–6. doi: 10.1681/ASN.2007080836
85. Garin EH, Mu W, Arthur JM, Rivard CJ, Araya CE, Shimada M, et al. Urinary CD80 is elevated in minimal change disease but not in focal segmental glomerulosclerosis. Kidney Int. (2010) 78:296–302. doi: 10.1038/ki.2010.143
86. Mishra OP, Kumar R, Narayan G, Srivastava P, Abhinay A, Prasad R. Toll-like receptor 3 (TLR-3), TLR-4 and CD80 expression in peripheral blood mononuclear cells and urinary CD80 levels in children with idiopathic nephrotic syndrome. Pediatr Nephrol. (2017) 32:1355–61. doi: 10.1007/s00467-017-3613-8
87. Fiorina P, Vergani A, Bassi R, Niewczas MA, Altintas MM, Pezzolesi MG. Role of podocyte B7-1 in diabetic nephropathy. J Am Soc Nephrol. (2014) 25:1415–29. doi: 10.1681/ASN.2013050518
88. Yu CC, Fornoni A, Weins A, Hakroush S, Maiquel D, Sageshima J. Abatacept in B7-1-positive proteinuric kidney disease. N Engl J Med. (2013) 369:2416–23. doi: 10.1056/NEJMoa1304572
89. Delville M, Baye E, Durrbach A, Audard V, Kofman T, Braun L. B7-1 blockade does not improve post-transplant nephrotic syndrome caused by recurrent FSGS. J Am Soc Nephrol. (2016) 27:2520–7. doi: 10.1681/ASN.2015091002
90. Quezada SA, Jarvinen LZ, Lind EF, Noelle RJ. CD40/CD154 interactions at the interface of tolerance and immunity. Annu Rev Immunol. (2004) 22:307–28. doi: 10.1146/annurev.immunol.22.012703.104533
91. Hassan GS, Merhi Y, Mourad W. CD40 ligand: a neo-inflammatory molecule in vascular diseases. Immunobiology (2012) 217:521–32. doi: 10.1016/j.imbio.2011.03.010
92. Kairaitis L, Wang Y, Zheng L, Tay YC, wang YD, Harris C. Blockade of CD40-CD40 ligand protects against renal injury in chronic proteinuric renal disease. Kidney Int. (2003) 64:1265–72. doi: 10.1046/j.1523-1755.2003.00223.x
93. Doublier S, Zennaro C, Musante L, Spatola T, Candiano G, Bruschi M. Soluble CD40 ligand directly alters glomerular permeability and may act as a circulating permeability factor in FSGS. PLoS ONE (2017) 12:e0188045. doi: 10.1371/journal.pone.0188045
94. Ruth AJ, Kitching AR, Semple TJ, Tipping PG, Holdsworth SR. Intrinsic renal cell expression of CD40 directs Th1 effectors inducing experimental crescentic glomerulonephritis. J Am Soc Nephrol. (2003) 14:2813–22. doi: 10.1097/01.ASN.0000091381.60059.FB
95. Reynolds J, Khan SB, Allen AR, Benjamin CD, Pusey CD. Blockade of the CD154-CD40 costimulatory pathway prevents the development of experimental autoimmune glomerulonephritis. Kidney Int. (2004) 66:1444–52. doi: 10.1111/j.1523-1755.2004.00907.x
96. Lee VW, Qin X, Wang Y, Zheng G, Wang Y, Wang Y. The CD40-CD154 co-stimulation pathway mediates innate immune injury in adriamycin nephrosis. Nephrol Dial Transplant. (2010) 25:717–30. doi: 10.1093/ndt/gfp569
97. Delville M, Sigdel TK, Wei C, Li J, Hsieh SC, Fornoni A. A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med. (2014) 6:256ra136. doi: 10.1126/scitranslmed.3008538
98. Sahali D, Sendeyo k, Mangier M, Audard V, Zhang SY, Lang P. Immunopathogenesis of idiopathic nephrotic syndrome with relapse. Semin Immunopathol. (2014) 36:421–9. doi: 10.1007/s00281-013-0415-3
99. Zhang SY, Kamal M, Dahan K, Pawlak A, Ory VD. Desvaux c-mip impairs podocyte proximal signaling and induces heavy proteinuria. Sci Signal. (2010) 3:ra39. doi: 10.1126/scisignal.2000678
100. Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol. (2009) 6:569–79. doi: 10.1038/nrclinonc.2009.130
101. Izzedine H, Massard C, Spano JP, Goldwasser F, Khayat D, Soria JC. VEGF signalling inhibition-induced proteinuria: mechanisms, significance and management. Eur J Cancer (2010) 46:439–48. doi: 10.1016/j.ejca.2009.11.00
102. Echeverria V, Burgess S, Gamble-George J, Zeitlin R, Lin X, Cao CG, et al. Sorafenib inhibits nuclear factor kappa B, decreases inducible nitric oxide synthase and cyclooxygenase-2 expression, and restores working memory in APPswe mice. Neuroscience (2009) 162:1220–31. doi: 10.1016/j.neuroscience.2009.05.019
103. Kamal M, Valanciute A, Dahan K, Ory V, Pawlak A, Lang P. C-mip interacts physically with RelA and inhibits nuclear factor kappa B activity. Mol Immunol. (2009) 46:991–8. doi: 10.1016/j.molimm.2008.09.034
104. Sendeyo K, Audard V, Zhang SY, Fan Q, Bouachi K, Ollero M. Upregulation of c-mip is closely related to podocyte dysfunction in membranous nephropathy. Kidney Int. (2013) 83:414–25. doi: 10.1038/ki.2012.426
105. Patrakka J, Tryggvason K. Nephrin–a unique structural and signaling protein of the kidney filter. Trends Mol Med. (2007) 13:396–403. doi: 10.1016/j.molmed.2007.06.006
106. Ossowski L, Aguirre-Ghiso JA. Urokinase receptor and integrin partnership: coordination of signaling for cell adhesion, migration and growth. Curr Opin Cell Biol. (2000) 12:613–20. doi: 10.1016/S0955-0674(00)00140-X
107. Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat Med. (2011) 17:952–60. doi: 10.1038/nm.2411
108. Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S. Modification of kidney barrier function by the urokinase receptor. Nat Med. (2008) 14:55–63. doi: 10.1038/nm1696
109. Hayek SS, Sever S, Ko YA, Trachtman H, Awad M, Wadhwani S. Soluble urokinase receptor and chronic kidney disease. N Engl J Med. (2015) 373:1916–25. doi: 10.1056/NEJMoa1506362
110. Hayek SS, Koh KH, Grams ME, Wei C, Ko YA, Li J. A tripartite complex of suPAR, APOL1 risk variants and alphavbeta3 integrin on podocytes mediates chronic kidney disease. Nat Med. (2017) 23:945–53. doi: 10.1038/nm.4362
111. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. (2011) 22:2129–37. doi: 10.1681/ASN.2011040388
112. Chugh SS, Clement LC, Mace C. New insights into human minimal change disease: lessons from animal models. Am J Kidney Dis. (2012) 59:284–92. doi: 10.1053/j.ajkd.2011.07.024
113. Clement LC, Avila-Casado C, Macé C, Soria E, Bakker WW, Kersten S, et al. Podocyte-secreted angiopoietin-like-4 mediates proteinuria in glucocorticoid-sensitive nephrotic syndrome. Nat Med. (2011) 17:117–22. doi: 10.1038/nm.2261
114. Immenschuh S, Song DX, Satoh H, Muller-Eberhard U. The type II hemopexin interleukin-6 response element predominates the transcriptional regulation of the hemopexin acute phase responsiveness. Biochem Biophys Res Commun. (1995) 207:202–8.
115. Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem. (1995) 41:1819–28.
116. Lennon R, Singh A, Welsh GI, Coward RJ, Satchell S, Ni L. Hemopexin induces nephrin-dependent reorganization of the actin cytoskeleton in podocytes. J Am Soc Nephrol. (2008) 19, 2140–9. doi: 10.1681/ASN.2007080940
117. Bakker WW, van Dael CM, Pierik LJ, van Wijk JA, Nauta J, Borghuis T, et al. Altered activity of plasma hemopexin in patients with minimal change disease in relapse. Pediatr Nephrol. (2005) 20:1410–5. doi: 10.1007/s00467-005-1936-3
118. Ravani P, Magnasco A, Edefonti A, Murer L, Rossi R, Ghio L. Short-term effects of rituximab in children with steroid- and calcineurin-dependent nephrotic syndrome: a randomized controlled trial. Clin J Am Soc Nephrol. (2011) 6:1308–15. doi: 10.2215/CJN.09421010
Keywords: minimal change disease, nephrotic syndrome, proteinuria, experimental models, focal segmental glomerulosclerosis
Citation: Bertelli R, Bonanni A, Caridi G, Canepa A and Ghiggeri GM (2018) Molecular and Cellular Mechanisms for Proteinuria in Minimal Change Disease. Front. Med. 5:170. doi: 10.3389/fmed.2018.00170
Received: 28 November 2017; Accepted: 15 May 2018;
Published: 11 June 2018.
Edited by:
Sandra Merscher, University of Miami, United StatesReviewed by:
Matthew Michael James Edey, Hull and East Yorkshire Hospitals NHS Trust, United KingdomCopyright © 2018 Bertelli, Bonanni, Caridi, Canepa and Ghiggeri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: G. M. Ghiggeri, Z21hcmNvZ2hpZ2dlcmlAZ2FzbGluaS5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.