 Knut A. Mosevoll
Knut A. Mosevoll Silje Johansen1
Silje Johansen1 Øystein Bruserud
Øystein Bruserud- 1Department of Medicine, Haukeland University Hospital, Bergen, Norway
- 2Section for Hematology, Institute of Clinical Science, University of Bergen, Bergen, Norway
The inflammatory response is a well-established part of, and a prerequisite for, venous thrombosis. To better understand the pathophysiology of venous thrombosis and to identify improved diagnostic biomarkers, further studies of the relationship between inflammation and coagulation are needed. We review previous studies concerning inflammatory biomarkers in venous thromboembolism, in particular cytokines, soluble adhesion molecules and matrix metalloproteases as predisposing, diagnostic and prognostic factors in venous thrombosis. Elevated cytokines and genetic alterations coding for cytokines are found in several patient cohorts which indicate that cytokines are involved as predisposing factors in venous thrombosis development. Increased levels of pro-inflammatory cytokines are detected both in animal models and in patients with acute venous thrombosis and clinical trials, although currently without evident diagnostic value. Adhesion molecules are crucial in the development of venous thrombosis, especially P-selectin seems important in initiating leukocyte accumulation and adhesion to endothelium for subsequent platelet accumulation. Several studies have demonstrated increased soluble P-selectin levels in patients with venous thrombosis, emphasizing its potential role as diagnostic marker and also as a therapeutic target. Matrix metalloproteases are essential effectors during venous thrombosis resolution and may impact vessel wall fibrosis, and together with their natural occurring inhibitors are crucial in acute and chronic thrombosis pathophysiology. Furthermore, studies in animal models of venous thrombosis have demonstrated anti-inflammatory treatment to be effective in terms of thrombus resolution and reduction of vessel wall damage, without increase in bleeding risk during the course of treatment. Thus, soluble mediators should be further investigated both as possible biomarkers and therapeutic targets in venous thromboembolic disease.
Introduction
Venous thromboembolism (VTE) is one of the most common haematological conditions, associated with increased morbidity and mortality, accounting for over 500,000 deaths per year in the European Union (1). Deep vein thrombosis (DVT) and pulmonary embolism (PE) could be difficult to diagnose, with undiagnosed VTE therefore representing an increased risk of death (1, 2). Improved risk stratification and diagnostic tools are important measures for VTE treatment and prevention (3).
Established risk factors for VTE include familial thrombophilia and acquired factors such as malignancies, previous VTE, reduced mobility, trauma or surgery, old age, pregnancy, heart failure, myocardial infarction, ischaemic stroke, obesity and use of oral contraceptives (4–6). Several of these risk factors represent inflammatory conditions (7). Emerging evidence suggests infection as a more important risk factor for VTE than previously recognized, and the coagulation system may play a major role in immune defence (8, 9). To better understand the pathophysiology of VTE and to identify improved diagnostic biomarkers for venous thrombosis, further studies on the relationship between inflammation and coagulation are needed (10). The term inflammation, derived from the Latin word inflammatio, is defined as a complex biological response of body tissues to harmful stimuli such as pathogens, damaged cells or irritants (9, 11–13). It is regarded as a protective response involving the innate and adaptive immune system, various blood cells, vessel wall, and a wide range of molecular mediators derived from the various cells involved in the inflammatory process. The main role of inflammation is to eliminate the initial cause of cell injury, to mediate clearance of necrotic cells and damaged tissues from the original insult and inflammatory process, and to initiate tissue repair. The classical local signs of inflammation are redness, heat, swelling, pain, and loss of function. In addition, severe inflammation also elicits systemic effects, probably mediated, at least in part, by circulating soluble mediators originating from the local inflammatory process.
The triad of increased coagluability, blood flow disturbance and vessel wall changes was postulated already by Rudolf Virchow (1821–1902) (14). However, increasing knowledge regarding the biological process bridging inflammation and coagulation have been established since Virchow's initial postulate. In this review we seek to further discuss the complex relationship between inflammation and VTE.
Pathophysiology of VTE
Endothelial cells are key regulators of the inflammatory response, as they: (i) form a physical barrier for blood cells and regulate the vascular permeability for immune cells, soluble proteins, electrolytes, and water; (ii) regulate the intravascular coagulation; (iii) regulate the vascular tone and blood pressure through initiation of vasoconstriction/vasodilatation; and finally (iv) release hormones and other soluble mediators, such as cytokines, that initiate and regulate the inflammatory process (15).
Endothelial cells activate, control, and direct leukocytes mainly through their cell surface expression of adhesion molecules and the release of chemotactic chemokines after activation. This enables immunocompetent cells to adhere to the endothelial cells and consequently cross the vessel wall by transendothelial migration (TEM), thus resulting in accumulation of immunocompetent cells at the inflammation site (16). Rapid endothelial activation (i.e., within minutes) is induced by stimuli like histamine and platelet-activating factor (PAF), initiating the expression of preformed adhesion molecules. In contrast, pro-inflammatory cytokines, such as interleukin (IL)-1β and Tumour Necrosis Factor (TNF)-α, induce a slower endothelial activation (i.e., within hours), involving transcriptional activation of adhering molecules and chemoattractants (16). The pathogenesis of VTE differs from that of arterial thrombosis, as venous thrombosis is initiated by intravascular events without exposure of the subendothelial structures (17). In a murine model of DVT inflamed endothelium increased the expression of a wide range of adhesion molecules that attach neutrophils and monocytes to the vessel wall, as an initial step in the formation of thrombus mainly consisting of leukocytes and few platelets (18). A complex interplay between monocytes, neutrophils, platelets, and the coagulation cascade leads to the formation of a thrombin-rich thrombus. Activated monocytes express TF that initiates the extrinsic pathway of the coagulation cascade. Thus, findings from the study suggest that TF expression by monocytes appears to be more important than endothelial expression of TF in triggering the coagulation cascade in DVT.
Both the leukocyte-endothelium and leukocyte-platelet interaction is a prerequisite for thrombosis initiation, shown in mouse-model studies of leukocyte integrin Mac-1, platelet C-type lectin-like receptor (CLEC-2), and vWF (19, 20, 21). Leukocyte integrin Mac-1 mediates adhesion of leukocytes-endothelium via the intracellular adhesion molecule-1 (ICAM-1), and adhesion of platelets via GPIbα, and targeting this adhesion-molecule could represent a possible therapeutic target (21). CLEC-2 is a receptor for podoplanin released from the endothelium during venous stenosis, and subsequently trigger thrombosis formation (20). CLEC-2 deficient mice are protected against thrombosis, and treatment of anti-podoplanin neutralizing antibodies reduces thrombosis size in animal models.
Moreover, neutropenia, genetic knockout of factor XII, and neutrophil extracellular traps (NETs) disintegration protected against the formation of DVT (22). This indicating neutrophil activation as a prerequisite for DVT formation, since neutrophils bind coagulation factor XII and release NETs, which together with platelets activate the coagulation cascade both through the intrinsic and extrinsic pathways (18). The host might prevent vascular occlusion through DNases that helps dissolve the NETs during an inflammatory response, thus control and prevent thrombus formation (23).
Soluble Mediators During Inflammation
Cytokines, adhesion molecules, and matrix metalloproteases (MMPs) are some of the key components involved in inflammation, see Figure 1 (12, 16, 24). Cytokines are important for cell-cell communication during inflammation, and they are highly heterogeneous and can be classified based on their function, structure, or based on their mechanism of action in the immune system, i.e., pro-inflammatory, anti-inflammatory or adaptive (12). The main pro-inflammatory cytokines belong to the IL-1, IL-6, IL-17, interferon, and TNF families. The IL-1 family is essential for initiation of the inflammatory cascade (25, 26), and cytokines in the IL-6 family have both immunoregulatory, as well as other systemic effects (27). The main anti-inflammatory cytokines include those in the IL-10 (28) and IL-12 families (29–32).
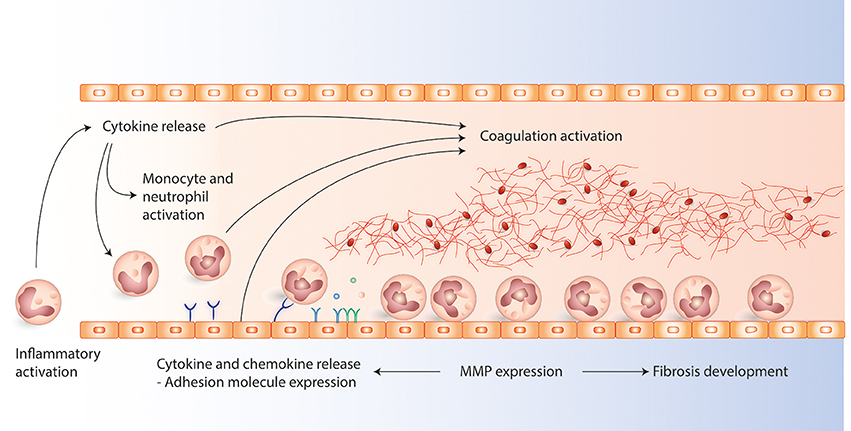
Figure 1. The figure present the main interactions between cytokines, chemokines, adhesion molecules, matric metalloproteases (MMPs), and coagulation activation involved in the pathophysiology during venous thrombosis development. Cytokines are early initiators of inflammation, ameliorating the interactions between leukocytes and endothelial cells. Activated leukocytes and endothelial cells express adhesion molecules which promotes leukocyte attachment to the endothelium which is important in the initiation and development of venous thrombosis. MMPs are involved in fibrosis of the vein walls modulation. In addition MMPs are important modulators of cytokines and adhesion molecules during inflammation as they can alter cytokines and contribute to adhesion molecule shedding.
Adhesion molecules are important mediators of cellular adhesion between leukocytes and endothelial cells and can exist in the membrane-bound as well as the biologically active soluble forms (16). The adhesion molecule family includes selectins (L-, E-, and P-selectins) and various immunoglobulin (Ig) family members [e.g., Vascular Cell Adhesion Molecule 1 (VCAM-1), ICAM-1/2]. E-selectin is expressed by endothelial cells, L-selectin by leukocytes, and P-selectin by endothelial cells and platelets, and they are all induced by pro-inflammatory mediators. Selectins are important for leukocyte rolling, and P-selectin is also involved in thrombus formation and intravascular coagulation during infection (9, 16). Ig family members are of particular importance in firm adhesion and trans-endothelial migration of leukocytes, and both ICAM-1 and VCAM-1 are expressed by endothelial cells (33).
Finally, MMPs have emerged not only as molecules involved in modelling extracellular tissue, but also as important regulators of inflammatory responses, e.g., through their activation and modulation of pro-inflammatory cytokines (24). Matrix metalloproteases (MMPs) and protease inhibitors both interact with cytokines and adhesion molecules at various levels. Hence, one may argue that these mediators should be regarded as functional parts of the cytokine network. MMPs are zinc-dependent enzymes belonging to the metzincin superfamily of zinc endopeptidases that, to date, comprise 24 mammalian proteases (24, 34, 35).
Cytokines and VTE
A range of risk factors for VTE include inflammatory conditions, gives rise to the hypothesis that elevated inflammatory mediators are risk factors for DVT and post thrombotic syndrome (PTS). Levels of several cytokines are found elevated as part of VTE pathobiology (Table 1), and findings from selected human and animal studies showing the importance of adhesion molecules in VTE are presented in Table 1.
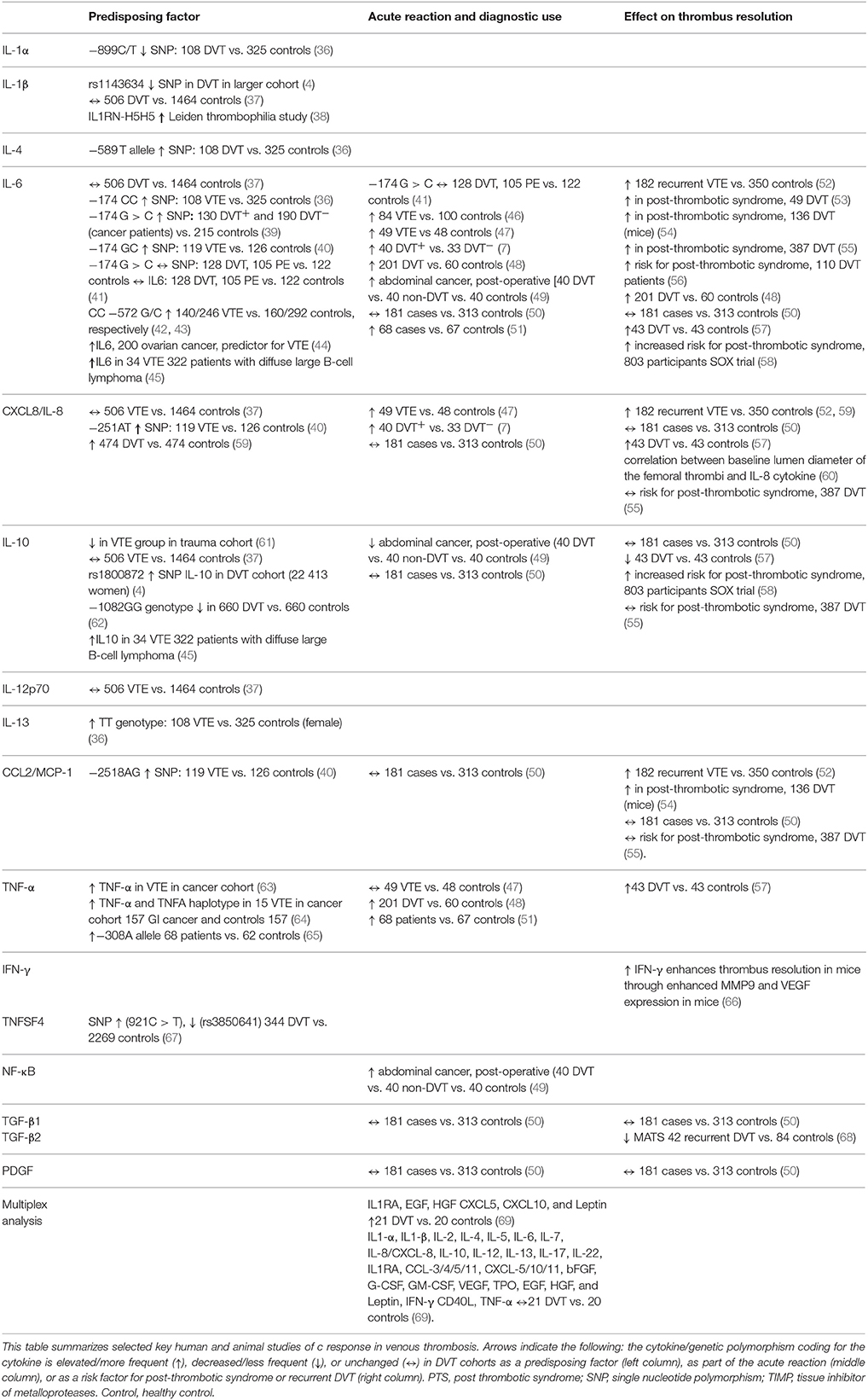
Table 1. Cytokines as predisposing factors, diagnostic markers, and prognostic markers in venous thrombosis.
Cytokines in Inflammation and Predisposition to VTE
Single nucleotide polymorphisms (SNPs) affecting cytokine genes are associated with an increased risk, and others with a reduced risk, of VTE (Table 1) (4, 67, 36, 40, 39). IL-1 cytokines are early inflammatory mediators. SNPs in IL-1α and IL-1β have shown to be associated with reduced risk of DVT, while another SNP for IL-1β is associated with increased risk of DVT (38). IL-6 is an inflammatory mediator later in the cascade, and is maybe the most studied cytokine during inflammation. Also certain SNPs of IL-6 has been associated as a risk factor for VTE (36).
A number of small case-control studies demonstrated increased levels of pro-inflammatory, or decreased levels of anti-inflammatory, cytokines in patients at risk for DVT (61, 63, 45). Although in a larger, prospective population-based case-control study, no such associations were found (37).
Increased inflammation is also a common feature of most of the epidemiologically proven risk factors (70). As previously genetic factors affecting the coagulation system (e.g., factor V, Leiden, and prothrombin mutations) are predisposing risk factors for VTE (4, 41, 71, 72). However, family history in itself is an independent risk factor that could be more useful than genetic testing when assessing the risk for thrombosis (73). This indicates that genetic predispositions are multifactorial and a complex combinations of several genetic traits, in which inflammation could represent a part of the explanation.
Cytokines as Part of the Acute Reaction and Their Possible Diagnostic Use
Increased levels of pro-inflammatory cytokines were detected in animal models of acute venous thrombosis (74). Several inflammatory cytokines like IL-6, IL-8/CXCL-8, and TNF-α are increased during acute thrombosis, although with no evident diagnostic value (7, 40, 59, 47, 46, 49). The focus of most of the studies has been the main cytokines IL-1, IL-6, IL-8/CXCL-8, and IL-10. Increased inflammatory mediators during the clinical course of thrombosis are in line with clinical signs as redness and swelling. However, even broader inflammatory profiles have limited diagnostic value as most differential diagnoses also show extensive inflammatory activity (69).
Cytokines and Thrombus Resolution
Inflammation is considered as an essential response during venous thrombosis formation and resolution, and increased levels of inflammatory cytokines seem to have prognostic impact (56, 55, 53). IL-6 could represent a therapeutic target in the prevention of post-thrombotic syndrome, as suggested by a previous study using an animal model of DVT (54).
In conclusion, cytokines and play a role as predisposing factors for VTE, however they are not established as important risk factors. Elevated cytokines are found in several studies of VTE patients, although it is difficult to draw clear conclusion of causality of the cytokines role in venous thrombosis in these patient cohorts.
Adhesion Molecules and VTE
Adhesion molecules are part of the formation of the inflammatory response (Figure 1). The P-selectin stored in platelets and endothelial cells, has been proposed as a key molecule in thrombosis and haemostasis (75, 76). P-selectin has been found upregulated as early as 6 h after thrombus induction (77). The binding of P-selectin to its receptor P-selectin ligand-1(PSGL-1) present on the surface of leukocytes and platelets, initiates pro-coagulatory mechanisms (77, 78). Findings from selected human and animal studies showing the importance of adhesion molecules in DVT are presented in Table 2.
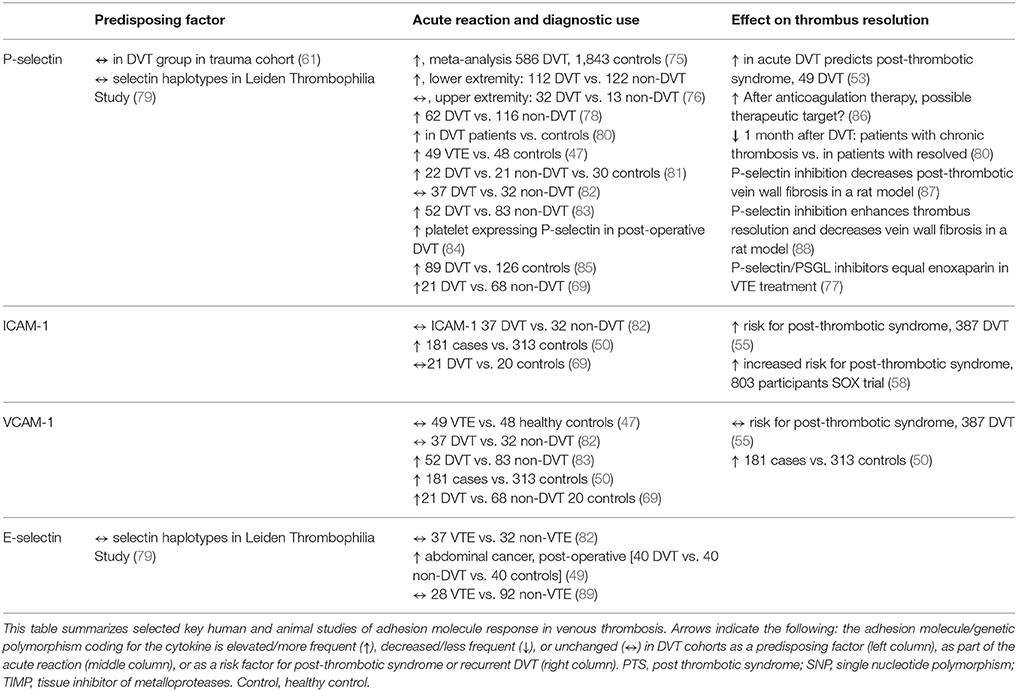
Table 2. Adhesion molecules as predisposing factors, diagnostic markers and prognostic markers in venous thrombosis.
Predisposing Factors for VTE
The gene for coagulation factor V is genetically closely linked to the genes encoding the selectins. In a study by Uitte de Willge et al. examination of the haplotypes of E-selectin and P-selectin demonstrated no association with DVT risk (79). Proctor et al. observed that serum P-selectin (sP-selectin) and IL-10 had a consistent association, during the first week after an injury, with the presence or absence of VTE. The sP-selectin level difference was not statistically significant between the groups. Those patients who developed VTE had a higher sP/IL-10 ratio, where lower levels of IL-10 accounted for the ratio-difference, statistically significant to those without VTE (61).
The Acute Phase Reaction and Diagnostic Use
Currently the biomarker used in VTE diagnostic is D-dimer, which is a non-specific biomarker of VTE, being a fibrin degradation product (76). sP-selectin has been a candidate as replacement of D-dimer as a diagnostic marker. An early study published in 2000 demonstrated increased sP-selectin levels in thrombosis patients (85). The two most promising studies demonstrated high positive predicative value (PPV) of 91/100%, and a specificity of 97.5/96% for establishing the diagnosis of DVT, with Wells score ≥ and sP-selectin ≥90 ng/ml (76, 78). The first study suggested that P-selectin may be both more specific and sensitive than D-dimer as a diagnostic marker at least in specific subsets of patients (78). The subsequent study also demonstrated higher specificity for sP-selectin, however D-dimer in combination with Well's score performed as well as sP-selectin combined with Well's score in ruling out DVT (76).
Changes in platelet expression of P-selectin may also be used as a diagnostic marker of susceptibility to DVT in high risk surgical procedures, demonstrated in a study of female patients undergoing a total knee arthroplasty (84). A small prospective study published in 2003 examined ICAM-1, VCAM-1, E-selectin, and P-selectin in the diagnostic of DVT, and no significant difference were found between the concentrations in patients with DVT vs. controls (82). Increased levels of E-selectin have also been found as an independent risk factor associated DVT in postoperative patients with abdominal malignancy (49), and VCAM levels seem to be higher in DVT patients (83).
Adhesion Molecules and Thrombus Resolution
Post-deep vein thrombosis venous insufficiency is characterized by vein wall fibrosis, and is central in the development of PTS, where the long term outcome is influenced by early resolution of the thrombus (80). The inflammatory response has been found to affect both thrombus resolution and PTS development (55, 77, 87). P-selectin and D-dimer have been found elevated in the acute phase of DVT, and decrease significantly during the first month (86). The recanalization is inversely related to coagulation markers and fibrinolytic inhibition (53). Increased levels of P-selectin may represent a hypercoagulable state, although it remains unclear if it precedes and promotes thrombosis or is a consequence of thrombosis (86). In patients with a more serve inflammatory response, the endogenic fibrinolysis is inhibited and coagulation stimulated and therefore thrombus resolution is delayed. This is demonstrated by an increased level of IL-6 and P-selectin in the chronic phase, indicating a worse prognosis (53). However, sP-selectin levels are higher 1 month after a thrombosis in patients who resolved their DVT, implying a more active thrombus metabolism in these patients compared to those with chronic thrombosis (80).
A cross-sectional study in a chronic phase after pregnancy-related DVT demonstrated higher levels of ICAM-1 and VCAM-1 in cases compared to controls. However, after univariate analyses these markers where not significantly associated with PTS (50). Though, patients who develop PTS appear to have higher ICAM-1 level then those patients who do not develop PTS (55).
P-selectin is strongly influenced by treatment of oral anticoagulation, illustrated by increased levels within few months after discontinuation of vitamin-K antagonists. Two rodent model studies showed that P-selectin inhibition decreases post DVT vein wall fibrosis (87, 88). It is assumed that the effect is associated with decreased profibrotic mediators, independent of changes in thrombus masses (87). A meta-analysis of non-human primate models confirmed a significant difference in the effect of vein-reopening and inflammation when comparing P-selectin antagonism and saline (78). Observations that thrombus formation and extension may be inhibited, and enhanced resolution when a DVT is treated with P-selectin antagonists highlight the importance of sP-selectin in thrombosis (75, 88).
In conclusion, adhesion molecules are vital for thrombus formation. P-selectin is a possible diagnostic marker for DVT, and might also represent a therapeutic target.
Matrix Metalloproteases and VTE
MMPs are important in inflammatory responses through their regulation of inflammatory mediators, as well as in maintenance of the function and integrity of physical barriers (Figure 1) (24). Animal models have suggested that MMPs are important effectors during VTE resolution and reduce vessel wall fibrosis (66, 90), thus indicating that MMPs represent potential therapeutic targets (54). Findings from selected human and animal studies showing the importance of MMPs in DVT are presented in Table 3.
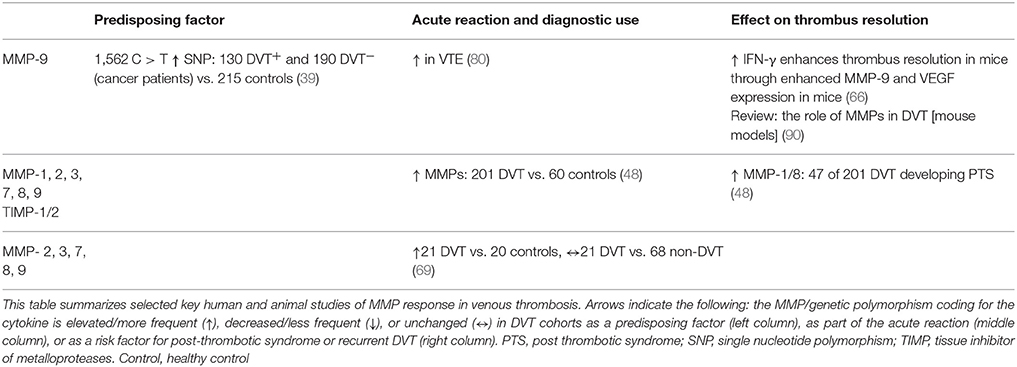
Table 3. Matrix metalloproteases as predisposing factors, diagnostic markers and prognostic markers in venous thrombosis.
The Role of MMPs as Predisposing Factors for VTE
SNPs affecting MMP-9 and IL-6 have been evaluated as predisposing factors for DVT in cancer patients (39). When comparing healthy controls with cancer patients with DVT, both SNPs were associated to DVT. Distribution of the SNPs was similar between healthy controls and DVT negative cancer cases. The results indicate that the GG and CC genotypes, respectively for both SNPs, are associated with increased risk of DVT in cancer patients by inducing the release of IL-6 with subsequent increment of MMP-9.
MMPs in Acute VTE and Post Thrombotic Syndrome
Studying PTS, one study aimed to quantify the change in vein wall thickness in patients who failed to resolve DVT within 6 months, and whether differences in levels of inflammatory proteins associated with venous remodelling (80). Results demonstrated increased levels of MMP-9 antigen in thrombosed patients compared with controls. Other biomarkers such as D-dimer and P-selectin were higher in thrombosed patients at diagnosis, although not significantly different at 6 months. P-selectin gene expression were higher in leukocytes from patients with chronic DVT compared with those who resolved within 1 month after diagnosis.
In another study the expression of inflammatory biomarkers in the early phase of DVT and their correlation with the onset of PTS was evaluated (48). Samples were harvested from 201 patients after their first episode of DVT over a period for 18 months, and from 60 patients without DVT. Analyses of inflammatory biomarkers demonstrated high plasma levels of MMPs and cytokines during the acute phase after DVT. Patients with PTS demonstrated higher levels of MMP-1 and MMP-8 than patients without PTS, implying a close relationship between DVT, the individual risk of PTS and specific biomarkers such as MMPs.
In conclusion, few studies exploring the role of MMPs in VTE are performed. MMPs are elevated during VTE and PTS, although also during other inflammatory conditions, and so far not proven value as diagnostic markers (69).
Conclusions and Future Perspective
Inflammatory mediators are part of several cascades in the pathophysiology of VTE; predisposing factors, initiation and prolongation of acute thrombosis and resolution. Especially cytokines have been implicated as a predisposing factor in several studies, while adhesion molecules and MMPs are to a lesser degree studied (Tables 1–3). High quality studies demonstrating the effect of inflammatory mediators as predisposing factors of thrombosis are difficult to design. However, the performed studies combined with the knowledge that most risk factors of VTE involves inflammation, indicate an important role of inflammatory mediators as risk factors for thrombosis. The complexity of the genetic predisposition of most human disease hampers to decipher the exact impact of different genetic and environmental factors (91). The focus on family history instead of genetic analysis in clinical assessment is a reasonable approach given this genetic complexity (73, 92). Until further studies have given utterly information, genetic tests involving inflammatory mediators should not be part of routine clinical practice (92).
Several case-control studies and selected animal studies clearly demonstrate the role of the inflammatory response in VTE (Tables 1–3). Despite this, few inflammatory mediators have shown utility in diagnostics compared with traditional biomarkers; D-dimer and CRP being the most important ones. The only inflammatory marker shown almost as helpful as D-dimer in diagnostic work up is sP-selectin, although has so far not proven to add definitive diagnostic value. As prognostic factors, the studies presented in Tables 1–3 are indicating higher inflammatory mediator levels can be used to identify patients of risk of PTS. Larger thrombotic burden is a reasonable explanation of these finding.
The intertwined effect of inflammation and coagulation is an important aspect of treatment of VTE. Inflammation as a target is responsible for parts of the effects of low molecular heparin as it is reducing TNF-α expression (93). Furthermore, statins have anti-inflammatory effects believed to be favourable for thrombosis resolution (94–97). Other modulations of the inflammatory response are explored in animal models as possible treatments. Increased levels of cytokines such as IL-8, CCL2 are favourable for thrombus resolution (98, 99), and impaired inflammatory response by cytokine receptor deletion or neutropenia also reduces thrombus resolution (100, 101). This leads to the assumption that the inflammatory response is a prerequisite not only for the thrombus formation, but also the resolution, although the results from using cytokine stimulation are conflicting (102). However, the resolution is also dependent on the leukocytes as neutropenia seems to reduce the capability for thrombus resolution (87).
Future research, both basic laboratory research including studies in murine models, in addition to translational and clinical research, are important to completely bridging the association between inflammation and VTE. This includes a better understanding of the inflammatory aspects of thrombosis and the pathophysiology role of mediator molecules, herein their potential role as diagnostic and prognostic biomarkers. The later will be of special interest for selective patient groups, there biomarkers have demonstrated to be of limited values, including patients with malignant disease and patients with other inflammatory comorbidities. Until then, good clinical judgments combined with established biomarkers and radiological examinations, are still the basic tools in diagnostic approaches for patients with VTE.
Author Contributions
KM: main responsibility for writing; SJ: responsibility for writing of the parts regarding Adhesion Molecules; ØW: responsibility for writing of the parts regarding Matrix Metalloproteases; IN: introduction and illustrations; ØB: main Idea of the publication; HR: corresponding author.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The manuscript is partly based on the Thesis Cytokine profiles in inflammation, by KM, published at the University of Bergen in 2017.
References
1. Cohen AT, Agnelli G, Anderson FA, Arcelus JI, Bergqvist D, Brecht JG etal. Venous thromboembolism (VTE) in Europe. The number of VTE events and associated morbidity and mortality. Thromb Haemost. (2007) 98:756–64. doi: 10.1160/TH07-03-0212
2. Vaitkus PT, Leizorovicz A, Cohen AT, Turpie AG, Olsson CG, Goldhaber SZ etal. Mortality rates and risk factors for asymptomatic deep vein thrombosis in medical patients. Thromb Haemost. (2005) 93:76–9. doi: 10.1160/TH04-05-0323
3. Lester W, Freemantle N, Begaj I, Ray D, Wood J, Pagano D. Fatal venous thromboembolism associated with hospital admission: a cohort study to assess the impact of a national risk assessment target. Heart (2013) 99:1734–9. doi: 10.1136/heartjnl-2013-304479
4. Zee RY, Glynn RJ, Cheng S, Steiner L, Rose L, Ridker PM. An evaluation of candidate genes of inflammation and thrombosis in relation to the risk of venous thromboembolism: The Women's Genome Health Study. Circ Cardiovasc Genet. (2009) 2:57–62. doi: 10.1161/CIRCGENETICS.108.801969
5. Barbar S, Noventa F, Rossetto V, Ferrari A, Brandolin B, Perlati M etal. A risk assessment model for the identification of hospitalized medical patients at risk for venous thromboembolism: the Padua Prediction Score. J Thromb Haemost. (2010) 8:2450–7. doi: 10.1111/j.1538-7836.2010.04044.x
6. Kahn SR, Lim W, Dunn AS, Cushman M, Dentali F, Akl EA etal. Prevention of VTE in nonsurgical patients: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest (2012) 141:e195S−226S. doi: 10.1378/chest.11-2296
7. Roumen-Klappe EM, Den Heijer M, Van Uum SH, Van Der Ven-Jongekrijg J, Van Der Graaf F, Wollersheim H. Inflammatory response in the acute phase of deep vein thrombosis. J Vasc Surg. (2002) 35:701–6. doi: 10.1067/mva.2002.121746
8. Duan Q, Gong Z, Song H, Wang L, Yang F, Lv W etal. Symptomatic venous thromboembolism is a disease related to infection and immune dysfunction. Int J Med Sci. (2012) 9:453–61. doi: 10.7150/ijms.4453
9. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
10. Lambert MP, Sachais BS, Kowalska MA. Chemokines and thrombogenicity. Thromb Haemost. (2007) 97:722–9. doi: 10.1160/TH07-01-0046
11. Mahindra A, Hideshima T, Anderson KC. Multiple myeloma: biology of the disease. Blood Rev. (2010) 24(Suppl. 1):S5–11. doi: 10.1016/S0268-960X(10)70003-5
12. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta (2014) 1843:2563–82. doi: 10.1016/j.bbamcr.2014.05.014
13. Wiersinga WJ, Leopold SJ, Cranendonk DR, Van Der Poll T. Host innate immune responses to sepsis. Virulence (2014) 5:36–44. doi: 10.4161/viru.25436
14. Samuels PB, Webster DR. The role of venous endothelium in the inception of thrombosis. Ann Surg. (1952) 136:422–38.
15. Khakpour S, Wilhelmsen K, Hellman J. Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun. (2015) 21:827–46. doi: 10.1177/1753425915606525
16. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity (2014) 41:694–707. doi: 10.1016/j.immuni.2014.10.008
17. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. (2008) 359:938–49. doi: 10.1056/NEJMra0801082
18. Von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M etal. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
19. Brill A, Fuchs TA, Chauhan AK, Yang JJ, De Meyer SF, Kollnberger M etal. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood (2011) 117:1400–7. doi: 10.1182/blood-2010-05-287623
20. Payne H, Ponomaryov T, Watson SP, Brill A. Mice with a deficiency in CLEC-2 are protected against deep vein thrombosis. Blood (2017) 129:2013–20. doi: 10.1182/blood-2016-09-742999
21. Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, A Soloviev D etal. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbalpha. Nat Commun. (2017) 8:15559. doi: 10.1038/ncomms15559
22. Manfredi AA, Ramirez GA, Rovere-Querini P, Maugeri N. The Neutrophil's choice: phagocytose vs make neutrophil extracellular traps. Front Immunol. (2018) 9:288. doi: 10.3389/fimmu.2018.00288
23. Jimenez-Alcazar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT etal. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science (2017) 358:1202–6. doi: 10.1126/science.aam8897
24. Nissinen L, Kahari VM. Matrix metalloproteinases in inflammation. Biochim Biophys Acta (2014) 1840:2571–80. doi: 10.1016/j.bbagen.2014.03.007
25. Dinarello CA, Van Der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. (2013) 25:469–84. doi: 10.1016/j.smim.2013.10.008
26. Narayanan KB, Park HH. Toll/interleukin-1 receptor (TIR) domain-mediated cellular signaling pathways. Apoptosis (2015) 20:196–209. doi: 10.1007/s10495-014-1073-1
27. Schaper F, Rose-John S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. (2015) 26:475–87. doi: 10.1016/j.cytogfr.2015.07.004
28. Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. (2012) 13:722–8. doi: 10.1038/ni.2366
29. Dennis KL, Blatner NR, Gounari F, Khazaie K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr Opin Oncol. (2013) 25:637–45. doi: 10.1097/CCO.0000000000000006
30. Mackenzie KF, Pattison MJ, Arthur JS. Transcriptional regulation of IL-10 and its cell-specific role in vivo. Crit Rev Immunol. (2014) 34:315–45. doi: 10.1615/CritRevImmunol.2014010694
31. Walter MR. The molecular basis of IL-10 function: from receptor structure to the onset of signaling. Curr Top Microbiol Immunol. (2014) 380:191–212. doi: 10.1007/978-3-662-43492-5_9
32. Ni G, Wang T, Walton S, Zhu B, Chen S, Wu X etal. Manipulating IL-10 signalling blockade for better immunotherapy. Cell Immunol. (2015) 293:126–9. doi: 10.1016/j.cellimm.2014.12.012
33. Zonneveld R, Martinelli R, Shapiro NI, Kuijpers TW, Plotz FB, Carman CV. Soluble adhesion molecules as markers for sepsis and the potential pathophysiological discrepancy in neonates, children and adults. Crit Care (2014) 18:204. doi: 10.1186/cc13733
34. Hatfield KJ, Reikvam H, Bruserud O. The crosstalk between the matrix metalloprotease system and the chemokine network in acute myeloid leukemia. Curr Med Chem. (2010) 17:4448–61. doi: 10.2174/092986710794183033
35. Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. (2014) 13:904–27. doi: 10.1038/nrd4390
36. Beckers MM, Ruven HJ, Haas FJ, Doevendans PA, Ten Cate H, Prins MH etal. Single nucleotide polymorphisms in inflammation-related genes are associated with venous thromboembolism. Eur J Intern Med. (2010) 21:289–92. doi: 10.1016/j.ejim.2010.04.001
37. Christiansen SC, Naess IA, Cannegieter SC, Hammerstrom J, Rosendaal FR, Reitsma PH. Inflammatory cytokines as risk factors for a first venous thrombosis: a prospective population-based study. PLoS Med. (2006) 3:e334. doi: 10.1371/journal.pmed.0030334
38. Van Minkelen R, De Visser MC, Houwing-Duistermaat JJ, Vos HL, Bertina RM, Rosendaal FR. Haplotypes of IL1B, IL1RN, IL1R1, and IL1R2 and the risk of venous thrombosis. Arterioscler Thromb Vasc Biol. (2007) 27:1486–91. doi: 10.1161/ATVBAHA.107.140384
39. Malaponte G, Polesel J, Candido S, Sambataro D, Bevelacqua V, Anzaldi M etal. IL-6-174 G > C and MMP-9-1562 C > T polymorphisms are associated with increased risk of deep vein thrombosis in cancer patients. Cytokine (2013) 62:64–9. doi: 10.1016/j.cyto.2013.02.017
40. Matos MF, Lourenco DM, Orikaza CM, Bajerl JA, Noguti MA, Morelli VM. The role of IL-6, IL-8 and MCP-1 and their promoter polymorphisms IL-6−174GC, IL-8−251AT and MCP-1−2518AG in the risk of venous thromboembolism: a case-control study. Thromb Res. (2011) 128:216–20. doi: 10.1016/j.thromres.2011.04.016
41. Vormittag R, Hsieh K, Kaider A, Minar E, Bialonczyk C, Hirschl M etal. Interleukin-6 and interleukin-6 promoter polymorphism (-174) G > C in patients with spontaneous venous thromboembolism. Thromb Haemost. (2006) 95:802–6. doi: 10.1160/TH05-12-0816
42. Mahemuti A, Abudureheman K, Aihemaiti X, Hu XM, Xia YN, Tang BP etal. Association of interleukin-6 and C-reactive protein genetic polymorphisms levels with venous thromboembolism. Chin Med J. (2012) 125:3997–4002. doi: 10.3760/cma.j.issn.0366-6999.2012.22.016
43. Yadav U, Mahemuti A, Hu X, Abudureheman K, Xia Y, Tang B etal. Single nucleotide polymorphisms in interleukin-6 and their association with venous thromboembolism. Mol Med Rep. (2015) 11:4664–70. doi: 10.3892/mmr.2015.3248
44. Matsuo K, Hasegawa K, Yoshino K, Murakami R, Hisamatsu T, Stone RL etal. Venous thromboembolism, interleukin-6 and survival outcomes in patients with advanced ovarian clear cell carcinoma. Eur J Cancer (2015) 51:1978–88. doi: 10.1016/j.ejca.2015.07.012
45. Lim SH, Woo SY, Kim S, Ko YH, Kim WS, Kim SJ. Cross-sectional study of patients with diffuse large b-cell lymphoma: assessing the effect of host status, Tumor Burden, and inflammatory activity on venous thromboembolism. Cancer Res Treat. (2016) 48:312–21. doi: 10.4143/crt.2014.266
46. Matos MF, Lourenco DM, Orikaza CM, Gouveia CP, Morelli VM. Abdominal obesity and the risk of venous thromboembolism among women: a potential role of interleukin-6. Metab Syndr Relat Disord. (2013) 11:29–34. doi: 10.1089/met.2012.0077
47. Jezovnik MK, Poredos P. Idiopathic venous thrombosis is related to systemic inflammatory response and to increased levels of circulating markers of endothelial dysfunction. Int Angiol. (2010) 29:226–31.
48. De Franciscis S, Gallelli L, Amato B, Butrico L, Rossi A, Buffone G etal. Plasma MMP and TIMP evaluation in patients with deep venous thrombosis: could they have a predictive role in the development of post-thrombotic syndrome? Int Wound J. (2016) 13:1237–45. doi: 10.1111/iwj.12489
49. Du T, Tan Z. Relationship between deep venous thrombosis and inflammatory cytokines in postoperative patients with malignant abdominal tumors. Braz J Med Biol Res. (2014) 47:1003–7. doi: 10.1590/1414-431X20143695
50. Wik HS, Jacobsen AF, Mowinckel MC, Sandset PM. The role of inflammation in post-thrombotic syndrome after pregnancy-related deep vein thrombosis: a population-based, cross-sectional study. Thromb Res. (2016) 138:16–21. doi: 10.1016/j.thromres.2015.12.014
51. Bittar LF, Mazetto Bde M, Orsi FL, Collela MP, De Paula EV, Annichino-Bizzacchi JM. Long-term increased factor VIII levels are associated to interleukin-6 levels but not to post-thrombotic syndrome in patients with deep venous thrombosis. Thromb Res. (2015) 135:497–501. doi: 10.1016/j.thromres.2014.12.024
52. Van Aken BE, Den Heijer M, Bos GM, Van Deventer SJ, Reitsma PH. Recurrent venous thrombosis and markers of inflammation. Thromb Haemost. (2000) 83:536–9. doi: 10.1055/s-0037-1613858
53. Jezovnik MK, Poredos P. Factors influencing the recanalisation rate of deep venous thrombosis. Int Angiol. (2012) 31:169–75.
54. Wojcik BM, Wrobleski SK, Hawley AE, Wakefield TW, Myers D. D. Jr., Diaz JA. Interleukin-6: a potential target for post-thrombotic syndrome. Ann Vasc Surg. (2011) 25:229–39. doi: 10.1016/j.avsg.2010.09.003
55. Shbaklo H, Holcroft CA, Kahn SR. Levels of inflammatory markers and the development of the post-thrombotic syndrome. Thromb Haemost. (2009) 101:505–12. doi: 10.1160/TH08-08-0511
56. Roumen-Klappe EM, Janssen MC, Van Rossum J, Holewijn S, Van Bokhoven MM, Kaasjager K etal. Inflammation in deep vein thrombosis and the development of post-thrombotic syndrome: a prospective study. J Thromb Haemost. (2009) 7:582–7. doi: 10.1111/j.1538-7836.2009.03286.x
57. Jezovnik MK, Fareed J, Poredos P. Patients With a History of Idiopathic Deep Venous thrombosis have long-term increased levels of inflammatory markers and markers of endothelial damage. Clin Appl Thromb Hemost. (2017) 23:124–31. doi: 10.1177/1076029616670259
58. Rabinovich A, Cohen JM, Cushman M, Wells PS, Rodger MA, Kovacs MJ etal. Inflammation markers and their trajectories after deep vein thrombosis in relation to risk of post-thrombotic syndrome. J Thromb Haemost. (2015) 13:398–408. doi: 10.1111/jth.12814
59. Van Aken BE, Reitsma PH, Rosendaal FR. Interleukin 8 and venous thrombosis: evidence for a role of inflammation in thrombosis. Br J Haematol. (2002) 116:173–7. doi: 10.1046/j.1365-2141.2002.03245.x
60. Montes-Worboys A, Arellano E, Elias T, Leon J, Rodriguez-Portal JA, Otero R. Residual thrombosis after a first episode of proximal deep venous thrombosis. Blood Coagul Fibrinolysis (2013) 24:335–60. doi: 10.1097/MBC.0b013e32835c32ef
61. Proctor MC, Sullivan V, Zajkowski P, Wolk SW, Pomerantz RA, Wakefield TW etal. A role for interleukin-10 in the assessment of venous thromboembolism risk in injured patients. J Trauma (2006) 60:147–51. doi: 10.1097/01.ta.0000197180.79965.bc
62. Tang B, Chen YK, Luo WJ, Fu J, Sun JM. Association between interleukin-10−1082A/G,−819C/T and−592C/A polymorphisms with deep venous thrombosis. Hum Immunol. (2014) 75:203–7. doi: 10.1016/j.humimm.2013.12.013
63. Ferroni P, Riondino S, Portarena I, Formica V, La Farina F, Martini F etal. Association between increased tumor necrosis factor alpha levels and acquired activated protein C resistance in patients with metastatic colorectal cancer. Int J Colorectal Dis. (2012) 27:1561–7. doi: 10.1007/s00384-012-1493-8
64. Roselli M, Ferroni P, Rolfo C, Peeters M, Palmirotta R, Formica V etal. TNF-alpha gene promoter polymorphisms and risk of venous thromboembolism in gastrointestinal cancer patients undergoing chemotherapy. Ann Oncol. (2013) 24:2571–5. doi: 10.1093/annonc/mdt251
65. Horakova K, Chylkova A, Kolorz M, Bartosova L, Pechacek V, Starostka D etal. Polymorphism G-308A in the promoter of the tumor necrosis factor-alpha gene and its association with the risk of venous thromboembolism. Blood Coagul Fibrinolysis (2012) 23:316–9. doi: 10.1097/MBC.0b013e3283527506
66. Nosaka M, Ishida Y, Kimura A, Kuninaka Y, Inui M, Mukaida N etal. Absence of IFN-gamma accelerates thrombus resolution through enhanced MMP-9 and VEGF expression in mice. J Clin Invest. (2011) 121:2911–20. doi: 10.1172/JCI40782
67. Malarstig A, Eriksson P, Rose L, Diehl KA, Hamsten A, Ridker PM etal. Genetic variants of tumor necrosis factor superfamily, member 4 (TNFSF4), and risk of incident atherothrombosis and venous thromboembolism. Clin Chem. (2008) 54:833–40. doi: 10.1373/clinchem.2007.096479
68. Memon AA, Sundquist K, Wang X, Svensson PJ, Sundquist J, Zoller B. Transforming growth factor (TGF)-beta levels and unprovoked recurrent venous thromboembolism. J Thromb Thrombolysis (2014) 38:348–54. doi: 10.1007/s11239-013-1047-0
69. Mosevoll KA, Lindas R, Tvedt TH, Bruserud O, Reikvam H. Altered plasma levels of cytokines, soluble adhesion molecules and matrix metalloproteases in venous thrombosis. Thromb Res. (2015) 136:30–9. doi: 10.1016/j.thromres.2015.04.002
70. Heit JA, Spencer FA, White RH. The epidemiology of venous thromboembolism. J Thromb Thrombolysis (2016) 41:3–14. doi: 10.1007/s11239-015-1311-6
71. Morange PE, Oudot-Mellakh T, Cohen W, Germain M, Saut N, Antoni G etal. KNG1 Ile581Thr and susceptibility to venous thrombosis. Blood (2011) 117:3692–4. doi: 10.1182/blood-2010-11-319053
72. Croles FN, Nasserinejad K, Duvekot JJ, Kruip MJ, Meijer K, Leebeek FW. Pregnancy, thrombophilia, and the risk of a first venous thrombosis: systematic review and bayesian meta-analysis. BMJ (2017) 359:j4452. doi: 10.1136/bmj.j4452
73. Bezemer ID, Van Der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. The value of family history as a risk indicator for venous thrombosis. Arch Intern Med. (2009) 169:610–5. doi: 10.1001/archinternmed.2008.589
74. Wakefield TW, Greenfield LJ, Rolfe MW, Delucia A. III, Strieter RM, Abrams GD etal. Inflammatory and procoagulant mediator interactions in an experimental baboon model of venous thrombosis. Thromb Haemost. (1993) 69:164–72.
75. Antonopoulos CN, Sfyroeras GS, Kakisis JD, Moulakakis KG, Liapis CD. The role of soluble P selectin in the diagnosis of venous thromboembolism. Thromb Res. (2013) 133:17–24. doi: 10.1016/j.thromres.2013.08.014
76. Vandy FC, Stabler C, Eliassen AM, Hawley AE, Guire KE, Myers DD etal. Soluble P-selectin for the diagnosis of lower extremity deep venous thrombosis. J Vasc Surg Venous Lymphat Disord. (2013) 1:117–25. doi: 10.1016/j.jvsv.2012.09.001
77. Ramacciotti E, Myers DD Jr, Wrobleski SK, Deatrick KB, Londy FJ, Rectenwald JE etal. P-selectin/ PSGL-1 inhibitors versus enoxaparin in the resolution of venous thrombosis: a meta-analysis. Thromb Res. (2010) 125:e138–42. doi: 10.1016/j.thromres.2009.10.022
78. Ramacciotti E, Blackburn S, Hawley AE, Vandy F, Ballard-Lipka N, Stabler C etal. Evaluation of soluble P-selectin as a marker for the diagnosis of deep venous thrombosis. Clin Appl Thromb Hemost. (2011) 17:425–31. doi: 10.1177/1076029611405032
79. Uitte De Willige S, De Visser MC, Vos HL, Houwing-Duistermaat JJ, Rosendaal FR, Bertina RM. Selectin haplotypes and the risk of venous thrombosis: influence of linkage disequilibrium with the factor V Leiden mutation. J Thromb Haemost. (2008) 6:478–85. doi: 10.1111/j.1538-7836.2007.02879.x
80. Deatrick KB, Elfline M, Baker N, Luke CE, Blackburn S, Stabler C etal. Postthrombotic vein wall remodeling: preliminary observations. J Vasc Surg. (2011) 53:139–46. doi: 10.1016/j.jvs.2010.07.043
81. Rectenwald JE, Myers DD Jr, Hawley AE, Longo C, Henke PK, Guire KE etal. D-dimer, P-selectin, and microparticles: novel markers to predict deep venous thrombosis. A pilot study. Thromb Haemost (2005) 94:1312–7. doi: 10.1160/TH05-06-0426
82. Bucek RA, Reiter M, Quehenberger P, Minar E, Baghestanian M. The role of soluble cell adhesion molecules in patients with suspected deep vein thrombosis. Blood Coagul Fibrinolysis (2003) 14:653–7. doi: 10.1097/00001721-200310000-00006
83. Bozic M, Blinc A, Stegnar M. D-dimer, other markers of haemostasis activation and soluble adhesion molecules in patients with different clinical probabilities of deep vein thrombosis. Thromb Res. (2002) 108:107–14. doi: 10.1016/S0049-3848(03)00007-0
84. Yang LC, Wang CJ, Lee TH, Lin FC, Yang BY, Lin CR etal. Early diagnosis of deep vein thrombosis in female patients who undergo total knee arthroplasty with measurement of P-selectin activation. J Vasc Surg. (2002) 35:707–12. doi: 10.1067/mva.2002.121852
85. Blann AD, Noteboom WM, Rosendaal FR. Increased soluble P-selectin levels following deep venous thrombosis: cause or effect? Br J Haematol. (2000) 108:191–3. doi: 10.1046/j.1365-2141.2000.01813.x
86. Gremmel T, Ay C, Seidinger D, Pabinger I, Panzer S, Koppensteiner R. Soluble p-selectin, D-dimer, and high-sensitivity C-reactive protein after acute deep vein thrombosis of the lower limb. J Vasc Surg. (2011) 54:48S−55S. doi: 10.1016/j.jvs.2011.05.097
87. Thanaporn P, Myers DD, Wrobleski SK, Hawley AE, Farris DM, Wakefield TW etal. P-selectin inhibition decreases post-thrombotic vein wall fibrosis in a rat model. Surgery (2003) 134:365–71. doi: 10.1067/msy.2003.249
88. Myers DD Jr, Henke PK, Wrobleski SK, Hawley AE, Farris DM, Chapman AM etal. P-selectin inhibition enhances thrombus resolution and decreases vein wall fibrosis in a rat model. J Vasc Surg. (2002) 36:928–38. doi: 10.1067/mva.2002.128636
89. Mosevoll KA, Lindas R, Wendelbo O, Bruserud O, Reikvam H. Systemic levels of the endothelium-derived soluble adhesion molecules endocan and E-selectin in patients with suspected deep vein thrombosis. Springerplus (2014) 3:571. doi: 10.1186/2193-1801-3-571
90. Henke PK. Plasmin and matrix metalloproteinase system in deep venous thrombosis resolution. Vascular (2007) 15:366–71. doi: 10.2310/6670.2007.00050
91. Goodwin S, Mcpherson JD, Mccombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. (2016) 17:333–51. doi: 10.1038/nrg.2016.49
92. Connors JM. Thrombophilia testing and venous thrombosis. N Engl J Med. (2017) 377:1177–87. doi: 10.1056/NEJMra1700365
93. Carr JA, Cho JS. Low molecular weight heparin suppresses tumor necrosis factor expression from deep vein thrombosis. Ann Vasc Surg. (2007) 21:50–5. doi: 10.1016/j.avsg.2006.07.003
94. Moaveni DK, Lynch EM, Luke C, Sood V, Upchurch GR, Wakefield TW etal. Vein wall re-endothelialization after deep vein thrombosis is improved with low-molecular-weight heparin. J Vasc Surg. (2008) 47:616–24. doi: 10.1016/j.jvs.2007.11.040
95. Rodriguez AL, Wojcik BM, Wrobleski SK, Myers D. D. Jr., Wakefield TW, Diaz JA. Statins, inflammation and deep vein thrombosis: a systematic review. J Thromb Thrombolysis (2012) 33:371–82. doi: 10.1007/s11239-012-0687-9
96. Zolcinski M, Ciesla-Dul M, Potaczek DP, Undas A. Atorvastatin favorably modulates proinflammatory cytokine profile in patients following deep vein thrombosis. Thromb Res. (2013) 132:e31–5. doi: 10.1016/j.thromres.2013.04.026
97. Feng Y, Lei B, Zhang F, Niu L, Zhang H, Zhang M. Anti-inflammatory effects of simvastatin during the resolution phase of experimentally formed venous thrombi. J Investig Med. (2017) 65:999–1007. doi: 10.1136/jim-2017-000442
98. Henke PK, Wakefield TW, Kadell AM, Linn MJ, Varma MR, Sarkar M etal. Interleukin-8 administration enhances venous thrombosis resolution in a rat model. J Surg Res. (2001) 99:84–91. doi: 10.1006/jsre.2001.6122
99. Wang ZW, Wang JJ, Zhang JZ, Xue ZJ, Miao J, Li L etal. Thrombolysis of deep vein thrombosis and inhibiting chemotaxis of macrophage by MCP-1 blockage. Eur Rev Med Pharmacol Sci. (2017) 21:1695–701.
100. Varma MR, Varga AJ, Knipp BS, Sukheepod P, Upchurch GR, Kunkel SL etal. Neutropenia impairs venous thrombosis resolution in the rat. J Vasc Surg. (2003) 38:1090–8. doi: 10.1016/S0741-5214(03)00431-2
101. Henke PK, Pearce CG, Moaveni DM, Moore AJ, Lynch EM, Longo C etal. Targeted deletion of CCR2 impairs deep vein thombosis resolution in a mouse model. J Immunol. (2006) 177:3388–97. doi: 10.4049/jimmunol.177.5.3388
Keywords: venous thrombosis, inflammation, cytokines, adhesion molecules, matrix metalloproteases
Citation: Mosevoll KA, Johansen S, Wendelbo Ø, Nepstad I, Bruserud Ø and Reikvam H (2018) Cytokines, Adhesion Molecules, and Matrix Metalloproteases as Predisposing, Diagnostic, and Prognostic Factors in Venous Thrombosis. Front. Med. 5:147. doi: 10.3389/fmed.2018.00147
Received: 02 February 2018; Accepted: 30 April 2018;
Published: 22 May 2018.
Edited by:
Ana Kasirer-Friede, University of California, San Diego, United StatesReviewed by:
Coen Maas, University Medical Center Utrecht, NetherlandsJonas Emsley, University of Nottingham, United Kingdom
Copyright © 2018 Mosevoll, Johansen, Wendelbo, Nepstad, Bruserud and Reikvam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Håkon Reikvam, aHJlMDAyQHVpYi5ubw==